
Submitted:
04 September 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
Bloodstream infection is a major health problem worldwide, with extremely high mortality. Detecting infection in the early stage is challenging due to the extremely low concentration of bacteria in the blood. Digital PCR provides unparalleled sensitivity and can achieve absolute quantification, but it is time-consuming. Moreover, the presence of unavoidable background signals in negative controls poses a significant challenge for single-molecule detection. Here, we propose a novel strategy called “Ultrafast flexible thin tube-based droplet digital PCR (utPCR)” that can shorten the digital PCR process from 2 hours to only 5 minutes, with primer annealing/extension time reduced from minutes to only 5 seconds. Importantly, the ultrafast PCR eliminates the nonspecific amplification and thus enables single-molecular detection. The utPCR enabled sensitive detection and digital quantification of E. coli O157 in the high background of 106-fold excess of E. coli K12 cells. Moreover, this method also displayed the potential to detect rare pathogens in blood samples, and the limit of detection (LOD) could be as low as 10 CFU per mL of blood without false positive results. Considering ultrafast (<5 min) and highly sensitive (single-molecule detection), the utPCR holds excellent prospects in the next generation of molecular diagnosis.
Keywords:
Ultrafast PCR
; droplet digital PCR (ddPCR)
; single-molecule detection
; bloodstream infection
; absolute quantification
; molecular diagnosis
1. Introduction
Bloodstream infection is a significant global health issue, with a mortality rate of up to 20%[1,2,3,4]. Rapid detection of bacteria, especially in the early stages of infection, is crucial. However, the traditional culture method is complex and time-consuming, taking several days to yield results, thus making it unsuitable for prompt diagnosis[5,6,7,8]. Recently, amplification-based molecular diagnostic methods, such as polymerase chain reaction (PCR), have been developed to reduce the assay time from days to hours[9,10,11,12,13,14]. Nonetheless, conventional PCR lacks the sensitivity required to detect bacteria present in low concentrations in the bloodstream (1~100 CFU per ml). Additionally, it tends to preferentially amplify short fragments and generate short chimeric molecules, which pose a significant challenge for single-molecule analysis when the template concentration is low[15].
Presently, digital PCR has emerged as a novel nucleic acid quantitative technique[16,17,18]. In digital PCR, DNA templates are randomly distributed into millions of small chambers or monodisperse droplets prior to amplification[14,19,20]. Theoretically, reaction compartments containing template molecules will exhibit a fluorescence signal after amplification, while those without template molecules will remain dark. Digital PCR offers unparalleled sensitivity and enables absolute quantification detection. However, most digital PCR processes still require several hours to complete[21,22]. Furthermore, in practical applications, there is typically some inevitable fluorescence present in negative controls during digital PCR detection, significantly interfering with result interpretation, particularly for trace detection[23,24,25]. Consequently, there is an urgent need for a faster, more specific, and extremely sensitive strategy for single-molecule amplification and analysis in the presence of a high background substrate.
In this study, we have developed a novel strategy called “Ultrafast flexible thin tube-based droplet digital PCR (utPCR)” that can be completed in less than 5 minutes. This approach utilizes the high surface-to-volume ratio of droplets, enabling rapid temperature elevation and reduction. Additionally, we have designed a rapid thermocycling strategy by shuttling the thin tube containing droplets between low and high-temperature zones at high speed to achieve amplification. During ultrafast thermocycling, the primer annealing time has been significantly reduced from minutes to seconds, effectively eliminating potential nonspecific amplification induced by primers. Theoretically, all droplets in negative samples will remain nonfluorescent, allowing for single-molecule detection. Furthermore, by dispersing individual targets into millions of droplets, the background interference, such as DNA from white cells, can be simultaneously distributed among the droplets, thereby relatively reducing background interferences in ultrafast ddPCR. Based on these advancements, our proposed system enables single-molecule detection and absolute quantification. It is ultrafast, highly sensitive, and capable of accurately quantifying trace samples in the presence of high background interference, making it highly promising for biomedical applications.
Scheme 1.
Schematic of ultrafast and accurate quantitation of rare target from the high background with the proposed utPCR method. Briefly, Targets with background DNA were randomly distributed into millions of monodisperse droplets. All droplets were transferred into flexible thin tube to make ultrafast PCR thermal cycling. After reaction, the droplets were observed under confocal or read by droplet reader.
Scheme 1.
Schematic of ultrafast and accurate quantitation of rare target from the high background with the proposed utPCR method. Briefly, Targets with background DNA were randomly distributed into millions of monodisperse droplets. All droplets were transferred into flexible thin tube to make ultrafast PCR thermal cycling. After reaction, the droplets were observed under confocal or read by droplet reader.
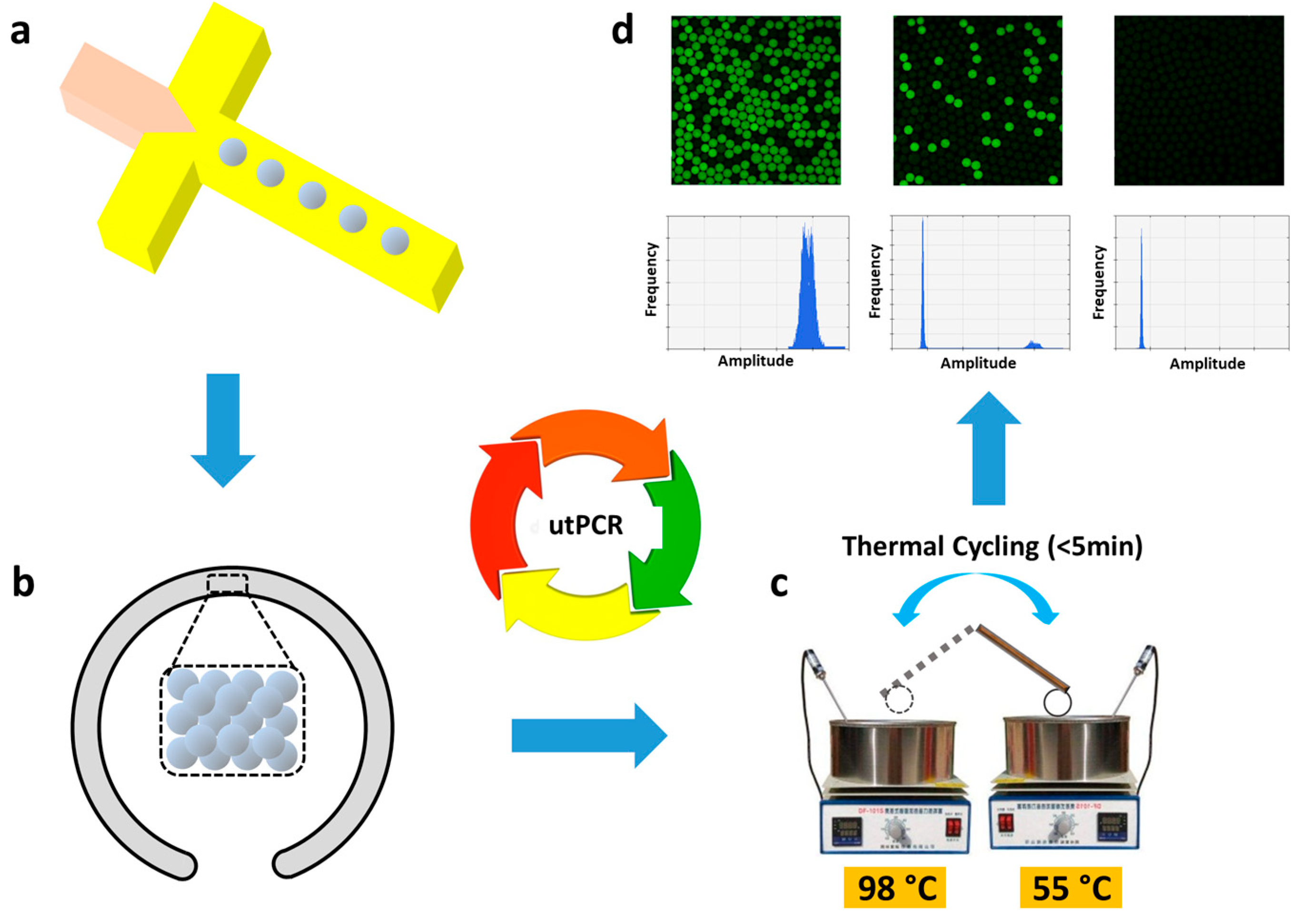
2. Materials and Methods
2.1. Reagents and Chemicals
Strains of E. coli O157 (ATCC 700728) and E. coli K12 (ATCC 700926) were generously provided by Harvard University (MA, USA). Fresh whole blood was obtained from Research Blood Components, LLC. (MA, USA).
Nucleic acid extraction kit was obtained from QIAGEN Inc. (MD, USA). For DNA amplification and analysis, all primers were synthesized by Integrated DNA Technologies, Inc. (IL, USA). TaKaRa SpeedSTAR™ polymerase was purchased from Takara Bio Inc. (CA, USA). The EvaGreen® Dye was obtained from Biotium, Inc. (CA, USA). The UltraPure™ Agarose was provided by Thermo Fisher Scientific Inc. (MA, USA). The GenElute™ Gel Extraction Reagent was bought from Sigma-Aldrich, Inc. (MO, USA). The DNA amplicons were sequenced by GENEWIZ LLC. (NJ, USA). Bovine serum albumin (BSA) was bought from Thermo Fisher Scientific Inc. (MA, USA). Tween 20 was purchased from Sigma-Aldrich, Inc. (MO, USA). The surfactant was purchased from RAN Biotechnologies, Inc. (MA, USA). HFE-7500 3MTM NovecTM Engineered fluid was provided by 3M Corp. (MN, USA). Carrier oil used for droplet generation, HFE-7500, contained 5% (w/w) surfactant.
For microfluidic device fabrication, silicon wafers were supported by University Wafer, Inc. (MA, USA). SU-8 regents were provided by MicroChem Corp. (MA, USA). Isopropyl Alcohol and BAKER BTS-220 were obtained from Avantor Performance Materials, LLC. (PA, USA). Sylgard 184 Silicone Encapsulant Clear was provided by Dow Corning Corp. (MI, USA).
Bacterial culture was performed with Bacto™ Tryptone, Bacto™ Yeast Extract, and Difco™ Agar that obtained from Becton, Dickinson, and Company (MD, USA). Phosphate-Buffered Saline (PBS) was purchased from Corning Inc. (NY, USA), and sodium chloride (NaCl) was obtained from Sigma-Aldrich, Inc. (MO, USA).
For digital PCR amplification, micro medical tubing PE/2 was purchased from Scientific Commodities, Inc. (AZ, USA). All water baths and thermo blocks were obtained from Thermo Fisher Scientific Inc. (MA, USA). The CFX96 fluorescent System, QX200™ Droplet Reader, and ChemiDoc XRS+ System were purchased from Bio-Rad Laboratories, Inc. (CA, USA). The NanoDrop ND-1000 was obtained from Thermo Fisher Scientific Inc. (MA, USA). The Leica TCS SP5 Confocal microscope was purchased from Leica Microsystems Inc. (IL, USA). Syringe pumps were provided by KD Scientific Inc. (MA, USA).
For device fabrication, the 2105C2 Illumination Controller was obtained from Radiation Power Systems, Inc. (CA, USA), and the Plasma Prep2 was purchased from Diener Electronic GmbH +Co. KG (Ebhausen, GERMANY).
2.2. Design and fabrication of the microfluidic device
The PDMS microfluidic devices were fabricated using soft lithographic techniques[26]. The design of the microfluidic channels in PDMS was created using AutoCAD. In preparation, the channel architectures were transferred onto high-resolution photomasks. Different photoresist SU-8 regents were spin-coated onto silicon wafers, taking into account the desired thickness for each channel. After a soft bake, the silicon wafers were patterned through UV exposure using the photomask. The patterned silicon wafers were then baked at 65 °C, followed by 95 °C. Subsequently, the silicon wafers underwent development by immersion in BAKER BTS-220. Once sufficiently washed and dried, the silicon wafers were placed in Petri dishes and filled with a mixture of pre-polymer and cross-linker (w/w, 10:1). After degassing in a vacuum desiccator chamber, the Petri dishes were heated at 65 °C for 2 hours to form structured microfluidic substrates. The resulting PDMS casts were then peeled off and punched to create inlet and outlet ports. The channel side of the PDMS cast was subsequently attached to a clean glass slide through plasma bonding. The specific details of the reaction chambers can be found in Table 1 , which indicate different channel lengths and widths. Following bonding, the microfluidic channels were treated with aquapel to render them hydrophobic and dried using nitrogen gas.
2.3. Bacterial culture and spiked sample preparation
Luria-Bertani (LB) medium was prepared prior to bacterial culture. The liquid LB medium was composed of 1% (w/v) Tryptone, 1% (w/v) NaCl and 0.5% (w/v) Yeast Extract. The LB agar plates additionally contained 1.5% (w/v) agar. All LB media were sterilized at 121 °C for 20 minutes, then placed at 4 °C before use.
Two strains of bacteria, namely E. coli O157 and E. coli K12, were cultured individually in 5 mL of liquid LB medium overnight at 37 °C. Subsequently, the cultured bacteria were separately collected after being washed three times with sterilized PBS, then centrifuged at 5000 rpm for 20 minutes. The bacterial pellets were placed in 5 mL PBS and gradually diluted ten-fold for subsequent spiking assays.
The concentration of bacteria in each dilution was determined in triplicate by measuring the UV absorption at 600 nm. In parallel, 50 μL of each bacterial dilution was spread on LB plates for colony counting after overnight incubation. The bacterial concentration measured via UV absorption was further calibrated using the bacterial colony counting method.
For rare pathogens detection in the presence of high background interference, E. coli O157 and E. coli K12 were uniformly mixed, with E. coli O157 concentration ranging from 5×104 to 5 CFU/mL, while E. coli K12 was maintained at a concentration of 5×106 CFU/mL. Each mixture was divided into six aliquots. Following DNA extraction, three aliquots from each mixture were analyzed using utPCR, while the remaining aliquots were subjected to standard droplet PCR as a control.
In the spiking assay using blood samples, 100 μL of serial 10-fold dilutions of E. coli O157 suspension were mixed with 900 μL of whole blood samples, respectively. Each spiking concentration was performed in six replicates and thoroughly mixed. After DNA extraction, three samples from each spiking concentration were analyzed using utPCR, while the remaining three samples were tested using standard droplet PCR as a control.
2.4. Nucleic acid extraction
Before nucleic acid extraction, all samples were centrifuged at 12000 rpm for 2 minutes with supernatant discarded. Then, nucleic acid were extracted with QIAamp® DNA Mini Kit. DNA templates were dissolved in sterile water, adjusted to a final volume of 20 μL. For each PCR assay, 10 μL of DNA was used as the template, and the total PCR reaction volume was 25 μL.
2.5. The utPCR and standard PCR amplification
The forward primer (5’-TGGCAGGAAGAGAGTGACAGG-3’) and reverse primer (5’-GGCCTTACCCGTGAACAGTA-3’) targeting the E. coli O157 gene were designed using Primer Premier 5.0. The reaction conditions for utPCR and standard droplet PCR were compared in Table S1. To achieve ultrafast nucleic acid amplification, the concentration of DNA polymerase in utPCR was ten times that of standard PCR. It was performed by hot-start at 98 °C for 30 s , then carried out 40 cycles of denaturation (98 °C for 1 s / 2 s / 5 s / 30 s) and annealing/extension (55 °C for 2 s / 3 s / 5 s / 30 s), respectively. The standard droplet PCR was carried out with hot-start at 95 °C for 3 min, then 40 cycles including denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 1 min, with a final extension at 72 °C for 5 min.
2.6. Data acquisition and analysis
After utPCR and standard droplet PCR amplification, the droplets were observed on a confocal platform. Fluorescent images were captured using a 10×objective under 470 nm fluorescence excitation. All images were analyzed using Image J and MATLAB. Droplets with a diameter of 120 μm underwent direct fluorescence flow cytometry analysis using the commercial QX200™ Droplet Reader. The concentration of template was calculated using the Poisson equation: P (X=k) = e-λλk(k!)-1, and N = -ln(1-a/b)b, where λ represents the average pathogen concentration in each droplet, a and b are the numbers of positive and total droplets in the sample, and N is the absolute number of pathogens in the sample.
3. Results
3.1. Monodisperse droplets generation and stability evaluation
First of all, to evaluate the stability of droplets in flexible thin tube during utPCR amplification, four types of droplets with diameter of 120 μm, 80 μm, 37 μm and 15 μm, respectively, were employed to simulate thermo cycling process. All droplets were generated with PDMS devices with different diameters. Most of the droplet generation time was less than 5 min (Table 1, video). The generated droplets were uniform under microscope before thermal cycling (Figure 1) . Then, they were transferred into flexible thin tube and carried out thermal cycling by hot start at 98 °C for 30 s, then 40 cycles of 98 °C for 30 s and 55 °C for 30 s. The droplets were observed and photographed under microscope after thermal cycling process. Results showed that after thermo cycling, all types of droplets still kept uniform, demonstrating strong tolerance to the shuttling process. The droplet sizes were almost the same before and after PCR thermal cycling. We assumed that the surfactant in the carrier oil strongly prevented droplet merging and the ultrafast thermal cycling process greatly lowered the rate of droplet expansion. Therefore, results verified that it is potential to achieve utPCR with the help of flexible thin tube. Considering droplets generation time and detection accuracy, we chose droplets with 37 μm diameter for the following assay.

Table 1.
The parameters of the device and relative flow rates to generate monodisperse droplets with different diameters.
Table 1.
The parameters of the device and relative flow rates to generate monodisperse droplets with different diameters.
| Device | Oil phase width (μm) |
Water phase width (μm) |
Channel Depth (μm) |
Oil phase speed (μL/h) |
Water phase speed (μL/h) | Droplet diameter (μm) |
Total reaction volume (μL) |
Total time (min) |
|---|---|---|---|---|---|---|---|---|
| 1 | 50 | 100 | 50 | 800 | 400 | 120 | 25 | 3.75 |
| 2 | 50 | 100 | 25 | 1200 | 400 | 80 | 25 | 3.75 |
| 3 | 15 | 30 | 25 | 800 | 400 | 37 | 25 | 3.75 |
| 4 | 8 | 8 | 8 | 200 | 100 | 15 | 25 | 15 |
3.2. Ultrfast droplet PCR built up
Then, we explored the optimal thermal cycling time for utPCR reaction in flexible thin tube. The detail of reaction conditions could be seen in Table S1. We tracked the temperature in flexible thin tube during PCR thermal cycling with thermocouple. Result indicated that the maximum heating/cooling ramp rates inside flexible thin tube could be above 150°C/s (Figure 2a,b). Therefore, the heat conductivity of the flexible thin tube is extremely high, which ensured the feasibility of ultrafast PCR thermal cycling. Then, we tested whether it was possible to successfully amplify templates with ultrafast thermal cycling process. As shown in Figure 2c, prior to the utPCR, no fluorescence was observed in any of the droplets. When the utPCR was performed with a 3-second cycle duration, some droplets exhibited fluorescence, but it was challenging to distinguish positive droplets from negative ones. However, when the cycle duration was extended to 5 seconds, droplets containing the DNA target displayed bright fluorescence, while the remaining droplets showed no fluorescence. Increasing the cycle duration from 5 seconds to 60 seconds resulted in all positive droplets emitting green fluorescence, clearly distinguishable from the negative ones. However, beyond 5 seconds per cycle, there was no significant increase in fluorescence intensity of the positive droplets, indicating that 5 seconds per cycle was sufficient for primer annealing and enzyme extension during thermal cycling. Importantly, no bright droplets were generated in the utPCR amplification with no template added as a control (NTCs) (Figure S1). This demonstrated that there was no background signal when no template was present in the ultrafast amplification processes. To minimize the overall reaction time, we selected the 5-second per cycle process for the subsequent utPCR assay.
3.3. Rare target detection from high-background interference
We further extended the utPCR system to quantify pathogenic E. coli O157 in the presence of a high background of normal E. coli K12 cells. The E. coli K12 concentration was kept at 5×106 CFU per mL, while the E. coli O157 density varied from 5×104 to 5 CFU per mL, corresponding to a range of 1:102 to 1:106 E. coli O157 to E. coli K12 ratios, challenging the sensitivity of the utPCR method. The sample containing only E. coli K12 at a concentration of 5×106 CFU/mL served as the negative control. After the utPCR amplification, the results were analyzed using a fluorescent confocal microscope, with standard droplet PCR amplification as the control.
Results demonstrated that, with the decrease of target E. coli O157 in each sample, the number of bright droplets reduced gradually (Figure 3a-e,g-j). Most importantly, for utPCR amplification, there was no bright droplet in the negative control, though the non-template interference was at a high level. However, for standard droplet PCR, there were some fluorescent droplets displayed in the negative control, which would greatly affect the final judgment of limit-of-detection (LOD). We assumed that nonspecific amplification would happen when non-template DNA existed in large amounts. Also, the long annealing time would increase the mismatch of primers to generate nonspecific amplification. Thus, utPCR illustrated higher detection accuracy and specificity than standard droplet PCR, especially for rare sample detection from high background interference. Because there was no false-positive for utPCR amplification, the initial template in the sample could be quantified by the quantity of positive droplets. In contrast, second-round PCR or further DNA sequencing was essential for standard droplet PCR when detecting rare targets from high background interference. Therefore, we primarily concluded that, with the ultrafast annealing time, utPCR demonstrated an advantage for rare template identification from high background interference.
3.4. Detection of blood stream infection with the proposed utPCR method
One step further, we challenged the system by detecting traces of E. coli O157 in blood samples. As is known, in early blood stream infection, it may carry as little as 1-100 pathogens against a high background of 107 white blood cells per milliliter of blood. To simulate this condition, we spiked blood samples with 10-fold gradient diluted E. coli O157 from 106 to 1 CFU per milliliter of blood, with no E. coli O157 spiked blood sample as the negative control (details could be seen in Figure 4). DNA templates from each sample were extracted with Qiagen mini DNA extraction kit and suspended in 20 μL of sterile water. For each utPCR reaction, 10 μL of DNA was added as a template, and the total reaction volume was 25 μL. Each sample was tested by dividing the template into two aliquots for utPCR amplification, and then the copy numbers were added together. To match the requirement of fluorescence flow cytometry analysis by the commercial QX200TM Droplet Reader, the droplets generated in this assay were 120 μm in diameter. The utPCR amplification was carried out in a flexible tube within 5 minutes with standard droplet PCR amplification (~2h) as control. After amplification, all droplets were read individually by QX200TM Droplet Reader. The copy number of each reaction was calculated by the number and fraction of positive droplets according to Poisson distribution.
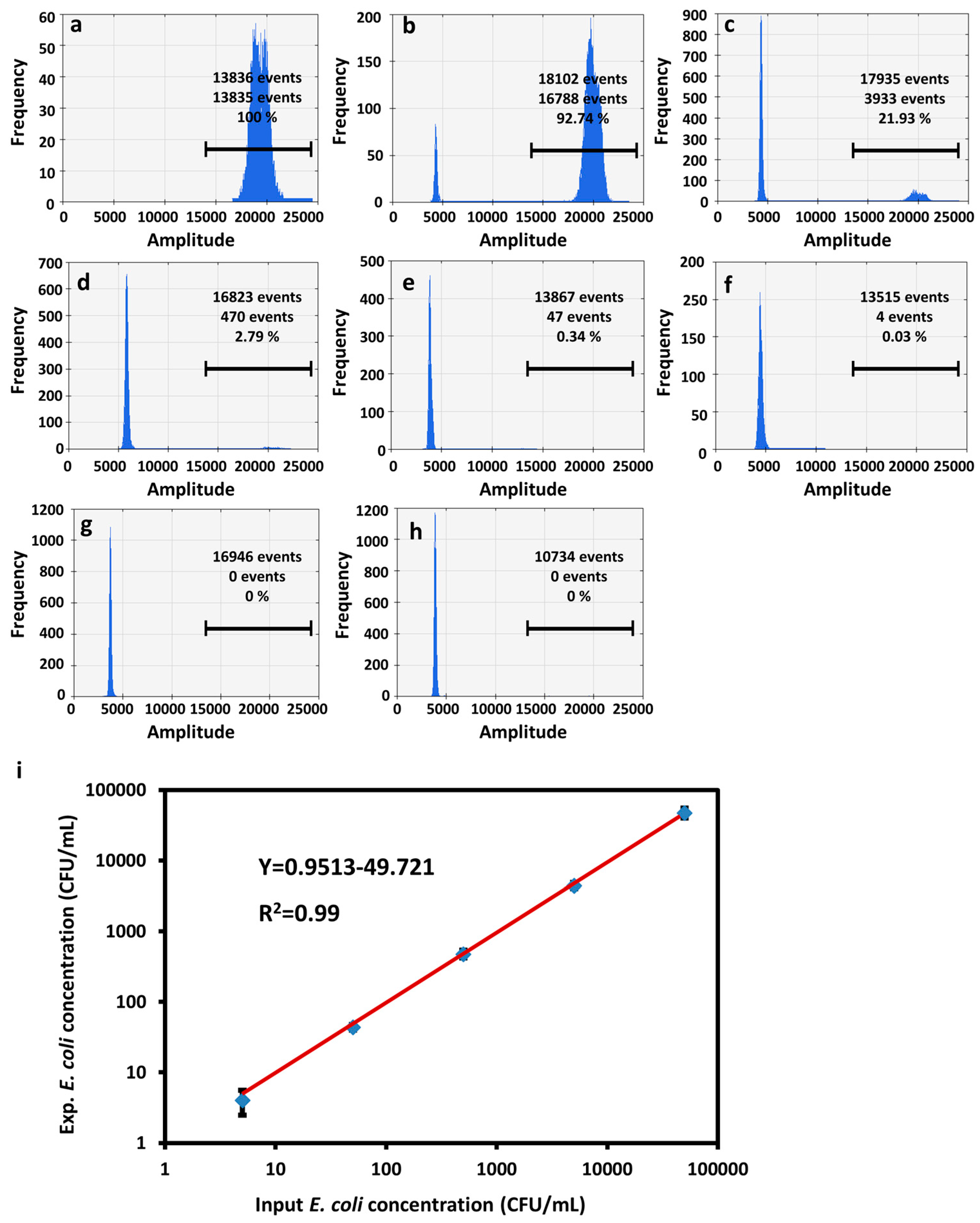
As expected, for utPCR, the number of positive droplets was 10-fold decreased with the reduction of E. coli O157 rate in each sample (Figure 5a-g, Figure S2). Most importantly, there were no positive events for the unspiked blood samples as the negative control (Figure 5h). Furthermore, we analyzed the number of E. coli O157 and compared the value with a theoretically added concentration in each sample. Results in Figure 5i displayed the background corrected calibration of E. coli O157 detection. Results showed a linear relationship between the input and measured E. coli O157 concentration ranging from 10 CFU/mL to 105 CFU/mL with 99% confidence. The standard droplet PCR amplification process also displayed a linear relationship between theoretical and practical E. coli O157 concentration when the spiked concentration was within the range of 105-102 CFU/mL (Figure S3, S4). When the concentration of E. coli O157 reduced to less than 100 CFU/mL, the detection result demonstrated a higher value
than the theoretically spiked concentration. Moreover, for unspiked blood samples, there was also bright fluorescence existing in droplets. We assume that for standard droplet PCR amplification, because of the long annealing time, in addition to the high concentration of white cells as background, the standard reaction process may amplify nonspecific DNA that does not match the desired sequence. Therefore, utPCR demonstrated non-background, high amplification efficiency and detection accuracy, and the LOD could be as low as 10 CFU per milliliter of blood. Moreover, the large dynamic range of 5 magnitude orders should make utPCR a powerful tool for accurate analysis of rare pathogens from high backgrounds.
4. Discussion
PCR is considered as the gold standard and plays a significant role in molecular diagnostics. Over the past four decades, it has evolved from end-point PCR to real-time PCR, and ultimately to absolute quantitative digital PCR[27,28]. To enable ultrafast and single-molecule detection, our focus centered on three crucial steps of digital PCR: sample dispersion, amplification, and quantification.
In terms of sample dispersion, the droplet-based method offers distinct advantages compared to microwell-based, channel-based, and printing-based methods. It allows for easy control of droplet size and composition, ensuring uniformity and rapid dispersion. In our study, we successfully generated monodisperse droplets with four different diameters, with most of the droplet generation process completed within 5 minutes for 25 μL reaction volume. Precise control of droplet diameter was achieved by adjusting channel size and liquid flow rate. Additionally, the stability of droplets was greatly influenced by the surfactant ratio. In our assay, we utilized a 5% surfactant concentration, resulting in uniformly stable droplets after PCR thermocycling. Similar findings have been reported in the literature, stating that surfactant ratio of no lower than 5% effectively maintains monodisperse droplet uniformity during heating incubation[29,30].
Amplification and quantification in digital PCR typically require several hours, which significantly hampers its application, particularly for rapid detection purposes. Additionally, the presence of unavoidable background signals in negative controls poses a significant challenge, particularly in single-molecule detection scenarios. Our preliminary results of gel imaging demonstrated that rapid PCR amplification exhibited no detectable background, unlike the standard amplification process[10]. We hypothesize that this is attributed to the prolonged annealing and extension times employed in the standard digital PCR amplification protocol. To address these limitations, we leveraged the high specific surface area of microscaled droplet systems and facilitated ultrafast heat transfer, resulting in a remarkable reduction in digital PCR amplification time from hours to just 5 minutes. Crucially, by shortening the primer annealing and extension time from minutes to seconds, we achieved the complete absence of fluorescence signals in samples without the target. This significantly enhanced the differentiation of positive droplets and rendered our method highly suitable for single-molecule detection applications.
In future studies, it is essential to address the potential issue of liquid barrier disruption among aqueous droplets during transfer and thermal cycling processes. To achieve this, we aim to enhance the sophistication and multifunctionality of our approach, particularly in terms of device design, optimization, fabrication, and system integration.
5. Conclusions
In conclusion, we have developed a novel utPCR strategy that enables highly sensitive, specific, and ultrafast detection of single molecules in the presence of high background interference. By capitalizing on the advantages of ultrafast annealing/extension, this streamlined method demonstrates remarkably high amplification specificity. As a result, it enables the sensitive detection and digital quantification of E. coli O157, even in the presence of a 106-fold excess of E. coli K12 cells. Furthermore, the method exhibits promising potential for the detection of rare pathogens in blood samples, achieving a remarkable limit of detection as low as 10 CFU/mL of blood with no background signals detected in negative controls. In addition, the proposed method also facilitates ultrafast and high-throughput droplet generation and analysis, which holds significant application potential in various areas such as the detection of rare pathogens in blood samples, elucidating drug-resistant mechanisms for drug development, and enumerating rare circulating tumor cells for early diagnosis. The versatility and capabilities of this method make it a promising tool for the next-generation of molecular diagnosis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Confocal images of negative samples after utPCR amplification; Figure S2: Fluorescence flow cytometry analysis of E. coli O157 in blood samples after utPCR amplification. Figure S3: Fluorescence flow cytometry analysis of E. coli O157 in blood samples after standard droplet PCR amplification. Figure S4: Plot of target pathogen concentration vs. measured pathogen concentration for E. coli O157 detection by standard droplet PCR amplification; Table S1: Reaction conditions of utPCR and standard droplet PCR amplification.
Author Contributions
Conceptualization, R.W.; methodology, Y.L.; software, S.C.; validation, L.B. and K.G.; formal analysis, Y.P.; resources, L.D.; data curation, S.C. and K.G.; writing—original draft preparation, R.W.; writing-review and editing, F.Q.; supervision, Y. F., L. D. and Y.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (32001786), Special Project for Experimental Animal Research (23141900300), Shanghai Rising-Star Program (23QA1404300), Special Project for Medical Innovation Research (22Y11909200), the National Natural Science Foundation of China (82103268, 82070258), Shanghai Sailing Program (21YF1459100), the Science and Technology Commission of Shanghai Municipality for International Partnership Project (20490780100), the National Key Research and Development Program of China (2021YFA0910602, 2021YFC2701103), Open Research Fund of State Key Laboratory of Genetic Engineering, Fudan University (No.SKLGE-2104), Science and Technology Research Program of Shanghai (19DZ2282100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data are contained within the article or the Supplementary Materials.
Acknowledgments
In this section, you can acknowledge any support given which is not covered by the author contribution or funding sections. This may include administrative and technical support, or donations in kind (e.g., materials used for experiments).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nestor, D.; Andersson, H.; Kihlberg, P.; Olson, S.; Ziegler, I.; Rasmussen, G.; Källman, J.; Cajander, S.; Mölling, P.; Sundqvist, M. Early prediction of blood stream infection in a prospectively collected cohort. BMC Infect. Dis. 2021, 21, 316. [Google Scholar] [CrossRef]
- Bullock, B.; Benham, M. D. Bacterial Sepsis, StatPearls 2023. https://www.ncbi.nlm.nih.gov/books/NBK537054/.
- Feng, X.; Yang, W.; Huang, L.; Cheng, H.; Ge, X.; Zan, G.; Tan, Y.; Xiao, L.; Liu, C.; Chen, X.; et al. Causal Effect of Genetically Determined Blood Copper Concentrations on Multiple Diseases: A Mendelian Randomization and Phenome-Wide Association Study. Phenomics 2022, 2, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, H.; Yan, J.; Zhang, T. Literature review on the distribution characteristics and antimicrobial resistance of bacterial pathogens in neonatal sepsis. J. Matern.-Featal Neo. M. 2022, 35, 861–870. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, F.; Wang, Y.; Ji, J.; Xu, Y.; Huang, Y.; Zhang, M.; Li, M.; Xia, J.; Wang, B. Methodological comparison of bronchoalveolar lavage fluid-based detection of respiratory pathogens in diagnosis of bacterium/fungus-associated pneumonia in critically ill patients. Front. public health 2023, 11, 1168812. [Google Scholar] [CrossRef] [PubMed]
- Yi, E. J.; Kim, A. J. Antimicrobial and Antibiofilm Effect of Bacteriocin-Producing Pediococcus inopinatus K35 Isolated from Kimchi against Multidrug-Resistant Pseudomonas aeruginosa. Antibiotics 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Damerum, A.; Malka, S.; Lofgren, N.; Vecere, G.; Krumbeck, J. A. Next-generation DNA sequencing offers diagnostic advantages over traditional culture testing. Am. J. Vet. Res. 2023, 1–9. [Google Scholar] [CrossRef]
- Zhou, Y.; Shi, W.; Wen, Y.; Mao, E.; Ni, T. Comparison of pathogen detection consistency between metagenomic next-generation sequencing and blood culture in patients with suspected bloodstream infection. Sci. Rep. 2023, 13, 9460. [Google Scholar] [CrossRef]
- Al-Bayati, L.; Fasaei, B. N.; Merat, S.; Bahonar, A.; Ghoddusi, A. Quantitative analysis of the three gut microbiota in UC and non-UC patients using real-time PCR. Microb. Pathogenesis 2023, 106198. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, F.; Qian, C.; Wu, C.; Ye, Z.; Wang, L.; Qian, W.; Ping, J.; Wu, J.; Ying, Y. Counting DNA molecules with visual segment-based readouts in minutes. Chem. Comm. 2018, 54, 1105–1108. [Google Scholar] [CrossRef]
- N. Khehra, I.S. N. Khehra, I.S. Padda, C.J. Swift, Polymerase Chain Reaction, StatPearls, 2023. https://pubmed.ncbi.nlm.nih.gov/36943981.
- Wang, R.; Li, Y.; Pang, Y.; Zhang, F.; Li, F.; Luo, S.; Qian, C. VIR-CRISPR: Visual in-one-tube ultrafast RT-PCR and CRISPR method for instant SARS-CoV-2 detection. Anal. Chim. Acta 2022, 1212, 339937. [Google Scholar] [CrossRef]
- Zhang, L.; Su, W.; Liu, S.; Huang, C.; Ghalandari, B.; Divsalar, A.; Ding, X. Recent Progresses in Electrochemical DNA Biosensors for MicroRNA Detection. Phenomics 2022, 2, 18–32. [Google Scholar] [CrossRef]
- Wang, R.; Chen, R.; Qian, C.; Pang, Y.; Wu, J.; Li, F. Ultrafast visual nucleic acid detection with CRISPR/Cas12a and rapid PCR in single capillary. Sensor. Actuat. B-Chem. 2021, 326, 128618. [Google Scholar] [CrossRef]
- Zhu, Z.; Jenkins, G.; Zhang, W.; Zhang, M.; Guan, Z.; Yang, C. J. Single-molecule emulsion PCR in microfluidic droplets. Anal. Bioanal. Chem. 2012, 403, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Kojabad, A. A.; Farzanehpour, M.; Galeh, H. E. G.; Dorostkar, R.; Jafarpour, A.; Bolandian, M.; Nodooshan, M. M. Droplet digital PCR of viral DNA/RNA, current progress, challenges, and future perspectives. J. Med. Virol. 2021, 93, 4182–4197. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yu, M.; Dong, G.; Chen, B.; Zhang, B. Digital PCR as an Emerging Tool for Monitoring of Microbial Biodegradation. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Jiang, Y.; Cao, X.; Liu, C.; Zhang, N.; Shi, D. Droplet digital PCR as an emerging tool in detecting pathogens nucleic acids in infectious diseases. Clin. Chim. Acta 2021, 517, 156–161. [Google Scholar] [CrossRef]
- Feng, X. J.; Yi, H. M.; Ren, X. X.; Ren, J. L.; Ge, J. R.; Wang, F. G. Digital PCR and its application in biological detection. Hereditas 2020, 42, 363–373. [Google Scholar] [CrossRef]
- Cao, L.; Cui, X.; Hu, J.; Li, Z.; Choi, J. R.; Yang, Q.; Lin, M.; Ying Hui, L.; Xu, F. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosens. Bioelectron. 2017, 90, 459–474. [Google Scholar] [CrossRef]
- Zaytseva, M.; Usman, N.; Salnikova, E.; Sanakoeva, A.; Valiakhmetova, A.; Chervova, A.; Papusha, L.; Novichkova, G.; Druy, A. Methodological Challenges of Digital PCR Detection of the Histone H3 K27M Somatic Variant in Cerebrospinal Fluid. Pathol. Oncol. Res. 2022, 28, 1610024. [Google Scholar] [CrossRef]
- Mavridis, K.; Michaelidou, K.; Vontas, J. Highly sensitive droplet digital PCR-based diagnostics for the surveillance of malaria vector populations in low transmission and incipient resistance settings. Expert Rev. Mol. Diagn. 2021, 21, 1105–1114. [Google Scholar] [CrossRef]
- Trypsteen, W.; Vynck, M.; De Neve, J.; Bonczkowski, P.; Kiselinova, M.; Malatinkova, E.; Vervisch, K.; Thas, O.; Vandekerckhove, L.; De Spiegelaere, W. ddpcRquant: threshold determination for single channel droplet digital PCR experiments. Anal. Bioanal. Chem. 2015, 407, 5827–5834. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. S.; Park, J. H.; Bae, Y. K. Comparison of two digital PCR methods for EGFR DNA and SARS-CoV-2 RNA quantification. Clin. Chim. Acta 2021, 521, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wolter, M.; Felsberg, J.; Malzkorn, B.; Kaulich, K.; Reifenberger, G. Droplet digital PCR-based analyses for robust, rapid, and sensitive molecular diagnostics of gliomas. Acta Neuropathol. Com. 2022, 10, 42. [Google Scholar] [CrossRef]
- Whitesides, G. M.; Ostuni, E.; Takayama, S.; Jiang, X.; Ingber, D. E. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 2001, 3, 335–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Parvin, R.; Fan, Q.; Ye, F. Emerging digital PCR technology in precision medicine. Biosens. Bioelectron. 2022, 211, 114344. [Google Scholar] [CrossRef]
- Koçer, İ.; Karsligil, T.; Sağlam, M.; Arslanyürekli, U.; Deveci, İ.; Şahin, E. Evaluation of real-time PCR and flow cytometry efficiency in rapid detection of carbapenemase-producing Enterobacteriales. J. Infect. Dev. Countr. 2023, 17, 635–642. [Google Scholar] [CrossRef]
- Wu, C.; Liu, L.; Ye, Z.; Gong, J.; Hao, P.; Ping, J.; Ying, Y. TriD-LAMP: A pump-free microfluidic chip for duplex droplet digital loop-mediated isothermal amplification analysis. Anal. Chim. Acta 2022, 1233, 340513. [Google Scholar] [CrossRef]
- Wu, H.; Cao, X.; Meng, Y.; Richards, D.; Wu, J.; Ye, Z.; deMello, A. J. DropCRISPR: A LAMP-Cas12a based digital method for ultrasensitive detection of nucleic acid. Biosens. Bioelectron. 2022, 211, 114377. [Google Scholar] [CrossRef]
Figure 1.
The actual image (a) and cross-sectional schematic view (b) of a PDMS microfluidic device for monodisperse droplets generation. c) Microscope images of droplets before (top) and after (bottom) PCR thermal cycling. From left to right, the diameter of droplets were 120 μm, 80 μm and 37 μm, respectively. Scale Bar: 50 μm.
Figure 1.
The actual image (a) and cross-sectional schematic view (b) of a PDMS microfluidic device for monodisperse droplets generation. c) Microscope images of droplets before (top) and after (bottom) PCR thermal cycling. From left to right, the diameter of droplets were 120 μm, 80 μm and 37 μm, respectively. Scale Bar: 50 μm.
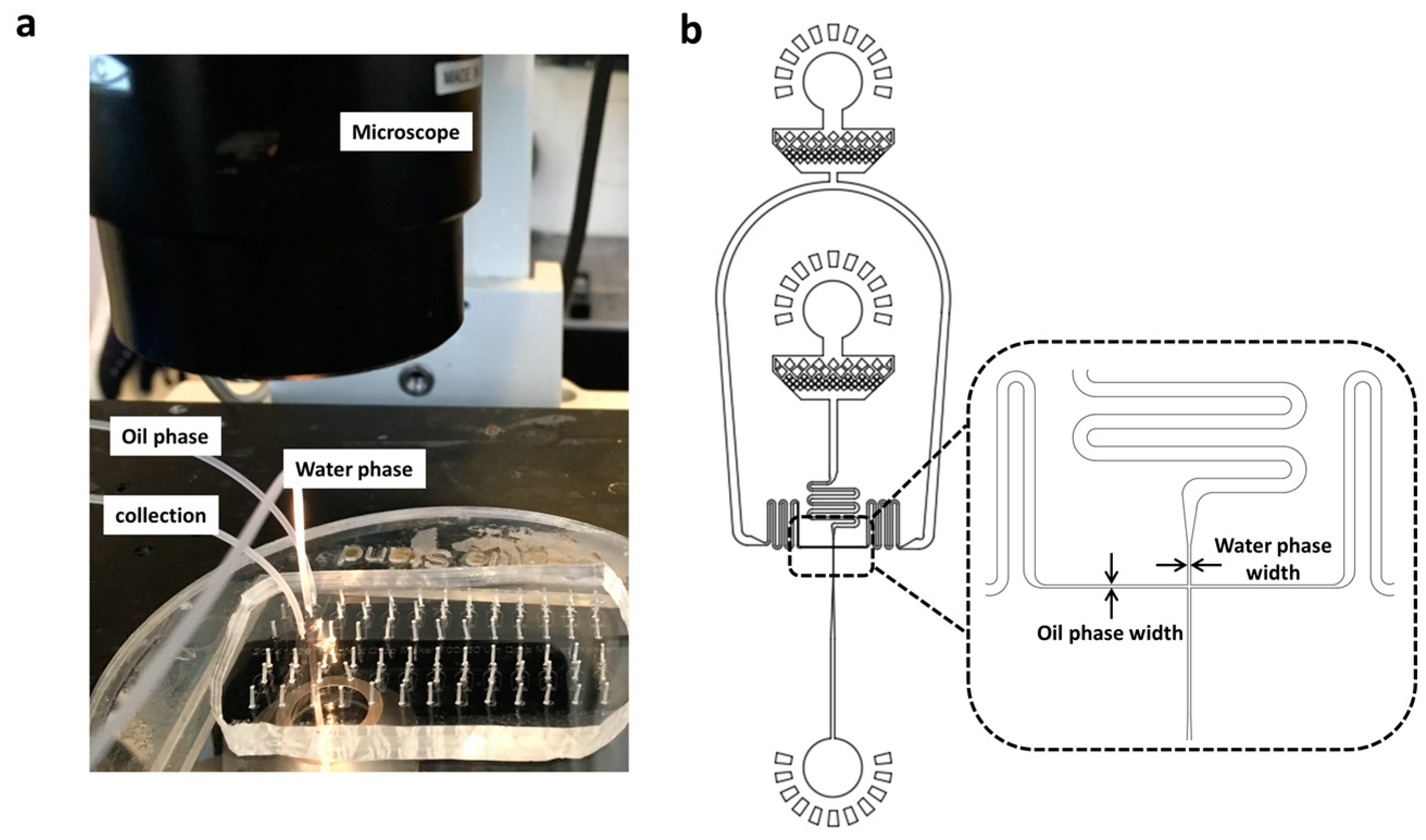

Figure 2.
a) The real-time temperature (a) and heating/cooling rate (b) in flexible thin tube with PCR thermal cycling process of 98 °C for 2 s and 55 °C for 3 s. c) Confocal images of droplets before and after utPCR amplification with standard droplet PCR as control ( Scale bar: 50 μm).
Figure 2.
a) The real-time temperature (a) and heating/cooling rate (b) in flexible thin tube with PCR thermal cycling process of 98 °C for 2 s and 55 °C for 3 s. c) Confocal images of droplets before and after utPCR amplification with standard droplet PCR as control ( Scale bar: 50 μm).
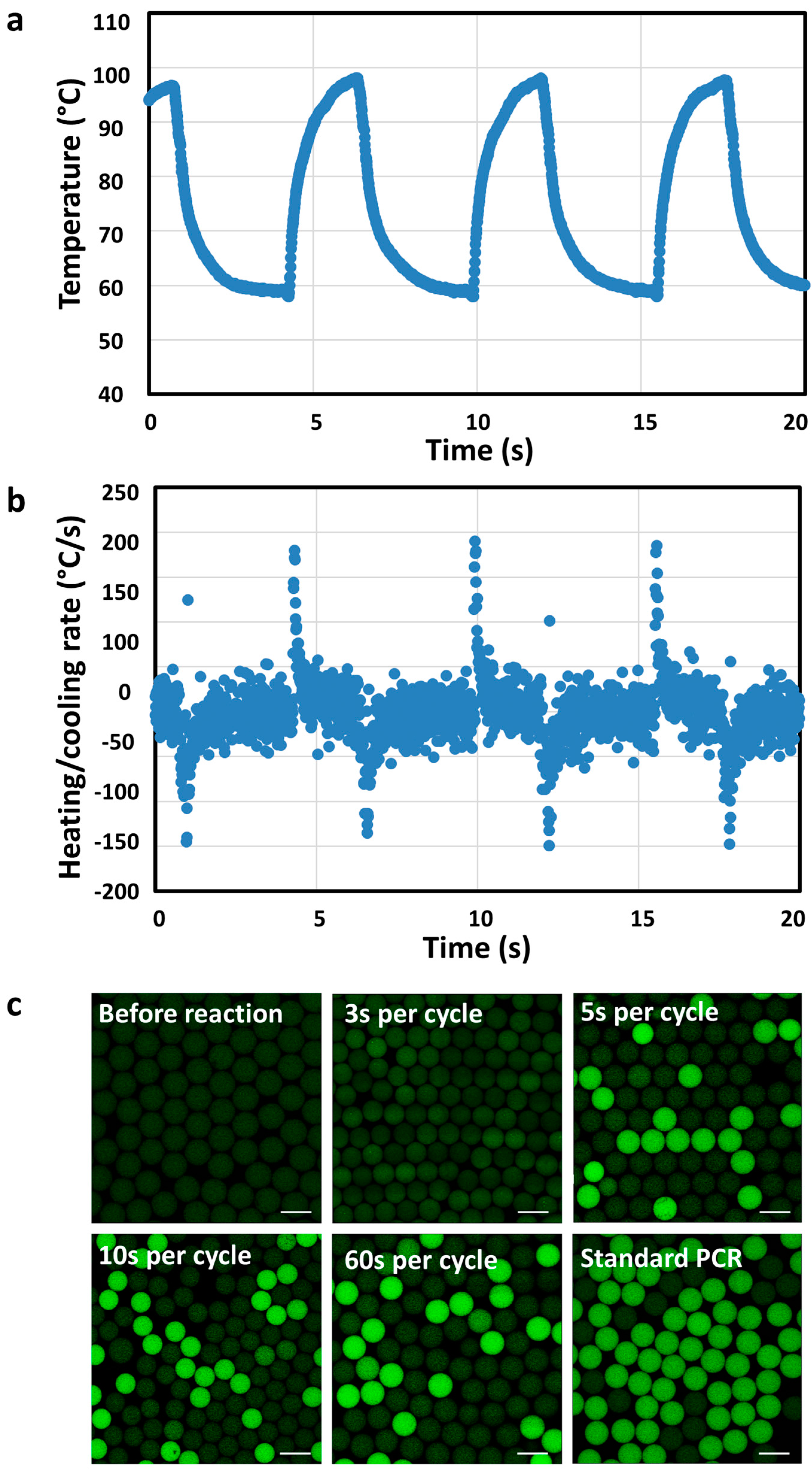
Figure 3.
Comparison of utPCR (a-f) and standard droplet PCR (g-l) for E. coli O157 detection from high background of E. coli K12. From a to e (g to k), the ratio of E. coli to E. coli K12 were 1:102, 1:103,1:104,1:105, 1:106 with only E. coli K12 added as the negative control (f, l).
Figure 3.
Comparison of utPCR (a-f) and standard droplet PCR (g-l) for E. coli O157 detection from high background of E. coli K12. From a to e (g to k), the ratio of E. coli to E. coli K12 were 1:102, 1:103,1:104,1:105, 1:106 with only E. coli K12 added as the negative control (f, l).

Figure 4.
Workflow of blood sample spiked with E. coli O157 dilutions and pathogen detection with utPCR and standard droplet PCR amplification.
Figure 4.
Workflow of blood sample spiked with E. coli O157 dilutions and pathogen detection with utPCR and standard droplet PCR amplification.
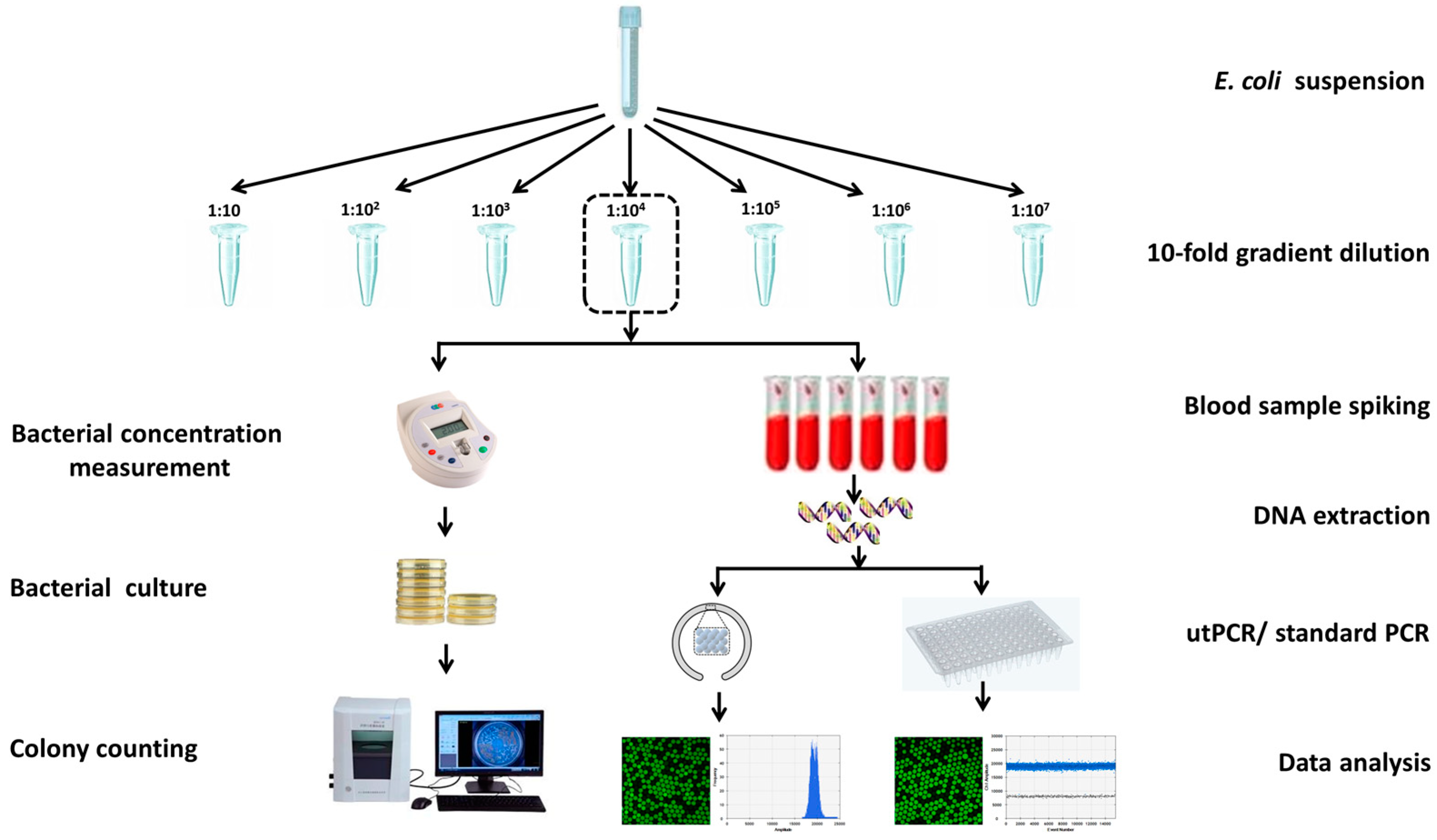
Figure 5.
Fluorescence flow cytometry analysis of E. coli O157 in blood samples after utPCR amplification. The E. coli O157 concentration was was gradually reduced by 10-fold from 106 to 100 CFU per milliliter of blood (a-g) with unspiked blood samples serving as the control (h). (i) plot of the target pathogen concentration versus the measured pathogen concentration for E. coli O157 in blood samples using utPCR detection.
Figure 5.
Fluorescence flow cytometry analysis of E. coli O157 in blood samples after utPCR amplification. The E. coli O157 concentration was was gradually reduced by 10-fold from 106 to 100 CFU per milliliter of blood (a-g) with unspiked blood samples serving as the control (h). (i) plot of the target pathogen concentration versus the measured pathogen concentration for E. coli O157 in blood samples using utPCR detection.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



