
Submitted:
05 September 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
The hydrogen evolution reaction (HER) has garnered attention to access sustainable energy generation with a pollutant-free combustion product in the form of H2 gas. Despite the utility of this pathway in the context of growing climate concerns, stable HER electrocatalysts which avoid the use of rare-earth platinum while retaining high catalytic performance and efficiency remains a challenge. In this work, we present the application of a binuclear nickel (II/III) Schiff base complex [Ni]2[L]2 2 for HER electrocatalysis. The design of the electrocatalyst provides optimal HER activity as the presence of electronegative heteroatoms in proximity to the metal centers augments proton affinity. As a result, the catalyst displays excellent HER activity with 100% faradaic efficiency (FE) at an onset potential of 0.4 V vs. reverse hydrogen electrode (RHE), sustained catalytic activity during prolonged electrolysis for greater than 17 hours, and provided a turn over number (TON) of 0.0006 s-1.
Keywords:
nickel
; electrolysis
; catalysis
; binuclear complex
; hydrogen evolution
1. Introduction
The growing climate and energy crises emphasize the necessity to reduce modern reliance on fossil fuels and rather focus efforts on the development of sustainable energy generation methodology [1]. For this reason, electrocatalytic technologies are highly coveted for their ability to manipulate chemical pathways in order to convert earth abundant feedstock to energy rich products [2]. Of these chemical pathways, electrochemical water splitting through the hydrogen evolution reaction (HER) is of particular interest to cleanly produce H2 gas. The use of H2 gas as an alternative energy is becoming increasingly attractive for its high energy density and pollutant free combustion products [3]. A critical limitation of H2 gas generation is due to production costs, as the use of electrochemical earth-abundant transition metal catalysts struggle to display activity similar to costly platinum containing materials [4,5]. Circumventing this gap between rare-earth and earth-abundant transition metal catalysts is a key step towards the advancement of industrial scale production of H2 gas for renewable energy.
To approach the high electrochemical performance of platinum, informed and rational design of earth-abundant transition metal catalysts is required. Electrocatalytic transition metal complexes such as cobalt, copper, molybdenum, and nickel have all shown promise in their ability to generate H2 gas at desirable working potentials [6,7,8,9]. Nickel shows particular promise for HER catalysis as DFT calculations reveal its free energy of hydrogen absorption (ΔGH) to be optimal of all non-noble transition metals [10]. These calculations are validated as previous work using nickel nanoparticles acquire current densities of 10 mA cm-2 with a low overpotential of 98 mV [11]. Despite the near platinum-like performance of nickel nanoparticles, limitations arise with stability as the production of nickel hydride dampen catalytic lifespan of the system. To avoid this limitation, recent progress in nickel HER catalysis involves the use of carefully designed ligands to yield organometallic electrocatalysts.
A shared feature among previous work utilizing molecular catalysts is the tuning of electronic properties via ancillary ligands. Providing electron deficient heteroatoms (O, N, S) near metal centers through redox-active ligands can promote the absorption of protons to the heteroatom sites, leading to an increase in surface area saturation of hydrogen [12]. In the underlying work, we present a novel binuclear nickel (II/III) HER electrocatalyst complexed from a Schiff base ligand. This electrocatalyst features two quaternary nickel centers surrounded by electronegative oxygen and nitrogen heteroatoms. The catalyst design enhances HER performance reaching nearly 100% Faradaic efficiency with an onset overpotential of 300 mV in 0.5 M sulfuric acid solution.
2. Results and Discussion
Synthesis of the [Ni]2[L]2 complex is both cheap and facile (Scheme 1). Preparation of the Schiff base ligand 1 originates from commercially available hydroquinone. Hydroquinone was treated with 1-bromooctane in alkaline conditions to obtain the 4-(octyloxy)phenol. Subsequent reaction with MgCl2, paraformaledhyde and triethylamine in THF at 70°C afforded the 2-hydroxy-5-(octyloxy)benzaldehyde in excellent yields. Then the 4-(octyloxy)phenol was nitrated in benzene and 56% aqueous nitric acid and 2-nitro-4-(octyloxy)phenol was obtained in 54 % yield. Hydrogenation of 2-nitro-4-(octyloxy)phenol on Pd/C gave 2-amino-4-(octyloxy)phenol in 90 % yield. The synthesis of (Z)-2-((2-hydroxy-5-(octyloxy)benzylidene)amino)-4-(octyloxy)phenol was then performed with the two precursors, 2-amino-4-(octyloxy)phenol and 2-hydroxy-5-(octyloxy)benzaldehyde, in dry EtOH at 80°C. The Schiff base ligand 1 was obtained in 70% yield.
The subsequent metalation of ligand 1 with nickel (II) acetate in ethanol at reflux temperature proceeds easily to afford the [Ni]2[L]2 complex. Solid state 13C NMR spectroscopy, MALDI-TOF mass spectrometry and CHN elemental analysis confirm the proposed composition of [Ni]2[L]2 . Solid state C13 NMR confirms presence of alkyl chain carbons (δ= 33.18-16.14) and aromatic carbons (δ=113.35-109.45) (Figure 1a). The MALDI-TOF mass spectrum shows the [M+H+]+ at m/z = 1051.165. In agreement with previous literature and our DFT frequency calculations, FT-IR spectra band at 1622 cm-1 characteristic of an imine group ν(C=N) in the Schiff base ligand was shifted to 1600 cm-1 after metalation for the complex (Figures S8 and S9) [13]. The utility of this synthetic pathway provides tunability for ligand side chains as evident in previous literature using Schiff base ligands for complexation [14]. The inherent nature of the catalyst structure leaves it prone to π–π stacking interactions, limiting solubility [15] (Figure 2a). For this reason, [Ni]2[L]2 makes for an excellent candidate for heterogenous electrocatalysis in aqueous mediums, lowering catalyst loading through adhesion to carbon paper electrode.
The drawback in the structural design of the catalyst is caused by issues of surface layer morphology as aggregation hinders electrochemical performance [16]. To amend this, we employ the –O-C8H17 side-chains to the ligand and catalyst structure providing steric bulk and greater spatial conformation on the surface layer of electrodes. Through computational modeling via DFT calculations, the optimized geometry as well as the HOMO and LUMO orbital configurations are both visualized (Figure 2b). The model displays a complete planar geometry of the catalyst with two open axes for coordination of hydrogen producing substrates. Furthermore, orbital depictions show ideal configurations for substrate binding thus giving molecular insight to the enhanced HER activity of the catalyst design.
XPS analyses detailing the elemental and electronic states of [Ni]2[L]2 2 are displayed in Figure 3a-c. Figure 3a shows the broad region of the XPS survey spectra of the pure [Ni]2[L]2 2 catalyst as a powder prior to electrolysis. The spectra are in agreement with elemental analysis characterization as the presence of Ni, O, N, and C are present.
Figure 3b shows the narrow scans of Ni2p1/2 and Ni2p3/2 in the 850 eV to 875 eV range. More specifically, the 856 eV peak can be assigned as NiIII to one of the nuclear nickel centers while the 874 eV peak can be assigned as NiII to the other accompanying nickel center. Satellite peaks at 879 eV and 862 eV provide evidence for the presence of both NiII and NiIII ions respectively [17]. This is in agreement with previous work characterizing the electrochemical behavior of molecular nickel centered complexes [18]. Narrow scans detailing the bond energies of the various carbon-heteroatom linkages are present in Figure 3c. Two distinct peaks at 288.89 eV (C-O) and 286.67 eV (C=N) can be assigned to the heteroatom bond linkages present near the nickel centers [19].
The electrocatalytic activity of [Ni]2[L]2 was initially determined by conducting cyclic voltammetry (CV) (Figure 4a). A standard three electrode configuration using Pt as a counter electrode (CE), [Ni]2[L]2 is loaded on carbon paper as working electrode (WE) and Ag/AgCl as a reference electrode in argon saturated 0.5 M H2SO4 (pH 1.8) electrolyte at room temperature. Figure 4a reveals a small anodic peak at 0.02 V vs. reversible hydrogen electrode (RHE) and an obvious anodic peak at an onset potential of 0.4 V vs. RHE, showing hydrogen evolution activity. The linear sweep voltammogram (LSV) presented in in Figure 4b and Figure S11 further support the excellent HER performance of [Ni]2[L]2.
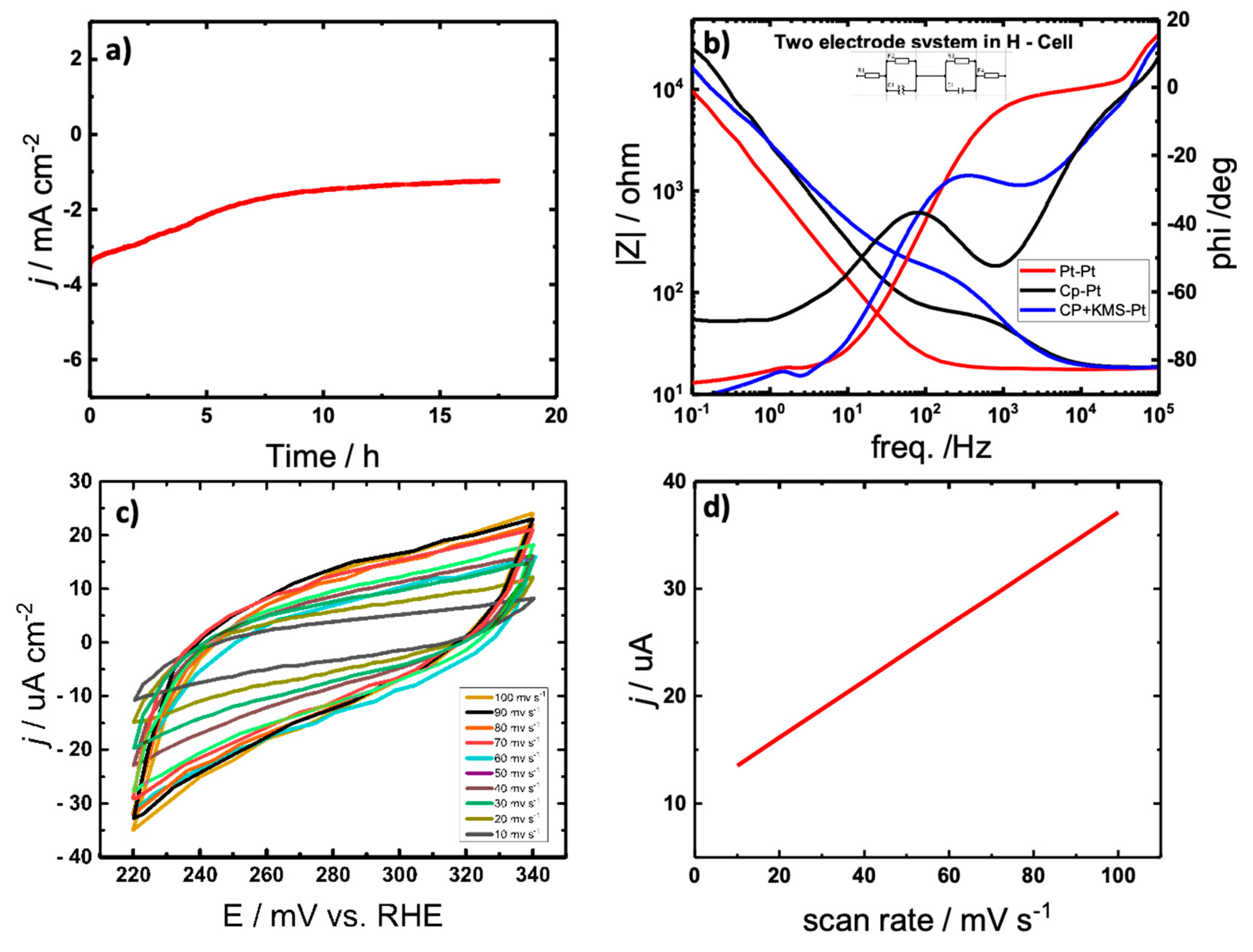
To probe the stability of [Ni]2[L]2 2, a carbon paper spray coated with the electrocatalyst is characterized via chronoamperometric studies in 0.5 M H2SO4 purged with argon at room temperature. Figure 5a reveals long-term durability as catalytic activity is retained after 17.5 hours of continued electrolysis. The electrochemical cell parameters in addition to the transfer resistance RCT are determined using electrochemical impedance spectroscopy (EIS) (Figure 5b). According to the measurements, there is negligible resistances presented by the cell and electrolyte. The corresponding resistances are summarized in the Supporting Information in Table S1. The electrochemically active surface area (ECSA) is calculated according to the electrochemical double-layer capacitance (Cdl), based on the correlation between the ECSA and Cdl and Cs, where Cs is the specific capacitance, by the equation ECSA = Cdl/Cs, resulting in 0.00002167 cm2[20]. The double layer capacitance is determined using CV in 0.5 M H2SO4 in a range of potentials from 220 to 340 V at varying scan rates beginning at 10 mV s-1 to 100 mV s-1 (Figure 5c). The value of Cdl was by extracting the value from the slope of the current density plotted as a function of scan rate (Figure 5d).
After characterization of [Ni]2[L]2 electrocatalytic capability, chronoamperometry was conducted for hour long periods at varying potentials to assess the volume and efficiency of H2 production (Table S2). From the results, we see that the catalyst achieved 100% FE after 1 hour of electrolysis at the onset potential of 400 mV. While electrolysis at both lower and higher potentials yielded H2 production, 400 mV was determined to be the optimal working potential. From the volume of H2 produced, the TOF and TON values were determined to be 0.214 and 0.0006 s-1 respectively.
3. Conclusions
To conclude, we report on a highly efficient and unique binuclear nickel complex [Ni]2[L]2 for HER electrolysis. Metalation of a Schiff base ligand as a precursor allows for a facile and highly tunable synthetic pathway. The design of the electrocatalyst involves electronegative heteroatoms in direct adjacency to the nickel centers to garner a high affinity to protons in aqueous electrolyte. Characterization of the electrocatalytic ability of the catalyst revealed promising results as an onset potential of 400 mV vs. RHE is comparable to highly efficient platinum containing materials for HER. When applied in HER electrolysis via chronoamperometric measurements, the catalyst performed with high efficiency providing H2 product with 100% FE at the onset potential of 400 mV vs. RHE. Prolonged electrolysis provided evidence for the long-term stability of the catalyst as well. As the standard for highly efficient HER catalyst for industrial use rely on rare earth platinum, we provide an environmentally friendly alternative with comparable efficiency and performance for HER electrolysis.
4. Materials and Methods
4.1. Synthesis and Characterization
Chemicals were purchased from Fluka, Alfa Aesar, Sigma-Aldrich, and Merck. Commercially available hydroquinone was purchased from Sigma-Aldrich. 4-(octyloxy)phenol[1], 2-hydroxy-5-(octyloxy)benzaldehyde[2], 2-nitro-4-(octyloxy)phenol[3], 2-amino-4-(octyloxy)phenol[3], (Z)-2-((2-hydroxy-5-(octyloxy)benzylidene)amino)-4-(octyloxy)phenol[4], and [Ni]2[L]2[4,5] were synthesized according to reported literature procedures. DCM was obtained using a M. Braun Inert Gas System GmBH where it is stored over molecular sieve MB-SPS-7 under argon atmosphere. NMR solvents were purchased from Sigma-Aldrich. TLC was performed on Macherey-Nagel silica gel 60 (0.20 mm) with fluorescent indicator UV254 on aluminium plates and on Merck aluminium oxide 60 (0.20 mm) with fluorescent indicator UV254 on aluminium plates. For chromatography, silica-gel columns were prepared with silica-gel 60 (0.070-0.20 mesh) from Grace and aluminium oxide 60, basic, activity level II from Acros. 1H and 13C NMR spectra were recorded on a Bruker DRX 500 MHz spectrometer and a Bruker Advance 300 MHz NMR spectrometer. Chemical shifts are given in parts per million (ppm) on the delta scale (δ) and are referenced to the used deuterated solvent for 1H-NMR. High resolution mass spectra were obtained using an Agilent 6520 Q- TOF mass spectrometer with an ESI source and an Agilent G1607A coaxial sprayer and a Thermo Fisher Scientific LTQ Orbitrap XL with an Ion Max API Source. MALDI-TOF was measured on a . UV-Vis absorption spectra were collected on a Varian CARY 300 Bio spectrophotometer from 200 to 900 nm.
4.1.1. Synthesis of 4-(octyloxy)phenol
Hydroquinone (4.0 g, 36.3 mmol) and potassium carbonate (5.0 g, 36.3 mmol) were added to a flame dried round bottom flask with a magnetic stir bar and dissolved in acetonitrile (100 mL) before being left to stir at room temperature for 30 minutes. After, 1-bromooctane (7.0 g, 36.3 mmol) was added drop wise and refluxed at 82°C for 24 h. Reaction mixture was then left to cool to room temperature before filtered through glass frit and washed with dichloromethane. The crude product was evaporated to dryness before purification via column chromatography (silica, EtOAc/heptane, [1:4]) to provide 4-(octyloxy)phenol (3.23 g, 40%) as a dark brown solid. Rf=0.5, 1H-NMR (300 MHz, CDCl3, 25°C): δ= 6.82 (m, 4H, Ar-H), 4.68(s, 1H, Ar-OH), 3.90 (t, J = 6.6 Hz, 2H, -OCH2-), 1.75 (m, 2H, -OCH2CH2-), 1.38-1.25 (m, 10H, -OCH2CH2(CH2)5), 0.90 (m, 3H, -OCH2CH2(CH2)5CH3).
4.1.2. Synthesis of 2-hydroxy-5-(octyloxy)benzaldehyde
Magnesium chloride (1.7 g 17.9 mmol) and paraformaldehyde (0.9 g, 30.0 mmol) were added to a flame dried round bottom flask with a magnetic stir bar and dissolved in THF (100 mL). Triethylamine (1.8 g, 17.8 mmol) was then added, and solution was left to stir at room temperature for 10 minutes. After, 4-(octyloxy)phenol (1.1 g, 5 mmol) was added and refluxed at 66˚C for 24 h. Reaction mixture was then left to cool before diluted with ether, and washed with 1M HCl. Mixture as dried with magnesium sulfate and evaporated under reduced pressure to afford 2-hydroxy-5-(octyloxy)benzaldehyde (1.1 g, 88%) as a brown oil. 1H-NMR (300 MHz, CDCl3, 25°C): δ= 10.62 (s, 1H, -OH), 9.82 (s, 1H, -CHO), 7.12 (dd, J = 9.0, 3.1 Hz, 1H, Ar-H), 6.98 (d, J = 3.1 Hz, 1H, Ar-H), 6.90 (d, J = 9.1 Hz, 1H, Ar-H), 3.92 (t, J = 6.6 Hz, 2H, -OCH2-), 3.90 (t, 2H, -OCH2-), 1.75 (m, 2H, -OCH2CH2-), 1.38 - 1.25 (m, 10H, -OCH2CH2(CH2)5), 0.90(m, 3H, -OCH2CH2(CH2)5CH3).
4.1.3. Synthesis of 2-nitro-4-(octyloxy)phenol
4-(octyloxy)phenol (3.0 g, 13.0 mmol) was added to benzene (100 mL) in a flame dried round bottom flask with a magnetic stir bar. Once cooled to 0˚C, 56% aqueous nitric acid solution (13 mmol) was added slowly and vigorously stirred for 2 minutes. The reaction mixture was then quenched with water after which the organic phase was dried with magnesium sulfate and evaporated to reveal the crude product as an orange powder. Impurities were removed via column chromatography (silica, EtOAc/heptane, [1:20]) to provide 2-nitro-4-(octyloxy)phenol (1.94 g, 56%) as an orange solid. Rf=0.4, 1H-NMR (300 MHz, CDCl3, 25°C): δ= 10.33 (s, 1H, -OH), 7.50 (d, J = 3.07, 1H, Ar-H), 7.22 (dd, J1 = 3.13, J2 = 3.04, 1H, Ar-H), 7.08 (d, J = 9.17, 1H, Ar-H), 3.94 (t, 2H, -OCH2-), 1.75 (m, 2H, -OCH2CH2-), 1.50-1.25 (m, 10H, -OCH2CH2(CH2)5), 0.90(m, 3H, -OCH2CH2(CH2)5CH3).
4.1.4. Synthesis of 2-amino-4-(octyloxy)phenol
In a flame dried round bottom flask equipped with a stir bar, a solution of 2-nitro-4-(octyloxy)phenol (1.3 g, 4.8 mmol) and 10% Pd/C (51 mg) in MeOH (50 mL) were placed under hydrogen atmosphere. The reaction mixture was left to stir for 24 h. After, the catalyst was filtered off and evaporated to reveal 2-amino-4-(octyloxy)phenol (0.99 g, 89%) as a dark red solid. (1H-NMR (300 MHz, CDCl3, 25°C): δ= 6.65 (d, J = 8.47, 1H, Ar-H), 6.35 (d, J = 2.77, 1H, Ar-H), 6.21 (dd, J1 = 2.79, J2 = 2.78, 1H, Ar-H), 3.87 (m, 2H, -NH2), 3.94 (t, 2H, -OCH2-), 1.75 (m, 2H, -OCH2CH2), 1.50-1.25 (m, 10H, -OCH2CH2(CH2)5), 0.90(m, 3H, -OCH2CH2(CH2)5CH3).
4.1.5. Synthesis of (Z)-2-((2-hydroxy-5-(octyloxy)benzylidene)amino)-4-(octyloxy)phenol 1
2-amino-4-(octyloxy)phenol (0.4 g, 1.68 mmol) was dissolved in ethanol (20 mL) in a flame dried round bottom flask equipped with a magnetic stir bar. Then, 2-hydroxy-5-(octyloxy)benzaldehyde (0.42 g, 1.68 mmol) was added dropwise to the reaction mixture and refluxed for 1 h at 80˚C. After, the reaction was left to cool and evaporated to dryness before purification via column chromatography (silica, EtOAc/heptane, [1:5]) (Z)-2-((2-hydroxy-5-(octyloxy)benzylidene)amino)-4-(octyloxy)phenol (0.55 g, 69%) as a black solid. (1H-NMR s(300 MHz, CDCl3, 25°C): δ= 8.61 (s, 1H, -CH=N-), 7.03 (dd, J1 = 9.0, J2 = 2.8 Hz, 1H, Ar-H), 7.00 (d, J = 7.5 Hz, 1H, Ar-H), 6.90 (m, 2H, Ar-H), 6.79 (dd, J = 8.8, J2 = 2.8 Hz, 1H, Ar-H), 6.72 (d, J = 2.8 Hz, 1H, Ar-H), 3.92 (m, 4H, -OCH2 -), 1.75 (m, 4H, -OCH2CH2 -), 1.50-1.25 (m, 20H, -OCH2CH2(CH2)5), 0.90(m, 6H, -OCH2CH2(CH2)5CH3). λmax nm: 386. FTIR (cm-1): 2985, 2940, 2895, 2830, 1622, 1492, 1454, 1299, 1241, 1126, 1036, 808, 735, 563, 464.
4.1.6. Synthesis of [Ni]2[L]2 Complex 2
(Z)-2-((2-hydroxy-5-(octyloxy)benzylidene)amino)-4-(octyloxy)phenol (0.1 g, .2 mmol) and triethyl amine (1.5 equiv.) are dissolved in ethanol (20 mL) in a flame dried round bottom flask equipped with a magnetic stir bar. After, a solution of nickel (II) acetate hexahydrate (50 mg, 0.2 mmol) in ethanol (20 mL) is added dropwise to the round bottom flask and refluxed for 1 h at 65˚C. The reaction mixture was cooled before filtering to obtain the [Ni]2[L]2 as a dark red precipitate. The crude, solid complex was washed with hot ethanol twice to remove any impurities, revealing the pure [Ni]2[L]2 complex (70 mg, 33%). MALDI-TOF m/z: calcd. For C58H82N2Ni2O8+ 1051.478; found [M+H]+: 1051.165. FTIR (cm-1): 2920, 2852, 1600, 1545, 1489, 1468, 1262, 1202, 1151, 1046, 809, 512. Elemental Analysis: Anal. Calcd: C: 66.18, H: 7.85, N: 2.66%. Found: C: 65.26, H: 7.27, N: 2.55%.
DFT Calculations
All calculations were performed using the Gaussian 09 package version EM64L- G09RevC.01. Electronic structure calculations were based on Kohn-Sham density functional theory (KS-DFT) with Becke’s three-parameter hybrid functional (B3LYP) and a compound basis set, where the Pople’s 6–311+G(d,p) basis sets were used for C, H, N, O, Ni. For our system, we first performed a tight structural optimization, followed by a frequency calculation to confirm that the optimized structure was indeed a minimum (with no imaginary frequencies).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, including characterization and spectral data of all intermediate compounds and Ni2L2 2 (1H NMR, 13C NMR, UV-vis, FT-IR), all heterogeneous electrochemical data acquired for Ni2L2 2.
Author Contributions
Conceptualization: W.S.; methodology: K.S., H.A., A.A., F.Y., and W.S.; validation: W.S.; formal analysis: K.S., H.A., A.A., F.Y. and W.S.; investigation: K.S., H.A., F.Y., A.A., and W.S.; writing—original draft preparation: K.S., A.A., and W.S.; writing—review and editing: K.S. and W.S.; visualization: K.S., A.A., and W.S.; supervision: W.S.; funding acquisition: K.S. and W.S. All authors have read and agreed to the published version of the manuscript.
Funding
W.S acknowledges the financial support of the Austrian Science Fund (FWF Standalone Projects P28167 “Heterogeneous catalysis for water oxidation and hydrogen evolution” and P32045 “Catalysts for biomass valorization”) and of the Austrian Research Promotion Agency FFG (FFG Project Nr.: 883671). The NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, “RERI-uasb”). K.S. acknowledges the financial support of the Fulbright-Austrian Marshall Plan Foundation Award in Science and Technology.
Data Availability Statement
Not applicable.
Acknowledgments
We gratefully thank Dr. Thomas Bögl from the Department of Analytical Chemistry and Prof. Dr. Clemens Schwarzinger from the Institute for Chemical Technology of Organic Materials at the JKU for carrying out ESI HR-MS and MALDI-TOF MS measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dresselhaus, M.S.; Thomas, I.L. Alternative Energy Technologies. 2001, 414.
- Gu, S.; Xu, B.; Yan, Y. Electrochemical Energy Engineering: A New Frontier of Chemical Engineering Innovation. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 429–454. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Majumdar, A. Opportunities and Challenges for a Sustainable Energy Future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Geng, B.; Yan, F.; Zhang, X.; He, Y.; Zhu, C.; Chou, S.; Zhang, X.; Chen, Y. Conductive CuCo-Based Bimetal Organic Framework for Efficient Hydrogen Evolution. Adv. Mater. 2021, 33, 2106781. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhao, S.; Zhao, K.; Muqsit, A.; Tang, H.; Chang, L.; Zhao, H.; Gao, Y.; Tang, Z. Ultrathin Platinum Nanowires Grown on Single-Layered Nickel Hydroxide with High Hydrogen Evolution Activity. Nat. Commun. 2015, 6, 6430. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bigi, J.P.; Piro, N.A.; Tang, M.L.; Long, J.R.; Chang, C.J. Molecular Cobalt Pentapyridine Catalysts for Generating Hydrogen from Water. J. Am. Chem. Soc. 2011, 133, 9212–9215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, M.; Yang, Y.; Yao, T.; Sun, L. A Molecular Copper Catalyst for Electrochemical Water Reduction with a Large Hydrogen-Generation Rate Constant in Aqueous Solution. Angew. Chem. Int. Ed. 2014, 53, 13803–13807. [Google Scholar] [CrossRef] [PubMed]
- Karunadasa, H.I.; Chang, C.J.; Long, J.R. A Molecular Molybdenum-Oxo Catalyst for Generating Hydrogen from Water. Nature 2010, 464, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, M.; Yang, Y.; Zheng, D.; Han, K.; Sun, L. Highly Efficient Molecular Nickel Catalysts for Electrochemical Hydrogen Production from Neutral Water. Chem Commun 2014, 50, 14153–14156. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational High-Throughput Screening of Electrocatalytic Materials for Hydrogen Evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Ji, S.; Wang, H.; Linkov, V.; Gai, H.; Liu, F.; Liu, Q.; Wang, R. N-Doped 3D Porous Ni/C Bifunctional Electrocatalysts for Alkaline Water Electrolysis. ACS Sustain. Chem. Eng. 2019, 7, 3974–3981. [Google Scholar] [CrossRef]
- Karunadasa, H.I.; Montalvo, E.; Sun, Y.; Majda, M.; Long, J.R.; Chang, C.J. A Molecular MoS2 Edge Site Mimic for Catalytic Hydrogen Generation. Science 2012, 335, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Abdel Aziz, A.A.; Salem, A.N.M.; Sayed, M.A.; Aboaly, M.M. Synthesis, Structural Characterization, Thermal Studies, Catalytic Efficiency and Antimicrobial Activity of Some M(II) Complexes with ONO Tridentate Schiff Base N-Salicylidene-o-Aminophenol (SaphH2). J. Mol. Struct. 2012, 1010, 130–138. [Google Scholar] [CrossRef]
- Kasumov, V.T. Oxovanadium(IV), Nickel(II) and Palladium(II) Complexes of Tridentate Salicylaldiminates Derived from 2,4-Di-Ter-Butyl-6-Aminophenol. Z. Für Naturforschung B 2001, 56, 263–270. [Google Scholar] [CrossRef]
- Setia, S.; Pal, S.K. Unsymmetrically Substituted Room Temperature Discotic Liquid Crystals Based on Hexa–Peri–Hexabenzocoronene Core. ChemistrySelect 2016, 1, 880–885. [Google Scholar] [CrossRef]
- Kong, L.; Wang, L.; Sun, D.; Meng, S.; Xu, D.; He, Z.; Dong, X.; Li, Y.; Jin, Y. Aggregation-Morphology-Dependent Electrochemical Performance of Co3O4 Anode Materials for Lithium-Ion Batteries. Molecules 2019, 24, 3149. [Google Scholar] [CrossRef] [PubMed]
- Nestke, S.; Stubbe, J.; Koehler, R.; Ronge, E.; Albold, U.; Vioel, W.; Jooss, C.; Sarkar, B.; Siewert, I. A Binuclear Cobalt Complex in the Electrochemical Water Oxidation Reaction. Z. Für Anorg. Allg. Chem. 2022, 648, e202200119. [Google Scholar] [CrossRef]
- Patrício, S.; Cruz, A.I.; Biernacki, K.; Ventura, J.; Eaton, P.; Magalhães, A.L.; Moura, C.; Hillman, A.R.; Freire, C. Novel Layer-by-Layer Interfacial [Ni(Salen)]−Polyelectrolyte Hybrid Films. Langmuir 2010, 26, 10842–10853. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.; Deng, F.; Li, J. Electropolymerization and Electrochemical Behavior of Nickel Schiff Base Complexes with Different Groups between Imine Linkages. RSC Adv. 2016, 6, 79894–79899. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, X.; Zhang, R.; Yu, S.; Levell, Z.H.; Wang, C.; Ma, S.; Zou, P.; Han, L.; Qin, J.; et al. Highly Selective Oxygen Reduction to Hydrogen Peroxide on a Carbon-Supported Single-Atom Pd Electrocatalyst. ACS Catal. 2022, 12, 4156–4164. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis procecure of binuclear [Ni]2[L]2 catalyst 2 for hydrogen evolution reaction (HER) from the octyloxy Schiff base ligand 1 precursor.
Scheme 1.
Synthesis procecure of binuclear [Ni]2[L]2 catalyst 2 for hydrogen evolution reaction (HER) from the octyloxy Schiff base ligand 1 precursor.
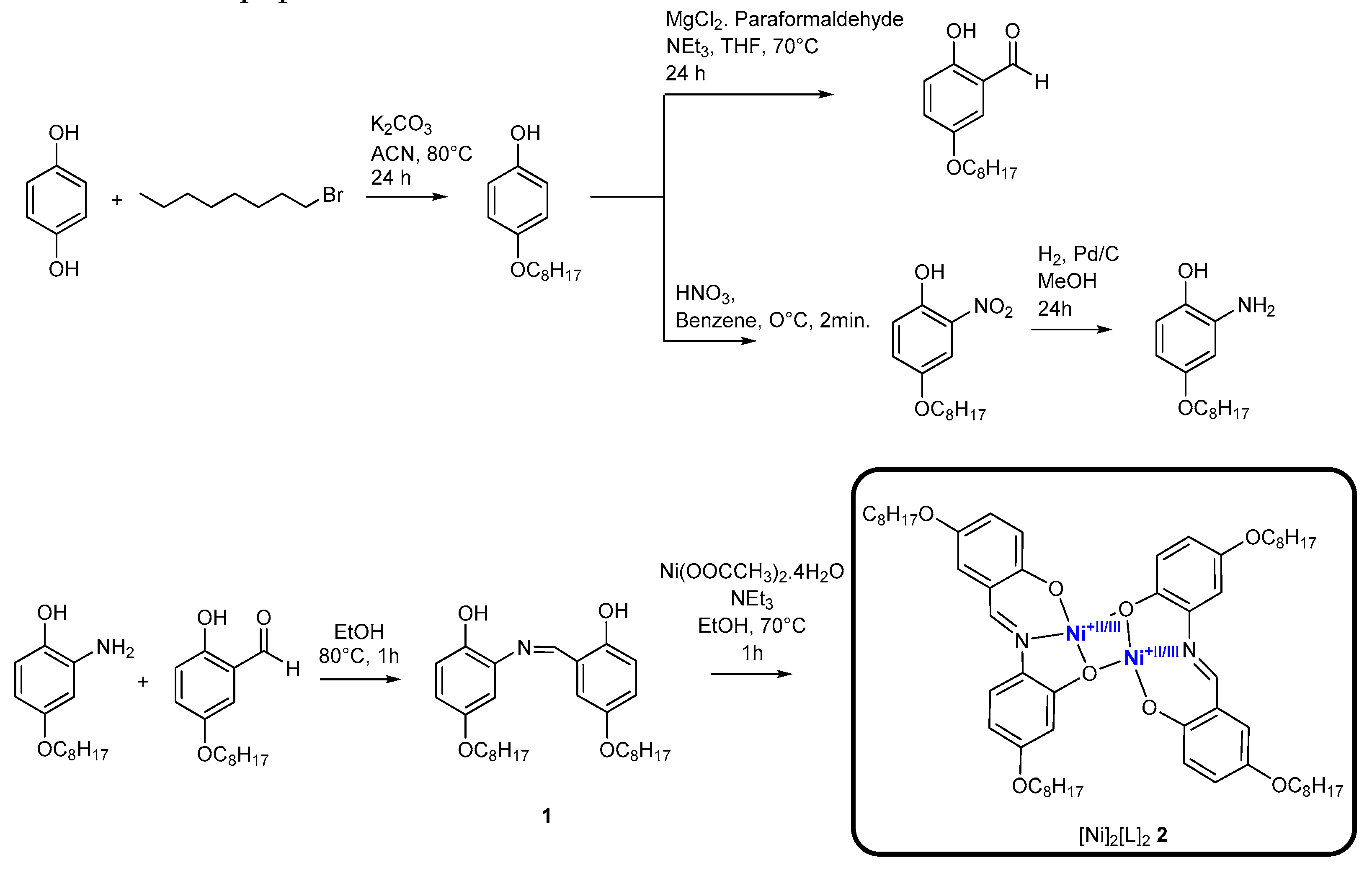
Figure 1.
a) Solid state 13C NMR spectrum and MALDI-TOF mass spectrum of [Ni]2[L]2 2.
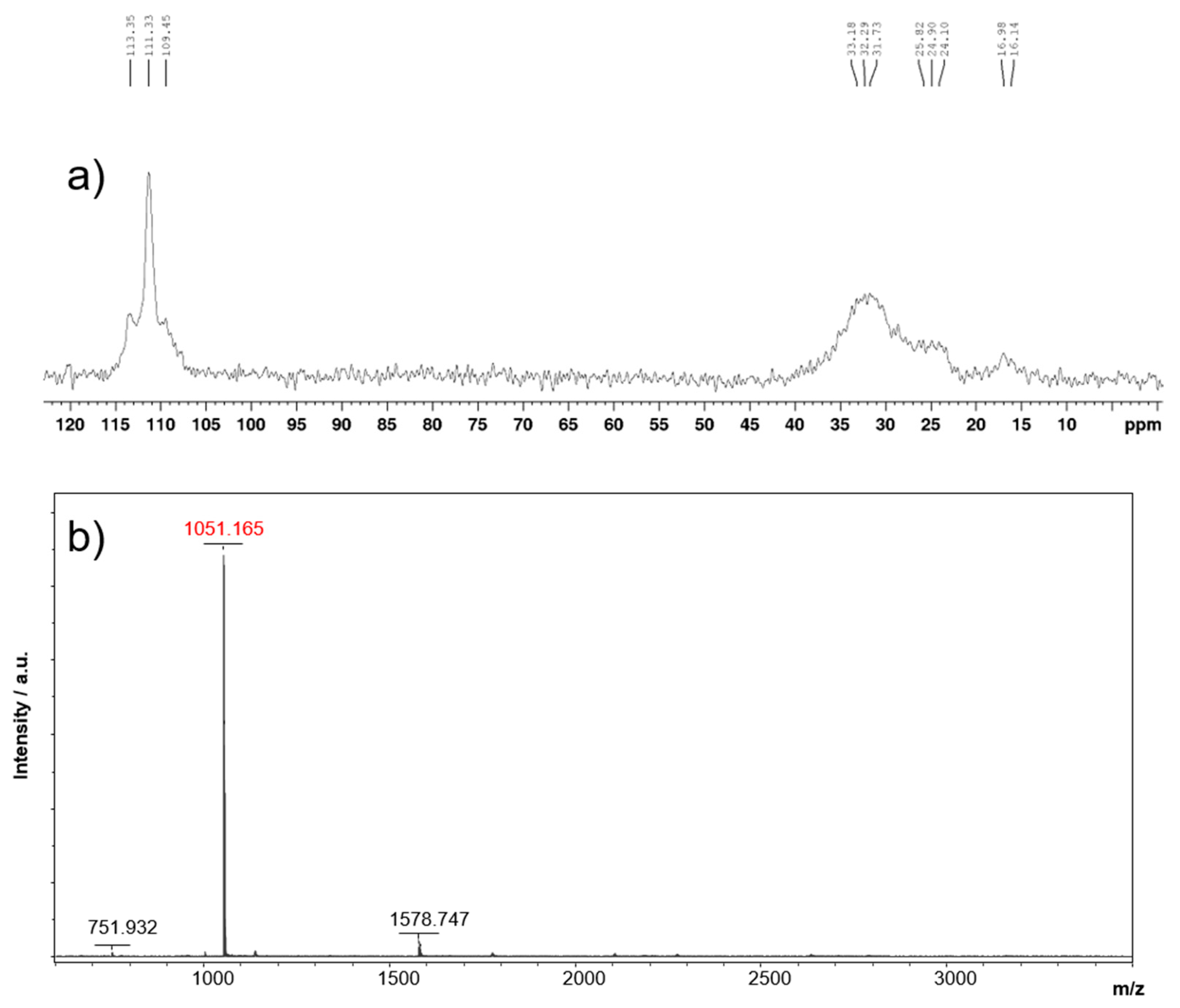
Figure 2.
a) Molecular structure (top and side view) of [Ni]2[L]2 2. b) frontier orbital maps of [Ni]2[L]2 2 obtained from DFT calculations.
Figure 2.
a) Molecular structure (top and side view) of [Ni]2[L]2 2. b) frontier orbital maps of [Ni]2[L]2 2 obtained from DFT calculations.
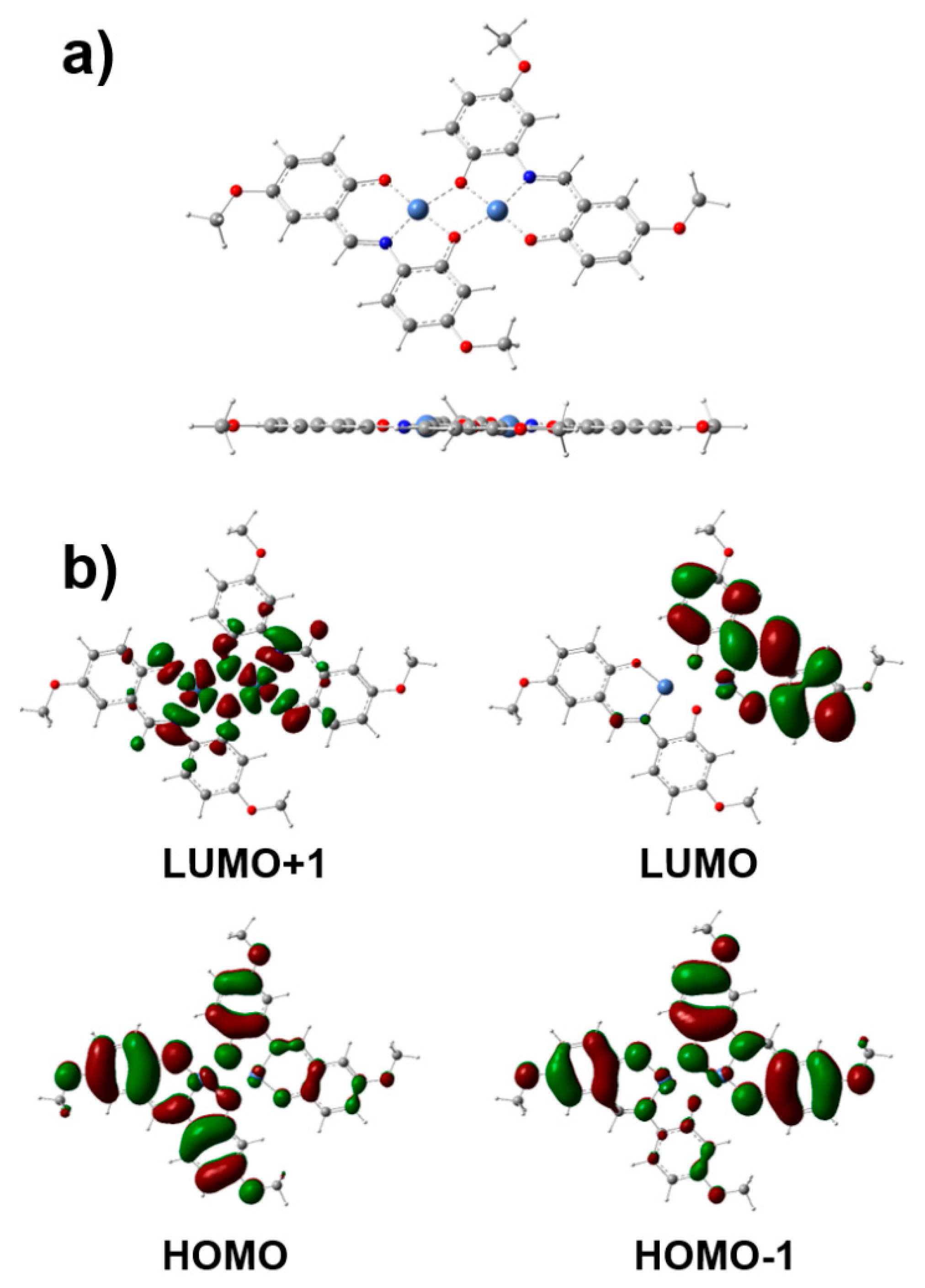
Figure 3.
a) Broad XPS survey scan of [Ni]2[L]2 2 as a pure, powder sample. c) Narrow scans for Ni2p3/2 and Ni2p1/2 present in [Ni]2[L]2 2. d) Narrow scans for carbon (C1) present in [Ni]2[L]2 2.
Figure 3.
a) Broad XPS survey scan of [Ni]2[L]2 2 as a pure, powder sample. c) Narrow scans for Ni2p3/2 and Ni2p1/2 present in [Ni]2[L]2 2. d) Narrow scans for carbon (C1) present in [Ni]2[L]2 2.
Figure 4.
a) Cyclic voltammogram (CV) and b) linear sweep voltammogram (LSV) of [Ni]2[L]2 is loaded on carbon paper as working electrode (WE) and Ag/AgCl as a reference electrode in argon saturated 0.5 M H2SO4 (pH 1.8) electrolyte at room temperature.
Figure 4.
a) Cyclic voltammogram (CV) and b) linear sweep voltammogram (LSV) of [Ni]2[L]2 is loaded on carbon paper as working electrode (WE) and Ag/AgCl as a reference electrode in argon saturated 0.5 M H2SO4 (pH 1.8) electrolyte at room temperature.
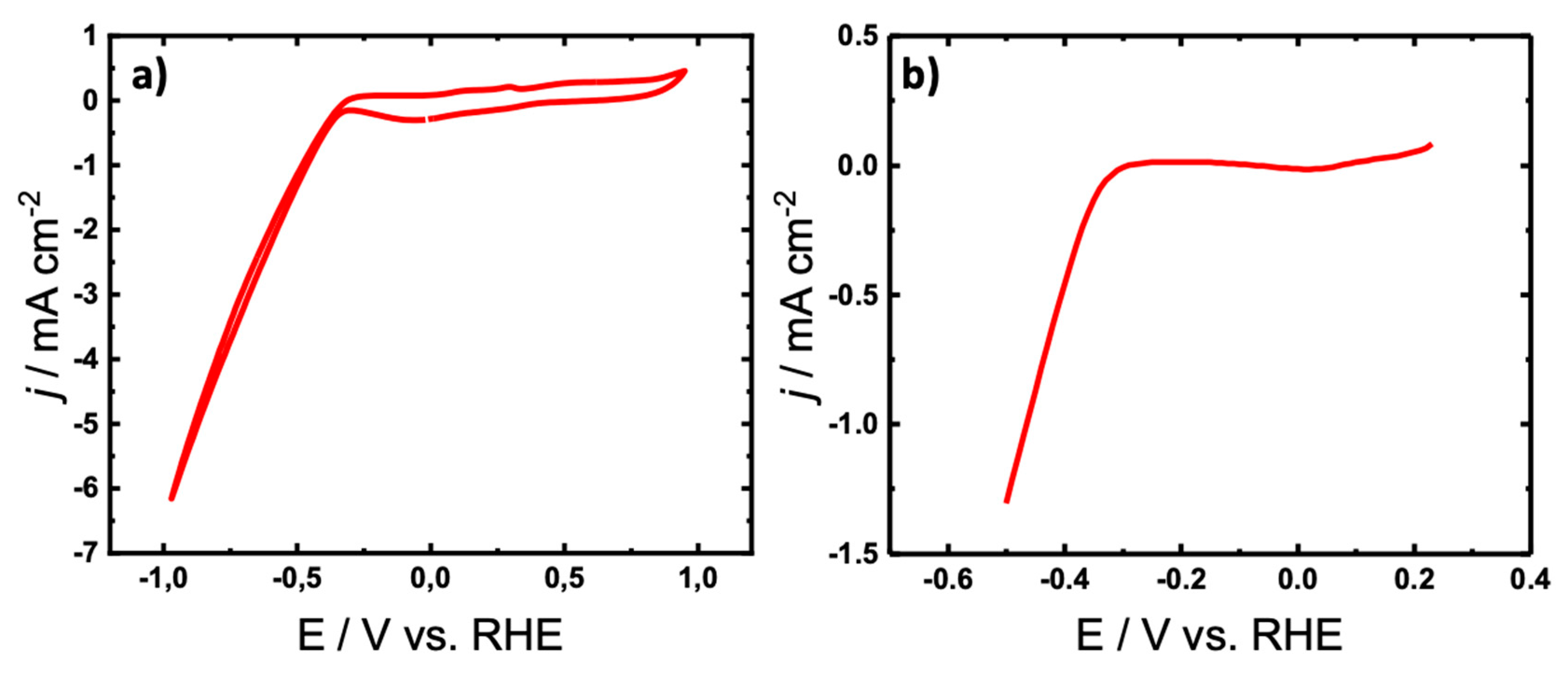
Figure 5.
a) Prolonged electrolysis for 17.5 hr via chronoamperomtry to determine long-term stability of [Ni]2[L]2 2 b) Bode plot recorded via electrochemical impedance spectroscopy in the frequency range of 1x10-1 Hz to 1x105 with a perturbation amplitude of 10 mV Hz c) Double-layer capacitance measurements for determining electrochemically active surface area for [Ni]2[L]2 2 catalyst in 0.5 M H2SO4 d) Linear dependence of the cathodic peak for current versus scan rate.
Figure 5.
a) Prolonged electrolysis for 17.5 hr via chronoamperomtry to determine long-term stability of [Ni]2[L]2 2 b) Bode plot recorded via electrochemical impedance spectroscopy in the frequency range of 1x10-1 Hz to 1x105 with a perturbation amplitude of 10 mV Hz c) Double-layer capacitance measurements for determining electrochemically active surface area for [Ni]2[L]2 2 catalyst in 0.5 M H2SO4 d) Linear dependence of the cathodic peak for current versus scan rate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



