
Submitted:
01 September 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
Pyrolysis is a thermochemical conversion process designed for biomass decomposition in an oxygen-free environment. It typically operates within a temperature window of 200 to 800°C and generates various byproducts, including gases, liquid fractions (both aqueous and organic) such as bio-oil, and solids like char. Bio-oil is a significant product of pyrolysis. It is characterised by a complex blend of organic compounds. The acidic nature of bio-oil primarily originates from volatile acids, such as acetic acid. The recovery of acetic acid from bio-oil facilitates the use of the extracted substance as a precious resource, hence enhancing its significance in the framework of a circular bioeconomy. Acetic acid finds widespread application in manufacturing commercially important chemicals like vinyl acetate monomers and formulating phytosanitary products due to its inherent herbicidal and antifungal properties. However, to be used, acetic acid has to be separate from the raw bio-oil. Separation methodologies, including nanofiltration and reverse osmosis, demonstrate potential in recovering acidic constituents from bio-oil due to their high selectivity and effective system management. This article offers a concise review about acetic production from lignocellulosic biomass from pyrolysis and its recovery through various membrane separation processes.
Keywords:
Lignocellulosic biomass
; pyrolysis
; pyrolytic oil
; acetic acid
; nanofiltration
; reverse osmosis
1. Introduction
Due to the growing concerns regarding greenhouse gas emissions and energy security issues, Quebec government has opted for a strategy that is more reliant on the harnessing of renewable biomass and biorefinery sector with a major focus on a sustainable bioeconomy [1]. Biorefineries utilise eco-friendly technologies in conjunction with biomass to generate biofuels and valuable products like bioenergies (biogas, bioethanol, biodiesel, etc.) and byproducts (bio-oil, syngas, etc.) while minimising their environmental footprint [2]. The lignocellulosic biomass is a promising and abundant renewable source. It consists of three primary components: cellulose, hemicellulose, and lignin. It is mainly generated from forest residues (slash, dead wood, maintenance cuttings, waste wood) and agricultural residues (straws, stalks, tops) [3]. Thermochemical processes, such as gasification and pyrolysis, are frequently employed in various biorefinery operations for biomass conversion into valuable products [4,5,6]. Gasification involves the partial oxidation of all the components of the biomass. It takes place in the presence of steam, air, or pure oxygen at temperatures ranging from 1023.15 to 1523.15 K and generates characteristics gases including carbon monoxide (CO), hydrogen (H2) and methane (CH4) [4]. Fast pyrolysis process yields mainly pyrolytic oil (also called bio-oil) which is about 40 to 75%, non-condensable gases and solid residues called biochar. This process is conducted under extreme temperature of approximately 773.15 K and fast condensing gas cooling [7]. The pyrolytic oil comprises two distinct phases: (i) an aqueous phase known as wood vinegar, which is abundant in decomposition byproducts of cellulose, hemicellulose, and small lignin molecules, and (ii) an organic discontinuous phase primarily composed of macromolecules with phenolic moieties resulting from the degradation of lignin [8]. Wood vinegar is another important byproduct of pyrolysis. It consists of a diverse array of chemical molecules, which encompass acetic acid, cyclopentanone, methoxyphenol, acetone, methanol, furfural, phenol, formic acid, levoglucosan, vanillin, and many others. The presence and abundance of these molecules are influenced by both the pyrolysis conditions and the composition of the biomass in terms of relative proportions of cellulose, hemicellulose and lignin [9,10].
The projected growth of the global acetic acid market is significant, with an estimated value of USD 15.5 billion expected by 2027, compared to approximately USD 10.5 billion in 2021. In fact, the utilisation of acetic acid covers a wide spectrum. It is extensively utilised in the production of vinyl acetate, which is further employed in the manufacturing of vinyl plastic, adhesives, textile finishes, and latex paints. [11]. In addition, it is used in agriculture due to its herbicidal activity against lamb’s-quarters, corn spurry and wild buckwheat and its antifungal activity against blue rot (Monilinia fructicola) and Aspergillus flavus responsible for the secretion of highly toxic and carcinogenic aflatoxins [12,13,14]. Acetic acid is traditionally produced through chemical processes using non-renewable raw materials such as n-butane oxidation, methanol carbonylation, and acetaldehyde oxidation (Kumbhar et al., 2019). But it can also be generated through biological processes through acetogenesis, acetic fermentation and yeast fermentation [15,16]. Chemical processes are characterised by their complexity and high costs, while biological approaches are known for being slow and relatively inefficient in terms of productivity [17,18]. A number of methods are currently available to extract acetic acid from bio-oil such as ion exchange resin (IER), activated carbon adsorption process and vacuum evaporation [19]. However, the latter methods, such as vacuum evaporation, are unsuitable for industrial implementation due to the phase change involved in the process, resulting in a significant energy expenditure during operation [20]. Thus, a paradigm shift was implemented in the industry with the democratisation of membrane processes. The advantages of this new perspective are the simple adjustment of operational variables, relatively easy scaling up and better energy management and consequently it is considered an environmentally sustainable treatment [21]. Regarding acetic acid, nanofiltration (NF) and reverse osmosis (RO) are recognised as the most selective processes among the pressure membrane techniques. The active layers of nanofiltration (NF) membranes exhibit a distinct pore size distribution ranging from 0.5 to 2 nm, providing selective retention capabilities for compounds with molecular weights spanning 150 to 1000 g/mol. Consequently, acetic acid, possessing a molecular weight of 60 g/mol, resides below the defined cut-off value of the NF membrane, thereby enabling efficient separation [22]. The permeation characteristics of solutes are governed by intricate mechanisms encompassing solute-membrane affinity, steric effects, electrostatic interactions, as well as operational parameters including applied pressure, feed composition, pH, and temperature [23,24]. To enhance comprehension regarding the distribution of acetic acid between the aqueous and organic phases produced during pyrolysis, the first chapter will involve a thorough analysis and subsequent discussion. Also, the effect of the chemical composition of the lignocellulosic biomass and the treatment conditions (pretreatment, temperature, residence time, heating rate, particle size, catalysts) on the yield will be studied through information available in the literature. The operation of nanofiltration and reverse osmosis for acetic acid separation will be presented in the second chapter.
2. Acetic acid production from lignocellulosic biomass pyrolysis
2.1. Pyrolysis reactions and multiphase behaviour of bio-oil
Biomass pyrolysis is most often considered as the combination of three fundamental states occurring simultaneously: (1) char formation, (2) depolymerisation and (3) fragmentation followed by secondary reactions (Collard and Blin, 2014). When exposed to elevated temperatures (773.15 K) for extended periods, the volatile compounds released become unstable and undergo additional reactions, specifically cracking, resulting in the creation of molecules with reduced molecular weights [25]. At temperatures below 673.15 K, secondary reactions can facilitate the recombination of volatile compounds, leading to the production of complex molecules with high molecular weights (secondary carbon) [26]. The prevalence of polycyclic aromatic hydrocarbons typifies the favourable conditions for recombination reactions (Table 1) [27]. To produce high yields of pyrolytic oil on a large scale, there are some parameters that must to be controlled: i) biomass particles less than 200 μm, (ii) a minimum heating of 80 K.min-1, (iii) external resistance (Biot number, Bi < 1) as it is the main factor affecting heat transfer. Considering the significance of both external and internal conduction, and (iv) moisture content less than 10 wt% in order to control the water content of the bio-oil [28]. A fast pyrolysis of mallee woody biomass between 623.15 and 853.15 K showed that 20 wt% of the biomass undergoes conversion, resulting in the formation of a water-soluble fraction consisting of 10 – 15 wt% water, 8 – 15 wt% organic compounds (including hydroxyacetaldehyde, acetol, acetic acid, formic acid, and methanol), 5 –10 wt% monophenols and furans, 6 – 15 wt% hydrolysable sugars probably derived from cellulose, and 6 – 15 wt% lignin oligomers [29]. The dissimilarities in both polarity and concentration levels observed among these distinct compound groups play a crucial role in the intricate multiphase characteristics of the bio-oil, resulting in the formation of a dense, opaque black oil phase and a less viscous, transparent aqueous phase [30]. As reported by Mohan, Pittman Jr and Steele [31], the upper aqueous phase exhibits a notable abundance of polar compounds derived from carbohydrates, while the lower, more viscous oil phase is primarily characterised by the presence of less polar chemicals derived from lignin. The multiphase behaviour of the bio-oil can also be explained by the condensing temperature. High condensation temperatures under short resistance time lead to biogas retention, low water content and high viscosity of the obtained bio-oil. In contrast, condensers running at lower temperatures contain a water phase with low molecular weight compounds [8]. From this aqueous phase, acetic acid can be effectively extracted due to its elevated polarity [32].
2.2. Chemical Conversion of the Main Biomass Constituents
The lignocellulosic biomass used in biorefinery is mainly produced from forestry waste (softwood and hardwood chips) and agricultural residues including gramineous lignocellulose (rice, wheat straw, corn straw, corn stalks, sugarcane bagasse, miscanthus and switchgrass) [34,35]. Cellulose establishes hydrogen bonds with hemicellulose or lignin molecules, whereas the interconnections between hemicellulose and lignin encompass both hydrogen and covalent bonds, thereby giving rise to resilient microfibres that serve as the structural framework of the cell wall, as described by Konan, Koffi [36] and Vorwerk, Somerville and Somerville [37]. The thermal degradation of biomass can be discerned in four distinct stages: the initiation of extract decomposition (< 490 K), hemicellulose decomposition (490 – 650 K), cellulose and lignin decomposition (650 – 780 K), and lignin decomposition (> 780 K) [8].
Hemicellulose is an important component of the biomass with a proportion that varies from 20% to 35% of which xylan is the most abundant compound. Xylan is a heteropolysaccharide classified as linear homoxylan, arabinoxylan, glucuronoxylan and glucuronoarabinoxylan. The backbone may consist of O-acetyl, α-L-arabinofuranosyl, α-1,2-glucuronic or 4-O-methylglucuronic acid substituents [38]. Degradation of xylan occurs mostly at temperatures between 493.15 – 588.15 K in an exothermic manner [39]. In the process of pyrolysis, acetic acid originates from the dissociation of acetyl groups that were initially linked to the xylose unit. Moreover, the main chain component of xylan can undergo decomposition, yielding intermediates featuring a hexatomic ring, such as xylan 4-O-methylglucuronic acid, which further decomposes due to the cleavage of the carboxyl group. Subsequently, these intermediates with hexatomic rings transform into a linear carbon chain that ultimately fragments into acetic acid [40]. When lignin is subjected to temperatures exceeding 473.15 K, it undergoes a preliminary radical initiation reaction that creates a phenoxy radical and a secondary alkyl aromatic radical due to the severing of weaker C – O bonds. During the propagation reaction, the typical initial radicals promote the release of OH radicals and thus an initial dehydration of the lignin structures with a corresponding increase in the degree of unsaturation and the formation of coumaryl and synaphylic alcohols. The phenoxy radicals can stabilise resonantly due to their antioxidant capacity. Upon surpassing 773.15 K in the de-volatilisation phases, the generation of polycyclic aromatic hydrocarbons (PAH) becomes prominent and progressively increases. This is primarily through radical mechanisms, leading to a continuous output of volatile components and a gradual decrease in oxygen and hydrogen content. The consistent interlinking and growth of these aromatic structures symbolise the creation of char residue. The concluding reactions involve all present radicals, including hydroxyl, alkyl, and alkyl aromatic radicals [41]. The kinetics of lignin de-volatilisation demonstrates the presence of acetic acid among other different components [42]. Chen, Li [43] pointed that acetic acid can also be generated by the deacetylation of 4-hydroxyphenylacetate in the lignin of sweet sorghum bagasse (Figure 1).
2.3. Operating conditions of pyrolysis influencing the yield of acetic acid
2.3.1. The composition of the raw materials
The quantity of acetic acid produced can vary according to the type and its holocellulose content (Table 2). In their investigation on Softwood Pinus armandii Franch, Wang, Lin [44] showed that the amount of acetic acid coming from hemicellulose is significantly higher than that from cellulose. Agricultural residues contain more hemicelluloses than woody biomass and therefore pyrolysis of various agricultural residues yields higher acetic acid than those produced from wood. The production of acetic acid (wt%) during fast pyrolysis of pine, eucalyptus, barley straw, timothy hay and reed canary grass has been demonstrated to be respectively about 2.2, 4.7, 3.2, 4.9, and 3.8. However, the organic liquid yields were higher for woody biomass and lower for alkali metal rich agro-biomass [30]. Garcia, Wang [45] studied the effect of feedstock variation on acetic acid yield during fast pyrolysis of woody biomass from a mallee and pine pellets at 773.15 K and reported yields (% by mass of bio-oil) of 5.73 and 2.66, respectively. As for Duman, Okutucu [46] they compared acetic acid production of cherry seed hull and cherry seeds. The results show that the cherry seed hull gives higher acetic acid yields than cherry seeds. The reason was that the former contains higher hemicellulose content than the second.
2.3.2. Biomass Particle Size
Biomass particle size significantly affects the apparent kinetics of biomass decomposition as well as the composition of the pyrolysates. Larger particle size causes a low rate of heat transfer across the particle diameter, resulting in a reduction in maximum bio-oil yield, while smaller size heats uniformly and optimises residence time [53]. An enhanced yield of acetic acid from larger particles is due to cleavage of the C3 unit in lignin consisting of phenylpropane (C6 – C3) units and to secondary reactions of primary volatile [54]. Bridgeman, Darvell [55] conducted a study which revealed that larger particle size fractions tend to accumulate more substantial, complex organic molecules such as cellulose. In contrast, the fractions with smaller particle sizes primarily contained a combination of smaller, soluble organic molecules and inorganic matter. The result is that the larger particle sizes have a higher acetic acid content. However, the yield of water from carbohydrate dehydration increases significantly with particle diameter and causes a significant reduction in the yield of all carbohydrate-derived compounds including acetic acid Garcia, Wang [45]. Yang, Wu and Wu [8] studied the effects of particle size with rice straw in a fluidized bed. Larger particle sizes (2 – 5 m) of rice straw fail to integrate homogeneously with the bed materials. Consequently, the likelihood of pyrolysis occurring is reduced due to the existence of smaller and more unevenly heated areas within the fluidized bed. Particles with diameters greater than 200 μm require 0.38 s to heat constituting nearly 15% of the proposed reaction duration of 2 – 2.5 seconds [28]. A proposed solution to solve this problem and have a high yield of acetic acid is the use of an ablative pyrolysis reactor.It offers the possibility of using large pieces of wood instead of only small particles due to the reactive process taking place at the surface layer of the biomass rather than the whole particle. Therefore, the reaction rates are not limited by the heat transfer through the entire particle [56].
2.3.3. Moisture Content
The correlation between the moisture content of biomass and its thermal degradation is substantial and significantly impact the physical properties and the final quality of the derived pyrolytic liquid. In an experiment involving spruce wood, it was found that a bio-oil yield of 39.7%, which was the highest recorded, corresponded with an initial moisture content of 60.5% and a reactor operating at a temperature of 689 K. This combination of conditions also produced the lowest viscosity [10]. In contrast, an increase in moisture content during the flash pyrolysis of beech wood (Fagus sylvatica) at 623.15 K from 0 to 26.2% resulted in a noticeable decrease in acetic acid yield from 6.64% to 6.06%. These results can be attributed to the obstruction caused by the additional thermal energy required for moisture evaporation, which in turn hinders particle heating [57].
2.3.4. Pretreatment of biomass
2.3.4.1. Chemical pretreatments
Acetic acid yields decreases when chemical pretreatment is applied to the biomass prior to pyrolysis. Chemical treatment of corn cobs with 1 wt% or 2 wt% H2SO4 and NaOH solutions at 373.15 K or 1 wt% H2O2 solution conducted at 353.15 K prior to fast pyrolysis caused passivation of alkali and alkaline earth metals. Results also showed the removal of hemicellulose and the decrease of the acetic acid yield, which is mainly derived from the deacetylation reaction of hemicellulose [58]. An evaluation of the impact of acid pretreatment, ranging from 0.5 M to 1.5 M, assisted by microwave heating between 373.15 K and 473.15 K, on the pyrolysis efficiency of moso bamboo sawdust revealed remarkable findings. As temperature, reaction time, and acid concentration escalated, the content of acetic acid significantly diminished. This outcome was primarily due to the detachment of acetyl groups from hemicellulose and the fragmentation of cellulose [59]. Another factor that affects the pyrolysis is the crystallinity of cellulose. The degree of polymerisation of cellulose is an important factor in determining the extent of its decomposition. It negatively influences the production of acetic acid derived from the disruption of the glucopyranose ring in cellulose. The more polymerised is the cellulose, the less acetic yield is obtained [60]. Although during chemical treatments amorphous components are broken down including hemicellulose, lignin, and non-crystalline regions of cellulose, the crystalline segments of cellulose predominantly remain unscathed and persist in a solid state [58].
2.3.4.2. Physical Pretreatments
The torrefaction of biomass at 473.15 – 573.15 K generates a less oxygenated and more stable bio-oil than that produced from conventional pyrolysis feedstocks. Above 473.15 K, the biomass browns and significant amounts of hemicellulose decompose generating carbon dioxide, acetic acid and additional moisture. At 553.15 K, carbon monoxide and hydrocarbons are generated, as well as phenols, cresols, and other heavier products [61]. Boateng and Mullen [62] examined the utility of torrefaction of wood and switchgrass pellets in a fluidized bed reactor as a feedstock for pyrolysis oil production. This study suggests that the acetic acid yield in pyrolysis oils produced from torrefaction-pretreated biomass is lower compared to those produced from raw biomass. Pyrolysis of cotton stalks after torrefaction pretreatment at 493.15 K and 553.15 K for 30 min produced a low aqueous phase yield and a decrease in acetic acid content from 29.35% to 15.56% for 493.15 K and 553.15 K, respectively, compared to 36.92% in the aqueous phase of untreated biomass [63]. The effect of hydrothermal pretreatment of beech wood performed at a gravity factor of 3.55 results in a decrease of acetic acid in the bio-oil derived from the pretreated biomass (3.85 wt%) compared to the untreated biomass (14.34 wt%). This is due to the solubilisation of a significant fraction of hemicellulose and part of the lignin by this pretreatment [64]. Physical pretreatment with the extruder of Douglas fir residues also showed that the crystallinity index increased slightly, attributed to the effects of heat and moisture content [65]. To overcome this limitation, many authors investigated biological pretreatment specially fungal pretreatment that has the potential to selectively degrade cellular components of biomass in a way that significantly influences the pyrolysis behaviour of biomass and the production of acetic acid.
2.3.4.3. Biological Pretreatments
Pretreatment of corn stover with Phanerochaete chrysosporium SHBCC D22643 showed interesting results. Within the initial two weeks of treatment, relative crystallinity escalates to a maximum of 50.24%, while a concomitant decrease in relative acetic acid content to 18.95% occurs, a significant reduction from the untreated control measure of 28.85%. Conversely, when the pretreatment duration extends to five weeks, the destruction of crystalline cellulose incurs an upsurge in relative acetic acid content to 31.25%, coupled with a decline in the relative crystallinity of the cellulose [66]. Notably, the use of white rot fungi in the fungal pretreatment of corn stover for fast catalytic pyrolysis yielded a substantial increase in acetic acid (151.8%), contrasting favourably with the yield from untreated biomass (106%) [67]. The phenomenon can be attributed to the depolymerisation and mineralisation of lignin polymers, facilitated by the secretion of two primary enzymes by white rot fungi: phenol oxidases (laccases) and peroxidases (manganese peroxidase and lignin peroxidase). Additionally, some of these white-rot fungi secrete accessory enzymes like cellobiose dehydrogenase, which help the degradation of cellulose and hemicellulose [68]. Moreover, the production of low molecular weight organic acids, predominantly oxalic acid, by a white rot fungus serves a dual purpose. It not only lowers the pH but also supplies the requisite H2O2 for hemicellulose degradation [69]. In summary, the application of pyrolysis can exploit this biological process to liberate the cellulose trapped within lignin and hemicellulose. This could lead to higher quality biomass, increased acetic acid yields, and a more favourable environmental impact, considering that biological processes are inherently more energy efficient than their chemical or thermal counterparts [70].
2.3.5. Catalysts
There are several methods of upgrading and improving the quality of bio-oil, including adding catalysts to the pyrolysis process. Fast pyrolysis of a corn cob sample mechanically mixed with 20 wt% ZnCl2 maximises the acetic acid yield with a value of 13.1 wt% [71]. The type of zeolite catalyst is important in the selection of the final products. Zeolite catalysts are responsible for enhanced dehydration reactions that cause the increase in the water content of the bio-oil as well as deoxygenation reactions that reduce the concentration of oxygenates in the bio-oil and potentially decrease the yield of the organic phase of the bio-oil in favour of hydrocarbon gases and coke. ZSM-5 zeolite increases the aromatic species as it diminishes the molecular weight of the bio-oil [64]. The removal of acetic acids present in bio-oil is facilitated during either in situ or ex situ upgrading by pyrolysis. This operation is achieved via the application of catalysts, either acidic (such as ZSM-5) or basic (like MgO). The process involves a distinct pathway that deoxygenates acetic acid through atomisation, subsequently yielding acetone, CO2, and water [72]. Additionally, deploying a NaY-type zeolite catalyst in the pyrolysis of bamboo allows the extraction of a light brown, single-phase liquid, which differs starkly from the dark brown pyrolytic oil produced in the absence of catalysis. This technique results in an increased yield of pyrolytic oil content, ranging between 64.1 – 68.8 wt%, contrasting with the 19.7 – 53.3 wt% obtained from direct pyrolysis. Following catalysis, the derived pyrolytic oil contains markedly fewer species, predominantly carboxylic and carbonyl compounds, among which acetic acid emerges as the main constituent. Its content is more than double compared to the results of non-catalytic pyrolysis [73]. The transformation of spruce wood pyrolysis vapours at 773.15 K in the presence of the Al-MCM-41 mesoporous catalyst, which carries a Si/Al ratio of 20, is distinguished by the abundant production of acetic acid [74]. It is also interesting that the presence of ash or alkaline earth metals, even at levels measured in ppm, can significantly influence the spectrum of pyrolysis products. These elements are particularly responsible for promoting the generation of low molecular weight species from cellulose, as opposed to anhydrous sugars [75].
2.3.6. Reactors
The type of reactor affects the composition of the bio-oils. Reactors for fast pyrolysis of biomass include the bubbling fluidized bed reactor, circulating fluidized bed reactor, cone reactors, screw reactors, and ablative reactors (Figure 2) [7]. With ablative pyrolysis, biomass undergoes fusion and/or sublimation reactions when it comes in direct contact with a hot solid surface [76]. Fast pyrolysis of whole wood chips (particle size < 1 mm) showed that the yield of acetic acid obtained by the ablative reactor was slightly lower compared to the yield obtained by a fluidized bed reactor. These results can be explained by the fact that the temperature in the fluidized bed reactor chamber was much higher (773.15 K) than that of the ablative reactor chamber (573.15 K). Therefore the fluidized bed reactor has an increasing selectivity for acetic acid [56]. Pyrolysis of acacia sawdust in an auger pyrolysis reactor system yielded 25% organic acid at 673.15 K with acetic acid being the majority compound [77]. Pyrolysis of cherry seed hulls as showed that the amount of acetic acid in aqueous phases in fixed bed reactors is respectively 80.82 mg/g and 78.88 mg/g for 573.15 K and 773.15 K. However, it was 73.32 mg/g, 80.57 mg/g and 96.25 mg/g respectively for 673.15 K, 773.15 K and 873.15 K during pyrolysis in fluidized bed reactors [46].
2.3.7. Temperature of pyrolysis
The determination of pyrolysis temperature exerts a substantial impact on the spatial distribution of acetic acid. As temperature escalates from 573.15 to 673.15 K, there is an observable amplification in the yields of acetic acid. Nevertheless, an appreciable drop in yield occurs at 873.15 K [46]. These empirical findings are aligned with Mabrouki, Abbassi [78] research, which illustrates that an increased temperature yield up to a specific limit exerts a propitious effect on yield. However, the occurrence of secondary cracking of volatile due to excessively high-temperature results in a surge in gas yield. Furthermore, pyrolysis vapours possessing a high molecular weight are triggered at temperatures exceeding 773.15 K, provoking secondary reactions [79]. When the temperature goes beyond 550 K, the yield of the oil phase rise up, reaching its peak at 700 K. Subsequently, the yield diminishes due to secondary reactions upon surpassing 700 K. Conversely, the yield of the aqueous phase displays an inverse relationship with temperature [8]. Employing distinct temperature ranges to recuperate the aqueous phase serves as an efficacious methodology for prefractionation and engendering a variety of chemicals. The yield of the aqueous phase diverges across the different temperature intervals: 11.44%, 31.49%, and 1.45% for the respective ranges of 363.15 to 503.15 K, 503.15 to 643.15 K, and 643.15 to 723.15 K. GC-MS analysis of the derived crude pyroligneous acids revealed that the organic acid content within the pyroligneous acid collected between 503.15 to 643.15 K accounted for 30.78% of the total mixture. Among these, acetic acid emerged as the major constituent, accounting for 22.62% [80].
According to Qi, Hu [73], the generation of the liquid phase during the pyrolysis of Neosinocalamus affinis was minimal at temperatures beneath 623 K. Within 623 and 773 K, an increase in pyrolysis temperature corresponded to an enhancement in yield, achieving an optimum yield of 34.1 wt% at 773 K. Beyond this threshold, the yield of the pyrolytic fluid declined significantly. This investigation also reveals that the acetic acid content was dominant among the diverse components extracted, especially within 623 K and 673 K. This elevated proportion was consistent with the high concentration of hydrogen gas in the gaseous products derived from pyrolysis due to the oxygen transfer to the acetic acid molecule. Additionally, the content of cyclopropanol acquired from the pyrolysis of Bambusa rigida was predominant at 773 K among the diverse components, while the 2,6-dimethoxyphenol content derived from Pubescens was maximal at 873 K [73].
2.3.8. Carrier Gas Flow Rate
The rate of flow is one of the multiple factors that contribute to the products distribution in fast biomass pyrolysis and has a more prominent impact on the oil phase than on the water phase [8]. Nitrogen plays a role in this process as a carrier gas, crafting an inert atmosphere that aids in the transfer of pyrolysis gases. This subsequently helps to bypass side reactions and vapour depolymerisation [81]. Specifically, a diminished nitrogen flow rate can lead to the sluggish transportation of pyrolysis vapours within the reactor, which potentially instigates secondary reactions [82]. Concurrently, an elevated flow rate can decrease the bio-oil yield due to some condensable vapours exiting the cooling unit prior to condensing [83].
2.3.9. Residence Time
In the context of fast pyrolysis reactions, time and temperature parameters are critical for the yield and composition of the resultant products. Fast pyrolysis necessitates a time frame of 2 – 2.5 seconds [28]. When examined under slow heating conditions of 3600 seconds, the yield of acetic acid remained unaffected by increasing temperature, providing a stark contrast to the trends observed under swift heating conditions (1 – 2 seconds). Here, the acetic acid content experiences a consistent reduction with the escalating pyrolysis temperature [46] (Table 3). Additionally, experimental studies conducted on pistachio shell pyrolysis at 773.15 K uncovered that a decreased nitrogen flow rate coupled with prolonged reaction times led to a decrease in pyroligneous acid yield. This reduction can be attributed to the secondary reactions of pyrolysis vapours, which yield non-condensable gaseous products [84].
2.3.10. Heating Rate
A high heating rate tends to favour the yield of the aqueous phase. Thermogravimetric analysis of the reaction of pyrolysis of cedar sawdust, coffee bean residue showed that hemicellulose mass loss at temperatures between 550-650k increased in parallel with increasing heating rates (1 -10 K/min) and consequently an increase in the yield of acetic acid [8,46]. The yields of raw wood vinegar (%) were 23.27, 24.45, 24.91, 27.45, 29.01 for heating rates (°C /min) 0.6, 0.7, 0.9, 1.4, 2.0 respectively [85].
3. Membrane filtration
Lignocellulosic biomass is a potentially promising feedstock to produce bio-oil pyrolysis. Bio-oil is a precursor for the production of biofuels and value-added chemicals. The extraction of acids from bio-oil may thus afford opportunities for improved applications and economic value. There are several methods to extract acetic acid from wood vinegar. Supercritical fluid extraction, liquid-liquid extraction and membrane processes are of the most common. However, since acetic acid is polar, the use of non-polar supercritical fluids such as carbon dioxide (CO2) is not recommended. Liquid-liquid extraction presents the disadvantage of expensive solvent regeneration and complex product recovery [86]. As for membrane processes, they are well known to be more economically and energy efficient. They also offer higher selectivity and improved system control.
3.1. Overview of Membrane Processes
Based on transmembrane pressure, filtration membranes can be classified into (i) low-pressure membranes generally below 100 – 200 kPa, and (ii) high-pressure membranes operating at transmembrane pressures above 200 kPa. The former includes microfiltration (0.1 – 10 μm) and ultrafiltration membranes (2 – 100 nm) while the second involves nanofiltration (0.5 – 2 nm) and reverse osmosis membranes. The variation in transmembrane pressure allows membrane systems to operate at constant permeate flow [87]. Darcy’s law describes the laminar flow through the pores of the porous membrane and the proportionality between the resulting flux density and the applied pressure difference (transmembrane pressure) [88] :
With J = membrane flux, Lp = hydrodynamic permeability of the solvent, µ = solution viscosity, and ∆P = transmembrane pressure.
The difference in concentration between the permeate and retentate leads to osmotic pressure that partially oppose the applied pressure Δp. The osmotic pressure represents the transfer of compounds from the less concentrated compartment (permeate) to the more concentrated one (retentate). The osmotic model used to calculate the permeation flux is given by the following equation [89]:
With ∆π = the osmotic pressure, ∆P = Transmembrane pressure, Rm = the hydraulic resistance of the membrane and µ = the dynamic viscosity.
Since the mass transport is convective in ultrafiltration (UF) membranes and diffusive in reverse osmotic (RO) membranes and the nanofiltration (NF) is between the two processes, the transfer of solutes in NF membranes can be convective or diffusive [90]. During membrane filtration and before reaching a steady state, components are transferred with a convective flux to the membrane surface. The flux is more important than the diffusion backflow to the solution which generates the effect of concentration polarisation [91]. In the polarisation layer, the larger the concentration gradient, the larger the back-diffusive transport. The permeation flux is described by the film model [92,93]:
With J = permeation flux, D = diffusion coefficient of the species considered, δ = thickness of the polarisation layer, = concentration at the membrane surface, = concentration in the permeate and = concentration in the feed solution, and = matter transfer coefficient.
The material transfer coefficient can be determined from the characteristics of the feed canal and the hydrodynamics within the unit cell according to the expression [94]:
With = = λSc1/3Re1/2, Sc = ν/D and Re = 2hQf/(νSf) are the dimensionless Sherwood, Schmidt and Reynolds numbers respectively, Qf = feed rate, Sf = cross-sectional area of the cell feed water channel, h = feed water channel height, ν = feed solution viscosity, D = diffusion coefficient of the species under consideration, and λ = dimensionless coefficients.
3.2. Operating conditions for the separation of acetic acid
3.2.1. Pressure
In nanofiltration (NF) and reverse osmosis (RO) processes, solute transportation across membranes is propelled by pressure, which in turn impacts the chemical release, permeate flux, and membrane fouling. Conventionally, as the transmembrane pressure escalates, so do flux and solute retention. This phenomenon was substantiated by experimental investigations using single solute solutions of acetic acid (7 wt%), which demonstrated a decrease in the flux of acetic acid solution through the membranes in the order DK > CE > MPF 34 > AG. Conversely, acetic acid retention adhered to the reverse order [95]. This occurrence aligns with the solution diffusion model predicated on thermodynamic equilibrium where the solvent flux (water) augments more rapidly than the solute flux (acetic acid) as the pressure rises, thus enhancing retention [90]. At elevated transmembrane pressures, an opposing effect is observed due to the reduction in the average pore size of the active layer of the membrane, thereby inducing a compression effect. This causes a delay in the transport rate of the component by bolstering the rejection, and thus decreases the permeation flux. Additionally, higher pressures necessitate larger investment in equipment such as pumps, thus amplifying operational costs [91]. About acetic separation from particular molecules in the bulk pyrolysate, it has been reported that the separation of acetic acid from glucose can be successfully achieved at high transmembrane pressures (40 and 58 bar). During the AFBO (aqueous fraction of bio-oil) model filtration with Desal DK and FMP 34 nanofiltration membranes, negative acetic acid retention was recorded, ranging between - 14 and - 7.7 [95]. Conversely, for the separation of acetic acid from monophenols and monosaccharides utilising a model solution that mirrors the composition of typical HTL (hydrothermal liquefaction)-derived hydrolysates from rice straw, a relatively low pressure (10 bar) sufficed to achieve a considerable rejection of monosaccharides and a minor rejection of acetic acid. Various membrane models showcased varying levels of rejection percentages. Reverse osmosis membranes could also be employed if the objective is the separation of acetic acid from monophenols, utilising a relatively low pressure to secure separation performance and high acetic acid recovery. Furthermore, aside from transmembrane pressure, osmotic pressure serves as an essential force that should be regulated to allow the permeate to traverse the membrane from the retentate side to the permeate side. Factors such as the friction of the permeate flow at the membrane or the osmotic pressure of the solution can result in a pressure drop between the inlet and outlet of a membrane system. The osmotic pressure is, in turn, dependent on solute concentrations and temperature. When sugar and acetic acid concentrations peak in a sugar maple hydrolysate solution, the osmotic pressure rises significantly, necessitating the application of higher pressure to sustain the same permeate flow rate [96]. Overall, to determine the final rejection of membranes, one must consider the two competing processes exhibiting the effect of pressure; On the one side, increasing pressure causes higher permeation of the solvent but solute transport is hindered by steric and electrical effects and therefore the rejection is increased. On the other side, solutes are transported to the membrane surface by increasing the pressure which causes a greater concentration polarisation effect and the solute tends to be transported across the membrane under a high concentration slope [97]. The retention of acetic acid increased from 9.99% to 15.78% in the pressure range of 220 kPa – 259 kPa due to the hydrophilic nature of the PA2-4040 membrane. In those conditions, water becomes more permeable (permeate flux increases) than solutes resulting in higher acetic acid retention [98].
3.2.2. Feed Solution
The efficiency of NF membrane separation is dictated by the concentration of the feed solution, which results in a decrease in permeation flux as the concentration rises [22,91]. Studies conducted using a model aqueous bio-oil fraction with concentrations up to 34 wt% of acetic acid, formic acid, hydroxy acetone, furfural, guaiacol, catechol, and glucose demonstrated that all nanofiltration and reverse osmosis membranes underwent irreversible damage due to the presence of guaiacol [95]. Given the same mass concentration, smaller molecules exert a greater osmotic pressure, as illustrated by the equivalent osmotic pressure (200 psi) of a 9.5 wt% glucose solution and a 16 wt% sucrose solution. Assessing the effect of the concentration factor — which is the quotient of the total solution volume in the feed tank before separation to the total feed volume as separation continues — on the permeate flow rate revealed that the permeate flow rate diminished with an increasing concentration factor. This led to a rise in osmotic pressure in direct proportion to the concentration of solutes remaining in the raffinate stream. The solutes included solids, polymers, sugars, acetic acid, and inorganic compounds. Initially, the feed tank contained an extract volume of 66 litres, with an osmotic pressure of 63.2 psi and an estimated acetic acid concentration of 4.62 g/L. Upon completion of the experiment, the remaining total volume in the feed tank (Vf) was reduced to 28 litres. The estimated acetic acid concentration was 5.19 g/L, and the osmotic pressure rose up to 150 psi. The permeate flow rate was reduced from 500 ml/min to 50 ml/min [96]. As for the separation of sugars and acetic acid (HAc) from the prehydrolysis liquor (PHL) of the kraft dissolving pulp process, it resulted in a concentration of sugars from 48 to 227 g/L at a volume reduction factor (VRF) of 5. Moreover about 80% to 90% of the acetic acid was recovered in the permeate. The concentration of acetic acid amplified from 10 to 50 g/L using the RO process, and an acetic acid retention of approximately 65.27% was achieved at 500 psi with a VRF of 12.56 [99].
3.2.3. Boundary Layer
The phenomenon of concentration polarisation significantly affects solute rejection, particularly by attenuating the effects of membrane charging [100]. This effect is further accentuated at the juncture of the membrane and the fluid, where a boundary layer exists. In this region, the fluid velocity is drastically reduced, and the flow regime is predominantly laminar, characterised by a low component transfer coefficient. To curtail the thickness of this boundary layer, particularly in solutions characterised by high viscosity and substantial mass transfer resistance, the rising of the feed rate is particularly effective. This increase in the feed rate amplifies the tangential velocity of the fluids, fostering a turbulent flow regime. The turbulent flow disrupts the boundary layer, facilitating an increased permeation flow rate. When such adjustments were implemented with an aqueous acetic acid solution, there was a marked increase in the permeation flux from 84.5 to 87 L.m2.h-1. This observation implies the existence of a mass transfer resistance, although its impact appears to be nominal. It further suggested that the conditions within the boundary layer were not substantially enough to be attributed to the reduced viscosity of the solution at the relatively low concentration of 2% [91].
In the context of separating furans and carboxylic acids from sugars within diluted acidic hydrolysates of rice straw, it was observed that a rise in the concentrations of xylose and arabinose in the permeate occurred because of the intensification of the concentration polarisation (CP) layer near the membrane surface. This intensification can be ascribed to the sustained rising of charge concentration over time. While applying a pressure of 34.3 bar momentarily disrupted this layer, the conclusion of the experiment revealed an enhanced concentration of xylose in the permeate. This increase was due to the reinforced convective transport towards the membrane under high operating pressure [20].
3.2.4. Effect of Time on Membrane Separation
Separation of monomers and oligomers of sugars (hexoses and pentoses), acetic acid, methanol, aromatics from aqueous biomass hydrolysate using an NF membrane with a molecular weight cutoff of 100 shows that the concentrations of solutes in the concentrate and permeate streams vary according to time. The concentrations of xylose in the concentrate stream rose from 5.9 g/L to 15 g/l after 360 min and from 0.04 to 0.1 g/L in the permeate stream. In addition, the rejection efficiency of acetate, methanol, furfural and hydroxymethylfurfural (HMF) from the permeate stream was less than 0.5. But the rejection rate increased with time. This reflected the increase in concentration in the retentate as an example of the acetate rejection rate which rose from 120 min and increased from 0.05 to 0.3 after 280 min [96].
3.2.5. Membranes
NF and RO membrane are polyamide (PA) based composite membranes that can be classified according to the structural monomer group into fully aromatic PAs with a rough surface and semi-aromatic PAs based on poly(piperazinamide) with a smooth surface (Figure 3) [101].
The semi-aromatic piperazine (NE40/70) and aromatic m-phenylene diamine (MPD) NF membranes were assessed for their chemical and physical properties using an acidic aqueous solution (15 wt% sulfuric acid) as a wastewater analogue from the melting process. Results from this investigation highlighted that the semi-aromatic membrane exhibited less chemical stability than its aromatic counterpart. Evidence for this was obtained through scanning electron microscopy (SEM) analysis, which identified surface detachment of the NE40/70 membranes from their support layer after 14 days of acid immersion. Further, these membranes exhibited smoother or more granulated surfaces than the NE90 membrane, thereby suggesting lower solubility/diffusivity in organic solutions. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra revealed a decrease in the C = O stretching amide bond peak with prolonging acid immersion time, a result that corroborates the surface morphology findings. Acid-catalysed hydrolysis was identified as the main agent responsible for converting amide groups into carboxyl and amine groups, leading to a decrease in contact angle and an increase in the absolute value of the zeta potential at the surfaces of NE40 and NE70 membranes. This study proposed a complex sequence of reactions for the acid-catalysed hydrolysis of the amide group, including spontaneous protonation of oxygen and nitrogen. The rate-determining step (RDS) was identified as the dissociative adsorption of the water molecule at the amide bond, leading to the degradation mechanism and deformation of the C – N bond, an exothermic reaction [102]. The treatment of carboxylic acid-rich agricultural wastewater digestates by the nanofiltration process demonstrated that the LF10 membrane (polyvinyl alcohol/polyamide, Nitto Denko) provided the maximum yield in terms of percent recovery of acetic and butyric acids when compared to piperazine-based polyamide membranes such as NF 270 (Polyamide, Dow Filmtec), HL, DL, DK (Polyamide/polysulfone, GE Osmonics) [103].
The ideal membrane is characterised by both high-water permeability and high selectivity. Even though the separation performance of NF and RO membranes varies, acetic acid rejection behaviour (98.6%) at 27.5 bar was observed for BW30XFR and NF90 membranes due to their active layers being composed of fully aromatic polyamide [94]. An interaction between acetic acid and the polyamide membrane through hydrogen bonding increases acid uptake [104]. However, polyamide membranes are sensitive to high concentrations of phenol compounds, such as guaiacol (∼1.5 wt%) and phenol (∼5 wt%), due to deprotonation of phenolic compounds to phenoxide ions. These ions could react with an amide group on the membrane, leading to ester formation. Conversely, cellulose acetate RO CE membranes turned pink after filtration of an AFBO model solution without guaiacol [95]. CD-polysulfone composite NF membrane showed minimal effect from high acetic acid concentration, resulting in a stable permeation flux (83.9 L.m-2.h-1) and high acetic acid rejection (99%) [91]. Furthermore, two commercial RO membranes (SWC5 and SWC6), made of polyamide, were evaluated for their resistance after six months of exposure to acid solutions. SEM analysis of the surface morphology revealed an increasing degree of surface roughness with the acid solution concentration, indicating a more severe degradation of the membranes under higher acid concentrations. These results were substantiated by FT-IR and XPS analysis, which demonstrated an increase in free carboxylic groups due to the hydrolysis of the amide bond and a high atomic percentage of carbon due to the absorption of acetic acid and acrylic acid. Consequently, in a strongly acidic environment, the increased electronegativity of oxygen causes a loss of electron density of carbon, making it much more sensitive to nucleophilic attack and leading to hydrolysis of the amide bond to NH2 and COOH [105,106]. Using an interfacial polymerisation process, hydrophilic SiO2 nanoparticles (HGPN-SiO2) acting as a modifying inorganic monomer were dispersed in an aqueous phase and then incorporated into the polyamide layer of thin-film nanocomposite (TFN) nanofiltration (NF) membranes through the ester bonds formed between hydroxyl groups (HGPN-SiO2) and acyl chloride groups (TMC). The SiO2-modified TFN NF membrane was able to increase permeate flux (2 times higher than that of the pure NF membrane) and the acid resistance. This consequently increased the SiO2-modified TFN NF membrane lifetime compared to of the NF membranes. These results can be attributed to the conversion of –Si–OH groups of HGPN –SiO2 into [Si (OH2)+] hydration state by capturing hydrogen ions to weaken their attacks on the carbonyl groups, on the one hand, and the formation of organic acid molecules after the hydrolysis of ester bonds, on the other hand, which would inhibit the hydrolysis of amide bonds [106]. In the range of experimental ethanol concentrations 0 g/L – 7.50 g/L, increasing the ethanol concentration resulted in a decrease in acetic acid retention. The weakening of chain segment entanglement increased the distance between polymer chains, and the phenomenon of membrane “swelling” when ethanol enters the polyamide network under pressure [98].
3.2.6. Temperature
The influence of temperature on the operational efficiency of filtration membranes is significant. iT primarily impacts solute transport, which includes convective, electromigratory, and diffusive mechanisms. These effects are largely attributed to cumulative changes in membrane parameters, with minor contributions (approximately 5%) stemming from alterations in the solvent viscosity and ion diffusivity [107]. A rising of the temperature may induce a direct effect. This phenomenon is described as the Arrhenius relationship that reveals enhanced mass transfer of both water and solutes, as well as an increased diffusion coefficient. Alternatively, an indirect effect might be observed, characterised by a less rigid polymer structure with an inclination for reorientation. Consequently, this alteration can enhance the impact of pH and KCl on membrane swelling [108]. Experimental investigations utilising DL (Osmonics, Minnetonka, MN) and TFCS (Koch Fluid Systems, San Diego, CA) to evaluate pure water permeability and rejection of several hydrophilic neutral organic solutes across a temperature range of 5 – 41°C exhibited an increase in the average pore radius and molecular weight cutoff. This trend suggests that thermal expansion of the polymer leads to modifications in their structure and morphology. Furthermore, the activation energies exhibited a similar positive trend with Stokes’s radius and molecular weight. The diffusion was then hindered in line with the free volume theory of activated gas transport [109].
Lyu, Fang [100] demonstrated that, an upsurge in temperature enhanced the separation efficiency of acetic acid from monosaccharides. On the contrary, a reduction in temperature was beneficial for NF membrane separation. In fact, there was a negligible impact of temperature on glucose rejection for RO membranes and tight NF membranes, such as DK and NF90. However, within a temperature range of 15 – 45°C, the rejection rate for glucose decreased for various membranes. Elevated temperatures have been proved to be advantageous for RO membrane separation, as the rejection of acetic acid significantly decreased for several membranes [104]. For the NTR729 membrane, temperature fluctuations did not affect the rejection of acetate, despite its ideal characteristics of high glucose flux and rejection paired with low acetate rejection [110]. This synthesis of findings underscores the need for further investigation into the dynamic and multifaceted influence of temperature on membrane performance.
3.2.7. Charge Characteristics
The Donnan exclusion principle, underpinned by charge separation of molecules, presents an interesting approach for molecules separation during Nanofiltration (NF) operations. The principle is based on the superior retention of charged molecules relative to their uncharged counterparts [20].
Feed solution pH is a paramount determinant in membrane filtration efficacy, specifically in the context of acetic acid separation. Despite the diameter of acetic acid (0.38 nm) being approximately a third of the NF membrane pores size (around 1 nm), reliance on steric exclusion alone does not guarantee acetic acid low retention. Its retention could exceed that of xylose with identical membranes when charge exclusion prevails [22]. As a monoprotic acid, acetic acid has a pKa value of 4.756 (In, 2006). The pH modifications have a pronounced influence on the dissociation degree of acetic acid. For instance, at pH 3, acetic acid predominantly exists in its neutral form (98.3%), with a mere 1.7% deprotonated into acetate. However, at pH 9, negatively charged acetate drastically increases to 99.994%, while the neutral acetic acid plummets to 0.006%. When the membrane isoelectric point falls below the solution pH, the membrane acquires a negative charge, thereby eliciting the Donnan effect which is based on electrical interactions. This induces a repulsion of the negatively charged molecules (such as acetate) in the solution by the negatively charged membrane. The result of this phenomenon can be observed in the increased retention of acetic acid, which escalates from -3% at pH 2.9 to over 90% at pH 9.1 [22]. For effective acetic acid separation from sugars, it becomes critical to counteract the exclusion or retention of inhibitors by the Donnan effect. One strategy involves employing pH 3 during the separation of furans and carboxylic acids from sugars in dilute acidic hydrolysates. This encourages the predominance of the sieving mechanism during NF, with the resultant low acid retention [20]. An examination of pH impact on acetic acid retention with a synthetic solution during RO process at 375 kPa revealed an upsurge in acetic acid retention from 22.46% to 98.33% within the pH range of 3.5 – 9.7, and so without altering the permeate flux [98]. The synthetic solution was composed of glucose, furfural, acetic acid, and ethanol at concentrations of 2.20 g/L, 0.40 g/L, 1.35 g/L, and 4.10 g/L respectively. Given that acetic acid gets deprotonated into negatively charged acetate above pH 4.756, and the RO PA2-4040 membrane also assumes a negative charge, the Donnan effect emerges as the critical determinant in the separation of charged solutes such as acetate.
4. Conclusion
The fibre content of lignocellulosic biomass namely higher cellulose and hemicellulose content is the main source contributing to the production of higher acetic acid yields. Biological pretreatment of biomass seems to improve acetic acid yield in contrast to thermal and chemical pretreatment. Lower temperature, rapid heating rates and fluidized bed reactors in the presence of some metal catalysts give the maximum acetic acid content. As for the separation of acetic acid from the aqueous phase, using acid separation NF/RO membranes seems feasible with the implementation of two important parameters: (i) a strong polymer formulation to concentrate the organic compounds namely phenols, sugars and furans in the retentate, and (ii) a relatively high transmembrane pressure which is required to achieve low acetic acid retention and high permeate flux. Acetic acid retention was significantly influenced by pH for all membranes due to the dominant role of the Donnan effect.
Author Contributions
Conceptualisation, Writing - Original Draft: Ghita Bennani; Writing - Review & Editing: Adama Ndao, Delon Konan; Review & Editing: Patrick Brassard, Etienne Leroux, Stéphane Godbout; Funding Acquisition, Supervision: Kokou Adjallé.
Funding
This project has been funded by the Ministère de l’Agriculture, des Pêcheries et de l’Alimentation (MAPAQ) under the project number IA119519.
Acknowledgments
The authors would like to acknowledge the Institut National de la Recherche Scientifique (INRS) and the Ministère de l’Agriculture, des Pêcheries et de l’Alimentation (MAPAQ) for it financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rafione, T., Développement d’une bioraffinerie forestière intégrée et verte. 2014, École Polytechnique de Montréal.
- Laurent, P., et al., Le bioraffinage, une alternative prometteuse à la pétrochimie. BASE, 2011.
- Ballerini, D., Les biocarburants: répondre aux défis énergétiques et environnementaux des transports. 2011: Editions Technip.
- Studer, M. and P. Poldervaart, Nouvelles voies dans le bioraffinage du bois.
- Alauddin, Z.A.B.Z., P. Lahijani, M. Mohammadi, and A.R. Mohamed, Gasification of lignocellulosic biomass in fluidized beds for renewable energy development: A review. Renewable and Sustainable Energy Reviews, 2010. 14(9): p. 2852-2862. [CrossRef]
- Jones, M., A. Gandia, S. John, and A. Bismarck, Leather-like material biofabrication using fungi. Nature Sustainability, 2021. 4(1): p. 9-16. [CrossRef]
- Venderbosch, R. and W. Prins, Fast pyrolysis technology development. Biofuels, bioproducts and biorefining, 2010. 4(2): p. 178-208. [CrossRef]
- Yang, S., M. Wu, and C. Wu, Application of biomass fast pyrolysis part I: Pyrolysis characteristics and products. Energy, 2014. 66: p. 162-171. [CrossRef]
- Grams, J., Chromatographic analysis of bio-oil formed in fast pyrolysis of lignocellulosic biomass. Reviews in Analytical Chemistry, 2020. 39(1): p. 65-77. [CrossRef]
- Demirbas, A., Biorefineries: Current activities and future developments. Energy conversion and management, 2009. 50(11): p. 2782-2801. [CrossRef]
- Vidra, A. and Á. Németh, Bio-produced acetic acid: A review. Periodica Polytechnica Chemical Engineering, 2018. 62(3): p. 245-256.
- Joachim, U.d.M.b., M.N. Thaddée, and M. Lelo, Inhibition du développement de l’Aspergillus flavus par l’acide acétique: Analyse de trois expériences réalisées à Kinshasa-RD Congo. 2020.
- Ivany, J.A., Acetic acid for weed control in potato (Solanum tuberosum L.). Canadian Journal of Plant Science, 2010. 90(4): p. 537-542. [CrossRef]
- Chu, C.-L., W.-T. Liu, and T. Zhou, Fumigation of sweet cherries with thymol and acetic acid to reduce postharvest brown rot and blue mold rot. Fruits, 2001. 56(2): p. 123-130. [CrossRef]
- Merli, G., A. Becci, A. Amato, and F. Beolchini, Acetic acid bioproduction: The technological innovation change. Science of the Total Environment, 2021. 798: p. 149292. [CrossRef]
- Kumbhar, G., et al., DIFFERENT ROUTES FOR PRODUCTION OF ACETIC ACID–A CASE STUDY. 2019.
- Hidalgo, C., et al., Effect of barrel design and the inoculation of Acetobacter pasteurianus in wine vinegar production. International Journal of Food Microbiology, 2010. 141(1-2): p. 56-62. [CrossRef]
- Oh, S.-J., G.-G. Choi, and J.-S. Kim, Production of acetic acid-rich bio-oils from the fast pyrolysis of biomass and synthesis of calcium magnesium acetate deicer. Journal of Analytical and Applied Pyrolysis, 2017. 124: p. 122-129. [CrossRef]
- Huang, H.-J., S. Ramaswamy, U. Tschirner, and B. Ramarao, A review of separation technologies in current and future biorefineries. Separation and purification technology, 2008. 62(1): p. 1-21. [CrossRef]
- Weng, Y.-H., et al., Separation of furans and carboxylic acids from sugars in dilute acid rice straw hydrolyzates by nanofiltration. Bioresource technology, 2010. 101(13): p. 4889-4894. [CrossRef]
- Pinelo, M., G. Jonsson, and A.S. Meyer, Membrane technology for purification of enzymatically produced oligosaccharides: molecular and operational features affecting performance. Separation and Purification Technology, 2009. 70(1): p. 1-11. [CrossRef]
- Weng, Y.-H., et al., Separation of acetic acid from xylose by nanofiltration. Separation and Purification Technology, 2009. 67(1): p. 95-102. [CrossRef]
- Baker, R.W., Membrane technology and applications. 2012: John Wiley & Sons.
- Bellona, C., J.E. Drewes, P. Xu, and G. Amy, Factors affecting the rejection of organic solutes during NF/RO treatment—a literature review. Water research, 2004. 38(12): p. 2795-2809. [CrossRef]
- Bridgwater, A., Principles and practice of biomass fast pyrolysis processes for liquids. Journal of analytical and applied pyrolysis, 1999. 51(1-2): p. 3-22. [CrossRef]
- Bridgwater, A.V., D. Meier, and D. Radlein, An overview of fast pyrolysis of biomass. Organic geochemistry, 1999. 30(12): p. 1479-1493. [CrossRef]
- Morf, P., P. Hasler, and T. Nussbaumer, Mechanisms and kinetics of homogeneous secondary reactions of tar from continuous pyrolysis of wood chips. Fuel, 2002. 81(7): p. 843-853. [CrossRef]
- Van de Velden, M., et al., Fundamentals, kinetics and endothermicity of the biomass pyrolysis reaction. Renewable energy, 2010. 35(1): p. 232-242.
- Garcia-Perez, M., et al., Effects of temperature on the formation of lignin-derived oligomers during the fast pyrolysis of Mallee woody biomass. Energy & Fuels, 2008. 22(3): p. 2022-2032. [CrossRef]
- Oasmaa, A., et al., Pyrolysis oil multiphase behavior and phase stability: a review. Energy & Fuels, 2016. 30(8): p. 6179-6200. [CrossRef]
- Mohan, D., C.U. Pittman Jr, and P.H. Steele, Pyrolysis of wood/biomass for bio-oil: a critical review. Energy & fuels, 2006. 20(3): p. 848-889. [CrossRef]
- Vitasari, C.R., G.W. Meindersma, and A.B. De Haan, Water extraction of pyrolysis oil: The first step for the recovery of renewable chemicals. Bioresource technology, 2011. 102(14): p. 7204-7210. [CrossRef]
- Collard, F.-X. and J. Blin, A review on pyrolysis of biomass constituents: Mechanisms and composition of the products obtained from the conversion of cellulose, hemicelluloses and lignin. Renewable and Sustainable Energy Reviews, 2014. 38: p. 594-608. [CrossRef]
- Bentsen, N.S., C. Felby, and B.J. Thorsen, Agricultural residue production and potentials for energy and materials services. Progress in energy and combustion science, 2014. 40: p. 59-73. [CrossRef]
- Lauri, P., et al., Woody biomass energy potential in 2050. Energy policy, 2014. 66: p. 19-31. [CrossRef]
- Konan, D., et al., An Overview of Extrusion as a Pretreatment Method of Lignocellulosic Biomass. Energies, 2022. 15(9). [CrossRef]
- Vorwerk, S., S. Somerville, and C. Somerville, The role of plant cell wall polysaccharide composition in disease resistance. Trends in plant science, 2004. 9(4): p. 203-209. [CrossRef]
- Aspinall, G.O., Chemistry of cell wall polysaccharides, in Carbohydrates: Structure and function. 1980, Elsevier. p. 473-500.
- Yang, H., et al., Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel, 2007. 86(12-13): p. 1781-1788. [CrossRef]
- Wang, S.-r., T. Liang, B. Ru, and X.-j. Guo, Mechanism of xylan pyrolysis by Py-GC/MS. Chemical research in Chinese universities, 2013. 29: p. 782-787.
- Faravelli, T., A. Frassoldati, G. Migliavacca, and E. Ranzi, Detailed kinetic modeling of the thermal degradation of lignins. Biomass and bioenergy, 2010. 34(3): p. 290-301. [CrossRef]
- Jakab, E., O. Faix, and F. Till, Thermal decomposition of milled wood lignins studied by thermogravimetry/mass spectrometry. Journal of Analytical and Applied Pyrolysis, 1997. 40: p. 171-186. [CrossRef]
- Chen, D., et al., Advancing biomass pyrolysis by torrefaction pretreatment: Linking the productions of bio-oil and oxygenated chemicals to torrefaction severity. Fuel, 2022. 330: p. 125514. [CrossRef]
- Wang, S., et al., Structural characterization and pyrolysis behavior of cellulose and hemicellulose isolated from softwood Pinus armandii Franch. Energy & Fuels, 2016. 30(7): p. 5721-5728. [CrossRef]
- Garcia, P.M., et al., Fast pyrolysis of oil mallee woody biomass: effect of temperature on the yield and quality of pyrolysis products. Industrial & engineering chemistry research, 2008. 47(6): p. 1846-1854.
- Duman, G., et al., The slow and fast pyrolysis of cherry seed. Bioresource technology, 2011. 102(2): p. 1869-1878. [CrossRef]
- Casoni, A.I., et al., Pyrolysis of sunflower seed hulls for obtaining bio-oils. Bioresource Technology, 2015. 177: p. 406-409. [CrossRef]
- Lyu, G., S. Wu, and H. Zhang, Estimation and comparison of bio-oil components from different pyrolysis conditions. Frontiers in Energy Research, 2015. 3: p. 28. [CrossRef]
- Lv, G. and S. Wu, Analytical pyrolysis studies of corn stalk and its three main components by TG-MS and Py-GC/MS. Journal of Analytical and Applied Pyrolysis, 2012. 97: p. 11-18. [CrossRef]
- Maschio, G., C. Koufopanos, and A. Lucchesi, Pyrolysis, a promising route for biomass utilization. Bioresource Technology;(United Kingdom), 1992. 42(3). [CrossRef]
- Jung, S.-H., B.-S. Kang, and J.-S. Kim, Production of bio-oil from rice straw and bamboo sawdust under various reaction conditions in a fast pyrolysis plant equipped with a fluidized bed and a char separation system. Journal of Analytical and Applied Pyrolysis, 2008. 82(2): p. 240-247. [CrossRef]
- Martins, J.P.G., et al., Study of pequi peel pyrolysis: Thermal decomposition analysis and product characterization. Biomass and Bioenergy, 2021. 149: p. 106095. [CrossRef]
- Isahak, W.N.R.W., M.W. Hisham, M.A. Yarmo, and T.-y.Y. Hin, A review on bio-oil production from biomass by using pyrolysis method. Renewable and sustainable energy reviews, 2012. 16(8): p. 5910-5923. [CrossRef]
- Suriapparao, D.V. and R. Vinu, Effects of biomass particle size on slow pyrolysis kinetics and fast pyrolysis product distribution. Waste and biomass valorization, 2018. 9: p. 465-477. [CrossRef]
- Bridgeman, T., et al., Influence of particle size on the analytical and chemical properties of two energy crops. Fuel, 2007. 86(1-2): p. 60-72. [CrossRef]
- Luo, G., et al., Pyrolysis of whole wood chips and rods in a novel ablative reactor. Fuel, 2017. 194: p. 229-238. [CrossRef]
- Beaumont, O. and Y. Schwob, Influence of physical and chemical parameters on wood pyrolysis. Industrial & Engineering Chemistry Process Design and Development, 1984. 23(4): p. 637-641.
- Zheng, A., et al., Quantitative comparison of different chemical pretreatment methods on chemical structure and pyrolysis characteristics of corncobs. Journal of the Energy Institute, 2018. 91(5): p. 676-682. [CrossRef]
- Wang, Y., et al., Properties and pyrolysis behavior of moso bamboo sawdust after microwave-assisted acid pretreatment. Journal of Analytical and Applied Pyrolysis, 2018. 129: p. 86-92. [CrossRef]
- Piskorz, J., D.S.A. Radlein, D.S. Scott, and S. Czernik, Pretreatment of wood and cellulose for production of sugars by fast pyrolysis. Journal of Analytical and Applied Pyrolysis, 1989. 16(2): p. 127-142. [CrossRef]
- Ciolkosz, D. and R. Wallace, A review of torrefaction for bioenergy feedstock production. Biofuels, Bioproducts and Biorefining, 2011. 5(3): p. 317-329. [CrossRef]
- Boateng, A. and C. Mullen, Fast pyrolysis of biomass thermally pretreated by torrefaction. Journal of Analytical and Applied Pyrolysis, 2013. 100: p. 95-102. [CrossRef]
- Chen, D., et al., Torrefaction of biomass stalk and its effect on the yield and quality of pyrolysis products. Fuel, 2015. 159: p. 27-32. [CrossRef]
- Stephanidis, S., et al., Catalytic upgrading of lignocellulosic biomass pyrolysis vapours: Effect of hydrothermal pre-treatment of biomass. Catalysis Today, 2011. 167(1): p. 37-45. [CrossRef]
- Fu, Y., et al., Novel micronized woody biomass process for production of cost-effective clean fermentable sugars. Bioresource technology, 2018. 260: p. 311-320. [CrossRef]
- Sun, Z., et al., Effect of pretreatment with Phanerochaete chrysosporium on physicochemical properties and pyrolysis behaviors of corn stover. Bioresource Technology, 2022. 361: p. 127687. [CrossRef]
- Yu, Y., et al., Improving the conversion of biomass in catalytic fast pyrolysis via white-rot fungal pretreatment. Bioresource technology, 2013. 134: p. 198-203. [CrossRef]
- Zhu, N., et al., Comparative analysis of the secretomes of Schizophyllum commune and other wood-decay basidiomycetes during solid-state fermentation reveals its unique lignocellulose-degrading enzyme system. Biotechnology for biofuels, 2016. 9: p. 1-22. [CrossRef]
- Xu, P., et al., Cadmium induced oxalic acid secretion and its role in metal uptake and detoxification mechanisms in Phanerochaete chrysosporium. Applied microbiology and biotechnology, 2015. 99: p. 435-443. [CrossRef]
- Yang, X., et al., Effect of biopretreatment on thermogravimetric and chemical characteristics of corn stover by different white-rot fungi. Bioresource technology, 2010. 101(14): p. 5475-5479. [CrossRef]
- Oh, S.-J., S.-H. Jung, and J.-S. Kim, Co-production of furfural and acetic acid from corncob using ZnCl2 through fast pyrolysis in a fluidized bed reactor. Bioresource technology, 2013. 144: p. 172-178. [CrossRef]
- Psarras, A., et al., Acetic acid conversion reactions on basic and acidic catalysts under biomass fast pyrolysis conditions. Molecular Catalysis, 2019. 465: p. 33-42. [CrossRef]
- Qi, W., et al., Catalytic pyrolysis of several kinds of bamboos over zeolite NaY. Green chemistry, 2006. 8(2): p. 183-190. [CrossRef]
- Adam, J., et al., Pyrolysis of biomass in the presence of Al-MCM-41 type catalysts. Fuel, 2005. 84(12-13): p. 1494-1502. [CrossRef]
- Patwardhan, P.R., J.A. Satrio, R.C. Brown, and B.H. Shanks, Influence of inorganic salts on the primary pyrolysis products of cellulose. Bioresource technology, 2010. 101(12): p. 4646-4655. [CrossRef]
- Lede, J., J. Panagopoulos, and J. Villermaux, Experimental Measurement of ablation rate of wood pieces, undergoing fast pyrolysis by contact with a heated wall. Prepr. Pap., Am. Chem. Soc., Div. Fuel Chem.;(United States), 1983. 38(CONF-830814-).
- Ahmed, A., et al., Sawdust pyrolysis from the furniture industry in an auger pyrolysis reactor system for biochar and bio-oil production. Energy Conversion and Management, 2020. 226: p. 113502. [CrossRef]
- Mabrouki, J., et al., Simulation of biofuel production via fast pyrolysis of palm oil residues. Fuel, 2015. 159: p. 819-827. [CrossRef]
- Heo, H.S., et al., Bio-oil production from fast pyrolysis of waste furniture sawdust in a fluidized bed. Bioresource technology, 2010. 101(1): p. S91-S96. [CrossRef]
- Wei, Q., X. Ma, and J. Dong, Preparation, chemical constituents and antimicrobial activity of pyroligneous acids from walnut tree branches. Journal of analytical and applied pyrolysis, 2010. 87(1): p. 24-28. [CrossRef]
- Uzun, B.B., A.E. Pütün, and E. Pütün, Fast pyrolysis of soybean cake: Product yields and compositions. Bioresource technology, 2006. 97(4): p. 569-576. [CrossRef]
- Laougé, Z.B., A.S. Çığgın, and H. Merdun, Optimization and characterization of bio-oil from fast pyrolysis of Pearl Millet and Sida cordifolia L. by using response surface methodology. Fuel, 2020. 274: p. 117842.
- Gupta, G.K. and M.K. Mondal, Bio-energy generation from sagwan sawdust via pyrolysis: product distributions, characterizations and optimization using response surface methodology. Energy, 2019. 170: p. 423-437. [CrossRef]
- Açıkalın, K., F. Karaca, and E. Bolat, Pyrolysis of pistachio shell: Effects of pyrolysis conditions and analysis of products. Fuel, 2012. 95: p. 169-177. [CrossRef]
- Ratanapisit, J., et al., Preliminary evaluation of production and characterization of wood vinegar from rubberwood. Songklanakarin Journal of Science & Technology, 2009. 31(3).
- Sarchami, T., N. Batta, and F. Berruti, Production and separation of acetic acid from pyrolysis oil of lignocellulosic biomass: a review. Biofuels, Bioproducts and Biorefining, 2021. 15(6): p. 1912-1937. [CrossRef]
- Guo, W., H.-H. Ngo, and J. Li, A mini-review on membrane fouling. Bioresource technology, 2012. 122: p. 27-34. [CrossRef]
- Giorno, L., E. Drioli, and H. Strathmann, Characterization of Porous and Dense Membranes. Encyclopedia of Membranes, 2016: p. 362-372. [CrossRef]
- Espinasse, B., Approche théorique et expérimentale de la filtration tangentielle de colloïdes: flux critique et colmatage. 2003, Université Paul Sabatier-Toulouse III.
- Pontalier, P.-Y., A. Ismail, and M. Ghoul, Mechanisms for the selective rejection of solutes in nanofiltration membranes. Separation and purification technology, 1997. 12(2): p. 175-181. [CrossRef]
- Baruah, K. and S. Hazarika, Separation of acetic acid from dilute aqueous solution by nanofiltration membrane. Journal of Applied Polymer Science, 2014. 131(15). [CrossRef]
- Blatt, W.F., A. Dravid, A.S. Michaels, and L. Nelsen, Solute polarization and cake formation in membrane ultrafiltration: causes, consequences, and control techniques, in Membrane Science and Technology: Industrial, Biological, and Waste Treatment Processes. 1970, Springer. p. 47-97. [CrossRef]
- In, C., Handbook of Chemistry and Physics, ; Lide, DR, Ed. 2006, Taylor and Francis Group LLC: New York.
- Wu, H., et al., Performance characterization of nanofiltration, reverse osmosis, and ion exchange technologies for acetic acid separation. Separation and Purification Technology, 2021. 265: p. 118108. [CrossRef]
- Teella, A., G.W. Huber, and D.M. Ford, Separation of acetic acid from the aqueous fraction of fast pyrolysis bio-oils using nanofiltration and reverse osmosis membranes. Journal of Membrane Science, 2011. 378(1-2): p. 495-502. [CrossRef]
- Liu, S., T.E. Amidon, and C. David Wood, Membrane filtration: concentration and purification of hydrolyzates from biomass. Journal of biobased materials and bioenergy, 2008. 2(2): p. 121-134. [CrossRef]
- Gherasim, C.-V., J. Cuhorka, and P. Mikulášek, Analysis of lead (II) retention from single salt and binary aqueous solutions by a polyamide nanofiltration membrane: Experimental results and modelling. Journal of membrane science, 2013. 436: p. 132-144. [CrossRef]
- Chen, C., et al., A method for concentration of monosaccharide and removal of inhibitors during hydrolysate pretreatment for improved bioethanol production. Journal of Cleaner Production, 2020. 260: p. 120999. [CrossRef]
- Ahsan, L., M.S. Jahan, and Y. Ni, Recovering/concentrating of hemicellulosic sugars and acetic acid by nanofiltration and reverse osmosis from prehydrolysis liquor of kraft based hardwood dissolving pulp process. Bioresource Technology, 2014. 155: p. 111-115. [CrossRef]
- Lyu, H., et al., Monophenols separation from monosaccharides and acids by two-stage nanofiltration and reverse osmosis in hydrothermal liquefaction hydrolysates. Journal of Membrane Science, 2016. 504: p. 141-152. [CrossRef]
- Pervez, M.N., et al., Factors influencing pressure-driven membrane-assisted volatile fatty acids recovery and purification-A review. Science of The Total Environment, 2022. 817: p. 152993.
- Jun, B.-M., S.H. Kim, S.K. Kwak, and Y.-N. Kwon, Effect of acidic aqueous solution on chemical and physical properties of polyamide NF membranes. Applied Surface Science, 2018. 444: p. 387-398. [CrossRef]
- Zacharof, M.-P., S.J. Mandale, P.M. Williams, and R.W. Lovitt, Nanofiltration of treated digested agricultural wastewater for recovery of carboxylic acids. Journal of Cleaner Production, 2016. 112: p. 4749-4761. [CrossRef]
- Liu, Q., et al., Study on the concentration of acrylic acid and acetic acid by reverse osmosis. Membranes, 2020. 10(7): p. 142. [CrossRef]
- Hoseinpour, H., M. Peyravi, A. Nozad, and M. Jahanshahi, Static and dynamic assessments of polysulfonamide and poly (amide-sulfonamide) acid-stable membranes. Journal of the Taiwan Institute of Chemical Engineers, 2016. 67: p. 453-466. [CrossRef]
- Wei, X., et al., SiO2-modified nanocomposite nanofiltration membranes with high flux and acid resistance. Journal of Applied Polymer Science, 2019. 136(18): p. 47436. [CrossRef]
- Roy, Y. and D.M. Warsinger, Effect of temperature on ion transport in nanofiltration membranes: Diffusion, convection and electromigration. Desalination, 2017. 420: p. 241-257. [CrossRef]
- Nilsson, M., G. Trägårdh, and K. Östergren, The influence of pH, salt and temperature on nanofiltration performance. Journal of Membrane Science, 2008. 312(1-2): p. 97-106.
- Sharma, R.R., R. Agrawal, and S. Chellam, Temperature effects on sieving characteristics of thin-film composite nanofiltration membranes: pore size distributions and transport parameters. Journal of membrane science, 2003. 223(1-2): p. 69-87. [CrossRef]
- Han, I. and M. Cheryan, Nanofiltration of model acetate solutions. Journal of membrane science, 1995. 107(1-2): p. 107-113. [CrossRef]
Figure 1.
Chemical decomposition pathways of lignocellulosic biomass during pyrolysis [43].
Figure 1.
Chemical decomposition pathways of lignocellulosic biomass during pyrolysis [43].
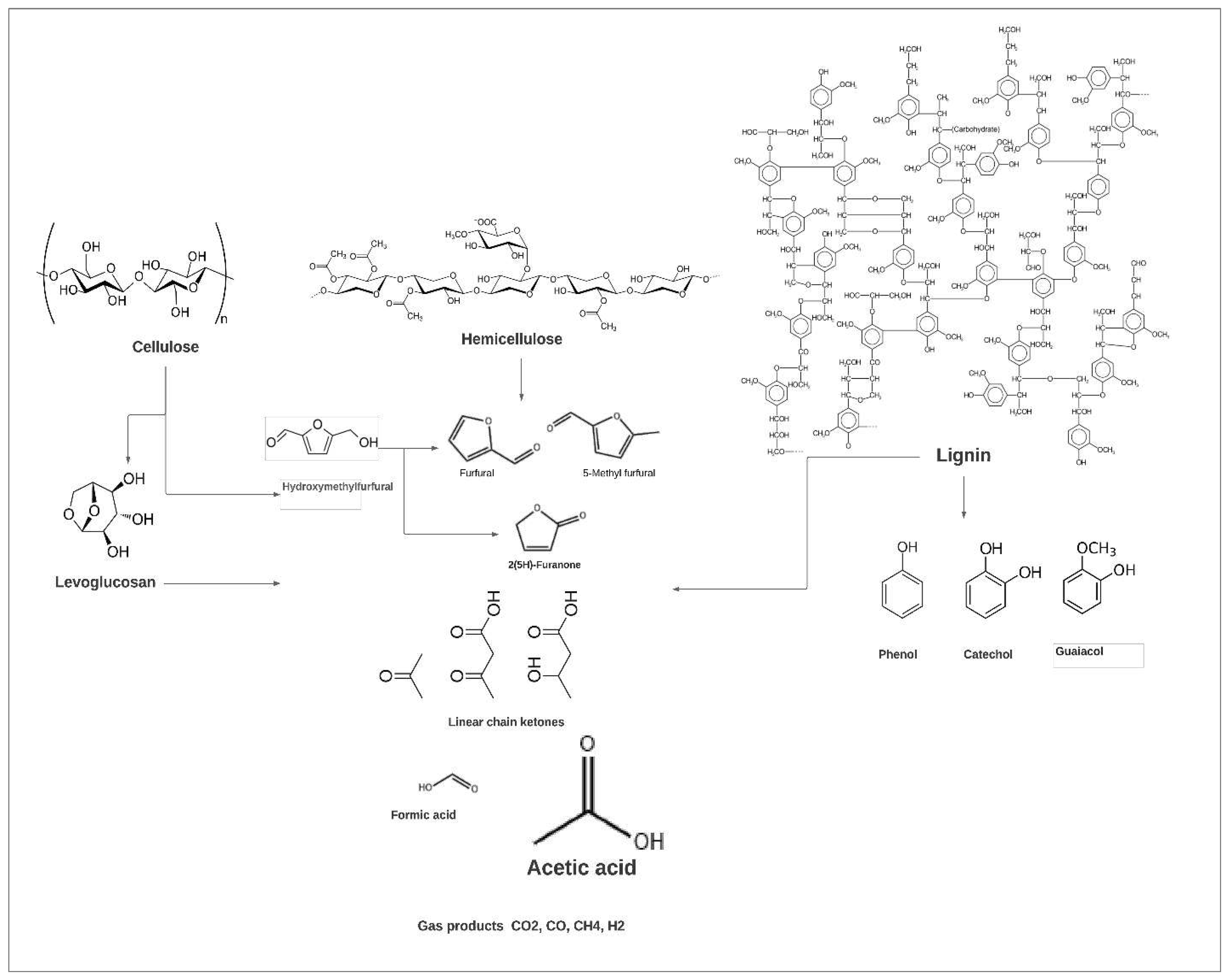
Figure 2.
(a) Ablative (b) Fluidized bed (c) Circulating fluidized bed (d) Screw (auger) (e) Vacuum (f) Rotary cone technologies [7].
Figure 2.
(a) Ablative (b) Fluidized bed (c) Circulating fluidized bed (d) Screw (auger) (e) Vacuum (f) Rotary cone technologies [7].
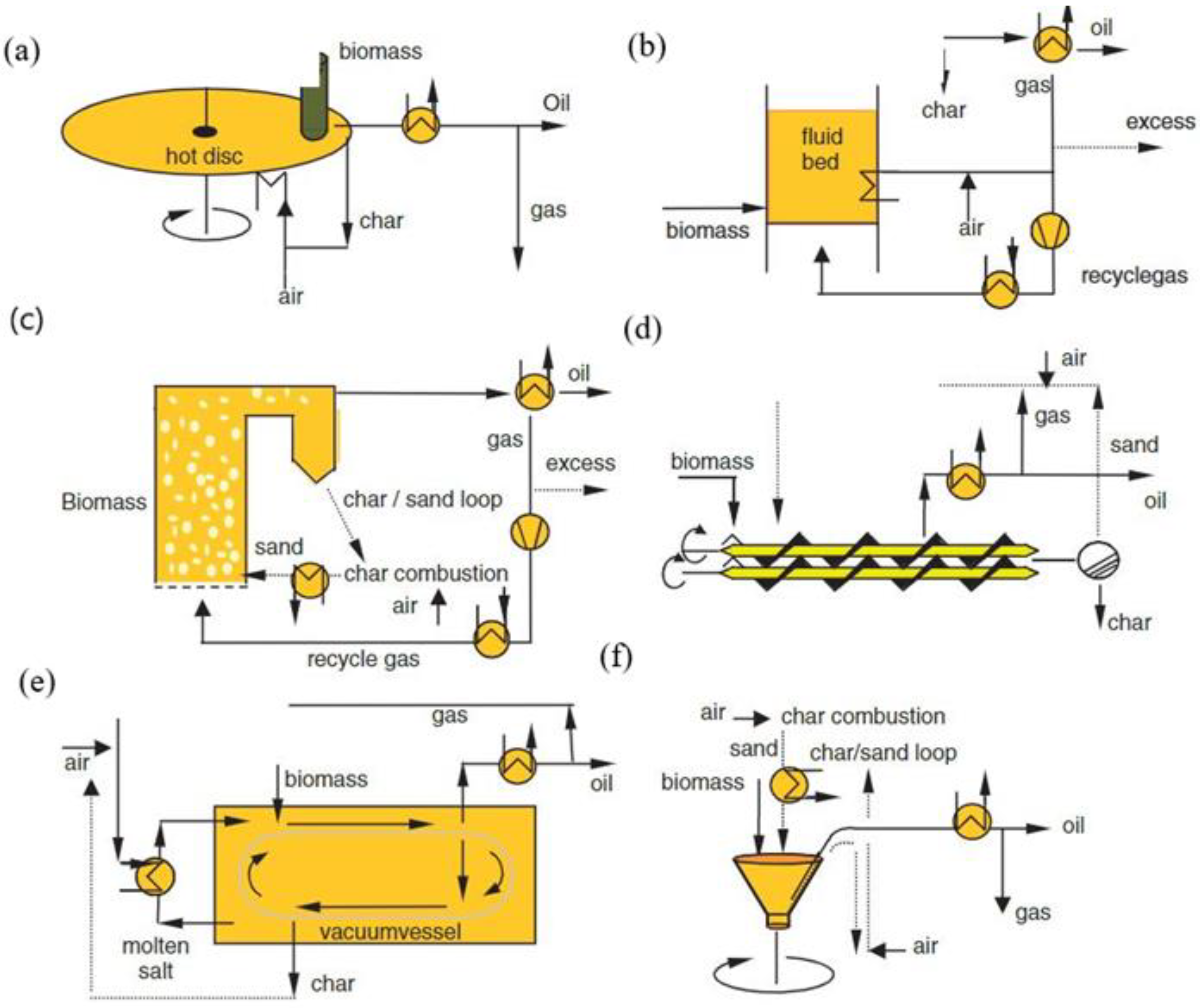
Figure 3.
Schematic of a composite polyamide-based membrane and typical characteristics of each layer [101].
Figure 3.
Schematic of a composite polyamide-based membrane and typical characteristics of each layer [101].
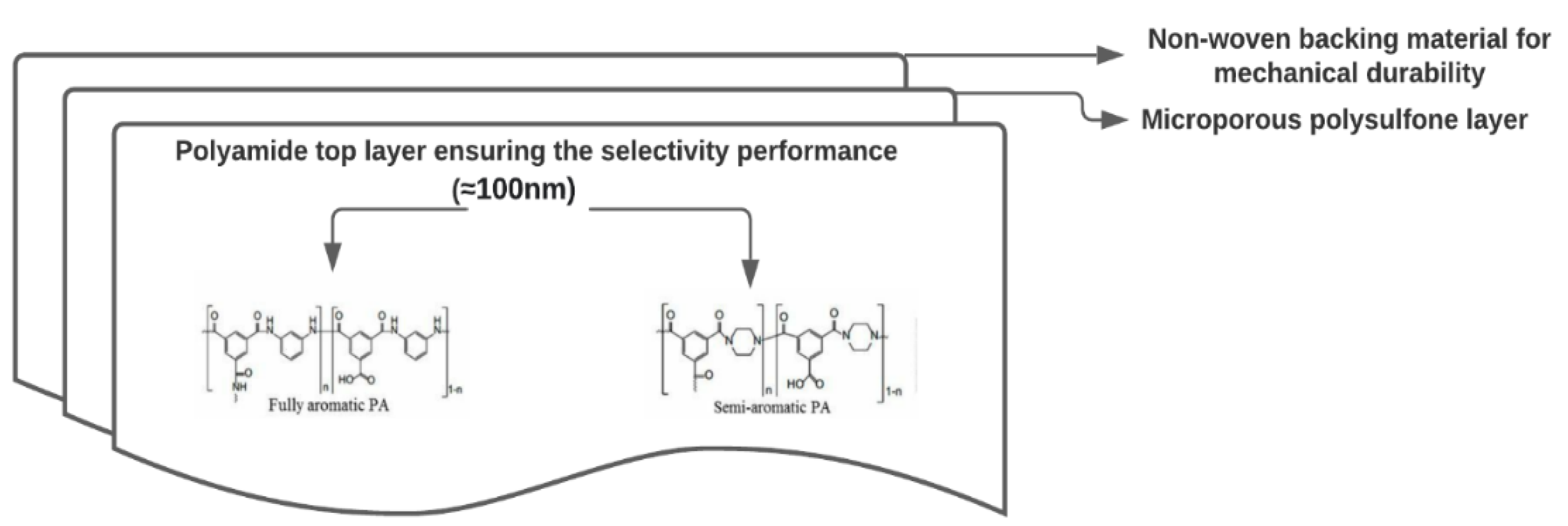

Table 1.
Pathways involved in the primary reactions of the biomass conversion constituents [33].
Table 1.
Pathways involved in the primary reactions of the biomass conversion constituents [33].
| Step | Temp. | Comment |
|---|---|---|
| Char formation | < 773.15 k: | Generation of benzene rings and their subsequent amalgamation into a polycyclic structure referred to as ‘Char’ |
| Depolymerisation | 523.15 and 773.15 k | The cleavage of chemical bonds connecting the monomeric units within the polymer matrix. Production of volatile molecules |
| Fragmentation | > 873.15 k | Linkage of multiple covalent bonds of the polymer and the monomer units. Production of non-condensable gases and various low-molecular-weight organic compounds (CH3COOH) |
Table 2.
Variation in acetic acid yield as a function of biomass nature and its holocellulose content.
Table 2.
Variation in acetic acid yield as a function of biomass nature and its holocellulose content.
| Biomass | Cellulose (wt%) | Hemicellulose (%) | Lignin (%) | Acetic acid yield (%) | References |
|---|---|---|---|---|---|
| Sunflower seed hulls | 40 | 18 | 20 | 60 | Casoni, Bidegain [47] |
| Bagasse | 45.9 | 28 | 20.7 | 4.8 wt. | Lyu, Wu and Zhang [48] |
| Spruce | 47.2 | 13.3 | 36 | 2.3 wt. | |
| Pine | 47 | 21.7 | 27.7 | 2.3 wt. | |
| Corn stalk | 42.4 | 29.6 | 21.7 | 35.9 | Lv and Wu [49] |
| Wood | 47.5 | 19.4 | 24 | Tar fraction : 4 – 5 wt. Aqueous fraction : 9.4 – 11.3 wt. |
Maschio, Koufopanos and Lucchesi [50] |
| Hazelnut shells | 27.5 | 24.1 | 40.7 | ||
| Corncobs | 51.2 | 31.8 | 14.8 | ||
| Olive husks | 22.2 | 21.1 | 45 | ||
| Wheat straw | 24.0 | 40 | 21 | ||
| Lucerne pressed cake | 13.7 | 45.5 | 21.3 | ||
| Rice straw | 37.8 | 25.3 | 23.3 | Upper phase: 5.73 | Jung, Kang and Kim [51] |
| Bamboo | 41 | 26.5 | 25.3 | Upper phase: 3.46 Lower phase: 1.07 |
|
| Pequi peel | 24.35 | 13.75 | 10.50 – 12.40 | Martins, Setter [52] | |
Table 3.
Acetic acid yield obtained with different pyrolysis reactors and different operating conditions (temperature, heating rate, carrier gas, particle size, residence time).
Table 3.
Acetic acid yield obtained with different pyrolysis reactors and different operating conditions (temperature, heating rate, carrier gas, particle size, residence time).
| Biomass | Reactor | Temp. | Carrier gas flow | Residence time | Heating rate | Particle size | Composition | Acetic acid yield (%) | References | ||
| Lignin | Cellulose | Hemicellulose | |||||||||
| Betula pubescens, Bambusa rigida and Dendrocalamus latiflorus | Fixed bed reactor | 773 K and 873 K | 28 ml.min-1 | 10 h | 10 K.min-1 | - | 11.9 – 21.7 | 28.8 – 44.8 | 10.5 – 21.8 | 15 – 38 (a) 54.6 – 100 (b) |
Qi, Hu [73] |
| Cedar sawdust | Fluidized bed reactor (40 × 400 mm) | 700 K | 30 L.min-1 | < 2 s | 10 K.min-1 | 0.5 – 1 mm | 31.45 | 48.34 | 11.3 | O.P: 0.25 (1)A.P : 0.39 (2) | Yang, Wu and Wu [8] |
| Cherry seed | Fixed bed | 300°C, 500°C | 25 ml.min-1 | 1 | 5°C.min-1 | < 2 mm | 29.08 | 32.06 | 28.59 | 26.51, 26.35 | Duman, Okutucu [46] |
| Cherry seed | Fluidized bed | 400°C, 600°C | 25 ml.min-1 | 1 – 2 | 5°C.min-1 | < 2 mm | 29.08 | 32.06 | 28.59 | 91.74, 5.63 | Duman, Okutucu [46] |
| Cherry seed shell (CSS) | Fixed bed | 300°C, 500°C | 25 ml.min-1 | 1 | 5°C.min-1 | < 2 mm | 36.90 | 27.19 | 31.93 | 42.47, 39.28, 74.5 | Duman, Okutucu [46] |
| Cherry seed shell (CSS) | Fluidized bed | 400°C, 500°C, 600°C | 25 ml.min-1 | 1 – 2 | 5°C.min-1 | < 2 mm | 36.90 | 27.19 | 31.93 | 64.07, 36.25 | Duman, Okutucu [46] |
| Coffee bean residue | Fluidized bed reactor (40 × 400 mm) | 700 K | 30 L.min-1 | < 2 s | 10 K.min-1 | 0.3 – 0.9 mm | 30.92 | 19.72 | 30.32 | O.P: 0.16 (1)A.P: 2.10 (2) | Yang, Wu and Wu [8] |
| Mallee woody biomass (eucalyptus Loxophleba, subspecies Lissophloia) | Fluidized-bed reactor | 350 °C – 580°C | 53 and 38 L.min-1 | 1.4 s and 0.7 s | - | 180 – 450 mm | - | - | - | 5.73 - 8.71 | Garcia, Wang [45] |
| Mixed wood sawdust | Pyroprobe® 5200 | 600 °C | - | 30 s | 10 000°C.s-1 | 26.5 µm – 925 µm | - | - | - | 0.49 – 3.76 | Suriapparao and Vinu [54] |
| Pearl Millet (PM) | - | 400 °C | 200 ml.min-1 | - | - | - | 15.75 | 48.93 | 3.16 | 7.04 |
Laougé, Çığgın and Merdun [82] |
| Pine pellets | Fluidized-bed reactor | 600°C | 53 and 38 L.min-1 | 1.4 s and 0.7 s | - | 450 – 600 mm | - | - | - | 2.66 | Garcia, Wang [45] |
| Pistachio shell | Fixed bed (102 ×4 cm) | 500 °C | 250 ml.min-1 | 40°C.min-1 | 1 – 2 mm | - | - | - | 9.68 |
Açıkalın, Karaca and Bolat [84] | |
| Rice straw | Fluidized bed reactor (40 × 400 mm) | 700 K | 30 L.min-1 | < 2 s | 10 K.min-1 | 2 – 5 mm | 12.66 | 36.78 | 31.67 | O.P 0.18 (1)A.P 2.92 (2) | Yang, Wu and Wu [8] |
| Rubberwood (Hevea brasiliensis) | Horizontal-cylindrical furnace (45×80 cm) | 400°C | - | - | 2.0°C.min-1 | 35 cm | - | - | - | 4.524 | Ratanapisit, Apiraksakul [85] |
| Rubberwood (Hevea brasiliensis) | Horizontal-cylindrical furnace (45×80 cm) | > 400°C | - | - | 2.0°C.min-1 | 35 cm | - | - | - | 4.259 | Ratanapisit, Apiraksakul [85] |
| Sagwan sawdust | Réacteur à lit fixe | 639.45°C | 181.59 ml.min-1 | - | 10°C.min-1 | 60 and 80 mesh | 24.7 | 51.1 | 12.9 | 3.611 |
Gupta and Mondal [83] |
| Sida cordifolia L. (Sida) | - | 400°C | 200 ml.min-1 | - | - | 1.5 mm | 12.04 | 49.17 | 21.91 |
7.73 |
Laougé, Çığgın and Merdun [82] |
| Walnut tree branches | Vertical pyrolytic (130 mm × 270 mm) | 450°C | - | - | 1°C.min-1 | 3 × 10 cm | 28.43 | 45.36 |
18.26 | 22.62 | Wei, Ma and Dong [80] |
- : Not identified; (a) Compositions of the main components of the liquid from the pyrolysis of bamboo at 773 K and 873 K (%); (b) Compositions of the main components of the liquid resulting from the catalytic pyrolysis of bamboo on the NaY catalyst at 773 K and 873 K (%). (1) Oily phase; (2) Aqueous phase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



