Submitted:
03 September 2023
Posted:
05 September 2023
You are already at the latest version
Abstract
Interstitial lung disease (ILD) is one of the most serious extra-articular complications of rheu-matoid arthritis (RA), which increases the mortality of RA. Because the pathogenesis of RA-ILD remains poorly understood, appropriate therapeutic strategies and biomarkers have not yet been identified. Thus, the goal of this review was to summarize and analyze the reported data on the etiology and pathogenesis of RA-ILD. The incidence of RA-ILD increases with age, and is also generally higher in men than in women and in patients with specific genetic variations and eth-nicity. Lifestyle factors associated with an increased risk of RA-ILD include smoking and exposure to pollutants. The presence of an anti-cyclic citrullinated peptide antibody, high RA disease activity, and rheumatoid factor positivity also increase the risk of RA-ILD. We also explored the roles of biological processes (e.g., fibroblast–myofibroblast transition, epithelial–mesenchymal transition, and immunological processes), signaling pathways (e.g., JAK/STAT and PI3K/Akt), and the his-topathology of RA involved in RA-ILD pathogenesis based on published preclinical and clinical models of RA-ILD in animal and human studies.
Keywords:
Rheumatoid arthritis
; interstitial lung disease
; pathogenesis
1. Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease that is primarily accompanied by articular manifestations, including continuous inflammation and joint deterioration of joints [1]. RA can also affect organs and systems beyond joints. The extra-articular manifestations of RA involve several organ systems, including the skin, eyes, heart, lungs, kidneys, nervous system, and gastrointestinal system [2]. The most common extra-articular manifestation is pulmonary involvement, which affects up to 60% of patients with RA [3,4]. RA-related pulmonary diseases include interstitial lung disease (ILD), bronchiectasis, and pleural diseases, which are generally detected using chest computed tomography (CT) imaging [5].
ILD, which is a pulmonary manifestation of RA, was first described by Ellman and Ball in 1948 [6]. ILD occurs in approximately 10% of the patients [7,8]. The major histopathological patterns observed in patients with RA-ILD include nonspecific interstitial pneumonia (NSIP) and usual interstitial pneumonia (UIP) patterns [9,10,11]. Other less commonly observed patterns include organizing pneumonia (OP) and obliterative bronchiolitis [12,13]. ILD causes lung dysfunction and is the leading cause of death in patients with RA after cardiovascular disease [14,15,16,17]. According to several studies, RA-ILD is a life-shortening disease. Raimundo et al. found that 35.9% of patients died five years after being first diagnosed with RA-ILD, and the median survival was 7.8 years in the United States [18]. Up to 7% of RA-related deaths are associated with RA-ILD [8]. Complications of ILD greatly affect the prognosis of RA because RA-ILD is generally progressive and only restricted therapeutic options are available [19]. Due to a shortage of definitive prognostic indicators or gold standard therapeutic strategies for RA-ILD, high mortality rates have been observed in these patients [20,21]. Therefore, annual screening for ILD is highly recommended for patients with RA [22].
Management of RA-ILD is challenging. Although several therapeutic agents have been proposed, large randomized controlled trials have not yet been conducted, and appropriate guidelines for clinical practice are not available [19]. Antifibrotic medications, including nintedanib, have been shown to inhibit the progression of RA-ILD; however, more clinical studies are required to verify these findings [23,24]. Moreover, antirheumatic medications such as methotrexate are not recommended for the treatment of RA-ILD because they can cause pulmonary toxicity [25,26]. Therefore, it is necessary to develop drugs that can be used to treat both arthritis and pulmonary fibrosis.
The development of novel treatments for RA-ILD requires a better understanding of the molecular mechanisms underlying its pathogenesis. Therefore, an understanding of the risk factors and pathogenesis of RA-ILD is required. Thus, in this review, we describe and analyze the previous and current research on RA-ILD. We provide a thorough overview of several risk factors and mechanisms that contribute to the development of ILD in patients with RA.
2. Risk Factors Associated with RA-ILD
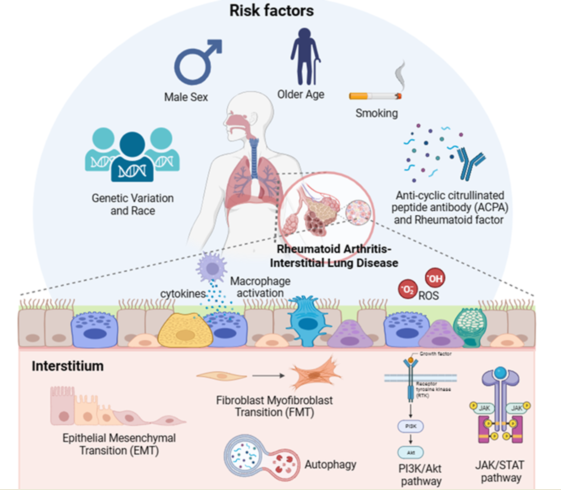
Although the exact mechanism by which RA-ILD occurs remains unknown, genetic factors such as age, sex, race, smoking, pollutants, and autoantibodies, especially rheumatoid factors (RF) and anti-cyclic citrullinated peptides (CCP) antibody (ACPA), have been proposed as risk factors.
2.1. Genetic factors
In an exome sequencing study of patients with RA-ILD, associations between RA-ILD and mutations in familial pulmonary fibrosis (FPF)-linked genes (TERT, RTEL1, PARN, or SFTPC) were identified. These mutations are more frequently observed in patients with RA-ILD than in controls [27]. In a large RA cohort study, the MUC5B promoter variant was associated with higher odds of RA-ILD by more than two-fold. As this variant is less common in African-American patients, its existence in this population causes a >4-fold increased odds of RA-ILD [28]. This association has been reported in previous studies. Evidence that the MUC5B promoter variant rs35705950 can serve as a strong risk factor for RA-ILD, especially in patients with UIP patterns, has been demonstrated [29]. A large observational cohort study demonstrated that the risk of ILD was 16.8% for MUC5B carriers and 6.1% for MUC5B non-carriers among patients with RA. This difference between risks began to appear at 65 years of age, with an increased risk among men [30]. Hayashi et al. demonstrated that the rs6578890 single nucleotide polymorphism (SNP) in PPFIA-binding protein 2 (PPFIBP2) gene was significantly associated with the occurrence of RA-ILD in a genome-wide association study [31]. Furukawa et al. conducted a study on the allelic association of human leukocyte antigen (HLA) with RA-ILD. In 450 Japanese patients with RA, HLA-DRB1*04 (a shared epitope) and DQB1*04 were significantly associated with a reduced risk of ILD. In contrast, DRB1*16, DR2 serological group (DRB1*15 and *16 alleles), and DQB1*06 were significantly associated with the risk of ILD [32]. Moreover, in the Japanese population, HLA-DRB1*1502 was significantly associated with RA-related pulmonary fibrosis [33]. Le Guen et al. reported the case of a 37-year-old female patient with a heterozygous NKX2.1 mutation associated with RA-ILD and a histological pattern of lymphocytic interstitial pneumonia [34]. From a genome-wide association study (GWAS) meta-analysis, variants of RPA3-UMAD1 were identified as a novel risk factor for RA-ILD in the Japanese population [35]. rs2609255G is a risk allele for UIP and ILD in Japanese patients with RA. FAM13A rs2609255 was significantly associated with UIP in male and older patients with RA [36]. Similarly, in patients with RA in northern Sweden, FAM13A was associated with ILD, as analyzed using GWAS [37].
2.2. Age
Physiological changes occur in the lungs with aging and induce deterioration of lung function and increased susceptibility to diseases. Age-related declines, such as immunosenescence and infammaging can reduce the regenerative capacity of the lung and trigger lung fibrosis [38,39]. Older age serves as a critical risk factor for RA because: 1) patients with RA live longer with better medical management and 2) more people are being diagnosed with RA at an older age. Because elderly patients with RA generally have other age-related diseases, they require different treatments than younger patients with RA [40]. An inception cohort of patients with RA with a 20-year follow-up reported that the incidence of RA-ILD was associated with older age [41]. Lai et al. found that the average age of patients with RA-ILD was higher than that of patients with RA without ILD, implying that RA-ILD is associated with advanced age [42]. RA-ILD is most commonly diagnosed at age 50–59. Age has been found to be an independent risk factor for the development of ILD in previous cohort studies [43,44].
2.3. Sex
In multivariate analysis, male sex was identified as a variable significantly associated with RA-ILD [45,46]. Male sex has also been identified as an independent predictor of co-occurring RA and lung diseases such as ILD, bronchiectasis, and nodules [47]. Men have a more than two-fold higher risk of RA-ILD compared to women [48]. Male sex was also significantly associated with unfavorable outcomes in patients with RA-ILD, with an HR of 2.52 [49]. In a retrospective study of patients with RA-ILD, although most patients with RA were female, the ratio of males in the patients with RA-ILD was significantly higher than that in patients with RA without ILD [42]. Most patients with RA are women; thus, these findings can be considered paradoxical but are not explained by potential confounding factors, such as cigarette smoking [50].
However, Olson et al. reported that RA-ILD-associated mortality rates decreased in men but increased in women between 1988 and 2004 [8]. Female sex was also an independent risk factor for the development of RA-ILD in a multi-ethnic Malaysian cohort study [51].
In a large cohort study, the early onset of menarche and irregular menstrual cycles were significantly associated with an increased risk of RA, whereas breastfeeding for >12 months was protective against RA risk [52]. In a population-based cohort study conducted in Norway, the total time of lactation was associated with reduced RA mortality, which was close to a dose-response relationship [53]. These results could partly explain why the RA risk differs according to sex.
2.4. Race
Jeganathan et al. demonstrated that RA- and RA-ILD-associated mortality rates vary substantially depending on the patient race. In general, RA-ILD-associated mortality was highest in Hispanic individuals (26% higher than white individuals), followed by white individuals, and lowest in black individuals (27% lower than white individuals) in the United States between 2005 and 2018 [54]. Native American individuals are more susceptible to RA than Caucasian individuals [55]. In a multi-ethnic Malaysian cohort study, Indian patients with RA demonstrated a significantly increased risk of developing RA-ILD [51]. Moreover, drug-induced interstitial pneumonia was significantly more frequent in the Japanese population than in people of other ethnicities, and acute exacerbation of IPF also occurred more frequently in the Japanese population than in other ethnic backgrounds [56,57].
2.5. Smoking
Previous reports have identified smoking as a predictive factor for RA-ILD [46,58,59,60,61]. Saag et al. demonstrated that the odds ratio (OR) for RA-ILD in smokers (>25 packs/year) relative to nonsmokers was 3.8 [62]. Smoking ≥30 packs/year significantly increased the risk of RA-ILD, while a lower level of smoking did not increase this risk [63]. The largest study on RA-ILD in the UK demonstrated that smoking was highly associated with ILD in males, which could explain the higher incidence of RA-ILD in men than in women [48].
RA-ILD is associated with smoking among patients with HLA–DRB1 shared epitope (SE) [45]. A population-based case-control study in Sweden showed that a history of smoking and carrying two copies of HLA-DR SE genes increased the risk of RA by 21-fold when compared to non-smokers without SE genes. It has been suggested that smoking triggers an immune response to citrulline-modified proteins by interacting with HLA-DR SE genes [64]. The chemical constituents of cigarette smoke can also trigger an immune response, and the resultant serum autoantibodies against citrullinated proteins accumulate in the lungs, causing inflammation and injury to epithelial cells, finally resulting in ILD [65]. Tobacco smoking upregulates the expression of peptidylarginine deiminase 2 (PAD2) in the bronchial mucosal and alveolar compartments with subsequent production of citrullinated proteins in pulmonary tissues [66].
However, contradictory results have been reported. Mori et al. showed that the significant association between smoking history and ILD acquired from univariate analysis in patients with RA with lung problems (ILD or airway disease) was not verified by multinomial logistic regression analysis [67].
2.6. Pollutants
A recent study revealed that higher levels of ambient air pollutants, including PM2.5, PM10, sulfur dioxide (SO2), and nitrogen dioxide (NO2), increase the risk of hospitalization in patients with RA-ILD [68]. Silica exposure in the workplace environment is also suggested to be associated with an increased risk of developing RA and related lung manifestations [69,70,71]. Air pollutant exposure is positively associated with the development of several pulmonary diseases, especially ILDs [72]. Few studies have shown that O3 exposure causes T cell reaction dysregulation and that quenching effects have an inverse association with the incidence of ILDs [73,74,75]. By analyzing newly onset patients with RA-ILD from the United States health care insurance database (MarketScan) (2011–2018), it was identified that exposure to elements of PM2.5, especially ammonium, mineral dust, and black carbon, caused a higher ILD risk than the other PM2.5 [76].
2.7. Anti-cyclic citrullinated peptide antibody
Anti-cyclic citrullinated peptide antibody (ACPA) is a group of auto-antibodies that bind to citrullinated epitopes specific to RA, occurring years before the clinically evident disease appears [77]. ACPA is commercially available and typically used in clinical applications [78]. Previous studies have shown that the specificity and sensitivity of ACPA detection for the diagnosis of RA are 96–99% and 47–88%, respectively, depending on the features of the RA population [79,80]. ACPA titers are significantly increased in patients with RA-ILD compared to those in patients with RA without ILD [16,81,82], suggesting that abnormal citrullination may contribute to the development of lung fibrosis in patients with RA. Klester et al. found that the ACPA titer was the most strongly associated risk factor for RA-ILD in a univariate analysis [46]. In addition, patients with high-positive ACPA titers (>15 U/mL) showed higher risk of prevalent ILD (OR, 1.91; 95% confidence interval [CI], 1.04–3.49) than that in ACPA-negative patients [83].
A systematic review and meta-analysis showed that the existence of ACPA was significantly associated with RA-ILD incidence, and autoantibody levels was significantly increased in patients with RA-ILD compared to patients with RA alone. Nevertheless, the authors declared that their findings may be limited in application because of the heterogeneity of the included studies [78]. In a previous study performed in the UK with 230 patients with RA-ILD, ACPA was identified as the strongest predictor of RA-ILD [48]. Giles et al. found that the levels of all specific ACPA were higher in patients with radiographic evidence of ILD than in those without RA-ILD. In contrast, the levels of antibodies against non-citrullinated proteins were not higher in patients with ILD [84]. A meta-analysis reported that serum ACPA positivity is strongly associated with the risk of RA-related pulmonary diseases, especially RA-related ILD and idiopathic pulmonary fibrosis (IPF) [85]. ACPA is highly specific or independently related to the development of extra-articular manifestations such as ischemic heart disease [86], type 1 diabetes [87], inflammation of a serous membrane [88], and subclinical atherosclerosis in patients with RA [89]. In several cohorts, ACPA were positively associated with IPF and ectopic lymphoid aggregates in the lungs [90]. Higher ACPA titers have also been demonstrated as predictors of ILD in a multi-ethnic Malaysian cohort study [51]. Moreover, in Korean patients with RA, the ACPA titer ratio was higher in the airway disease and ILD groups than that in the control group [91].
In addition, a significantly higher positive association of ACPA has been reported in patients with RA-ILD than in patients with RA alone [92]. Citrullinated proteins accumulate in bronchoalveolar lavage fluid of patients with RA-ILD and IPF [93]. A previous study showed that increased anti-CCP2 antibody levels were an independent risk factor for the co-occurrence of RA and lung diseases, including ILD, bronchiectasis, and nodules [47]. Moreover, the ACPA titer is closely associated with joint damage and airway lesions in RA [94,95,96]. ACPA was highly associated with RA-ILD in both sexes [48]. Restrepo et al. found that variables including ACPA and RF were significantly associated with RA-ILD in multivariable analysis. These associations became stronger when the concentrations of ACPA and RF increased [45]. Citrullination not only exists in the synovial tissue of patients with RA but is also present in the affected extra-articular tissue. Bongartz et al. demonstrated that nearly 50% of the patients with RA-associated interstitial pneumonia showed evidence of citrullination in their lung tissue [97]. Samara et al. identified that the citrullination pathway was upregulated in bronchoalveolar lavage cells of patients with RA-ILD. In particular, the mRNA and protein expression levels of PAD4, which catalyzes citrullination, are elevated in RA-ILD. Moreover, the levels of citrullinated proteins were positively correlated with the levels of PAD4 and ACPAs in RA-ILD compared to the control group [93]. Moreover, the citrullinating enzyme PAD2 is upregulated in the lungs and associated fibroblasts of patients with RA-ILD when compared to the control group. PAD2 serves as a pro-fibrotic mediator in fibroblasts of patients with RA-ILD, and its suppression reduces fibroblast–myofibroblast transition (FMT) and extracellular matrix formation [98].
The anti-mutated citrullinated vimentin antibody (MCV) titer in patients with RA-ILD was significantly higher than that in patients with RA without ILD. However, there was no significant difference in ACPA levels between the two groups [99].
However, controversy over the association of anti-cyclic citrullinated peptide and RF with the development of RA-ILD still exists [47,100,101]. In a previous study, no significant differences were observed between the levels of ACPA and RA-associated pulmonary involvement, including ILD and bronchiolitis [100]. This discrepancy can be attributed to the obscure distinction between ILD and idiopathic pulmonary fibrosis owing to their unclear definitions and the relatively small number of studies included in the systematic review [85].
2.8. Disease activity of RA
It is well established that low disease activity or remission of disease activity is correlated with better therapeutic results for RA [102,103]. Disease activity has been identified as a risk factor for RA-ILD using the Disease Activity Score in 28 joints (DAS28) [104] or Clinical Disease Activity Index (CDAI) [105] as estimation methods.
Zhuo et al. showed that patients with RA-ILD had a greater disease burden than those with RA without ILD. RA disease activity may worsen after ILD diagnosis compared to the preclinical ILD status and patients with RA alone [106]. Sparks et al. found that moderate and high disease activity was associated with a two-fold elevation in RA-ILD risk compared to remission/low disease activity. Reducing systemic inflammation by treating RA symptoms may prevent the manifestation of RA-ILD [104]. Moderate or high joint disease activity was considered as an independent factor associated with RA-ILD, with an OR of 3.03 [107]. Patients with RA-ILD with high disease activity during follow-up showed decreased survival compared to those with moderate and low disease activity [108]. Moreover, in a nested case-control study that matched incident RA-ILD to RA without ILD, moderate to high disease activity was associated with an increased risk of RA-ILD, implying that the regulation of disease activity may reduce risk [63]. Another case-control study demonstrated that a CDAI score >28 was associated with the incidence of RA-ILD [105]. These results suggest that controlling systemic inflammation is important for preventing RA-ILD development. Thus, regular measurement of clinical disease activity may help identify patients with RA at a higher risk for complications [106].
2.9. RF
The RF titers showed a monotonic relationship with the prevalence of ILD. Only patients with RF concentrations >90 IU/mL are at increased risk of incident RA-ILD [83]. Smoking and high RF titers (hazard ratio [HR], 1.09; 95% CI, 1.001–1.11) have been identified as risk factors for RA-ILD development [46]. Moreover, RF titers are closely related to joint injuries and RA airway lesions [94,95,96].
RF positivity has been identified as a risk factor for RA-ILD [61]. RF positivity was also identified as an independent predictor of RA-ILD development in a multiethnic Malaysian cohort study [51]. In a retrospective cohort study of patients with RA-ILD, high-titer RF seropositivity was significantly correlated with mediastinal lymph node (MLN) enlargement, CT honeycombing, and reduced transplant-free survival [109].
It was demonstrated that RF-immunoglobulin (Ig)A levels >200 RU/mL (HR, 3.17, P = 0.012) were regarded as a predicting factor for poor prognosis of RA-ILD from the multivariate analysis [49]. In addition, the positive ratio of RF-IgA was higher in patients with RA-ILD than that in patients with RA without ILD, whereas the positive ratios of RF-IgG, RF-IgM, anti-AKA antibodies, anti-APF antibodies, and ACPA were not significantly different between the two groups [99].
2.10. Combination of factors
A combination of age, sex, smoking status, RF, and ACPA was highly associated with the incidence of RA-ILD (areas under the curve are 0.88 for subjects enrolled in the Brigham and Women’s Hospital Rheumatoid Arthritis Sequential Study [BRASS] and 0.89 for American College of Rheumatology [ACR] cohorts) [82]. Patients with a combination of RF/ACPA seropositivity had a higher possibility of ILD than seronegative patients (OR, 2.90; 95% CI, 1.24–6.78). Nonetheless, combined RF/ACPA seropositivity was not associated with an increased risk of incident ILD [83].
3. Pathogenesis of RA-ILD
Although several risk factors associated with the development of RA-ILD have been identified, the pathophysiological connections between these factors and the pulmonary changes remain unclear.
The pathogenesis of RA-ILD is primarily related to aberrant tissue reactions in the alveolar wall and lung parenchyma [110]. The first step in the pathogenesis of RA-ILD is the deterioration of airway and alveolar epithelial cells [111]. Continuous damage to epithelial cells causes the activation of immune cells, including neutrophils, dendritic cells, and macrophages, leading to the excessive accumulation of extracellular matrix (ECM) components in lung tissues [111].
Only a few studies have identified the mechanism of RA-ILD, which is mainly due to the limitations in establishing an animal model. As the construction of an animal model of RA representing the pathology of both articular and pulmonary manifestations is challenging, experimental studies using these models have been restricted [112,113]. Distinct from bleomycin (BLM)- or collagen-induced arthritis (CIA)-induced pulmonary fibrosis animal models, the murine model established by the combination of CIA and BLM showed significant interstitial fibrosis, involving dilatation of air gaps and thickening of interstitial walls. However, the CIA+BLM animal model is characterized by combined features of arthritis and pulmonary fibrosis and can be utilized to better study the progression of RA-ILD [114].
3.1. FMT
The activation of fibroblasts to become myofibroblasts and escape the quiescent state is defined as FMT and serves as a key step in fibrotic pathogenesis [115]. FMT is generally divided into two stages. After the fibroblast is activated to become a proto-myofibroblast phenotype, it enters the second stage to complete cell phenotype transition [116,117]. Myofibroblasts are derived from the excessive activation and aberrant differentiation of fibroblasts by FMT and are known to be responsible for fibrosis [118,119]. Lung epithelial cellular senescence orchestrates lung injury by stimulating FMT [120,121].
Continuous transforming growth factor (TGF)-β signaling is the major underlying mechanism of FMT and fibrosis [122]. Dysregulation of TGF-β1 and TGF-β2 signaling has been associated with the development of fibrotic diseases, including IPF [123] and systemic sclerosis (SSc)-ILD [122]. TGF-β1-induced FMT causes aberrant hyperplasia of collagen in lung interstitial tissues, which results in decline of lung function [124,125]. Smad3, a downstream effector of TGF-β1, is a key mediator of fibrotic pathogenesis [126]. Thus, FMT inhibition plays a preventive role against ILD progression [127]. Following stimulation with TGF-β1 on human lung fibroblasts (HLF)-1, myofibroblast transition was increased compared to the control group, as determined by a transwell assay. Additionally, staining of mouse embryonic fibroblast (MEF) cells showed the elevated productions of collagen, fibronectin, and α-smooth muscle actin compared to the control group [128]. Moreover, in HLFs, TGF-β induced upregulation of dedicator of cytokinesis 2 (DOCK2) expression at both transcriptional and post-translational levels. DOCK2 upregulation promotes FMT and plays a role in the development of pulmonary fibrosis [129].
In murine lung fibroblasts, signal transducers and activators of transcription (STAT)3 contributed to the lung fibrosis by stimulating interleukin (IL)-6- and TGF-β1-mediated myofibroblast differentiation [130]. Furthermore, inhibition of STAT3 in fibroblasts showed reductions in TGFβ1-induced FMT and alleviated skin fibrosis in mouse models of systemic sclerosis [131]. It was also demonstrated that fibroblasts with high STAT3 activity were largely found in the dense fibrotic areas of IPF lungs, and showed impaired expression of α-SMA at baseline, accompanied by upregulation of ECM proteins and TGF-β-mediated signaling and downregulation of several key adhesion molecules [132]. Milara et al. also found that the dual inhibition of p-STAT3 and p-JAK2 in lung fibroblasts of patients with IPF could alleviate TGF-β1 and IL-6/IL-13-induced FMT [133].
Wu et al. demonstrated that pirfenidone, a therapeutic agent for IPF, suppresses FMT in lung fibroblasts from patients with RA-ILD by downregulating activating transcription factor 3 (ATF3) [134]. It was also found that downregulated expression of cyclooxygenase-2 (COX-2) and prostaglandin E2 (PGE2) in human lung myofibroblasts is associated with FMT and epithelial–mesenchymal transition (EMT) and may have key roles in IPF development [125].
3.2. EMT
EMT is a cellular process of which epithelial cells acquire mesenchymal features while losing the epithelial features [135]. EMT has significant roles in the development and progression of several pulmonary diseases [136,137,138]. Tubular epithelial cells can be transformed into fibroblasts via EMT in adult kidneys [139]. EMT is involved in the accumulation of myofibroblasts and subsequent deposition of the ECM, which are associated with ILD progression [140]. Mechanisms related to EMT have been suggested to contribute to the development of Leflunomide-induced ILD [141].
From a recent study using a RA-ILD mice model established by CIA combined with BLM-induced pulmonary fibrosis, EMT was upregulated through the TGF-β-SMAD2/3 signaling pathway in lung tissues [142]. In D1CC×D1BC transgenic mice of the RA-ILD model, the expression of Pad4 and citrullinated peptides in the lungs was upregulated, which led to an increase of EMT in epithelial cell and fibrosis [143]. In addition, M1 macrophages promote myofibroblast apoptosis and degrade the ECM via matrix metalloproteinase (MMP) activation. On the other hand, M2 macrophages activate fibroblasts by the secretion of TGF-β1 and platelet derived growth factor (PDGF) and secrete tissue inhibitors (TIMPs) and inhibit ECM degradation [144]. An imbalance in Th1/Th2 cells has an impact on pulmonary fibrosis. Specifically, Th1 and Th22 cells inhibit fibrosis, whereas Th2, Th9, Th17, and Tregs worsen fibrosis [144].
MMPs, which play key roles in EMT, are proteases involved in ECM degradation and are cell surface receptors [145]. Serum MMP-3 protein levels are correlated with RA disease activity [146]. MMP-3 promotes fibrogenesis by EMT activation [147]. MMP-3 levels are primarily upregulated in bronchial and alveolar epithelial cells, interstitial fibroblasts, alveolar macrophages, and leukocytes in IPF lungs [147,148]. Thus, it has been well described that MMP-3 is involved in the development of RA and lung fibrosis [149]. It has also been demonstrated that MMP-7 can be used as a biomarker of RA-ILD [150]. Another study revealed that MMP-7 is associated with RA-ILD, and risk factors (age, sex, smoking, RF, and ACPA) are known to be related to RA [82]. In a cohort of Korean patients with RA-ILD, serum MMP-7 levels were negatively correlated with the diffusing capacity for carbon monoxide (DLCO) and forced vital capacity (FVC) measured using the pulmonary function test (PFT). Additionally, MMP-7 levels correlated with the semi-quantitative grade of CT. Thus, the authors suggested that MMP-7 can be used to measure the functional and anatomical status of lungs in patients with RA-ILD [151].
3.3. Immunological pathways for production of different cytokines
Chronic and continuous damage to the airway mucosa and distal lung tissue during the preclinical stages of RA can trigger innate immune and inflammatory reactions [152]. Furthermore, the constant immune stimulation and inflammation induced by RA can accelerate abnormal fibroproliferative responses [153].
Several cytokines are involved in RA-ILD pathogenesis. From the cytokine profile analysis, MCP-1/CCL2 and SDF-1α were associated with RA-ILD, and IL-18 levels were associated with RA-ILD and more prominent progressed lung disease [154]. Tumor necrosis factor (TNF)-α is a major proinflammatory cytokine associated with the pathogenesis of interstitial lung involvement. TNF-α triggers fibroblast proliferation and the secretions of PDGF-β, TGF-β, cytokines, and chemokines [16]. TNF-α inhibitors are often used to treat RA [155]. Nonetheless, an increasing number of studies have suggested that TNF-α inhibitors are involved in ILD pathogenesis and may cause pulmonary toxicity [156]. Zhang et al. found that IL-23 serves as an initiator of fibrogenesis in RA-ILD by stimulating EMT in transitioning alveolar epithelial type I cells. IL-23 induces cells to acquire invasive properties, deposit ECM, and resist apoptosis, leading to the production of fibroblast foci in fibrotic ILD, particularly in RA [157]. Microarray profiling of primary adult pulmonary fibroblasts cultured from patients with scleroderma-associated ILD and IPF showed that IL-11 was upregulated [158].
Serum levels of IL-11 are significantly upregulated in patients with RA, which is associated with the development of ILD and disease activity, suggesting that IL-11 may be involved in the pathogenesis of RA and/or RA-ILD. It was also shown that serum IL-11 levels were positively correlated with DAS28 in patients with RA [159]. Similar results have been reported by Chung et al. who showed that patients with RA in the remission stage showed lower serum levels of IL-11, which correlated with DAS28 [160].
An animal study demonstrated that IL-23p19, IL-17A, and IL-17RA signaling mediate pulmonary inflammation. In addition, γδ T cells was identified as the key source of early IL-17A that may be involved in the mechanism by which IL-1 and IL-23 mediate pulmonary fibrosis [161]. It was also reported that the histone deacetylase HDAC3 triggered the development of RA-ILD fibrosis by upregulating miR-19a-3p-mediated IL17RA in an RA-ILD mouse model induced by zymosan [162]. From a RA-ILD mice model developed by CIA with BLM-induced pulmonary fibrosis, expressions of TNF-α and IL-6 in the ankle joints of mice were upregulated compared to the control group [142]. Yang et al. observed the significantly increased levels of TNF-α, IL-6, and IL-1β and decreased levels of IL-10 in serum and lung tissues of an RA-ILD rat model [163].
Through proteomic profiling, 234 proteins were differentially expressed between the RA-ILD and RA-without-ILD groups. Gene set enrichment analysis (GSEA) proteins affecting these gene sets in RA-ILD included several proteins involved in lung fibrosis: cytokines (CCL18 and IL-17), chemokines (CXCL12 and CCL5), FGF family members (FGF4 and FGF7), and S-type lectin galectin-3 (LGALS3) [164]. The level of CXCL13, a B cell chemoattractant, correlates with the degree of inducible bronchial-associated lymphoid tissue (iBALT) in RA-ILD. Nonetheless, the exact mechanism underlying these chemokines are involved in the development of RA-ILD has not been clarified [16]. Sendo et al. recently found CD11b+Gr-1dim tolerogenic dendritic cell-like cells, which are distinctive suppressive myeloid cells that differentiate from monocytic myeloid-derived suppressor cells (MDSCs) in SKG mice with ILD [165]. In addition, the authors demonstrated that tofacitinib triggered the expansion of MDSCs in the lungs and inhibited ILD progression in SKG mice [166].
Xu et al. showed that the serum level of soluble programmed death molecule-1 (sPD-1), a common immunosuppressive member on the surface of T cells, in patients with RA-ILD were significantly elevated compared to that in patients with RA without ILD and healthy controls [167]. Other studies have also reported that sPD-1 level are upregulated in patients with RA and correlated with disease activity [168,169].
Furthermore, heat shock proteins (HSPs) such as Hsp60, Hsp70, Hsp90, and gp96, are involved in immune reactions including antigen presentation and activation of macrophages, lymphocytes, and dendritic cells [170]. Heat shock proteins have been reported to have inhibitory effects on the pathogenesis of ILD, as demonstrated in a mouse model of BLM-induced pulmonary fibrosis [171,172]. It was shown that anti-citrullinated HSP90β induced significantly increased interferon (IFN)-γ production in patients with RA-ILD compared to patients with RA without ILD [173]. Previous studies have also reported that anti-citHSP90 antibody positivity can identify RA-ILD with high specificity (>90%) [174]. Sibinska et al. revealed that HSP90 has a direct effect on the TGF-β1 signaling pathway and that inhibition of HSP90 decreases lung fibrogenesis and fibrosis progress in mice model [175].
3.4. Oxidative stress
Autoimmune diseases are also associated with oxidative stress [176]. The levels of oxidative damage markers, including malondialdehyde (MDA) and thiobarbituric acid reactive substances (TBARS) in patients with RA was higher than those in healthy individuals [177,178,179]. The lungs are particularly vulnerable to oxidative stress because of their anatomy, location, and function [180,181,182]. Oxidants can affect the ECM features [183]. Both reactive oxygen species (ROS) and reactive nitrogen species (RNS) affect the degradation and turnover of the ECM [184,185]. In the lung tissues of patients with IPF, inflammatory cells, fibroblasts, and myofibroblasts also produce high levels of ROS and RNS in response to cytokines and growth factors and contribute to the development of fibrosis [186,187]. Mitochondrial DNA mutations, consecutive respiratory chain dysfunction, and the resulting ROS accumulation have also been shown to be involved in the pathogenesis of ILD [188].
Terasaki et al. showed that oxidative stress was induced in the fibrotic lung lesions of a RA-ILD model in DICC mice, which was similar to the findings in patients with RA-ILD. In a mouse model of RA-ILD, oxidative stress was induced by increased levels of serum lipid peroxide (LPO) and 8-hydroxydeoxyguanosine (8-OhdG)-positive cells. Chronic and inflammatory interstitial pneumonia was observed with increased levels of TNF-α, IL-6, and TGF-β in the lungs [189]. Wang et al. found that the serum nuclear factor erythroid 2-related factor 2 (Nrf2) levels in patients with RA was significantly associated with ILD, and that the Nrf2-related antioxidant pathway was involved in the development of oxidative stress-mediated RA-ILD [190].
3.5. Autophagy
Bao et al. found that in RA-ILD, the level of autophagy was increased in the early stages, whereas it was suppressed in the later stages in fibrotic lungs using a CIA + BLM RA-ILD mouse model [114]. Vasarmidi et al. revealed that the expression of BECLIN1, which helps in the initiation of autophagosome formation, was significantly upregulated in bronchoalveolar lavage fluid (BALF) cells from patients with RA-ILD compared to that in patients with IPF. Nonetheless, other major molecules involved in the autophagy pathway were comparable between patients with IPF and RA-ILD [191].
Several studies have shown that autophagy is dysregulated in IPF lungs, highlighting the need for further research [192]. Araya et al. found that patients with IPF show insufficient levels of autophagy, which may result in the senescence of alveolar epithelial cells and differentiation of FMT [193]. Suppression of autophagy caused by reduced expression of forkhead transcription factor O subfamily member 3a (FoxO3a) increases the viability of IPF fibroblasts. Thus, the dysregulation of autophagy induced by FoxO3a suppression may mediate IPF development [194]. Similarly, Patel et al. revealed that autophagy was not induced by IPF and that several activators of autophagy were significantly induced. The authors’ in vitro experiments demonstrated that the pro-fibrotic mediator, TGF-β1, may have roles in suppressing autophagy [195].
Autophagy was inhibited by upregulation of the TGF-β1-Smad3/ERK/P38 signaling pathway, which lead to an increase of activated myoblasts and collagen accumulation in BLM-induced pulmonary fibrosis mice model [196,197]. It was also demonstrated that the levels of autophagy-related markers such as LC3I/II and beclin-1 were downregulated in the lungs of rats with BLM-induced fibrosis [133]. It has also been demonstrated that suppression of miR-33 in macrophages alleviates lung fibrosis by augmenting autophagy in in vivo and ex vivo models of IPF [198].
3.6. Janus kinase/signal transducers and activators of transcription pathway
Previous studies have demonstrated that inhibition of the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway was effective in protecting against RA [199]. RA-ILD has also been associated with the JAK/STAT pathway [200]. JAK/STAT signaling is upregulated in fibrotic disorders of several organs, including the lungs, and it has been reported that tofacitinib, a JAK inhibitor, is effective in a murine model of ILD [166]. In a post hoc analysis of data from 21 tofacitinib clinical trials, the incidence rate of ILD was 0.18 per 100 patient-years after tofacitinib treatment [201]. Using a CIA mouse model, it has been reported that the administration of tofacitinib, a potent and selective inhibitor of JAK, significantly attenuates the development of arthritis [202,203,204].
The JAK/STAT pathway is involved in lung fibrosis [205]. Protein expression of JAK, STAT, and receptor activator of nuclear factors κB ligand (RANKL) were upregulated in the lung tissues of a RA-ILD rat model, suggesting that the JAK/STAT/RANKL signaling pathway is involved in RA-ILD pathogenesis [163]. The RA-ILD mouse model showed upregulated protein expression of JAK2, STAT3, and phosphorylated STAT3 in the ankle joints compared to the control group [142]. Clinical evidence for the inhibitory roles of JAK inhibition in RA-ILD has been continuously reported [206]. Numerous clinical and experimental studies have demonstrated the protective effects of JAK inhibitors against connective tissue disease (CTD)-ILD [207]. JAK2 is phosphorylated in the lung tissue of patients [133]. It was also found that JAK2 served as a regulator of fibrogenesis in RA-UIP and IPF as well as TGF-β signaling in both normal and lung fibrosis [208]. It has been suggested that inhibition of JAK1 and JAK2 could relieve pain in patients with RA. The JAK-signal transducer and activation of the JAK/STAT pathway are involved in the regulation of numerous inflammatory cytokines [209]. In a CIA mouse model, angiogenesis was increased by activation of the JAK/STAT signaling pathway [210].
The JAK/STAT pathway is activated by several cytokines, including IL-4, IL-13, IL-6, IL-11, and IL-31, which are involved in the pathogenesis of ILD [200]. Because of their key roles in immune reactions and connections with several cytokine receptors [211], the inhibition of JAKs is regarded as a potential therapeutic strategy for autoimmune diseases [212]. IL-11 has been shown to participate in lung fibrosis through STAT3 activation in a BLM-induced fibrosis mouse model [213,214]. Moreover, IL-11 induces STAT3 phosphorylation. However, the effect of IL-11-induced STAT activation on transcriptional alterations and its relevance to fibrogenesis has not been clearly identified [215]. JAK inhibitors can regulate the activation, proliferation, and differentiation of B cells, indicating a key role of B cells in the development of RA from early disease onset [211].
3.7. Phosphoinositide-3-kinase/protein kinase B pathway
The upregulated phosphoinositide-3-kinase (PI3K)/protein kinase B (Akt) signaling pathway facilitates the repair and development of ILD [216]. It was reported that inhibition of PI3Kγ could relieve chronic inflammatory disorders including RA by using a collagen-induced RA mice model [217]. Blockade of PI3Kδ was also demonstrated to be effective in treating an autoimmune disease in which neutrophil infiltration assists tissue injury. In a K/BxN serum transfer model of arthritis, genetic deletion or selective inhibition of PI3Kδ could ameliorate joint erosion [218]. PI3Kγ specifically has effects on the antigen-induced arthritis (AIA) during the early stage of inflammation, when innate immune cells play crucial roles in pathogenesis. PI3Kγ affects the migration and activation of macrophages, and macrophage and neutrophil infiltration into the knee joints is also involved. However, deletion or inhibition of PI3Kγ did not affect T cell function in the AIA model [219]. Upregulation of the citrullinating enzyme PAD2 in the lung tissues of patients with RA-ILD is controlled by syndecan-2 (SDC2) via regulation of the PI3K/Akt/Sp1 pathway, which is dependent on CD148 [98].
Global mRNAs and microRNAs were profiled from the lung tissues of patients with several types of ILD, and differentially expressed genes were analyzed. Several apoptosis-associated genes, including PI3K, receptor-interacting serine-threonine kinase 1 (RIP1), and bcl2-associated X protein (BAX), are downregulated, and this anti-apoptosis signaling may facilitate the survival of myofibroblasts in the lungs of patients with ILD [220]. In addition, serum IL-11 levels are substantially increased in patients with RA, which was associated with the development of ILD [159]. After IL-11 binds to its receptor, it activates the JAK/STAT3, ERK, and PI3K/AKT/mTORC1 signaling pathways to exert its effects [221,222].
4. Histopathological type of RA
Although the overall RA-related mortality is decreasing, RA-ILD with a UIP pattern shows an increased risk of disease progression and mortality [113]. UIP is generally characterized by a straight edge, anterior upper lobe, and exuberant honeycombing [223].
Chen et al. suggested that the UIP pattern is a significant risk factor for RA-ILD progression [49]. RA-ILD with a UIP histological pattern is associated with an increased risk of disease progression and mortality [13]. A prospective study demonstrated that two of three patients with RA showing a prominent ground-glass pattern on high-resolution computed tomography (HRCT) scans experienced spontaneous regression of the disease compared with the group with a reticular pattern [224]. Park et al. found that patients with RA-ILD with UIP had a worse survival rate than patients with NSIP [225].
Patients with RA-ILD with UIP have a worse prognosis than those with OP [226,227]. Suhara et al. identified several proteins that may be involved in the development of differences between UIP and OP patterns in RA-ILD pathogenesis. The concentrations of gelsolin and Ig κ chain C regions were significantly upregulated in the UIP pattern compared to the OP pattern. In contrast, the levels of α-1 antitrypsin, CRP, haptoglobin β, and surfactant protein A were significantly increased in the OP pattern than those in the UIP pattern. In addition, the gelsolin levels were significantly higher in the UIP group than those in the OP pattern [228]. These findings suggest that distinguishing the UIP pattern from the other patterns in patients with RA-ILD may lead to better therapeutic outcomes.
5. Conclusions
While ILD affects a substantial proportion of patients with RA and is associated with a decreased survival rate, reliable therapeutic agents and screening biomarkers are not currently available because of the lack of understanding of the mechanism underlying the development and progression of RA-ILD. Thus, in this review, we attempted to analyze the previously reported literature and evidence of the etiology and pathogenesis of RA-ILD, as shown in Figure 1. Based on previously reported data, older age, male sex, smoking, and exposure to specific pollutants (e.g., PM2.5, PM10, SO2, and NO2) are associated with an increased risk of RA-ILD. Specific genetic variations are independent risk factors for RA-ILD. Regarding the underlying molecular mechanisms of RA-ILD, the activation of biological processes (FMT, EMT, and immunological pathways) and signaling pathways (JAK/STAT and PI3K/Akt) could be deleterious and may contribute to the development of RA-ILD. Upregulated autophagy and oxidative stress are also involved in the pathogenesis of RA-ILD. Future research may provide more concrete evidence regarding the underlying mechanisms of RA-ILD. Further experimental studies are needed to investigate the pathogenesis of RA-ILD. Moreover, patients with RA should undergo HRCT and pulmonary function tests to determine whether they have certain pulmonary problems at an early stage and to provide proper treatment.
Author Contributions
Conceptualization, K.K.; writing—original draft preparation, Y.K.; writing—review and editing, K.K. and H.Y.; supervision, K.K.; funding acquisition, H.Y. and K.K All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science, and Technology (Grant number 2018R1D1A1B07048706 and 2021R1F1A1048536).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| RA | rheumatoid arthritis |
| ILD | interstitial lung disease |
| CT | computed tomography |
| NSIP | nonspecific interstitial pneumonia |
| UIP | usual interstitial pneumonia |
| OP | organizing pneumonia |
| FPF | familial pulmonary fibrosis |
| SNP | single nucleotide polymorphism |
| PPFIBP2 | PPFIA binding protein 2 |
| HLA | human leukocyte antigen |
| GWAS | genome-wide association studies |
| OR | odds ratio |
| SE | shared epitope |
| PAD | peptidylarginine deiminase |
| AD | airway disease |
| SO2 | sulfur dioxide |
| NO2 | nitrogen dioxide |
| ACPA | anti-cyclic citrullinated peptide antibody |
| CI | confidence interval |
| IPF | idiopathic pulmonary fibrosis |
| CCP | cyclic citrullinated peptide |
| RF | rheumatoid factor |
| MCV | mutated citrullinated vimentin antibody |
| DAS28 | disease activity score in 28 joints |
| CDAI | clinical disease activity index |
| HR | hazard ratio |
| Ig | immunoglobulin |
| BRASS | Brigham and Women’s Hospital Rheumatoid Arthritis Sequential Study |
| ACR | American college of rheumatology |
| ECM | extracellular matrix |
| BLM | bleomycin |
| CIA | collagen-induced arthritis |
| FMT | fibroblast to myofibroblast transition |
| TGF | transforming growth factor |
| SSc | systemic sclerosis |
| HLF | human lung fibroblasts |
| MEF | mouse embryonic fibroblast |
| DOCK2 | dedicator of cytokinesis 2 |
| STAT | signal transducers and activators of transcription |
| ATF3 | activating transcription factor 3 |
| COX-2 | cyclooxygenase-2 |
| PGE2 | and prostaglandin E2 |
| EMT | epithelial-mesenchymal transition |
| PDGF | platelet derived growth factor |
| TIMP | tissue inhibitor |
| MMP | matrix metalloproteinase |
| DLCO | diffusing capacity for carbon monoxide |
| FVC | forced vital capacity |
| PFT | pulmonary function test |
| IL | interleukin |
| TNF | tumor necrosis factor |
| GSEA | gene set enrichment analysis |
| iBALT | inducible bronchial-associated lymphoid tissue |
| MDSCs | myeloid-derived suppressor cells |
| sPD-1 | soluble programmed death molecule-1 |
| HSPs | heat shock proteins |
| IFN | interferon |
| MDA | malondialdehyde |
| TBARS | thiobarbituric acid reactive substances |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| LPO | lipid peroxide |
| 8-OhdG | 8-hydroxydeoxyguanosine |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| BALF | bronchoalveolar lavage fluid |
| FoxO3a | forkhead transcription factor O subfamily member 3a |
| JAK | janus kinase |
| RANKL | receptor activator of nuclear factors κB ligand |
| CTD | connective tissue disease |
| PI3K | phosphoinositide-3-kinase |
| Akt | protein kinase B |
| AIA | antigen-induced arthritis |
| SDC2 | syndecan-2 |
| RIP1 | receptor-interacting serine-threonine kinase 1 |
| BAX | bcl2-associated X protein |
| HRCT | high resolution computed tomography |
References
- Cassone, G.; Manfredi, A.; Vacchi, C.; Luppi, F.; Coppi, F.; Salvarani, C.; Sebastiani, M. Treatment of rheumatoid arthritis-associated interstitial lung disease: lights and shadows. J. Clin. Med. 2020, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, M.; Cojocaru, I.M.; Silosi, I.; Vrabie, C.D.; Tanasescu, R. Extra-articular manifestations in rheumatoid arthritis. Maedica 2010, 5, 286. [Google Scholar]
- Wilsher, M.; Voight, L.; Milne, D.; Teh, M.; Good, N.; Kolbe, J.; Williams, M.; Pui, K.; Merriman, T.; Sidhu, K. Prevalence of airway and parenchymal abnormalities in newly diagnosed rheumatoid arthritis. Respir. Med. 2012, 106, 1441–1446. [Google Scholar] [CrossRef]
- Norton, S.; Koduri, G.; Nikiphorou, E.; Dixey, J.; Williams, P.; Young, A. A study of baseline prevalence and cumulative incidence of comorbidity and extra-articular manifestations in RA and their impact on outcome. Rheumatology 2013, 52, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Chansakul, T.; Dellaripa, P.F.; Doyle, T.J.; Madan, R. Intra-thoracic rheumatoid arthritis: Imaging spectrum of typical findings and treatment related complications. Eur. J. Radiol. 2015, 84, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Ellman, P.; Ball, R. “Rheumatoid disease” with joint and pulmonary manifestations. BMJ 1948, 2, 816. [Google Scholar] [CrossRef]
- Bongartz, T.; Nannini, C.; Medina-Velasquez, Y.F.; Achenbach, S.J.; Crowson, C.S.; Ryu, J.H.; Vassallo, R.; Gabriel, S.E.; Matteson, E.L. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2010, 62, 1583–1591. [Google Scholar] [CrossRef]
- Olson, A.L.; Swigris, J.J.; Sprunger, D.B.; Fischer, A.; Fernandez-Perez, E.R.; Solomon, J.; Murphy, J.; Cohen, M.; Raghu, G.; Brown, K.K. Rheumatoid arthritis–interstitial lung disease–associated mortality. Am. J. Respir. Crit. Care Med. 2011, 183, 372–378. [Google Scholar] [CrossRef]
- Yoshinouchi, T.; Ohtsuki, Y.; Fujita, J.; Yamadori, I.; Bandoh, S.; Ishida, T.; Ueda, R. Nonspecific interstitial pneumonia pattern as pulmonary involvement of rheumatoid arthritis. Rheumatol. Int. 2005, 26, 121–125. [Google Scholar] [CrossRef]
- Tanaka, N.; Kim, J.S.; Newell, J.D.; Brown, K.K.; Cool, C.D.; Meehan, R.; Emoto, T.; Matsumoto, T.; Lynch, D.A. Rheumatoid arthritis–related lung diseases: CT findings. Radiology 2004, 232, 81–91. [Google Scholar] [CrossRef]
- Lee, H.-K.; Kim, D.S.; Yoo, B.; Seo, J.B.; Rho, J.-Y.; Colby, T.V.; Kitaichi, M. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest 2005, 127, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K. Roger S. Mitchell Lecture. Rheumatoid Lung Disease. Proc. Am. Thorac. Soc. 2007, 4, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Collard, H.R.; King Jr, T.E. Rheumatoid arthritis-associated interstitial lung disease: the relevance of histopathologic and radiographic pattern. Chest 2009, 136, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.; Collins, B.F.; Ho, L.A.; Raghu, G. Rheumatoid arthritis-associated lung disease. Eur. Respir. Rev. 2015, 24, 1–16. [Google Scholar] [CrossRef]
- Marigliano, B.; Soriano, A.; Margiotta, D.; Vadacca, M.; Afeltra, A. Lung involvement in connective tissue diseases: a comprehensive review and a focus on rheumatoid arthritis. Autoimmun. Rev. 2013, 12, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Cavagna, L.; Monti, S.; Grosso, V.; Boffini, N.; Scorletti, E.; Crepaldi, G.; Caporali, R. The multifaceted aspects of interstitial lung disease in rheumatoid arthritis. Biomed Res. Int. 2013, 2013. [Google Scholar] [CrossRef]
- de Lauretis, A.; Veeraraghavan, S.; Renzoni, E. Review series: aspects of interstitial lung disease: connective tissue disease-associated interstitial lung disease: how does it differ from IPF? How should the clinical approach differ? Chron Respir Dis 2011, 8, 53–82. [Google Scholar] [CrossRef]
- Raimundo, K.; Solomon, J.J.; Olson, A.L.; Kong, A.M.; Cole, A.L.; Fischer, A.; Swigris, J.J. Rheumatoid arthritis–interstitial lung disease in the United States: prevalence, incidence, and healthcare costs and mortality. J. Rheumatol. 2019, 46, 360–369. [Google Scholar] [CrossRef]
- Iqbal, K.; Kelly, C. Treatment of rheumatoid arthritis-associated interstitial lung disease: a perspective review. Ther. Adv. Musculoskelet. Dis. 2015, 7, 247–267. [Google Scholar] [CrossRef]
- Hyldgaard, C.; Hilberg, O.; Pedersen, A.B.; Ulrichsen, S.P.; Løkke, A.; Bendstrup, E.; Ellingsen, T. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: comorbidity and mortality. Ann. Rheum. Dis. 2017, 76, 1700–1706. [Google Scholar] [CrossRef]
- Johnson, C. Recent advances in the pathogenesis, prediction, and management of rheumatoid arthritis-associated interstitial lung disease. Curr. Opin. Rheumatol. 2017, 29, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Minhas, R.; Shankar, S.; Taha, O. Rheumatoid arthritis-associated interstitial lung disease. QJM 2019, 112, 815–816. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Kaneko, Y. Pathogenesis, clinical features, and treatment strategy for rheumatoid arthritis-associated interstitial lung disease. Autoimmun. Rev. 2022, 21, 103056. [Google Scholar] [CrossRef] [PubMed]
- Redente, E.F.; Aguilar, M.A.; Black, B.P.; Edelman, B.L.; Bahadur, A.N.; Humphries, S.M.; Lynch, D.A.; Wollin, L.; Riches, D.W. Nintedanib reduces pulmonary fibrosis in a model of rheumatoid arthritis-associated interstitial lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L998–L1009. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, G.E.; Conway, R.; Nikiphorou, E. Methotrexate and interstitial lung disease: controversies and questions. A narrative review of the literature. Rheumatology 2019, 58, 1900–1906. [Google Scholar] [CrossRef]
- Juge, P.-A.; Lee, J.S.; Lau, J.; Kawano-Dourado, L.; Serrano, J.R.; Sebastiani, M.; Koduri, G.; Matteson, E.; Bonfiglioli, K.; Sawamura, M. Methotrexate and rheumatoid arthritis associated interstitial lung disease. Eur. Respir. J. 2021, 57. [Google Scholar] [CrossRef]
- Juge, P.-A.; Borie, R.; Kannengiesser, C.; Gazal, S.; Revy, P.; Wemeau-Stervinou, L.; Debray, M.-P.; Ottaviani, S.; Marchand-Adam, S.; Nathan, N. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef]
- Wheeler, A.M.; Baker, J.F.; Poole, J.A.; Ascherman, D.P.; Yang, Y.; Kerr, G.S.; Reimold, A.; Kunkel, G.; Cannon, G.W.; Wysham, K.D. Genetic, social, and environmental risk factors in rheumatoid arthritis-associated interstitial lung disease. In Proceedings of the Seminars in arthritis and rheumatism; 2022; p. 152098. [Google Scholar]
- Juge, P.-A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med 2018, 379, 2209–2219. [Google Scholar] [CrossRef]
- Palomäki, A.; Palotie, A.; Koskela, J.; Eklund, K.K.; Pirinen, M.; Ripatti, S.; Laitinen, T.; Mars, N.; Group, F.R.C.E. Lifetime risk of rheumatoid arthritis-associated interstitial lung disease in MUC5B mutation carriers. Ann. Rheum. Dis. 2021, 80, 1530–1536. [Google Scholar] [CrossRef]
- Hayashi, S.; Matsubara, T.; Fukuda, K.; Maeda, T.; Funahashi, K.; Hashimoto, M.; Takashima, Y.; Kikuchi, K.; Fujita, M.; Matsumoto, T. A genome-wide association study identifying single nucleotide polymorphisms in the PPFIBP2 gene was predictive for interstitial lung disease in rheumatoid arthritis patients. Rheumatol. Adv. Pract. 2022, 6, rkac088. [Google Scholar] [CrossRef]
- Furukawa, H.; Oka, S.; Shimada, K.; Sugii, S.; Ohashi, J.; Matsui, T.; Ikenaka, T.; Nakayama, H.; Hashimoto, A.; Takaoka, H. Association of human leukocyte antigen with interstitial lung disease in rheumatoid arthritis: a protective role for shared epitope. PloS one 2012, 7, e33133. [Google Scholar] [CrossRef]
- Migita, K.; Nakamura, T.; Koga, T.; Eguchi, K. HLA-DRB1 alleles and rheumatoid arthritis-related pulmonary fibrosis. J. Rheumatol. 2010, 37, 205–207. [Google Scholar] [CrossRef]
- Le Guen, P.; Borie, R.; Legendre, M.; Dupin, C.; Dunogeant, L.; Ottaviani, S.; Debray, M.-P.; Cazes, A.; Dieudé, P.; Kannengiesser, C. NKX2. 1 mutation revealed by a lymphoid interstitial pneumonia in an adult with rheumatoid arthritis. ERJ Open Research 2023, 9. [Google Scholar] [CrossRef]
- Shirai, Y.; Honda, S.; Ikari, K.; Kanai, M.; Takeda, Y.; Kamatani, Y.; Morisaki, T.; Tanaka, E.; Kumanogoh, A.; Harigai, M. Association of the RPA3-UMAD1 locus with interstitial lung diseases complicated with rheumatoid arthritis in Japanese. Ann. Rheum. Dis. 2020, 79, 1305–1309. [Google Scholar] [CrossRef]
- Higuchi, T.; Oka, S.; Furukawa, H.; Shimada, K.; Tsunoda, S.; Ito, S.; Okamoto, A.; Katayama, M.; Saisho, K.; Shinohara, S. Association of a FAM13A variant with interstitial lung disease in Japanese rheumatoid arthritis. RMD open 2023, 9, e002828. [Google Scholar] [CrossRef]
- Jönsson, E.; Ljung, L.; Norrman, E.; Freyhult, E.; Ärlestig, L.; Dahlqvist, J.; Rantapää-Dahlqvist, S. Pulmonary fibrosis in relation to genetic loci in an inception cohort of patients with early rheumatoid arthritis from northern Sweden. Rheumatology 2022, 61, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Murtha, L.A.; Morten, M.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Ngo, D.T.; Sverdlov, A.L.; Knight, D.A. The role of pathological aging in cardiac and pulmonary fibrosis. Aging Dis. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Innala, L.; Berglin, E.; Möller, B.; Ljung, L.; Smedby, T.; Södergren, A.; Magnusson, S.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S. Age at onset determines severity and choice of treatment in early rheumatoid arthritis: a prospective study. Arthrit. Res. Ther. 2014, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Koduri, G.; Norton, S.; Young, A.; Cox, N.; Davies, P.; Devlin, J.; Dixey, J.; Gough, A.; Prouse, P.; Winfield, J. Interstitial lung disease has a poor prognosis in rheumatoid arthritis: results from an inception cohort. Rheumatology 2010, 49, 1483–1489. [Google Scholar] [CrossRef]
- Lai, N.-L.; Jia, W.; Wang, X.; Luo, J.; Liu, G.-Y.; Gao, C.; Li, X.-F.; Xie, J.-F. Risk factors and changes of peripheral NK and T cells in pulmonary interstitial fibrosis of patients with rheumatoid arthritis. Can. Respir. J. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Assayag, D.; Lubin, M.; Lee, J.S.; King, T.E.; Collard, H.R.; Ryerson, C.J. Predictors of mortality in rheumatoid arthritis-related interstitial lung disease. Respirology 2014, 19, 493–500. [Google Scholar] [CrossRef]
- Saag, K.G.; Cerhan, J.R.; Kolluri, S.; Ohashi, K.; Hunninghake, G.W.; Schwartz, D.A. Cigarette smoking and rheumatoid arthritis severity. Ann. Rheum. Dis. 1997, 56, 463–469. [Google Scholar] [CrossRef]
- Restrepo, J.F.; Del Rincón, I.; Battafarano, D.F.; Haas, R.W.; Doria, M.; Escalante, A. Clinical and laboratory factors associated with interstitial lung disease in rheumatoid arthritis. Clin. Rheumatol. 2015, 34, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Klester, E.; Klester, K.; Shoykhet, Y.; Elykomov, V.; Yarkova, V.; Berdyugina, A.; Mukhtarova, E. Risk factors of interstitial lung diseases in patients with rheumatoid arthritis. Eur Respiratory Soc 2019, 54. [Google Scholar]
- Aubart, F.; Crestani, B.; Nicaise-Roland, P.; Tubach, F.; Bollet, C.; Dawidowicz, K.; Quintin, E.; Hayem, G.; Palazzo, E.; Meyer, O. High levels of anti-cyclic citrullinated peptide autoantibodies are associated with co-occurrence of pulmonary diseases with rheumatoid arthritis. J. Rheumatol. 2011, 38, 979–982. [Google Scholar] [CrossRef]
- Kelly, C.A.; Saravanan, V.; Nisar, M.; Arthanari, S.; Woodhead, F.A.; Price-Forbes, A.N.; Dawson, J.; Sathi, N.; Ahmad, Y.; Koduri, G. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics—a large multicentre UK study. Rheumatology 2014, 53, 1676–1682. [Google Scholar] [CrossRef]
- Chen, N.; Diao, C.-Y.; Gao, J.; Zhao, D.-B. Risk factors for the progression of rheumatoid arthritis-related interstitial lung disease: clinical features, biomarkers, and treatment options. In Proceedings of the Seminars in Arthritis and Rheumatism; 2022; p. 152004. [Google Scholar]
- Huang, S.; Kronzer, V.L.; Dellaripa, P.F.; Deane, K.D.; Bolster, M.B.; Nagaraja, V.; Khanna, D.; Doyle, T.J.; Sparks, J.A. Rheumatoid arthritis–associated interstitial lung disease: current update on prevalence, risk factors, and pharmacologic treatment. Curr. Treat. Options Rheumatol. 2020, 6, 337–353. [Google Scholar] [CrossRef]
- Gaik, O.S.; Jen, D.H.; Hamid, Z.A.; Aziz, A.A.; Wong, N.I. Predictors and radiological characteristics of rheumatoid arthritis-associated interstitial lung disease in a multi-ethnic Malaysian cohort. Med J Malaysia 2022, 77, 293. [Google Scholar]
- Karlson, E.W.; Mandl, L.A.; Hankinson, S.E.; Grodstein, F. Do breast-feeding and other reproductive factors influence future risk of rheumatoid arthritis?: Results from the Nurses' Health Study. Arthritis Rheum. 2004, 50, 3458–3467. [Google Scholar] [CrossRef]
- Brun, J.; Nilssen, S.; Kvåle, G. Breast feeding, other reproductive factors and rheumatoid arthritis. A prospective study. Rheumatology 1995, 34, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, N.; Nguyen, E.; Sathananthan, M. Rheumatoid arthritis and associated interstitial lung disease: mortality rates and trends. Ann. Am. Thorac. Soc. 2021, 18, 1970–1977. [Google Scholar] [CrossRef] [PubMed]
- Peschken, C.A.; Hitchon, C.A.; Robinson, D.B.; Smolik, I.; Barnabe, C.R.; Prematilake, S.; El-Gabalawy, H.S. Rheumatoid arthritis in a north american native population: longitudinal followup and comparison with a white population. J. Rheumatol. 2010, 37, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Azuma, A.; Kudoh, S. High prevalence of drug-induced pneumonia in Japan. Japan Med. Assoc. J. 2007, 50, 405. [Google Scholar]
- Jo, H.E.; Corte, T.J. Nintedanib for idiopathic pulmonary fibrosis in the Japanese population. Respirology 2017, 22, 630–631. [Google Scholar] [CrossRef]
- Tekaya, A.B.; Mokaddem, S.; Athimini, S.; Kamoun, H.; Mahmoud, I.; Abdelmoula, L. Risk factors for rheumatoid arthritis-associated interstitial lung disease: a retrospective study. Multidiscip. Respir. Med. 2022, 17. [Google Scholar]
- Sparks, J.A.; Karlson, E.W. The roles of cigarette smoking and the lung in the transitions between phases of preclinical rheumatoid arthritis. Curr. Rheumatol. Rep. 2016, 18, 15. [Google Scholar] [CrossRef]
- Gizinski, A.M.; Mascolo, M.; Loucks, J.L.; Kervitsky, A.; Meehan, R.T.; Brown, K.K.; Holers, V.M.; Deane, K.D. Rheumatoid arthritis (RA)-specific autoantibodies in patients with interstitial lung disease and absence of clinically apparent articular RA. Clin. Rheumatol. 2009, 28, 611–613. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.; Wu, N.; Dong, X.; Zheng, Y. Retrospective study of the clinical characteristics and risk factors of rheumatoid arthritis-associated interstitial lung disease. Clin. Rheumatol. 2017, 36, 817–823. [Google Scholar] [CrossRef]
- Saag, K.G.; Kolluri, S.; Koehnke, R.K.; Georgou, T.A.; Rachow, J.W.; Hunninghake, G.W.; Schwartz, D.A. Rheumatoid arthritis lung disease. Determinants of radiographic and physiologic abnormalities. Arthritis Rheum. 1996, 39, 1711–1719. [Google Scholar] [CrossRef]
- Kronzer, V.L.; Huang, W.; Dellaripa, P.F.; Huang, S.; Feathers, V.; Lu, B.; Iannaccone, C.K.; Gill, R.R.; Hatabu, H.; Nishino, M. Lifestyle and clinical risk factors for incident rheumatoid arthritis-associated interstitial lung disease. J. Rheumatol. 2021, 48, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Erlandsson Harris, H.; Ulfgren, A.K.; Rantapää-Dahlqvist, S. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA–DR (shared epitope)–restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006, 54, 38–46. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, W.; Yu, Y.; Hu, S. Rheumatoid arthritis–associated interstitial lung disease: an overview of epidemiology, pathogenesis and management. Clin. Rheumatol. 2021, 40, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Makrygiannakis, D.; Hermansson, M.; Ulfgren, A.-K.; Nicholas, A.P.; Zendman, A.J.; Eklund, A.; Grunewald, J.; Skold, C.M.; Klareskog, L.; Catrina, A.I. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann. Rheum. Dis. 2008, 67, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Koga, Y.; Sugimoto, M. Different risk factors between interstitial lung disease and airway disease in rheumatoid arthritis. Respir. Med. 2012, 106, 1591–1599. [Google Scholar] [CrossRef]
- Liu, B.; Sun, G.; Liu, Y.; Hou, Y. Observational studies: Ambient air pollution and hospitalization for RA-ILD in a heavily polluted city in China. Medicine 2022, 101. [Google Scholar] [CrossRef]
- Yahya, A.; Bengtsson, C.; Larsson, P.; Too, C.L.; Mustafa, A.N.; Abdullah, N.A.; Muhamad, N.A.; Klareskog, L.; Murad, S.; Alfredsson, L. Silica exposure is associated with an increased risk of developing ACPA-positive rheumatoid arthritis in an Asian population: evidence from the Malaysian MyEIRA case–control study. Mod. Rheumatol. 2014, 24, 271–274. [Google Scholar] [CrossRef]
- Stolt, P.; Yahya, A.; Bengtsson, C.; Källberg, H.; Rönnelid, J.; Lundberg, I.; Klareskog, L.; Alfredsson, L.; Group, E.S. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1072–1076. [Google Scholar] [CrossRef]
- Stolt, P.; Källberg, H.; Lundberg, I.; Sjögren, B.; Klareskog, L.; Alfredsson, L. Silica exposure is associated with increased risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann. Rheum. Dis. 2005, 64, 582–586. [Google Scholar] [CrossRef]
- Chen, H.-H.; Yong, Y.-M.; Lin, C.-H.; Chen, Y.-H.; Chen, D.-Y.; Ying, J.-C.; Chao, W.-C. Air pollutants and development of interstitial lung disease in patients with connective tissue disease: a population-based case–control study in Taiwan. Bmj Open 2020, 10, e041405. [Google Scholar] [CrossRef]
- Sack, C.; Vedal, S.; Sheppard, L.; Raghu, G.; Barr, R.G.; Podolanczuk, A.; Doney, B.; Hoffman, E.A.; Gassett, A.; Hinckley-Stukovsky, K. Air pollution and subclinical interstitial lung disease: the Multi-Ethnic Study of Atherosclerosis (MESA) air–lung study. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Conti, S.; Harari, S.; Caminati, A.; Zanobetti, A.; Schwartz, J.D.; Bertazzi, P.A.; Cesana, G.; Madotto, F. The association between air pollution and the incidence of idiopathic pulmonary fibrosis in Northern Italy. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.B.; Li, W.; Schwartz, J.; Di, Q.; Kloog, I.; Koutrakis, P.; Gold, D.R.; Hallowell, R.W.; Zhang, C.; O'connor, G. Ambient air pollution exposure and risk and progression of interstitial lung abnormalities: the Framingham Heart Study. Thorax 2019, 74, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Al-Aly, Z.; Zheng, B.; van Donkelaar, A.; Martin, R.V.; Pineau, C.A.; Bernatsky, S. Fine particulate matter components and interstitial lung disease in rheumatoid arthritis. Eur. Respir. J. 2022, 60. [Google Scholar] [CrossRef] [PubMed]
- Van Venrooij, W.J.; Van Beers, J.J.; Pruijn, G.J. Anti-CCP antibodies: the past, the present and the future. Nat. Rev. Rheumatol. 2011, 7, 391–398. [Google Scholar] [CrossRef]
- Kamiya, H.; Panlaqui, O.M. Systematic review and meta-analysis of the risk of rheumatoid arthritis-associated interstitial lung disease related to anti-cyclic citrullinated peptide (CCP) antibody. BMJ open 2021, 11, e040465. [Google Scholar] [CrossRef]
- Zeng, X.; Ai, M.; Tian, X.; Gan, X.; Shi, Y.; Song, Q.; Tang, F. Diagnostic value of anti-cyclic citrullinated Peptide antibody in patients with rheumatoid arthritis. J. Rheumatol. 2003, 30, 1451–1455. [Google Scholar]
- Rodríguez-Mahou, M.; López-Longo, F.J.; Sánchez-Ramón, S.; Estecha, A.; García-Segovia, A.; Rodríguez-Molina, J.J.; Carreño, L.; Fernández-Cruz, E. Association of anti–cyclic citrullinated peptide and anti-Sa/citrullinated vimentin autoantibodies in rheumatoid arthritis. Arthritis Care Res 2006, 55, 657–661. [Google Scholar] [CrossRef]
- Yang, J.A.; Lee, J.S.; Park, J.K.; Lee, E.B.; Song, Y.W.; Lee, E.Y. Clinical characteristics associated with occurrence and poor prognosis of interstitial lung disease in rheumatoid arthritis. Korean J Med 2019, 34, 434. [Google Scholar] [CrossRef]
- Doyle, T.J.; Patel, A.S.; Hatabu, H.; Nishino, M.; Wu, G.; Osorio, J.C.; Golzarri, M.F.; Traslosheros, A.; Chu, S.G.; Frits, M.L. Detection of rheumatoid arthritis–interstitial lung disease is enhanced by serum biomarkers. Am. J. Respir. Crit. Care Med. 2015, 191, 1403–1412. [Google Scholar] [CrossRef]
- Natalini, J.G.; Baker, J.F.; Singh, N.; Mahajan, T.D.; Roul, P.; Thiele, G.M.; Sauer, B.C.; Johnson, C.R.; Kawut, S.M.; Mikuls, T.R. Autoantibody seropositivity and risk for interstitial lung disease in a prospective male-predominant rheumatoid arthritis cohort of US veterans. Ann. Am. Thorac. Soc. 2021, 18, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.T.; Danoff, S.K.; Sokolove, J.; Wagner, C.A.; Winchester, R.; Pappas, D.A.; Siegelman, S.; Connors, G.; Robinson, W.H.; Bathon, J.M. Association of fine specificity and repertoire expansion of anticitrullinated peptide antibodies with rheumatoid arthritis associated interstitial lung disease. Ann. Rheum. Dis. 2014, 73, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhou, Y.; Chen, X.; Li, J. A metaanalysis of the increased risk of rheumatoid arthritis-related pulmonary disease as a result of serum anticitrullinated protein antibody positivity. J. Rheumatol. 2014, 41, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- López-Longo, F.J.; Oliver-Miñarro, D.; de la Torre, I.; González-Díaz de Rábago, E.; Sánchez-Ramón, S.; Rodríguez-Mahou, M.; Paravisini, A.; Monteagudo, I.; González, C.M.; GarCía-Castro, M. Association between anti–cyclic citrullinated peptide antibodies and ischemic heart disease in patients with rheumatoid arthritis. Arthritis Care Res 2009, 61, 419–424. [Google Scholar] [CrossRef]
- Liao, K.P.; Gunnarsson, M.; Källberg, H.; Ding, B.; Plenge, R.M.; Padyukov, L.; Karlson, E.W.; Klareskog, L.; Askling, J.; Alfredsson, L. Specific association of type 1 diabetes mellitus with anti–cyclic citrullinated peptide–positive rheumatoid arthritis. Arthritis Rheum. 2009, 60, 653–660. [Google Scholar] [CrossRef]
- Alexiou, I.; Germenis, A.; Koutroumpas, A.; Kontogianni, A.; Theodoridou, K.; Sakkas, L.I. Anti-cyclic citrullinated peptide-2 (CCP2) autoantibodies and extra-articular manifestations in Greek patients with rheumatoid arthritis. Clin. Rheumatol. 2008, 27, 511–513. [Google Scholar] [CrossRef]
- Gerli, R.; Bocci, E.B.; Sherer, Y.; Vaudo, G.; Moscatelli, S.; Shoenfeld, Y. Association of anti-cyclic citrullinated peptide antibodies with subclinical atherosclerosis in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 724–725. [Google Scholar] [CrossRef]
- Solomon, J.J.; Matson, S.; Kelmenson, L.B.; Chung, J.H.; Hobbs, S.B.; Rosas, I.O.; Dellaripa, P.F.; Doyle, T.J.; Poli, S.; Esposito, A.J. IgA antibodies directed against citrullinated protein antigens are elevated in patients with idiopathic pulmonary fibrosis. Chest 2020, 157, 1513–1521. [Google Scholar] [CrossRef]
- Kim, J.-W.; Lee, H.; Choe, J.-Y.; Hwang, J.H.; Park, S.-H.; Lee, H.-S.; Kim, S.-K. Factors associated with airway disease and interstitial lung disease in rheumatoid arthritis. J Rheum Dis 2016, 23, 101–108. [Google Scholar] [CrossRef]
- Yin, Y.; Liang, D.; Zhao, L.; Li, Y.; Liu, W.; Ren, Y.; Li, Y.; Zeng, X.; Zhang, F.; Tang, F. Anti-cyclic citrullinated peptide antibody is associated with interstitial lung disease in patients with rheumatoid arthritis. PloS one 2014, 9, e92449. [Google Scholar] [CrossRef]
- Samara, K.D.; Trachalaki, A.; Tsitoura, E.; Koutsopoulos, A.V.; Lagoudaki, E.D.; Lasithiotaki, I.; Margaritopoulos, G.; Pantelidis, P.; Bibaki, E.; Siafakas, N.M. Upregulation of citrullination pathway: from autoimmune to idiopathic lung fibrosis. Respir. Res. 2017, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-X.; Du, C.-G. A retrospective study of clinical characteristics of interstitial lung disease associated with rheumatoid arthritis in Chinese patients. Med Sci Monit 2015, 21, 708. [Google Scholar] [PubMed]
- Turesson, C.; Jacobsson, L.; Sturfelt, G.; Matteson, E.; Mathsson, L.; Rönnelid, J. Rheumatoid factor and antibodies to cyclic citrullinated peptides are associated with severe extra-articular manifestations in rheumatoid arthritis. Ann. Rheum. Dis. 2007, 66, 59–64. [Google Scholar] [CrossRef]
- Zrour, S.H.; Touzi, M.; Bejia, I.; Golli, M.; Rouatbi, N.; Sakly, N.; Younes, M.; Tabka, Z.; Bergaoui, N. Correlations between high-resolution computed tomography of the chest and clinical function in patients with rheumatoid arthritis: prospective study in 75 patients. Jt. Bone Spine 2005, 72, 41–47. [Google Scholar] [CrossRef]
- Bongartz, T.; Cantaert, T.; Atkins, S.; Harle, P.; Myers, J.; Turesson, C.; Ryu, J.; Baeten, D.; Matteson, E. Citrullination in extra-articular manifestations of rheumatoid arthritis. Rheumatology 2007, 46, 70–75. [Google Scholar] [CrossRef]
- Tsoyi, K.; Esposito, A.J.; Sun, B.; Bowen, R.G.; Xiong, K.; Poli, F.; Cardenas, R.; Chu, S.G.; Liang, X.; Ryter, S.W. Syndecan-2 regulates PAD2 to exert antifibrotic effects on RA-ILD fibroblasts. Sci. Rep. 2022, 12, 2847. [Google Scholar] [CrossRef]
- Tian, F.; Li, J.; Tuo, H.; Ling, Q.; Zeng, S.; Wen, Z.; Luo, X. The anti-mutated citrullinated vimentin antibody as a potential predictor for rheumatoid arthritis associated interstitial lung diseases. Int J Clin Exp Med 2016, 9, 6813–6818. [Google Scholar]
- Inui, N.; Enomoto, N.; Suda, T.; Kageyama, Y.; Watanabe, H.; Chida, K. Anti-cyclic citrullinated peptide antibodies in lung diseases associated with rheumatoid arthritis. Clin. Biochem. 2008, 41, 1074–1077. [Google Scholar] [CrossRef]
- Jearn, L.-H.; Kim, T.-Y. Level of anticitrullinated peptide/protein antibody is not associated with lung diseases in rheumatoid arthritis. J. Rheumatol. 2012, 39, 1493–1494. [Google Scholar] [CrossRef]
- Aletaha, D.; Smolen, J.S. Diagnosis and management of rheumatoid arthritis: a review. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef]
- Solomon, D.; Reed, G.; Kremer, J.; Curtis, J.; Farkouh, M.; Harrold, L.; Hochberg, M.; Tsao, P.; Greenberg, J. Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheumatol. 2015, 67, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A.; He, X.; Huang, J.; Fletcher, E.A.; Zaccardelli, A.; Friedlander, H.M.; Gill, R.R.; Hatabu, H.; Nishino, M.; Murphy, D.J. Rheumatoid arthritis disease activity predicting incident clinically apparent rheumatoid arthritis–associated interstitial lung disease: a prospective cohort study. Arthritis Rheumatol. 2019, 71, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Paulin, F.; Doyle, T.J.; Mercado, J.F.; Fassola, L.; Fernández, M.; Caro, F.; Alberti, M.L.; Espíndola, M.E.C.; Buschiazzo, E. Development of a risk indicator score for the identification of interstitial lung disease in patients with rheumatoid arthritis. Reumatol. Clin. 2021, 17, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.; Lama, S.; Knapp, K.; Gutierrez, C.; Lovett, K.; Thai, S.; Craig, G.L. Epidemiology and clinical characteristics of interstitial lung disease in patients with rheumatoid arthritis from the JointMan database. Sci. Rep. 2023, 13, 11678. [Google Scholar] [CrossRef] [PubMed]
- Severo, C.R.; Chomiski, C.; Valle, M.B.d.; Escuissato, D.L.; Paiva, E.d.S.; Storrer, K.M. Assessment of risk factors in patients with rheumatoid arthritis-associated interstitial lung disease. J Bras Pneumol 2022, 48. [Google Scholar]
- Rojas-Serrano, J.; Mejía, M.; Rivera-Matias, P.A.; Herrera-Bringas, D.; Pérez-Román, D.I.; Pérez-Dorame, R.; Mateos-Toledo, H. Rheumatoid arthritis-related interstitial lung disease (RA-ILD): a possible association between disease activity and prognosis. Clin. Rheumatol. 2022, 41, 1741–1747. [Google Scholar] [CrossRef]
- Tyker, A.; Ventura, I.B.; Lee, C.T.; Strykowski, R.; Garcia, N.; Guzy, R.; Jablonski, R.; Vij, R.; Strek, M.E.; Chung, J.H. High-titer rheumatoid factor seropositivity predicts mediastinal lymphadenopathy and mortality in rheumatoid arthritis-related interstitial lung disease. Sci. Rep. 2021, 11, 22821. [Google Scholar] [CrossRef]
- Kadura, S.; Raghu, G. Rheumatoid arthritis-interstitial lung disease: manifestations and current concepts in pathogenesis and management. Eur Respir Rev 2021, 30. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.-I.; Kim, H.-O. Recent advances in basic and clinical aspects of rheumatoid arthritis-associated interstitial lung diseases. J Rheum Dis 2022, 29, 61–70. [Google Scholar] [CrossRef]
- Wu, E.K.; Ambrosini, R.D.; Kottmann, R.M.; Ritchlin, C.T.; Schwarz, E.M.; Rahimi, H. Reinterpreting evidence of rheumatoid arthritis-associated interstitial lung disease to understand etiology. Curr. Rheumatol. Rev. 2019, 15, 277–289. [Google Scholar] [CrossRef]
- Doyle, T.J.; Lee, J.S.; Dellaripa, P.F.; Lederer, J.A.; Matteson, E.L.; Fischer, A.; Ascherman, D.P.; Glassberg, M.K.; Ryu, J.H.; Danoff, S.K. A roadmap to promote clinical and translational research in rheumatoid arthritis-associated interstitial lung disease. Chest 2014, 145, 454–463. [Google Scholar] [CrossRef]
- Bao, L.; Ye, J.; Liu, N.; Shao, Y.; Li, W.; Fan, X.; Zhao, D.; Wang, H.; Chen, X. Resveratrol Ameliorates Fibrosis in Rheumatoid Arthritis-Associated Interstitial Lung Disease via the Autophagy–Lysosome Pathway. Molecules 2022, 27, 8475. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Gabbiani, G. Cell-matrix and cell-cell contacts of myofibroblasts: role in connective tissue remodeling. Thromb. Haemost. 2003, 90, 993–1002. [Google Scholar]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Conforti, F.; Hill, C.; Bell, J.; Drawater, L.; Li, J.; Liu, D.; Xiong, H.; Alzetani, A.; Chee, S.J. Paracrine signalling during ZEB1-mediated epithelial–mesenchymal transition augments local myofibroblast differentiation in lung fibrosis. Cell Death Differ. 2019, 26, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Hansel, C.; Jendrossek, V.; Klein, D. Cellular senescence in the lung: the central role of senescent epithelial cells. Int. J. Mol. Sci. 2020, 21, 3279. [Google Scholar] [CrossRef]
- Györfi, A.H.; Matei, A.-E.; Distler, J.H. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68, 8–27. [Google Scholar] [CrossRef]
- Guillotin, D.; Taylor, A.R.; Platé, M.; Mercer, P.F.; Edwards, L.M.; Haggart, R.; Miele, G.; McAnulty, R.J.; Maher, T.M.; Hynds, R.E. Transcriptome analysis of IPF fibroblastic foci identifies key pathways involved in fibrogenesis. Thorax 2021, 76, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev 2015, 24, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Gabasa, M.; Royo, D.; Molina-Molina, M.; Roca-Ferrer, J.; Pujols, L.; Picado, C.; Xaubet, A.; Pereda, J. Lung myofibroblasts are characterized by down-regulated cyclooxygenase-2 and its main metabolite, prostaglandin E2. PloS one 2013, 8, e65445. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Yettella, R.R.; Kim, J.; Kwon, K.; Kim, M.; Min, D.B. Effects of grilling and roasting on the levels of polycyclic aromatic hydrocarbons in beef and pork. Food Chem. 2011, 129, 1420–1426. [Google Scholar] [CrossRef]
- Scotton, C.J.; Chambers, R.C. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest 2007, 132, 1311–1321. [Google Scholar] [CrossRef]
- Tanner, L.; Bergwik, J.; Bhongir, R.K.; Egesten, A. Zoledronic acid targets the mevalonate pathway causing reduced cell recruitment and attenuation of pulmonary fibrosis. bioRxiv 2021. 2021.2012. 2001.470755. [Google Scholar]
- Guo, X.; Adeyanju, O.; Sunil, C.; Mandlem, V.; Olajuyin, A.; Huang, S.; Chen, S.-Y.; Idell, S.; Tucker, T.A.; Qian, G. DOCK2 contributes to pulmonary fibrosis by promoting lung fibroblast to myofibroblast transition. Am. J. Physiol. Cell Physiol. 2022, 323, C133–C144. [Google Scholar] [CrossRef]
- Pedroza, M.; Le, T.T.; Lewis, K.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016, 30, 129. [Google Scholar] [CrossRef]
- Chakraborty, D.; Šumová, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Prêle, C.M.; Wong, J.; Hogaboam, C.M.; McAnulty, R.J.; Laurent, G.J.; Zhang, S.S.-M.; Selman, M.; Mutsaers, S.E.; Knight, D.A. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am. J. Pathol. 2012, 180, 1398–1412. [Google Scholar] [CrossRef]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escrivá, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respiratory research 2018, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lin, H.; Zhang, X. Inhibitory effects of pirfenidone on fibroblast to myofibroblast transition in rheumatoid arthritis-associated interstitial lung disease via the downregulation of activating transcription factor 3 (ATF3). Int. Immunopharmacol. 2019, 74, 105700. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.H.; Li, T.; Wang, H.; Wu, Y.N.; Wang, S.P.; Zhao, Y.Y.; Zhang, G.Q.; Duan, J. IL-17A-producing T cells exacerbate fine particulate matter-induced lung inflammation and fibrosis by inhibiting PI3K/Akt/mTOR-mediated autophagy. J. Cell. Mol. Med. 2020, 24, 8532–8544. [Google Scholar] [CrossRef] [PubMed]
- Konen, J.M.; Rodriguez, B.L.; Padhye, A.; Ochieng, J.K.; Gibson, L.; Diao, L.; Fowlkes, N.W.; Fradette, J.J.; Peng, D.H.; Cardnell, R.J. Dual inhibition of MEK and AXL targets tumor cell heterogeneity and prevents resistant outgrowth mediated by the epithelial-to-mesenchymal transition in NSCLC. Cancer Res. 2021, 81, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Salazar, Y.; Zheng, X.; Brunn, D.; Raifer, H.; Picard, F.; Zhang, Y.; Winter, H.; Guenther, S.; Weigert, A.; Weigmann, B. Microenvironmental Th9 and Th17 lymphocytes induce metastatic spreading in lung cancer. J. Clin. Investig. 2020, 130, 3560–3575. [Google Scholar] [CrossRef]
- Zeisberg, M.; Kalluri, R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 2004, 82, 175–181. [Google Scholar] [CrossRef]
- Marmai, C.; Sutherland, R.E.; Kim, K.K.; Dolganov, G.M.; Fang, X.; Kim, S.S.; Jiang, S.; Golden, J.A.; Hoopes, C.W.; Matthay, M.A. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L71–L78. [Google Scholar] [CrossRef]
- Namba, T.; Tanaka, K.; Ito, Y.; Hoshino, T.; Matoyama, M.; Yamakawa, N.; Isohama, Y.; Azuma, A.; Mizushima, T. Induction of EMT-like phenotypes by an active metabolite of leflunomide and its contribution to pulmonary fibrosis. Cell Death Differ. 2010, 17, 1882–1895. [Google Scholar] [CrossRef]
- Ba, X.; Wang, H.; Huang, Y.; Yan, J.; Han, L.; Lin, W.; Shen, P.; Huang, Y.; Yang, S.; Qin, K. Simiao pill attenuates collagen-induced arthritis and bleomycin-induced pulmonary fibrosis in mice by suppressing the JAK2/STAT3 and TGF-β/Smad2/3 signalling pathway. J. Ethnopharmacol. 2023, 309, 116274. [Google Scholar] [CrossRef]
- Miura, Y.; Ohkubo, H.; Niimi, A.; Kanazawa, S. Suppression of epithelial abnormalities by nintedanib in induced-rheumatoid arthritis-associated interstitial lung disease mouse model. ERJ Open Res. 2021, 7. [Google Scholar] [CrossRef]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune mechanisms in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef]
- Przybylo, J.A.; Radisky, D.C. Matrix metalloproteinase-induced epithelial–mesenchymal transition: tumor progression at Snail's pace. Int. J. Biochem. Cell Biol. 2007, 39, 1082–1088. [Google Scholar] [CrossRef]
- Kollar, B.; Uffing, A.; Borges, T.J.; Shubin, A.V.; Aoyama, B.T.; Dagot, C.; Haug, V.; Kauke, M.; Safi, A.-F.; Talbot, S.G. MMP3 is a non-invasive biomarker of rejection in skin-bearing vascularized composite allotransplantation: a multicenter validation study. Front. immunol. 2019, 10, 2771. [Google Scholar] [CrossRef]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol 2011, 179, 1733–1745. [Google Scholar] [CrossRef]
- McKeown, S.; Richter, A.G.; O'Kane, C.; McAuley, D.F.; Thickett, D. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur. Respir. J. 2009, 33, 77–84. [Google Scholar] [CrossRef]
- Xue, J.; Hu, W.; Wu, S.; Wang, J.; Chi, S.; Liu, X. Development of a risk nomogram model for identifying interstitial lung disease in patients with rheumatoid arthritis. Front. immunol. 2022, 13, 823669. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Doyle, T.J.; Liu, Y.; Aggarwal, R.; Wang, X.; Shi, Y.; Ge, S.X.; Huang, H.; Lin, Q.; Liu, W. Biomarkers of rheumatoid arthritis–associated interstitial lung disease. Arthritis Rheumatol. 2015, 67, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Lee, J.S.; Im Yoon, Y.; Chang, S.H.; Lee, Y.A.; Ha, Y.-J.; Kang, E.H.; Park, Y.-B.; Lee, H.; Choe, J.-Y. Association of serum biomarkers with pulmonary involvement of rheumatoid arthritis interstitial lung disease: from KORAIL cohort baseline data. J Rheum Dis 2021, 28, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef]
- O'Dwyer, D.N.; Armstrong, M.E.; Cooke, G.; Dodd, J.D.; Veale, D.J.; Donnelly, S.C. Rheumatoid arthritis (RA) associated interstitial lung disease (ILD). Eur. J. Intern. Med. 2013, 24, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Mena-Vázquez, N.; Godoy-Navarrete, F.J.; Lisbona-Montañez, J.M.; Redondo-Rodriguez, R.; Manrique-Arija, S.; Rioja, J.; Mucientes, A.; Ruiz-Limón, P.; Garcia-Studer, A.; Ortiz-Márquez, F. Inflammatory Biomarkers in the Diagnosis and Prognosis of Rheumatoid Arthritis–Associated Interstitial Lung Disease. Int. J. Mol. Sci. 2023, 24, 6800. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xu, S. TNF inhibitor therapy for rheumatoid arthritis. Biomed Rep 2013, 1, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lin, W.; Chen, Z.; Wang, Y.; Huang, Y.; Tu, S. Effect of tumor necrosis factor inhibitors on interstitial lung disease in rheumatoid arthritis: angel or demon? Drug Des Devel Ther 2019, 2111–2125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, S.; Lau, J.; Roden, A.C.; Matteson, E.L.; Sun, J.; Luo, F.; Tschumperlin, D.J.; Vassallo, R. IL-23 amplifies the epithelial-mesenchymal transition of mechanically conditioned alveolar epithelial cells in rheumatoid arthritis-associated interstitial lung disease through mTOR/S6 signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L1006–L1022. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, G.E.; Stock, C.J.; Shi-Wen, X.; Leoni, P.; Sestini, P.; Howat, S.L.; Bou-Gharios, G.; Nicholson, A.G.; Denton, C.P.; Grutters, J.C. Microarray profiling reveals suppressed interferon stimulated gene program in fibroblasts from scleroderma-associated interstitial lung disease. Respir. Res. 2013, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, G.; Ren, Q.; Wu, J.; Gu, B.; Su, D.; Shen, M. Increased interleukin-11 associated with disease activity and development of interstitial lung disease in patients with rheumatoid arthritis. Clin. Exp. Rheumatol 2021. [Google Scholar] [CrossRef]
- Chung, S.-J.; Kwon, Y.-J.; Park, M.-C.; Park, Y.-B.; Lee, S.-K. The correlation between increased serum concentrations of interleukin-6 family cytokines and disease activity in rheumatoid arthritis patients. Yonsei Med. J. 2011, 52, 113–120. [Google Scholar] [CrossRef]
- Gasse, P.; Riteau, N.; Vacher, R.; Michel, M.-L.; Fautrel, A.; Di Padova, F.; Fick, L.; Charron, S.; Lagente, V.; Eberl, G. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PloS one 2011, 6, e23185. [Google Scholar] [CrossRef]
- Yuan, H.; Jiao, L.; Yu, N.; Duan, H.; Yu, Y.; Bai, Y. Histone deacetylase 3-mediated inhibition of microRNA-19a-3p facilitates the development of rheumatoid arthritis-associated interstitial lung disease. Front. physiol. 2020, 11, 549656. [Google Scholar] [CrossRef]
- Yang, G.; Lyu, L.; Wang, X.; Bao, L.; Lyu, B.; Lin, Z. Systemic treatment with resveratrol alleviates adjuvant arthritis-interstitial lung disease in rats via modulation of JAK/STAT/RANKL signaling pathway. Pulm. Pharmacol. Ther. 2019, 56, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Jeong, Y.; de Frías, S.P.; Easthausen, I.; Hoffman, K.; Oromendia, C.; Taheri, S.; Esposito, A.J.; Arias, L.Q.; Ayaub, E.A. Serum proteomic profiling of rheumatoid arthritis–interstitial lung disease with a comparison to idiopathic pulmonary fibrosis. Thorax 2022, 77, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Sendo, S.; Saegusa, J.; Okano, T.; Takahashi, S.; Akashi, K.; Morinobu, A. CD 11b+ Gr-1dim Tolerogenic Dendritic Cell–Like Cells Are Expanded in Interstitial Lung Disease in SKG Mice. Arthritis Rheumatol. 2017, 69, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Sendo, S.; Saegusa, J.; Yamada, H.; Nishimura, K.; Morinobu, A. Tofacitinib facilitates the expansion of myeloid-derived suppressor cells and ameliorates interstitial lung disease in SKG mice. Arthritis Res. Ther. 2019, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Jiang, L.; Nie, L.; Zhang, S.; Liu, L.; Du, Y.; Xue, J. Soluble programmed death molecule 1 (sPD-1) as a predictor of interstitial lung disease in rheumatoid arthritis. BMC Immunol. 2021, 22, 1–10. [Google Scholar] [CrossRef]
- Greisen, S.; Rasmussen, T.; Stengaard-Pedersen, K.; Hetland, M.L.; Hørslev-Petersen, K.; Hvid, M.; Deleuran, B. Increased soluble programmed death-1 (sPD-1) is associated with disease activity and radiographic progression in early rheumatoid arthritis. Scand. J. Rheumatol. 2014, 43, 101–108. [Google Scholar] [CrossRef]
- Guo, Y.; Walsh, A.M.; Canavan, M.; Wechalekar, M.D.; Cole, S.; Yin, X.; Scott, B.; Loza, M.; Orr, C.; McGarry, T. Immune checkpoint inhibitor PD-1 pathway is down-regulated in synovium at various stages of rheumatoid arthritis disease progression. PloS one 2018, 13, e0192704. [Google Scholar] [CrossRef]
- Tsan, M.-F.; Gao, B. Heat shock proteins and immune system. J. Leukoc. Biol. 2009, 85, 905–910. [Google Scholar] [CrossRef]
- Tanaka, K.-I.; Tanaka, Y.; Namba, T.; Azuma, A.; Mizushima, T. Heat shock protein 70 protects against bleomycin-induced pulmonary fibrosis in mice. Biochem. Pharmacol. 2010, 80, 920–931. [Google Scholar] [CrossRef]
- Namba, T.; Tanaka, K.-I.; Hoshino, T.; Azuma, A.; Mizushima, T. Suppression of expression of heat shock protein 70 by gefitinib and its contribution to pulmonary fibrosis. PloS one 2011, 6, e27296. [Google Scholar] [CrossRef]
- Chen, J.; Song, S.; Liu, Y.; Liu, D.; Lin, Y.; Ge, S.; Ascherman, D.P. Autoreactive T cells to citrullinated HSP90 are associated with interstitial lung disease in rheumatoid arthritis. Int. J. Rheum. Dis. 2018, 21, 1398–1405. [Google Scholar] [CrossRef]
- Harlow, L.; Rosas, I.O.; Gochuico, B.R.; Mikuls, T.R.; Dellaripa, P.F.; Oddis, C.V.; Ascherman, D.P. Identification of citrullinated Hsp90 isoforms as novel autoantigens in rheumatoid arthritis–associated interstitial lung disease. Arthritis Rheum. 2013, 65, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Sibinska, Z.; Tian, X.; Korfei, M.; Kojonazarov, B.; Kolb, J.S.; Klepetko, W.; Kosanovic, D.; Wygrecka, M.; Ghofrani, H.A.; Weissmann, N. Amplified canonical transforming growth factor-β signalling via heat shock protein 90 in pulmonary fibrosis. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef]
- Ramani, S.; Pathak, A.; Dalal, V.; Paul, A.; Biswas, S. Oxidative stress in autoimmune diseases: an under dealt malice. Curr. Protein Pept. Sci. 2020, 21, 611–621. [Google Scholar] [CrossRef]
- Mishra, R.; Singh, A.; Chandra, V.; Negi, M.P.; Tripathy, B.C.; Prakash, J.; Gupta, V. A comparative analysis of serological parameters and oxidative stress in osteoarthritis and rheumatoid arthritis. Rheumatol. Int. 2012, 32, 2377–2382. [Google Scholar] [CrossRef]
- Aryaeian, N.; Djalali, M.; Shahram, F.; Jazayeri, S.; Chamari, M.; Nazari, S. Beta-carotene, vitamin E, MDA, glutathione reductase and arylesterase activity levels in patients with active rheumatoid arthritis. Iran. J. Public Health 2011, 40, 102. [Google Scholar] [PubMed]
- Veselinovic, M.; Barudzic, N.; Vuletic, M.; Zivkovic, V.; Tomic-Lucic, A.; Djuric, D.; Jakovljevic, V. Oxidative stress in rheumatoid arthritis patients: relationship to diseases activity. Mol. Cell. Biochem. 2014, 391, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Crapo, J. Oxidative stress as an initiator of cytokine release and cell damage. Eur. Respir. J. 2003, 22, 4s–6s. [Google Scholar] [CrossRef] [PubMed]
- Kinnula, V.L.; Fattman, C.L.; Tan, R.J.; Oury, T.D. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am. J. Respir. Crit. Care Med. 2005, 172, 417–422. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Javad-Moosavi, S.A.; Reiter, R.J.; Yarahmadi, R.; Ghaznavi, H.; Mehrzadi, S. Oxidative/nitrosative stress, autophagy and apoptosis as therapeutic targets of melatonin in idiopathic pulmonary fibrosis. Expert Opin. Ther. Targets 2018, 22, 1049–1061. [Google Scholar] [CrossRef]
- Larios, J.M.; Budhiraja, R.; Fanburg, B.L.; Thannickal, V.J. Oxidative Protein Cross-linking Reactions Involvingl-Tyrosine in Transforming Growth Factor-β1-stimulated Fibroblasts. J. Biol. Chem. 2001, 276, 17437–17441. [Google Scholar] [CrossRef]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J. Biol. Chem. 2003, 278, 28403–28409. [Google Scholar] [CrossRef]
- Nelson, K.K.; Melendez, J.A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004, 37, 768–784. [Google Scholar] [CrossRef]
- Bergeron, A.; Soler, P.; Kambouchner, M.; Loiseau, P.; Milleron, B.; Valeyre, D.; Hance, A.; Tazi, A. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-β and IL-10. Eur. Respir. J. 2003, 22, 69–76. [Google Scholar] [CrossRef]
- Waghray, M.; Cui, Z.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, V.K.; Lebrecht, D.; Nicholson, A.G.; Wells, A.; Bhayani, H.; Gazdhar, A.; Tamm, M.; Venhoff, N.; Geiser, T.; Walker, U.A. Mitochondrial DNA mutations and respiratory chain dysfunction in idiopathic and connective tissue disease-related lung fibrosis. Sci. Rep. 2019, 9, 5500. [Google Scholar] [CrossRef]
- Terasaki, Y.; Terasaki, M.; Kanazawa, S.; Kokuho, N.; Urushiyama, H.; Kajimoto, Y.; Kunugi, S.; Maruyama, M.; Akimoto, T.; Miura, Y. Effect of H2 treatment in a mouse model of rheumatoid arthritis-associated interstitial lung disease. J. Cell. Mol. Med. 2019, 23, 7043–7053. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xia, G.; Guan, X.; Wang, L.; Qin, L.; Fu, M. Expression of Nrf2 protein in serum of patients with rheumatoid arthritis: A novel indicator for disease activity and disease prognosis. Clin. Biochem. 2023, 113, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vasarmidi, E.; Sarantoulaki, S.; Trachalaki, A.; Margaritopoulos, G.; Bibaki, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K. Investigation of key autophagy-and mitophagy-related proteins and gene expression in BALF cells from patients with IPF and RA-ILD. Mol. Med. Rep. 2018, 18, 3891–3897. [Google Scholar] [CrossRef]
- Hill, C.; Wang, Y. Autophagy in pulmonary fibrosis: friend or foe? GENES DISs 2022, 9, 1594–1607. [Google Scholar] [CrossRef]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed]
- Im, J.; Hergert, P.; Nho, R.S. Reduced FoxO3a expression causes low autophagy in idiopathic pulmonary fibrosis fibroblasts on collagen matrices. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L552–L561. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PloS one 2012, 7, e41394. [Google Scholar] [CrossRef] [PubMed]
- Tieyuan, Z.; Ying, Z.; Xinghua, Z.; Huimin, W.; Huagang, L. Piceatannol-mediated JAK2/STAT3 signaling pathway inhibition contributes to the alleviation of oxidative injury and collagen synthesis during pulmonary fibrosis. Int. Immunopharmacol. 2022, 111, 109107. [Google Scholar] [CrossRef]
- Sheng, H.; Lin, G.; Zhao, S.; Li, W.; Zhang, Z.; Zhang, W.; Yun, L.; Yan, X.; Hu, H. Antifibrotic mechanism of Piceatannol in bleomycin-induced pulmonary fibrosis in mice. Front. pharmacol. 2022, 13, 771031. [Google Scholar] [CrossRef]
- Ahangari, F.; Price, N.L.; Malik, S.; Chioccioli, M.; Bärnthaler, T.; Adams, T.S.; Kim, J.; Pradeep, S.P.; Ding, S.; Cosmos Jr, C. microRNA-33 deficiency in macrophages enhances autophagy, improves mitochondrial homeostasis, and protects against lung fibrosis. JCI insight 2023, 8. [Google Scholar] [CrossRef]
- Hodge, J.A.; Kawabata, T.T.; Krishnaswami, S.; Clark, J.D.; Telliez, J.-B.; Dowty, M.E.; Menon, S.; Lamba, M.; Zwillich, S. The mechanism of action of tofacitinib-an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 2016, 34, 318–328. [Google Scholar]
- Montero, P.; Milara, J.; Roger, I.; Cortijo, J. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int. J. Mol. Sci. 2021, 22, 6211. [Google Scholar] [CrossRef]
- Citera, G.; Mysler, E.; Madariaga, H.; Cardiel, M.H.; Castañeda, O.; Fischer, A.; Richette, P.; Chartrand, S.; Park, J.K.; Strengholt, S. Incidence rates of interstitial lung disease events in tofacitinib-treated rheumatoid arthritis patients: post hoc analysis from 21 clinical trials. J. Clin. Rheumatol. 2021, 27, e482. [Google Scholar] [CrossRef]
- Dowty, M.E.; Jesson, M.I.; Ghosh, S.; Lee, J.; Meyer, D.M.; Krishnaswami, S.; Kishore, N. Preclinical to clinical translation of tofacitinib, a Janus kinase inhibitor, in rheumatoid arthritis. J. Pharmacol. Exp. Ther. 2014, 348, 165–173. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Lee, J.L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-Tharp, S.M.; Gadina, M. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Milici, A.J.; Kudlacz, E.M.; Audoly, L.; Zwillich, S.; Changelian, P. Cartilage preservation by inhibition of Janus kinase 3 in two rodent models of rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, R.; Liu, X.; Xie, J.; Si, K.; Duan, L. Effects of matrine on JAK-STAT signaling transduction pathways in bleomycin-induced pulmonary fibrosis. Afr J Tradit Complement Altern Med 2013, 10, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Garton, M.; Kelly, C. Is Janus Kinase Inhibition the Future of the Management of Rheumatoid Arthritis-associated Interstitial Lung Disease? touchREVIEWS in Respiratory & Pulmonary Diseases 2022, 7. [Google Scholar]
- Florescu, A.; Gherghina, F.L.; Mușetescu, A.E.; Pădureanu, V.; Roșu, A.; Florescu, M.M.; Criveanu, C.; Florescu, L.-M.; Bobircă, A. Novel Biomarkers, Diagnostic and Therapeutic Approach in Rheumatoid Arthritis Interstitial Lung Disease—A Narrative Review. Biomedicines 2022, 10, 1367. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, M.; Li, X.; Zhang, J.; Wang, F.; Zhang, C.; Roden, A.; Ryu, J.H.; Warrington, K.J.; Sun, J. Canonical and noncanonical regulatory roles for JAK2 in the pathogenesis of rheumatoid arthritis-associated interstitial lung disease and idiopathic pulmonary fibrosis. FASEB J. 2022, 36, e22336. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.S.; Taylor, P.C.; Choy, E.H.; Sebba, A.; Quebe, A.; Knopp, K.L.; Porreca, F. The Jak/STAT pathway: A focus on pain in rheumatoid arthritis. In Proceedings of the Semin. Arthritis Rheum. 2021; pp. 278–284. [Google Scholar]
- He, L.; Qin, Q.; He, J.; Wang, H.; Hu, Y.; He, W.; Xu, B.; Zhou, G.; Shan, H.; Yang, B. ErMiao San inhibits angiogenesis in rheumatoid arthritis by suppressing JAK/STAT signaling pathways. Evid. Based Complementary Altern. Med. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Moura, R.A.; Fonseca, J.E. JAK inhibitors and modulation of B cell immune responses in rheumatoid arthritis. Front. Med. 2021, 7, 607725. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Javadian, M.; Babaloo, Z.; Baradaran, B. Janus kinase inhibitors: A therapeutic strategy for cancer and autoimmune diseases. J. Cell. Physiol. 2020, 235, 5903–5924. [Google Scholar] [CrossRef]
- Ng, B.; Dong, J.; D’Agostino, G.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Ko, N.S.; Tan, J.; Chothani, S.P.; Huang, B. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaaw1237. [Google Scholar] [CrossRef]
- Ng, B.; Dong, J.; Viswanathan, S.; Widjaja, A.A.; Paleja, B.S.; Adami, E.; Ko, N.S.; Wang, M.; Lim, S.; Tan, J. Fibroblast-specific IL11 signaling drives chronic inflammation in murine fibrotic lung disease. FASEB J. 2020, 34, 11802–11815. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.; Cook, S.A.; Schafer, S. Interleukin-11 signaling underlies fibrosis, parenchymal dysfunction, and chronic inflammation of the airway. Exp. Mol. Med. 2020, 52, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Camps, M.; Rückle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Françon, B. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef]
- Randis, T.M.; Puri, K.D.; Zhou, H.; Diacovo, T.G. Role of PI3Kδ and PI3Kγ in inflammatory arthritis and tissue localization of neutrophils. Eur. J. Immunol. 2008, 38, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Gruen, M.; Rose, C.; König, C.; Gajda, M.; Wetzker, R.; Bräuer, R. Loss of phosphoinositide 3-kinase γ decreases migration and activation of phagocytes but not T cell activation in antigen-induced arthritis. BMC Musculoskelet. Disord. 2010, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.-H.; Gelinas, R.; Wang, K.; Etheridge, A.; Piper, M.G.; Batte, K.; Dakhlallah, D.; Price, J.; Bornman, D.; Zhang, S. Systems biology of interstitial lung diseases: integration of mRNA and microRNA expression changes. BMC Medical Genom. 2011, 4, 1–20. [Google Scholar] [CrossRef]
- Nguyen, P.M.; Abdirahman, S.M.; Putoczki, T.L. Emerging roles for Interleukin-11 in disease. Growth Factors 2019, 37, 1–11. [Google Scholar] [CrossRef]
- Xu, D.H.; Zhu, Z.; Wakefield, M.R.; Xiao, H.; Bai, Q.; Fang, Y. The role of IL-11 in immunity and cancer. Cancer Lett. 2016, 373, 156–163. [Google Scholar] [CrossRef]
- Chung, J.H.; Cox, C.W.; Montner, S.M.; Adegunsoye, A.; Oldham, J.M.; Husain, A.N.; Vij, R.; Noth, I.; Lynch, D.A.; Strek, M.E. CT features of the usual interstitial pneumonia pattern: differentiating connective tissue disease–associated interstitial lung disease from idiopathic pulmonary fibrosis. Am. J. Roentgenol. 2018, 210, 307–313. [Google Scholar] [CrossRef]
- Dawson, J.; Fewins, H.; Desmond, J.; Lynch, M.; Graham, D. Predictors of progression of HRCT diagnosed fibrosing alveolitis in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2002, 61, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, D.S.; Park, I.-N.; Jang, S.J.; Kitaichi, M.; Nicholson, A.G.; Colby, T.V. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease–related subtypes. Am. J. Respir. Crit. Care Med. 2007, 175, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Takayanagi, N.; Sugiura, H.; Miyahara, Y.; Tokunaga, D.; Kawabata, Y.; Sugita, Y. Lung diseases directly associated with rheumatoid arthritis and their relationship to outcome. Eur. Respir. J. 2011, 37, 1411–1417. [Google Scholar] [CrossRef]
- Nakamura, Y.; Suda, T.; Kaida, Y.; Kono, M.; Hozumi, H.; Hashimoto, D.; Enomoto, N.; Fujisawa, T.; Inui, N.; Imokawa, S. Rheumatoid lung disease: prognostic analysis of 54 biopsy-proven cases. Respir. Med. 2012, 106, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Suhara, K.; Miyazaki, Y.; Okamoto, T.; Ishizuka, M.; Tsuchiya, K.; Inase, N. Fragmented gelsolins are increased in rheumatoid arthritis-associated interstitial lung disease with usual interstitial pneumonia pattern. Allergol. Int. 2016, 65, 88–95. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic diagram of risk factors that induce rheumatoid arthritis-interstitial lung disease (RA-ILD) and proposed pathogenesis. Although the exact pathophysiological mechanisms in which RA-ILD is developed remains unknown, several risk factors and plausible mechanisms have been proposed. Risk factors associated with RA-ILD include genetic variations, male sex, older age, race, smoking, exposure to pollutants, and anti-cyclic citrullinated peptide antibody (ACPA). Reported plausible pathophysiological mechanisms involve biological processes (e.g., fibroblast–myofibroblast transition [FMT], epithelial–mesenchymal transition [EMT], and immunological processes), signaling pathways (e.g., JAK/STAT and PI3K/Akt), and RA histopathology.
Figure 1.
Schematic diagram of risk factors that induce rheumatoid arthritis-interstitial lung disease (RA-ILD) and proposed pathogenesis. Although the exact pathophysiological mechanisms in which RA-ILD is developed remains unknown, several risk factors and plausible mechanisms have been proposed. Risk factors associated with RA-ILD include genetic variations, male sex, older age, race, smoking, exposure to pollutants, and anti-cyclic citrullinated peptide antibody (ACPA). Reported plausible pathophysiological mechanisms involve biological processes (e.g., fibroblast–myofibroblast transition [FMT], epithelial–mesenchymal transition [EMT], and immunological processes), signaling pathways (e.g., JAK/STAT and PI3K/Akt), and RA histopathology.
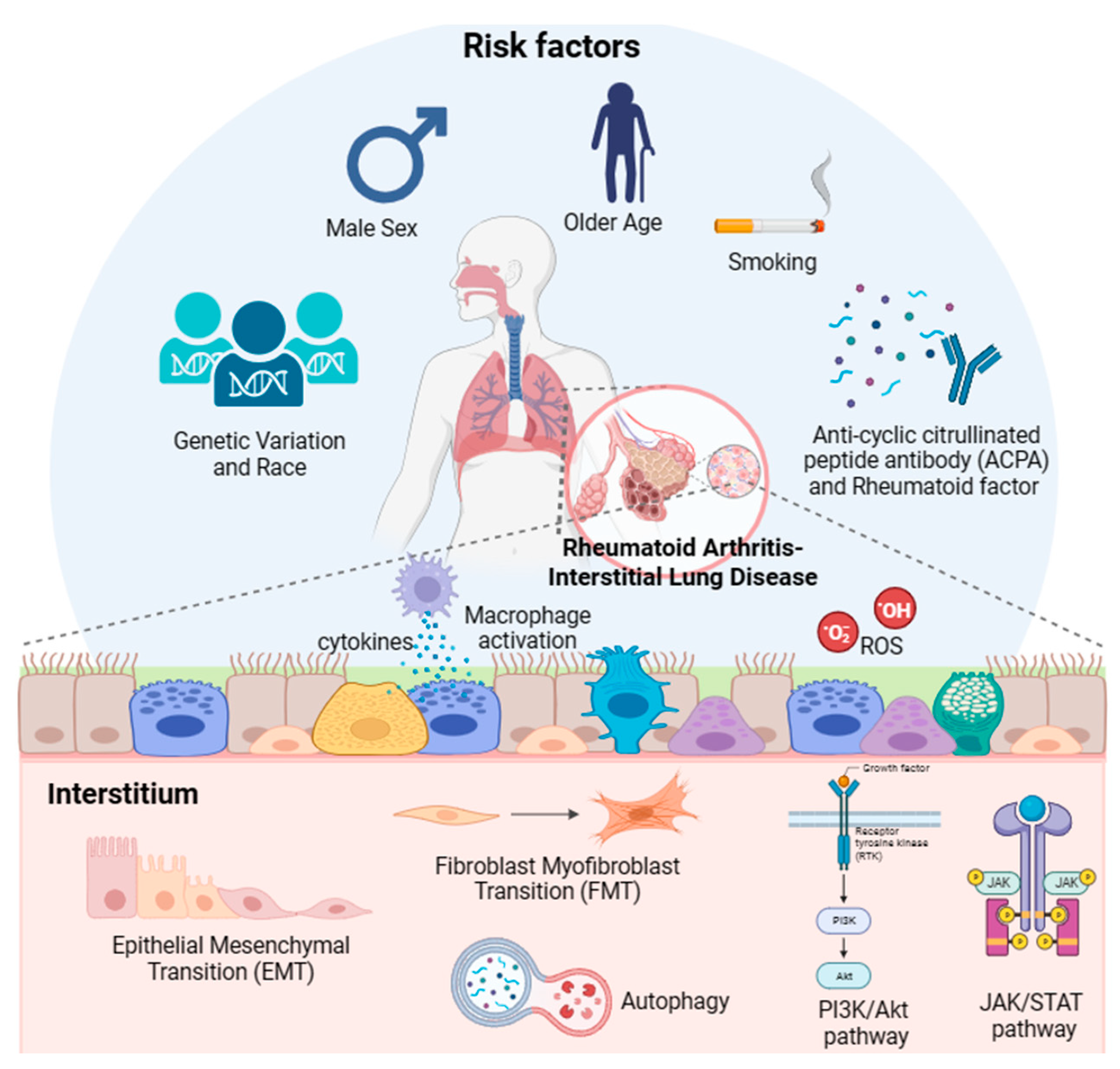
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



