
Submitted:
04 September 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
Parasitic diseases are among the most serious public health problems in developing countries, causing high mortality and economic burdens. The Trypanosomatidae family includes Leishmania spp. and Trypanosoma cruzi, which cause leishmaniasis and Chagas disease, respectively. Both these parasites are among the major agents of neglected tropical diseases (NTDs). Leishmaniasis is currently the second most widespread vector-borne parasitic disease after malaria. The World Health Organization (WHO) records approximately 0.7-1 million newly diagnosed leishmaniasis cases each year, resulting in approximately 20,000-30,000 deaths. Also, 25 million people worldwide are at risk of Chagas disease and an estimated 6 million people are infected with Trypanosoma cruzi. Despite the similarity of the parasites, the evolution of the disease and the clinical indexes are explicitly different in African, Asian, and American forms of infection. Clinical therapies are associated with drug resistance and side effects. As a result, the discovery of novel therapeutic agents has emerged as a top priority and a promising alternative. Peptide-based drugs are an attractive class of therapeutic agents. They are recognized for being highly selective and effective, as well as safe and well tolerated. Moreover, they are now being explored in the development of novel therapeutics for a variety of health disorders, including oncology, endocrinology, metabolic, cardiovascular, and bone diseases. In addition, they are being explored in tropical diseases such as leishmaniasis and Chagas disease. We will discuss current knowledge regarding the parasite life cycle, signs and symptoms, diagnosis, and treatment options for leishmaniasis and Chagas disease.
Keywords:
Neglected tropic diseases
; Parasites
; Chagas disease
; Leishmaniasis
; Life cycle
; Drug target
; Biochemical mechanisms
; Pharmacological therapeutics
; Peptides
1. Introduction
Chagas disease (CD), leishmaniasis, and human African trypanosomiasis (HAT) are neglected tropical diseases (NTDs) caused by insect vector-borne protozoan parasites. Over 100,000 people die each year from NTDs worldwide, most living on less than $2 per day [1]. Chagas disease (aka American trypanosomiasis) is an infectious disease caused by the parasite Trypanosoma cruzi (T. cruzi), African Trypanosomiasis (aka “sleeping sickness”),is caused by Trypanosoma brucei (T. brucei), and leishmaniasis is caused by a protozoa parasite from over 20 Leishmania species[2,3]. Leishmaniasis is ranked second among all protozoan diseases in mortality after malaria [4]. The current drugs used against NTDs are suboptimal. For example, the only US Food and Drug Administration (FDA)-approved medications for leishmaniasis are intravenous liposomal amphotericin B (L-AmB) for visceral leishmaniasis (VL) and oral miltefosine for cutaneous leishmaniasis (CL), mucosal leishmaniasis (ML), and VL caused by particular species. Lipid formulations of amphotericin B are extremely expensive (the estimated cost of treatment with amphotericin B deoxycholate is about USD 1,522 [5]), and it is well known for its severe and potentially lethal side effects (e.g., fever, shaking, chills, flushing, loss of appetite, dizziness, nausea, vomiting, headache, and many more). Because of this the recommended course of treatment is often not completed, resulting in considerable scope for the resistance development [6,7,8]. Moreover, there are no effective vaccines for leishmaniasis [8]. Similar challenges are also associated with Chagas disease drugs. The safety profile of currently available drugs is far from ideal, with frequent adverse events and high drug discontinuation rates. There are only two drugs available for treating Chagas disease: benznidazole (BZN) and nifurtimox (NFX). Although these drugs are effective in the acute phase, they have limitations for chronic infections, and both drugs are restricted due to their toxic effects. For example, Nifurtimox is no longer used in many places since it causes neurological disorders or psychiatric episodes. Therefore, there is an urgent need to develop effective, safe, and affordable drugs [9].
Drug discovery aims to identify novel compounds that specifically and significantly change the course of the diseases they are intended to treat. There is a pressing need to identify new therapeutics for NTDs and novel approaches to drug design and development are needed [10,11]. For example, the virtual screening approach used for anti-trypanosomal and anti-leishmanial diseases is an example of how computational medicinal chemistry can be used for NTD drug discovery [12]. Nevertheless, computational methods should be validated experimentally [13]. Other examples are target-based or phenotype-based drug discovery, which is probably the most effective strategy for drug discovery for NTDs [14]. Another appealing approach is repurposing existing drugs, which is not a novel concept for NTDs. Drug repurposing discovers new disease indications for previously approved drugs that benefit from accessible compound libraries. This has yielded some promising compounds for various NTDs. Yet, repurposing an existing drug poses a major challenge in identifying an appropriate therapeutic indication for it, and it involves multiple factors, such as technology, patents, investment, and clinical trials. Considering this, peptide-based drugs are currently regarded as an appealing class of therapeutic agents, which are compounds used in the development of novel therapeutics for NTDs. Peptides have gained increased interest as therapeutics in recent years due to advances in production, modification, and analytical technologies [15,16]. Due to the lack of commercial benefits, pharmaceutical companies are discouraged from investing in NTD product development.
2. Leishmaniasis and Chagas disease
2.1. Life cycle of Trypanosomatida
Leishmaniasis and Chagas disease are caused by trypanosomatid parasites. In mammals, leishmaniasis and Chagas disease are transmitted through the sandfly and the triatomine bug. The differentiation of these trypanosomatids occurs partially in the vector and the host. This is done with different developmental stages giving rise to the infective stage of the parasite that infects mammals [17]. We discuss the stages of development and differentiation of the parasites in the vector and host involved in the life cycles of both leishmaniasis and Chagas disease (Figure 1).
2.1.1. Life cycle of Leishmania species
Leishmaniasis is caused by the protozoan parasite of the genus Leishmania belonging to the Trypanosomatidae family. It is spread through female sandflies of the genera Phlebotomus and Lutzomyia [18]. The parasite's life cycle is complex as it includes the vector and mammalian reservoirs for its morphogenesis, hence it resembles a digenetic life cycle [19]. The parasite entry is during the blood meal event, where motile metacyclic promastigotes are transferred into the blood through sandfly saliva [20]. Next, the metacyclic promastigotes migrate to healthy macrophages. Proteins on the parasite flagella interact with the macrophagic cell membrane and gain entry through phagocytosis by forming pseudopods infecting macrophages [21,22]. In the macrophage, promastigote forms differentiate from motile forms, which have elongated cell shapes and long flagellums, to amastigote forms, which have short flagellums [1]. Following, the host lysosomes fuse with the parasitophorous vacuole, creating an environment for the parasite to replicate. Finally, the macrophages rupture, and the amastigotes are released into the bloodstream [20].
The blood meal is digested in the sandfly midgut where the amastigotes cluster together forming nest cells. The nest cells are surrounded by an enclosed structure called the peritrophic matrix which protects the amastigotes from the insect’s gut environment [20,23]. The amastigotes transform into elongated flagellated procyclic promastigotes inside the peritrophic matrix. It's their first replicative stage. After 48-72 hours the procyclic promastigotes differentiate into long motile nectomonad promastigotes that break open the peritrophic matrix and enter the midgut lumen. Nectomonad promastigotes transform into short motile nectomonad promastigotes called leptomonad promastigotes and adhere to the microvilli of the midgut epithelium of the insect [24,25]. Here, the leptomonad promastigotes migrate and concentrate at the anterior stomodeal valve where they differentiate into metacyclic promastigotes that infect mammalian hosts. During the transformation to metacyclic stages, they secrete a gel called promastigote secretory gel (PSG) creating a “block fly” which forces the sandfly to feed on the blood and transfer metacyclic promastigotes from the vector to the mammalian host thereby starting the cycle all over [26].
2.1.2. Life cycle of Trypanosoma cruzi
Chagas disease is a protozoan disease caused by T. cruzi of the genus Trypanosoma belonging to the Trypanosomatidae family. It is caused by the parasitic defecation of a blood-sucking vector called the triatomine bug (aka the kissing bug). As a result of the undetermined spread of the feces on the wound by human actions, the parasite enters the host [27]. The first step upon entry into the host is adhesion. This is where metacyclic trypomastigotes attach to host cellular receptors with their flagella. Parasite flagella consists of molecules belonging to the Apicomplexa family that adhere to the host cell membrane. By binding glycoproteins to the cell membrane, metacyclic trypomastigotes mobilize calcium necessary for entry into the cytoplasm [28]. Next, the adhered parasite recognizes and alters ligands such as lectin-like molecules on the surface for safe internalization via endocytosis mediated by clathrin and caveolar lipids [28]. Also, internalization by phagocytosis, micropinocytosis, and circular dorsal ruffle formation has been reported [29]. The parasite resides safely within the parasite phosphorus vacuole formed by the plasma membrane and then fusses with the lysosome. When lysosomes fuse, parasites can differentiate from trypomastigotes to short globular organisms with small flagella, known as amastigotes, whereas some remain trypomastigotes [30]. After or during differentiation the amastigotes secrete hemolysin also called Tc-Tox and trypomastigotes secrete trans-sialidase/ neuraminidase required for fragmentation of the parasite phosphorus vacuole and enter the cytoplasm. As this process occurs during the differentiation of metacyclic trypomastigotes into amastigotes both forms are observed in the cytoplasm. The intracellular amastigote form further differentiates to form bloodstream trypomastigotes. Bloodstream trypomastigotes rupture the host cell and enter the bloodstream infecting other cells [30,31,32].
During the blood meal of an infected individual, trypomastigotes enter the triatomine bug's gut. Here, most bloodstream trypomastigotes are digested. Surviving parasites differentiate and transform into small spherical structures called spheromastigotes with a small extended flagellum. In the midgut, the spheromastigotes elongate and further extend their flagella, differentiating them into epimastigotes [33]. The long flagellated epimastigotes migrate to the hindgut and anchor to the perimicrovillar membranes of the hindgut intestine. This anchorage initiates the non-infective epimastigotes differentiation to infective metacyclic trypomastigotes which detach from the intestinal membranes and are migrated to the rectum where they are excreted in the feces and urine of the triatomine bug infecting mammalian hosts [34].
2.2. Epidemiology of Trypanosomatids
Trypanosomatids are a family of parasites causing infectious diseases categorized into trypanosomiasis and leishmaniasis. Trypanosomiasis is further subdivided into American and African diseases, also known as Chagas disease and African sleeping sickness respectively. Leishmaniasis is also further classified based on its prevalent geographical location. Trypanosomiasis and leishmaniasis are neglected diseases widely observed in underdeveloped countries, mainly in Southern Asian countries, African countries, and South American countries [34]. Various factors influence trypanosomiasis and leishmaniasis species and their development, including carrier vectors, vertebrate hosts, biochemical enzyme patterns, parasite phylogeny, and geographical distribution [35,36]. Table 1 lists the main Leishmania and Trypanosoma species.
2.3. Clinical complications and conditions of leishmaniasis and Chagas disease
Leishmaniasis and Chagas disease cover a broad disease spectrum that affects humans and other mammals. Clinical manifestations and treatment options for leishmaniasis and Chagas disease are limited due to various factors, including the complexity of the parasitic species and subtypes. This makes it very difficult for doctors to determine the disease's type and choose the optimal treatment plan [53].
2.3.1. Leishmaniasis
2.3.1.1. Cutaneous leishmaniasis (CL)
Cutaneous leishmaniasis (CL) is the most typical clinical symptom of Leishmania infection. Initial signs include single or multiple skin lesions localized to exposed body parts such as the face and may result in social stigma [54]. Numerous painless brownish erythematous papules with multiple nodules that resemble plague are seen after an incubation period of between two and seven months. It is followed by itchy and painful fluid discharge, warmth, swelling, and fever, which further develop into volcanic plagues associated with lymphangitis that differentiate from other ulcerative plagues (Figure 2). Bacterial and fungal infections are common on exposed cutaneous lesions causing secondary infections [55]. There are findings of co-infection with diseases like helminthiasis, leprosy, Human African trypanosomiasis, and Chagas disease [56]. Few reports of leishmaniasis co-infection with immunosuppressed patients under Human immunodeficiency virus (HIV) treatment were also observed among patients. There were also granulomatous and tumorous skin diseases, including subcutaneous mycosis, deep mycosis, cutaneous lymphoma, pseudolymphoma, basal cell carcinoma, and squamous cell carcinoma among the patients (Figure 2) [57]. Due to these clinical complications, doctors cannot use a specific treatment regimen to treat the disease.
2.3.1.2. Diffuse cutaneous leishmaniasis (DCL)
Diffuse cutaneous leishmaniasis (DCL) is widely observed in patients with a suppressed cell-mediated immune response against the invading parasite, also termed anergy [58]. DCL is characterized in patients with a low count of CD4 T-cells and immediately after antiretroviral drug therapy [59]. Several Leishmania (L.) species such as L. aethiopica, L. major, L. amazonensis, L. Mexicana, and L. braziliensis cause DCL [60]. An initial symptom is erythematous lesions with a well-defined periphery that extend to mucocutaneous junctions including nodules, ulcers, and plaques [61]. As the lesions progress, they spread to the face, buttocks, and extremities, and ulcerate the entire surface. The nodules across the nasopharynx and oropharynx cause airway obstructions and may be associated with lymphedema and lymphadenopathy where the lesions are observed as sporotrichoid in morphology [62,63]. The lesions also resemble other chronic lesions [64]. DCL is associated with immunocompromised diseases such as acquired immunodeficiency syndrome (AIDS) and lepromatous leprosy [65]. These findings are a major clinical observation in defining Leishmania infection.
2.3.1.3. Mucocutaneous leishmaniasis (ML)
The majority of mucosal/mucocutaneous leishmaniasis (ML) is transmitted by L. braziliensis, L. amazonensis, L. panamensis, L. infantum, and L. guyanensis [66]. It is observed as the serious outcome of post-cutaneous leishmaniasis in immunosuppressed patients and a few cases associated with cutaneous leishmaniasis were diagnosed as chronic disease [3]. It is initially painless, but ulcerative purulent lesions develop. They continue as erythema, edema, posterior nasal septal granulomas, and ulceration of the nares [67,68]. It spreads to the oral cavity of the oropharynx and larynx. This causes perforation of the nasal septum, periodontitis, palatal ulcers, and gingivitis damaging the vocal cartilage and resulting in speech abnormalities (Figure 2) [69]. Lymphedema leads to systemic infection causing fever and liver inflammation. Secondary complications include airway obstruction, fungal and bacterial infections [70]. The associated infections include epidermoid carcinoma, rhinosporidiosis or sinusitis, and AIDS where a differential diagnosis is required (Figure 2) [67]. ML is associated with cross- and co-infections, which can worsen both the patient's physical and psychological condition.
2.3.1.4. Visceral leishmaniasis (VL)
Visceral leishmaniasis (VL) is usually caused by L. donovani and L. infantum. These species are commonly found across North and East Africa, the Indian subcontinent, and Southern American nations. Infection is prevalent in all age groups but most prevalent in immune suppressant patients and children [9]. The parasite incubates for about two to six months and leads to chronic infection lasting for several years. The initial clinical observations include fever, weight loss, loss of appetite, and a malaise that progresses to a few months and is followed by enlarged lymph nodes, splenomegaly, and hepatomegaly [70]. The disease is also called Kala Azar due to hyperpigmentation of the skin and visceral regions. These hyperpigmentation patches are observed as dark patches stretching the skin surface associated with abdominal congestion and pain as the infection continues. The patient also suffers from bone marrow suppression, hemolysis, low albumin levels, jaundice, thrombocytopenia, cachexia, fluid accumulation, and bleeding from the mucosal cavities [71]. This may be followed by hemophagocytic lymph cytosis and intravascular coagulation [72].
Infections of the oral mucosa, nasal, and gastrointestinal tracts are prone to bacterial and fungal contamination. This leads to sepsis and pneumonia which is fatal in children and geriatrics. It could cause abortion or congenital leishmaniasis in pregnant patients [73]. HIV infection, tuberculosis, anemia, and jaundice cases are frequently reported as co-infections with VL and increase VL mortality in southern Asian and African countries [74,75]. Post-kala azar dermal leishmaniasis is also common after recovery from VL, which may lead to other medical complications.
2.3.1.5. Post Kala Azar Dermal Leishmaniasis (PKDL)
Post-kala azar dermal leishmaniasis (PKDL) is mostly observed in post-VL treatment. It could be defined as the final stage of VL infection before complete recovery from the disease. It is mostly observed in Asia and African countries as VL is a concern in these regions [76]. African and Asian PKDL can be differentiated by macular rashes in Asian countries whereas papular rashes in African countries [77]. The onset of PKDL symptoms usually starts after a few months post-VL treatment. This mimics a chronic skin infection that lasts for three to four years if untreated. The initial clinical presentations include dispersed hyperpigmented maculopapular and nodular lesions on the face which spread to the chest and arms. These lesions cover the entire length of the skin with scaly skin and dense nodular skin rashes (Figure 2). The lesions or rashes may be ulcerative, erythematous, or keloidal [78]. The nodules and papules are hypopigmented, photosensitive, and edematous, with a succulent nature, which irritates the skin. They appear warty, papillomatous, and fibroid with spontaneous ulcerations in the peritoneal area, chin, and parts of the face [79]. As the infection spreads to sensitive parts such as the tongue, groin, genitalia, and axilla it can cause permanent deformity in the patient. Treatment approaches should be enhanced against infection and active surveillance should be carried out for a better understanding of how diseases spread.
2.3.2. Chagas disease
Clinical outcomes of T. cruzi infection are divided into two categories: acute and chronic stages. Before the infection progresses to the chronic stage, it incubates in the intermediate stage, which mediates chronic infection. We discuss each clinical complication and condition of Chagas disease in detail below.
2.3.2.1. Acute Chagas disease
The acute stage of Chagas disease is mostly asymptomatic, and unnoticed by the patient. Symptoms are rarely observed after 30 days of incubation. During this stage, invasive trypomastigotes can be detected in fresh blood smears [80]. Less than 5% of patients in their acute stage of infection display symptoms such as malaise, fever, gastric imbalance, cardiac imbalances, inflammation at the inoculation site, periorbital swelling, epidermal eruptions, and edema with conjunctivitis indicating cutaneous infection (Figure 3) [81]. It might be accompanied by anemia, thrombocytopenia, hepatomegaly, and splenomegaly [82]. Death during this stage of infection is very rare and is primarily due to congestive heart failure, bronchopneumonia, myocarditis, and meningoencephalitis [83]. Children are more prone to death and are affected by myocarditis [84]. The acute stage persists for three-four months and then the infection enters the intermediate stage which lasts for six-eight weeks.
2.3.2.2. Chronic Chagas disease
The immediate phase after the acute stage is asymptomatic. Clinically, the patient has a normal electrocardiogram (ECG), gastrointestinal findings, and healthy physical condition, but positive serological findings for infection. The intermediate phase of infection is followed by the chronic phase of infection, which is fatal [85]. Around 20-30% of patients with acute and intermediate infections progress to the chronic phase of Chagas disease, which can have highly symptomatic symptoms. This includes thromboembolism, conduction abnormalities, arrhythmias, and heart failure due to dilated cardiomyopathy (Figure 3) [80]. The severity of the disease depends on cardiac lesions duration, location, and nature. The parasite resides in the cardiac tissues and causes immune activation in the myocardium. This causes denervation, myocardial fibrosis, microvascular disturbances, and myocardial injury [86,87]. Cardiomyopathies in Chagas disease manifest as ventricular remodeling, dysautonomia, and imbalances in cardiac perfusion [88,89]. Chronic Chagas cardiomyopathy is defined as a left ventricle aneurysm. This leads to an increase in left atrial pressure resulting in systolic dysfunction and reduced left ventricular filling [90]. The right bundle branch block is a common ECG found in chronic Chagas disease and is mostly found in association with an anterior fascicular block [82]. The chances of sudden death under this condition are very high due to ventricular tachyarrhythmia [91].
Damage to the brain and stroke is very common in Chagas disease due to hypoxia and reduced blood supply to the brain [82]. Brain embolism is a clinically recognized event associated with stroke in asymptomatic patients [80]. Other clinical manifestations include mega syndrome which is a result of the denervation of tubular structures of the enteric nerves that modulate normal gastrointestinal function directed towards acute infection in the rectum, colon, and esophagus (aka oesophagus). In the colon and rectum, it leads to bowel dysmotility, constipation, and fecal impaction. In the esophagus it causes achalasia in Chagas-infected patients. Also, it causes dysphagia, odynophagia, weight loss, idiopathic achalasia, regurgitation, cough, and chronic aspiration [92].
2.4. Pathways involved in leishmaniasis and Chagas disease
Trypanosomatids survival and differentiation involve various pathways. The parasite cannot synthesize all the metabolites needed for its survival, so it interferes with the host's biochemical mechanisms to fulfill its nutritional needs. There are enzymes that resemble host intermediates and are salvaged into the parasite to create a safe environment, escape the immune system, and develop for their own purposes [93]. Some of the biochemical pathways affected by trypanosomatids infection are discussed below.
Sterols serve as a key metabolic element for Leishmania and T. curzi survival. Both these parasites increase ergosterol production by interfering with host sterol biosynthesis [93]. Leishmania and T. curzi parasites infest macrophagic sterol levels for parasitic differentiation, cellular shape, and division. This results in lowering cholesterol levels in macrophage plasma membranes [93,94]. In Chagas disease, cholesterol is employed in the endocytic vesicle and cytostome of amastigotes [95]. It leads to CD40 signaling pathway disturbances and alters Interleukin 12 (IL-12) production. It also increases arginase I expression which inhibits NO synthesis and favors parasite survival and differentiation [96,97].
Both Leishmania and T. curzi are incapable of synthesizing the purines required for their differentiation and survival; hence they salvage the purine molecules used by the host for their living [98]. Purine phosphoribosyl transferase assists Leishmania and T. curzi salvage purines from the phosphorus vacuole. It helps in catalyzing the formation of guanosine monophosphate and inosine monophosphate which is required for parasite survival [99,100]. Typically, hypoxanthine-Guanine Phosphoribosyltransferase (HGPRT) and xanthine Phosphoribosyltransferase (XPRT) enzymes are involved in purine salvage [101]. Other enzymes such as adenine phosphoribosyl transferase (APRT), hypoxanthine-guanine-xanthine phosphoribosyl transferase (HGXPRT), adenosine kinase (AK), and nucleoside hydrolase (NH) were also identified to play a major role in acquiring purines from the host in cases of both Leishmania and T. curzi infections [102,103].
Cell surface proteins are anchored by glycosylphosphatidylinositol (GPI) molecules. GPI molecules, including Lipophosphoglycan (LPG) and Gylcoinositol phospholipids (GIPL) are synthesized by Leishmania and T. curzi, which help them anchor to macrophages and internalize them [104]. Most GPI anchors require trimannose backbones and dolichol-phosphate-mannose (DPM) that act as mannose donors [105]. These GPI molecules help host and parasite communication and immune invasion [106]. Mucins also play a role in anchoring parasite development. Trans-sialidase is one such mucin responsible for active anchorage and infectivity [107]. Both Leishmania and T. curzi contain mucin, but T. curzi contains more than Leishmania [108]. Molecules such as phosphatidylinositol, inositol phosphorylceramide, glycoproteins 63, glycoprotein 82, and glycoprotein 30 also assist in parasite anchorage and internalization of both Leishmania and T. curzi parasites. Also, these molecules facilitate the mobilization of intracellular calcium and the exocytosis of lysosomes, leading to a cascade of signaling between the host and the parasite for cellular invasion [109].
Folic acid and pteridines play a major role in Leishmania and T. curzi metabolic interventions [110]. Tetrahydrofolate is essential for purine, thymidylate, and pantothenate biosynthesis. Also, it is required for RNA protein formation and is involved in various signaling cascades [111]. Dihydrofolate reductase (DHFR) and Thymidine synthase (TS) act as bifunctional enzymes as they are both connected to N- and C-terminals respectively and separated by a linker peptide [112]. DHFR-TS helps protect dihydrofolate and drives cellular colocalization, and parasite survival [113]. Pteridine reductase I (PTR-1) also plays a role in the conversion of biopterin to tetrahydrobiopterin when DHFR-TS inhibitors are used. PTR-I is activated when DHFR-TS is inhibited or malfunctioning and is responsible for parasite survival and a proper survival environment for their growth [114]. Trypanothione (TSH2), is a principal metabolite that protects Leishmania and T. curzi parasites from oxidative stress and is responsible for parasite differentiation. The formation of TSH2 is mediated by glutathionyl spermidine synthase (GSPS) and trypanothine synthase (TRYS), TSH2 is involved in the reduction of host glutathione and decreases macrophage oxidative stress during infection [115]. TSH2 is maintained at reduced levels as required by the parasites by trypanothione reductase (TR) [116]. TR shares 67% similarities among Leishmania species and 80% similarities among trypanosome species [117]. In Leishmania, TSH2 catalyzes the hydroperoxide reduction with the assistance of two proteins namely tryparedoxin (TXN) and tryparedoxin dependent peroxidase (TDPX). This reduces oxidative stress and enhances parasite survival rates [118]. TSH2 also possesses the properties of xenobiotics, and endobiotic neutralization, and enhances the reduction of ascorbate and iron-sulfur complexes [119].
N-(2-amino2-hyroxymethyl) lysine, also called hypusine, is found in two eukaryotic proteins eukaryotic translation initiation factor (eIF)-5A1 and eIF5A2 [120]. eIF5A is responsible for cell cycle regulation, apoptosis, translation, elongation, and termination [121]. The post-translational modification leads to the binding of hypusine with lysine residues of eIF5A proteins by the transfer of 4-aminobutyl moiety from spermidine to lysine chain which is aided by enzymes deoxyhypusine synthase (DHPS) and deoxyhypusine hydroxylase (DOHH) is known as hypusination [122]. Arginine serves as a precursor for eIF5A which is essential in hypusine synthesis [123] and determines Leishmania and T. curzi infection rates. Arginine is translocated to parasites using amino acid permease 3 (AAP3) [124]. Arginine plays a role in the polyamine pathway. In Leishmania the promastigotes accumulate arginine, utilize it in polyamine biosynthesis and regulate arginine transport under conditions of arginine deprivation [125].
3. Drug discovery strategies and insights into current therapeutics
3.1. Approaches of drug development against leishmaniasis and Chagas disease
Drug discovery strategies for leishmaniasis and Chagas disease have devolved based on various approaches, such as structural biology, genetic engineering, molecular modeling, and high-content screening platforms [126,127]. Identification of functional molecular targets is crucial to target-based drug discovery [128]. Molecules are then designed to interrupt specific targets essential for parasite survival [14]. Targets extracted from T. cruzi genome data analysis have been investigated for the development of high throughput target-based drug screening assays [129]. In drug development, two different mechanisms are assumed for compound identification such as target-based drug discovery (TDD) or phenotype-based drug discovery (PDD) [14]. Identifying new drugs for inadequately treated medical conditions relies on TDD [128]. However, PDD involves evaluating diverse chemicals in contradiction to the pathogen’s phenotype in a biological system and animal model. For example, benzothiophene analogs emerged as novel agents from a phenotypic screen against GSK’s kinetoboxes [130,131]. However, the increased cost and high rates of failure of traditional drug discovery and development approaches have provoked the community to explore alternative approaches, such as computer-aided drug discovery which comprises structure-based, ligand-based, and system-based approaches to drug discovery [132,133].
Structure-based-drug-discovery (SBDD) is the use of 3D structures of molecular targets to increase ligand-receptor complementarity. It is mainly obtained by biophysical techniques and used in research for the determination of pharmacological targets [134]. For example, virtual screening approaches have recently been undertaken using imidazole-pyridine as a reference structure [12]. In addition, the identification of oxidosqualene cyclase as a novel molecular target in Leishmania could lead to promising programs to discover new agents for leishmaniasis treatment [36]. Finally, the ligand-based drug design (LBDD) approach also identifies key characteristics that contribute to biological activity while improving or identifying new chemotypes [135,136,137].
3.2. Current drugs for leishmaniasis and Chagas disease
The chemotherapy currently used for leishmaniasis and Chagas disease includes a small spectrum of drugs, used as monotherapy or in combination [53,138]. Provided that there is no vaccine available for the prevention of leishmaniasis and Chagas disease, the control of the diseases depends mainly on drugs. The therapeutic scheme depends on the type of disease, parasite species, and geographical location. The drug discovery process for leishmaniasis and Chagas disease has been ongoing with thousands of compounds tested annually before finding a promising candidate. High throughput screening (HTS) campaigns, which represent a valuable approach to identifying novel sets of leads for treatment, have been carried out [139]. Drugs for Neglected Diseases Initiative (DNDi) is developing a portfolio of early hits and leads series with collections of natural products and synthetic compounds to find new drug candidates for leishmaniasis and Chagas disease. In 2015, the NTD Drug Discovery Booster was launched with several pharmaceutical companies and this project is ongoing [140]. In 2019, DNDi launched the ‘Chagas Hit-to-lead’ project with the objective of identifying promising leads with activity in animal models with the disease and establishing an innovative strategy[140,141].
The majority of the mechanisms of action of drugs used today to treat leishmaniasis and Chagas diseases are unknown. Furthermore, the ineffectiveness of such drugs, and emerging resistance, combined with severe side effects, encourage the development of alternatives for NTDs [142,143]. Therefore, the development of novel drug discovery programs for anti-leishmanial and anti-trypanosomal compounds is essential [144,145].
3.2.1. Leishmaniasis
Sodium stibogluconate, meglumine antimonate, and sodium stibogluconate have been used extensively to treat visceral, cutaneous, and mucocutaneous leishmaniasis for decades [146]. Yet their parenteral therapy is long, the drugs are toxic, and resistance is on the rise. Therefore, other medications such as pentamidine, paromomycin, fluconazole, ketoconazole, miltefosine, and amphotericin B, have been repurposed (Figure 4) [147]. Additionally, it has been noted that amphotericin B therapy does not result in a sterile cure and is linked to rising kala azar leishmaniasis rates as well as the emergence of resistant parasites in clinical settings [148]. Although repositioning efforts have shown promise in discovering new treatments for this disease, the identification of novel therapeutic targets and the development of effective leishmanicidal drugs should be prioritized [149].
3.2.2. Chagas disease
Nifurtimox (NFX) and Benznidazole (BZN) are the two main drugs used to treat Chagas disease, and both were developed more than 50 years ago (Figure 5). The treatment is lengthy (60-90 days) and ineffective for chronic patients, in addition to having a toxic effect and the potential for evolving resistance [150,151,152]. Although the exact mechanism underlying Benznidazole's activity is unknown, it is activated by NADH-dependent trypanosomal reductases and produces reductive metabolites that are thought to have several negative consequences, including DNA damage and protein synthesis inhibition [153]. The FDA approved benznidazole for the treatment of Chagas disease in children aged 2 to 12 years old in 2017. It was the first medicine licensed in the United States for Chagas disease. A new drug that is both safe and effective for Chagas disease's acute and chronic stages is required. However, several factors impediment the development of new candidate drugs, including the lack of biomarkers for the two stages of the disease and for evaluating treatment success, as well as the genetic diversity of T. cruzi strains [154,155].
Current treatments for leishmaniasis and Chagas disease have several problems. Most of them have side effects, treatment failures, and relapses caused by drug resistance. These anti-trypanosomal and anti-leishmanial medications also have drawbacks that exclude their use in specific population cohorts or under specific conditions. For example, several of them are teratogenic and inappropriate for pregnant women and newborns. Another concern is the poor oral bioavailability of most of these drugs [156]. Chagas disease and leishmaniasis treatments differ depending on the endemic location. Table 2 summarizes the WHO-recommended treatment regimens for major Chagas disease and leishmaniasis endemic sites. Treatment alternatives are generally insufficient, and new medications are desperately needed. Most drugs used to treat Chagas disease and leishmaniasis are on the 19th edition of the WHO Model List of Essential Medicines, including pentavalent antimonials (SbV), miltefosine, amphotericin B deoxycolate or formulated in liposomal formulations, paromomycin, and pentamidine (Figure 4 and Figure 5).
4. Novel strategies for leishmaniasis and Chagas disease treatments
There is an urgent need to discover and develop compounds effective against leishmaniasis and Chagas disease, which demonstrate high bioactivity, low toxicity, and acceptable cost and administration profile [14,177]. In the past two decades, numerous assays have been designed for use in screening programs targeting the various lifecycle stages of Leishmania and Trypanosoma parasites. As soon as active compounds are developed against the intracellular amastigote form of the parasite, they are tested in vivo, following a pharmacokinetic and pharmacodynamic assessment [139]. Barriers to the development of new drugs include the lack of well-characterized and validated targets, the absence of diagnostic biomarkers resulting in diagnostic failures, the variety of parasite strains, issues with the standardization of methodologies, such as various in vitro assays, different host cell culture lines, and issues with the translation process [140]. In addition, the treatment of these deadly diseases is still a global challenge due to limited drug regimens, the emergence of resistance, toxicities issues, co-infection cases, and the low investment in the novel drug discovery/development [178,179]. Various strategies must be explored and implemented to combat these NTDs to overcome conventional drugs' drawbacks. The following section summarizes recent developments and upcoming insights into traditional anti-trypanosomal and anti-leishmanial therapies.
4.1. Host-directed therapy (HDT)
Host-directed therapy (HDT) aims to achieve a clinical cure by manipulating the host immune system rather than using drugs to treat the parasite [180]. HDT uses various molecules (e.g., repurposed drugs, immuno-modulators, synthetic nucleic acids, cytokines, cellular therapy, and micronutrients) that can boost the immune function of the host body or control the inflammatory response. Both have a long-lasting healing effect and lower mortality and morbidity [181]. A better understanding of the immune response to Leishmania has revealed the crucial fact that infection depends on the successful downregulation of the T-helper cell type 1 (Th1) immune response and the development of the Th2 responses [182]. Since the immune response profile in Chagas disease is similar to that in Leishmania [183], host-directed therapies relevant to Leishmania might also benefit patients with Trypanosome infection [183,184].
For example, simvastatin inhibits the pathway that produces cholesterol and causes macrophages to mature their phagosomes, increasing parasite clearance [185]. Similarly, lovastatin, which is chemically similar to simvastatin, is essential for lowering parasite internalization and infectivity in the J774A.1 macrophage cell line due to cholesterol depletion [186]. Eugenol (Syzygium aromaticum) and its derivatives have been hailed as promising treatments for inducing immunostimulatory responses that benefit the host. The growth of promastigotes and amastigotes of Leishmania was reportedly inhibited by eugenol derivative [187]. In conclusion, immuno-modulatory compounds may prove valuable candidates for treating leishmaniasis and Chagas disease.
4.2. Multi-drug or combination therapy
The primary goals of the combinational or multi-drug approach are to shorten the treatment period, lower the relapse rate, reduce dosage, avoid fatal side effects, and combat drug resistance problems [157]. HIV, malaria, and tuberculosis have all been successfully treated using this approach [188,189]. Itraconazole, ezetimibe, and miltefosine were some of the drugs used in a multi-drug therapy approach to evaluate the anti-leishmanial efficacy against the VL model of infection in BALB/c mice [182]. Due to its capacity to maximize the efficacy of currently available medications while lowering dosage requirements and treatment times, the combination therapeutic approach has recently seen significant use in the treatment of leishmaniasis and Chagas disease. A wide range of trustworthy combinations to treat leishmaniasis is possible. In the search for the most effective leishmaniasis treatment regimen, novel drug combinations can now be further investigated.
4.3. Drug repurposing
Drug repurposing (aka reprofiling, drug repositioning, or re-tasking) is a method that identifies novel applications for clinically approved drugs that were originally developed for another medical treatment [190]. This method is associated with a shorter drug development timeline and lower investment costs because the drugs have already undergone clinical studies [191]. Numerous medications have been repurposed for leishmaniasis and have shown promising results, such as Amp B, miltefosine, and azoles (fluconazole, itraconazole, and posaconazole) [192]. Numerous antidepressants, including Ketanserin, imipramine, clomipramine, nitroimipramine, sertraline, etc., have shown promising results when applied as repurposed anti-leishmanial medications [192,193]. A recent study using transmission electron microscopy and metabolomics platforms identified the mechanism of sertraline's leishmanicidal effect on Leishmania parasites [194].
4.4. Promising natural products
The Drug Discovery Research Program, and the Tropical Diseases Program of the TDR (Tropical Disease Research) WHO have discussed making plant pharmacology a priority. An article with a complete list of plants and natural products that demonstrated anti-leishmanial and anti-trypanosomal activities was published in 2015 [195]. Several studies have been conducted since then that demonstrate the pursuit of novel products derived from microbial or marine sources. For example, a glycoprotein isolated from the sponge Pachymatisma johnstonii showed strong in vitro activity against L. donovani, L. braziliensis, and L. Mexicana [196]. Essential oil's anti-leishmanial activity has also been studied [197]. Advanced research has also evaluated additional potential substances isolated from natural sources and showed anti-leishmanial and anti-trypanosomal activities [198].
4.5. Nanotechnology
Drug delivery and drug design can use nanomedicine, a branch of science that deals with particles at the nanoscale [199]. Low permeability, insolubility, painful injections, prolonged hospitalization, and unfavorable side effects are just a few of the limitations of the conventional delivery system. In the search for the most effective leishmaniasis treatment, researchers have used nanoparticles (NPs)-based approach. NPs are employed in drug delivery due to their biocompatibility, improved drug solubility, on-target drug delivery to the target organ, immuno-compatibility, and increased potential for a variety of administration routes [200]. Target-mediated drug delivery systems use metallic, inorganic, organic, and polymeric nanomaterials, such as carbon nanotubes, dendrimers, liposomes, and micelles [201]. Since phagocytic cells, particularly macrophages, are the target organs in leishmaniasis, liposomal derivatives, and polymeric NPs are used for drug delivery. Higher reactive oxygen species (ROS) production, an increase in immuno-modulatory response, DNA damage, disruption of mitochondrial membrane potential and electron transport chain, and inhibition of trypanothione reductase enzyme vital for the Leishmania parasite's anti-oxidation process are just a few of the mechanisms NPs encapsulated drugs/molecules use to kill Leishmania parasites [202]. Although there are countless benefits, nanoscience's use in practical life is limited. This is because it can sometimes have a significant adverse effect on the body due to its biological sensitivity [203]. Preparation and manipulation require specialized and engineered products, making it a cost-intensive task unsuitable for NTDs like leishmaniasis [204].
4.6. Nano vaccines
Nano vaccines are emerging as a novel approach to vaccination methodology, causing both humoral and cell-mediated immune responses. These positive results have led to a promising path to more suitable treatment for several diseases, including leishmaniasis [205]. In a 2011 study, recombinant superoxide dismutase (SODB1) was loaded onto chitosan nanoparticles by the ionotropic gelation method to develop an innovative nano vaccine evaluated on BALB/c mice. The results showed that in single and triple doses of SODB1 nanoparticles, IgG2a and IgG2a/IgG1 were significantly higher than in the other groups. This shows the efficiency of chitosan nanoparticles in developing a nano vaccine for leishmaniasis [206]. Another study examined the effectiveness of chimeric peptides containing HLA-restricted epitopes from three immunogenic L. infantum proteins, in poly (lactic-coglycolic) acid nanoparticles with or without the adjuvant monophosphorylate lipid A (MPLA) or surface modification. The nano vaccine induced dendritic cell maturation and promoted peptide-specific non-producing CD8+ T cells [207]. Additional study used synthetic peptide-based nano vaccines along with MPLA adjuvant co-encapsulated in PLGA nanoparticles. The results demonstrated a strong spleen lymphoproliferative response and high levels of IL-2, interferon gamma (IFN-γ), and tumour necrosis factor α (TNFα) versus low IL-4 and IL-10 secretion [208,209]. A 2019 study developed a process to prepare lipidic NPs loaded with plasmid pVAX1-NH36 for application as a leishmaniasis nano vaccine [197].
5. Peptide targeting for leishmaniasis and Chagas disease therapies
5.1. Peptide therapies
Peptides are short chains of amino acids that play critical roles in human physiology. They were first used in medicine in the first half of the 20th century. Therapeutic peptides are exceptional pharmaceutical tools with a molecular weight of 500-5000 Da [210]. As technology advances, peptide drug discovery has become established. This includes drug design, peptide drug discovery, peptide synthesis, structural modification, and the association between these developments and peptide bioactivities. Peptides have their unique intrinsic characteristics which are produced and modified through chemical and biochemical methods associated with novel design and delivery strategies. Currently, in addition to many preclinical studies, more than 170 peptides are actively in clinical developments [210,211] and sales total more than $70 billion [212]. Compared with biologics, therapeutic peptides show less immunogenicity and have lower production costs [213,214]. Compared to small molecules peptides typically demonstrate higher potency and selectivity for their targets [215].
While therapeutic peptides have two basic limitations: weak membrane permeability and poor in vivo stability. Yet, peptides as drugs have many advantages including high potency, high selectivity, and low toxicity. Thus, efforts using several models confirmed the effects of several peptides on various parasites (Table 3) [216].
5.2. Anti-microbial peptides
Anti-microbial peptides (AMPs) are small cationic molecules, amphipathic with a wide spectrum of activities against viruses, bacteria, fungi, and parasites [224]. Membrane depolarization, disruption of plasma membrane permeability, and programmed cell death are some major microbicidal mechanisms of action employed by these peptides [225]. Moreover, AMPs are considered first-line immune defense systems that instigate the production of specific cytokines molecules, leading to host immuno-modulatory responses [226]. AMPs are also called host defense peptides and have gained immense attention from researchers working in the field of parasites [227]. Asadi et al., assessed the role of murine cathelicidin or cathelin-derived AMP against L. major infection. In vitro, results showed enhanced expression of cathelin-derived AMP in the J774A.1 cell line compared to control. Similarly, up-regulated expression of cathelin-derived AMP was recorded at the inoculation site of the dermis layer of L. major infected BALB/c mice, unlike the control group [182,228]. Further, protein assay revealed the increase in the level of cathelin-related AMP in both infected macrophages and mice model confirming the role of cathelin-related AMP as a defensive immune response against L. major. Zahedifard et al., evaluated the leishmanicidal potential of Brevinin 2R (belonging to the class of defensins) alone and in conjugation with lauric acid against L. major parasites and found that mice treated with Brevinin 2R exhibited decreased parasite load in the lymph nodes [182,229].
Since AMPs have excellent leishmanicidal and immunomodulatory properties, they can serve as leads during drug discovery pipelines and vaccine design for leishmaniasis and Chagas disease. Currently, these AMPs get more attention due to different factors such as antibiotic-resistant microorganisms [230]. While AMP mechanisms remain unclear, many AMPs cause swelling in the promastigote membrane. This leads to loss of integrity and substantial cell death [187].
5.2.1. Anti-microbial peptides against Chagas disease and leishmaniasis
K777, a vinyl sulfone cysteine protease inhibitor, is considered a highly powerful and well-recognized cysteine peptidase inhibitor. It inhibits cruzain (aka Cruzipain), a key protease required for T. cruzi survival. It did not promote a parasitological cure, but significantly reduced parasite-induced heart damage in vivo [231]. Over the past few years, several AMPs have been evaluated for their effects on T. cruzi, and apidaecin, magainin II, melittin, and cecropin A have been identified as potential candidates for Chagas disease as they kill T. cruzi in low concentrations [232].
AMPs are an essential protection mechanism and part of vertebrates' native immunity. These peptides damage the protozoan’s cell membrane affecting membrane integrity and producing lethal pores [233]. Over 2,000 AMPs have been identified from a variety of organisms, including bacteria, insects, plants, amphibians, birds, reptiles, and mammals, including humans. Some of these AMPs demonstrate leishmanicidal activity, including halictine-2 from the poison of eusocial honey-bees [234]. Attacin, cecropin, and defensins from lutzomyia longipalpis respond to Leishmania infection [235], and Dragomide E., a linear lipopeptide isolated from the cyanobacteria Lyngbya majuscula demonstrated anti-leishmanial activity against L. donovani promastigotes. LZ1 peptide, derived from Snake cathelicidin, is an artificially designed and synthesized active polypeptide demonstrating high antimicrobial activity. It inhibits ATP production in parasite-infected erythrocytes [236]. Finally, Phylloseptin-1, a cationic peptide from the skin secretion of Phyllomedusa azurea, demonstrated high anti-parasitic activity and prevented cross-resistance because of its distinctive chemical structure [237].
The pharmaceutical industry recognizes that peptide-based drugs are a significant class of therapeutic agents. Anti-protozoal therapeutics can be derived from insects, bacteria, marine organisms, and amphibians [226]. The next figure describes some of the AMPs that have been identified and their functions against the different developmental forms of parasites (Figure 6) [233].
5.3. Protein-protein interactions as drug targets in leishmaniasis and Chagas disease
The human proteome contains approximately 30,000 proteins and significantly more protein-protein interactions (PPIs) that play critical roles in biological processes. Their dysregulation results in the onset and progression of a variety of diseases. PPIs thus represent a treasure trove of disease-modifying drug targets. However, targeting these is difficult when converting drug-like small molecules into therapeutics. When targeting PPIs, it is critical to strike a balance between the interacting proteins to elicit a therapeutic effect while avoiding a significant adverse effect [238]. Researchers have reported that PPIs as a drug discovery strategy target a variety of diseases [238]. However, there is limited information about PPIs for leishmaniasis and Chagas disease. Proteins 3B64, 1LML, and 4P4M have recently been identified as structural macromolecules that play a variety of roles in the Leishmania parasite. It is worth considering the importance of these proteins in the context of Leishmania, as well as their potential implications for future research into the parasite's metabolism and pathogenicity. Therefore, PPIs offer the potential for further research into drug discovery for leishmaniasis and Chagas disease.
6. Conclusions and Future Directions
Leishmaniasis and Chagas disease are NTDs and global burdens. As a result of recurrent failures in leishmaniasis and Chagas disease diagnosis, chemotherapy is delayed and eventually results in death. Despite the efforts of many research groups leishmaniasis and Chagas disease remain without an effective solution. Challenges regarding leishmaniasis and Chagas disease include accurate and timely disease diagnosis, which are crucial in identifying asymptomatic patients and co-infected cases. However, recent diagnostic procedures such as advanced molecular techniques, proteomics, and nano-diagnosis may provide revolutionary improvements to the prognosis of these diseases. Other key challenges are related to the currently available treatments, including the emergence of drug resistance and toxicities issues. These may be overcome by less traditional approaches, such as multi-drug and host-directed therapies, which have yielded some promising results in recent studies. In addition, clinical trials with innovative treatment strategies should be performed to explore the impact of novel approaches on improving treatment efficacy, decreasing side effects, and lowering treatment costs. The review discusses the advancement of novel treatment strategies, featuring peptides as potential therapeutics against NTDs. Since peptides have specific actions and reduced toxicity, they are promising for new drug discovery and development. It is evident that these targeted compounds are becoming increasingly popular within the pharmaceutical industry, proving their importance to the Pharmaceutical Sciences. It is our hope that we can contribute to the discovery of medicines in this relatively neglected area. This will greatly improve the health of many people suffering in the world's poorest countries.
Author Contributions
M.Z., M.K. H.B. and N.Q. conceptualized and wrote the review and prepared the tables and figures. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Israel Science Foundation (ISF)- CIHR-IDRC through a research grant (no. 2993/22) awarded to N.Q.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to apologize to those colleagues whose work could not be cited or discussed in sufficient detail due to space limitations. We thank those who were involved in the proofreading and editing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gürtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: related protozoan pathogens, different diseases. The Journal of clinical investigation 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Naula, C.; Parsons, M.; Mottram, J.C. Protein kinases as drug targets in trypanosomes and Leishmania. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 2005, 1754, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Abadías-Granado, I.; Diago, A.; Cerro, P.; Palma-Ruiz, A.; Gilaberte, Y. Cutaneous and mucocutaneous leishmaniasis. Actas Dermo-Sifiliográficas (English Edition) 2021, 112, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kashif, M.; Srivastava, P.; Manna, P.P. Recent Advances in Chemotherapeutics for Leishmaniasis: Importance of the Cellular Biochemistry of the Parasite and Its Molecular Interaction with the Host. Pathogens 2023, 12, 706. [Google Scholar] [CrossRef]
- Assis, T.S.M.d.; Rosa, D.C.P.; Teixeira, E.d.M.; Cota, G.; Azeredo-da-Silva, A.L.F.; Werneck, G.; Rabello, A. The direct costs of treating human visceral leishmaniasis in Brazil. Revista da Sociedade Brasileira de Medicina Tropical 2017, 50, 478–482. [Google Scholar] [CrossRef]
- Alvar, J.; Arana, B. I. Appraisal of Leishmaniasis Chemotherapy, Current Status and Pipeline Strategies Chapter 1 Leishmaniasis, Impact and Therapeutic Needs 3. Drug Discovery for Leishmaniasis doi 2018, 10, 9781788010177–9781788000001. [Google Scholar]
- Rajao, M.A.; Furtado, C.; Alves, C.L.; Passos-Silva, D.G.; De Moura, M.B.; Schamber-Reis, B.L.; Kunrath-Lima, M.; Zuma, A.A.; Vieira-da-Rocha, J.P.; Garcia, J.B.F. Unveiling benznidazole's mechanism of action through overexpression of DNA repair proteins in Trypanosoma cruzi. Environmental and molecular mutagenesis 2014, 55, 309–321. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Khan, F.A.K.; Kulkarni, A.A.; Arote, R.; Patil, R.H. Antileishmanial drug discovery: Comprehensive review of the last 10 years. Rsc Advances 2015, 5, 32376–32415. [Google Scholar] [CrossRef]
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nature reviews microbiology 2007, 5, 873–882. [Google Scholar] [CrossRef]
- Sundberg, S.A. High-throughput and ultra-high-throughput screening: solution-and cell-based approaches. Current opinion in biotechnology 2000, 11, 47–53. [Google Scholar] [CrossRef]
- Mayr, L.M.; Bojanic, D. Novel trends in high-throughput screening. Current opinion in pharmacology 2009, 9, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Canan, S.; Cao, Y.; Condroski, K.; Engkvist, O.; Itono, S.; Kaki, R.; Kimura, C.; Kogej, T.; Nagaoka, K. Collaborative virtual screening to elaborate an imidazo [1, 2-a] pyridine hit series for visceral leishmaniasis. RSC medicinal chemistry 2021, 12, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Nalini, S.; Lampa, S.; Saw, S.; Gleeson, M.P.; Ola, S.; Chanin, N. Towards reproducible computational drug discovery. Journal of Cheminformatics 2020, 12. [Google Scholar]
- Chatelain, E.; Ioset, J.-R. Phenotypic screening approaches for Chagas disease drug discovery. Expert opinion on drug discovery 2018, 13, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Zahedifard, F.; Rafati, S. Prospects for antimicrobial peptide-based immunotherapy approaches in Leishmania control. Expert review of anti-infective therapy 2018, 16, 461–469. [Google Scholar] [CrossRef]
- Rafferty, J.; Nagaraj, H. P McCloskey A, Huwaitat R, Porter S, Albadr A, et al. Peptide therapeutics and the pharmaceutical industry: barriers encountered translating from the laboratory to patients. Curr Med Chem 2016, 23, 4231–4259. [Google Scholar] [CrossRef]
- Díaz-Garrido, P.; Cárdenas-Guerra, R.E.; Martínez, I.; Poggio, S.; Rodríguez-Hernández, K.; Rivera-Santiago, L.; Ortega-López, J.; Sánchez-Esquivel, S.; Espinoza, B. Differential activity on trypanosomatid parasites of a novel recombinant defensin type 1 from the insect Triatoma (Meccus) pallidipennis. Insect Biochemistry and Molecular Biology 2021, 139, 103673. [Google Scholar] [CrossRef]
- Dostálová, A.; Volf, P. Leishmania development in sand flies: parasite-vector interactions overview. Parasites & vectors 2012, 5, 1–12. [Google Scholar]
- Killick-Kendrick, R. The biology and control of phlebotomine sand flies. Clinics in dermatology 1999, 17, 279–289. [Google Scholar] [CrossRef]
- Teixeira, D.E.; Benchimol, M.; Rodrigues, J.C.; Crepaldi, P.H.; Pimenta, P.F.; de Souza, W. The cell biology of Leishmania: how to teach using animations. PLoS pathogens 2013, 9, e1003594. [Google Scholar] [CrossRef] [PubMed]
- Sunter, J.; Gull, K. Shape, form, function and Leishmania pathogenicity: from textbook descriptions to biological understanding. Open biology 2017, 7, 170165. [Google Scholar] [CrossRef]
- Saada, E.A.; Kabututu, Z.P.; Lopez, M.; Shimogawa, M.M.; Langousis, G.; Oberholzer, M.; Riestra, A.; Jonsson, Z.O.; Wohlschlegel, J.A.; Hill, K.L. Insect stage-specific receptor adenylate cyclases are localized to distinct subdomains of the Trypanosoma brucei flagellar membrane. Eukaryotic cell 2014, 13, 1064–1076. [Google Scholar] [CrossRef]
- Secundino, N.; Eger-Mangrich, I.; Braga, E.; Santoro, M.; Pimenta, P. Lutzomyia longipalpis peritrophic matrix: formation, structure, and chemical composition. Journal of medical entomology 2005, 42, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.E.; Chance, M.; Bates, P. The role of promastigote secretory gel in the origin and transmission of the infective stage of Leishmania mexicana by the sandfly Lutzomyia longipalpis. Parasitology 2002, 124, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Gossage, S.M.; Rogers, M.E.; Bates, P.A. Two separate growth phases during the development of Leishmania in sand flies: implications for understanding the life cycle. International journal for parasitology 2003, 33, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.A. Revising Leishmania’s life cycle. Nature microbiology 2018, 3, 529–530. [Google Scholar] [CrossRef]
- Martín-Escolano, J.; Marín, C.; Rosales, M.J.; Tsaousis, A.D.; Medina-Carmona, E.; Martín-Escolano, R. An updated view of the trypanosoma cruzi life cycle: Intervention points for an effective treatment. ACS Infectious Diseases 2022, 8, 1107–1115. [Google Scholar] [CrossRef]
- Onyekwelu, K.C. Life Cycle of Trypanosoma cruzi in the Invertebrate and the Vertebrate Hosts. Biology of Trypanosoma cruzi 2019, 1–19. [Google Scholar]
- Rodríguez-Bejarano, O.H.; Avendaño, C.; Patarroyo, M.A. Mechanisms associated with Trypanosoma cruzi host target cell adhesion, recognition and internalization. Life 2021, 11, 534. [Google Scholar] [CrossRef]
- Teixeira, D.E.; Benchimol, M.; Crepaldi, P.H.; de Souza, W. Interactive multimedia to teach the life cycle of Trypanosoma cruzi, the causative agent of Chagas disease. 2012.
- Andreoli, W.; Taniwaki, N.; Mortara, R. Survival of Trypanosoma cruzi metacyclic trypomastigotes within Coxiella burnetii vacuoles: differentiation and replication within an acidic milieu. Microbes and infection 2006, 8, 172–182. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. The EMBO journal 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.R.; Albuquerque-Cunha, J.M.; Mello, C.; Garcia, E.d.S.; Nogueira, N.; Bourguingnon, S.C.; De Souza, W.; Azambuja, P.; Gonzalez, M.S. Trypanosoma cruzi: attachment to perimicrovillar membrane glycoproteins of Rhodnius prolixus. Experimental Parasitology 2007, 116, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.S.; Ratcliffe, N.A.; Whitten, M.M.; Gonzalez, M.S.; Azambuja, P. Exploring the role of insect host factors in the dynamics of Trypanosoma cruzi–Rhodnius prolixus interactions. Journal of Insect Physiology 2007, 53, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Roldan, J.A.; Votýpka, J.; Bandi, C.; Epis, S.; Modrý, D.; Tichá, L.; Volf, P.; Otranto, D. Leishmania tarentolae: A new frontier in the epidemiology and control of the leishmaniases. Transboundary and Emerging Diseases 2022, 69, e1326–e1337. [Google Scholar] [CrossRef]
- Ferreira, L.C.; Quintella, L.P.; Schubach, A.d.O.; Miranda, L.d.F.C.; Madeira, M.d.F.; Pimentel, M.I.F.; Vasconcellos, É.d.C.F.e.; Lyra, M.R.; Oliveira, R.d.V.C.d.; Menezes, R.C. Comparison between Colorimetric In Situ Hybridization, Histopathology, and Immunohistochemistry for the Diagnosis of New World Cutaneous Leishmaniasis in Human Skin Samples. Tropical Medicine and Infectious Disease 2022, 7, 344. [Google Scholar] [CrossRef]
- Hadermann, A.; Heeren, S.; Maes, I.; Dujardin, J.-C.; Domagalska, M.A.; Van den Broeck, F. Genome diversity of Leishmania aethiopica. Frontiers in Cellular and Infection Microbiology 2023, 13, 406. [Google Scholar] [CrossRef]
- Bezemer, J.M.; Freire-Paspuel, B.P.; Schallig, H.D.; de Vries, H.J.; Calvopiña, M. Leishmania species and clinical characteristics of Pacific and Amazon cutaneous leishmaniasis in Ecuador and determinants of health-seeking delay: a cross-sectional study. BMC Infectious Diseases 2023, 23, 395. [Google Scholar] [CrossRef]
- Preativatanyou, K.; Chinwirunsirisup, K.; Phumee, A.; Khositharattanakool, P.; Sunantaraporn, S.; Depaquit, J.; Siriyasatien, P. Species diversity of phlebotomine sand flies and sympatric occurrence of Leishmania (Mundinia) martiniquensis, Leishmania (Leishmania) donovani complex, and Trypanosoma spp. in the visceral leishmaniasis focus of southern Thailand. Acta Tropica 2023, 244, 106949. [Google Scholar] [CrossRef]
- Dinç, M.; Yalçın, T.; Çavuş, İ.; Özbilgin, A. Comparative proteomic analysis of Leishmania parasites isolated from visceral and cutaneous leishmaniasis patients. Parasitology 2022, 149, 298–305. [Google Scholar] [CrossRef]
- Gramiccia, M.; Gradoni, L. The current status of zoonotic leishmaniases and approaches to disease control. International journal for parasitology 2005, 35, 1169–1180. [Google Scholar] [CrossRef]
- Schriefer, A.; Guimarães, L.H.; Machado, P.R.; Lessa, M.; Lessa, H.A.; Lago, E.; Ritt, G.; Góes-Neto, A.; Schriefer, A.L.; Riley, L.W. Geographic clustering of leishmaniasis in northeastern Brazil. Emerging infectious diseases 2009, 15, 871. [Google Scholar] [CrossRef]
- Montalvo Alvarez, A.M.; Nodarse, J.F.; Goodridge, I.M.; Fidalgo, L.M.; Marin, M.; Van Der Auwera, G.; Dujardin, J.-C.; Velez Bernal, I.D.; Muskus, C. Differentiation of Leishmania (Viannia) panamensis and Leishmania (V.) guyanensis using Bcc I for hsp 70 PCR-RFLP. Transactions of the Royal Society of Tropical Medicine and Hygiene 2010, 104, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Ducharme, O.; Simon, S.; Ginouves, M.; Prévot, G.; Couppie, P.; Demar, M.; Blaizot, R. Leishmania naiffi and lainsoni in French Guiana: Clinical features and phylogenetic variability. PLoS Neglected Tropical Diseases 2020, 14, e0008380. [Google Scholar] [CrossRef]
- Espada, C.R.; Ferreira, B.A.; Ortiz, P.A.; Uliana, S.R.; Coelho, A.C. Full nucleotide sequencing of ribosomal DNA internal transcribed spacer of Leishmania species causing cutaneous leishmaniasis in Brazil and its potential for species typing. Acta Tropica 2021, 223, 106093. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Morales, D.; Rodríguez, A.L.; Silva-Caso, W.; Suarez-Ognio, L.; Pons, M.J.; del Valle Mendoza, J. An atypical case of disseminated cutaneous leishmaniasis due to Leishmania peruviana in the valleys of Ancash-Peru. Asian Pacific journal of tropical medicine 2017, 10, 1101–1103. [Google Scholar] [CrossRef]
- Passero, L.F.D.; Carvalho, A.K.; Bordon, M.L.; Bonfim-Melo, A.; Carvalho, K.; Kallás, E.G.; Santos, B.B.; Toyama, M.H.; Paes-Leme, A.; Corbett, C.E. Proteins of Leishmania (Viannia) shawi confer protection associated with Th1 immune response and memory generation. Parasites & vectors 2012, 5, 1–10. [Google Scholar]
- Sukhumavasi, W.; Kaewamatawong, T.; Somboonpoonpol, N.; Jiratanh, M.; Wattanamethanont, J.; Kaewthamasorn, M.; Leelayoova, S.; Tiwananthagorn, S. Liver-and spleen-specific immune responses in experimental leishmania martiniquensis infection in BALB/c mice. Frontiers in Veterinary Science 2021, 8, 794024. [Google Scholar] [CrossRef]
- Supsrisunjai, C.; Kootiratrakarn, T.; Puangpet, P.; Bunnag, T.; Chaowalit, P.; Wessagowit, V. Case report: Disseminated autochthonous dermal leishmaniasis caused by Leishmania siamensis (PCM2 Trang) in a patient from central Thailand infected with human immunodeficiency virus. The American journal of tropical medicine and hygiene 2017, 96, 1160. [Google Scholar]
- Ramírez, J.D.; Hernández, C.; León, C.M.; Ayala, M.S.; Flórez, C.; González, C. Taxonomy, diversity, temporal and geographical distribution of Cutaneous Leishmaniasis in Colombia: A retrospective study. Scientific reports 2016, 6, 28266. [Google Scholar] [CrossRef]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. Clinical epidemiology 2014, 257–275. [Google Scholar]
- Schmunis, G.A.; Yadon, Z.E. Chagas disease: a Latin American health problem becoming a world health problem. Acta tropica 2010, 115, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Kourbeli, V.; Chontzopoulou, E.; Moschovou, K.; Pavlos, D.; Mavromoustakos, T.; Papanastasiou, I.P. An overview on target-based drug design against kinetoplastid protozoan infections: Human African trypanosomiasis, Chagas disease and leishmaniases. Molecules 2021, 26, 4629. [Google Scholar] [CrossRef]
- Bilgic-Temel, A.; Murrell, D.F.; Uzun, S. Cutaneous leishmaniasis: a neglected disfiguring disease for women. International journal of women's dermatology 2019, 5, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Kaluarachchi, T.J.; Campbell, P.M.; Wickremasinghe, R.; Ranasinghe, S.; Yasewardene, S.; De Silva, H.; McBain, A.J.; Weerasekera, M. Possible clinical implications and future directions of managing bacterial biofilms in cutaneous leishmaniasis wounds. Tropical Medicine and Health 2022, 50, 58. [Google Scholar] [CrossRef]
- Martínez, D.Y.; Verdonck, K.; Kaye, P.M.; Adaui, V.; Polman, K.; Llanos-Cuentas, A.; Dujardin, J.-C.; Boelaert, M. Tegumentary leishmaniasis and coinfections other than HIV. PLoS neglected tropical diseases 2018, 12, e0006125. [Google Scholar] [CrossRef]
- Meireles, C.B.; Maia, L.C.; Soares, G.C.; Teodoro, I.P.P.; Gadelha, M.d.S.V.; da Silva, C.G.L.; de Lima, M.A.P. Atypical presentations of cutaneous leishmaniasis: a systematic review. Acta Tropica 2017, 172, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: a review. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Sinha, S.; Fernández, G.; Kapila, R.; Lambert, W.C.; Schwartz, R.A. Diffuse cutaneous leishmaniasis associated with the immune reconstitution inflammatory syndrome. International journal of dermatology 2008, 47, 1263–1270. [Google Scholar] [CrossRef]
- Hashiguchi, Y.; Gomez, E.L.; Kato, H.; Martini, L.R.; Velez, L.N.; Uezato, H. Diffuse and disseminated cutaneous leishmaniasis: clinical cases experienced in Ecuador and a brief review. Tropical medicine and health 2016, 44, 1–9. [Google Scholar] [CrossRef]
- Goihman-Yahr, M. American mucocutaneous leishmaniasis. Dermatologic clinics 1994, 12, 703–712. [Google Scholar] [CrossRef]
- Vera-Izaguirre, D.S.; Vega-Memije, E.; Quintanilla-Cedillo, M.R.; Arenas, R. Leishmaniasis. A review. Dermatología Cosmética, Médica Y Quirúrgica 2006, 4, 252–260. [Google Scholar]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in leishmaniasis. The Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- CONVIT, J.; REYES, O.; KERDEL, F. Disseminated Anergic American Leishmaniasis: Report of Three Cases of a Type Clinically Resembling Lepromatous Leprosy. AMA archives of dermatology 1957, 76, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.G.; Bilimoria, F.E.; Katare, S. Diffuse cutaneous leishmaniasis: co-infection with human immunodeficiency virus (HIV). Indian journal of dermatology, venereology and leprology 2008, 74, 641. [Google Scholar] [CrossRef]
- David, C.V.; Craft, N. Cutaneous and mucocutaneous leishmaniasis. Dermatologic therapy 2009, 22, 491–502. [Google Scholar] [CrossRef]
- Garrido-Jareño, M.; Sahuquillo-Torralba, A.; Chouman-Arcas, R.; Castro-Hernández, I.; Molina-Moreno, J.M.; Llavador-Ros, M.; Gómez-Ruiz, M.D.; López-Hontangas, J.L.; Botella-Estrada, R.; Salavert-Lleti, M. Cutaneous and mucocutaneous leishmaniasis: experience of a Mediterranean hospital. Parasites & vectors 2020, 13, 1–7. [Google Scholar]
- Ahluwalia, S.; Lawn, S.D.; Kanagalingam, J.; Grant, H.; Lockwood, D.N. Mucocutaneous leishmaniasis: an imported infection among travellers to central and South America. Bmj 2004, 329, 842–844. [Google Scholar] [CrossRef]
- Marra, F.; Chiappetta, M.C.; Vincenti, V. Ear, nose and throat manifestations of mucocutaneous Leishmaniasis: a literature review. Acta Biomed 2014, 85, 3–7. [Google Scholar]
- Bern, C. Visceral leishmaniasis: clinical manifestations and diagnosis. U: UpToDate, Post TW ur. UpToDate [Internet]. Waltham, MA: UpToDate, 2021. [Google Scholar]
- Baba, C.S.; Makharia, G.K.; Mathur, P.; Ray, R.; Gupta, S.D.; Samantaray, J. Chronic diarrhea and malabsorption caused by Leishmania donovani. Indian Journal of Gastroenterology 2006, 25, 309. [Google Scholar]
- Boukhris, I.; Azzabi, S.; Chérif, E.; Kéchaou, I.; Mahjoub, S.; Kooli, C.; Aoun, K.; Khalfallah, N. Hemophagocytosis and disseminated intravascular coagulation in visceral leishmaniasis in adults: three new cases. The Pan African Medical Journal 2015, 22, 96–96. [Google Scholar]
- Pagliano, P.; Carannante, N.; Rossi, M.; Gramiccia, M.; Gradoni, L.; Faella, F.S.; Gaeta, G.B. Visceral leishmaniasis in pregnancy: a case series and a systematic review of the literature. Journal of Antimicrobial Chemotherapy 2005, 55, 229–233. [Google Scholar] [CrossRef]
- Herrero, M.; Orfanos, G.; Argaw, D.; Mulugeta, A.; Aparicio, P.; Parreño, F.; Bernal, O.; Rubens, D.; Pedraza, J.; Lima, M.A. Natural history of a visceral leishmaniasis outbreak in highland Ethiopia. The American journal of tropical medicine and hygiene 2009, 81, 373–377. [Google Scholar] [CrossRef]
- Mueller, Y.; Mbulamberi, D.B.; Odermatt, P.; Hoffmann, A.; Loutan, L.; Chappuis, F. Risk factors for in-hospital mortality of visceral leishmaniasis patients in eastern Uganda. Tropical Medicine & International Health 2009, 14, 910–917. [Google Scholar]
- Zijlstra, E.E. Biomarkers in post-kala-azar dermal leishmaniasis. Frontiers in cellular and infection microbiology 2019, 9, 228. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E. The immunology of post-kala-azar dermal leishmaniasis (PKDL). Parasites & vectors 2016, 9, 1–9. [Google Scholar]
- Zijlstra, E.; Musa, A.; Khalil, E.; El Hassan, I.; El-Hassan, A. Post-kala-azar dermal leishmaniasis. The Lancet infectious diseases 2003, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Chatterjee, M.; Das, N.K. Post kala-azar dermal leishmaniasis: Clinical features and differential diagnosis. Indian Journal of Dermatology 2021, 66, 24. [Google Scholar]
- Suárez, C.; Nolder, D.; García-Mingo, A.; Moore, D.A.; Chiodini, P.L. Diagnosis and clinical management of Chagas disease: An increasing challenge in non-endemic areas. Research and Reports in Tropical Medicine 2022, 25–40. [Google Scholar] [CrossRef]
- Nunes, M.C.P.; Dones, W.; Morillo, C.A.; Encina, J.J.; Ribeiro, A.L.; Cardiology, C.o.C.D.o.t.I.S.o. Chagas disease: an overview of clinical and epidemiological aspects. Journal of the American College of Cardiology 2013, 62, 767–776. [Google Scholar] [CrossRef]
- Hemmige, V.; Tanowitz, H.; Sethi, A. Trypanosoma cruzi infection: a review with emphasis on cutaneous manifestations. International journal of dermatology 2012, 51, 501–508. [Google Scholar] [CrossRef]
- Teixeira, A.; Nitz, N.; Guimaro, M.; Gomes, C.; Santos-Buch, C. Chagas disease. Postgraduate medical journal 2006, 82, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Organization, W.H. Control of Chagas disease: second report of the WHO expert committee; World Health Organization: 2002; Vol. 2.
- Malik, L.H.; Singh, G.D.; Amsterdam, E.A. The epidemiology, clinical manifestations, and management of chagas heart disease. Clinical cardiology 2015, 38, 565–569. [Google Scholar] [CrossRef] [PubMed]
- de Lourdes Higuchi, M.; Fukasawa, S.; De Brito, T.; Parzianello, L.C.; Bellotti, G.; Ramires, J.A.F. Different microcirculatory and interstitial matrix patterns in idiopathic dilated cardiomyopathy and Chagas’ disease: a three dimensional confocal microscopy study. Heart 1999, 82, 279–285. [Google Scholar] [CrossRef]
- Benvenuti, L.; Roggério, A.; Freitas, H.; Mansur, A.; Fiorelli, A.; Higuchi, M. Chronic American trypanosomiasis: parasite persistence in endomyocardial biopsies is associated with high-grade myocarditis. Annals of Tropical Medicine & Parasitology 2008, 102, 481–487. [Google Scholar]
- Marin-Neto, J.A.; Cunha-Neto, E.; Maciel, B.C.; Simões, M.V. Pathogenesis of chronic Chagas heart disease. Circulation 2007, 115, 1109–1123. [Google Scholar] [CrossRef]
- Rossi, M.A.; Tanowitz, H.B.; Malvestio, L.M.; Celes, M.R.; Campos, E.C.; Blefari, V.; Prado, C.M. Coronary microvascular disease in chronic Chagas cardiomyopathy including an overview on history, pathology, and other proposed pathogenic mechanisms. PLoS neglected tropical diseases 2010, 4, e674. [Google Scholar] [CrossRef]
- Rocha, M.O.C.; Nunes, M.C.P.; Ribeiro, A.L. Morbidity and prognostic factors in chronic chagasic cardiopathy. Memórias do Instituto Oswaldo Cruz 2009, 104, 159–166. [Google Scholar] [CrossRef]
- Rassi Jr, A.; Rassi, S.G.; Rassi, A. Sudden death in Chagas' disease. Arquivos brasileiros de cardiologia 2001, 76, 86–96. [Google Scholar] [CrossRef]
- Nunes, M.; Dones, W.; Morillo, C.; Encina, J.; Ribeiro, A. Council on Chagas disease of the InterAmerican Society of Cardiology. J Am Coll Cardiol 2013, 62, 767–776. [Google Scholar] [CrossRef]
- Tripodi, K.E.; Menendez Bravo, S.M.; Cricco, J.A. Role of heme and heme-proteins in trypanosomatid essential metabolic pathways. Enzyme research 2011, 2011. [Google Scholar] [CrossRef]
- Friggeri, L.; Hargrove, T.Y.; Rachakonda, G.; Blobaum, A.L.; Fisher, P.; De Oliveira, G.M.; Da Silva, C.F.; Soeiro, M.d.N.C.; Nes, W.D.; Lindsley, C.W. Sterol 14α-demethylase structure-based optimization of drug candidates for human infections with the protozoan Trypanosomatidae. Journal of medicinal chemistry 2018, 61, 10910–10921. [Google Scholar] [CrossRef] [PubMed]
- de Souza, W.; Rodrigues, J.C.F. Sterol biosynthesis pathway as target for anti-trypanosomatid drugs. Interdisciplinary perspectives on infectious diseases 2009, 2009. [Google Scholar] [CrossRef]
- Marathe, C.; Bradley, M.N.; Hong, C.; Lopez, F.; de Galarreta, C.M.R.; Tontonoz, P.; Castrillo, A. The arginase II gene is an anti-inflammatory target of liver X receptor in macrophages. Journal of Biological Chemistry 2006, 281, 32197–32206. [Google Scholar] [CrossRef]
- Gallardo-Soler, A.; Gómez-Nieto, C.; Campo, M.L.; Marathe, C.; Tontonoz, P.; Castrillo, A.; Corraliza, I. Arginase I induction by modified lipoproteins in macrophages: a peroxisome proliferator-activated receptor-γ/δ-mediated effect that links lipid metabolism and immunity. Molecular endocrinology 2008, 22, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Silva-Jardim, I.; Thiemann, O.H.; Anibal, F.F. Leishmaniasis and Chagas disease chemotherapy: a critical review. Journal of the Brazilian Chemical Society 2014, 25, 1810–1823. [Google Scholar] [CrossRef]
- McConville, M.J.; Naderer, T. Metabolic pathways required for the intracellular survival of Leishmania. Annual review of microbiology 2011, 6, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Sanchez, M.A.; Pierce, S.; Herrmann, T.; Kimblin, N.; Archie Bouwer, H.; Landfear, S.M. Molecular genetic analysis of purine nucleobase transport in Leishmania major. Molecular microbiology 2007, 64, 1228–1243. [Google Scholar] [CrossRef]
- Figueroa-Villar, J.D.; Sales, E.M. The importance of nucleoside hydrolase enzyme (NH) in studies to treatment of Leishmania: A review. Chemico-Biological Interactions 2017, 263, 18–27. [Google Scholar] [CrossRef]
- Majumder, H.K. Drug targets in kinetoplastid parasites; Springer Science & Business Media: 2008; Vol. 625.
- Berg, M.; Van der Veken, P.; Goeminne, A.; Haemers, A.; Augustyns, K. Inhibitors of the purine salvage pathway: a valuable approach for antiprotozoal chemotherapy? Current medicinal chemistry 2010, 17, 2456–2481. [Google Scholar] [CrossRef]
- Ilgoutz, S.C.; Zawadzki, J.L.; Ralton, J.E.; McConville, M.J. Evidence that free GPI glycolipids are essential for growth of Leishmania mexicana. The EMBO journal 1999, 18, 2746–2755. [Google Scholar] [CrossRef]
- Menon, A.K.; Mayor, S.; Schwarz, R.T. Biosynthesis of glycosyl-phosphatidylinositol lipids in Trypanosoma brucei: involvement of mannosyl-phosphoryldolichol as the mannose donor. The EMBO Journal 1990, 9, 4249–4258. [Google Scholar] [CrossRef]
- Horn, D. Antigenic variation in African trypanosomes. Molecular and biochemical parasitology 2014, 195, 123–129. [Google Scholar] [CrossRef]
- Alves, M.J.M.; Colli, W. Role of the gp85/trans-sialidase superfamily of glycoproteins in the interaction of Trypanosoma cruzi with host structures. Molecular Mechanisms of Parasite Invasion: Subcellular Biochemistry, 2008; 58–69. [Google Scholar]
- Giorgi, M.E.; Lederkremer, R.M.d. The glycan structure of T. cruzi mucins depends on the host. Insights on the chameleonic galactose. Molecules 2020, 25, 3913. [Google Scholar] [CrossRef]
- Barreto de Albuquerque, J.; Silva dos Santos, D.; Stein, J.V.; de Meis, J. Oral versus intragastric inoculation: Similar Pathways of Trypanosoma cruzi Experimental infection? From target tissues, parasite evasion, and immune response. Frontiers in immunology 2018, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays in biochemistry 2011, 51, 63–80. [Google Scholar]
- Shulpekova, Y.; Nechaev, V.; Kardasheva, S.; Sedova, A.; Kurbatova, A.; Bueverova, E.; Kopylov, A.; Malsagova, K.; Dlamini, J.C.; Ivashkin, V. The concept of folic acid in health and disease. Molecules 2021, 26, 3731. [Google Scholar] [CrossRef]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. Examination of multiple Trypanosoma cruzi targets in a new drug discovery approach for Chagas disease. Bioorganic & medicinal chemistry 2022, 58, 116577. [Google Scholar]
- Robello, C.; Navarro, P.; Castanys, S.; Gamarro, F. A pteridine reductase gene ptr1 contiguous to a P-glycoprotein confers resistance to antifolates in Trypanosoma cruzi. Molecular and biochemical parasitology 1997, 90, 525–535. [Google Scholar] [CrossRef]
- Bello, A.R.; Nare, B.; Freedman, D.; Hardy, L.; Beverley, S.M. PTR1: a reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proceedings of the National Academy of Sciences 1994, 91, 11442–11446. [Google Scholar] [CrossRef] [PubMed]
- Krauth-Siegel, R.L.; Comini, M.A. Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochimica et Biophysica Acta (BBA)-General Subjects 2008, 1780, 1236–1248. [Google Scholar] [CrossRef]
- Colotti, G.; Ilari, A. Polyamine metabolism in Leishmania: from arginine to trypanothione. Amino acids 2011, 40, 269–285. [Google Scholar] [CrossRef] [PubMed]
- BAILEY, S.; SMITH, K.; FAIRLAMB, A.H.; HUNTER, W.N. Substrate interactions between trypanothione reductase and N1-glutathionylspermidine disulphide at 0.28-nm resolution. European journal of biochemistry 1993, 213, 67–75. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO journal 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Manta, B.; Comini, M.; Medeiros, A.; Hugo, M.; Trujillo, M.; Radi, R. Trypanothione: a unique bis-glutathionyl derivative in trypanosomatids. Biochimica et Biophysica Acta (BBA)-General Subjects 2013, 1830, 3199–3216. [Google Scholar] [CrossRef]
- Jain, S.; Sahu, U.; Kumar, A.; Khare, P. Metabolic pathways of Leishmania parasite: source of pertinent drug targets and potent drug candidates. Pharmaceutics 2022, 14, 1590. [Google Scholar] [CrossRef]
- Moreira, D.d.S.; Duarte, A.P.; Pais, F.S.M.; Silva-Pereira, R.A.d.; Romanha, A.J.; Schenkman, S.; Murta, S.M.F. Overexpression of eukaryotic initiation factor 5A (eIF5A) affects susceptibility to benznidazole in Trypanosoma cruzi populations. Memórias do Instituto Oswaldo Cruz 2018, 113, e180162. [Google Scholar] [CrossRef]
- Nakanishi, S.; Cleveland, J.L. Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer. Amino acids 2016, 48, 2353–2362. [Google Scholar] [CrossRef]
- Goldman-Pinkovich, A.; Balno, C.; Strasser, R.; Zeituni-Molad, M.; Bendelak, K.; Rentsch, D.; Ephros, M.; Wiese, M.; Jardim, A.; Myler, P.J. An arginine deprivation response pathway is induced in Leishmania during macrophage invasion. PLoS pathogens 2016, 12, e1005494. [Google Scholar] [CrossRef]
- da Silva, M.F.L.; Floeter-Winter, L.M. Arginase in leishmania. Proteins and proteomics of Leishmania and Trypanosoma 2013, 103–117. [Google Scholar]
- Darlyuk, I.; Goldman, A.; Roberts, S.C.; Ullman, B.; Rentsch, D.; Zilberstein, D. Arginine homeostasis and transport in the human pathogen Leishmania donovani. Journal of biological chemistry 2009, 284, 19800–19807. [Google Scholar] [CrossRef] [PubMed]
- Maddison, J.E.; Page, S.W.; Church, D.B. Small animal clinical pharmacology; Elsevier Health Sciences: 2008; Vol. 5.
- Marchal, B.; Van Dormael, M.; Pirard, M.; Cavalli, A.; Kegels, G.; Polman, K. Neglected tropical disease (NTD) control in health systems: the interface between programmes and general health services. Acta tropica 2011, 120, S177–S185. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nature reviews Drug discovery 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Myler, P.J. Searching the Tritryp genomes for drug targets. Drug Targets in Kinetoplastid Parasites 2008, 133–140. [Google Scholar]
- Paradela, L.S.; Wall, R.J.; Carvalho, S.; Chemi, G.; Corpas-Lopez, V.; Moynihan, E.; Bello, D.; Patterson, S.; Güther, M.L.S.; Fairlamb, A.H. Multiple unbiased approaches identify oxidosqualene cyclase as the molecular target of a promising anti-leishmanial. Cell chemical biology 2021, 28, 711–721. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Prema, K.; Balaji, S. Machine learning models for drug–target interactions: current knowledge and future directions. Drug Discovery Today 2020, 25, 748–756. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of health economics 2016, 47, 20–33. [Google Scholar] [PubMed]
- Thomas, D.W. Clinical development success rates 2006–2015. BIO Industry Anal. 2016, 1, 16. [Google Scholar]
- Surade, S.; Blundell, T.L. Structural biology and drug discovery of difficult targets: the limits of ligandability. Chemistry & biology 2012, 19, 42–50. [Google Scholar]
- Gamo, F.-J.; Sanz, L.M.; Vidal, J.; De Cozar, C.; Alvarez, E.; Lavandera, J.-L.; Vanderwall, D.E.; Green, D.V.; Kumar, V.; Hasan, S. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef]
- Don, R.; Ioset, J.-R. Screening strategies to identify new chemical diversity for drug development to treat kinetoplastid infections. Parasitology 2014, 141, 140–146. [Google Scholar] [CrossRef]
- Guiguemde, W.A.; Shelat, A.A.; Bouck, D.; Duffy, S.; Crowther, G.J.; Davis, P.H.; Smithson, D.C.; Connelly, M.; Clark, J.; Zhu, F. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [Google Scholar] [CrossRef]
- De Rycker, M.; Wyllie, S.; Horn, D.; Read, K.D.; Gilbert, I.H. Anti-trypanosomatid drug discovery: progress and challenges. Nature Reviews Microbiology 2023, 21, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Peinado, N.; Cortes-Serra, N.; Losada-Galvan, I.; Alonso-Vega, C.; Urbina, J.A.; Rodríguez, A.; VandeBerg, J.L.; Pinazo, M.-J.; Gascon, J.; Alonso-Padilla, J. Emerging agents for the treatment of Chagas disease: what is in the preclinical and clinical development pipeline? Expert Opinion on Investigational Drugs 2020, 29, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Mansoldo, F.R.P.; Carta, F.; Angeli, A.; Cardoso, V.d.S.; Supuran, C.T.; Vermelho, A.B. Chagas disease: perspectives on the past and present and challenges in drug discovery. Molecules 2020, 25, 5483. [Google Scholar] [CrossRef]
- Francisco, E.C.; de Almeida Junior, J.N.; Queiroz-Telles, F.; Aquino, V.R.; Mendes, A.V.A.; de Oliveira Silva, M.; Castro, P.d.T.O.e.; Guimarães, T.; Ponzio, V.; Hahn, R.C. Correlation of Trichosporon asahii genotypes with anatomical sites and antifungal susceptibility profiles: data analyses from 284 isolates collected in the last 22 years across 24 medical centers. Antimicrobial Agents and Chemotherapy 2021, 65, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Schiavuzzo, J.; Teixeira, J.; Melo, B.; Dos Santos, D.d.S.; Jorge, C.; Oliveira-Fusaro, M.; Parada, C. Muscle hyperalgesia induced by peripheral P2X3 receptors is modulated by inflammatory mediators. Neuroscience 2015, 285, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Mackey, T.K.; Liang, B.A.; Cuomo, R.; Hafen, R.; Brouwer, K.C.; Lee, D.E. Emerging and reemerging neglected tropical diseases: a review of key characteristics, risk factors, and the policy and innovation environment. Clinical microbiology reviews 2014, 27, 949–979. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, P.A.; Santos, M.M. Recent progress in the development of indole-based compounds active against malaria, trypanosomiasis and leishmaniasis. Molecules 2022, 27, 319. [Google Scholar] [CrossRef]
- Reimão, J.Q.; Scotti, M.T.; Tempone, A.G. Anti-leishmanial and anti-trypanosomal activities of 1, 4-dihydropyridines: In vitro evaluation and structure–activity relationship study. Bioorganic & medicinal chemistry 2010, 18, 8044–8053. [Google Scholar]
- Yadav, C.L.; Manar, K.K.; Yadav, M.K.; Tiwari, N.; Singh, R.K.; Drew, M.G.; Singh, N. Synthesis, crystal structures and properties of new homoleptic Ni (II)/Pd (II) β-oxodithioester chelates. Journal of Molecular Structure 2018, 1160, 488–496. [Google Scholar] [CrossRef]
- Sahu, A.; Kumar, D.; Agrawal, R.K. Antileishmanial drug discovery: synthetic methods, chemical characteristics, and biological potential of quinazolines and its derivatives. Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Inflammatory and Anti-Allergy Agents) 2017, 16, 3–32. [Google Scholar]
- Charlton, R.L.; Rossi-Bergmann, B.; Denny, P.W.; Steel, P.G. Repurposing as a strategy for the discovery of new anti-leishmanials: the-state-of-the-art. Parasitology 2018, 145, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Reguera, R.M.; Pérez-Pertejo, Y.; Gutiérrez-Corbo, C.; Domínguez-Asenjo, B.; Ordóñez, C.; García-Estrada, C.; Martínez-Valladares, M.; Balaña-Fouce, R. Current and promising novel drug candidates against visceral leishmaniasis. Pure and Applied Chemistry 2019, 91, 1385–1404. [Google Scholar] [CrossRef]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi Jr, A.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C. Randomized trial of benznidazole for chronic Chagas’ cardiomyopathy. New England Journal of Medicine 2015, 373, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Junior, P.A.S.; Molina, I.; Murta, S.M.F.; Sánchez-Montalvá, A.; Salvador, F.; Corrêa-Oliveira, R.; Carneiro, C.M. Experimental and clinical treatment of Chagas disease: a review. The American journal of tropical medicine and hygiene 2017, 97, 1289. [Google Scholar] [CrossRef]
- Caldas, I.S.; Santos, E.G.; Novaes, R.D. An evaluation of benznidazole as a Chagas disease therapeutic. Expert opinion on pharmacotherapy 2019, 20, 1797–1807. [Google Scholar] [CrossRef]
- Patterson, S.; Wyllie, S. Nitro drugs for the treatment of trypanosomatid diseases: past, present, and future prospects. Trends in parasitology 2014, 30, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, E.; Scandale, I. Animal models of Chagas disease and their translational value to drug development. Expert Opinion on Drug Discovery 2020, 15, 1381–1402. [Google Scholar] [CrossRef] [PubMed]
- Vermelho, A.B.; Rodrigues, G.C.; Supuran, C.T. Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opinion on Drug Discovery 2020, 15, 145–158. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Sur, D.; Karbwang, J. Childhood visceral leishmaniasis. Indian Journal of Medical Research 2006, 123, 353. [Google Scholar]
- Sundar, S.; Singh, A. Chemotherapeutics of visceral leishmaniasis: present and future developments. Parasitology 2018, 145, 481–489. [Google Scholar] [CrossRef]
- Dorlo, T.P.; Kip, A.E.; Younis, B.M.; Ellis, S.J.; Alves, F.; Beijnen, J.H.; Njenga, S.; Kirigi, G.; Hailu, A.; Olobo, J. Visceral leishmaniasis relapse hazard is linked to reduced miltefosine exposure in patients from Eastern Africa: a population pharmacokinetic/pharmacodynamic study. Journal of Antimicrobial Chemotherapy 2017, 72, 3131–3140. [Google Scholar] [CrossRef]
- Mbui, J.; Olobo, J.; Omollo, R.; Solomos, A.; Kip, A.E.; Kirigi, G.; Sagaki, P.; Kimutai, R.; Were, L.; Omollo, T. Pharmacokinetics, safety, and efficacy of an allometric miltefosine regimen for the treatment of visceral leishmaniasis in Eastern African children: an open-label, phase II clinical trial. Clinical Infectious Diseases 2019, 68, 1530–1538. [Google Scholar] [CrossRef]
- Musa, A.M.; Younis, B.; Fadlalla, A.; Royce, C.; Balasegaram, M.; Wasunna, M.; Hailu, A.; Edwards, T.; Omollo, R.; Mudawi, M. Paromomycin for the treatment of visceral leishmaniasis in Sudan: a randomized, open-label, dose-finding study. PLoS neglected tropical diseases 2010, 4, e855. [Google Scholar] [CrossRef]
- Jamil, K.M.; Haque, R.; Rahman, R.; Faiz, M.A.; Bhuiyan, A.T.M.R.H.; Kumar, A.; Hassan, S.M.; Kelly, H.; Dhalaria, P.; Kochhar, S. Effectiveness study of paromomycin IM injection (PMIM) for the treatment of visceral leishmaniasis (VL) in Bangladesh. PLoS Neglected Tropical Diseases 2015, 9, e0004118. [Google Scholar] [CrossRef] [PubMed]
- Diro, E.; Ritmeijer, K.; Boelaert, M.; Alves, F.; Mohammed, R.; Abongomera, C.; Ravinetto, R.; De Crop, M.; Fikre, H.; Adera, C. Long-term clinical outcomes in visceral leishmaniasis-HIV co-infected patients during and after pentamidine secondary prophylaxis in Ethiopia: a single-arm clinical trial Authors and affiliations. Clinical Infectious Diseases 2017. [Google Scholar]
- Rahman, A.-E.; Mabrouk, R. The Impact of First-line Nurse Managers Leadership Development Training Program on Workgroup Climate and Performance. Diss: 2017.
- Balasegaram, M.; Ritmeijer, K.; Lima, M.A.; Burza, S.; Ortiz Genovese, G.; Milani, B.; Gaspani, S.; Potet, J.; Chappuis, F. Liposomal amphotericin B as a treatment for human leishmaniasis. Expert opinion on emerging drugs 2012, 17, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Deray, G. Amphotericin B nephrotoxicity. Journal of antimicrobial chemotherapy 2002, 49 (Suppl. 1), 37–41. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.; Boelaert, M. Leishmaniasis Lancet 392: 951–970. 2018.
- Sundar, S.; Chakravarty, J. Antimony toxicity. International journal of environmental research and public health 2010, 7, 4267–4277. [Google Scholar] [CrossRef]
- Arce, A.; Estirado, A.; Ordobas, M.; Sevilla, S.; García, N.; Moratilla, L.; De La Fuente, S.; Martínez, A.; Pérez, A.; Aránguez, E. Re-emergence of leishmaniasis in Spain: community outbreak in Madrid, Spain, 2009 to 2012. Eurosurveillance 2013, 18, 20546. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J.; Agarwal, D.; Rai, M.; Murray, H.W. Single-dose liposomal amphotericin B for visceral leishmaniasis in India. New England Journal of Medicine 2010, 362, 504–512. [Google Scholar] [CrossRef]
- Pereira, I.R.; Vilar-Pereira, G.; Silva, A.A.d.; Lannes-Vieira, J. Severity of chronic experimental Chagas' heart disease parallels tumour necrosis factor and nitric oxide levels in the serum: models of mild and severe disease. Memórias do Instituto Oswaldo Cruz 2014, 109, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Molina, I.; Gómez i Prat, J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L. Randomized trial of posaconazole and benznidazole for chronic Chagas' disease. New England Journal of Medicine 2014, 370, 1899–1908. [Google Scholar] [CrossRef]
- Lima, Á.A.; Soares-Sobrinho, J.L.; Silva, J.L.; Corrêa-Júnior, R.A.; Lyra, M.A.; Santos, F.L.; Oliveira, B.G.; Hernandes, M.Z.; Rolim, L.A.; Rolim-Neto, P.J. The use of solid dispersion systems in hydrophilic carriers to increase benznidazole solubility. Journal of pharmaceutical sciences 2011, 100, 2443–2451. [Google Scholar] [CrossRef]
- Ben Beard, C. Control of Chagas Disease. Second Report of the WHO Expert Committee. WHO Technical Report Series, No. 905. Geneva: World Health Organization, 2002. vi+ 109pp. Price Sw. fr. 23/US $20.70 (in developing countries Sw. fr. 16.10). ISBN 92-4-120905.4. Royal Society of Tropical Medicine and Hygiene: 2002.
- Organization, W.H. Sustaining the drive to overcome the global impact of neglected tropical diseases: second WHO report on neglected diseases; World Health Organization: 2013.
- de Mecca, M.M.; Fanelli, S.L.; Bartel, L.C.; de Castro, C.R.; Díaz, E.G.; Castro, J.A. Nifurtimox nitroreductase activity in different cellular fractions from male rat pancreas. Biochemical and ultrastructural alterations. Life sciences 2007, 81, 144–152. [Google Scholar] [CrossRef]
- Mecca, M.M.d.; Bartel, L.C.; Castro, C.R.d.; Castro, J.A. Benznidazole biotransformation in rat heart microsomal fraction without observable ultrastructural alterations: comparison to Nifurtimox-induced cardiac effects. Memorias do Instituto Oswaldo Cruz 2008, 103, 549–553. [Google Scholar] [CrossRef]
- Bhargava, P.; Singh, R. Developments in diagnosis and antileishmanial drugs. Interdisciplinary perspectives on infectious diseases 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Alcântara, L.M.; Ferreira, T.C.; Gadelha, F.R.; Miguel, D.C. Challenges in drug discovery targeting TriTryp diseases with an emphasis on leishmaniasis. International Journal for Parasitology: Drugs and Drug Resistance 2018, 8, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Roatt, B.M.; de Oliveira Cardoso, J.M.; De Brito, R.C.F.; Coura-Vital, W.; de Oliveira Aguiar-Soares, R.D.; Reis, A.B. Recent advances and new strategies on leishmaniasis treatment. Applied Microbiology and Biotechnology 2020, 104, 8965–8977. [Google Scholar] [CrossRef]
- Zahid, M.S.H.; Johnson, M.M.; Tokarski II, R.J.; Satoskar, A.R.; Fuchs, J.R.; Bachelder, E.M.; Ainslie, K.M. Evaluation of synergy between host and pathogen-directed therapies against intracellular Leishmania donovani. International Journal for Parasitology: Drugs and Drug Resistance 2019, 10, 125–132. [Google Scholar] [CrossRef]
- Novais, F.O.; Amorim, C.F.; Scott, P. Host-Directed Therapies for Cutaneous Leishmaniasis. Frontiers in Immunology 2021, 12, 660183. [Google Scholar] [CrossRef]
- Kumari, D.; Perveen, S.; Sharma, R.; Singh, K. Advancement in leishmaniasis diagnosis and therapeutics: An update. European Journal of Pharmacology 2021, 910, 174436. [Google Scholar] [CrossRef]
- Rassi Jr, A.; Rassi, A.; Marin-Neto, J.A. Chagas disease. The Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. The Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Parihar, S.P.; Hartley, M.-A.; Hurdayal, R.; Guler, R.; Brombacher, F. Topical simvastatin as host-directed therapy against severity of cutaneous leishmaniasis in mice. Scientific reports 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Shivam, P.; Mandal, S.; Prasanna, P.; Kumar, S.; Prasad, S.R.; Kumar, A.; Das, P.; Ali, V.; Singh, S.K. Synthesis, characterization, and mechanistic studies of a gold nanoparticle–amphotericin B covalent conjugate with enhanced antileishmanial efficacy and reduced cytotoxicity. International Journal of Nanomedicine 2019, 14, 6073. [Google Scholar] [CrossRef] [PubMed]
- Raja, M.R.C.; Velappan, A.B.; Chellappan, D.; Debnath, J.; Mahapatra, S.K. Eugenol derived immunomodulatory molecules against visceral leishmaniasis. European journal of medicinal chemistry 2017, 139, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Tindana, P.; de Haan, F.; Amaratunga, C.; Dhorda, M.; van der Pluijm, R.W.; Dondorp, A.M.; Cheah, P.Y. Deploying triple artemisinin-based combination therapy (TACT) for malaria treatment in Africa: ethical and practical considerations. Malaria Journal 2021, 20, 1–7. [Google Scholar] [CrossRef]
- Larkins-Ford, J.; Greenstein, T.; Van, N.; Degefu, Y.N.; Olson, M.C.; Sokolov, A.; Aldridge, B.B. Systematic measurement of combination-drug landscapes to predict in vivo treatment outcomes for tuberculosis. Cell Systems 2021, 12, 1046–1063. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: progress, challenges and recommendations. Nature reviews Drug discovery 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Breckenridge, A.; Jacob, R. Overcoming the legal and regulatory barriers to drug repurposing. Nature reviews Drug discovery 2019, 18, 1–2. [Google Scholar] [CrossRef]
- Braga, S.S. Multi-target drugs active against leishmaniasis: A paradigm of drug repurposing. European Journal of Medicinal Chemistry 2019, 183, 111660. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.H.; Neves, B.J.; Melo-Filho, C.C.; Rodrigues, J.; Silva, D.C.; Braga, R.C.; Cravo, P.V. In silico chemogenomics drug repositioning strategies for neglected tropical diseases. Current Medicinal Chemistry 2019, 26, 4355–4379. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, E.A.M.; Lang, K.L. Drug repositioning: concept, classification, methodology, and importance in rare/orphans and neglected diseases. Journal of Applied Pharmaceutical Science 2018, 8, 157–165. [Google Scholar]
- Jantan, I.; Bukhari, S.N.A.; Mohamed, M.A.S.; Wai, L.K.; Mesaik, M.A. The evolving role of natural products from the tropical rainforests as a replenishable source of new drug leads. Drug Discovery and Development-From Molecules to Medicine, 2005; 3–38. [Google Scholar]
- García, M.; Monzote, L. Marine products with anti-protozoal activity: a review. Current Clinical Pharmacology 2014, 9, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, R.; Jain, N. CURRENT STRATEGIES AND RECENT ADVANCES IN NANOSTRUCTURED DELIVERY SYSTEMS FOR MANAGING LEISHMANIASIS-A REVIEW. Journal of Advanced Scientific Research 2022, 13, 43–55. [Google Scholar] [CrossRef]
- Santos, S.S.; de Araujo, R.V.; Giarolla, J.; El Seoud, O.; Ferreira, E.I. Searching for drugs for Chagas disease, leishmaniasis and schistosomiasis: a review. International journal of antimicrobial agents 2020, 55, 105906. [Google Scholar] [CrossRef]
- Gharpure, S.; Ankamwar, B. Use of nanotechnology in combating coronavirus. 3 Biotech 2021, 11, 358. [Google Scholar] [CrossRef]
- Karuppusamy, C.; Venkatesan, P. Role of nanoparticles in drug delivery system: a comprehensive review. Journal of Pharmaceutical sciences and Research 2017, 9, 318. [Google Scholar]
- Kim, D.Y.; Kadam, A.; Shinde, S.; Saratale, R.G.; Patra, J.; Ghodake, G. Recent developments in nanotechnology transforming the agricultural sector: a transition replete with opportunities. Journal of the Science of Food and Agriculture 2018, 98, 849–864. [Google Scholar] [CrossRef]
- Celis-Giraldo, C.T.; López-Abán, J.; Muro, A.; Patarroyo, M.A.; Manzano-Román, R. Nanovaccines against animal pathogens: The latest findings. Vaccines 2021, 9, 988. [Google Scholar] [CrossRef]
- Javed, R.; Ghonaim, R.; Shathili, A.; Khalifa, S.A.; El-Seedi, H.R. Phytonanotechnology: A greener approach for biomedical applications. In Biogenic Nanoparticles for Cancer Theranostics, Elsevier: 2021; pp. 43-86.
- Sangar, O.S.; Patil, A.C.; Payghan, S.A. Nanoparticles: As a Nano based Drug Delivery System. 2022.
- Nafari, A.; Cheraghipour, K.; Sepahvand, M.; Shahrokhi, G.; Gabal, E.; Mahmoudvand, H. Nanoparticles: New agents toward treatment of leishmaniasis. Parasite epidemiology and control 2020, 10, e00156. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, P.; Kumar, P.; Kumar, S.; Rajana, V.K.; Kant, V.; Prasad, S.R.; Mohan, U.; Ravichandiran, V.; Mandal, D. Current status of nanoscale drug delivery and the future of nano-vaccine development for leishmaniasis–a review. Biomedicine & Pharmacotherapy 2021, 141, 111920. [Google Scholar]
- Athanasiou, E.; Agallou, M.; Tastsoglou, S.; Kammona, O.; Hatzigeorgiou, A.; Kiparissides, C.; Karagouni, E. A poly (lactic-co-glycolic) acid nanovaccine based on chimeric peptides from different Leishmania infantum proteins induces dendritic cells maturation and promotes peptide-specific IFNγ-producing CD8+ T cells essential for the protection against experimental visceral leishmaniasis. Frontiers in immunology 2017, 8, 684. [Google Scholar] [PubMed]
- Agallou, M.; Margaroni, M.; Athanasiou, E.; Toubanaki, D.K.; Kontonikola, K.; Karidi, K.; Kammona, O.; Kiparissides, C.; Karagouni, E. Identification of BALB/c immune markers correlated with a partial protection to Leishmania infantum after vaccination with a rationally designed multi-epitope cysteine protease a peptide-based nanovaccine. PLoS Neglected Tropical Diseases 2017, 11, e0005311. [Google Scholar] [CrossRef] [PubMed]
- Assolini, J.P.; Carloto, A.C.M.; da Silva Bortoleti, B.T.; Gonçalves, M.D.; Pellissier, F.T.; Feuser, P.E.; Cordeiro, A.P.; de Araújo, P.H.H.; Sayer, C.; Sapla, M.M.M. Nanomedicine in leishmaniasis: A promising tool for diagnosis, treatment and prevention of disease-An update overview. European Journal of Pharmacology 2022, 923, 174934. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: back to the future? Journal of medicinal chemistry 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduction and Targeted Therapy 2022, 7, 48. [Google Scholar] [CrossRef]
- Derda, R.; Jafari, M.R. Synthetic cross-linking of peptides: molecular linchpins for peptide cyclization. Protein and Peptide Letters 2018, 25, 1051–1075. [Google Scholar] [CrossRef]
- Kang, N.J.; Jin, H.-S.; Lee, S.-E.; Kim, H.J.; Koh, H.; Lee, D.-W. New approaches towards the discovery and evaluation of bioactive peptides from natural resources. Critical Reviews in Environmental Science and Technology 2020, 50, 72–103. [Google Scholar] [CrossRef]
- Davda, J.; Declerck, P.; Hu-Lieskovan, S.; Hickling, T.P.; Jacobs, I.A.; Chou, J.; Salek-Ardakani, S.; Kraynov, E. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. Journal for immunotherapy of cancer 2019, 7, 1–9. [Google Scholar] [CrossRef]
- Nishioka, K.; Hayashi, S.; Farrer, M.J.; Singleton, A.B.; Yoshino, H.; Imai, H.; Kitami, T.; Sato, K.; Kuroda, R.; Tomiyama, H. Clinical heterogeneity of α-synuclein gene duplication in Parkinson's disease. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 2006, 59, 298–309. [Google Scholar] [CrossRef]
- Weng, H.-B.; Chen, H.-X.; Wang, M.-W. Innovation in neglected tropical disease drug discovery and development. Infectious diseases of poverty 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, T.; Bruhn, H.; Gaworski, I.; Fleischer, B.; Leippe, M. NK-lysin and its shortened analog NK-2 exhibit potent activities against Trypanosoma cruzi. Antimicrobial agents and chemotherapy 2003, 47, 607–613. [Google Scholar] [CrossRef]
- Souza, A.L.; Faria, R.X.; Calabrese, K.S.; Hardoim, D.J.; Taniwaki, N.; Alves, L.A.; De Simone, S.G. Temporizin and temporizin-1 peptides as novel candidates for eliminating Trypanosoma cruzi. PLoS One 2016, 11, e0157673. [Google Scholar] [CrossRef]
- Kleschenko, Y.E.; Karpenko, L.; Villalta, F. Effects of human defensin-α1 on Trypanosoma cruzi trypomastigotes in vitro. Bulletin of experimental biology and medicine 2010, 149, 731. [Google Scholar] [CrossRef]
- Pinto, E.G.; Pimenta, D.C.; Antoniazzi, M.M.; Jared, C.; Tempone, A.G. Antimicrobial peptides isolated from Phyllomedusa nordestina (Amphibia) alter the permeability of plasma membrane of Leishmania and Trypanosoma cruzi. Experimental parasitology 2013, 135, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Brand, G.D.; Leite, J.R.S.; Silva, L.P.; Albuquerque, S.; Prates, M.V.; Azevedo, R.B.; Carregaro, V.; Silva, J.S.; Sá, V.C.; Brandao, R.A. Dermaseptins from Phyllomedusa oreades andPhyllomedusa distincta: Anti-Trypanosoma Cruzi Activity Without Cytotoxicity To Mammalian Cells. Journal of Biological Chemistry 2002, 277, 49332–49340. [Google Scholar] [CrossRef]
- Adade, C.M.; Oliveira, I.R.; Pais, J.A.; Souto-Padrón, T. Melittin peptide kills Trypanosoma cruzi parasites by inducing different cell death pathways. Toxicon 2013, 69, 227–239. [Google Scholar] [CrossRef]
- Freire, K.A.; Torres, M.D.T.; Lima, D.B.; Monteiro, M.L.; Martins, A.M.C.; Oliveira Jr, V.X. Wasp venom peptide as a new antichagasic agent. Toxicon 2020, 181, 71–78. [Google Scholar] [CrossRef] [PubMed]
- El-Dirany, R.; Shahrour, H.; Dirany, Z.; Abdel-Sater, F.; Gonzalez-Gaitano, G.; Brandenburg, K.; Martinez de Tejada, G.; Nguewa, P.A. Activity of anti-microbial peptides (AMPs) against Leishmania and other parasites: an overview. Biomolecules 2021, 11, 984. [Google Scholar] [CrossRef] [PubMed]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A new era of antibiotics: the clinical potential of antimicrobial peptides. International journal of molecular sciences 2020, 21, 7047. [Google Scholar] [CrossRef]
- Robles-Loaiza, A.A.; Pinos-Tamayo, E.A.; Mendes, B.; Teixeira, C.; Alves, C.; Gomes, P.; Almeida, J.R. Peptides to tackle leishmaniasis: Current status and future directions. International Journal of Molecular Sciences 2021, 22, 4400. [Google Scholar] [CrossRef] [PubMed]
- Lewies, A.; Wentzel, J.F.; Jacobs, G.; Du Plessis, L.H. The potential use of natural and structural analogues of antimicrobial peptides in the fight against neglected tropical diseases. Molecules 2015, 20, 15392–15433. [Google Scholar] [CrossRef]
- Price, R.L. Determining the effect of treatment with an exogenous host defence peptide on Mycobacterium marinum-zebrafish infection. 2017.
- Zahedifard, F.; Lee, H.; No, J.H.; Salimi, M.; Seyed, N.; Asoodeh, A.; Rafati, S. Anti-leishmanial activity of Brevinin 2R and its Lauric acid conjugate type against L. major: In vitro mechanism of actions and in vivo treatment potentials. PLoS neglected tropical diseases 2019, 13, e0007217. [Google Scholar]
- Rivas, L.; Luque-Ortega, J.R.; Andreu, D. Amphibian antimicrobial peptides and Protozoa: lessons from parasites. Biochimica et Biophysica Acta (BBA)-Biomembranes 2009, 1788, 1570–1581. [Google Scholar] [CrossRef]
- Salas-Sarduy, E.; Niemirowicz, G.T.; José Cazzulo, J.; Alvarez, V.E. Target-based screening of the Chagas box: setting up enzymatic assays to discover specific inhibitors across bioactive compounds. Current Medicinal Chemistry 2019, 26, 6672–6686. [Google Scholar] [CrossRef]
- Fieck, A.; Hurwitz, I.; Kang, A.S.; Durvasula, R. Trypanosoma cruzi: synergistic cytotoxicity of multiple amphipathic anti-microbial peptides to T. cruzi and potential bacterial hosts. Experimental parasitology 2010, 125, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Sabia Junior, E.F.; Menezes, L.F.S.; de Araújo, I.F.S.; Schwartz, E.F. Natural occurrence in venomous arthropods of antimicrobial peptides active against protozoan parasites. Toxins 2019, 11, 563. [Google Scholar] [CrossRef]
- Richter, A.; Sutherland, D.; Ebrahimikondori, H.; Babcock, A.; Louie, N.; Li, C.; Coombe, L.; Lin, D.; Warren, R.L.; Yanai, A. Associating Biological Activity and Predicted Structure of Antimicrobial Peptides from Amphibians and Insects. Antibiotics 2022, 11, 1710. [Google Scholar] [CrossRef]
- Telleria, E.L.; Tinoco-Nunes, B.; Leštinová, T.; de Avellar, L.M.; Tempone, A.J.; Pitaluga, A.N.; Volf, P.; Traub-Csekö, Y.M. Lutzomyia longipalpis antimicrobial peptides: Differential expression during development and potential involvement in vector interaction with microbiota and leishmania. Microorganisms 2021, 9, 1271. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; He, X.; Zhang, P.; Shen, C.; Mwangi, J.; Xu, C.; Mo, G.; Lai, R.; Zhang, Z. In vitro and in vivo antimalarial activity of LZ1, a peptide derived from snake cathelicidin. Toxins 2019, 11, 379. [Google Scholar] [CrossRef]
- Adade, C.M.; Souto-Padrón, T. Venoms as sources of novel anti-parasitic agents. Toxins and Drug Discovery 2015, 1–31. [Google Scholar]
- Rapposelli, S.; Gaudio, E.; Bertozzi, F.; Gul, S. Protein–protein interactions: Drug discovery for the future. Frontiers in Chemistry 2021, 9, 811190. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The life cycle of Leishmania species and Trypanosoma cruzi. The complete life cycle is illustrated with a comparison of the life cycle of the parasites inside the vector and the host. BioRender.com. was used to generate this Figure.
Figure 1.
The life cycle of Leishmania species and Trypanosoma cruzi. The complete life cycle is illustrated with a comparison of the life cycle of the parasites inside the vector and the host. BioRender.com. was used to generate this Figure.
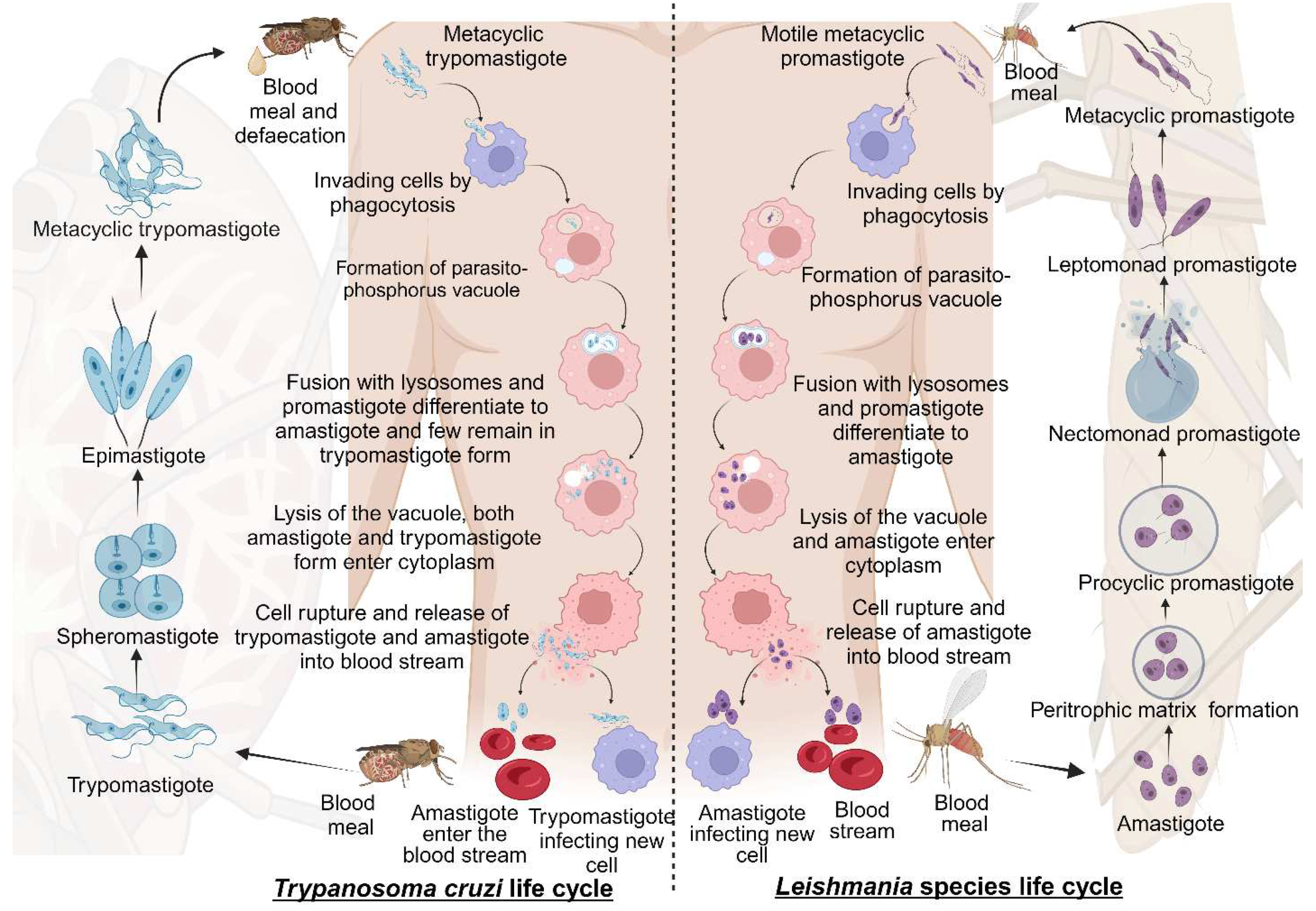
Figure 2.
Clinical presentation of leishmaniasis. Clinical manifestations of individual leishmaniasis types during infection. It is characterized by early and later stages of infection and possible conditions. BioRender.com. was used to generate this Figure.
Figure 2.
Clinical presentation of leishmaniasis. Clinical manifestations of individual leishmaniasis types during infection. It is characterized by early and later stages of infection and possible conditions. BioRender.com. was used to generate this Figure.
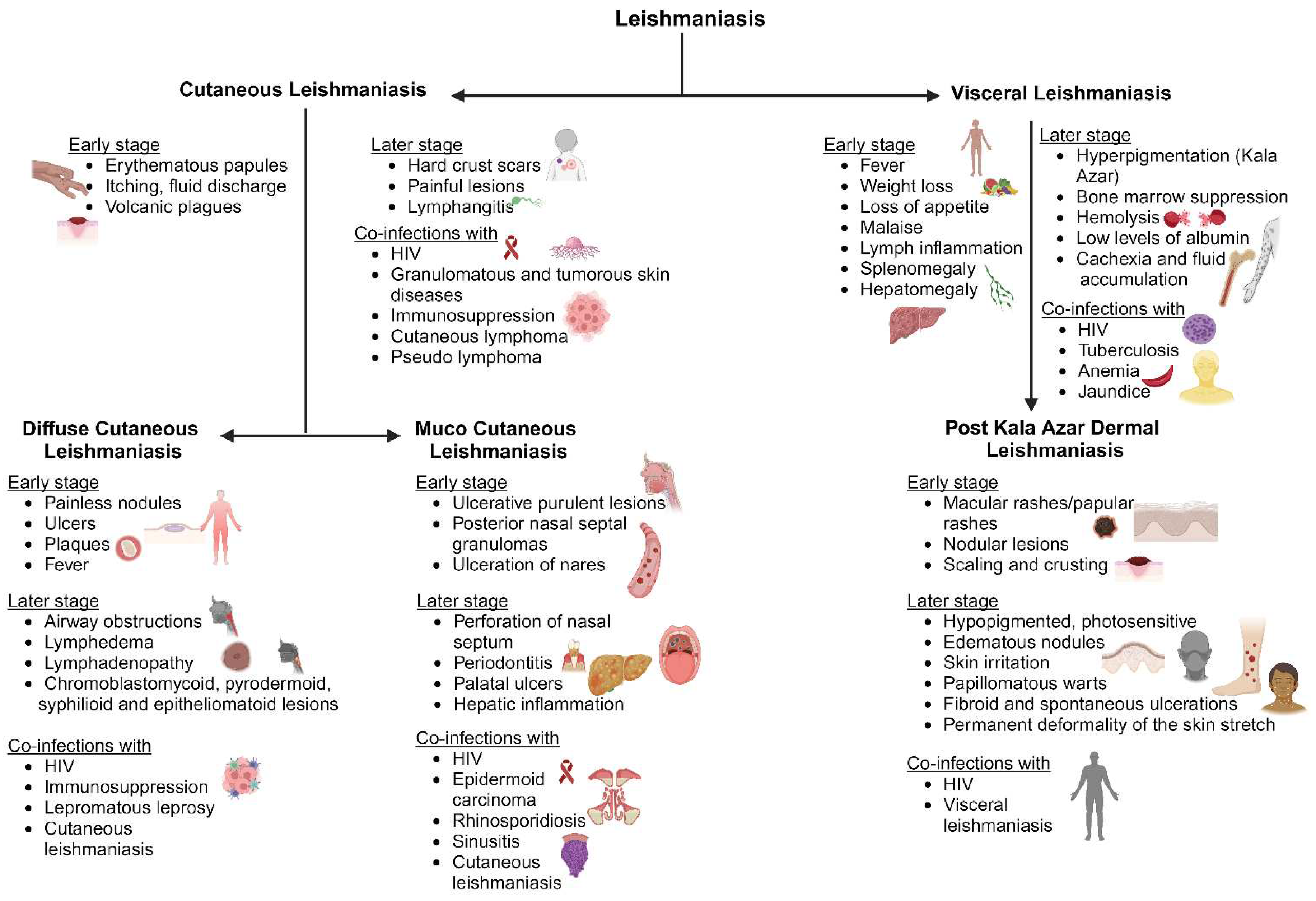
Figure 3.
Diagram of a clinical representation of Chagas disease. Chagas disease has two stages or clinical phases: an acute and a chronic phase. Most infected people (between 70 and 80 percent) remain asymptomatic their entire lives, but in 20 to 30 percent of cases, the disease progresses to cause chronic symptoms. BioRender.com. was used to generate this Figure.
Figure 3.
Diagram of a clinical representation of Chagas disease. Chagas disease has two stages or clinical phases: an acute and a chronic phase. Most infected people (between 70 and 80 percent) remain asymptomatic their entire lives, but in 20 to 30 percent of cases, the disease progresses to cause chronic symptoms. BioRender.com. was used to generate this Figure.
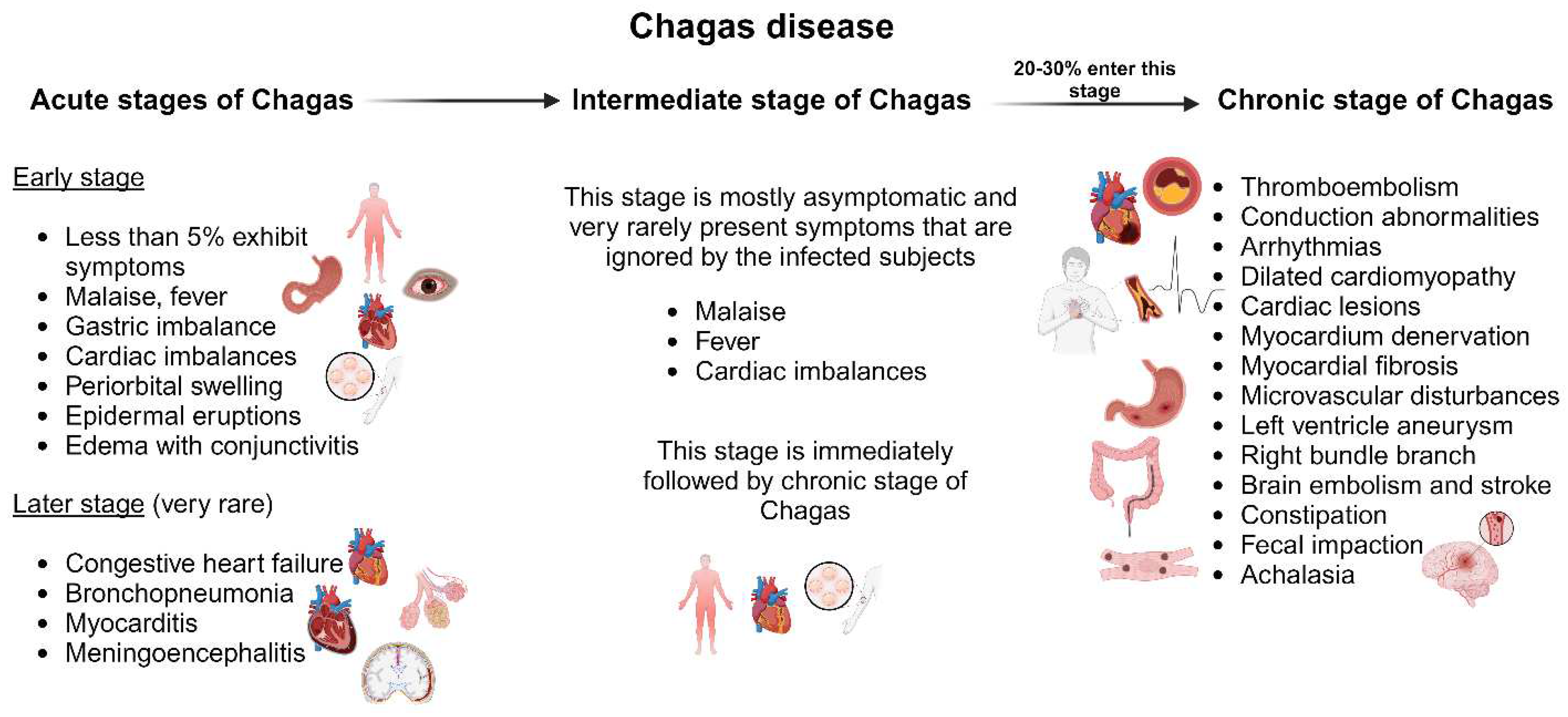
Figure 4.
Chemical structures of currently used drugs against leishmaniasis.
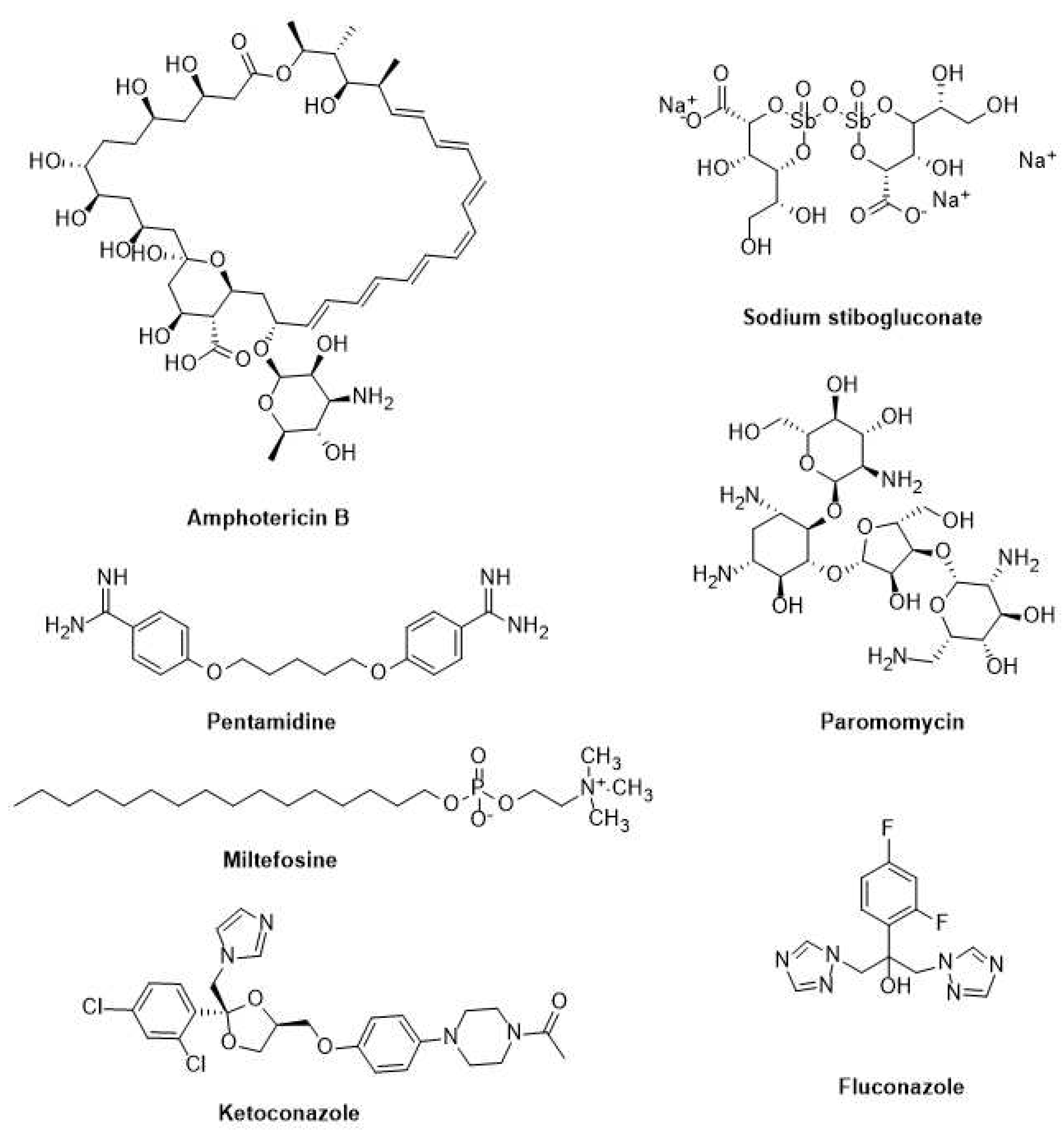
Figure 5.
The chemical structures of currently used drugs against Chagas disease.
Figure 6.
Schematic representations of T. cruzi and Leishmania life cycle associated anti-microbial peptides (AMPs) against each specific developmental form of the parasite. BioRender.com. was used to generate this Figure.
Figure 6.
Schematic representations of T. cruzi and Leishmania life cycle associated anti-microbial peptides (AMPs) against each specific developmental form of the parasite. BioRender.com. was used to generate this Figure.
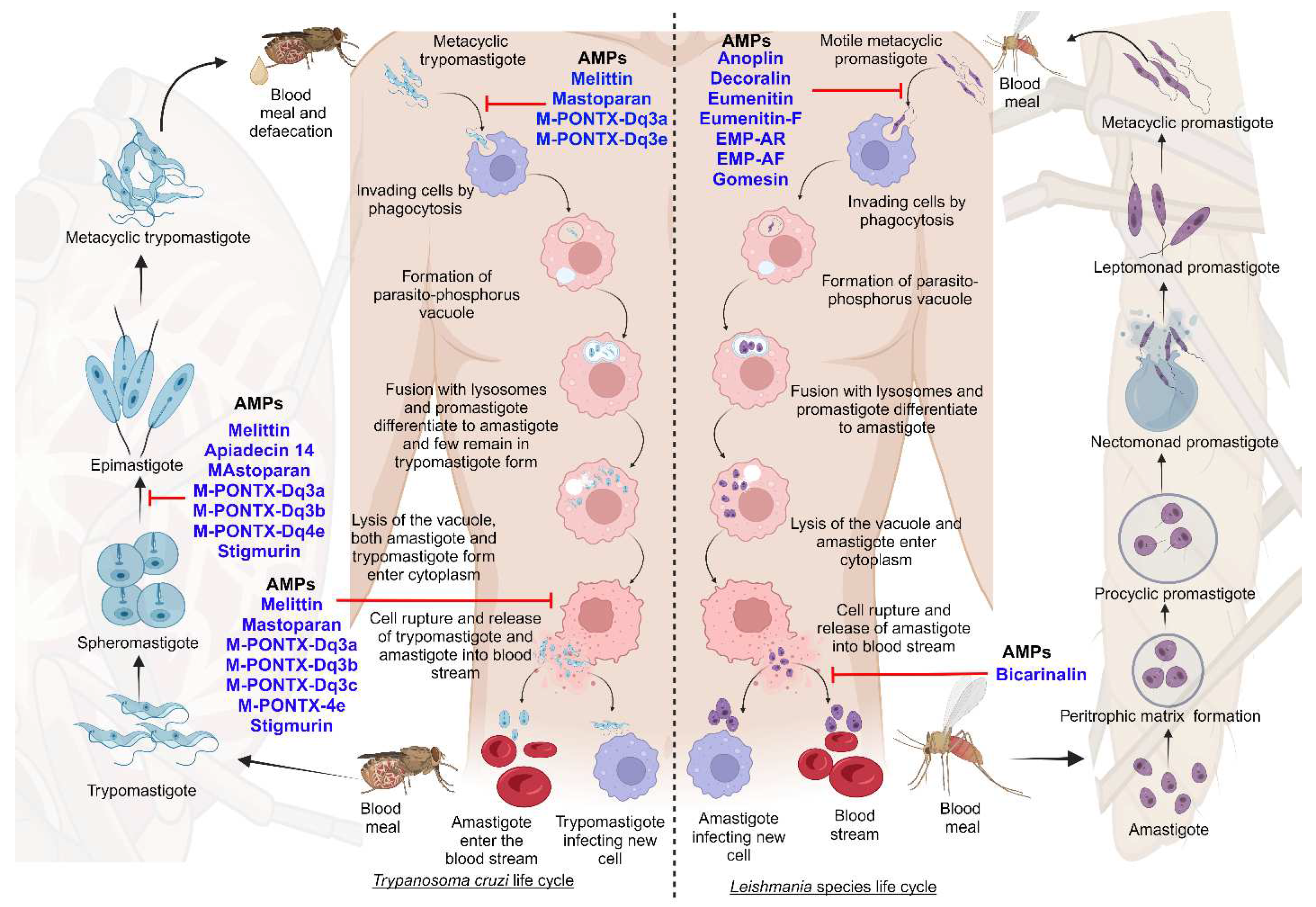

Table 1.
Geographical and species distribution of leishmaniasis and Chagas disease.
| No | Diseases | Species | Types | Geographic distributions | References |
|---|---|---|---|---|---|
| 1 |
Leishmaniasis |
Leishmania (L.) Aethiopica | Cutaneous and mucocutaneous leishmaniasis | Ethiopia, Kenya | [37] |
| 2 | L. Tropica | Visceral and cutaneous leishmaniasis | Eastern and Northern India, Central Asia, East and South Africa, Middle East | [37] | |
| 3 | L. Amazonensis | Cutaneous and mucocutaneous leishmaniasis | Brazil, Bolivia, Venezuela | [38] | |
| 4 | L. Infantum | Visceral and cutaneous leishmaniasis | Mexico, Brazil, Bolivia, Venezuela, Northern Africa, Middle East, Mediterranean regions, Southern Europe, Central Asia | [39] | |
| 5 | L. Donovani | Post Kala azar dermal leishmaniasis, visceral and cutaneous leishmaniasis | India, Myanmar, Nepal, Sri Lanka, Middle East, China, Ethiopia, Kenya, Sudan | [39] | |
| 6 | L. Major | Cutaneous and mucocutaneous leishmaniasis | Central Asia, Middle East, and Central, Northern and West Africa | [40] | |
| 7 | L. Mexicana | Cutaneous and visceral leishmaniasis | United States of America, Venezuela, Ecuador, Peru, Brazil | [38] | |
| 8 | L. Venezuelensis | Cutaneous leishmaniasis | Northern and Southern America, Venezuela | [41] | |
| 9 | L. Braziliensis | Cutaneous and mucocutaneous leishmaniasis | Amazon stretch, Brazil, Southern America, Bolivia, Peru, Venezuela | [42] | |
| 10 | L. Guyanensis | Cutaneous and mucocutaneous leishmaniasis | Southern America, French Guiana, Suriname, Brazil | [43] | |
| 11 | L. Panamensis | Cutaneous and mucocutaneous leishmaniasis | Panama, South and Northern America, Brazil, Ecuador, Columbia, Venezuela | [43] | |
| 12 | L. Lainsoni | Cutaneous leishmaniasis | French Guiana, Peru, Bolivia, Brazil | [44] | |
| 13 | L. Naffi | Cutaneous leishmaniasis | French Guiana, Brazil | [44] | |
| 14 | L. Lindenberg | Cutaneous leishmaniasis | Brazil | [45] | |
| 15 | L. Peruviana | Cutaneous and mucocutaneous leishmaniasis | Peru, Bolivia, Amazon | [46] | |
| 16 | L. Shawi | Cutaneous leishmaniasis | Brazil | [47] | |
| 17 | L. Martiniquensis | Visceral and cutaneous leishmaniasis | Martinique, Thailand, France, Germany, Switzerland, Myanmar | [48] | |
| 18 | L. Siamensis | Visceral and cutaneous leishmaniasis | Central Europe, Thailand, United States of America | [49] | |
| 19 | L. Colombiensis | Visceral and cutaneous leishmaniasis | Columbia | [50] | |
| 20 | African Trypanosomiasis (aka Sleeping Sickness) | T. brucei | Acute and chronic infections | Eastern, Western, Southern and Central Africa | [51] |
| 21 | Chagas disease (aka American trypanosomiasis) | T. cruzi | Acute and chronic infections | Bolivia, Argentina, Paraguay, Ecuador, El Salvador, and Guatemala | [52] |
Table 2.
Current pharmacological treatments against Chagas disease and leishmaniasis.
| Disease | Drug | Route | Advantages | Disadvantages | Toxicity | References |
|---|---|---|---|---|---|---|
| Leishmaniasis | Miltefosine | Oral 5-100 mg/kg/day for 28 days | No hospitalization | Need allometric administration in children | Gastrointestinal complications, teratogenic | [157,158,159] |
| Leishmaniasis | Paromomycin | Parenteral (im) 15 mg/kg/ day for 21 days | Low cost | Poor results against African VL as monotherapy | Pain in the injection site, hepatotoxicity | [160,161] |
| Leishmaniasis | Pentamidine | Slow infusion (iv) 4 mg/kg monthly for 12 months | Use in HIV positive co-infections | Multiple adverse effects | Insulin-dependent diabetes, myocarditis, nephrotoxicity | [162] |
| Leishmaniasis | SbV - paromomycin combination |
Parenteral (im) SbV 20 mg/ kg/day + paromomycin for 17 days | Reduce the number of injections | Require hospitalization | Problems regarding SbV administration | [157,163] |
| Leishmaniasis | Amphotericine B deoxycholate | Slow infusion (iv) 1 mg/kg/day for 30 days |
Effective against SbV resistant strains |
Require hospitalization | Nephrotoxicity | [164,165] |
| Leishmaniasis | SbV -based drugs | Parenteral (im) 20 mg/kg/day for 28-30 days | Low cost | Drug resistance in Bihar (India), PKDL | Pain in the injection site, cardiotoxicity, pancreatitis | [166,167] |
| Leishmaniasis | AmBisome | Slow infusion (iv) 10 mg/kg single dose | Effective at single dose |
Costly, chemically unstable | Fever during infusion, back pain, nephrotoxicity | [168,169] |
| Chagas disease | Benznidazole (BZL) | It is given for 60 days on daily basis at 5-7 mg/kg, and 10 mg/kg for adults and children, respectively | Lower side effects than Nifurtimox, better tolerance by children, and more effective during the acute phase of the disease. | Low solubility, toxic and several side effects | Low bioavailability and drug effectiveness, chronic effects | [170,171,172,173] |
| Chagas disease | Nifurtimox (NFX) | 8-10 mg/Kg daily in three divided doses for adults, and 15-20 mg/kg daily in four divided doses for children during 60 to 90 days | Better tolerance by children and more effectiveness during the acute phase of the disease. | Toxic and have side effects, causes gastrointestinal, maladies (nausea, vomiting, abdominal pain) effects |
Have higher toxicity and adverse effect than BZL, and it affects the pancreases and heart via increasing of oxidative stress | [174.175.176] |
Abbreviations: im - intramuscular, iv - intravenous,.
Table 3.
Studies involving advanced peptides against leishmaniasis and Chagas disease.
| Peptide | Source | Sequence | Study model | IC50 µg/mL | Reference |
| NK-2 | Synthetic peptide | KILRGVCKKIMRTFLRRISKDILTGKK | In vitro | - | [217] |
| Temporizin-1 | Synthetic peptide | FLPLWLWLWLWLWKLK | In vitro | - | [218] |
| Defensin-α1 | Human | ACYCRIPACIAGERRYGTCIYQGRLWAFCC | In vitro | - | [219] |
| Phylloseptin 7 | Phyllomedusa nordestina (Frog) | FLSLIPHAINAVSAIAKHF | In vitro | 0.34 | [220] |
| DS 01 | Phyllomedusa oreades (Frog) | GLWSTIKQKGKEAAIAAAKAAGQAALGAL | In vitro | - | [221] |
| Melittin | Apis mellifera (Bee) | - | In vitro | 2.44 | [222] |
| Polybia-CP | Polybia paulista (Wasp) | ILGTILGLLSKL | In vitro | - | [223] |
| Hmc 364–382 | Penaeus monodon (Shrimp) | NVQYYGALHNTAHIVLGRQ | In vitro | 4.79 | [223] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



