
Submitted:
05 September 2023
Posted:
07 September 2023
You are already at the latest version
Abstract
Neuroinflammation is considered to be a significant component in a range of neuropathologies. Unfortunately, whilst its role is well recognized, the options for therapeutic intervention are limited. As such, there is a need to identify novel targets in order to increase treatment options. Given its role as both a neurotransmitter and an immune modulator, substance P and its NK1 receptor have been widely studied as a potential therapeutic target. There is evidence that NK1 receptor antagonists may exert beneficial effects in a range of conditions, including traumatic brain injury and stroke. Blocking the NK1 receptor has been shown to reduce blood-brain barrier dysfunction, reduce cerebral oedema, and reduce the levels of pro-inflammatory cytokines. These actions are associated with improved survival and functional outcomes. The NK1 receptor has also been shown to be involved in the inflammatory reaction to CNS infection, and hence antagonist may have some benefit in reducing infection-driven inflammation. However, the NK1 receptor may also play a role in the host immune response to infection, and so here, the potential beneficial and detrimental effects need to be carefully balanced. As such, the purpose of this review is to provide a summary of the involvement of substance P in acute inflammation, particularly in the context of traumatic brain injury and stroke.
Keywords:
NK1 receptor
; Substance P
; Neuroinflammation
; Blood-brain barrier
; Traumatic brain injury
; Stroke
; CNS infection
; NK1 receptor antagonist
1. Introduction
A major advance in our understanding of central nervous system disorders has been the recognition that inflammation plays a significant role in many acute and chronic conditions. The role of inflammation is clearly seen in relation to acute traumatic and ischaemic insults, as well as infection, but it also an important factor in many chronic and neurodegenerative conditions, including multiple sclerosis, epilepsy, Alzheimer’s and Parkinson’s disease. Collectively, neuroinflammatory conditions represent a major healthcare challenge, both in terms of their social and economic impact. Unfortunately, the common anti-inflammatory agents, namely steroids and non-steroidal anti-inflammatory drugs (NSAIDs), are of limited value in the clinical management of these conditions, due to limited efficacy, potentially significant side effects, or both [1-3]. Hence, there is a need to develop novel, targeted and more effective anti-inflammatory therapies that may help manage these significant and prevalent conditions [4]. Given the role that substance P and the NK1 receptor appear to play in neurogenic inflammation in peripheral tissues, there has been an interest in seeing whether the NK1 receptor may represent a novel target to manage acute neuroinflammatory responses in the CNS [5].
2. Inflammation in the Central Nervous System
Both naturally, and clinically, there is a need to maintain the beneficial aspects of inflammation (e.g. defense against pathogens,tissue healing etc,) whilst, at the same time, limiting the detrimental aspects (i.e. tissue injury, chronic inflammation) [6-8]. However, balancing these opposing effects of inflammation within the CNS this is even more critical. As in the periphery, uncontrolled activation of CNS inflammatory responses can progress to detrimental, chronic inflammation. There is growing evidence to suggest that excessive or inappropriate neuroinflammation plays an active role in neurodegenerative conditions, such as Alzheimer’s and Parkinson’s disease [9]. Whilst excessive acute inflammatory responses are well documented to contribute to the secondary injury processes, such as cerebral oedema and raised intracranial pressure, associated with traumatic brain injury and stroke [10,11].
Historically, the CNS was viewed as being immunoprivileged, with the notion that it was shielded from the peripheral immune system by the blood-brain barrier (BBB) [9]. Allied to that, within the CNS glial cells provide dedicated immune function, engaging in inflammatory responses that serve to defend the CNS against pathogens, as well as aid in its recovery from insult and injury. In terms of that resident immune function, astrocytes, the most numerous cell type in the CNS, are key immune-active cells [12], whilst microglia and perivascular macrophages represent the phagocytic cells of the CNS [13]. The immune responses mediated by these glial cells are commonly referred to as neuroinflammation. However, it is now well understood that the brain CNS is not protected from the effects of peripheral immune activation, and that resident microglia may also play deleterious roles within the CNS.
Whilst the BBB has been viewed as a physical barrier between the CNS and peripheral immune system, in reality it is a dynamic, functional barrier that serves as an interface between the two. As a functional barrier, it is subject to complex control mechanisms as part of the neurovascular unit (NVU). The concept of the NVU encompasses the anatomical and functional relationship between brain cells, both neurons and glial cells, and the cerebrovasculature (Figure 1). The NVU is considered to exert control over such functions as cerebral blood flow and immune responses [14]. The concept of the BBB serving as a functional immune interface emphasizes the range of cell types that may contribute to normal immune/inflammatory function or dysfunction (in the case of disease or injury), in the CNS. Positioned at the interface between the systemic circulation and brain parenchyma, the NVU and BBB can monitor and respond to immune and inflammatory signals in both the systemic circulation and the CNS [15]. Specifically, the endothelial cells of the cerebrovasculature mediate changes in vascular permeability, as well as the expression and function of adhesion molecules, allowing for leukocyte transmigration [16-18]. Pericytes, lying in close contact with capillary endothelial cells, also serve important roles in regulating immunoregulatory functions and formation of new blood vessels [15]. In addition to providing dedicated immune protection within the CNS, they have close structural and functional relationships with the BBB [19], enabling the integration of CNS and peripheral immune and inflammatory responses. Other vessel-associated immune cells include perivascular macrophages and vessel-associated microglia [20]. The numerous microglia that perform immune surveillance within the environment of the NVU are capable of producing a wide range of pro-inflammatory mediators that influence NVU function as well as BBB integrity [21]. Pericytes also play an important role in maintaining BBB integrity, and controlling a number of aspects of CNS immune responses, including leukocyte extravasation [15,19].
Figure 1 The cells associated with the NVU. SP expression is associated with neurons involved in neurovascular coupling, as well as with the vascular endothelium and glial cells, including astrocytes and microglia. The NK1-R has also been shown to be expressed on neurons, endothelial cells, astrocytes and microglia. Whilst NK1-Rs have not been explicitly shown to be associated with pericytes, their embryological origin makes this a possibility.
In terms of CNS inflammation, there appears to be a two-way relationship between BBB dysfunction and neuroinflammation. Altered BBB function may serve as a precursor for neuroinflammation, whilst neuroinflammation and altered NVU function, will impact on BBB permeability and function [15]. BBB dysfunction is implicated in some of the most prevalent and significant neurological conditions, ranging from acute insults, such as trauma, ischaemia and infection, through to chronic and neurodegenerative conditions, such as epilepsy, multiple sclerosis, Alzheimer’s and Parkinson’s disease [22]. Studies of the systemic circulation indicate that perivascular nerves may play a role in mediating vascular inflammatory responses [23,24]. As a result, there was an interest in whether similar mechanisms may be involved in regulating BBB integrity and central inflammation [25-27].
3. Perivascular Innervation and Neurogenic Inflammation
The systemic vasculature is innervated by the peripheral nervous system, encompassing both autonomic and sensory nerves. The perivascular sensory nerves include the neuropeptide-containing nociceptive nerves, which release a range of neurotransmitters, including the amino acid transmitter, glutamate, and the neuropeptides, substance P (SP) and calcitonin gene-related peptide (CGRP) [23], and also characterized by the expression of TRPV1 ion channels [28]. The peptide neurotransmitters are synthesized in the cell bodies of these neurons located in the dorsal root ganglia of the spinal cord, from where they are transported both centrally, and peripherally to the free nerve endings [29]. Activation of nociceptive nerves leads to peptide release from both the central and peripheral nerve terminals, conferring these nerves with a sensory-motor function involving local axon reflexes. The peripheral peptide release can provoke a potent vasodilator effect mediated by CGRP, as well as SP-mediated plasma extravasation [30]. This motor response may underlie the cutaneous “flare” reaction seen following injury, as well as hyperaemia and oedema in other tissues. It has been postulated that the sensory-motor function of these nerves could represent a mechanism to respond to tissue ischaemia, but it may also have a broader role in initiating immune and inflammatory responses following the detection of a noxious event [24,30].
The term neurogenic inflammation is used to denote the role of sensory nerves in inflammatory responses [23,24]. There is considerable evidence to support substance P and the NK1 receptor playing a key role in mediating the vascular changes associated with neurogenic inflammation [31-33], as well as them playing a broader role in the innate immune response [34-36]. SP increases vascular permeability, and promotes oedema, as a result of endothelial cell retraction and the formation of gaps between cells [35,37,38]. The NK1-mediated effects of SP on vascular endothelium appear to occur at the level of venules and capillaries [39]. SP also enhances leukocyte migration, through its effects on vascular and intracellular adhesion molecules, as well as matrix metalloproteinase secretion [40-42]. In addition to its direct vascular effects, SP can also activate mast cells, leading to the release of other vasoactive mediators [43], with mast cells have been shown to exist in close proximity to SP-containing nerve terminals [44]. Indeed, the literature also supports a broader role for SP in inflammation, such that SP is one of the earliest activators of nuclear factor kappa B in response to injury, and can increase the secretion of other pro-inflammatory mediators, including interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNF-α), elaborating the inflammatory response [34]. Beyond its neuronal expression, substance P and the NK1 receptor are expressed in a range of immune cells, including T lymphocytes, macrophages and eosinophils, suggesting the potential for cross-talk between the immune and nervous systems [40,44,45]. For example, both in vitro and in vivo studies suggest a role for SP in neutrophil migration and activation at the site of inflammation [45].
The wealth of data supporting the role of perivascular sensory nerves, and in particular substance P, in modulating immune and inflammatory responses in the periphery, subsequently drove interest as to whether the same neuropeptides may be exerting a similar pro-inflammatory action within the CNS [26]. However, whilst there are similarities, there are also clear distinctions between the innervation of the cerebrovascular and systemic circulations. The superficial, or extracerebral vessels of the brain, are extrinsically innervated, including receiving sensory innervation by the trigeminal nerve [46]. Indeed, there is good evidence to support the involvement of these nerves in migraine, with a particular focus on the role of CGRP [47]. However, the cerebrovasculature itself is “intrinsically” innervated by the central neurons that contribute to the function of the NVU [46]. Hence, for substance P to be influencing the function of the cerebrovasculature and BBB, it would either have to be of central origin, or potentially in the circulation, although that would rely on SP having a luminal influence on BBB integrity, for which there is little supporting evidence [27,48-50].
4. Distribution of SP and the NK1 receptor in the CNS
Whilst the initial characterization of SP’s biological activity focused on its actions in peripheral tissues, it was soon recognized that SP and the NK1 receptor were also both abundant and widespread within the CNS [51-54]. Whilst some of that localization, for example the dorsal horn of the spinal cord, is consistent with SP being associated with primary sensory nerve signaling, there is both anatomical and functional evidence that SP is involved in other CNS pathways [55]. Whilst the presence of SP has been demonstrated in structures such as the cerebral cortex and cerebellum, intense immunostaining has been observed in regions such as the amygdala, locus ceruleus and hypothalamus [56]. There is also a reasonable correlation between the distribution of SP and the neuronal expression of the NK1 receptor, again, with some of the highest levels of constitutive NK1 receptor expression being associated with the putamen and caudate nuclei, and the limbic system. However, there is also some anatomical mismatch in terms of the co-localization of SP and NK1 receptors in the CNS, raising the suggestion that SP may be involved in non-synaptic signaling, relying on local diffusion in the manner of an autocrine signal [57]. The high levels of expression of SP and the NK1 receptor in the limbic and reticular activating systems led to the notion that SP may be involved in stress and emotional responses [58], which has been validated in preclinical studies demonstrating that NK1 receptor antagonists may exert anxiolytic and antidepressant activity [59,60]. The potential for SP and the NK1 receptor to play a role in depression is of interest, given the recognition that inflammation is associated with the development of depression [61]. However, the best clinical evidence to support central activity of SP is the effective antiemetic action of the NK1 receptor antagonists [62,63], which appears to correlate with the brain stem distribution of SP and the NK1 receptor [64].
Within the CNS, there is a significant and widespread distribution of the NK1 receptor, with distinct labeling of neuronal dendrites and soma [57]. However, whilst some initial studies suggested that expression of the NK1 receptor in the CNS was almost exclusively neuronal, there is strong evidence to suggest that it is also associated with non-neuronal cells, including endothelial cells, astrocytes and microglia [65]. It is known that there are two main, naturally occurring isoforms of the NK1 receptor, namely the full-length (407 amino acids) and the truncated isoform (311 amino acids) [66], with the central NK1 receptors shown to be primarily the full-length isoform. Whilst the receptor binding pocket is the same for both isoforms, it is the carboxyl tail of the receptor that is affected by truncation, impacting upon receptor-effector coupling. In the periphery, the truncated isoform of the NK1 receptor is primarily has been shown to be associated with immune cells, such as macrophages and lymphocytes. The truncated isoform of the receptor requires higher SP concentrations in order to generate responses due to reduced agonist efficacy [53,55], although, perhaps more importantly, unlike the full-length receptor, stimulation does not result in NFκB activation [67]. There is evidence that both receptor isoforms are expressed in the CNS [68]. In the study performed by Liu and colleagues [57], the antibody used was directed against carboxyl terminal of the NK1 receptor, which would only label the full-length receptor isoform, and as demonstrated, these are associated with the neurons [69]. However, another factor to consider when comparing studies is that a pro-inflammatory state, or pathologies, can significantly influence SP and NK1 receptor expression [53,70].
In addition to the neuronal expression of the NK1 receptor, there is also constitutive and induced expression of NK1 receptors associated with non-neuronal cells linked to CNS immune function. Whilst their association with the NVU has not been explicitly documented, the expression of NK1 receptors in the cells that constitute the NVU has been clearly demonstrated.
5. Non-neuronal NK1 receptor activity in the CNS
As in the periphery, NK1 receptors have been shown to be associated with vascular endothelium in the cerebrovasculature, and particularly with post-capillary venules [16,70,71]. Through the use of selective NK1 antagonists, substance P has been shown to mediate the increased endothelial permeability induced by pro-inflammatory mediators, such as TNF-α, as well as viral proteins [16,72]. In the same manner, NK1-R receptor antagonism can reduce the expression of endothelial adhesion molecules, thereby reducing recruitment of inflammatory cells and their transmigration into the brain [42]. In addition, an autocrine feedback mechanism may be associated with these SP-mediated changes in endothelial function. Pro-inflammatory cytokines, such as TNF-α and IL-1β, can stimulate the expression and release of SP from endothelial cells, which can then activate endothelial NK1 receptors [16,73]. In addition, the expression of endothelial NK1 receptors may also be influenced by an inflammatory state [70] which could establish a pro-inflammatory positive feedback mechanism.
In terms of glial cells, NK1 receptors have been shown to be constitutively expressed by astrocytes, including human astrocytes [65,74]. Whilst more contentious following early studies, there is now a growing body of evidence to support the involvement of the NK1-R in the activation of microglia [65,70,75]. Stimulation of the NK1 receptor results in the activation of NFκB and an elaboration of inflammatory mediators [76,77]. importantly, since we are considering immune and inflammatory responses, it is the full-length isoform of the receptor that is expressed in these cells [65,66]. However, a maximal pro-inflammatory response may rely on co-stimulation involving SP / NK1-R together with other pro-inflammatory factors [75].
Whilst we are primarily concerned with the potential role of SP and the NK1-R in promoting neuroinflammation, there is evidence that SP-containing interneurons can play a role in normal neurovascular coupling [78,79]. However, it is the association of the NK1-R with the cells that maintain and control BBB function that is of interest in terms of the potential to ameliorate both vasogenic oedema and neuroinflammatory responses [16]. Whilst the intravenous administration of capsaicin, and activation of TRPV1, affected vascular permeability in meningeal, but not cerebral vessels [48], activation of TRPV1 associated with the cerebral microvasculature does increase vessel permeability via SP. It has also been shown to play a role in BBB disruption following ischaemia-reperfusion injury [27]. The NK1 receptor also has the potential to generate a significant positive-feedback cycle in relation to neuroinflammation. The NK1 receptor has been shown to not only activate resident glial cells, but also stimulate infiltrating leukocytes, recruited through BBB disruption, in the same manner. Indeed, SP-mediated activation of NF-κB leading to the elaboration of inflammatory mediators, such as IL-1β [76,77], in turn can lead to the upregulation of SP and NK1-R expression [80,81]. Based on these observations, the NK1-R appears to represent an important novel target to manage acute neuroinflammatory responses in the CNS. Indeed, there is a growing list of neuropathologies where there is evidence to a beneficial action of NK-1R antagonists (Table 1).
It is also worth noting that a substantial number of in vivo studies examining the role of SP and the NK1 receptor have used N-acetyl-L-tryptophan (NAT) as an agent with assumed NK1 receptor antagonist activity, primarily based on the reported activities of analogues of L-tryptophan [100]. Although, specific receptor biding has not been characterized, with a recent publication demonstrating a lack of NK1-R binding by NAT [101], an in silico screening assay for NK1-R antagonist activity predicted NAT would have favourable binding characteristics [102]. In line with the concerns raised by Matalinska and Lipinski [101], it is appropriate that further studies of the role of the NK1-R in neuropathologies should utilize selective NK1-R antagonists. That said, there have also been in vivo studies where positive neuroprotective outcomes assigned to supposed NK1-mediated action of NAT have subsequently been fully replicated with highly selective NK1 antagonists [83,85,87,88,103].
6. Evidence for the Involvement of the NK1-R in Acute CNS Inflammation
Acute CNS inflammation is associated with infection (bacterial, viral & parasitic), as well as autoimmune reactions, trauma and ischaemia [9,53]. Whilst there are significant differences in relation to the primary insult, there is a broad evidence base to support the role of the NK1-R in the associated inflammatory response, as well as the potential benefit of antagonizing the receptor [5]. One other common factor is that across these diverse conditions, there is a common pattern of increased expression of SP and the NK1-R, which could fuel that cycle of inflammation [82,98,104]. Interestingly, in a murine model of West Nile neuroinvasive disease, there was a positive correlation between SP levels and mortality [98].
Stroke
In a similar vein, data from stroke patients has shown that there is a significant increase in serum SP levels following cerebral ischaemia [105]. One of the earliest reports of NK1-R antagonists having a neuroprotective action was in a rat model of cerebral ischaemia [82]. The selective antagonist, SR140333, was shown to reduce infarct volume and improve functional outcomes. This valuable observation has been replicated by multiple groups using different models. Turner and colleagues, using NAT in a rodent model of transient middle cerebral artery occlusion, demonstrated the potential to restore BBB integrity and reduced cerebral oedema, as well as significantly reduce functional deficits [83]. In a similar study, they also demonstrated that NAT was able to ameliorate tissue plasminogen activator-induced BBB dysfunction, in both naïve and stroke animals [84]. Whilst there are concerns about inferring a selective mode of action to NAT, Turner and colleagues also demonstrated the ability of a highly selective NK1 antagonist to post-stroke cerebral oedema and raised intracranial pressure (ICP) in their ovine model of stroke [85,106]. SP release associated with ischaemia and reperfusion, and the associated neurogenic inflammation, are considered to be involved in the pathogenesis of vascular dementia. Kaur and colleagues, using a rodent model of bilateral common carotid artery occlusion, have demonstrated the ability of the selective antagonist, aprepitant, to reduce the ischaemia-induced cerebrovasculature dysfunction [86].
Traumatic Brain Injury
In terms of traumatic brain injury (TBI), neuroinflammation is a significant secondary injury mechanism following the primary insult, and it is linked to the development of complications such as cerebral oedema and raised ICP [107-109]. Given the clinical need to better control cerebral oedema and raised ICP, there was an interest to see whether inflammatory neuropeptides, and particularly the NK1-R, may represent an effective target to manage the inflammation and oedema [26]. An initial study, using capsaicin as a pre-injury treatment to produce neuropeptide depletion in a rodent model of TBI, there was a significant reduction in post-traumatic BBB dysfunction and oedema formation, together with a significant decrease in the motor and cognitive deficits associated with injury [25]. From this initial observation, intravenously administered NAT was used in order to see whether this protective effect could be achieved using a post-injury treatment [87]. Administration of NAT 30 mins after trauma significantly attenuated vascular permeability and edema formation, as well as improved both motor and cognitive neurologic outcomes. Whilst a question mark remains around the mechanism of action of NAT, these results were fully replicated when a highly selective NK1 antagonist was administered in the same manner and model [88,103]. Indeed, there is evidence that a selective NK1 antagonist may exert a broader neuroprotectant effect than NAT. Studies on blast injury and chronic traumatic encephalopathy have shown that, in contrast to NAT, an NK1 antagonist can attenuate the injury-induced phosphorylation of tau by modulating the activity of several key kinases including Akt, ERK1/2 and JNK [89]. Li and colleagues have also shown that a selective NK1 antagonist, as well as SP deletion, can exert a neuroprotective effect in a murine model of TBI [90]. The use of a large animal model of severe TBI has demonstrated the ability of an NK1 antagonist to control raised ICP and the associated reduction in brain tissue oxygen levels [91]. A selective NK1-R antagonist, EU-C-001, is currently in a multi-national Phase 2 clinical trial for TBI (ACTRN 12619000074190).
Spinal Cord Injury
Like TBI, acute inflammation is also associated with traumatic spinal cord injuries [110], and there is evidence for the involvement of SP and the NK1-R [111]. Whilst a study using systemic i.v. administration of NAT did not demonstrate any significant benefit [112], a recent study using continuous subdural infusion of a selective NK1 antagonist, did exhibit some reduction in neuroinflammation together with some positive functional outcomes [92].
Bacterial Infections
Acute neuroinflammation is also associated with CNS infections. Infections can lead to inflammation of both the meningeal membranes (meningitis) and parenchyma of the brain (encephalitis) and spinal cord (myelitis) [113]. Bacterial infections of the CNS are of significant clinical concern and are associated with the proliferation of resident glial cells, as well as the recruitment of peripheral leukocytes to the site of infection. Using murine models of bacterial infection, the use of a selective NK1 antagonist (L703,606), as well as using NK1-R deficient mice, has provided evidence for the role of the NK1-R in altered BBB function and promotion of neuroinflammation [75,93]. Antagonism of the NK1-R can reduce the proliferation of pro-inflammatory cytokines, as well as decrease the severity of the disease. These beneficial effects were demonstrated against a range of clinically-relevant infectious agents, including Streptococcus pneumoniae and Neisseria meningitidis. These studies also demonstrated that the NK1-R was involved in bacterial-induced demyelination [93]. Infection was also associated with increased expression of the NK1-R on microglia and astrocytes, whilst SP was shown to have a broad pro-inflammatory action [75,94].
Viral Infections
A similar picture of neuroinflammation is associated with viral infections. In terms of CNS inflammation associated with human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) infection, perivascular macrophages are potentially the first cells infected in the CNS [104]. Studies of the brains in SIV-infected macaque monkeys have shown that there is intense expression of SP and the NK1-R in SIV infected cells and encephalitic lesions [104]. In addition, HIV infection has been associated with increased serum levels of substance P, and a similar picture is seen with SIV infection in monkeys [104,114,115]. Positive correlations between SP levels and viral load have suggested that the peptide may play a role in facilitating viral replication [97,116], potentially by increasing the expression of the receptors (e.g. CCR5) required for uptake of the virus. There is evidence that SP, acting via the NK1-R, can enhance HIV infection [117], whilst the NK1 antagonist, aprepitant, may potentially exert anti-HIV activity, and help prevent infection in microglia and macrophages [96]. Aprepitant may also produce synergistically beneficial effects when used in combination with other anti-retrovirals [97]. Another virus a neuroinvasive potential is West Nile virus. Those who develop neuroinvasive disease (WNND) will present with meningitis and encephalitis. In cases of encephalitis, pathology of brain tissue reveals inflammation, which is mostly mononuclear, with formed microglial nodules and perivascular clusters in both white and gray matter. In a mouse model Ronca and colleagues showed that SP was significantly upregulated in the brain during infection, which correlated with neuroinvasion and BBB dysfunction [98]. Blocking the NK1-R with a selective antagonist at disease onset modestly improved survival and prolonged time to death. Unlike HIV infection, the results indicated that SP does not appear to play a role in viral replication, but rather may mediate the immune response to infection.
Parasitic Infections
Finally, there is also evidence that the NK1-R contributes to neuroinflammation in response to parasitic infection. Cysticercosis is a parasitic infection associated the pork tapeworm, Taenia solium. If larval cysts infect the brain, this is referred to neurocysticercosis. Dying parasites trigger a granulomatous response, which is associated with the development of seizures. SP-positive cells are observed is association with these granulaomas, both in human biopsies and animal models [118]. There is evidence that SP and the NK1-R are associated with the seizure activity, granuloma development, and the cytokine response associated with granuloma formation [119,120]. SP and the NK1-R have been associated with other parasitic infections that lead to CNS involvement, including the Trypanosoma brucei brucei infection associated with sleeping sickness. Trypanosma infection is associated with a severe neuroinflammatory reaction and peripheral inflammatory infiltration. In mouse model of Trypanosoma infection associated with severe meningoencephalitis, the selective NK1-R antagonist, RP-67,580, was shown to significantly reduce the inflammatory response and reactive astrogliosis [99].
7. The double-edged sword of inflammation
As discussed, inflammation represents a double-edged sword, with both beneficial and detrimental aspects. The host immune response is central to dealing with infection, and there is significant evidence to support the role of the NK1-R in the host response to infection, viral, bacterial and parasitic [116,121,122]. SP and the NK1-R exert an immune-protective role at mucosal barriers, contributing to the host response to a diverse range of infections [123]. In a mouse model of herpes virus infection, increased expression of the NK1-R was found to correlate to decreased viral load when compared to mice who were NK-1 receptor-deficient [121]. Similarly, in relation to bacterial infection, demonstrate increased bacterial load in response to salmonella infection [124]. This reduced host response is also observed in parasitic infections, with mice treated with an NK1-R antagonist showing limited granuloma formation in response to infection with Schistosoma mansoni [123]. However, as we have seen, the NK1-R can also contribute to the pathogenesis of infection, and particularly the destructive inflammatory response [116], so both aspects need to be considered in relation to the potential application of NK1-R antagonists.
Whilst the purpose of this review is to examine the evidence surrounding the role of the NK1-R in acute neuroinflammation, given the clinical burden associated with neurodegeneration, the potential involvement of SP here should be mentioned. It is recognized that there is a link between excessive neuroinflammation and neurodegenerative disorders, such as Parkinson’s disease, Alzheimer’s disease and multiple sclerosis, and that these diseases have a significant inflammatory component [125-128]. In contrast to the acute inflammatory situation, there is evidence that SP and the NK1-R may exert a neuroprotective effect in some situations [53]. For example, in a rodent model of Alzheimer’s disease, SP administration was able to prevent β-amyloid peptide-induced cognitive deficits [129]. Analysis of human post-mortem samples from patients with confirmed multiple system atrophy (MSA) and Parkinson's disease showed a significant depletion of NK1-R-like-immunoreactive neurons in the ventrolateral medulla, with these changes being more marked in MSA patients, being suggestive of the broader system involvement [130]. On the reverse side, there is evidence that SP / NK1-R interaction may be necessary to maintain chronic inflammation in an experimental model of autoimmune encephalomyelitis [131]. However, an NK1-R antagonist, SR140333, an NK-1 antagonist, on its own was not sufficient to ameliorate symptoms. Again, whilst there may be a delicate balance between the neuroprotective effects of SP, and its pro-inflammatory effects, it is considered that the effective management of acute neuroinflammation may help prevent the development of chronic inflammation and neurodegeneration [132].
8. Concluding Remarks
Severe acute neuroinflammation is associated with significant mortality and morbidity in a range of clinical situations including CNS trauma, ischaemia and infection. Unfortunately, the therapeutic options for the effective management of these issues is limited [1]. As a result, there is significant interest is potential novel targets [4].
There is a large body of evidence to support the role that SP and the NK1-R may play in acute inflammation in both the periphery and the CNS [5]. In terms of neuroinflammation, the NK1-R may not only be involved in activating the resident immune cells in the CNS, such as astrocytes and microglia [65], but may also facilitate the entry of peripheral immune cells into the CNS [42]. The NK1-R on cerebrovascular endothelium may also be responsible for the development of cerebral oedema and raised intracranial pressure [91]. Given the availability of safe, effective antagonists of the NK1-R, there is the potential for expedited clinical translation of these agents [63].
There have been multiple in vivo studies to support the potential beneficial effects of NK1-R antagonists in TBI and stroke [82,83,85,87]. Whilst some of these studies did utilize NAT as a proposed NK-1R antagonist, the results have been fully replicated using highly potent and selective agents. Indeed, these agents may exhibit more benefit than initially exhibited by NAT [89,92].
There is also evidence that NK1-R antagonists can help ameliorate severe neuroinflammation associated with CNS infection, be it viral, bacterial or parasitic [75,98,120], as well as decrease viral load [95]. However, the double-edged sword associated with inflammation is apparent with the NK1-R’s role in infection, as it may also contribute the host immune response against infection [123]. Hence, the application of NK1-R antagonists to manage infection needs to be carefully considered. In a similar manner, the NK1-R may mediate a neuroprotective role in some situations [129], whilst helping to control the underlying inflammation in others [131]. In summary, there is significant foundational evidence to support the wider use of NK1-R antagonists in the clinic.
Acknowledgments
The authors would like to gratefully acknowledge Ms Shannon ??? for their assistance with the preparation of Figure 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beauchamp, K.; Mutlak, H.; Smith, W.R.; Shohami, E.; Stahel, P.F. Pharmacology of traumatic brain injury: where is the "golden bullet"? Mol Med 2008, 14, 731–740. [Google Scholar] [CrossRef]
- Cook, A.M.; Morgan Jones, G.; Hawryluk, G.W.J.; Mailloux, P.; McLaughlin, D.; Papangelou, A.; Samuel, S.; Tokumaru, S.; Venkatasubramanian, C.; Zacko, C.; et al. Guidelines for the Acute Treatment of Cerebral Edema in Neurocritical Care Patients. Neurocrit Care 2020, 32, 647–666. [Google Scholar] [CrossRef] [PubMed]
- Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer's Disease Anti-Inflammatory Prevention Trial (ADAPT). PLoS Clin Trials 2006, 1, e33. [CrossRef]
- Vink, R.; Nimmo, A.J. Multifunctional drugs for head injury. Neurotherapeutics 2009, 6, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, F.; Vink, R.; Turner, R.J. Inflammation in acute CNS injury: a focus on the role of substance P. Br J Pharmacol 2016, 173, 703–715. [Google Scholar] [CrossRef]
- Gordon, S. Phagocytosis: An Immunobiologic Process. Immunity 2016, 44, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. The Journal of Clinical Investigation 2018, 128, 2657–2669. [Google Scholar] [CrossRef]
- Liu, C.H.; Abrams, N.D.; Carrick, D.M.; Chander, P.; Dwyer, J.; Hamlet, M.R.J.; Macchiarini, F.; PrabhuDas, M.; Shen, G.L.; Tandon, P.; et al. Biomarkers of chronic inflammation in disease development and prevention: challenges and opportunities. Nature Immunology 2017, 18, 1175–1180. [Google Scholar] [CrossRef]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br J Pharmacol 2006, 147 Suppl 1, S232–240. [Google Scholar] [CrossRef]
- Kinoshita, K. Traumatic brain injury: pathophysiology for neurocritical care. J Intensive Care 2016, 4, 29. [Google Scholar] [CrossRef]
- Heiss, W.D. Malignant MCA Infarction: Pathophysiology and Imaging for Early Diagnosis and Management Decisions. Cerebrovasc Dis 2016, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bellaver, B.; Dos Santos, J.P.; Leffa, D.T.; Bobermin, L.D.; Roppa, P.H.A.; da Silva Torres, I.L.; Goncalves, C.A.; Souza, D.O.; Quincozes-Santos, A. Systemic Inflammation as a Driver of Brain Injury: the Astrocyte as an Emerging Player. Mol Neurobiol 2018, 55, 2685–2695. [Google Scholar] [CrossRef]
- Lapenna, A.; De Palma, M.; Lewis, C.E. Perivascular macrophages in health and disease. Nat Rev Immunol 2018, 18, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat Neurosci 2021, 24, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Jansson, D.; Smyth, L.C.; Dragunow, M. Brain Pericytes As Mediators of Neuroinflammation. Trends Pharmacol Sci 2017, 38, 291–304. [Google Scholar] [CrossRef]
- Annunziata, P.; Cioni, C.; Santonini, R.; Paccagnini, E. Substance P antagonist blocks leakage and reduces activation of cytokine-stimulated rat brain endothelium. J Neuroimmunol 2002, 131, 41–49. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harb Perspect Biol 2014, 7, a016345. [Google Scholar] [CrossRef]
- Hickey, W.F. Leukocyte traffic in the central nervous system: the participants and their roles. Semin Immunol 1999, 11, 125–137. [Google Scholar] [CrossRef]
- Jansson, D.; Rustenhoven, J.; Feng, S.; Hurley, D.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.; Dragunow, M. A role for human brain pericytes in neuroinflammation. J Neuroinflammation 2014, 11, 104. [Google Scholar] [CrossRef]
- Koizumi, T.; Kerkhofs, D.; Mizuno, T.; Steinbusch, H.W.M.; Foulquier, S. Vessel-Associated Immune Cells in Cerebrovascular Diseases: From Perivascular Macrophages to Vessel-Associated Microglia. Front Neurosci 2019, 13, 1291. [Google Scholar] [CrossRef]
- da Fonseca, A.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci 2014, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, E.A.; Bauer, B.; Fahlke, C.; Fricker, G.; Iadecola, C.; Janigro, D.; Leybaert, L.; Molnar, Z.; O'Donnell, M.E.; Povlishock, J.T.; et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci 2011, 12, 169–182. [Google Scholar] [CrossRef]
- Sousa-Valente, J.; Brain, S.D. A historical perspective on the role of sensory nerves in neurogenic inflammation. Semin Immunopathol 2018, 40, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; von Hehn, C.A.; Woolf, C.J. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci 2012, 15, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, A.J.; Cernak, I.; Heath, D.L.; Hu, X.; Bennett, C.J.; Vink, R. Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 2004, 38, 40–47. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Marcoionni, A.M.; Dempsey, E.R.; Woenig, J.A.; Turner, R.J. The Role of Neurogenic Inflammation in Blood-Brain Barrier Disruption and Development of Cerebral Oedema Following Acute Central Nervous System (CNS) Injury. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.E.; Easton, A.S.; Fraser, P.A. TRPV1 activation results in disruption of the blood-brain barrier in the rat. Br J Pharmacol 2005, 146, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. TRPV1 Modulators for the Treatment of Pain and Inflammation. ACS Medicinal Chemistry Letters 2019, 10, 143–144. [Google Scholar] [CrossRef]
- Harmar, A.; Keen, P. Synthesis, and central and peripheral axonal transport of substance P in a dorsal root ganglion-nerve preparation in vitro. Brain Res 1982, 231, 379–385. [Google Scholar] [CrossRef]
- Aalkjær, C.; Nilsson, H.; De Mey, J.G.R. Sympathetic and Sensory-Motor Nerves in Peripheral Small Arteries. Physiol Rev 2021, 101, 495–544. [Google Scholar] [CrossRef]
- Garret, C.; Carruette, A.; Fardin, V.; Moussaoui, S.; Peyronel, J.F.; Blanchard, J.C.; Laduron, P.M. Pharmacological properties of a potent and selective nonpeptide substance P antagonist. Proc Natl Acad Sci U S A 1991, 88, 10208–10212. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.V.; Helme, R.D.; Thomas, K.L. NK-1 receptor mediation of neurogenic plasma extravasation in rat skin. Br J Pharmacol 1989, 97, 1232–1238. [Google Scholar] [CrossRef]
- Lundberg, J.M.; Saria, A.; Brodin, E.; Rosell, S.; Folkers, K. A substance P antagonist inhibits vagally induced increase in vascular permeability and bronchial smooth muscle contraction in the guinea pig. Proc Natl Acad Sci U S A 1983, 80, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.D.; Leeman, S.E. Neurokinin-1 receptor: functional significance in the immune system in reference to selected infections and inflammation. Ann N Y Acad Sci 2011, 1217, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Holzer, P. Neurogenic vasodilatation and plasma leakage in the skin. Gen Pharmacol 1998, 30, 5–11. [Google Scholar] [CrossRef]
- Bekkemeyer, H.; Oehme, P.; Foreman, J.C. Accidental injection of substance P. J R Soc Med 1983, 76, 801–802. [Google Scholar] [CrossRef]
- van Hinsbergh, V.W.; van Nieuw Amerongen, G.P. Endothelial hyperpermeability in vascular leakage. Vascul Pharmacol 2002, 39, 171–172. [Google Scholar] [CrossRef]
- Baluk, P.; Hirata, A.; Thurston, G.; Fujiwara, T.; Neal, C.R.; Michel, C.C.; McDonald, D.M. Endothelial gaps: time course of formation and closure in inflamed venules of rats. American Journal of Physiology-Lung Cellular and Molecular Physiology 1997, 272, L155–L170. [Google Scholar] [CrossRef]
- Bowden, J.J.; Baluk, P.; Lefevre, P.M.; Vigna, S.R.; McDonald, D.M. Substance P (NK1) receptor immunoreactivity on endothelial cells of the rat tracheal mucosa. Am J Physiol 1996, 270, L404–414. [Google Scholar] [CrossRef]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell Mol Life Sci 2016, 73, 4249–4264. [Google Scholar] [CrossRef]
- Leroux, A.; Paiva Dos Santos, B.; Leng, J.; Oliveira, H.; Amedee, J. Sensory neurons from dorsal root ganglia regulate endothelial cell function in extracellular matrix remodelling. Cell Commun Signal 2020, 18, 162. [Google Scholar] [CrossRef]
- Nessler, S.; Stadelmann, C.; Bittner, A.; Schlegel, K.; Gronen, F.; Brueck, W.; Hemmer, B.; Sommer, N. Suppression of autoimmune encephalomyelitis by a neurokinin-1 receptor antagonist--a putative role for substance P in CNS inflammation. J Neuroimmunol 2006, 179, 1–8. [Google Scholar] [CrossRef]
- Foreman, J.C.; Jordan, C.C.; Oehme, P.; Renner, H. Structure-activity relationships for some substance P-related peptides that cause wheal and flare reactions in human skin. J Physiol 1983, 335, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, P. Mast Cells in Neuroimmune Interactions. Trends Neurosci 2019, 42, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Suvas, S. Role of Substance P Neuropeptide in Inflammation, Wound Healing, and Tissue Homeostasis. J Immunol 2017, 199, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol (1985) 2006, 100, 1059–1064. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies - successful translation from bench to clinic. Nat Rev Neurol 2018, 14, 338–350. [Google Scholar] [CrossRef]
- Markowitz, S.; Saito, K.; Moskowitz, M.A. Neurogenically mediated leakage of plasma protein occurs from blood vessels in dura mater but not brain. J Neurosci 1987, 7, 4129–4136. [Google Scholar] [CrossRef]
- Ashina, M.; Hansen, J.M.; Do, T.P.; Melo-Carrillo, A.; Burstein, R.; Moskowitz, M.A. Migraine and the trigeminovascular system-40 years and counting. Lancet Neurol 2019, 18, 795–804. [Google Scholar] [CrossRef]
- Saria, A.; Lundberg, J.M.; Skofitsch, G.; Lembeck, F. Vascular protein linkage in various tissue induced by substance P, capsaicin, bradykinin, serotonin, histamine and by antigen challenge. Naunyn Schmiedebergs Arch Pharmacol 1983, 324, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Hokfelt, T.; Kellerth, J.O.; Nilsson, G.; Pernow, B. Substance p: localization in the central nervous system and in some primary sensory neurons. Science 1975, 190, 889–890. [Google Scholar] [CrossRef] [PubMed]
- Mantyh, P.W.; Johnson, D.J.; Boehmer, C.G.; Catton, M.D.; Vinters, H.V.; Maggio, J.E.; Too, H.P.; Vigna, S.R. Substance P receptor binding sites are expressed by glia in vivo after neuronal injury. Proc Natl Acad Sci U S A 1989, 86, 5193–5197. [Google Scholar] [CrossRef]
- Johnson, M.B.; Young, A.D.; Marriott, I. The Therapeutic Potential of Targeting Substance P/NK-1R Interactions in Inflammatory CNS Disorders. Front Cell Neurosci 2016, 10, 296. [Google Scholar] [CrossRef]
- Mai, J.K.; Stephens, P.H.; Hopf, A.; Cuello, A.C. Substance P in the human brain. Neuroscience 1986, 17, 709–739. [Google Scholar] [CrossRef] [PubMed]
- Mantyh, P.W. Neurobiology of substance P and the NK1 receptor. J Clin Psychiatry 2002, 63 Suppl 11, 6–10. [Google Scholar]
- Dudas, B.; Merchenthaler, I. Substance P-Immunoreactive Fiber Varicosities Appear to Innervate Galaninergic Perikarya in the Human Hypothalamus. Brain Connect 2021, 11, 493–500. [Google Scholar] [CrossRef]
- Liu, H.; Brown, J.L.; Jasmin, L.; Maggio, J.E.; Vigna, S.R.; Mantyh, P.W.; Basbaum, A.I. Synaptic relationship between substance P and the substance P receptor: light and electron microscopic characterization of the mismatch between neuropeptides and their receptors. Proc Natl Acad Sci U S A 1994, 91, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, K.; Siddiq, A.; Baig, S.G.; Zehra, S. Substance P: A neuropeptide involved in the psychopathology of anxiety disorders. Neuropeptides 2020, 79, 101993. [Google Scholar] [CrossRef] [PubMed]
- Rupniak, N.M.J.; Kramer, M.S. NK1 receptor antagonists for depression: Why a validated concept was abandoned. J Affect Disord 2017, 223, 121–125. [Google Scholar] [CrossRef]
- Frick, A.; Ahs, F.; Linnman, C.; Jonasson, M.; Appel, L.; Lubberink, M.; Långström, B.; Fredrikson, M.; Furmark, T. Increased neurokinin-1 receptor availability in the amygdala in social anxiety disorder: a positron emission tomography study with [11C]GR205171. Translational Psychiatry 2015, 5, e597–e597. [Google Scholar] [CrossRef]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O'Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med 2013, 11, 200. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, F.D.; Rycroft, W.; Hill, R.G.; Hargreaves, R.J. Enantioselective inhibition of apomorphine-induced emesis in the ferret by the neurokinin1 receptor antagonist CP-99,994. Neuropharmacology 1994, 33, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Aapro, M.; Carides, A.; Rapoport, B.L.; Schmoll, H.J.; Zhang, L.; Warr, D. Aprepitant and fosaprepitant: a 10-year review of efficacy and safety. Oncologist 2015, 20, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Takano, Y.; Kamiya, H.O. Roles of substance P and NK(1) receptor in the brainstem in the development of emesis. J Pharmacol Sci 2003, 91, 87–94. [Google Scholar] [CrossRef]
- Burmeister, A.R.; Johnson, M.B.; Chauhan, V.S.; Moerdyk-Schauwecker, M.J.; Young, A.D.; Cooley, I.D.; Martinez, A.N.; Ramesh, G.; Philipp, M.T.; Marriott, I. Human microglia and astrocytes constitutively express the neurokinin-1 receptor and functionally respond to substance P. Journal of Neuroinflammation 2017, 14, 245. [Google Scholar] [CrossRef]
- Spitsin, S.; Pappa, V.; Douglas, S.D. Truncation of neurokinin-1 receptor-Negative regulation of substance P signaling. J Leukoc Biol 2018. [Google Scholar] [CrossRef]
- Lai, J.P.; Lai, S.; Tuluc, F.; Tansky, M.F.; Kilpatrick, L.E.; Leeman, S.E.; Douglas, S.D. Differences in the length of the carboxyl terminus mediate functional properties of neurokinin-1 receptor. Proc Natl Acad Sci U S A 2008, 105, 12605–12610. [Google Scholar] [CrossRef]
- Lai, J.P.; Cnaan, A.; Zhao, H.; Douglas, S.D. Detection of full-length and truncated neurokinin-1 receptor mRNA expression in human brain regions. J Neurosci Methods 2008, 168, 127–133. [Google Scholar] [CrossRef]
- Tuluc, F.; Lai, J.P.; Kilpatrick, L.E.; Evans, D.L.; Douglas, S.D. Neurokinin 1 receptor isoforms and the control of innate immunity. Trends Immunol 2009, 30, 271–276. [Google Scholar] [CrossRef]
- Stumm, R.; Culmsee, C.; Schafer, M.K.; Krieglstein, J.; Weihe, E. Adaptive plasticity in tachykinin and tachykinin receptor expression after focal cerebral ischemia is differentially linked to gabaergic and glutamatergic cerebrocortical circuits and cerebrovenular endothelium. J Neurosci 2001, 21, 798–811. [Google Scholar] [CrossRef]
- Cyrino, L.A.; Cardoso, R.C.; Hackl, L.P.; Nicolau, M. Effect of quercetin on plasma extravasation in rat CNS and dura mater by ACE and NEP inhibition. Phytother Res 2002, 16, 545–549. [Google Scholar] [CrossRef]
- Annunziata, P.; Cioni, C.; Toneatto, S.; Paccagnini, E. HIV-1 gp120 increases the permeability of rat brain endothelium cultures by a mechanism involving substance P. AIDS 1998, 12, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Cioni, C.; Renzi, D.; Calabro, A.; Annunziata, P. Enhanced secretion of substance P by cytokine-stimulated rat brain endothelium cultures. J Neuroimmunol 1998, 84, 76–85. [Google Scholar] [CrossRef]
- Marriott, D.R.; Wilkin, G.P. Substance P receptors on O-2A progenitor cells and type-2 astrocytes in vitro. J Neurochem 1993, 61, 826–834. [Google Scholar] [CrossRef]
- Chauhan, V.S.; Sterka, D.G., Jr.; Gray, D.L.; Bost, K.L.; Marriott, I. Neurogenic exacerbation of microglial and astrocyte responses to Neisseria meningitidis and Borrelia burgdorferi. J Immunol 2008, 180, 8241–8249. [Google Scholar] [CrossRef] [PubMed]
- Lieb, K.; Fiebich, B.L.; Berger, M.; Bauer, J.; Schulze-Osthoff, K. The neuropeptide substance P activates transcription factor NF-kappa B and kappa B-dependent gene expression in human astrocytoma cells. J Immunol 1997, 159, 4952–4958. [Google Scholar] [CrossRef] [PubMed]
- Rasley, A.; Bost, K.L.; Olson, J.K.; Miller, S.D.; Marriott, I. Expression of functional NK-1 receptors in murine microglia. Glia 2002, 37, 258–267. [Google Scholar] [CrossRef]
- Vo, T.T.; Im, G.H.; Han, K.; Suh, M.; Drew, P.J.; Kim, S.G. Parvalbumin interneuron activity drives fast inhibition-induced vasoconstriction followed by slow substance P-mediated vasodilation. Proc Natl Acad Sci U S A 2023, 120, e2220777120. [Google Scholar] [CrossRef]
- Vruwink, M.; Schmidt, H.H.; Weinberg, R.J.; Burette, A. Substance P and nitric oxide signaling in cerebral cortex: anatomical evidence for reciprocal signaling between two classes of interneurons. J Comp Neurol 2001, 441, 288–301. [Google Scholar] [CrossRef]
- Guo, C.J.; Douglas, S.D.; Gao, Z.; Wolf, B.A.; Grinspan, J.; Lai, J.P.; Riedel, E.; Ho, W.Z. Interleukin-1beta upregulates functional expression of neurokinin-1 receptor (NK-1R) via NF-kappaB in astrocytes. Glia 2004, 48, 259–266. [Google Scholar] [CrossRef]
- Weinstock, J.V.; Blum, A.; Metwali, A.; Elliott, D.; Arsenescu, R. IL-18 and IL-12 signal through the NF-kappa B pathway to induce NK-1R expression on T cells. J Immunol 2003, 170, 5003–5007. [Google Scholar] [CrossRef]
- Yu, Z.; Cheng, G.; Huang, X.; Li, K.; Cao, X. Neurokinin-1 receptor antagonist SR140333: a novel type of drug to treat cerebral ischemia. Neuroreport 1997, 8, 2117–2119. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Helps, S.C.; Thornton, E.; Vink, R. A substance P antagonist improves outcome when administered 4 h after onset of ischaemic stroke. Brain Res 2011, 1393, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Vink, R. Combined tissue plasminogen activator and an NK1 tachykinin receptor antagonist: an effective treatment for reperfusion injury following acute ischemic stroke in rats. Neuroscience 2012, 220, 1–10. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Leonard, A.V.; Hoving, J.W.; Yassi, N.; Vink, R.; Wells, A.J.; Turner, R.J. NK1-r Antagonist Treatment Comparable to Decompressive Craniectomy in Reducing Intracranial Pressure Following Stroke. Front Neurosci 2019, 13, 681. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Sharma, S.; Sandhu, M.; Sharma, S. Neurokinin-1 receptor inhibition reverses ischaemic brain injury and dementia in bilateral common carotid artery occluded rats: possible mechanisms. Inflammopharmacology 2016, 24, 133–143. [Google Scholar] [CrossRef]
- Donkin, J.J.; Nimmo, A.J.; Cernak, I.; Blumbergs, P.C.; Vink, R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab 2009, 29, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Torsten Hoffmann, A.N., Andrew Sleight, Pierre Vankan, Robert Vink METHOD OF TREATMENT AND/OR PREVENTION OF BRAIN, SPINAL OR NERVE INJURY. 2003, Hoffmann La Roche Inc.
- Corrigan, F.; Cernak, I.; McAteer, K.; Hellewell, S.C.; Rosenfeld, J.V.; Turner, R.J.; Vink, R. NK1 antagonists attenuate tau phosphorylation after blast and repeated concussive injury. Sci Rep 2021, 11, 8861. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, X.; Yang, Y.; Zhang, Y.; He, F.; Xu, X.; Zhang, Z.; Tao, L.; Luo, C. Tachykinin NK1 receptor antagonist L-733,060 and substance P deletion exert neuroprotection through inhibiting oxidative stress and cell death after traumatic brain injury in mice. Int J Biochem Cell Biol 2019, 107, 154–165. [Google Scholar] [CrossRef]
- Vink, R.; Gabrielian, L.; Thornton, E. The Role of Substance P in Secondary Pathophysiology after Traumatic Brain Injury. Front Neurol 2017, 8, 304. [Google Scholar] [CrossRef]
- Zheng, G.; Harms, A.K.; Tail, M.; Zhang, H.; Nimmo, A.; Skutella, T.; Kiening, K.; Unterberg, A.; Zweckberger, K.; Younsi, A. Effects of a neurokinin-1 receptor antagonist in the acute phase after thoracic spinal cord injury in a rat model. Front Mol Neurosci 2023, 16, 1128545. [Google Scholar] [CrossRef]
- Chauhan, V.S.; Kluttz, J.M.; Bost, K.L.; Marriott, I. Prophylactic and therapeutic targeting of the neurokinin-1 receptor limits neuroinflammation in a murine model of pneumococcal meningitis. J Immunol 2011, 186, 7255–7263. [Google Scholar] [CrossRef]
- Rasley, A.; Marriott, I.; Halberstadt, C.R.; Bost, K.L.; Anguita, J. Substance P augments Borrelia burgdorferi-induced prostaglandin E2 production by murine microglia. J Immunol 2004, 172, 5707–5713. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Ho, W.Z.; Zhan, G.X.; Yi, Y.; Collman, R.G.; Douglas, S.D. Substance P antagonist (CP-96,345) inhibits HIV-1 replication in human mononuclear phagocytes. Proc Natl Acad Sci U S A 2001, 98, 3970–3975. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Douglas, S.D.; Song, L.; Wang, Y.J.; Ho, W.Z. Neurokinin-1 receptor antagonist (aprepitant) suppresses HIV-1 infection of microglia/macrophages. J Neuroimmune Pharmacol 2008, 3, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Manak, M.M.; Moshkoff, D.A.; Nguyen, L.T.; Meshki, J.; Tebas, P.; Tuluc, F.; Douglas, S.D. Anti-HIV-1 activity of the neurokinin-1 receptor antagonist aprepitant and synergistic interactions with other antiretrovirals. AIDS 2010, 24, 2789–2796. [Google Scholar] [CrossRef]
- Ronca, S.E.; Gunter, S.M.; Kairis, R.B.; Lino, A.; Romero, J.; Pautler, R.G.; Nimmo, A.; Murray, K.O. A Potential Role for Substance P in West Nile Virus Neuropathogenesis. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.; Rodgers, J.; Jennings, F.W.; Murray, M.; Leeman, S.E.; Burke, J.M. A substance P antagonist, RP-67,580, ameliorates a mouse meningoencephalitic response to Trypanosoma brucei brucei. Proc Natl Acad Sci U S A 1997, 94, 4167–4170. [Google Scholar] [CrossRef]
- MacLeod, A.M.; Merchant, K.J.; Brookfield, F.; Kelleher, F.; Stevenson, G.; Owens, A.P.; Swain, C.J.; Casiceri, M.A.; Sadowski, S.; Ber, E.; et al. Identification of L-tryptophan derivatives with potent and selective antagonist activity at the NK1 receptor. J Med Chem 1994, 37, 1269–1274. [Google Scholar] [CrossRef]
- Matalinska, J.; Lipinski, P.F.J. Correcting a widespread error: Neuroprotectant N-acetyl-L-tryptophan does not bind to the neurokinin-1 receptor. Mol Cell Neurosci 2022, 120, 103728. [Google Scholar] [CrossRef]
- Satarker, S.; Maity, S.; Mudgal, J.; Nampoothiri, M. In silico screening of neurokinin receptor antagonists as a therapeutic strategy for neuroinflammation in Alzheimer's disease. Mol Divers 2022, 26, 443–466. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, A.J.; Vink, R. Recent patents in CNS drug discovery: the management of inflammation in the central nervous system. Recent Pat CNS Drug Discov 2009, 4, 86–95. [Google Scholar] [CrossRef]
- Vinet-Oliphant, H.; Alvarez, X.; Buza, E.; Borda, J.T.; Mohan, M.; Aye, P.P.; Tuluc, F.; Douglas, S.D.; Lackner, A.A. Neurokinin-1 receptor (NK1-R) expression in the brains of SIV-infected rhesus macaques: implications for substance P in NK1-R immune cell trafficking into the CNS. Am J Pathol 2010, 177, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Tega, F.; Bruno, A.; Graf, U.; Corelli, F.; Molfetta, R.; Barucco, M. The role of substance P in cerebral ischemia. Int J Immunopathol Pharmacol 2003, 16, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.; Bös, M.; Stadler, H.; Schnider, P.; Hunkeler, W.; Godel, T.; Galley, G.; Ballard, T.M.; Higgins, G.A.; Poli, S.M.; et al. Design and synthesis of a novel, achiral class of highly potent and selective, orally active neurokinin-1 receptor antagonists. Bioorg Med Chem Lett 2006, 16, 1362–1365. [Google Scholar] [CrossRef]
- Visser, K.; Koggel, M.; Blaauw, J.; van der Horn, H.J.; Jacobs, B.; van der Naalt, J. Blood-based biomarkers of inflammation in mild traumatic brain injury: A systematic review. Neuroscience & Biobehavioral Reviews 2022, 132, 154–168. [Google Scholar]
- Sahuquillo, J.; Dennis, J.A. Decompressive craniectomy for the treatment of high intracranial pressure in closed traumatic brain injury. Cochrane Database Syst Rev 2019, 12, Cd003983. [Google Scholar] [CrossRef]
- Corrigan, F.; Mander, K.A.; Leonard, A.V.; Vink, R. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. J Neuroinflammation 2016, 13, 264. [Google Scholar] [CrossRef] [PubMed]
- Hellenbrand, D.J.; Quinn, C.M.; Piper, Z.J.; Morehouse, C.N.; Fixel, J.A.; Hanna, A.S. Inflammation after spinal cord injury: a review of the critical timeline of signaling cues and cellular infiltration. J Neuroinflammation 2021, 18, 284. [Google Scholar] [CrossRef]
- Leonard, A.V.; Manavis, J.; Blumbergs, P.C.; Vink, R. Changes in substance P and NK1 receptor immunohistochemistry following human spinal cord injury. Spinal Cord 2014, 52, 17–23. [Google Scholar] [CrossRef]
- Leonard, A.V.; Thornton, E.; Vink, R. NK1 receptor blockade is ineffective in improving outcome following a balloon compression model of spinal cord injury. PLoS One 2014, 9, e98364. [Google Scholar] [CrossRef]
- Bystritsky, R.J.; Chow, F.C. Infectious Meningitis and Encephalitis. Neurol Clin 2022, 40, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.D.; Cnaan, A.; Lynch, K.G.; Benton, T.; Zhao, H.; Gettes, D.R.; Evans, D.L. Elevated substance P levels in HIV-infected women in comparison to HIV-negative women. AIDS Res Hum Retroviruses 2008, 24, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.D.; Ho, W.Z.; Gettes, D.R.; Cnaan, A.; Zhao, H.; Leserman, J.; Petitto, J.M.; Golden, R.N.; Evans, D.L. Elevated substance P levels in HIV-infected men. AIDS 2001, 15, 2043–2045. [Google Scholar] [CrossRef] [PubMed]
- Bost, K.L. Tachykinin-modulated anti-viral responses. Front Biosci 2004, 9, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Spitsin, S.V.; Meshki, J.; Tuluc, F.; Douglas, S.D.; Wolfe, J.H. Substance P enhances HIV-1 infection in human fetal brain cell cultures expressing full-length neurokinin-1 receptor. J Neurovirol 2013, 19, 219–227. [Google Scholar] [CrossRef]
- Robinson, P.; White, A.C.; Lewis, D.E.; Thornby, J.; David, E.; Weinstock, J. Sequential expression of the neuropeptides substance P and somatostatin in granulomas associated with murine cysticercosis. Infect Immun 2002, 70, 4534–4538. [Google Scholar] [CrossRef]
- Garza, A.; Tweardy, D.J.; Weinstock, J.; Viswanathan, B.; Robinson, P. Substance P signaling contributes to granuloma formation in Taenia crassiceps infection, a murine model of cysticercosis. J Biomed Biotechnol 2010, 2010, 597086. [Google Scholar] [CrossRef]
- Garza, A.; Weinstock, J.; Robinson, P. Absence of the SP/SP receptor circuitry in the substance P-precursor knockout mice or SP receptor, neurokinin (NK)1 knockout mice leads to an inhibited cytokine response in granulomas associated with murine Taenia crassiceps infection. J Parasitol 2008, 94, 1253–1258. [Google Scholar] [CrossRef]
- Elsawa, S.F.; Taylor, W.; Petty, C.C.; Marriott, I.; Weinstock, J.V.; Bost, K.L. Reduced CTL response and increased viral burden in substance P receptor-deficient mice infected with murine gamma-herpesvirus 68. J Immunol 2003, 170, 2605–2612. [Google Scholar] [CrossRef]
- Blum, A.M.; Metwali, A.; Kim-Miller, M.; Li, J.; Qadir, K.; Elliott, D.E.; Lu, B.; Fabry, Z.; Gerard, N.; Weinstock, J.V. The substance P receptor is necessary for a normal granulomatous response in murine schistosomiasis mansoni. J Immunol 1999, 162, 6080–6085. [Google Scholar] [CrossRef]
- Blum, A.M.; Elliott, D.E.; Metwali, A.; Li, J.; Qadir, K.; Weinstock, J.V. Substance P regulates somatostatin expression in inflammation. J Immunol 1998, 161, 6316–6322. [Google Scholar] [CrossRef]
- Kincy-Cain, T.; Bost, K.L. Increased susceptibility of mice to Salmonella infection following in vivo treatment with the substance P antagonist, spantide II. J Immunol 1996, 157, 255–264. [Google Scholar] [CrossRef]
- de Oliveira, A.C.; Candelario-Jalil, E.; Fiebich, B.L.; Santos Mda, S.; Palotas, A.; dos Reis, H.J. Neuroinflammation and Neurodegeneration: Pinpointing Pathological and Pharmacological Targets. Biomed Res Int 2015, 2015, 487241. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M.; Moore, Z.; Minter, M.R.; Crack, P.J. Type-I interferon pathway in neuroinflammation and neurodegeneration: focus on Alzheimer's disease. J Neural Transm (Vienna) 2018, 125, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Arena, G.; Sharma, K.; Agyeah, G.; Kruger, R.; Grunewald, A.; Fitzgerald, J.C. Neurodegeneration and Neuroinflammation in Parkinson's Disease: a Self-Sustained Loop. Curr Neurol Neurosci Rep 2022, 22, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Ellwardt, E.; Zipp, F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp Neurol 2014, 262 Pt A, 8–17. [Google Scholar] [CrossRef]
- Campolongo, P.; Ratano, P.; Ciotti, M.T.; Florenzano, F.; Nori, S.L.; Marolda, R.; Palmery, M.; Rinaldi, A.M.; Zona, C.; Possenti, R.; et al. Systemic administration of substance P recovers beta amyloid-induced cognitive deficits in rat: involvement of Kv potassium channels. PLoS One 2013, 8, e78036. [Google Scholar] [CrossRef]
- Benarroch, E.E.; Schmeichel, A.M.; Low, P.A.; Parisi, J.E. Depletion of ventromedullary NK-1 receptor-immunoreactive neurons in multiple system atrophy. Brain 2003, 126, 2183–2190. [Google Scholar] [CrossRef]
- Reinke, E.K.; Johnson, M.J.; Ling, C.; Karman, J.; Lee, J.; Weinstock, J.V.; Sandor, M.; Fabry, Z. Substance P receptor mediated maintenance of chronic inflammation in EAE. J Neuroimmunol 2006, 180, 117–125. [Google Scholar] [CrossRef]
- Stuckey, S.M.; Ong, L.K.; Collins-Praino, L.E.; Turner, R.J. Neuroinflammation as a Key Driver of Secondary Neurodegeneration Following Stroke? Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The expression of substance P and the NK1 reception in cells associated with the neurovascular unit.
Figure 1.
The expression of substance P and the NK1 reception in cells associated with the neurovascular unit.
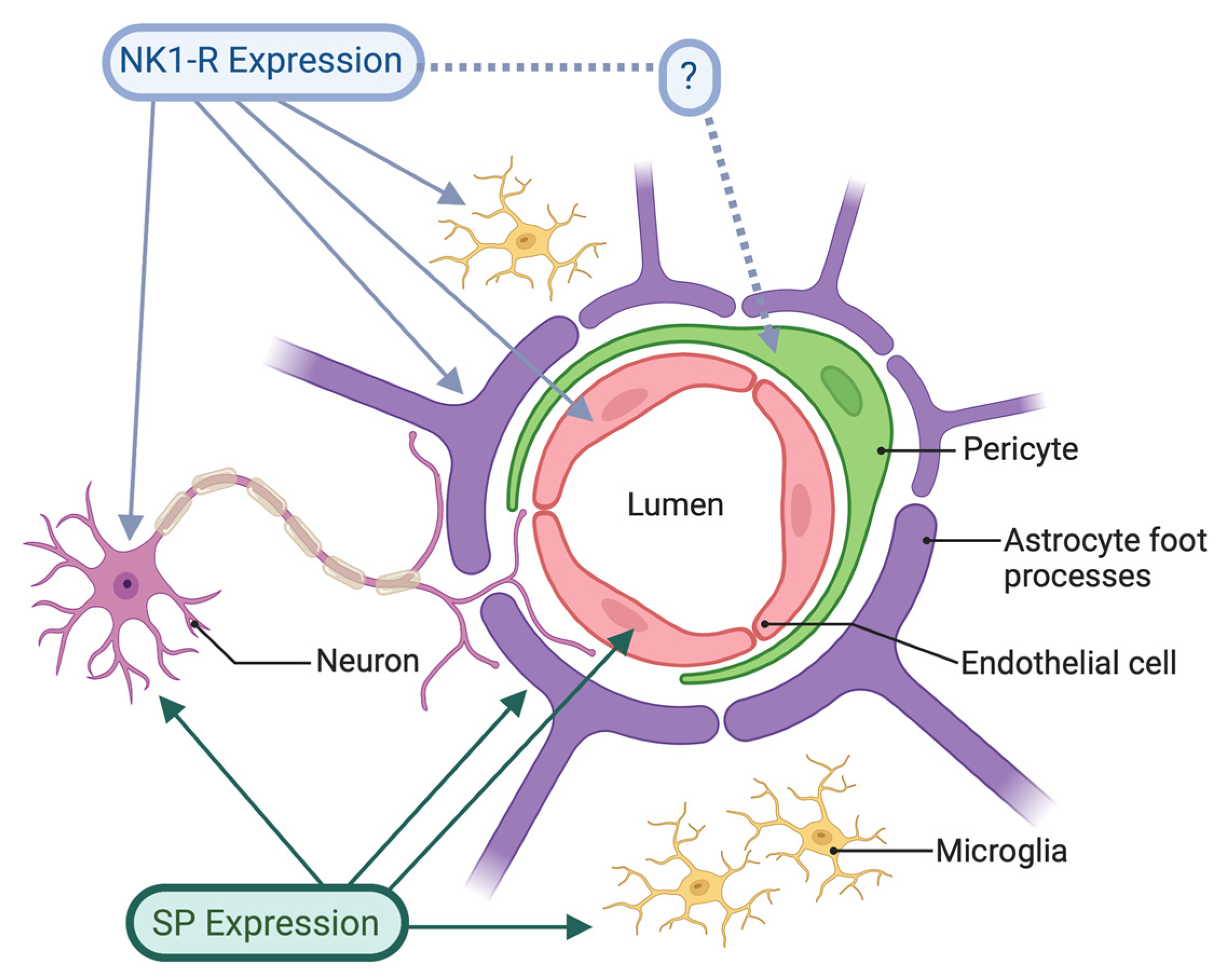

Table 1.
In vivo and cellular models where selective NK1 receptor antagonists or NAT have been shown to ameliorate neuroinflammation and its associated consequences.
Table 1.
In vivo and cellular models where selective NK1 receptor antagonists or NAT have been shown to ameliorate neuroinflammation and its associated consequences.
| CNS Pathology | Model | Drug | Reference |
|
Ischaemic Stroke |
Rat – internal carotid A Rat – transient middle cerebral (tMC) Rat – t MC + tPA Sheep – t MC Rat – bilateral common carotid |
SR140333 NAT NAT EU-C-001 Aprepitant |
Yu et al [82] Turner et al [83] Turner et al [84] Sorby-Adams et al [85] Kaur et al [86] |
|
Traumatic Brain Injury |
Rat – diffuse axonal injury Rat – diffuse axonal injury Rat – diffuse axonal injury Rat – Blast injury and CTE Mouse – Cortical impact Sheep – Diffuse axonal injury |
Capsaicin NAT EU-C-001 EU-C-001 L-733,060 EU-C-001 |
Nimmo et al [25] Donkin et al [87] Hoffmann et al [88] Corrigan et al [89] Li et al [90] Vink et al [91] |
|
Spinal Cord Injury |
Rat - contusion/compression SCI | EU-C-001 | Zheng et al [92] |
|
Bacterial Infection |
Mouse – N meningitidis & B burgdorferi Mouse – S pneumoniae Primary mouse microglia - B burgdorferi |
L 703,606 L 703,606 L 703,606 |
Chauhan et al [75] Chauhan et al [93] Rasley et al [94] |
|
Viral Infection |
Human mononuclear phagocytes – HIV Microglia/macrophages - HIV Peripheral blood monocytes – HIV Mouse – WNV neuroinvasive disease |
CP-96,345 Aprepitant Aprepitant Ro 67,5930 |
Lai et al [95] Wang et al [96] Manak et al [97] Ronca et al [98] |
|
Parasitic Infection |
Mouse – Trypanosoma brucei brucei | RP 67,580 | Kennedy et al [99] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



