
Submitted:
05 September 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
Human T-cell leukemia virus type-1 (HTLV-1) is the causative agent of adult T-cell leukemia/lymphoma (ATL). HTLV-1 carriers have a life-long asymptomatic balance between infected cells and host antiviral immunity, but 5–10% of carriers lose this balance and develop ATL. Coinfection with Strongyloides also promotes ATL development, suggesting that the immunological status of infected individuals is a determinant of viral pathogenicity. As CD4+ T cells play a central role in host immunity, deregulation of their function and differentiation by HTLV-1 promotes the immune evasion of infected T cells. During ATL development, the accumulation of genetic and epigenetic alterations in key host immunity-related genes further disturbs immunological conditions. Various therapeutic approaches have been developed to treat these abnormalities. Allogeneic hematopoietic stem cell transplantation is currently the only treatment with the potential to cure ATL; however, the patient's immune state may also contribute to its outcome. Additionally, the activity of the anti-CC chemokine receptor 4 antibody, mogamulizumab, also depends on immune functions, such as antibody-dependent cytotoxicity. In this review, we comprehensively summarize the immunological pathogenesis of HTLV-1 infection in ATL and integrate clinical findings to determine the factors to be considered for developing treatment strategies for ATL.
Keywords:
human T-cell leukemia virus type 1
; adult T-cell leukemia/lymphoma
; viral genes
; genetic alterations
; immune response
; host–pathogen interaction
; pathogenesis
; treatment
1. Introduction
Human T-cell leukemia virus type 1 (HTLV-1) is a retrovirus that causes diseases, such as adult T-cell leukemia/lymphoma (ATL), HTLV-1–associated myelopathy/tropical spastic paraparesis (HAM/TSP), and HTLV-1 uveitis (HU). HTLV-1 mainly infects CD4+ T cells and transforms the infected cells to cause ATL with a poor prognosis [1,2]. ATL is classified into indolent types (smoldering and chronic) and aggressive types (acute and lymphoma). Patients with aggressive ATL have a median survival time of less than one year, and those with indolent ATL often undergo a blast crisis after several years [3,4,5,6,7]. Approximately 5–10 million individuals are infected with HTLV-1 (carriers) worldwide, and they are geographically concentrated mainly in Japan, South America, Africa, and the Caribbean islands. The age of ATL onset varies regionally, ranging from 47–49 years in South America, to 68 years in Japan, with a slightly higher prevalence in men than in women [8,9,10]. The main routes of HTLV-1 infection differ across countries. In Japan, mother-to-child transmission occurs mostly via breast milk, whereas in South America and Africa, horizontal transmission is also common [11,12,13]. ATL cells have a mature T-cell phenotype with characteristic multi-lobulated nuclei, known as “flower cells,” and typical cell surface markers of ATL cells are the CD4+ CD25+ activated T cell phenotype [14].
The genomic structures of HTLV-1 and human immunodeficiency virus (HIV) are similar, with structural genes containing Gag, Pol, and Env, and regulatory genes, such as HIV-1 transactivator of transcription (Tat) and Rev of HIV; Tax and Rex of HTLV-1; accessory genes such as Vif, Vpr, Vpx, Vpu, and Nef of HIV; p12, p13, p30, and HBZ of HTLV-1, which are flanked by 5′ and 3′ long terminal repeats (LTR) [15]. Pathogenicity differs depending on gene differences in these accessory and regulatory regions. HIV mainly infects the CD4 receptor, whereas HTLV interacts with glucose transporter 1, neuropilin-1, and heparan sulfate proteoglycans to enter target cells. In addition to CD4+ T cells, both HIV and HTLV-1 infect non-lymphoid monocytic cells, such as macrophages and dendritic cells; however, both cause diseases originating from CD4+ helper T cells [16]. During HIV infection, direct cell destruction because of HIV infection and proliferation and destruction of infected cells by HIV antigen-specific CD8+ cytotoxic T cells (CTLs) decreases the CD4+ T cell number and consequently causes acquired immunodeficiency syndrome (AIDS) [17]. In contrast, HTLV-1 remains dormant in the host genome as a provirus for several decades, resulting in persistent infection, in which 1–2% (copies/100 peripheral blood mononuclear cells) of HTLV-1-infected lymphocytes are present in the peripheral blood [18,19]. These infected lymphocytes become cancerous in some HTLV-1 carriers, causing ATL. Therefore, the onset of ATL cannot be explained by viral infection alone, and host-side factors are speculated to be involved in this process.
HTLV-1 encodes the viral transcription transactivator, Tax, in the pX region of its genome, which promotes oncogenesis. Tax interacts with various host cell proteins, affects intracellular signaling pathways, regulates gene transcription, and contributes to HTLV-1-infected T cell proliferation. However, because Tax exhibits strong immunogenicity, infected T cells expressing Tax are actively eliminated by tax-specific CTLs [20,21]. At the onset of ATL, approximately 50% of cases do not show Tax expression due to methylation or deletion of the 5′ LTR region; thus, selective proliferation of clones that are not attacked by CTLs is likely to be promoted [22]. In addition, recent studies have revealed that sense-side transcription leading to Tax expression occurs in transient bursts even in the absence of genomic deletions or epigenetic changes in the provirus [23,24,25]. This may be a viral survival strategy that minimizes Tax expression and avoids CTL attacks. HTLV-1 encodes the oncogenic factor HBZ via antisense transcription of the pX region. HBZ promotes the transcription of forkhead box protein P3 (Foxp3), the master regulator of regulatory T cells, and confers regulatory T cell traits to HTLV-1-infected T cells [26]. As a result, the proliferation of HTLV-1-infected T cells is expected to promote immunosuppression through the secretion of inhibitory cytokines, such as interleukin (IL)-10. Changes in the innate immune system, such as macrophages and eosinophils, have been observed in patients with ATL [28,29]. Therefore, immunity may be a host-specific factor involved in ATL development. Age, HTLV-1 proviral load (PVL), human leukocyte antigen (HLA) haplotype, and strongyloidiasis are known risk factors for ATL development [19, 30–32]; this suggests a relationship between immunity and carcinogenesis.
Clinically, ATL patients such as those with AIDS develop profound immunodeficiency [33]. Furthermore, for patients with acute and lymphoma types, antibiotics are generally administered to prevent infections because immune function is weakened by treatments such as intense chemotherapy and transplantation. In addition, complications of hypercalcemia and opportunistic infections are associated with a poor prognosis of ATL [35]. Opportunistic infections, including Pneumocystis carinii, aspergillosis, and candidiasis, which are observed in patients with AIDS, cytomegalovirus pneumonia, and Strongyloides stercoralis infections have also been observed [36,37]. Opportunistic malignancies, including Kaposi’s sarcoma and Epstein-Barr virus-associated B-cell lymphoma, which are common in patients with AIDS, have also been reported in patients with ATL [38,39]. Importantly, opportunistic infections have also been reported in HTLV-1 carriers, and carriers of opportunistic infections may be more susceptible to ATL [40,41]. Thus, immunity is closely related to progression of HTLV-1 carriers and prognosis of patients with ATL. Therefore, elucidation of immune pathology will enhance the understanding of ATL onset mechanisms and the establishment of therapeutic methods for its treatment. However, few studies have comprehensively integrated and discussed the knowledge accumulated from basic research and clinical aspects. In this review, we focus on immune abnormalities in HTLV-1 infection and ATL onset, summarize the findings reported thus far from both basic and clinical studies, and discuss them in an integrated manner towards the establishment of ATL therapeutics.
2. Immune disruption because of HTLV-1 infection
Approximately 95% of HTLV-1 carriers remain asymptomatic without any clinical symptoms associated with HTLV-1 infection throughout their lives. Therefore, unlike HIV, which continues to proliferate actively even during the asymptomatic period, HTLV-1 is a latent infection, which allows the possibility that the HTLV-1 infection alone does not affect the infected host. However, as evidenced by several studies, the skillful ability of HTLV-1 to evade and disturb the host immune system is undeniable; these processes are described below.
2.1. T cell anergy induced by HTLV-1 infection
Sense-strand transcription of HTLV-1 is generally silent; however, it has been reported to be activated in ex vivo cultures under hypoxic stress or high-glucose conditions [42,43]. The plus strand is suggested to be silent in the peripheral blood but expressed in the lymph nodes, leading to de novo cell-to-cell infection [43]. Zinc-finger CCCH-type antiviral protein 1 (ZC3HAV1/ZAP) is a host antiviral factor that suppresses translation and promotes degradation of specific viral mRNAs via binding to CG-rich RNA sequences. HTLV-1 sequences are rich in CG dinucleotides and are targets of ZAP [44], which may be a mechanism underlying viral transcriptional repression.
HTLV-1 encodes a potent transcription factor, Tax, which strongly induces viral gene expression via enhancing the recruitment of enhancers such as p300 to the viral LTR [1]. Tax has also been shown to transform T cells and fibroblasts in vitro, indicating that it functions as an oncogene [45,46]. Tax interacts with intracellular signaling pathways and transcription factors in infected T cells to regulate the cell cycle, cell proliferation, and apoptosis. Tax induces expression of genes encoding various cytokines and chemokines. In in vitro experiments, Tax expression has been reported to promote the production of inflammatory cytokines such as IL-2, IL-6, tumor necrosis factor (TNF)-α, and the immunosuppressive cytokine IL-10 [47,48]. Furthermore, Tax ectopically upregulates HLA class II expression in HTLV-1 infected T cells [49]. CD4+ T cells infected with HTLV-1 are in a state of excessive activation and express HLA class II molecules, which are normally expressed by only a small fraction of T cells, and may present antigens as antigen-presenting cells (APCs). HTLV-1-infected T cells may become tolerogenic APCs and cause antigen-specific T-cell tolerance (anergy) [49].
2.2. Acquisition of regulatory T cell phenotype by HTLV-1 infection
HBZ is known to have low immunogenicity and is hence persistently expressed during the viral incubation period and ATL onset and progression [50,51]. HBZ is a viral factor essential for ATL oncogenesis and functions at the protein and RNA levels [52]. In particular, HBZ induces the expression of Foxp3 and regulatory T cell-related molecules, such as CC chemokine receptor 4 (CCR4) and T cell immunoreceptors with Ig and ITIM domains (TIGIT) [53]. HBZ has also been reported to induce inflammatory pathology in addition to T cell tumorigenesis in HBZ transgenic mice [54]. Furthermore, HBZ suppresses the expression or function of the inhibitory co-receptors TIGIT, programmed cell death 1 (PD-1), B and T lymphocyte attenuator, and leukocyte-associated Ig-like receptor 1, in HTLV-1 infected T cells, thereby enhancing T cell receptor signaling and promoting the proliferation of infected cells [55].
Additionally, Rex, p30, p12, and p13 are transcribed from the sense strand of the pX region HTLV-1 via alternative RNA splicing. CTLs recognize HLA class I presented with the Tax peptide, and p12 expression has been shown to reduce the plasma membrane expression of HLA class I [56], likely promoting CTL evasion. In contrast, p30 expression decreases the expression of PU.1, a factor essential for the development of granulocytes, monocytes, macrophages, and lymphoid cells [57]. PU.1 downregulation is reported to reduce Toll-like receptor 4 expression and suppresses the secretion of pro-inflammatory cytokines, including monocyte chemoattractant protein-1, TNF-α, and IL-8 in macrophages [57]. Thus, multiple mechanisms involving viral factors suppress the immune response caused by HTLV-1 infection, and infected cells continue to survive without being eliminated (Figure 1).
3. Immune abnormalities in ATL
3.1. Genetic alterations
A large-scale integrative genomic analysis of ATL revealed that approximately 90% of ATL cases were enriched with activating mutations in the T cell receptor-nuclear factor (NF)-κB pathway [58]. Furthermore, using comprehensive DNA methylation analysis, hypermethylation of CpG islands, also known as CpG island methylator phenotype hypermethylation (CIMP), has been observed in approximately 40% of ATL cases [58]. In addition, genetic abnormalities such as mutations and deletions accumulate in HLA class I genes [58]. Genomic aberrations targeting the 3′-untranslated region (UTR) of programmed death ligand 1 (PD-L1) have also been reported to accumulate in ATL [59]. Truncation of the 3′-UTR of the PD-L1 gene occurs as a result of structural abnormalities such as deletion, translocation, and inversion of the PD-L1 3′-UTR genomic region, resulting in increased PD-L1 mRNA expression [59]. These findings lead to the speculation that during the carcinogenic process of ATL, clones that accumulate genomic abnormalities that can evade tumor immunity, such as HLA class I and PD-L1 aberrations, selectively increase in number. ATL cases with CIMP or PD-L1 amplification have a high degree of malignant phenotypes [58,60].
In addition, single-cell analysis revealed that HTLV-1-infected cells that acquired clonal proliferation ability exhibited abnormal expression of various immune-related molecules [61]. In addition to the upregulation of the immunosuppressive molecules PD-L1, CD73, and CD39, the upregulation of activation markers such as CD71, CD25, and CD38 and co-stimulatory molecules such as CD99, CD28, and CD278 were observed [61]. In HTLV-1-infected T cells, before clonal expansion, the expression of HLA class II molecules increased, but their expression decreased during clonal expansion [61]. Therefore, ATL cells appear to evade antitumor immunity via expressing immunosuppressive molecules with activated phenotypes (Figure 1).
3.2. Tumor immune microenvironment
Tumor-associated macrophages (TAMs) are major constituents of tumor stromal cells and act as tumor promoters via promoting cancer cell proliferation, invasion, angiogenesis, and immunosuppression [62,63,64]. Examination of lymph node involvement in acute and lymphoma-type ATL has shown that TAM infiltration is frequently observed and associated with poor prognosis [65]. Phagocytosis by TAMs is suppressed when cells express CD47, a ligand for SIRPα of TAMs, which is known as a "don't eat me" signal [66]. Although CD47 is abundantly expressed in erythrocytes and platelets, tumor cells also express CD47 to prevent phagocytosis by macrophages [66]. Expression of CD47 on ATL cells is not shown to be associated with patient prognosis, and expression of SIRPα on stromal cells was associated with favorable prognosis, which is unexpected [67].
PD-L1 is a transmembrane protein that suppresses immune responses via binding to two inhibitory receptors, PD-1 and B7-1 (CD80), and is strongly expressed on the surface of tumor and non-transformed cells in the tumor microenvironment [68]. PD-L1 expression in ATL cells has also been observed in ATL lymph node lesions and has been shown to be a poor prognostic factor [69]. PD-L1 expression in stromal cells, including macrophages and dendritic cells, is associated with a favorable prognosis in ATL patients [69]. However, the biological role of PD-L1 expression in the tumor microenvironment remains unclear. Additionally, the proportions of invariant natural killer T, natural killer (NK), and dendritic cells are decreased in the peripheral blood of ATL patients [70].
Single-cell analyses have shown that ATL is accompanied by a decrease in B cells and an increase in myeloid cells [61]. Among the myeloid cells, dendritic cells and atypical monocytes are upregulated in patients with ATL. These myeloid cells are also associated with increased activation markers such as CD64 and immune checkpoint molecules such as PD-1 [61]. Although B cell numbers tend to decrease in patients with ATL, the interferon pathway is upregulated in myeloid cells [61]. Importantly, the CTL decline in patients was associated with genetic aberrations in PD-L1, and in such cases, PD-L1 is upregulated in B cells and myeloid cells in the immune microenvironment [61]. Furthermore, NK cells exhibit functional abnormalities in HTLV-1 carriers and patients with ATL [61]. These findings suggest that ATL carcinogenesis is accompanied by changes in the immune microenvironment [61] (Figure 1).
4. Clinical aspects of HTLV-1 infection and ATL related to immunological alterations
4.1. Mother-to-child Transmission
ATL occurs in less than 5% of HTLV-1 carriers after five-six decades of chronic infection. Moreover, risk factors for ATL transformation are HTLV-1 infection during childhood, specifically more than six months of breastfeeding (RR, 2.91; 95% confidence interval, 1.69–5.03) [71]. This type of transmission has been associated with increased maternal PVL after delivery [72] and a genetic predisposition to a major locus on chromosome 6q27 in children [73]. Moreover, an increase in APCs in breast milk may be responsible for HTLV-1 infection during extended breastfeeding [74]. In addition, pregnant women coinfected with Strongyloides stercoralis have a higher risk of transmission [75].
4.2. Coinfections
4.2.1. HIV
HIV and HTLV belong to the Retroviridae family and share genomic and epidemiological characteristics. Moreover, a recent meta-analysis showed that HIV and HTLV-I-coinfected subjects have accelerated HIV progression, worse survival, and a high frequency of developing HAM/TSP, peripheral neuropathy, encephalopathy, and opportunistic infections, such as scabies, candidiasis, and strongyloidiasis [76,77]. HTLV-1 activates CD4+ T cells, and Tax upregulates HIV-1 infection [78]. In contrast, coinfection of HIV with HTLV-2, another type of HTLV that is non-pathogenic but rarely associated with HAM/TSP, showed slow T cell depletion and AIDS progression, with higher HIV-1 viral control and lower mortality rates [79]. This effect is mediated via CTL expansion induced by HTLV-2, including a high rate of the effector memory phenotype and increased levels of granzymes A and B and perforin [79]. Although a case of ATL in an HIV-1-positive patient has been reported [80], more evidence is needed to determine the possibility of patients with HIV having an increased risk of ATL.
4.2.2. Strongyloides
S. stercoralis is an opportunistic nematode that can cause severely disseminated infections in immunocompromised individuals. It is typically found in tropical regions, and overlaps with the high prevalence distribution of HTLV-1 [81]. HTLV-1 infections have been linked to severe strongyloidiasis via the high production of interferon (IFN)-γ and downregulation of IL-4, IL-5, IL-13, and IgE, which are necessary for host defense against parasites [82]. Additionally, HTLV-1 PVLs are significantly higher in HTLV-1 carriers with strongyloidiasis than in those without strongyloidiasis [41]. Strongyloidiasis can induce polyclonal expansion of HTLV-1-infected T cells via upregulation of the IL-2/IL-2R pathway [83]. S. stercoralis infections may partially explain the clinical and genomic disparities in ATL cases between Japan and other regions. Particularly, early onset ATL has been associated with S. stercoralis infections outside Japan [84].
4.3. Prognosis
The prognosis of ATL is poor owing to chemotherapy resistance, high relapse rates, and opportunistic infections. Despite aggressive treatment, ATL prognosis remains poor, with a three-year overall survival rate of less than 25%. Multiagent chemotherapy, interferon-α plus zidovudina, mogamulizumab, and allogeneic hematopoietic stem cell transplantation (allo-HSCT) remain the main treatment formulations used. Mogamulizumab is a monoclonal antibody that targets CCR4 and is associated with a higher response rate in combination with chemotherapy; however, it may improve the transient responses before allo-HSCT, which remains the only curative treatment available for patients with aggressive ATL. However, not all patients are allo-HSCT candidates. In ideal clinical trials, only 7% of patients with aggressive ATL undergo allo-HSCT [85]. The 5-year survival was 40% compared to 12% in patients who did not receive allo-HSCT [85].
4.4. Prognostic factors in aggressive ATL-subtypes.
Several scoring methods have been developed worldwide using host-dependent and disease-burden factors. The ATL Prognostic Index was constructed retrospectively for patients with aggressive subtypes who did not undergo allo-HSCT [86]; host factors found were age (> 70 years), serum albumin (< 3.5g/dL) and poor performance status (ECOG score > 1); disease-burden factors were Ann Arbor stage (I–II vs III–IV) and soluble interleukin-2 receptor (sIL-2R, > 20 000U/mL). The Japan Clinical Oncology Group also developed a prognostic index that included only performance status and corrected calcium levels (2.75 mmol/L) based on a combined analysis of three consecutive prospective clinical trials; however, patients were relatively young, physically fit, and without organ dysfunction [85]. These prognostic indices indicate a median overall survival of less than eight months for high-risk patients.
A nationwide survey in Japan evaluated patients with aggressive ATL undergoing intensive treatment with potential indications for allo-HSCT. The independent prognostic factors identified were clinical subtype (acute, worse than lymphoma), poor performance status (ECOG score > 1), corrected calcium levels (≥12 mg/dL), C-reactive protein level ≥ 2.5 mg/dL, and sIL-2R level > 5,000 U/mL [87]. The stem cell source and disease status during transplantation are factors related to better outcomes in allo-HSCT [88].
4.5. Anti-HTLV-1 immunity
4.5.1. Tax -specific CTLs
Tumor-specific CTL induction is associated with the prognosis of solid tumors [89]. Tax-specific CTLs increase after allo-HSCT and mogamulizumab treatment in patients with ATL [90,91]. Tax-CTLs are found in long-term allo-HSCT survivors; posttransplant induction and increased Tax-CTLs may contribute to the graft-versus-leukemia effect in long-term survivors [91]. Skin rash induced by mogamulizumab is reportedly linked to better survival, and CD8+ T cell infiltration in skin lesions and Tax-specific CTL increase could explain this benefit [90]. The reversal of the CD4-to-CD8 ratio in peripheral mononuclear cells has been reported in long-term survivors [90]. Moreover, Tax-CTLs occur more frequently after severe herpes simplex virus infections during or after chemotherapy or mogamulizumab treatment [90]. A recent study showed preliminary positive results with varicella-zoster virus vaccination, including an increase in Tax-CTLs after mogamulizumab-based treatment in aggressive ATL subtypes [92]. Thus, herpes simplex virus and varicella zoster virus may induce CTL activation via cytokine and chemokine release. However, exhaustion of Tax-CTLs is reported to correlate with the number of HTLV-1+ T cells and CTL activity [93]. These findings suggest that functional Tax-CTLs are important prognostic factors for the treatment of patients with ATL. Therefore, considering that CTL decrease in patients with ATL is associated with genetic aberration of PD-L1 in ATL cells [70], interactions between the immune microenvironment and ATL cells may be important factors in CTL function, and thus, for post-transplant outcomes (Figure 2).
4.5.2. HBZ -specific CTLs
The graft-versus-leukemia effect of allo-HSCT has been explained primarily by Tax-specific CTLs, although Tax is expressed in approximately 50% of ATL cases. HBZ transcripts were detected in all ATL cases. HBZ-specific CTLs have also been induced in vitro without showing limited cytotoxic activity against ATL cells [94]. Additionally, Tax peptides have stronger binding characteristics than HBZ peptides. Consequently, HTLV-1-infected individuals have a higher frequency of Tax-specific CTLs [94]. However, HLA class I alleles related to high-affinity HBZ peptides may play a protective role in asymptomatic HTLV-1 carriers and HAM/TSP patients [95]. This effect has been explained by the correlation between a low PVL and the binding affinity of HBZ peptides for HLA class I [95]. Moreover, a study reported that HBZ could induce CD4+ T cell responses in some ATL cases but only after allo-HSCT [96]. Hence, the HBZ peptide RRRAEKKAADVA (HBZ114-125) sequence with HLA-DRB1*15:02 restriction was shown to be recognized by specific CD4 + T cells [96]. Further studies are required to determine the immunogenicity and protective efficacy of HBZ peptides against ATL.
5. Development of therapeutic drugs targeting immunity for ATL
5.1. Anti-CCR4 antibody (mogamulizumab)
Mogamulizumab, an anti-CCR4 antibody, was the first molecular-targeted drug approved for ATL treatment in Japan; its effects include antibody-dependent cellular cytotoxicity and enhancement of antitumor immunity via regulatory T cell suppression [97,98]. Mogamulizumab has been favored in clinical practice because of its ability to control disease activity in patients with aggressive ATL; however, its add-on effects to chemotherapy remain controversial [99,100,101]. In addition, in patients with ATL treated with mogamulizumab, death from acute graft-versus-host disease after transplantation is often associated with regulatory T cell suppression, and mogamulizumab should be used with caution in transplant-eligible patients [102,103] (Figure 2). Thus, mogamulizumab is expected to be effective in combination with other treatments.
5.2. Anti-PD-1 antibody (nivolumab)
Anti-PD-1 antibody exerts an antitumor effect via binding to PD-1 on T cells and inhibits the transduction of inhibitory signals via inhibiting PD-1 binding to PD-L1/PD-L2, resulting in T cell activation. It is effective in treating solid tumors such as malignant melanoma and lung cancer [104]. Although clinical trials using anti-PD-1 antibody have been conducted in patients with ATL, their effectiveness has not been proven; conversely, administration of anti-PD-1 antibody (nivolumab) has exacerbated disease conditions [105]. ATL cells have been reported have traits similar to those of tumor-infiltrating regulatory T cells after anti-PD-1 antibody treatment [106]. As various abnormalities occur in the immune environment of patients with ATL [70], the benefit of anti-PD-1 antibody administration may not be obtained in some patients with ATL, and further verification is required for clinical application.
5.3. Lenalidomide
Lenalidomide, a thalidomide derivative, is an immunomodulatory agent used in the treatment of multiple myeloma. Lenalidomide has been reported to have immunopotentiating activity against multiple myeloma, including a direct tumor cell-killing effect and enhanced T and NK cell function [107]. In ATL, the inhibition of zeste homolog 2 enhancer expression induces the phosphorylation of the signal transducer and activator of transcription 3, resulting in cell growth suppression [108]. In contrast, a phase II trial of lenalidomide in patients with recurrent or relapsing ATL showed clinically significant antitumor activity and acceptable toxicity [109]. The effect of lenalidomide on ATL requires further study; however, elucidation of its mechanism of action in ATL will enable the development of a more effective lenalidomide therapy.
5.4. Dendritic cell-based vaccine
Dendritic cell vaccine therapy efficiently induces tumor antigen-specific CTLs and exerts an antitumor effect via administering tumor antigen peptide-pulsed dendritic cells to patients with cancer [110]. A dendritic cell vaccine targeting Tax has been developed for ATL [111]. The vaccine is prepared via culturing and differentiating monocytes obtained from the patient blood into dendritic cells, followed by the addition of Tax antigen peptides to the dendritic cells [111]. As it is an autologous dendritic cell vaccine preparation, it can be expected to effectively activate CTLs against ATL cells. A phase I trial has reported good safety and efficacy [111], and this cancer immunotherapy is expected to be established as a future treatment for ATL. As in the case of allo-HSCT, the therapeutic effect of dendritic cell therapy depends on CTL function; therefore, establishing a therapeutic method in the future, taking into account the genetic abnormalities of ATL cells, such as PD-L1 is necessary (Figure 2).
6. Conclusions
Antiviral therapy, multidrug chemotherapy, and allo-HSCT are mainly used for the treatment of ATL. However, regardless of the treatment method used, controlling patient conditions is not easy, and further improvement is required. Among the therapeutic drugs for ATL, mogamulizumab, lenalidomide, and allo-HSCT are greatly affected by the patient's immune function (Figure 2). Recent basic research has revealed that the immune function of patients with ATL is weakened. Further elucidation of the pathology is expected to lead to the establishment of a curative treatment for ATL. While chimeric antigen receptor T-cell therapy is also being developed for various types of leukemia, scientifically understanding the patient’s immune status and correlating it with ATL treatment is important.
Author Contributions
Conceptualization, S.N. and Y.S.; writing—original draft preparation, S.N., D.E and Y.S; writing—review and editing S.N., D.E, I.J., K.S. and Y.S.; funding acquisition, S.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grant from the Kobayashi Foundation for Cancer Research, grant number 2021SK020 to SN; the Japan Agency for Medical Research and Development (AMED) under Grant Number JP23fk0410052 and JP23wm0325068 to YS; KAKENHI research grants from the Japan Society for the Promotion of Science (JSPS) under Grant Number JP21K08494 to KS; Intramural Grant of Collaborative Research on Infection, at Joint Research Center for Human Retrovirus Infection, Kumamoto University to SN and YS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We would like to thank Editage (www.editage.jp) for English language editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bangham, C.R. Human T Cell Leukemia Virus Type 1: Persistence and Pathogenesis. Annu. Rev. Immunol. 2018, 36, 43–71. [Google Scholar] [CrossRef] [PubMed]
- Hirons, A.; Khoury, G.; Purcell, D.F.J. Human T-cell lymphotropic virus type-1: a lifelong persistent infection, yet never truly silent. Lancet Infect. Dis. 2021, 1, e2–e10. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Marçais, A.; Nasr, R.; Kato, K.; Fukuda, T.; Hermine, O.; Bazarbachi, A. Diagnostic Approaches and Established Treatments for Adult T Cell Leukemia Lymphoma. Front. Microbiol. 2020, 11, 1207. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, H. Current and emerging therapeutic strategies in adult T-cell leukemia–lymphoma. Int. J. Hematol. 2023, 117, 512–522. [Google Scholar] [CrossRef] [PubMed]
- A Phillips, A. Advances in the treatment of HTLV-1-associated adult T-cell leukemia lymphoma. Curr. Opin. Virol. 2023, 58, 101289. [Google Scholar] [CrossRef]
- Malpica, L.; Enriquez, D.J.; Castro, D.A.; Peña, C.; Idrobo, H.; Fiad, L.; Prates, M.; Otero, V.; Biglione, M.; Altamirano, M.; et al. Real-World Data on Adult T-Cell Leukemia/Lymphoma in Latin America: A Study From the Grupo de Estudio Latinoamericano de Linfoproliferativos. JCO Glob. Oncol. 2021, 7, 1151–1166. [Google Scholar] [CrossRef]
- Legrand, N.; McGregor, S.; Bull, R.; Bajis, S.; Valencia, B.M.; Ronnachit, A.; Einsiedel, L.; Gessain, A.; Kaldor, J.; Martinello, M. Clinical and Public Health Implications of Human T-Lymphotropic Virus Type 1 Infection. Clin. Microbiol. Rev. 2022, 35, e0007821. [Google Scholar] [CrossRef]
- Nosaka, K.; Iwanaga, M.; Imaizumi, Y.; Ishitsuka, K.; Ishizawa, K.; Ishida, Y.; Amano, M.; Ishida, T.; Uike, N.; Utsunomiya, A.; et al. Epidemiological and clinical features of adult T-cell leukemia-lymphoma in Japan, 2010-2011: A nationwide survey. Cancer Sci. 2017, 108, 2478–2486. [Google Scholar] [CrossRef]
- Ramassamy, J.-L.; Tortevoye, P.; Ntab, B.; Seve, B.; Carles, G.; Gaquière, D.; Madec, Y.; Fontanet, A.; Gessain, A. Adult T-cell leukemia/lymphoma incidence rate in French Guiana: a prospective cohort of women infected with HTLV-1. Blood Adv. 2020, 4, 2044–2048. [Google Scholar] [CrossRef]
- Oliveira, P.D.; De Carvalho, R.F.; Bittencourt, A.L. Adult T-cell leukemia/lymphoma in South and Central America and the Caribbean: systematic search and review. Int. J. STD AIDS 2017, 28, 217–228. [Google Scholar] [CrossRef]
- Itabashi, K.; Miyazawa, T.; Uchimaru, K. How Can We Prevent Mother-to-Child Transmission of HTLV-1? Int J Mol Sci. 2023, 24, 6961. [Google Scholar] [CrossRef]
- Nunes, D.; Boa-Sorte, N.; Grassi, M.F.R.; Taylor, G.P.; Teixeira, M.G.; Barreto, M.L.; Dourado, I.; Galvão-Castro, B. HTLV-1 is predominantly sexually transmitted in Salvador, the city with the highest HTLV-1 prevalence in Brazil. PLOS ONE 2017, 12, e0171303. [Google Scholar] [CrossRef] [PubMed]
- Ramassamy, J.L.; Bilounga Ndongo, C. ; Nnuka, P; Antunes, M. ; Le Mener, M.; Betsem A Betsem, E.; Njouom, R.; Cassar, O.; Fontanet, A.; et al. Epidemiological Evidence of Nosocomial and Zoonotic Transmission of Human T-Cell Leukemia Virus-1 in a Large Survey in a Rural Population of Central Africa. J Infect Dis. 2023, 227, 752–760. [Google Scholar]
- Matutes, E. Adult T-cell leukaemia/lymphoma. J Clin Pathol. 2007, 60, 1373–1377. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, S.; Komkov, D.; Mazurov, D. HIV-1 and HTLV-1 Transmission Modes: Mechanisms and Importance for Virus Spread. Viruses 2022, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Mulherkar, T.H.; Gómez, D.J.; Sandel, G.; Jain, P. Co-Infection and Cancer: Host-Pathogen Interaction between Dendritic Cells and HIV-1, HTLV-1, and Other Oncogenic Viruses. Viruses 2022, 14, 2037. [Google Scholar] [CrossRef]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T.T.; Harab, R.C.; Leite, A.C.; Schor, D.; Araújo, A.; Andrada-Serpa, M.J. Human T Lymphotropic Virus Type 1 (HTLV-1) Proviral Load in Asymptomatic Carriers, HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis, and Other Neurological Abnormalities Associated with HTLV-1 Infection. Clin. Infect. Dis. 2007, 44, 689–692. [Google Scholar] [CrossRef]
- Iwanaga, M.; Watanabe, T.; Utsunomiya, A.; Okayama, A.; Uchimaru, K.; Koh, K.-R.; Ogata, M.; Kikuchi, H.; Sagara, Y.; Uozumi, K.; et al. Joint Study on Predisposing Factors of ATL Development investigators. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood 2010, 116, 1211–1219. [Google Scholar] [CrossRef]
- El Hajj, H.; Bazarbachi, A. Interplay between innate immunity and the viral oncoproteins Tax and HBZ in the pathogenesis and therapeutic response of HTLV-1 associated adult T cell leukemia. Front. Immunol. 2022, 13, 957535. [Google Scholar] [CrossRef]
- Kannagi, M.; Harashima, N.; Kurihara, K.; Ohashi, T.; Utsunomiya, A.; Tanosaki, R.; Masuda, M.; Tomonaga, M.; Okamura, J. Tumor immunity against adult T-cell leukemia. Cancer Sci. 2005, 96, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Yasunaga, J.-I.; Matsuoka, M. HTLV-1's Foxy Strategy for Survival and Transmission. Front. A J. Women Stud. 2022, 1, 792659. [Google Scholar] [CrossRef]
- Mahgoub, M.; Yasunaga, J.-I.; Iwami, S.; Nakaoka, S.; Koizumi, Y.; Shimura, K.; Matsuoka, M. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1269–E1278. [Google Scholar] [CrossRef]
- Kiik, H.; Ramanayake, S.; Miura, M.; Tanaka, Y.; Melamed, A.; Bangham, C.R.M. Time-course of host cell transcription during the HTLV-1 transcriptional burst. PLOS Pathog. 2022, 18, e1010387. [Google Scholar] [CrossRef] [PubMed]
- Ramanayake, S.; Moulding, D.A.; Tanaka, Y.; Singh, A.; Bangham, C.R.M. Dynamics and consequences of the HTLV-1 proviral plus-strand burst. PLOS Pathog. 2022, 18, e1010774. [Google Scholar] [CrossRef]
- Matsuoka, M.; Mesnard, J.-M. HTLV-1 bZIP factor: the key viral gene for pathogenesis. Retrovirology 2020, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Miyoshi, H.; Ohshima, K. Tumor microenvironment of adult T-cell leukemia/lymphoma. J. Clin. Exp. Hematop. 2021, 61, 202–209. [Google Scholar] [CrossRef]
- Komohara, Y.; Niino, D.; Saito, Y.; Ohnishi, K.; Horlad, H.; Ohshima, K.; Takeya, M. Clinical significance of CD163⁺ tumor-associated macrophages in patients with adult T-cell leukemia/lymphoma. Cancer Sci. 2013, 104, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Yamada, Y.; Kamihira, S.; Atogami, S.; Tsukasaki, K.; Momita, S.; Amagasaki, T.; Sadamori, N.; Tomonaga, M.; Kinoshita, K.-I.; et al. Frequency of eosinophilia in adult T-cell leukemia/lymphoma. Cancer 1992, 69, 966–971. [Google Scholar] [CrossRef]
- Yashiki, S.; Fujiyoshi, T.; Arima, N.; Osame, M.; Yoshinaga, M.; Nagata, Y.; Tara, M.; Nomura, K.; Utsunomiya, A.; Hanada, S.; et al. HLA-A*26, HLA-B*4002, HLA-B*4006, and HLA-B*4801 alleles predispose to adult T cell leukemia: the limited recognition of HTLV type 1 tax peptide anchor motifs and epitopes to generate anti-HTLV type 1 tax CD8(+) cytotoxic T lymphocytes. AIDS Res Hum Retroviruses. 2001, 17, 1047–1061. [Google Scholar] [CrossRef]
- Gillet, N.A.; Cook, L.; Laydon, D.J.; Hlela, C.; Verdonck, K.; Alvarez, C.; Gotuzzo, E.; Clark, D.; Farré, L.; Bittencourt, A.; et al. Strongyloidiasis and Infective Dermatitis Alter Human T Lymphotropic Virus-1 Clonality in vivo. PLOS Pathog. 2013, 9, e1003263. [Google Scholar] [CrossRef]
- Plumelle, Y.; Gonin, C.; Edouard, A.; Bucher, B.J.; Thomas, L.; Brebion, A.; Panelatti, G. Effect ofStrongyloides stercoralisInfection and Eosinophilia on Age at Onset and Prognosis of Adult T-Cell Leukemia. Am. J. Clin. Pathol. 1997, 107, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Hleihel, R.; Akkouche, A.; Skayneh, H.; Hermine, O.; Bazarbachi, A.; El Hajj, H. Adult T-Cell Leukemia: a Comprehensive Overview on Current and Promising Treatment Modalities. Curr. Oncol. Rep. 2021, 23, 141. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, D.U.; Proietti, F.A.; Ribas, J.G.R.; Araújo, M.G.; Pinheiro, S.R.; Guedes, A.C.; Carneiro-Proietti, A.B.F. Epidemiology, Treatment, and Prevention of Human T-Cell Leukemia Virus Type 1-Associated Diseases. Clin. Microbiol. Rev. 2010, 23, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Hermine, O.; Bazarbachi, A.; Ratner, L.; Ramos, J.C.; Harrington, W. Jr.; O'Mahony, D.; Janik, J.E.; Bittencourt, A.L.; Taylor, G.P. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009, 27, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Maeda, T.; Babazono, A.; Nishi, T.; Yasui, M.; Matsuda, S.; Fushimi, K.; Fujimori, K. The Impact of Opportunistic Infections on Clinical Outcome and Healthcare Resource Uses for Adult T Cell Leukaemia. PLOS ONE 2015, 10, e0135042. [Google Scholar] [CrossRef]
- White, J.D.; Zaknoen, S.L.; Kasten-Sportès, C.; Top, L.E.; Navarro-Roman, L.; Nelson, D.L.; Waldmann, T.A. Infectious complications and immunodeficiency in patients with human T-cell lymphotropic virus I-associated adult T-cell leukemia/lymphoma. Cancer 1995, 75, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.J.; Jaffe, E.S.; Ehrlich, G.D.; Korman, N.J.; Poiesz, B.J.; Waldmann, T.A. Kaposi's sarcoma in human T-cell leukemia virus type I-associated adult T-cell leukemia. Blood 1990, 76, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, K.; Shindo, T.; Miyahara, M.; Kitaura, K.; Akashi, M.; Shin, T.; Suzuki, R.; Oshima, K.; Kimura, S. Epstein–Barr virus-related diffuse large B-cell lymphoma in mogamulizumab-treated adult T-cell leukemia with incomplete T-cell reconstitution. Int. J. Hematol. 2019, 109, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Kawano, N.; Nagahiro, Y.; Yoshida, S.; Tahara, Y.; Himeji, D.; Kuriyama, T.; Tochigi, T.; Nakaike, T.; Shimokawa, T.; Yamashita, K.; et al. Clinical features and treatment outcomes of opportunistic infections among human T-lymphotrophic virus type 1 (HTLV-1) carriers and patients with adult T-cell leukemia-lymphoma (ATL) at a single institution from 2006 to 2016. J. Clin. Exp. Hematop. 2019, 59, 156–167. [Google Scholar] [CrossRef]
- Gabet, A.-S.; Mortreux, F.; Talarmin, A.; Plumelle, Y.; Leclercq, I.; Leroy, A.; Gessain, A.; Clity, E.; Joubert, M.; Wattel, E. High circulating proviral load with oligoclonal expansion of HTLV-1 bearing T cells in HTLV-1 carriers with strongyloidiasis. Oncogene 2000, 19, 4954–4960. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Taylor, G.P.; Klose, R.J.; Schofield, C.J.; Bangham, C.R. Histone H2A monoubiquitylation and p38-MAPKs regulate immediate-early gene-like reactivation of latent retrovirus HTLV-1. J. Clin. Investig. 2018, 3, e123196. [Google Scholar] [CrossRef]
- Kulkarni, A.; Mateus, M.; Thinnes, C.C.; McCullagh, J.S.; Schofield, C.J.; Taylor, G.P.; Bangham, C.R. Glucose Metabolism and Oxygen Availability Govern Reactivation of the Latent Human Retrovirus HTLV-1. Cell Chem. Biol. 2017, 24, 1377–1387.e3. [Google Scholar] [CrossRef]
- Miyazato, P.; Matsuo, M.; Tan, B.J.Y.; Tokunaga, M.; Katsuya, H.; Islam, S.; Ito, J.; Murakawa, Y.; Satou, Y. HTLV-1 contains a high CG dinucleotide content and is susceptible to the host antiviral protein ZAP. Retrovirology 2019, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, R.; Dengler, C.; Müller-Fleckenstein, I.; Fleckenstein, B.; McGuire, K.; Dokhelar, M.C.; Sodroski, J.G.; A Haseltine, W. Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type I X-region genes transduced by a Herpesvirus saimiri vector. Proc. Natl. Acad. Sci. USA 1989, 86, 3351–3355. [Google Scholar] [CrossRef]
- Tanaka, A.; Takahashi, C.; Yamaoka, S.; Nosaka, T.; Maki, M.; Hatanaka, M. Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 1071–1075. [Google Scholar] [CrossRef]
- Sugata, K.; Yasunaga, J.-I.; Mitobe, Y.; Miura, M.; Miyazato, P.; Kohara, M.; Matsuoka, M. Protective effect of cytotoxic T lymphocytes targeting HTLV-1 bZIP factor. Blood 2015, 126, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Portis, T.; Harding, J.C.; Ratner, L. The contribution of NF-kappa B activity to spontaneous proliferation and resistance to apoptosis in human T-cell leukemia virus type 1 Tax-induced tumors. Blood 2001, 98, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.J.; Sugata, K.; Reda, O.; Matsuo, M.; Uchiyama, K.; Miyazato, P.; Hahaut, V.; Yamagishi, M.; Uchimaru, K.; Suzuki, Y.; et al. HTLV-1 infection promotes excessive T cell activation and transformation into adult T cell leukemia/lymphoma. J. Clin. Investig. 2021, 131, e150472. [Google Scholar] [CrossRef]
- Shiohama, Y.; Naito, T.; Matsuzaki, T.; Tanaka, R.; Tomoyose, T.; Takashima, H.; Fukushima, T.; Tanaka, Y.; Saito, M. Absolute quantification of HTLV-1 basic leucine zipper factor (HBZ) protein and its plasma antibody in HTLV-1 infected individuals with different clinical status. Retrovirology 2016, 13, 29. [Google Scholar] [CrossRef]
- Satou, Y.; Yasunaga, J.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef]
- Toyoda, K.; Matsuoka, M. Functional and Pathogenic Roles of Retroviral Antisense Transcripts. Front. Immunol. 2022, 13, 875211. [Google Scholar] [CrossRef]
- Yasunaga, J.-I. Strategies of Human T-Cell Leukemia Virus Type 1 for Persistent Infection: Implications for Leukemogenesis of Adult T-Cell Leukemia-Lymphoma. Front. Microbiol. 2020, 11, 979. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Yasunaga, J.-I.; Zhao, T.; Yoshida, M.; Miyazato, P.; Takai, K.; Shimizu, K.; Ohshima, K.; Green, P.L.; Ohkura, N.; et al. HTLV-1 bZIP Factor Induces T-Cell Lymphoma and Systemic Inflammation In Vivo. PLOS Pathog. 2011, 7, e1001274. [Google Scholar] [CrossRef] [PubMed]
- Kinosada, H. ; Yasunaga, J-I. ; Shimura, K.; Miyazato, P.; Onishi, C.; Iyoda, T.; Inaba, K.; Matsuoka, M. HTLV-1 bZIP Factor Enhances T-Cell Proliferation by Impeding the Suppressive Signaling of Co-inhibitory Receptors. PLoS Pathog. 2017, 13, e1006120. [Google Scholar]
- Johnson, J.M.; Nicot, C.; Fullen, J.; Ciminale, V.; Casareto, L.; Mulloy, J.C.; Jacobson, S.; Franchini, G. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J Virol. 2001, 75, 6086–6094. [Google Scholar] [CrossRef]
- Datta, A.; Sinha-Datta, U.; Dhillon, N.K.; Buch, S.; Nicot, C. The HTLV-I p30 Interferes with TLR4 Signaling and Modulates the Release of Pro- and Anti-inflammatory Cytokines from Human Macrophages. J. Biol. Chem. 2006, 281, 23414–23424. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Iwanaga, M.; Yasunaga, J.-I.; Nagata, Y.; Kitanaka, A.; Kameda, T.; Yoshimitsu, M.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; et al. Prognostic relevance of integrated genetic profiling in adult T-cell leukemia/lymphoma. Blood 2018, 131, 215–225. [Google Scholar] [CrossRef]
- Koya, J.; Saito, Y.; Kameda, T.; Kogure, Y.; Yuasa, M.; Nagasaki, J.; McClure, M.B.; Shingaki, S.; Tabata, M.; Tahira, Y.; et al. Single-Cell Analysis of the Multicellular Ecosystem in Viral Carcinogenesis by HTLV-1. Blood Cancer Discov. 2021, 2, 450–467. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Saeed, A.F.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Troiano, G.; Caponio, V.C.A.; Adipietro, I.; Tepedino, M.; Santoro, R.; Laino, L.; Russo, L.L.; Cirillo, N.; Muzio, L.L. Prognostic significance of CD68+ and CD163+ tumor associated macrophages in head and neck squamous cell carcinoma: A systematic review and meta-analysis. Oral Oncol. 2019, 93, 66–75. [Google Scholar] [CrossRef]
- Shen, H.; Liu, J.; Chen, S.; Ma, X.; Ying, Y.; Li, J.; Wang, W.; Wang, X.; Xie, L. Prognostic Value of Tumor-Associated Macrophages in Clear Cell Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 657318. [Google Scholar] [CrossRef]
- Komohara, Y.; Niino, D.; Saito, Y.; Ohnishi, K.; Horlad, H.; Ohshima, K.; Takeya, M. Clinical significance of CD163⁺ tumor-associated macrophages in patients with adult T-cell leukemia/lymphoma. Cancer Sci. 2013, 104, 945–951. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Yanagida, E.; Miyoshi, H.; Takeuchi, M.; Yoshida, N.; Nakashima, K.; Yamada, K.; Umeno, T.; Shimasaki, Y.; Furuta, T.; Seto, M.; et al. Clinicopathological analysis of immunohistochemical expression of CD47 and SIRPα in adult T-cell leukemia/lymphoma. Hematol. Oncol. 2020, 38, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Kiyasu, J.; Kato, T.; Yoshida, N.; Shimono, J.; Yokoyama, S.; Taniguchi, H.; Sasaki, Y.; Kurita, D.; Kawamoto, K.; et al. PD-L1 expression on neoplastic or stromal cells is respectively a poor or good prognostic factor for adult T-cell leukemia/lymphoma. Blood 2016, 128, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Azakami, K.; Sato, T.; Araya, N.; Utsunomiya, A.; Kubota, R.; Suzuki, K.; Hasegawa, D.; Izumi, T.; Fujita, H.; Aratani, S.; et al. Severe loss of invariant NKT cells exhibiting anti–HTLV-1 activity in patients with HTLV-1–associated disorders. Blood 2009, 114, 3208–3215. [Google Scholar] [CrossRef]
- Itabashi, K.; Miyazawa, T. Mother-to-Child Transmission of Human T-Cell Leukemia Virus Type 1: Mechanisms and Nutritional Strategies for Prevention. Cancers 2021, 13, 4100. [Google Scholar] [CrossRef]
- Fuchi, N.; Miura, K.; Tsukiyama, T.; Sasaki, D.; Ishihara, K.; Tsuruda, K.; Hasegawa, H.; Miura, S.; Yanagihara, K.; Masuzaki, H. Natural Course of Human T-Cell Leukemia Virus Type 1 Proviral DNA Levels in Carriers During Pregnancy. J. Infect. Dis. 2018, 217, 1383–1389. [Google Scholar] [CrossRef]
- Plancoulaine, S.; Gessain, A.; Tortevoye, P.; Boland-Auge, A.; Vasilescu, A.; Matsuda, F.; Abel, L. A major susceptibility locus for HTLV-1 infection in childhood maps to chromosome 6q27. Hum. Mol. Genet. 2006, 15, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Takahashi, M.; Norose, Y.; Takeshita, T.; Fukunaga, Y.; Takahashi, H. Transformation of breast milk macrophages by HTLV-I: implications for HTLV-I transmission via breastfeeding. Biomed. Res. 2010, 31, 53–61. [Google Scholar] [CrossRef]
- Gotuzzo, E.; Moody, J.; Verdonck, K.; Cabada, M.M.; González, E.; Van Dooren, S.; Vandamme, A.-M.; Terashima, A.; Vermund, S.H. Frequent HTLV-1 infection in the offspring of Peruvian women with HTLV-1-associated myelopathy/tropical spastic paraparesis or strongyloidiasis. Rev Panam Salud Publica. 2007, 22, 223–230. [CrossRef]
- Montaño-Castellón, I.; Marconi, C.S.C.; Saffe, C.; Brites, C. Clinical and Laboratory Outcomes in HIV-1 and HTLV-1/2 Coinfection: A Systematic Review. Front Public Health. 2022, 10, 820727. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.J.; Julien-Serrette, K.; Edwards, J.; Boyce, G. HTLV-1 Coinfection among Patients Attending a Large HIV Treatment Centre in Trinidad. Microorganisms 2022, 10, 2207. [Google Scholar] [CrossRef]
- Geddes, V.E.V.; José, D.P.; Leal, F.E.; Nixon, D.F.; Tanuri, A.; Aguiar, R.S. HTLV-1 Tax activates HIV-1 transcription in latency models. Virology 2017, 504, 45–51. [Google Scholar] [CrossRef]
- Abad-Fernández, M.; Hernández-Walias, F.J.; de León, M.J.R.; Vivancos, M.J.; Pérez-Elías, M.J.; Moreno, A.; Casado, J.L.; Quereda, C.; Dronda, F.; Moreno, S.; et al. HTLV-2 Enhances CD8+ T Cell-Mediated HIV-1 Inhibition and Reduces HIV-1 Integrated Proviral Load in People Living with HIV-1. Viruses 2022, 14, 2472. [Google Scholar] [CrossRef] [PubMed]
- Richey, J.D.; Chen, B.J.; Deng, A.C. Indolent, waxing and waning cutaneous presentation of HTLV-1-associated adult T-cell leukemia/lymphoma in an HIV-1-positive patient. J Cutan Pathol. 2018, 45, 171–175. [Google Scholar] [CrossRef]
- Schär, F.; Trostdorf, U.; Giardina, F.; Khieu, V.; Muth, S.; Marti, H.; Vounatsou, P.; Odermatt, P. Strongyloides stercoralis: Global Distribution and Risk Factors. PLOS Negl. Trop. Dis. 2013, 7, e2288. [Google Scholar] [CrossRef]
- Carvalho, E.M.; Porto, A.D.F. Epidemiological and clinical interaction between HTLV-1 and Strongyloides stercoralis. Parasite Immunol. 2004, 26, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Toma, H.; Sugahara, K.; Etoh, K.; Shiroma, Y.; Kiyuna, S.; Takara, M.; Matsuoka, M.; Yamaguchi, K.; Nakada, K.; et al. Involvement of IL-2/IL-2R system activation by parasite antigen in polyclonal expansion of CD4(+)25(+) HTLV-1-infected T-cells in human carriers of both HTLV-1 and S. stercoralis. Oncogene 2002, 21, 2466–2475. [Google Scholar] [CrossRef] [PubMed]
- Dykie, A.; Wijesinghe, T.; Rabson, A.B.; Madugula, K.; Farinas, C.; Wilson, S.; Abraham, D.; Jain, P. Human T-cell Leukemia Virus Type 1 and Strongyloides stercoralis: Partners in Pathogenesis. Pathogens 2020, 9, 904. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Nomura, S.; Shimoyama, M.; Shibata, T.; Imaizumi, Y.; Moriuchi, Y.; Tomoyose, T.; Uozumi, K.; Kobayashi, Y.; Fukushima, N.; et al. Japan Clinical Oncology Group (JCOG) prognostic index and characterization of long-term survivors of aggressive adult T-cell leukaemia-lymphoma (JCOG0902A). Br. J. Haematol. 2014, 166, 739–748. [Google Scholar] [CrossRef]
- Katsuya, H.; Yamanaka, T.; Ishitsuka, K.; Utsunomiya, A.; Sasaki, H.; Hanada, S.; Eto, T.; Moriuchi, Y.; Saburi, Y.; Miyahara, M.; et al. Prognostic Index for Acute- and Lymphoma-Type Adult T-Cell Leukemia/Lymphoma. J. Clin. Oncol. 2012, 30, 1635–1640. [Google Scholar] [CrossRef]
- Fuji, S.; Yamaguchi, T.; Inoue, Y.; Utsunomiya, A.; Moriuchi, Y.; Uchimaru, K.; Owatari, S.; Miyagi, T.; Taguchi, J.; Choi, I.; et al. Development of a modified prognostic index for patients with aggressive adult T-cell leukemia-lymphoma aged 70 years or younger: possible risk-adapted management strategies including allogeneic transplantation. Haematologica 2017, 102, 1258–1265. [Google Scholar] [CrossRef]
- Ishida, T.; Hishizawa, M.; Kato, K.; Tanosaki, R.; Fukuda, T.; Taniguchi, S.; Eto, T.; Takatsuka, Y.; Miyazaki, Y.; Moriuchi, Y.; et al. Allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia-lymphoma with special emphasis on preconditioning regimen: a nationwide retrospective study. Blood 2012, 120, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2020, 18, 842–859. [Google Scholar] [CrossRef]
- Jo, T.; Noguchi, K.; Sakai, T.; Kubota-Koketsu, R.; Irie, S.; Matsuo, M.; Taguchi, J.; Abe, K.; Shigematsu, K. HTLV-1 Tax-specific memory cytotoxic T lymphocytes in long-term survivors of aggressive-type adult T-cell leukemia/lymphoma. Cancer Med. 2022, 11, 3238–3250. [Google Scholar] [CrossRef]
- Harashima, N.; Kurihara, K.; Utsunomiya, A.; Tanosaki, R.; Hanabuchi, S.; Masuda, M.; Ohashi, T.; Fukui, F.; Hasegawa, A.; Masuda, T.; et al. Graft-versus-Tax Response in Adult T-Cell Leukemia Patients after Hematopoietic Stem Cell Transplantation. Cancer Res 2004, 64, 391–399. [Google Scholar] [CrossRef]
- Jo, T.; Kubota-Koketsu, R.; Kaneko, Y.; Sakai, T.; Noguchi, K.; Irie, S.; Matsuo, M.; Taguchi, J.; Abe, K.; Shigematsu, K. Live attenuated VZV vaccination induces antitumor immunity in ATL patients. Cancer Immunol Immunother. 2023, 72, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Masaki, A.; Ishida, T.; Suzuki, S.; Ito, A.; Narita, T.; Kinoshita, S.; Ri, M.; Kusumoto, S.; Komatsu, H.; Inagaki, H.; et al. Human T-cell lymphotropic/leukemia virus type 1 (HTLV-1) Tax-specific T-cell exhaustion in HTLV-1-infected individuals. Cancer Sci. 2018, 109, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Ishida, T.; Masaki, A.; Suzuki, S.; Ito, A.; Mori, F.; Yamada, T.; Ri, M.; Kusumoto, S.; Komatsu, H.; et al. HTLV-1 bZIP Factor–Specific CD4 T Cell Responses in Adult T Cell Leukemia/Lymphoma Patients after Allogeneic Hematopoietic Stem Cell Transplantation. J. Immunol. 2014, 192, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Suemori, K.; Fujiwara, H.; Ochi, T.; Ogawa, T.; Matsuoka, M.; Matsumoto, T.; Mesnard, J.-M.; Yasukawa, M. HBZ is an immunogenic protein, but not a target antigen for human T-cell leukemia virus type 1-specific cytotoxic T lymphocytes. J. Gen. Virol. 2009, 90, 1806–1811. [Google Scholar] [CrossRef] [PubMed]
- Macnamara, A.; Rowan, A.; Hilburn, S.; Kadolsky, U.; Fujiwara, H.; Suemori, K.; Yasukawa, M.; Taylor, G.; Bangham, C.R.; Asquith, B. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. 2010, 6, e1001117. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Ishida, T.; Utsunomiya, A.; Inagaki, A.; Yano, H.; Komatsu, H.; Iida, S.; Imada, K.; Uchiyama, T.; Akinaga, S.; et al. Defucosylated Humanized Anti-CCR4 Monoclonal Antibody KW-0761 as a Novel Immunotherapeutic Agent for Adult T-cell Leukemia/Lymphoma. Clin. Cancer Res. 2010, 16, 1520–1531. [Google Scholar] [CrossRef]
- Saito, M.; Ishii, T.; Urakawa, I.; Matsumoto, A.; Masaki, A.; Ito, A.; Kusumoto, S.; Suzuki, S.; Takahashi, T.; Morita, A.; et al. Robust CD8+ T-cell proliferation and diversification after mogamulizumab in patients with adult T-cell leukemia-lymphoma. Blood Adv. 2020, 4, 2180–2191. [Google Scholar] [CrossRef]
- Ishida, T.; Jo, T.; Takemoto, S.; Suzushima, H.; Suehiro, Y.; Choi, I.; Yoshimitsu, M.; Saburi, Y.; Nosaka, K.; Utsunomiya, A.; et al. Follow-up of a randomised phase II study of chemotherapy alone or in combination with mogamulizumab in newly diagnosed aggressive adult T-cell leukaemia-lymphoma: impact on allogeneic haematopoietic stem cell transplantation. Br. J. Haematol. 2019, 184, 479–483. [Google Scholar] [CrossRef]
- Tanaka, T.; Inamoto, Y.; Ito, A.; Watanabe, M.; Takeda, W.; Aoki, J.; Kim, S.; Fukuda, T. Lenalidomide treatment for recurrent adult T-cell leukemia/lymphoma after allogeneic hematopoietic cell transplantation. Hematol. Oncol. 2022, 41, 389–395. [Google Scholar] [CrossRef]
- Shichijo, T.; Nosaka, K.; Tatetsu, H.; Higuchi, Y.; Endo, S.; Inoue, Y.; Toyoda, K.; Kikukawa, Y.; Kawakita, T. ; Yasunaga, J-I. ; et al. Beneficial impact of first-line mogamulizumab-containing chemotherapy in adult T-cell leukaemia-lymphoma. Br J Haematol. 2022, 198, 983–987. [Google Scholar]
- Kawano, N.; Kuriyama, T.; Yoshida, S.; Kawano, S.; Yamano, Y.; Marutsuka, K.; Minato, S.; Yamashita, K.; Ochiai, H.; Shimoda, K.; et al. The Impact of a Humanized CCR4 Antibody (Mogamulizumab) on Patients with Aggressive-Type Adult T-Cell Leukemia-Lymphoma Treated with Allogeneic Hematopoietic Stem Cell Transplantation. J. Clin. Exp. Hematop. 2017, 56, 135–144. [Google Scholar] [CrossRef]
- Kamada, Y.; Arima, N.; Hayashida, M.; Nakamura, D.; Yoshimitsu, M.; Ishitsuka, K. Prediction of the risk for graft versus host disease after allogeneic hematopoietic stem cell transplantation in patients treated with mogamulizumab. Leuk. Lymphoma 2022, 63, 1701–1707. [Google Scholar] [CrossRef]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef]
- Ratner, L.; Waldmann, T.A.; Janakiram, M.; Brammer, J.E. Rapid Progression of Adult T-Cell Leukemia–Lymphoma after PD-1 Inhibitor Therapy. New Engl. J. Med. 2018, 378, 1947–1948. [Google Scholar] [CrossRef]
- Rauch, D.A.; Conlon, K.C.; Janakiram, M.; Brammer, J.E.; Harding, J.C.; Ye, B.H.; Zang, X.; Ren, X.; Olson, S.; Cheng, X.; et al. Rapid progression of adult T-cell leukemia/lymphoma as tumor-infiltrating Tregs after PD-1 blockade. Blood 2019, 134, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Armoiry, X.; Aulagner, G.; Facon, T. Lenalidomide in the treatment of multiple myeloma: a review. J. Clin. Pharm. Ther. 2008, 33, 219–226. [Google Scholar] [CrossRef]
- Kondo, N.; Nagano, Y.; Hasegawa, A.; Ishizawa, M.; Katagiri, K.; Yoneda, T.; Masuda, T.; Kannagi, M. Involvement of EZH2 inhibition in lenalidomide and pomalidomide-mediated growth suppression in HTLV-1-infected cells. Biochem. Biophys. Res. Commun. 2021, 574, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Fujiwara, H.; Nosaka, K.; Taira, N.; Abe, Y.; Imaizumi, Y.; Moriuchi, Y.; Jo, T.; Ishizawa, K.; Tobinai, K.; et al. Multicenter Phase II Study of Lenalidomide in Relapsed or Recurrent Adult T-Cell Leukemia/Lymphoma: ATL-002. J Clin Oncol. 2016, 34, 4086–4093. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012, 12, 265–77. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, Y.; Hasegawa, A.; Iino, T.; Sasada, A.; Watanabe, N.; Matsuoka, M.; Takamori, A.; Tanosaki, R.; Utsunomiya, A.; Choi, I.; et al. Clinical outcomes of a novel therapeutic vaccine with Tax peptide-pulsed dendritic cells for adult T cell leukaemia/lymphoma in a pilot study. Br. J. Haematol. 2015, 169, 356–367. [Google Scholar] [CrossRef]
Figure 1.
Figure 1. A summary of the immune pathologies during HTLV-1 latency and onset of ATL. (A) In asymptomatic HTLV-1 carriers, the proliferation of HTLV-1-infected T cells and their clearance by CTLs are in equilibrium. HTLV-1 transiently expresses Tax and affects the expression and function of immune-related molecules to evade host immunity and promote survival of HTLV-1-infected cells. (B) Patients with ATL develop immunodeficiency; however, ATL cells also accumulate genetic abnormalities in immune-related genes and clones that are advantageous for escaping host immunity are selected for extensive proliferation. In addition to CD4+ T cells, decreased numbers of CTLs and B cells, increased numbers of myeloid cells, and decreased NK cell function are observed. HTLV-1, Human T-cell leukemia virus type 1; ATL, adult T-cell leukemia/lymphoma; CTL, cytotoxic T cells; NK, Natural killer.
Figure 1.
Figure 1. A summary of the immune pathologies during HTLV-1 latency and onset of ATL. (A) In asymptomatic HTLV-1 carriers, the proliferation of HTLV-1-infected T cells and their clearance by CTLs are in equilibrium. HTLV-1 transiently expresses Tax and affects the expression and function of immune-related molecules to evade host immunity and promote survival of HTLV-1-infected cells. (B) Patients with ATL develop immunodeficiency; however, ATL cells also accumulate genetic abnormalities in immune-related genes and clones that are advantageous for escaping host immunity are selected for extensive proliferation. In addition to CD4+ T cells, decreased numbers of CTLs and B cells, increased numbers of myeloid cells, and decreased NK cell function are observed. HTLV-1, Human T-cell leukemia virus type 1; ATL, adult T-cell leukemia/lymphoma; CTL, cytotoxic T cells; NK, Natural killer.
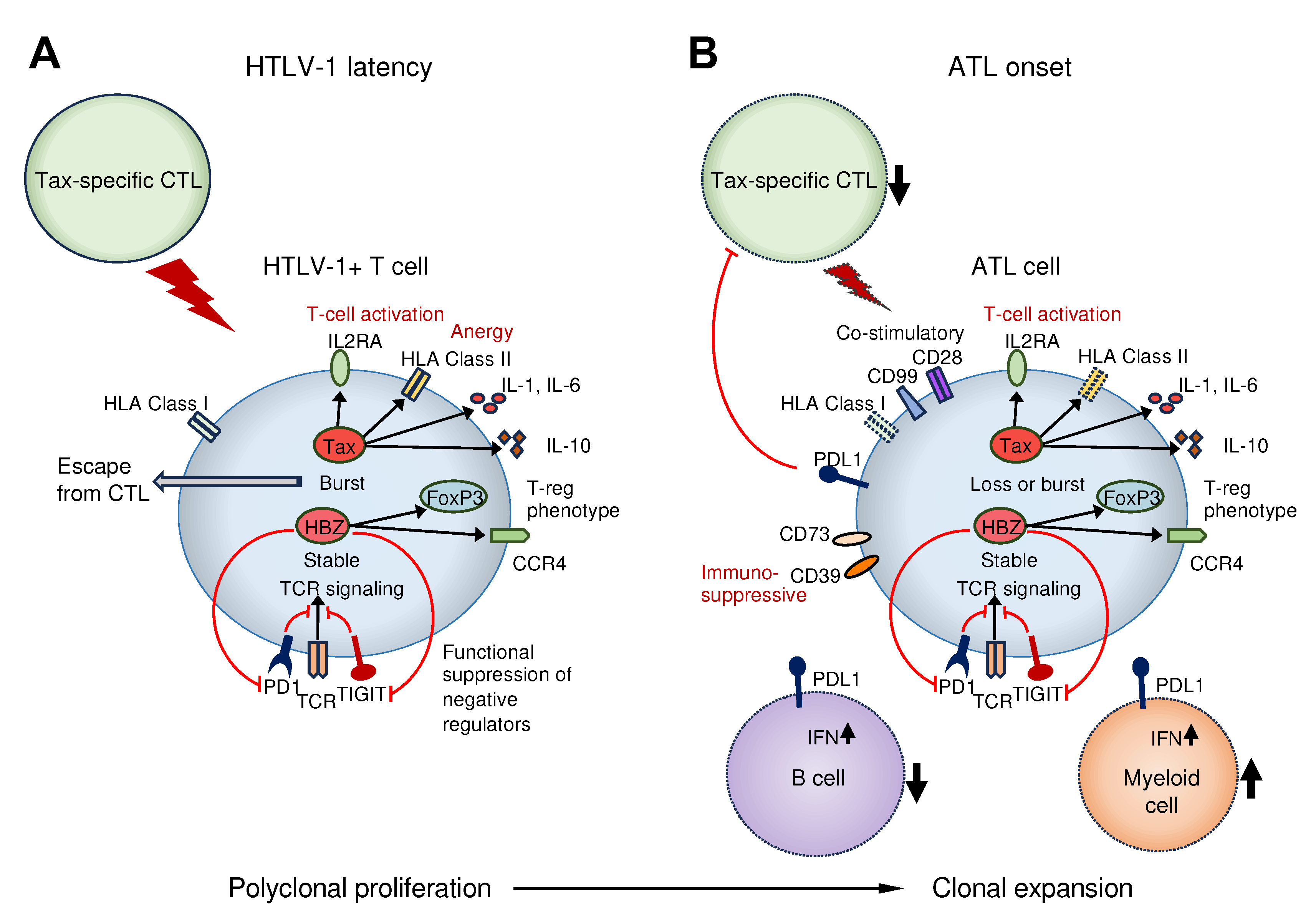
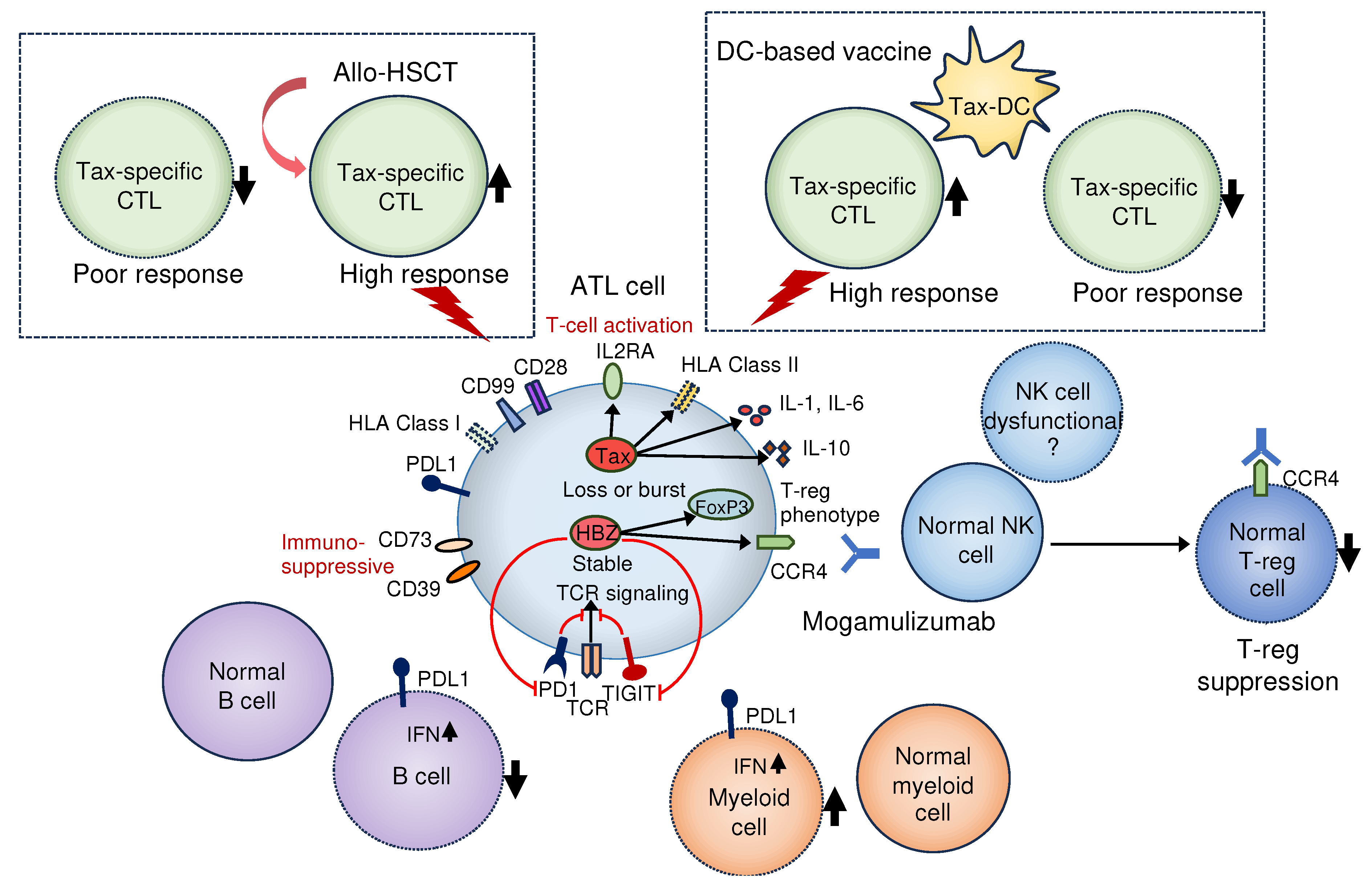
Figure 2.
Relationship between immune pathology and therapeutic efficacy in patients with ATL. As the therapeutic effects of allo-HSCT and Tax-dendritic cell vaccine therapy depend on the activity of Tax-specific CTLs, the presence of functional CTLs is indispensable. Characterization circulating ATL cells may be important, as CTL decline is also associated with genomic abnormalities in ATL cells, such as PD-L1 [70]. Mogamulizumab suppresses not only ATL cells, but also regulatory T cells (Treg). The antibody-dependent cellular cytotoxic effect of mogamulizumab depends on the activity of NK cells and may be less effective in some patients with ATL owing to decreased NK cell function [70]. Dotted lines in boxes indicate the hypotheses. ATL, adult T-cell leukemia/lymphoma; allo-HSCT, allogeneic hematopoietic stem cell transplantation; PD-L1, programmed death ligand 1; NK, Natural killer
Figure 2.
Relationship between immune pathology and therapeutic efficacy in patients with ATL. As the therapeutic effects of allo-HSCT and Tax-dendritic cell vaccine therapy depend on the activity of Tax-specific CTLs, the presence of functional CTLs is indispensable. Characterization circulating ATL cells may be important, as CTL decline is also associated with genomic abnormalities in ATL cells, such as PD-L1 [70]. Mogamulizumab suppresses not only ATL cells, but also regulatory T cells (Treg). The antibody-dependent cellular cytotoxic effect of mogamulizumab depends on the activity of NK cells and may be less effective in some patients with ATL owing to decreased NK cell function [70]. Dotted lines in boxes indicate the hypotheses. ATL, adult T-cell leukemia/lymphoma; allo-HSCT, allogeneic hematopoietic stem cell transplantation; PD-L1, programmed death ligand 1; NK, Natural killer
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



