
Submitted:
06 September 2023
Posted:
07 September 2023
You are already at the latest version
Abstract
Adenylosuccinic acid (ASA) is a small molecule dicarboxylate that could be a strong clinical development candidate for inherited myopathies involving dysregulated purine nucleotide metabo-lism. Currently, there are no published pharmacokinetic/dynamic or toxicology data available, albeit 10-year clinical trial data in Duchenne muscular dystrophy patients suggests a chronically safe drug. In this study, we tested the toxicity of ASA to cultured myoblasts in vitro and acute systemic toxicity in mice. ASA is a non-toxic small molecule with an LD50> 5000mg/kg. Some background necrotic foci in the liver, kidney and gastrointestinal tract were shown that are likely incidental but warrant follow-up sub-/chronic oral exposure studies.
Keywords:
adenylosuccinic acid
; Duchenne muscular dystrophy
; myopathy
; metabolic disease
; skeletal muscle
; toxicology
; drug development
1. Introduction
Adenylosuccinic acid (ASA) is a small molecule metabolite that was clinically evaluated for Duchenne muscular dystrophy (DMD) in the 1980-90’s [1]. Dystrophin-deficient muscles manifest extensive metabolic anomalies [2,3] and ASA had previously been shown in vitro to correct aberrant mitochondrial isocitrate dehydrogenase activity [4]. Although the trial showed ASA could attenuate clinical biomarkers of DMD (e.g., serum creatine kinase) [1], the research program was ultimately abandoned after a decade due to lack of funding and the discovery of the etiological DMD gene mutation, which shifted research focus toward gene-targeted therapeutic development. Nevertheless, ASA could be a powerful therapeutic for skeletal/striated muscle wasting due to its role in driving the purine nucleotide cycle (PNC). Within skeletal muscle, the PNC regulates energy balance during extreme metabolic demand (e.g., high intensity exercise, growth), purine and pyrimidine biosynthesis necessary for DNA, RNA and enzyme cofactor production (which ultimately enables biomass accretion), and importantly, the excretion of otherwise toxic ammonia [5] [6] (reviewed in [7]).
Outside of the 10-year clinical trial data indicating safety, there are no pharmacokinetic, pharmacodynamic or toxicological data available for ASA. During the ASA trial, biofluid markers of liver (bilirubin), kidney (BUN, creatinine) and haematological (RBC, WBC, haemoglobin) function were not affected by 1-200mg/kg daily ASA treatment [1]. The drug was administered initially via the subcutaneous route and later by intraperitoneal port when dose escalated beyond the capacity for effective delivery via injection or insulin pump. However, the maximum dose reached was 300mg/kg/day and higher dosages or more frequent delivery may be required to correct purine nucleotide biochemistry in disease, and especially, to normalise muscle ASA levels. For this reason, oral delivery may be favourable but with potential risk to the liver during first pass metabolism. At the conclusion of the ASA trial, bis in die oral delivery of 5mg ASA was administered to the few remaining patients but there are no corresponding biofluid data to indicate safety at this orally administered higher dose [8].
In this study, we aimed to establish acute toxicology data toward the clinical development of ASA for DMD and other muscle wasting conditions involving dysregulated PNC metabolism. In the first instance we assessed dose escalation in vitro using immortalised human myoblasts derived from a healthy paediatric patient, to provide application context. In the second part, we assessed acute toxicity in vivo using Organisation for Economic Co-operation and Development (OECD) testing guidelines in mice [9]. Our data indicate ASA has an LD50 >5000mg/kg consistent with a non-toxic compound. There were incidental non-dose-dependent lesions noted in the liver, kidney and gastrointestinal tract common to the mus musculis species [10]. However, it cannot be ruled out that ASA treatment was involved, especially at upper dosages where all these organs are subjected to high concentrations of the metabolite (gut and liver) or its excretion by-product (urea). Our data suggest follow up sub-chronic and chronic toxicology studies may be warranted.
2. Results
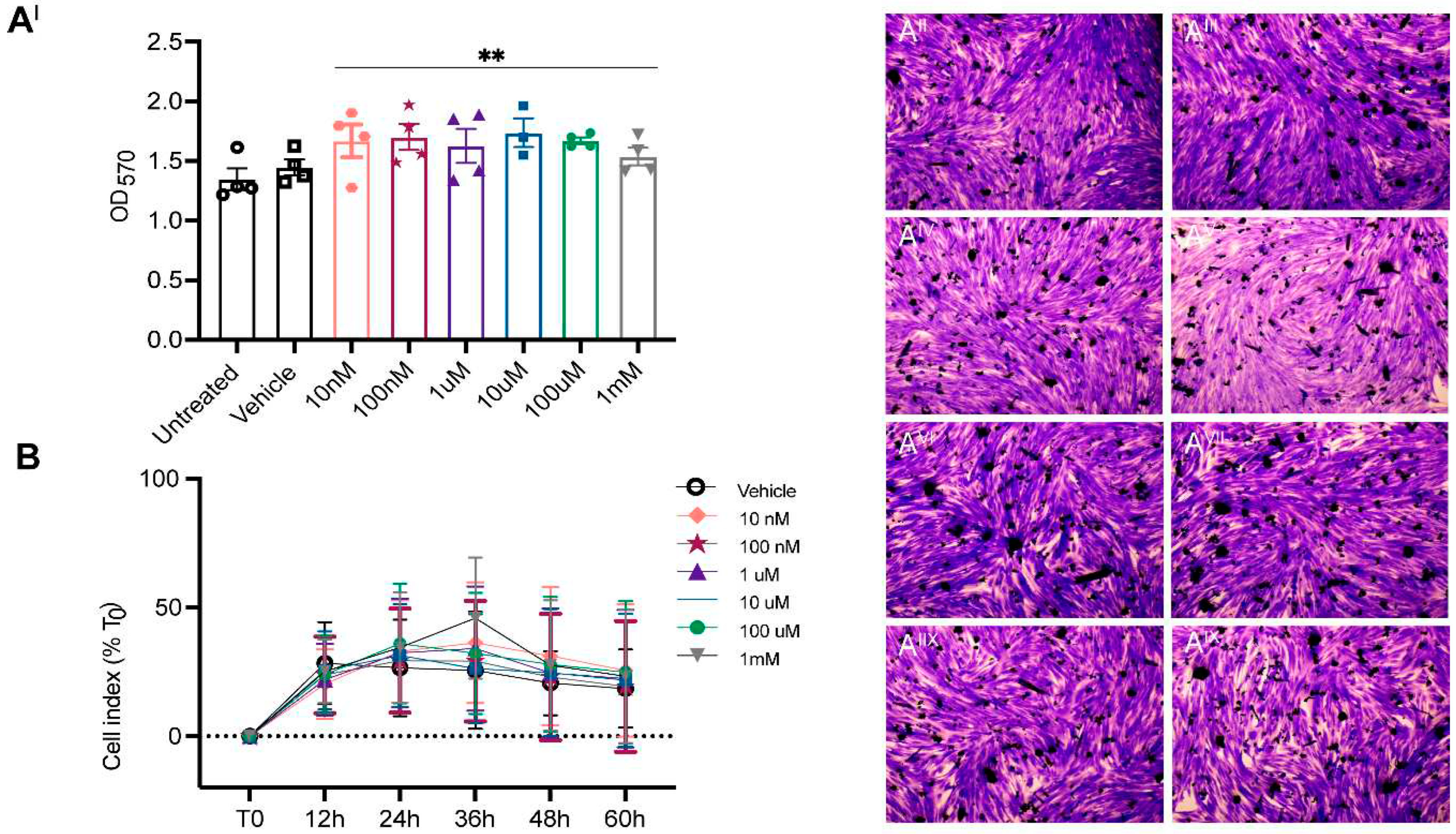
ASA is non-toxic to human myoblasts. The effect of ASA on myoblast viability was assessed across a dose range of 1nM-1mM using endpoint crystal violet staining and real-time Xcelligence bioimpedance monitoring. Rather than causing toxicity, ASA treatment increased the viability of myoblasts in a non-dose-dependent manner as indicated by crystal violet absorbance (Figure 1A). Cell index was monitored in real time for 60 hours post-treatment to assess the effect of ASA treatment across the same 1nM-1mM range on myoblast proliferation rate. There was no effect of ASA at any dose on real-time cell index over 60 hours (Figure 1B). Our data demonstrate that ASA is non-toxic when applied directly to cells up to a concentration of 1mM highlighting that the cytotoxic IC50 is >1mM
Figure 1.
Adenylosuccinic acid (ASA) is non-toxic to human myoblasts. Across the 10nM-1mM dose range, ASA increased viability of myoblasts as determined by crystal violet absorbance (AI). Representative images of crystal violet staining in untreated (AII) and vehicle (AIII) treated myoblasts and 10nM (AIV), 100nM (AV), 1µM (AVI), 10µM (AVII), 100µM (AIIX) and 1mM (AIX) ASA-treated myoblasts. The real-time cell index was unaffected by ASA treatment across the 10nM-1mM range over 60 hours (B).
Figure 1.
Adenylosuccinic acid (ASA) is non-toxic to human myoblasts. Across the 10nM-1mM dose range, ASA increased viability of myoblasts as determined by crystal violet absorbance (AI). Representative images of crystal violet staining in untreated (AII) and vehicle (AIII) treated myoblasts and 10nM (AIV), 100nM (AV), 1µM (AVI), 10µM (AVII), 100µM (AIIX) and 1mM (AIX) ASA-treated myoblasts. The real-time cell index was unaffected by ASA treatment across the 10nM-1mM range over 60 hours (B).
ASA is non-toxic to healthy mice. Mice were dosed once with ASA dissolved in milli Q at a concentration of either 175, 550, 1750 or 5000 mg/kg (Table 1). There were no mortalities observed with ASA treatment at any dose. Since all mice survived the upper limit test (i.e., 5000mg/kg ASA), a further two mice were administered 5000mg/kg ASA and survived. The LD50 of ASA was determined to be >5000mg/kg consistent with a non-toxic compound.

Table 1.
Dose preparation of adenylosuccinic acid (ASA).
| Dose | Post-fasted body weight (g) | ASA mass (mg) | Volume H2O/dose (ml) |
|---|---|---|---|
| 175 mg/kg | 20.4 | 3.57 | 0.204 |
| 550 mg/kg | 19.6 | 10.78 | 0.196 |
| 1750 mg/kg | 22.2 | 38.85 | 0.222 |
| 5000 mg/kg | 21.8 | 109 | 0.218 |
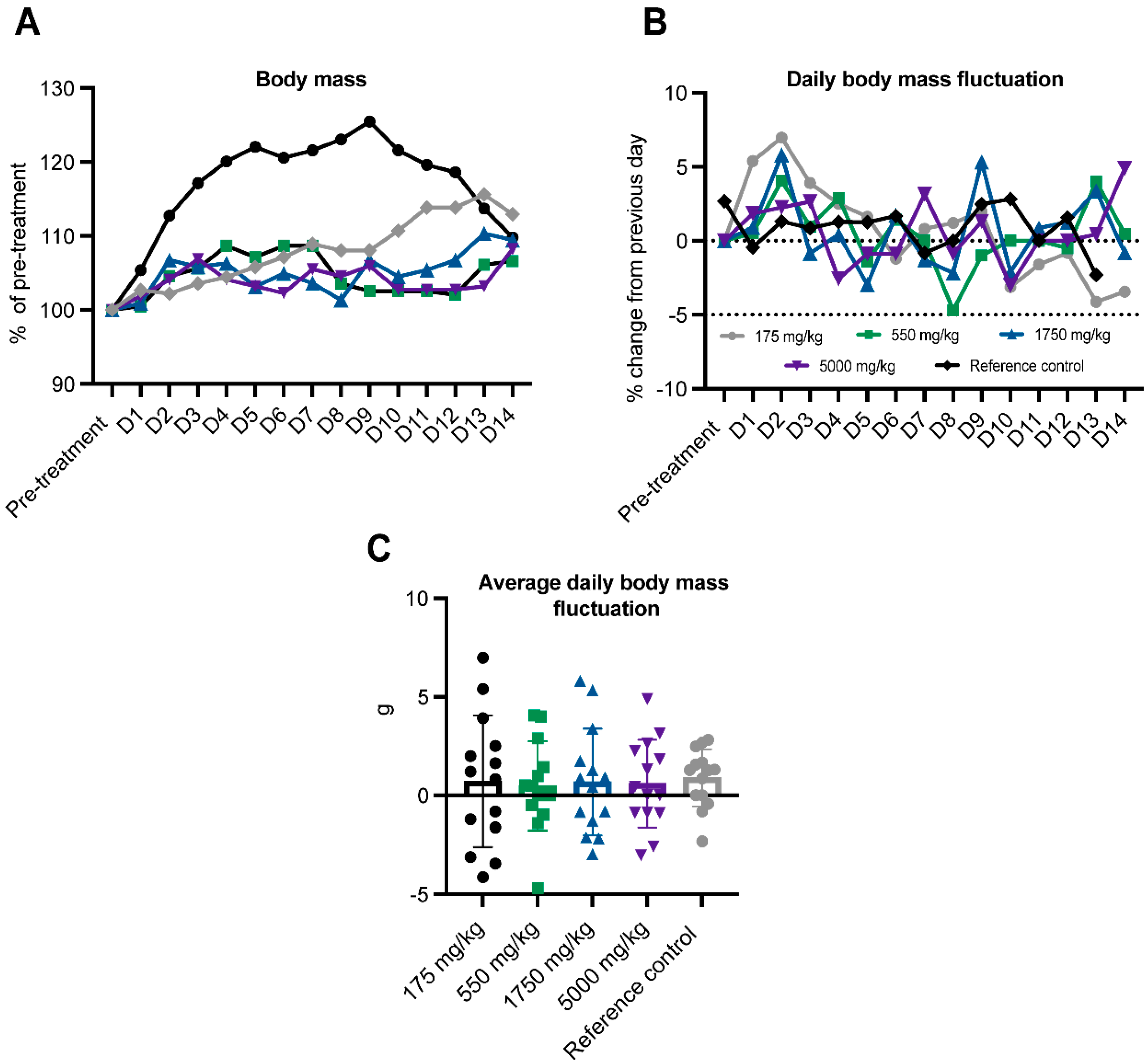
Body mass. Mice were weighed just prior to treatment (following a 4 h fasting period) and daily for 14 days after ASA administration. A 5% loss of body mass represents a toxicologically meaningful observation. No mouse fell below the pre-treatment body mass on any day of the 14-day observation period (Figure 2A). Day-to-day body mass changes were also plotted. While all mice (including the untreated control) lost body mass or failed to gain body mass over at least one 24-hour period during the clinical observation period (Figure 2B), no mouse lost >5% from the previous day in any one 24-hour period (Figure 2B and 2C).
Figure 2.
Adenylosuccinic acid (ASA) does not affect mouse body weight. Body weight was measured pre-treatment and up to 14 days post-treatment to observe if ASA was detrimental to body mass regulation (as indicated by 5% loss of body weight). Across the post-treatment period, no mouse lost >5% body weight following treatment with any ASA dose (175-5000mg/kg; A-C).
Figure 2.
Adenylosuccinic acid (ASA) does not affect mouse body weight. Body weight was measured pre-treatment and up to 14 days post-treatment to observe if ASA was detrimental to body mass regulation (as indicated by 5% loss of body weight). Across the post-treatment period, no mouse lost >5% body weight following treatment with any ASA dose (175-5000mg/kg; A-C).
Open field observations. Mice were observed for 14 days for changes in food and water consumption, as well as symptoms of toxicity in the neuromotor, respiratory, gastrointestinal, integumentary and pain systems (summarised in Table 2). There were no adverse symptoms observed at any dose below 5000mg/kg. However, increased motor activity was observed in the first 0.25 hours and transient diarrhea was observed from 8-12 hours following 5000mg/kg ASA treatment.
Table 2.
Effects of acute adenylosuccinic acid (ASA) exposure (175-5000mg/kg) on in-life observations. Key: - = no changes, + = mild changes.
Table 2.
Effects of acute adenylosuccinic acid (ASA) exposure (175-5000mg/kg) on in-life observations. Key: - = no changes, + = mild changes.
| Observations | ||||||
|---|---|---|---|---|---|---|
| Observations | Symptoms | 175mg/kg ASA n=1 |
550mg/kg ASA n=1 |
1750mg/kg ASA n=1 |
5000mg/kg ASA n=1 |
Untreated |
| Body weight | >5% loss of body weight | - | - | - | - | - |
| Food & water consumption | - | - | - | - | - | - |
| Motor activity | Home-cage activity | - | - | - | + (<30 m) |
- |
| Neurological system | Tremors, limb tone, ataxia | - | - | - | - | - |
| Respiratory system | Gasping, heaving, cyanosis | - | - | - | - | - |
| Gastrointestinal function | Abdominal griping, vomiting, diarrhoea | - | - | - | + (diarrhoea) |
- |
| Skin & mucous membranes | Secretions, excretions | - | - | - | - | - |
| Pain | Grimacing, altered social activity | - | - | - | - | |
Haematology and serum biochemistry. On post-treatment day 14, blood was removed from live mice via tail venipuncture and tested for biochemical and haematological abnormalities. Data are summarised in Table 3 relative to published reference ranges for female C57Bl/6J mice [11] and an untreated C57Bl/10 control. There was no specific effect of ASA dose escalation on haematological parameters. Segmented neutrophil concentration was higher than the reference range in the mice treated with 550 and 5000 mg/kg ASA but it was also higher in the untreated reference control suggesting this was not an effect of ASA per se. Lymphocyte concentration was higher than the reference range in the mouse treated with 175 mg/kg but was either normal or lower than the reference range in mice treated with ASA at all other doses. Eosinophil concentration was 7 times higher than the upper reference value in the mouse treated with 5000 mg/kg ASA, which may represent a drug-related effect.
There was no ASA treatment-specific effect on serum biochemistry. All mice tested had relatively low serum total protein (mice treated with 550mg/kg and 5000mg/kg ASA fell below the normal reference range for this parameter) and 3-fold lower urea concentration than the reference range. In contrast, all mice for which serum glucose concentration was available (e.g., mice treated with 175mg/kg, 550mg/kg and 1750mg/kg ASA) showed values above the reference range. These data appear related to metabolism of the C57Bl/10 strain and/or the 4 hour fast prior to ASA dosing that is stipulated by the testing guidelines, rather than an effect of ASA treatment per se. Mild changes to serum electrolytes were also observed. However again, this effect was not treatment-specific and was discerned an artefact of cardiocentesis by the veterinary pathologist. Liver function enzyme, ALT, was lower than the reference for the 175mg/kg and the 5000mg/kg ASA mice and was at the lowest end of the reference range for the untreated reference mouse (42 U/L). However, ALT was above the reference range for the 1750mg/kg ASA mouse, which also had ~2-fold higher serum AST concentration indicative of liver damage. Since 5000mg/kg ASA did not cause the same, or higher ALT and AST levels, this effect appears to be specific to the mouse rather than escalating ASA dosage. There was no effect of ASA at any dose on serum ALP or cholesterol concentrations.
Table 3.
Effects of acute adenylosuccinic acid (ASA) exposure (175-5000mg/kg) on serum biochemistry. Key: *= outside of reference range; a [12]; b [13]; c [12]; d [14]; IS = insufficient sample to measure analyte; MCH = mean corpuscular haemoglobin; MCHC = mean corpuscular haemoglobin concentration; MCV = mean corpuscular volume; WCC = white cell count.
Table 3.
Effects of acute adenylosuccinic acid (ASA) exposure (175-5000mg/kg) on serum biochemistry. Key: *= outside of reference range; a [12]; b [13]; c [12]; d [14]; IS = insufficient sample to measure analyte; MCH = mean corpuscular haemoglobin; MCHC = mean corpuscular haemoglobin concentration; MCV = mean corpuscular volume; WCC = white cell count.
| Parameters | Reference Range | ASA Dose | Reference control | |||
|---|---|---|---|---|---|---|
| 175 mg/kg | 550 mg/kg | 1750 mg/kg | 5000 mg/kg |
|||
| Haematological | ||||||
| Erythrocytes (x1012/L) | 3.47-11.73a | 10.5 | 11.06 | 10.66 | 11.0 | 11.02 |
| Haematocrit (L/L) | 0.16-0.58a | 0.48 | 0.54 | 0.54 | 0.54 | 0.54 |
| Haemoglobin (g/L) | 57.0-170.0a | 156 | 162 | 153 | 157 | 158 |
| MCV (fL) | 25.7-59.7b | 48 | 49 | 50 | 49 | 49 |
| MCH (pg) | 9.0-19.2b | 16 | 15 | 14 | 14 | 14 |
| MCHC (g/L) | 264.6-314.2b | 323* | 300 | 286 | 289 | 290 |
| Platelet (x109/L) | 520-1880b | 942 | 954 | 1040 | 1077 | 1188 |
| WCC (x109/L | 2.2-11.53a | 8.9 | 5.5 | 3.8 | 4.6 | 5.8 |
| Segmented Neutrophils (x109/L) | 0.12-0.30b | 0.3 | 0.7* | 0.2 | 0.4* | 0.5* |
| Lymphocytes (x109/L) | 4.36-5.68b | 8.5* | 4.6 | 3.4* | 3.4* | 4.8 |
| Monocytes (x109/L) | 0-0.3b | 0.18 | 0.06 | 0.23 | 0 | 0 |
| Eosinophils (x109/L) | 0-5.1d | 0 | 2 | 0 | 7* | 0.1 |
| Basophils (x109/L) | 0-0.1d | 0 | 0 | 0 | 0.1 | 0 |
| Biochemical | ||||||
| Total Protein (g/L) | 45-83a | 48 | 42* | 53 | 40* | 53 |
| Albumin (g/L) | 20-47a | 29 | 24 | 30 | 21 | 29 |
| Total globulin (g/L) | 18-21c | 19 | 18 | 23* | 19 | 24 |
| Glucose (mmol/L) | 5.2-12.2a | 18.1* | 13.8* | 15* | IS | IS |
| Sodium (mmol/L) | 149.0-281.4 a | 142* | IS | 145* | IS | IS |
| Potassium (mmol/L) | 4.0-14.0a | 10.8 | IS | 7.5 | IS | IS |
| Chloride (mmol/L) | 110.0-204.4a | 98* | IS | 101* | IS | IS |
| Calcium (mmol/L) | 2.3-3.5a | 2.64 | 2.15* | 2.57 | 1.94* | 2.66 |
| Phosphorous (mmol/L) | 2.0-3.1a | 3.32* | 2.23 | 3.95* | 2.83 | 3.69 |
| Urea (mmol/L) | 14.5-21.4a | 4.1* | 5.3* | 5.5* | 4.9* | 5.4* |
| Total bilirubin (µmol/L) | 3.4-14.3a | 9 | 1* | 4 | IS | IS |
| Alanine aminotransferase (ALT; U/L) | 42-73a | 39* | 50 | 75* | 25* | 42 |
| Aspartate aminotransferase (AST; U/L) | 51-122a | 97 | 76 | 220* | 62 | 86 |
| Alkaline phosphatase (ALP; U/L) | 103-217a | 140 | 155 | 193 | 137 | 178 |
| GammaGT (U/L) | 6.0-8.0a | 7 | <5* | <5* | IS | IS |
| Creatine kinase (U/L) | 105-649a | 100* | 43* | 290 | IS | IS |
| Total cholesterol (mmol/L) | 1.3-3.4a | 2.02 | 1.77 | 2.29 | 2.02 | 2.27 |
Macropathology. Mice were humanely killed and underwent a full necropsy. Splenic melanosis – a relatively common observation in mice – was observed in mice treated with 550 and1750 mg/kg ASA (Table 4) and are incidental. There were no gross macropathologic changes observed in any other organ harvested from these mice or in any organ from the 175 and 5000 mg/kg ASA mice or the reference control mouse (Table 4).
Table 4.
Effects of acute adenylosuccinic acid (ASA) exposure (175-5000 mg/kg) on macro- and histo-pathology. Key: - = no changes, + = mild changes, ++ = moderate changes, +++ = significant changes.
Table 4.
Effects of acute adenylosuccinic acid (ASA) exposure (175-5000 mg/kg) on macro- and histo-pathology. Key: - = no changes, + = mild changes, ++ = moderate changes, +++ = significant changes.
| Pathology | Symptoms | ASA dose | ||||
|---|---|---|---|---|---|---|
| 175 mg/kg | 550 mg/kg |
1750 mg/kg |
5000 mg/kg | Reference control | ||
| Macropathology | Splenic melanosis (background lesion) | - | + | + | - | - |
| Kidney | Rare interstitial lymphoplasmacytic infiltrate with fibrosis | - | + | + | + | - |
| Liver | Multiple random foci of necrosis with neutrophil aggregates | +++ | +++ | + | + | +++ |
| Mild anisocytosis with scant megalocytosis | + | + | + | ++ | - | |
| Stomach | Focal neutrophilic infiltrate into gastric mucosa; | - | - | + | ++ | + |
| Low-grade focal granulocytic gastritis | - | - | + | +++ | +++ | |
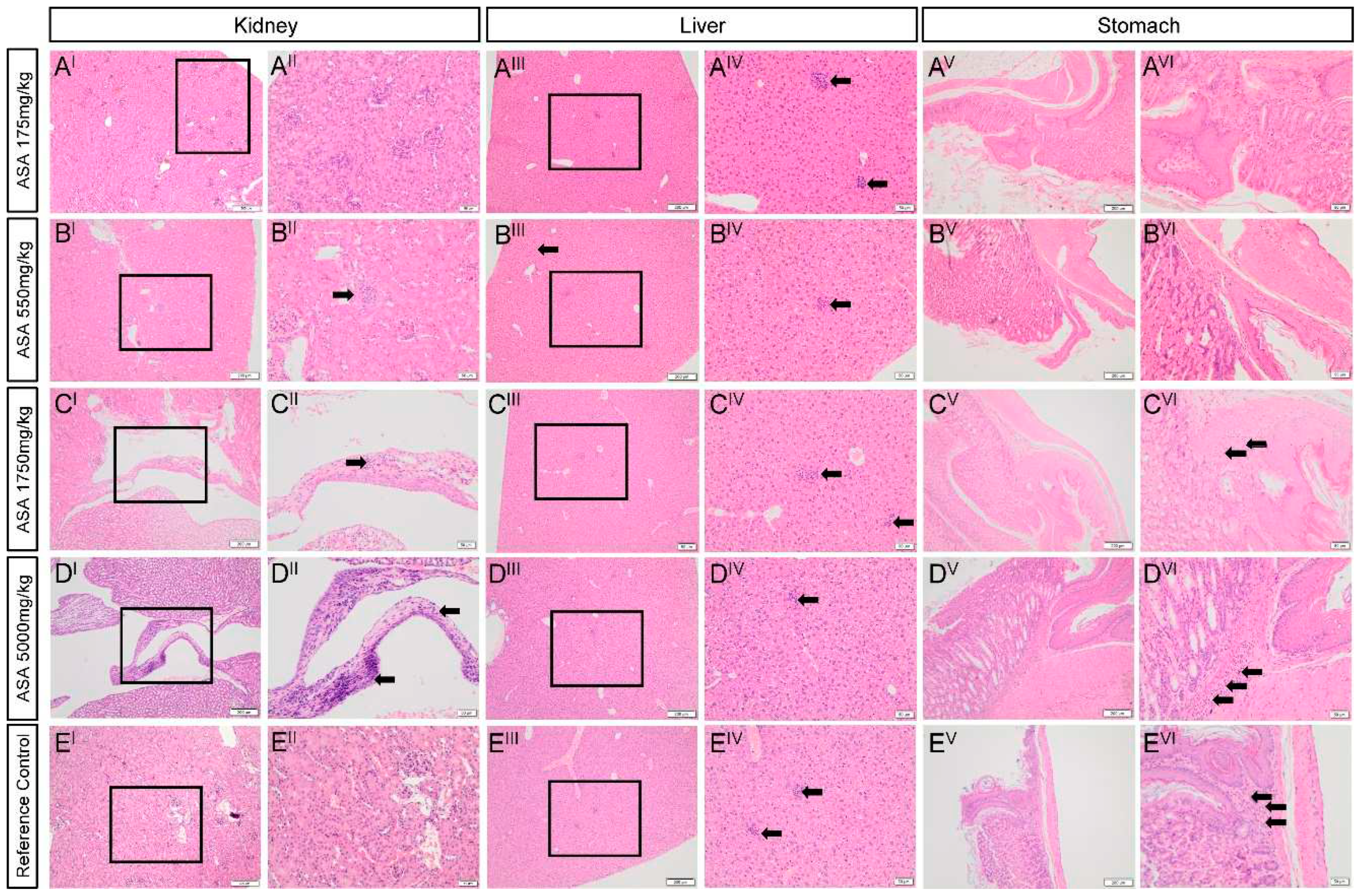
Histopathology. Rare lymphoplasmacytic infiltrate with mild fibrosis was observed in the kidneys of mice treated with 550-5000mg/kg ASA (1+) but not in the kidneys of the mouse treated with 175mg/kg ASA or the untreated mouse (Figure 3A-EI-II and Table 4). In the liver, multiple random foci of hepatocellular necrosis with neutrophil aggregates were observed in all mice to differing extents (Figure 3A-EIII-IV and Table 4) and was not associated with ASA treatment. In contrast, mild hepatocellular anisocytosis with scant megalocytosis was observed only in the ASA treated mice and escalated in severity in the mouse treated with 5000mg/kg ASA (Figure 3A-DIII-IV and Table 4). Isolated interstitial lymphoplasmacytic infiltrate of the kidney, and focal necrosis and mild anisocytosis of the liver are common background findings in laboratory mice [10].
No significant abnormalities were observed in the stomach of the mice treated with 175mg/kg or 550mg/kg ASA. However, mild to moderate focal neutrophilic infiltrate into the gastric mucosa at the glandular and non-glandular margin and mild focal granulocytic gastritis was observed in the stomach of the untreated mouse and the mice treated with 1750 and 5000 mg/kg ASA. Gastritis and neutrophilic infiltrate in the gastric mucosa are notable background findings in laboratory mice (personal observations, L. Rasmussen) as indicated by the fact that intensity of pathology was equivalent between the mouse treated with 5000mg/kg ASA and the untreated reference control.
Figure 3.
Effect of adenylosuccinic acid (ASA) on kidney, liver and stomach histopathology. In the kidney (A-EI-II), sporadic areas of mild focal interstitial lymphoplasmacytic infiltrate with fibrosis were observed in the kidneys (B-CII) but were not dependent on ASA. Areas of focal necrosis with neutrophil infiltrate were also observed in the liver (A-EIII-IV) and were more frequent in the mouse treated with 5000mg/kg ASA (DIV). In the stomach (A-EV-VI), low grade focal granulocytic gastritis was evident in the stomachs of mice treated with 1750mg/kg ASA (CVI) and 5000mg/kg ASA (DVI), and the untreated reference control (EVI). Images in A-EII, IV and VI are callouts from images in A-EI, III and V, respectively, as indicated by the black boxes. Images in A-EI, III and V are x4 magnification (scale bar = 50μm) and in A-EII, IV and VI are x10 magnification (scale bar = 200μm).
Figure 3.
Effect of adenylosuccinic acid (ASA) on kidney, liver and stomach histopathology. In the kidney (A-EI-II), sporadic areas of mild focal interstitial lymphoplasmacytic infiltrate with fibrosis were observed in the kidneys (B-CII) but were not dependent on ASA. Areas of focal necrosis with neutrophil infiltrate were also observed in the liver (A-EIII-IV) and were more frequent in the mouse treated with 5000mg/kg ASA (DIV). In the stomach (A-EV-VI), low grade focal granulocytic gastritis was evident in the stomachs of mice treated with 1750mg/kg ASA (CVI) and 5000mg/kg ASA (DVI), and the untreated reference control (EVI). Images in A-EII, IV and VI are callouts from images in A-EI, III and V, respectively, as indicated by the black boxes. Images in A-EI, III and V are x4 magnification (scale bar = 50μm) and in A-EII, IV and VI are x10 magnification (scale bar = 200μm).
3. Discussion
The first part of our study revealed ASA as a non-toxic compound to human myoblasts in vitro. ASA was non-toxic to cells at any dose within a 1nM-mM range and therefore has a cytotoxic IC50 > 1mM. In fact, in the endpoint cell density assay, ASA (any dose) treatment for 4 days resulted in a higher number of live myoblasts than in the untreated or vehicle-treated control cultures. Purine nucleotides are the fundamental building blocks of molecules that regulate cell life, including DNA, RNA, enzyme cofactors and high energy phosphates (ATP, ADP, AMP) [2]. They also function in signal transduction of critical cell survival responses [15,16,17,18,19]. Consistent with its function in the de novo purine biosynthesis and salvage/recycling pathways, ASA appears to support cell survival in vitro, but does not impact the rate of myoblast proliferation per se.
The second part of our study revealed ASA was non-toxic to mice in vivo at the upper acute toxicity testing limit of 5000mg/kg and caused no adverse clinical symptoms in the open field. For this reason, a limit test was applied at 5000mg/kg. Although drug testing in the GHS Category 5 range (2000-500mg/kg) is usually discouraged within the acute toxicity test, in the previous ASA clinical trial, dose escalation was required to lower clinical biomarkers of muscle degradation as patients attained muscle biomass during adolescence and disease progressed [1]. High ASA doses will likely be needed to biochemically correct primary and secondary PNC deficits in the clinical setting. Three mice survived the limit test and only mild, sporadic symptoms were observed. For example, in the 0.25 h immediately after treatment, 5000mg/kg ASA induced hyperactivity in the open field and transient diarrhea that resolved within the first 24 h period. These data suggest gastrointestinal discomfort could be symptomatic of acute, high-dose ASA treatment. Weight loss was observed at day 12 of the 14-day observational period and may be related or unrelated to ASA exposure. While there were no macro- or histo-pathological changes observed in the gastrointestinal tract due to ASA exposure, other similar metabolite small molecules are associated with gastrointestinal symptoms. ASA generates fumarate as a biproduct of its enzymatic degradation by adenylosuccinate lyase (ADSL) within the PNC [5]. Synthetic fumarate drugs used in the clinical management of multiple sclerosis (MS) and psoriasis, such as dimethyl- and monomethyl-fumarate [20], are known to aggravate the gastrointestinal tract resulting in diarrhea, nausea and stomach cramps and pain in up to 60% of patients [21,22,23]. At least in the context of MS, gastrointestinal complaints are usually mild in severity and resolve within the first 2 months of treatment [24]. However, they are the most frequent cause of treatment discontinuation [24]. If a proportion of ASA metabolism occurs in the gut, either within the gastric tissues or resident microbiome, endogenous fumarate production could be responsible for similar/symptoms and ultimately manifest as weight loss. Our findings suggest sub-/chronic studies should be pursued to understand whether ASA shares a similar side effect profile to fumarate compounds, and whether conventional clinical management strategies, such as dosing with food, bis in die, or formulated in slow-release packaging could minimise gut symptoms.
There were some haematological anomalies observed in the ASA treated mice that were deemed to be an artefact of cardiocentesis – particularly blood electrolyte concentration. Unfortunately, there was insufficient blood sample to detect blood electrolytes in the untreated reference control for comparison. Sodium and chloride concentration were both lower than the reference range in blood collected from the mice with sufficient sample available (the 175 and 1750 mg/kg ASA treated mice). Although the mechanism of ASA transport across cell membranes has not been established, we previously hypothesised that a likely transport mechanism is via the transmembrane solute carrier (SLC) as per fumarate and citrate [7]. Di- and tri-carboxylate SLC’s are sodium-dependent symporters while the glycine SLC is a sodium/chloride symporter [25]. If ASA is transported via SLC’s its administration could be associated with fluctuations in the sodium and/or chloride concentration of the extracellular and intracellular compartments. No changes in blood electrolyte levels were reported in the ASA clinical trial throughout its duration [1]. However, ASA was administered in the trial as a tetra-sodium salt formulation [26]. Sodium-based salt formulations are ideal for increasing the absorption of weak acids across the GI tract and their bioavailability to target tissues [27]. In future trials, formulation may need to consider ASA’s uptake route and the potential requirement of electrolytes to support maximal absorption.
Adverse drug reactions are a frequent and challenging aspect of modern medicine [28]. Ultimately, a drug’s benefit must be weighed against its side effect profile and provide significant advantage to human livelihood. Oral administration has benefits over injectable delivery via the subcutaneous, intraperitoneal and intravenous routes in that large quantities of drug can be sustainably delivered with minimal medical intervention [27]. However, a significant draw-back is that the liver is subjected to the highest drug concentration during first pass metabolism following drug absorption across the gastrointestinal tract. As a result, drug toxicity is commonly observed in the liver (second only to the cardiovascular system as a site of toxicology attrition [29]) as hepatocellular injury and release of enzymes, e.g., ALT, AST and ALP, into the blood circulation. In our study, it was difficult to discern random incidental histopathologic events from those caused specifically by ASA. Background lesions that are commonly observed in mice with no significance are splenic melanosis, interstitial lymphoplasmacytic infiltrates in the kidney, random foci of hepatocellular necrosis with neutrophil aggregates, and hepatocellular anisocytosis with megalocytosis [10]. Granulocytic gastritis is also frequently observed (Personal observations, L. Rasmussen). All of these pathologies were seen to some extent in our mice and appeared independent of ASA treatment. However, it cannot be ruled out that moderate to high dose ASA may cause focal injury to both the liver and kidney. Whereas dose escalation did not intensify kidney histopathology, high (5000mg/kg) dose ASA increased the severity of histopathologic symptoms in the liver. Since the bioavailability and tissue distribution of oral ASA treatment is currently unknown, it should be assumed that high dose delivery could be needed to normalise dysregulated purine biochemistry in relevant diseases and required on a chronic basis [7]. As such, our histopathology data warrant follow-up with sub-/chronic studies testing low-high dosages to establish longitudinal safety.
Idiosyncratic drug toxicities are generally unpredictable and can present as delayed onset. Thus, these toxicities are typically not revealed during acute toxicity testing. Their proposed mechanisms include via metabolic, haptenogenic, inflammogenic and/or danger signalling, or via reversible drug induction of the immunological response [29]. Our previous work investigating ASA’s mechanism(s) of action demonstrates regulation of high energy phosphate metabolism (e.g., ATP and the phosphocreatine pool) [30], suppression of inflammation and the immune systems similar to other dicarboxylate metabolites (e.g., β-hydroxybutyrate) [7], and activation of the danger signalling transcription factor, nuclear factor-erythroid factor 2-related factor 2 (Nrf2) [15]. These mechanisms of action could render ASA particularly prone to isolated toxicity due to nuances in inflammation and immune system function between individuals. A relatively rare complication of dimethyl fumarate treatment in the context of MS is lymphopenia (<2% of consumers) which is more frequent in older males [31,32]. Purportedly, lymphopenia persists in many of these patients after treatment discontinuation demonstrating that fumarate induces long-term changes to the immune system and that person-specific autoimmune factors may be involved. High dose ASA (1750 and 5000 mg/kg) was associated with lymphopaenia in our haematological observations and should be followed up in sub-/chronic toxicology studies. Ultimately, detailed toxicity surveillance within the clinical trial setting will be required, especially over the long-term, to determine if ASA treatment is safe for all individuals irrespective of underlying conditions.
4. Materials and Methods
In vitro toxicity testing
Potential cytotoxicity of ASA was assessed in in vitro cell culture. Immortalised skeletal muscle myoblasts were cultured in growth medium, seeded at a density of 90-100,000 cells per well and passaged every 3 days [30,33]. The maximal tolerable dose of ASA was assessed using two methods: end-point crystal violet staining was used to quantitate myoblast viability while an xCELLigence bioimpedance system was used to quantitate real-time myoblast cell index. For the crystal violet assay, myoblasts were grown in media only, or media supplemented with vehicle (milliQ H2O) or increasing concentrations of ASA (1nM-1mM) in vehicle. Following 4 days incubation, cells were stained with 0.1% crystal violet solution and absorbance was measured spectrophotometrically at 570nm [34]. For cell proliferation assay, myoblasts were seeded at 5000 cells/well and once adhered, were treated with 50μl media, media with vehicle or media with increasing concentrations of ASA (1nM-1mM). Cell proliferation was monitored in real-time using an xCELLigence RTCA MP system in which the cell index was measured every 30 mins for 4 days.
In vivo toxicity testing
Animals
Animal experimentation was approved by the Victoria University Animal Ethics Committee (17/006) and performed in accordance with the Australian Code of Practice for the Care and Use of Animals for Research Purposes. Female C57Bl/10 mice aged 8weeks were used in the in vivo studies and purchased from the Animal Resource Centre (Murdoch, Western Australia, AUS). Mice acclimatized for one week prior to testing in the Victoria University (Western Centre for Health Research and Education, Sunshine Hospital) vivarium. Animals were maintained on a 12:12 light: dark cycle at 23°C (21% humidity) with unrestricted access to standard rodent chow and water.
Drug details
ASA (CAS 19046-78-7; C14H18N5O11P; 463.29 m.w.; 95% purity) was purchased from Santa Cruz (sc-214511; Dallas, Texas). Doses were prepared in double-distilled milliQ H2O relative to body weight as described in Table 1.
Acute oral toxicity procedure
The OECD “Up-and-down” toxicity test was implemented to evaluate ASA [9]. Mice (n=1/dose) were sequentially dosed with increasing concentrations of ASA dissolved in milliQ water using an incremental scale of 3.2 (i.e., 175, 550, 1750 mg/kg in the first instance). The commencing dose of 175 mg/kg was selected since lower doses (corrected for the different metabolic rates of mice compared to humans) were demonstrably safe in humans in the ASA clinical trial [1]. The dose was only increased when no signs of toxicity were observed in the first 48 hours. In life observations included monitoring of body weight, motor activity, neurological, respiratory and gastrointestinal function, skin and mucous membrane changes, and pain. Mice were observed for 14 days post-treatment. The first mouse was dosed with 175mg/kg ASA via oral gavage (21G) and monitored for 48 hours. Since no signs of toxicity were observed, a second mouse was dosed with 550 mg/kg ASA and monitored for 48 hours. This was repeated for the 1750 mg/kg dose. Since the upper limit dosage for the up-and-down procedure is 2000 mg/kg, but since no signs of toxicity were observed at the relatively equivalent dose of 1750 mg/kg we added an upper limit test of 5000 mg/kg to the protocol as recommended by the OECD test. At the conclusion of the observation period, mice were transported to an independent commercial laboratory animal testing facility (Cerberus Sciences, Scorebsy, Victoria, AUS).
Haematology
Blood was removed from anaesthetised mice via cardiocentesis into EDTA-coated vacutainer tubes. Blood samples were transferred to a commercial veterinary pathology testing laboratory (ASAP Laboratories, Mulgrave, Victoria, AU) and tested for haematocrit, haemoglobin, total erythrocyte, platelet, leukocyte, neutrophil, lymphocyte, monocyte, eosinophil and basophil count using a Siemens ADVIA 2120® haematology system.
Biochemistry
Whole blood was centrifuged at 3000 g for 10 minutes at 4 °C. Serum was separated and analysed for routine chemistry including glucose, creatinine, urea, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, total cholesterol, triglycerides, uric acid, albumin and total proteins using a Beckman Coulter AU680 chemistry system.
Histopathology
Mice were killed via CO2 asphyxiation and a full necropsy was performed. Liver, kidney and stomach were further processed for histopathological analysis. Samples were fixed in 10% neutral buffered formalin, sectioned into histology cassettes and processed through to haematoxylin and eosin stain. Photomicrographs were made using an Olympus BX41 microscope and LC30 camera and software. Histological photomicrographs were graded by an independent, certified specialist veterinary pathologist (Dr Lorna Rasmussen, B. VSc., B. VSc. (Hons), M. MedVet(Path)., Dip. ACVP). An untreated, sex and age matched control mouse was included in the macro- and micro-pathology analysis as a phenotype and environmental control to validate reference values species/strain-related nuances.
5. Conclusions
Our acute toxicity study revealed ASA as a non-toxic compound without obvious impact on macropathology following oral administration. Some hepatic and haematological nuances were observed with high dose treatment which may be incidental or associated with first pass metabolism and the known immunologic effects of fumarate treatment, respectively. These observations require follow up with sub-/chronic toxicology studies.
Author Contributions
Conceptualization, E.R. and C.A.T.; methodology, C.A.T ., E.R., and L.R.; formal analysis, E.R., L.R. and C.A.T.; data curation, E.R. and L.R.; writing—original draft preparation, C.A.T., and E.R.; writing—review and editing C.A.T., E.R., and L.R.; visualization, E.R. and L.R.; project administration, E.R. and C.A.T.; funding acquisition, E.R. and C.A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by an Australian Institute for Musculoskeletal Science seed grant.
Institutional Review Board Statement
Data presented in this manuscript were collected in an animal study protocol approved by the Victoria University Animal Ethics Committee (AEEC 17-006).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data will be made available upon request.
Conflicts of Interest
E.R. has consulted for Santhera Pharmaceuticals and Epirium Bio previously. E.R.’s laboratory group is currently funded by the Estate of Charles A. Bonsett (USA), AFM Telethon (FRA), Muscular Dystrophy Association (USA), the Jack Brockhoff Foundation (AUS) and Parent Project Muscular Dystrophy (NL).
References
- Bonsett, C. and A. Rudman, The dystrophin connection—ATP? Medical Hypotheses, 1992. 38(2): p. 139-154.
- Timpani, C.A., A. Hayes, and E. Rybalka, Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Medical hypotheses, 2015. 85(6): p. 1021-1033. [CrossRef]
- Budzinska, M., A. Zimna, and M. Kurpisz, The role of mitochondria in Duchenne muscular dystrophy. J Physiol Pharmacol, 2021. 72(2). [CrossRef]
- Bonsett, C. and A. Rudman, Duchenne’s muscular dystrophy: a tissue culture perspective. Indiana medicine: the journal of the Indiana State Medical Association, 1984. 77(6): p. 446.
- Lowenstein, J.M. and M.N. Goodman, The purine nucleotide cycle in skeletal muscle. Fed Proc, 1978. 37(9): p. 2308-12.
- Arinze, I.J., Facilitating understanding of the purine nucleotide cycle and the one-carbon pool: Part I: The purine nucleotide cycle. Biochemistry and Molecular Biology Education, 2005. 33(3): p. 165-168. [CrossRef]
- Rybalka, E., et al., Adenylosuccinic Acid: An Orphan Drug with Untapped Potential. Pharmaceuticals, 2023. 16(6): p. 822. [CrossRef]
- Bonsett, C.A. and A. Rudman, ’Oil globules’ in Duchenne muscular dystrophy--history, demonstration, and metabolic significance. Med Hypotheses, 1994. 43(5): p. 327-38.
- OECD, Test No. 425: Acute Oral Toxicity: Up-and-Down Procedure. 2022.
- McInnes, E.F., Background Lesions in Laboratory Animals: A Color Atlas. 2012: Saunders/Elsevier.
- Mazzaccara, C., et al., Age-Related Reference Intervals of the Main Biochemical and Hematological Parameters in C57BL/6J, 129SV/EV and C3H/HeJ Mouse Strains. PLoS One, 2008. 3(11): p. e3772. [CrossRef]
- Zaias, J., et al., Reference values for serum proteins of common laboratory rodent strains. J Am Assoc Lab Anim Sci, 2009. 48(4): p. 387-90.
- Silva-Santana, G., et al., Clinical hematological and biochemical parameters in Swiss, BALB/c, C57BL/6 and B6D2F1 Mus musculus. Animal Models and Experimental Medicine, 2020. 3(4): p. 304-315. [CrossRef]
- Nemzek, J.A., et al., Differences in normal values for murine white blood cell counts and other hematological parameters based on sampling site. Inflamm Res, 2001. 50(10): p. 523-7. [CrossRef]
- Rybalka, E., et al., Adenylosuccinic acid: a novel inducer of the cytoprotectant Nrf2 with efficacy in Duchenne muscular dystrophy. Curr Med Res Opin, 2021. 37(3): p. 465-467. [CrossRef]
- Zou, G.L., et al., The role of Nrf2/PIWIL2/purine metabolism axis in controlling radiation-induced lung fibrosis. Am J Cancer Res, 2020. 10(9): p. 2752-2767.
- Furuhashi, M., New insights into purine metabolism in metabolic diseases: role of xanthine oxidoreductase activity. Am J Physiol Endocrinol Metab, 2020. 319(5): p. E827-E834. [CrossRef]
- Gooding, J.R., et al., Adenylosuccinate Is an Insulin Secretagogue Derived from Glucose-Induced Purine Metabolism. Cell Rep, 2015. 13(1): p. 157-167. [CrossRef]
- Cader, M.Z., et al., FAMIN Is a Multifunctional Purine Enzyme Enabling the Purine Nucleotide Cycle. Cell, 2020. 180(2): p. 278-295.e23.
- Kourakis, S., et al., Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility? Pharmaceuticals (Basel), 2020. 13(10): p. 306.
- Ormerod, A.D. and U. Mrowietz, Fumaric acid esters, their place in the treatment of psoriasis. Br J Dermatol, 2004. 150(4): p. 630-2. [CrossRef]
- Harries, M.J., R.J. Chalmers, and C.E. Griffiths, Fumaric acid esters for severe psoriasis: a retrospective review of 58 cases. Br J Dermatol, 2005. 153(3): p. 549-51. [CrossRef]
- Smith, D., Fumaric acid esters for psoriasis: a systematic review. Ir J Med Sci, 2017. 186(1): p. 161-177. [CrossRef]
- Palte, M.J., et al., Improving the Gastrointestinal Tolerability of Fumaric Acid Esters: Early Findings on Gastrointestinal Events with Diroximel Fumarate in Patients with Relapsing-Remitting Multiple Sclerosis from the Phase 3, Open-Label EVOLVE-MS-1 Study. Adv Ther, 2019. 36(11): p. 3154-3165. [CrossRef]
- Colas, C., P.M. Ung, and A. Schlessinger, SLC Transporters: Structure, Function, and Drug Discovery. Medchemcomm, 2016. 7(6): p. 1069-1081. [CrossRef]
- Bonsett, C.A. and A. Rudman, The dystrophin connection--ATP? Med Hypotheses, 1992. 38(2): p. 139-54.
- Alqahtani, M.S., et al., Advances in Oral Drug Delivery. Frontiers in Pharmacology, 2021. 12. [CrossRef]
- Coleman, J.J. and S.K. Pontefract, Adverse drug reactions. Clin Med (Lond), 2016. 16(5): p. 481-485.
- Guengerich, F.P., Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab Pharmacokinet, 2011. 26(1): p. 3-14. [CrossRef]
- Timpani, C.A., et al., Adenylosuccinic acid therapy ameliorates murine Duchenne Muscular Dystrophy. Sci Rep, 2020. 10(1): p. 1125. [CrossRef]
- Longbrake, E.E., et al., Dimethyl fumarate-associated lymphopenia: risk factors and clinical significance. Multiple Sclerosis Journal–Experimental, Translational and Clinical, 2015. 1: p. 2055217315596994. [CrossRef]
- Timpani, C.A. and E. Rybalka, Calming the (Cytokine) Storm: Dimethyl Fumarate as a Therapeutic Candidate for COVID-19. Pharmaceuticals, 2021. 14(1): p. 15. [CrossRef]
- Timpani, C.A., et al., Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go? Antioxidants, 2020. 9(12): p. 1268.
- Rybalka, E., et al., Chemotherapeutic agents induce mitochondrial superoxide production and toxicity but do not alter respiration in skeletal muscle in vitro. Mitochondrion, 2018. 42: p. 33-49. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



