
Submitted:
06 September 2023
Posted:
07 September 2023
You are already at the latest version
Abstract
Plant-derived diterpene synthases (PdiTPSs) play a critical role in the formation of structurally and functionally diverse diterpenoids. However, the specificity or promiscuity of PdiTPSs remains unclear. In order to gain more understanding of this, the sequences of 199 functionally characterized PdiTPSs and their corresponding 3D structures were collected and manually corrected. Then, the correlations among sequences, domains, structures and their corresponding products were comprehensively analyzed. However, those features alone was insufficient for ef-fective product-specific classification of PdiTPSs as these methods could not establish a clear mapping between the enzymes and products. Nevertheless, local structural analysis can identify residues that have been experimentally proven to influence product outcomes through mutagenesis, and these residues exhibit conservation in spatial positioning and physicochemical properties. And aromatic residues surrounding the substrate exhibited selectivity towards its chemical structure. Specifically, tryptophan (W) was preferentially located around the linear substrate geranylgeranyl pyrophosphate (GGPP), while phenylalanine (F) and tyrosine (Y) were preferentially located around the initial cyclized diterpene intermediate. This analysis revealed the functional space of residues surrounding the substrate of PdiTPSs, most of which have not been experimentally explored. These findings provide guidance for screening specific residues for mutation studies to change the catalytic products of PdiTPSs.
Keywords:
plant diterpene synthases
; functional annotation dataset
; product specificity analysis
1. Introduction
Diterpenoids are a class of widely distributed C20 isoprenoids of natural products, with more than 18,000 members identified in plants [1]. They play an important role in plant growth, development [2] and mediate complex plant-environment interactions [3]. They also have applications in medicine, flavor, and food industries [4,5,6,7]. All the discovered diterpenoids can be classified according to their core diterpene skeletons by removing all heteroatoms, stereocenters, and reducing unsaturated structures [1,8].
Diterpenoids are highly diversified and complex compounds derived from 5-carbon building blocks isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). GGPP synthase catalyzes the coupling of IPP and DMAPP in a processive head-to-tail fashion to generate linear hydrocarbon molecules. Then, diterpene synthases (diTPS) and cytochrome P450 monooxygenases (P450s) are responsible for synthesizing a variety of intermediates and modifying skeletons [2,9,10,11]. Particularly, diTPS catalyze remarkably complex cyclization cascades with structural and stereochemical precision and create chemical library of 20-carbon hydrocarbons. Based on the reaction mechanism, diTPS either employ ionization-induced carbocation formation (diTPS I), protonation-induced carbocation formation (diTPS II), or use both mechanisms by bifunctional enzymes (diTPS I/II) [12]. These diTPS make a significant contribution in synthesizing diverse range of diterpenoid skeletons. However, the limited knowledge on enzyme-substrate recognition and product distribution of diTPS hinder the identification of novel functional diTPS.
Classic multiple sequence alignment methods have been used to identify the functional motifs of diTPS. The identified functional motifs include DXDD [13], DDXXD [14,15,16], NSE\DTE [17], PIX [18] and LHS...PNV [19,20,21]. By examining the structure-function relationship of Selaginella moellendorffii miltiradiene synthase (SmMDS), specific residues around the substrate responsible for product specificity, such as E690, S717, and H721, were identified [22]. Structural analysis and catalytic mechanism also suggest that the cavity formed by the substrate surrounding residues can selectively choose the substrate [23]. Product-changing mutational studies and structural analysis provide valuable insights for investigating PdiTPSs function. However, these researches have only covered a small fraction of the characterized PdiTPSs. Furthermore, as of now, no research has comprehensively investigated the correlation between product of PdiTPSs and their sequence and structural features.
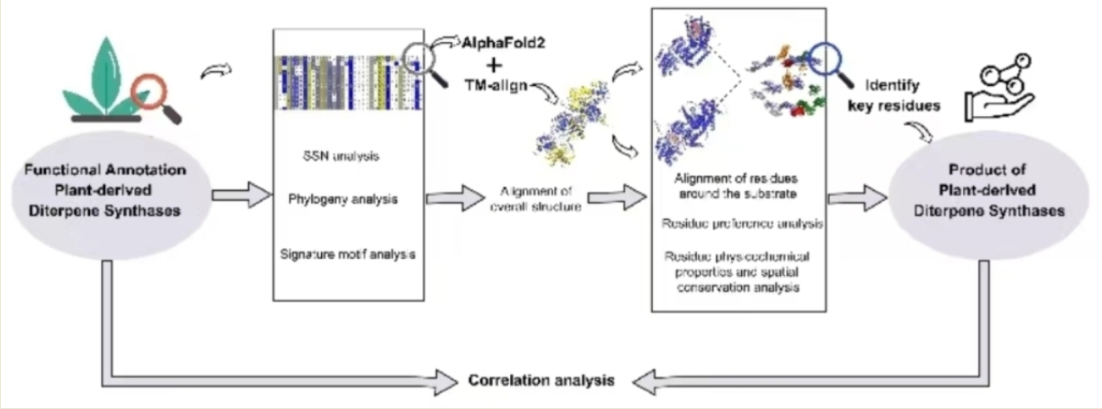
In this study, a manually curated and annotated database has been utilized to investigate the partitioning of PdiTPSs functions using SSN and phylogenetic tree analysis. The correlations were examined among various factors, including overall sequences, subsequences, overall structures, residues around the substrate, and product similarity. We counted the residue preferences surrounding the substrate and analyzed their spatial conservation to determine the range of residues that significantly affect substrate type and product outcome. The results of this comprehensive analysis will provide valuable insights into exploring product-specific residues in PdiTPSs and the mapping patterns between PdiTPSs and their functions.
2. Results and discussion
2.1. Overview of functional annotations of PdiTPSs
A manually curated database of 199 functional characterized PdiTPSs has been presented, including 27 bifunctional enzymes, 90 class I enzymes, and 82 class II enzymes (Table S1). These PdiTPSs were derived from 69 plant species belonging to 26 families and 52 genera, producing 16 diterpene intermediates and 63 diterpene precursors. Of these products, only a small fraction was found to be associated with multiple PdiTPSs, while the majority of products were primarily catalyzed by a single PdiTPSs, which affected the product-specific analysis.
To solve this problem, the existing terpenoid skeleton classification system [24] was employed to group these products into 16 different types. Products from PdiTPS I and PdiTPS I/II were classified into 15 skeleton types, while those from PdiTPS II were classified into only one single type (Table S1). The classification scheme allowed us to group multiple products from a single enzyme into the same category, such as SsSS synthase from Salvia sclarea [5], which could catalyze the dephosphorylation and minor rearrangement of 9 diterpene intermediates to produce 11 diterpene precursors, all of which fall into the same Labdane scaffold (SK1). However, this classification system is not always effective, for example, TrTPS13 from Tripterygium regelii produces five products belonging to SK4, SK5, and SK9 scaffolds (ntkrn, sndarpardn, ipsfdn, spsfdn, sdmon), and PdiTPS from Grindelia hirsutula produces three products (abedn, epmnlo, mnlo) belonging to SK1 and SK3 scaffolds, respectively. Therefore, in this study, we also attempt to explore the correlation between PdiTPSs in terms of sequence and structure with substrates or products’ similarity.
2.2. The sequence similarity network generates PdiTPSs clusters
The results of applying a skeleton classification system for functional mapping in SSN analysis was examined. This all-pairs local sequence-based comparison method could rapidly generate a network of nodes and edges using any expectation value (E) as a threshold. By appending annotation information, the sequence-function relationship profile of the enzyme could be quickly viewed.
Our results suggested that C-terminal, N-terminal, NC terminal subsequences and overall sequences could be used to classify the PdiTPS I and PdiTPS II. In general, the N-terminal (Figure 1a), C-terminal (Figure 1b) and NC-terminal (Figure 1c) networks generated by SSN resulted in mainly multiple backbone clustering. In particular, SK1, SK3, and SK4 skeletons were often clustered together, despite the clear differences in their product structures. In contrast, the product skeleton clusters obtained from multiple sequence comparison of N-terminal subsequences were more refined, with fewer outliers.
Multiple sequence comparisons also revealed that PdiTPSs from different species were clustered together, which might limit the grouping of PdiTPSs by product type. Additionally, it could be observed that products belonging to the SK1, SK3, SK4, and SK5 skeletons are predominantly found in early diverging plant lineages such as ferns and mosses (Figure 1). These skeletons, including labdane (SK1), abietane (SK3), pimarane (SK4), and kaurane (SK5), are the most abundant and widely distributed [25]. SSN analysis yielded additional insights into the relationship between product backbone and enzyme sequence. We find the SK1 and SK5 backbones serve as crucial linkage points for other backbone groups (Figure 1c). It is important to note that while the SK1 and SK5 skeletons may serve as connection points for other skeleton clusters, this does not necessarily imply that they are the fundamental skeletons driving the evolution and diversification of diterpene synthase products. Further experimental research is needed to confirm this.
2.3. Phylogenetic analysis of PdiTPSs
Multiple sequence alignment and evolutionary information in phylogenetic analysis can be used for comparing protein homology, providing insight into protein sequences, domains and motifs, specific conserved sites evolution, and functional variation. The phylogenetic tree of PdiTPSs was constructed to detect evolutionary relationships and identify lineages with similar features. By examining the changes in product skeleton, it could be able to identify potential correlations between product and PdiTPSs mapping. Terpene synthases commonly contain two conserved structural domains, the N-terminal and C-terminal domains. Therefore, it was also constructed for the phylogenetic trees for N-terminal, and C-terminal subsequences.
Unfortunately, the phylogenetic tree did not provide a clear division of PdiTPSs based on their functions. However, it has been found that SK1, SK2, SK5, SK4, and PdiTPS II-products were frequently present in the early diverging PdiTPSs products. While this phylogenetic tree exclusively illustrates the gene’s evolutionary relationships, similar patterns are also evident in the phylogenetic trees of the overall sequences (Figure 2) as well as the N-terminal (Figure S1a) and C-terminal (Figure S1b) domains. On the other hand, SK6, SK7, SK8, and SK9 were found in the late-emerging PdiTPSs products. It could also be observed for the trend of PdiTPSs product functions evolving towards multi-ring skeletons from the major branches of the trees. Moreover, the PdiTPSs that produced the SK10, SK11, SK12, SK13, and SK14 skeletons showed shorter evolutionary distances from the ancestral PdiTPSs. This provides valuable insights into how the function and evolution of PdiTPSs may contribute to species-specific adaptations to unique ecological niches. However, additional investigations are necessary to further explore the distribution patterns of diterpenoid compound types and their relationship with the evolutionary status of plants. Similar research has been carried out to investigate the distribution of terpenoid compounds and their biosynthetic pathways in various species of Isodon plants [26].
Our analysis of the domain composition of PdiTPSs showed that the γβα triple-domain structure and βα bi-domain structure alternately appeared in the phylogenetic tree, indicating a phenomenon of continual loss and acquisition of structural domain subsequences during the evolution of terpene synthases. In addition, the bi-domain βα structure was only found in the angiosperms (Figure 2), which was generated by the loss of the γ domain in ancestral terpene synthases that had the γβα structure [27,28].
The LHS and PNV motifs (Figure 2) were CPS-specific motifs [19], as confirmed by our computational analysis. These two motifs were conserved in PdiTPS II, but had undergone mutations in PdiTPS I. The histidine (H) residue in the FEHXW motif exerted cooperative GGPP/Mg2+ inhibition on CPS [29], but histidine was not always conserved in the FEHXW motif of PdiTPS II. Although the function of aromatic amino acids in this motif remained unclear, it had been observed in PdiTPS I that these residues were no longer predominantly composed of aromatic amino acids in this motif, but rather of aliphatic and uncharged amino acids. The PIX motif (Figure 2) displayed was related to ent-kaurene synthesis [18], and was lost in the PdiTPSs of angiosperms that produced primarily SK1 and SK3, as well as in that of polycyclic skeleton SK10, SK11, and SK13. This motif was present in the PdiTPSs of mosses that produce SK1 and SK3, but had undergone mutations. This indicates that there may be potential product-specific motifs in PdiTPSs, which have evolved through deletions and mutations that have resulted in enzyme sequences acquired new function. Therefore, motifs that are different from other PdiTPSs nd absent in other PdiTPSs may help uncover product-specific motifs.
2.4. Identify conserved and diverse subsequences via sequence similarity analysis
The sequence similarity features of PdiTPSs were examined, where the larger the upper quartile and lower quartile in the box plot, the higher the sequence similarity. Statistical analysis (Figure 3a) showed that the C-terminal was more conserved in PdiTPS I, while the N-terminal was more conserved in PdiTPS II. The main differences in sequence similarity between PdiTPS I and PdiTPS II were located in the C-terminal (Figure 3a), suggesting that the use of C-terminal subsequences might facilitate the divergence. The similarity distribution of PdiTPS I and PdiTPS I/II sequences was lower in the NC-terminus and overall regions than that of in the C- and N-terminal subsequences, while the similarity distribution of PdiTPS II and PdiTPS I/II sequences was lower in the C-terminus than that of in the N-, NC-, and overall regions (Figure 3a). Therefore, when distinguishing between PdiTPS I and PdiTPS I/II, it was necessary to consider the differences between the NC-terminus and overall sequences, while differences in the C-terminal subsequence might be helpful for distinguishing PdiTPS II from PdiTPS I/II. Additionally, it should be noted that PdiTPS I/II was relatively conserved in the N-, C-, NC-, and overall sequences, with sequence similarities mostly above 50%, especially in the N-terminal subsequence, which exhibited the highest fourth quartile of sequence similarity values. Thus, the N-terminal subsequence might be useful for clustering PdiTPS I/II.
After obtaining the basic conservation features of the PdiTPSs sequences, it had been comparatively analyzed for the sequence similarity in accepting the same or different substrates and producing the same or different products. Theoretically, the sequence similarity that recognize the same substrates or produce the same products should be higher than those that recognize different substrates or produce different products. The results showed that the N-terminal subsequence had the highest upper and lower quartiles of sequence similarity in identifying the same substrate and producing the same product, while the C-terminal subsequence had the lowest lower quartile of sequence similarity in identifying different substrates and producing different products. Additionally, the C-terminal subsequence had the fewest 100% sequence similarity values in identifying different substrates and producing different products (Figure 4a,b).
Similar results have been reported in the study of sesquiterpene synthases, where the phylogenetic tree constructed using the C-terminal subsequence can group enzymes based on their product types, and the addition of the N-terminal subsequence has little effect on the topological structure of tree [30]. This is also reflected in our SSN analysis, where the C-terminal subsequence generated more different clusters and distant outliers for different types of products than the N-terminal subsequence at the same threshold (Figure 1a,b).
2.5. PdiTPSs have a conservative structure and a flexible surrounding residue topology
Through sequence analysis, some clues have been obtained to predict the substrates and products of PdiTPSs, but more evidence still is needed to help us to understand product specificity. Therefore, it needs to further examine the structure of PdiTPSs and their residues around the substrates. AlphaFold2 was applied to help rebuild the structures of all collected PdiTPSs. Most amino acid residues had a high credible score for pLDDT, indicating that the predicted structures were highly accurate and close to X-ray resolutions.
It had been calculated for the distribution of topological similarity of the overall structure (Figure 3a) and the TM scores for topological similarity were mostly greater than 0.75. Furthermore, the median TM score of the overall structure when recognizing the same substrate and different substrates (Figure 4c), as well as producing the same and different products (Figure 4d), were also greater than 0.75 and closer to the upper quartile (Q3). These results suggested that PdiTPSs that perform different functions shared a similar TPS fold. Here, only the superimposed results of the representative structures of different types of PdiTPSs at the median (Q2) of TM score were shown (Figure 3b). Supplementary Figure S2 showed the structural superposition results of the representative structures of different types of structures representing PdiTPSs in Figures 4 with 2 extreme values and 3 quartiles of TM score. In addition, as the selected residue range around the substrate increased, the TM score also increased, indicating that the difference of topological structure formed by residues closer to the substrate was greater, while the conservation of the topological structure formed by residues further away from the substrate increased, but the increasing trend became flat (Figure 4c,d).
Both the structure formed by substrate-surrounding residues and the overall structure were significantly higher distributed in the TM score of the same substrate or same product than those of the different substrates and different products (Figure 4c,d). This trend was more significant than the overall difference trend contributed by sequence similarity (Figure 4a,b). Similar to the evaluation of sequence similarity, it was expected for the TM score between substrate-surrounding residues and overall structures that recognized the same substrate or produced the same product to be TM score > 0.5, and TM score of those that recognized different substrates and produced different products to be TM score < 0.5 in the analysis of structures. Therefore, the topological structures formed by residues within 6Å of the substrate appeared to be the best choice for determining substrate and product similarity.
2.6. N-terminal subsequence strongly correlates with overall sequence similarity
The above results have provided some insights for the determination of product types at both the sequence and structure levels. Hence, it would be interested to further explore methods for quantitative assessment the relationship between PdiTPSs sequences, structures and products. The correlation was evaluated using Pearson’s correlation coefficient (PCC) [31], with only the final coefficient shown here. The statistical results showed that the similarity between the C-terminal subsequence and the overall sequence (PCC = 0.46, p < 0.001) was significantly weaker than that between the N-terminal subsequence (PCC = 0.91, p < 0.001). And there was almost no correlation between the C-terminal and N-terminal subsequences (PCC = 0.26, p < 0.001).
The weak correlation of sequence similarity between these two domains indicates that their contributions to PdiTPSs specificity are indeed significantly different. However, the phylogenetic tree structure of C- terminal subsequence, N- terminal subsequence and full-length sequence is similar. Another noteworthy observation is that the sequence similarity between the N-terminal subsequence and the full-length sequence is highly correlated, while the C-terminal subsequence is not related to either the N- terminal or the full-length sequence. A study has suggested that the N-terminal subsequence has maintained conservation during evolution [32]. In contrast, the C-terminal subsequence likely to underwent functional selection and evolved at a faster rate to acquire new functions to adapt to the environment. Clues to this can be found in our statistics of the sequence divergence of the C-terminal subsequences than the N-terminal subsequences.
2.7. Substrate-surrounding residue topology in PdiTPSs is independent of the overall structure
The analysis based on sequence features provide limited insights into substrate recognition and functional diversification of PdiTPSs. Protein structures provide a higher resolution platform for understanding function, but acquiring protein structures is expensive and difficult. Therefore, the crystal structures of diterpene synthases are also limited, and the emergence of AlphaFold2 and its high accuracy is exciting, as it has been applied to understand the mechanisms of enzyme [33]. AlphaFold2 has been applied to obtain structural data for the PdiTPSs in this study. Blind docking using CB-Dock2 can be used to study the binding properties and molecular mechanisms between protein and substrate, to reveal key residues that are functionally relevant in the binding pocket [34,35,36]. Based on the structural data, it has been observed that the variable arrangement of the γ, β, and α domains in PdiTPSs (Figure 2) is an important strategy for expanding and diversifying diterpene synthases. Their combination, presence, and absence constitute the structural chemistry of diterpene synthases [32,37,38].
In addition, the correlation analysis showed that the similarity of overall structures increased with the similarity of overall sequences (PCC = 0.78, p < 0.001). The N-terminal subsequence showed high correlation with the overall sequence and moderate correlation with the TM score of the overall structure (PCC = 0.69, p < 0.001). Conversely, the C-terminal subsequence differed significantly from the N-terminal and overall sequences, and exhibited weak correlation with the overall structure (PCC = 0.33, p < 0.001). When analyzing the correlation between residues surrounding the substrate and the overall structure, the trend was completely opposite (Table S5). The average correlation between the C-terminal subsequence (0.54) and residues around the substrate was higher than that between the N-terminal subsequence (0.37). Combining the N- and C-terminal subsequence as the NC-terminal subsequence increased the average correlation with residues around the substrate to 0.58, which is understandable because both N- and C-terminal subsequences contain residues around the substrate. Furthermore, there was no correlation between residues around the substrate and the topology of the overall structure, as evidenced by the TM score distribution of residues around the substrate, which was significantly lower than that of the overall structure (Figure 4c,d). This indicates that the topological structure of PdiTPSs, formed by residues around the substrate, may have significantly different folding mechanisms compared to the overall structure.
To evaluate the relationships between sequence similarity, TM score of protein structures, and products, the impact of sequence similarity and TM score on products was indirectly reflected by measuring the strength of their correlation. The similarity between products was calculated, and the final correlation coefficients were summarized in Table 1. Surprisingly, the overall sequence of PdiTPSs had the highest correlation with the product, while the residues around the substrate and the overall topology had a weak correlation with the product. The phenomenon can be explained, as our research primarily centers around assessing the geometric compatibility between the substrate and the enzyme pocket. It’s evident that, in the case of plant diterpene synthase, the chosen approach of static structural analysis has its limitations, thereby hindering a comprehensive understanding of the enzyme-product relationship. Hence, we propose a deeper exploration of the conserved physicochemical properties of residues occupying the same spatial vicinity as the substrate. This avenue of investigation promises a more insightful elucidation of the intricate interplay between diterpene synthase and the process of product formation. Because subsequences containing residues around the substrate are more relevant to the product.
To further validate the impact of the correlation between product and sequence similarity on product grouping, the similarity mapping between product and conserved motifs in PdiTPSs was analyzed by MEME. The four signature motifs (LHS, PNV, FERLW, and PIX) located in one different long motifs (Figure 5) had been identified. The similarity of these motifs to product was then correlated with product similarity. The correlation between the similarity of motifs 1 (PNV and FERLW) and product was 0.51, while the correlation for motif 3 (PIX) and motif 2 (LHS) were 0.32 and 0.36, respectively. Motifs with higher product correlation have more mutated residues in non-functional motifs, and conversely, possess fewer mutations. This correlation might indirectly reflect the level of differentiation between enzyme motifs, where motifs with high product correlation likely represented functional domains of the enzyme, while positions with low product correlation might contain product-specific motifs.
2.8. Aromatic residues around PdiTPSs substrates effect substrate type
Generally, the probability of catalytic associated residues appearing elsewhere in the sequence should be lower than that of appearing at the catalytic center. The statistical analysis revealed that aromatic amino acids around substrates with different sizes were the most frequently observed in PdiTPSs (Figure 6a,b). This might be due to the fact that the aromatic rings in aromatic amino acids could donate electrons to stabilize the carbocation intermediates in the catalytic process of PdiTPSs, making it easier for the substrates to be converted into products. Specifically, tryptophan (W) was more likely to occur around the linear substrate of GGPP, while phenylalanine (F) and tyrosine (Y) were more likely to present around the substrate of the initial cyclization intermediate of PdiTPSs (Figure 6c,d).
Studies on the impact of aromatic residues on the function of diterpene synthases have only indicated that the active sites of Class I and II enzymes both contain at least 2-3 aromatic residues, which guide the intermediate involved in the reaction through spatial constraints and cation-π interactions. Moreover, the number of aromatic residues in the active site can be used to predict the promiscuity of the enzyme [39]. However, there is no report explaining the selectivity of substrate structure exhibited by the observed types of aromatic residues.
2.9. Residues within 8 Å of substrate have more impact on products
It had been analyzed for the residues surrounding the substrate of PdiTPSs that generated different diterpene scaffolds and intermediates. The residues whose mutation have been experimentally demonstrated to affect product formation were also examined. It had been found that the physicochemical properties and spatial orientations of these residues surrounding the substrate were highly conserved. Some frequently occurring residues, such as arginine (R), cysteine (C), tryptophan (W), aspartic acid (D), isoleucine (I), serine (S), threonine (T), valine (V), phenylalanine (F), tyrosine (Y), and methionine (M) (Figure 6b), were also conserved in their spatial positions (Figure 6c,d). Except for arginine (R), cysteine (C), phenylalanine (F), and tryptophan (W), the effects of the other residues on enzyme function and product had been demonstrated by mutagenesis experiments. For example, the PdiTPSs OsKSL5i: I664T and OsKSL5i: I718V from rice (Oryza sativa) specifically produced ent-pimara-8(14),15-diene and ent-isokaur-15-ene, respectively [40]; Six PdiTPSs from Tripterygium wilfordii independently evolved new functions by mutating specific residues, including TwKSL1v2: M607\T638A, TwKSL3: M608\I639, TwKSL2: A608\I639, TwCPS3: I115\N327\V328\H268, TwCPS5: T115\A327\T326, and TwCPS6: Y265 [41]. Moreover, mutation of glutamic acid (E) at position 690 to arginine (R), phenylalanine (F), lysine (K), proline (P), or aspartic acid (D) or mutation of serine (S) at position 721 to valine (V) in SmMDS resulted in product loss [22], respectively.
It had been also observed that some low-frequency residues, such as alanine (A) and histidine (H), contributed to product specificity. For example, the AgAS: A723S mutant of abietadiene synthase specifically produced pimaradienes, and the H268 residue in TwCPS3 also contributed to product specificity [41,42]. Additionally, these low-frequency residues around the substrate often appeared in spatially conserved but physicochemically lowly conserved positions (Figure 6c,d). Mutated residues affecting the product mainly occurred within 6 Å of the substrate in PdiTPS I and within 6 Å to 8 Å of that in PdiTPS II. (Figure 6c,d). For specific residue shapes and maps within 4 Å to 8 Å of representative PdiTPS I and PdiTPS I/II that produced SK1-SK15 skeleton types, as well as that of PdiTPS II, please referred to the supplementary data (Figures S3–S21).
The previous analysis of residues around the substrate can provide clues to the importance of residues, but the effect of the residue on the product outcome still needs to consider factors such as spatial position and interaction with adjacent residues around the substrate. Nevertheless, these valuable experimental results also provide important evidence for the important position pattern of residues located within 8 Å of the substrate in PdiTPSs proteins that determines the product outcome. Local structural analysis reveals that exploring and examining structural alignment to generate sequence-order independent structural site motifs [43,44] might offer us a perspective to unveil the remarkable chemical diversity of PdiTPSs.
3. Methods
3.1. Collect characterized PdiTPSs
To find potentially characterized PdiTPSs, we manually searched for evidence of experimental characterization of diterpenes from literatures up to May 2022 and collected their corresponding GenBank accession numbers (NCBI). HMM of the N-terminal domain (PF01397) and C-terminal domain (PF03936) sequences of terpene synthases were downloaded from the Pfam database. Hmmsearch v.3.1.2 was used to search for the N- and C-terminal sequences in each PdiTPSs sequence. When multiple N- or C-terminal sequences were identified, the result with the lowest e-value was retained.
3.2. Construction of sequence similarity networks
All-versus-all pairwise local sequence alignments were performed using SSNpipe v.1.0.0 for PdiTPSs C, N, NC domains, and overall sequences [46]. The BLAST result files were searched with E-value thresholds ranging from 10-5 to 10-140 at 5 log unit intervals. The network files were visualized using Cytoscape (http://www.cytoscape.org/).
3.3. Phylogenetic analysis and visualization
The protein sequences were aligned using MAFFT v.7.310 [47] with the following parameters: maxiterate 1000 –localpair –thread 30--maxiterate 1000 --genafpair --thread 30. --maxiterate 1000 --globalpair --thread 30. Manual inspection was performed to ensure proper alignment of known motifs such as the DDXXD and DXDD motif. Phylogenetic tree was inferred using IQTree v.2.0.3 [48] with the following parameters: -s Mafft_Sequence -m JTT+F+R7 (full-length)/JTT+F+R5 (C-terminal and N-terminal subsequences) -B 1000 -nt AUTO. Levopimaradiene synthase from the hornwort Phaeoceros carolinianus was specified as the outgroup, and the resulting tree was visualized by Chiplot (https://www.chiplot.online/tvbot.html).
3.4. Retrieve and visualize sequence motifs
199 PdiTPSs amino acid sequences were submitted to the MEME online tool (https://meme-suite.org/meme/tools/meme) to identify motifs. Based on the known functional widths, the number of motifs was set to 20, and the minimum width of the motifs was set to 3. Other parameters were set to default values.
3.5. Perform calculations of similarity and correlation between sequences, structures, and small molecules
The similarities between C-terminal, N-terminal, and overall sequences were compared by using TBtools [49] Protein Pairwise Similarity Matrix. TMalign [50] was used to compare the topology similarity between the residues around the substrate and the overall structure, with the command "TMalign Pdb-A Pdb-B -outfmt 2", and the structure similarity was expressed as the TM score. The Dice Similarity Coefficient (DSC) [51] was used to measure chemical similarity in extended connectivity fingerprint (ECFP/Morgan Fingerprint, radius 2). For each pair of PdiTPSs, their corresponding products were arranged and combined, and the similarity score was calculated using the RDKIT similarity matrix. For PdiTPSs that produce only one product, there is only one product pair and thus only one similarity value. However, for those that produce multiple products, several product pairs were obtained, and the average of their similarity scores was used to represent the product similarity values. The pearson correlation coefficient (PPC) was calculated using ggstatsplot in R. The correlation analysis involved the following factors: the overall sequences of 199 PdiTPSs, the sequence similarity of C-terminal, N-terminal, and NC terminal subsequences, as well as the structure formed by residues surrounding the substrate and the overall structure.
3.6. Perform structure prediction, molecular docking and visualization
153 structures of PdiTPSs were downloaded from the AlphaFold database (https://alphafold.com/), and the rest were predicted for their 3D structures using ColabFold [52,53]. The obtained structures were docked with substrates using the CB-Dock2 molecular docking program (https://cadd.labshare.cn/cb-dock2). Structures were docked with substrates using the CB-Dock2 molecular docking program (https://cadd.labshare.cn/cb-dock2). The docking postures outputted by CB-Dock2 were compared with the crystal structures of PdiTPSs, and the complex closest to the crystal structure was selected as the final docking result. The amino acids within 4-10Å of the substrate were selected using the command "select AA, byres all within 4 of sele" in PyMOL (version 2.0) for further analysis. The diagrams were made with PyMOL.
3.7. Calculate amino acid frequencies
Software iLearnPlus v.1.0.1 [54] was used to extract the AAC and GAAC features from the residues surrounding the substrates of different sizes and the overall sequences. The resulting features included the frequencies of the 20 amino acids and their categorization based on 5 physicochemical properties. The residue preferential values were then calculated the ratio of frequency of each residue around the substrate to its frequency in the overall sequence.
4. Conclusions
In this study, we have compiled a dataset of plant-derived diterpene synthase with sequences and structural information, which is the largest annotated collection of PdiTPSs to date. It covers mosses, ferns, gymnosperms, and angiosperms. The dataset can be applied for studies of sequence and structure analysis, as well as serving as a library of enzyme components for combinatorial biology experiments to obtain non-natural diterpene products and a wider range of diterpene derivatives [45]. We also attempt to categorize diterpene products by their skeletal structure. While this simplifies analysis and function comparison, it can become disorderly when one enzyme produces multiple diterpene skeletons. However, this method may be useful for researchers studying diterpene synthesis in plants. It has been discovered for the strong correlation between N-terminal subsequence and overall sequence, significant sequence differences between N- and C-terminal subsequences from this data. Structural conservation increases with increasing similarity between the N-terminal sequence and the overall sequence. Moreover, an independent topological structure exists between the residues around the substrate and the overall structure. Quantitative analysis showed that sequence similarity has a greater impact on product distribution than structural similarity or similarity in the topological structure of residues around the substrate. This could be related to the single structural factor we are focusing on. However, some residues within 8 Å of the substrate have a profound effect on product. Furthermore, it has been found that tryptophan (W) is specific to the binding cavity of PdiTPSs that recognize linear substrate GGPP, while phenylalanine (F) and tyrosine (Y) are specific to the binding cavity of PdiTPSs that recognize cyclic substrates. Our exploration of the features of PdiTPSs and their mapping to product outcomes can be applied to enzyme identification, engineering, and product prediction research. In summary, our curated dataset will help to understand the sequence and structural space of PdiTPSs and provide a starting point for the specificity analysis of enzyme-controlled product distribution.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Y. Z.; conceptualization, original draft preparation, data curation, analyses, visualization and writing. Y.L.; conceptualization, editing and supervision. Y.L., X.H. and M.W.: review and editing, supervision, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant (No. 2021KF011) from State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Yunnan University.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zeng, T.; Chen, Y. X. X.; Jian, Y. X.; Zhang, F.; Wu, R. B., Chemotaxonomic investigation of plant terpenoids with an established database (TeroMOL). New Phytol 2022, 235, 662–673. [CrossRef]
- Zerbe, P.; Bohlmann, J., Plant diterpene synthases: exploring modularity and metabolic diversity for bioengineering. Trends Biotechnol 2015, 33, 419–428. [CrossRef]
- Gershenzon, J.; Dudareva, N., The function of terpene natural products in the natural world. Nat Chem Biol 2007, 3, 408–414. [CrossRef]
- Jennewein, S.; Croteau, R., Taxol: biosynthesis, molecular genetics, and biotechnological applications. Appl Microbiol Biot 2001, 57, 13–19. [CrossRef]
- Caniard, A.; Zerbe, P.; Legrand, S.; Cohade, A.; Valot, N.; Magnard, J. L.; Bohlmann, J.; Legendre, L., Discovery and functional characterization of two diterpene synthases for sclareol biosynthesis in Salvia sclarea (L.) and their relevance for perfume manufacture. Bmc Plant Biol 2012, 12. [CrossRef]
- Schalk, M.; Pastore, L.; Mirata, M. A.; Khim, S.; Schouwey, M.; Deguerry, F.; Pineda, V.; Rocci, L.; Daviet, L., Toward a Biosynthetic Route to Sclareol and Amber Odorants. J Am Chem Soc 2012, 134, 18900–18903. [CrossRef]
- Philippe, R. N.; De Mey, M.; Anderson, J.; Ajikumar, P. K., Biotechnological production of natural zero-calorie sweeteners. Curr Opin Biotech 2014, 26, 155–161. [CrossRef] [PubMed]
- Bemis, G. W.; Murcko, M. A., The properties of known drugs. 1. Molecular frameworks. J Med Chem 1996, 39, 2887–2893. [CrossRef]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E., The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J 2011, 66, 212–229. [CrossRef]
- Banerjee, A.; Hamberger, B., P450s controlling metabolic bifurcations in plant terpene specialized metabolism. Phytochem Rev 2018, 17, 81–111. [CrossRef]
- Bathe, U.; Tissier, A., Cytochrome P450 enzymes: A driving force of plant diterpene diversity. Phytochemistry 2019, 161, 149–162. [CrossRef]
- Jia, Q. D.; Kollner, T. G.; Gershenzon, J.; Chen, F., MTPSLs: New Terpene Synthases in Nonseed Plants. Trends Plant Sci 2018, 23, 121–128. [CrossRef]
- Abbas, F.; Ke, Y.; Yu, R.; Yue, Y.; Amanullah, S.; Jahangir, M. M.; Fan, Y., Volatile terpenoids: multiple functions, biosynthesis, modulation and manipulation by genetic engineering. Planta 2017, 246, 803–816. [CrossRef] [PubMed]
- Starks, C. M.; Back, K. W.; Chappell, J.; Noel, J. P., Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science 1997, 277, 1815–1820. [CrossRef] [PubMed]
- Rynkiewicz, M. J.; Cane, D. E.; Christianson, D. W., Structure of trichodiene synthase from Fusarium sporotrichioides provides mechanistic inferences on the terpene cyclization cascade. P Natl Acad Sci USA 2001, 98, 13543–13548. [CrossRef]
- Whittington, D. A.; Wise, M. L.; Urbansky, M.; Coates, R. M.; Croteau, R. B.; Christianson, D. W., Bornyl diphosphate synthase: Structure and strategy for carbocation manipulation by a terpenoid cyclase. P Natl Acad Sci USA 2002, 99, 15375–15380. [CrossRef]
- Degenhardt, J.; Kollner, T. G.; Gershenzon, J., Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [CrossRef]
- Jia, M.; Zhou, K.; Tufts, S.; Schulte, S.; Peters, R. J., A Pair of Residues That Interactively Affect Diterpene Synthase Product Outcome. Acs Chem Biol 2017, 12, 862–867. [CrossRef] [PubMed]
- Potter, K.; Criswell, J.; Zi, J. C.; Stubbs, A.; Peters, R. J., Novel Product Chemistry from Mechanistic Analysis of ent-Copalyl Diphosphate Synthases from Plant Hormone Biosynthesis. Angew Chem Int Edit 2014, 53, 7198–7202. [CrossRef]
- Potter, K. C.; Jia, M. R.; Hong, Y. J.; Tantillio, D.; Peters, R. J., Product Rearrangement from Altering a Single Residue in the Rice syn-Copalyl Diphosphate Synthase. Org Lett 2016, 18, 1060–1063. [CrossRef]
- Potter, K. C.; Zi, J. C.; Hong, Y. J.; Schulte, S.; Malchow, B.; Tantillo, D. J.; Peters, R. J., Blocking Deprotonation with Retention of Aromaticity in a Plant ent-Copalyl Diphosphate Synthase Leads to Product Rearrangement. Angew Chem Int Edit 2016, 55, 634–638. [CrossRef] [PubMed]
- Tong, Y. R.; Ma, X. L.; Hu, T. Y.; Chen, K.; Cui, G. H.; Su, P.; Xu, H. F.; Gao, W.; Jiang, T.; Huang, L. Q., Structural and mechanistic insights into the precise product synthesis by a bifunctional miltiradiene synthase. Plant Biotechnol J 2022. [CrossRef]
- Tao, H.; Lauterbach, L.; Bian, G. K.; Chen, R.; Hou, A. W.; Mori, T.; Cheng, S.; Hu, B.; Lu, L.; Mu, X.; Li, M.; Adachi, N.; Kawasaki, M.; Moriya, T.; Senda, T.; Wang, X. H.; Deng, Z. X.; Abe, I.; Dickschat, J. S.; Liu, T. G., Discovery of non-squalene triterpenes. Nature 2022, 606, 414–419. [CrossRef]
- Hu, Z. M.; Liu, X. Y.; Tian, M.; Ma, Y.; Jin, B. L.; Gao, W.; Cui, G. H.; Guo, J.; Huang, L. Q., Recent progress and new perspectives for diterpenoid biosynthesis in medicinal plants. Med Res Rev 2021, 41, 2971–2997. [CrossRef] [PubMed]
- Johnson, S. R.; Bhat, W. W.; Bibik, J.; Turmo, A.; Hamberger, B.; Hamberger, B.; Genomics, E. M., A database-driven approach identifies additional diterpene synthase activities in the mint family (Lamiaceae). Journal of Biological Chemistry 2019, 294, 1349–1362. [CrossRef]
- LI H., P. J.X., LI J., Diterpenoids Chemodiversity of the Genus Isodon Spach from Lamiaceae. Plant Diversity 2013, 35, 81–88.
- Christianson, D. W., Structural and Chemical Biology of Terpenoid Cyclases. Chem Rev 2017, 117, 11570–11648. [CrossRef]
- Wang, Z. B.; Nelson, D. R.; Zhang, J.; Wan, X. Y.; Peters, R. J., Plant (di)terpenoid evolution: from pigments to hormones and beyond. Nat Prod Rep 2023, 40, 452–469. [CrossRef]
- Jia, Q. D.; Brown, R.; Kollner, T. G.; Fu, J. Y.; Chen, X. L.; Wong, G. K. S.; Gershenzon, J.; Peters, R. J.; Chen, F., Origin and early evolution of the plant terpene synthase family. P Natl Acad Sci USA 2022, 119. [CrossRef]
- Durairaj, J.; Di Girolamo, A.; Bouwmeester, H. J.; de Ridder, D.; Beekwilder, J.; van Dijk, A. D. J., An analysis of characterized plant sesquiterpene synthases. Phytochemistry 2019, 158, 157–165. [CrossRef]
- Riziotis, I. G.; Ribeiro, A. J. M.; Borkakoti, N.; Thornton, J. M., Conformational Variation in Enzyme Catalysis: A Structural Study on Catalytic Residues. J Mol Biol 2022, 434. [CrossRef]
- Faylo, J. L.; Ronnebaum, T. A.; Christianson, D. W., Assembly-Line Catalysis in Bifunctional Terpene Synthases. Accounts Chem Res 2021, 54, 3780–3791. [CrossRef]
- Zhai, G. Q.; Zhang, Z. Y.; Dong, C. J., Mutagenesis and functional analysis of SotB: A multidrug transporter of the major facilitator superfamily from Escherichia coli. Frontiers in Microbiology 2022, 13. [CrossRef]
- Chen, Z. R.; Lv, Q. L.; Peng, H. W.; Liu, X. Y.; Hu, W. L.; Hu, J. F., Drug screening against F13 protein, the target of tecovirimat, as potential therapies for monkeypox virus. J Infection 2023, 86, 195–198. [CrossRef]
- Liu, Y.; Yang, X. C.; Gan, J. H.; Chen, S.; Xiao, Z. X.; Cao, Y., CB-Dock2: improved protein ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res 2022, 50, W159–W164. [CrossRef]
- Alvarez, A. F.; Rodriguez, C.; Gonzalez-Chavez, R.; Georgellis, D., The Escherichia coli two-component signal sensor BarA binds protonated acetate via a conserved hydrophobic-binding pocket. J Biol Chem 2021, 297, 101383. [CrossRef]
- Koksal, M.; Jin, Y. H.; Coates, R. M.; Croteau, R.; Christianson, D. W., Taxadiene synthase structure and evolution of modular architecture in terpene biosynthesis. Nature 2011, 469, 116–U138. [CrossRef] [PubMed]
- Zhou, K.; Gao, Y.; Hoy, J. A.; Mann, F. M.; Honzatko, R. B.; Peters, R. J., Insights into Diterpene Cyclization from Structure of Bifunctional Abietadiene Synthase from Abies grandis. Journal of Biological Chemistry 2012, 287, 6840–6850. [CrossRef] [PubMed]
- Zhang, F.; An, T. Y.; Tang, X. W.; Zi, J. C.; Luo, H. B.; Wu, R. B., Enzyme Promiscuity versus Fidelity in Two Sesquiterpene Cyclases (TEAS versus ATAS). Acs Catal 2020, 10, 1470–1484. [CrossRef]
- Xu, M. M.; Wilderman, P. R.; Peters, R. J., Following evolution’s lead to a single residue switch for diterpene synthase product outcome. P Natl Acad Sci USA 2007, 104, 7397–7401. [CrossRef]
- Tu, L. C.; Cai, X. B.; Zhang, Y. F.; Tong, Y. R.; Wang, J.; Su, P.; Lu, Y.; Hu, T. Y.; Luo, Y. F.; Wu, X. Y.; Li, D.; Huang, L. Q.; Gao, W., Mechanistic analysis for the origin of diverse diterpenes in Tripterygium wilfordii. Acta Pharm Sin B 2022, 12, 2923–2933. [CrossRef]
- Wilderman, P. R.; Peters, R. J., A single residue switch converts abietadiene synthase into a pimaradiene specific cyclase. J Am Chem Soc 2007, 129, 15736–15737. [CrossRef]
- Sankar, S.; Chandran Sakthivel, N.; Chandra, N., Fast Local Alignment of Protein Pockets (FLAPP): A System-Compiled Program for Large-Scale Binding Site Alignment. J Chem Inf Model 2022, 62, 4810–4819. [CrossRef] [PubMed]
- Sankar, S.; Chandra, N., SiteMotif: A graph-based algorithm for deriving structural motifs in Protein Ligand binding sites. PLoS Comput Biol 2022, 18, e1009901. [CrossRef]
- Jia, M. R.; Mishra, S. K.; Tufts, S.; Jernigan, R. L.; Peters, R. J., Combinatorial biosynthesis and the basis for substrate promiscuity in class I diterpene synthases. Metab Eng 2019, 55, 44–58. [CrossRef] [PubMed]
- Viborg, A. H.; Terrapon, N.; Lombard, V.; Michel, G.; Czjzek, M.; Henrissat, B.; Brumer, H., A subfamily roadmap of the evolutionarily diverse glycoside hydrolase family 16 (GH16). J Biol Chem 2019, 294, 15973–15986. [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T., MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002, 30, 3059–66. [CrossRef] [PubMed]
- Nguyen, L. T.; Schmidt, H. A.; von Haeseler, A.; Minh, B. Q., IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 2015, 32, 268–274. [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H. R.; Frank, M. H.; He, Y.; Xia, R., TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant 2020, 13, 1194–1202. [CrossRef]
- Zhang, Y.; Skolnick, J., TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 2005, 33, 2302–2309. [CrossRef] [PubMed]
- Willett, P.; Barnard, J. M.; Downs, G. M., Chemical similarity searching. Journal of chemical information and computer sciences 1998, 38, 983–996. [CrossRef]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [CrossRef]
- Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022, 19, 679–682. [CrossRef]
- Chen, Z.; Zhao, P.; Li, C.; Li, F. Y.; Xiang, D. X.; Chen, Y. Z.; Akutsu, T.; Daly, R. J.; Webb, G. I.; Zhao, Q. Z.; Kurgan, L.; Song, J. N., iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res 2021, 49. [CrossRef]
Figure 1.
Similarity networks of PdiTPSs sequences. (a) Clusters of PdiTPSs product skeletons defined by N-terminal subsequence similarity (E-value threshold 10 -70); (b) Clusters of PdiTPSs product skeletons defined by C-terminal subsequence similarity (E-value threshold 10 -70); (c) Clusters of PdiTPSs product skeletons defined by NC-terminal subsequence similarity (E-value threshold 10 -120).
Figure 1.
Similarity networks of PdiTPSs sequences. (a) Clusters of PdiTPSs product skeletons defined by N-terminal subsequence similarity (E-value threshold 10 -70); (b) Clusters of PdiTPSs product skeletons defined by C-terminal subsequence similarity (E-value threshold 10 -70); (c) Clusters of PdiTPSs product skeletons defined by NC-terminal subsequence similarity (E-value threshold 10 -120).
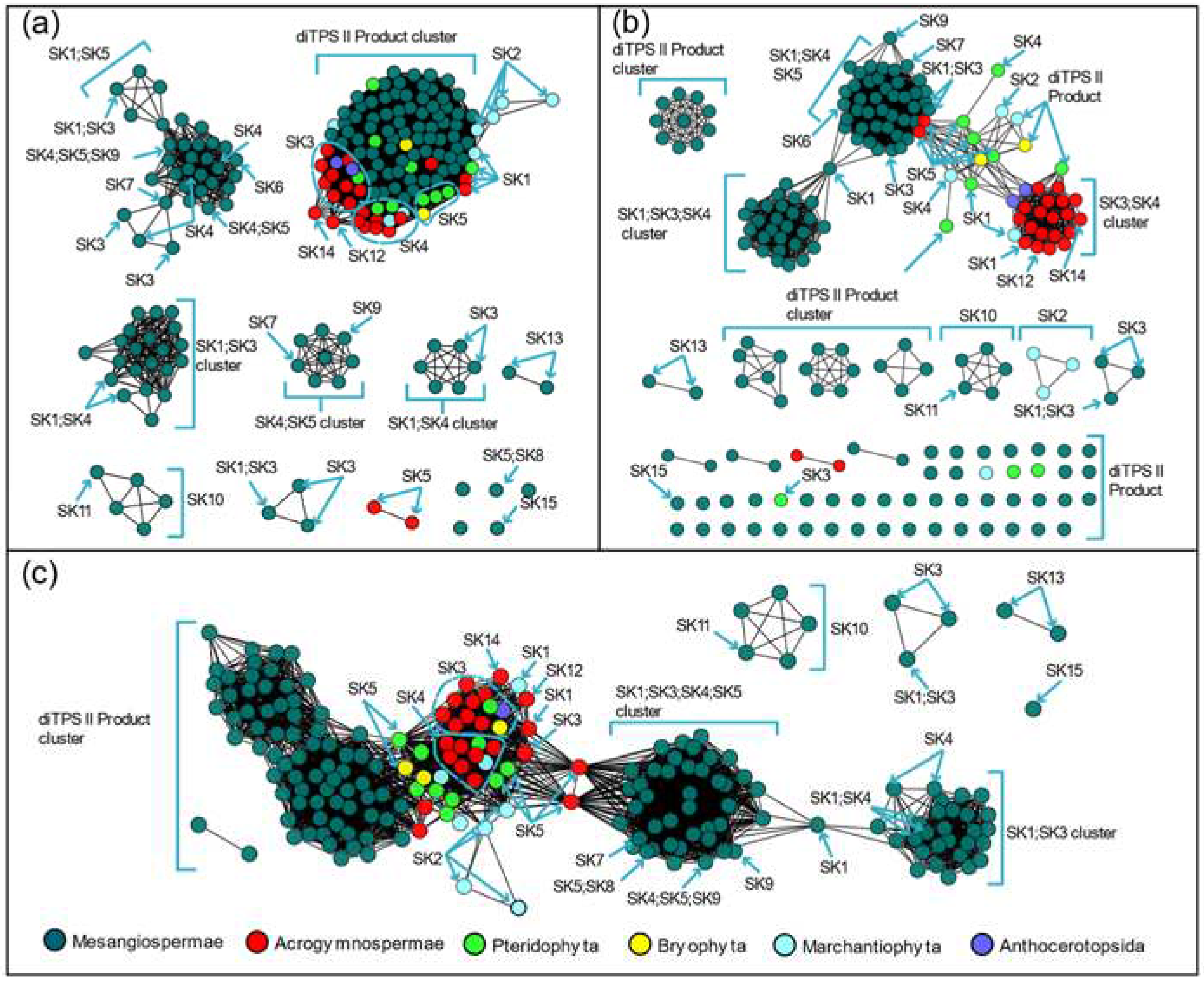
Figure 2.
The relationship between the overall sequence phylogeny of PdiTPSs, their product scaffolds, function-related motifs, and enzyme source classification. The phylogenetic tree labels each enzyme with its accession ID, product class, and scaffold classification. It also displays the skeleton structures of SK1-SK15, major product structures of diTPS II, function-verified motifs in some diterpene synthases, and the domain composition of each diterpene synthase.
Figure 2.
The relationship between the overall sequence phylogeny of PdiTPSs, their product scaffolds, function-related motifs, and enzyme source classification. The phylogenetic tree labels each enzyme with its accession ID, product class, and scaffold classification. It also displays the skeleton structures of SK1-SK15, major product structures of diTPS II, function-verified motifs in some diterpene synthases, and the domain composition of each diterpene synthase.
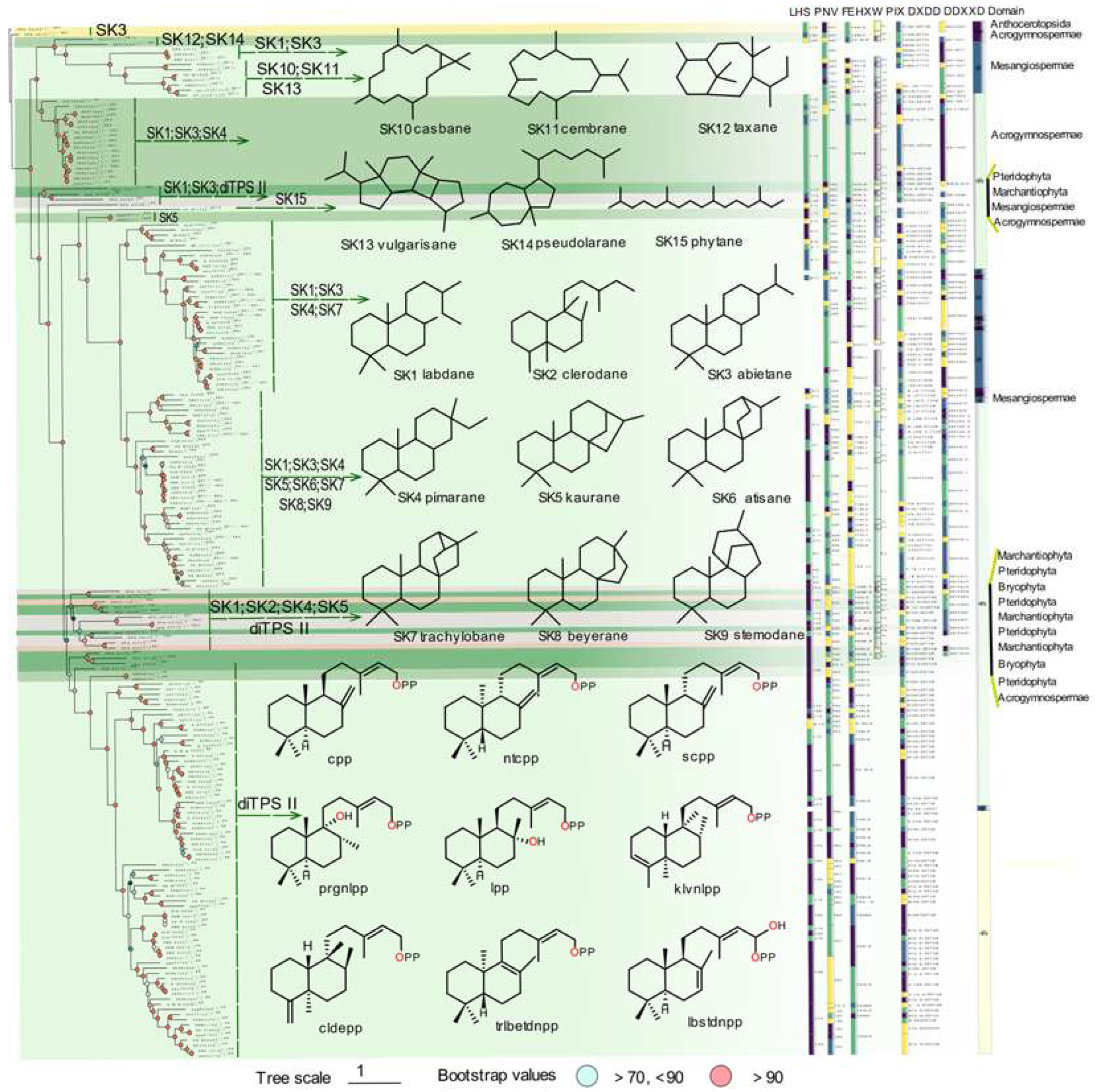
Figure 3.
Distribution of sequence and structural similarity data. (a) Distribution of sequence and structural similarity data among N-terminal, C-terminal, NC-terminal domains, overall sequences, and overall structures of diTPS I, II, and I/II. (b) Superimposition of representative structures at the Q2 position based on the TM-score.
Figure 3.
Distribution of sequence and structural similarity data. (a) Distribution of sequence and structural similarity data among N-terminal, C-terminal, NC-terminal domains, overall sequences, and overall structures of diTPS I, II, and I/II. (b) Superimposition of representative structures at the Q2 position based on the TM-score.
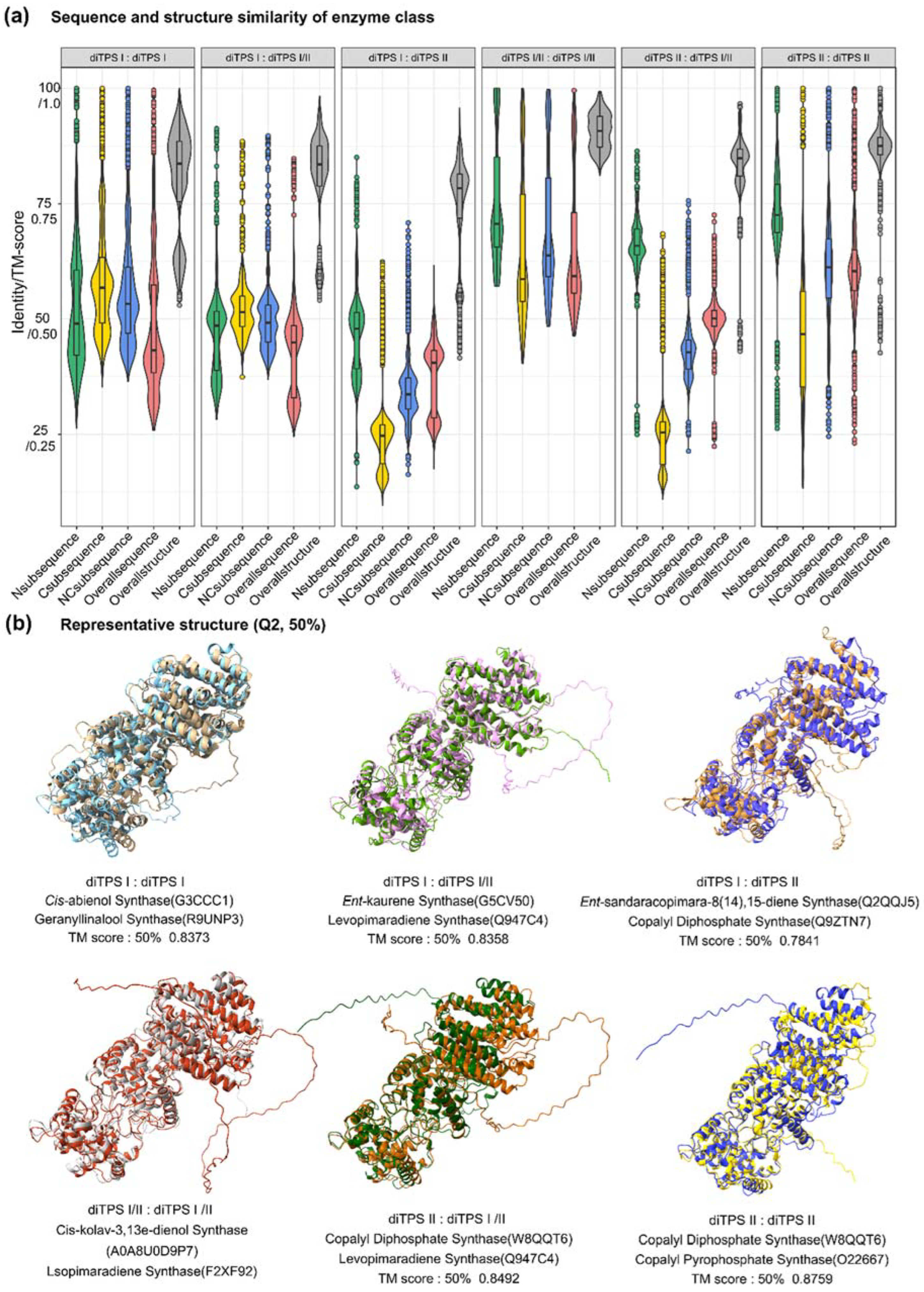
Figure 4.
Distribution of similarity data between N-terminal, C-terminal, NC-terminal subsequences, overall sequences, and overall structures of PdiTPSs for same and different substrates and products. (a) Comparison of sequence similarity data between N-terminal, C-terminal, and NC-terminal subsequences and overall sequences for the same and different substrates. (b) Comparison of sequence similarity data between N-terminal, C-terminal, and NC-terminal subsequences and overall sequences for the same and different products. (c) Comparison of sequence similarity data between the topology structures formed by residues within 4 Å, 6 Å, 8 Å, and 10 Å of the substrate with the overall structures for the same and different substrates. (d) Comparison of sequence similarity data between the topology structures formed by residues within 4 Å, 6 Å, 8 Å, and 10 Å of the substrate with the overall structures for the same and different products. Asterisks indicate statistical significance (*: p < 0.05, **: p < 0.01, ***: p < 0.001), and "NS" indicates no statistical difference.
Figure 4.
Distribution of similarity data between N-terminal, C-terminal, NC-terminal subsequences, overall sequences, and overall structures of PdiTPSs for same and different substrates and products. (a) Comparison of sequence similarity data between N-terminal, C-terminal, and NC-terminal subsequences and overall sequences for the same and different substrates. (b) Comparison of sequence similarity data between N-terminal, C-terminal, and NC-terminal subsequences and overall sequences for the same and different products. (c) Comparison of sequence similarity data between the topology structures formed by residues within 4 Å, 6 Å, 8 Å, and 10 Å of the substrate with the overall structures for the same and different substrates. (d) Comparison of sequence similarity data between the topology structures formed by residues within 4 Å, 6 Å, 8 Å, and 10 Å of the substrate with the overall structures for the same and different products. Asterisks indicate statistical significance (*: p < 0.05, **: p < 0.01, ***: p < 0.001), and "NS" indicates no statistical difference.
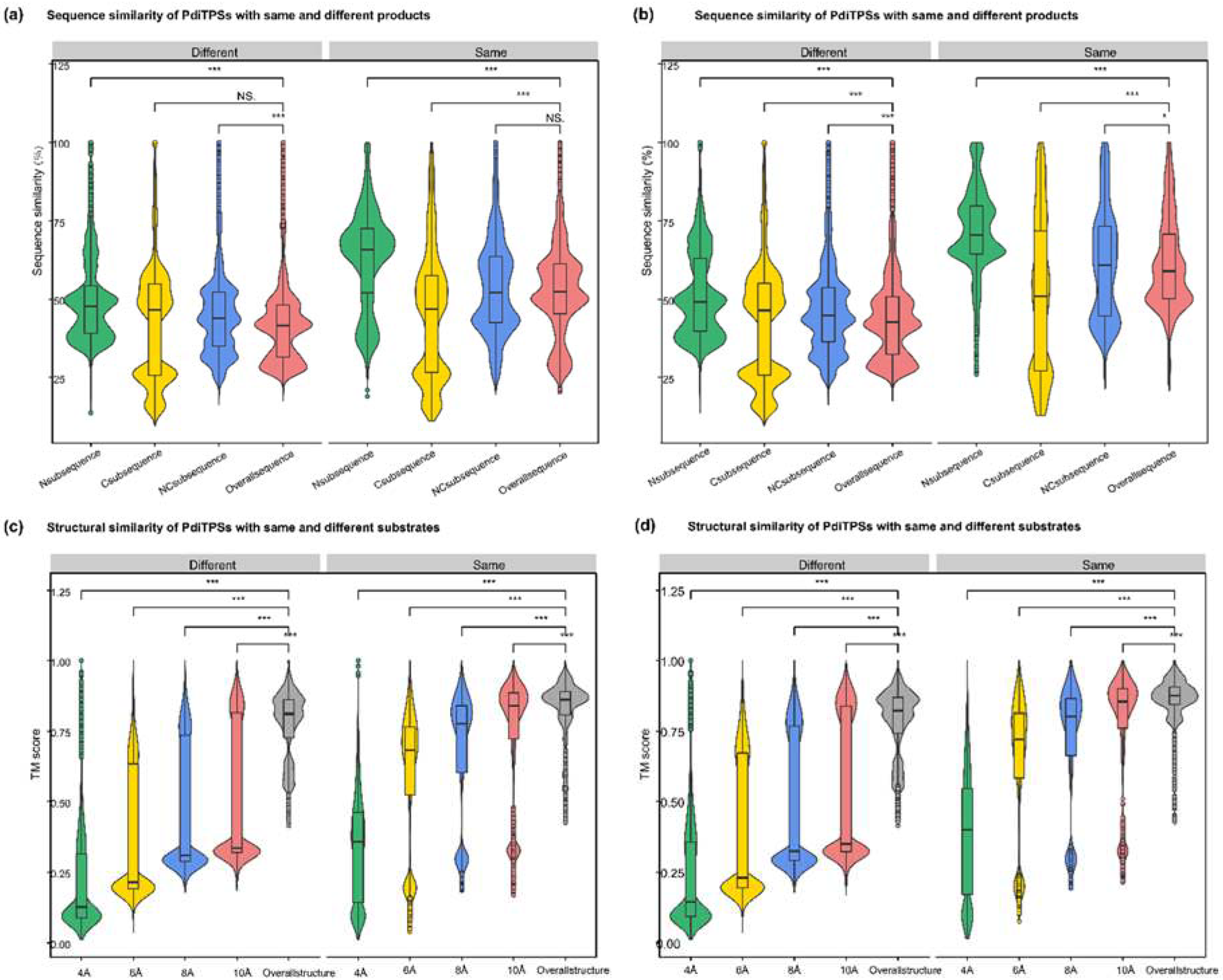
Figure 5.
The positions of validated functional motifs in protein structures, along with the number of motifs and specific residues near the motifs in the three classes of PdiTPSs. (a) The crystal structure of Taxadiene synthase (diTPS I); (b) The crystal structure of ent-copalyl diphosphate synthase (diTPS II); (c) The crystal structure of Abietadiene cyclase (diTPS I/II); (d) The crystal structure of Casbene synthase (diTPS I). (e) Seqlogo of the PNV and FERLW motifs and their neighboring residues; (f) Seqlogo of the LHS motif and its neighboring residues; (g) Seqlogo of the PIX motif and its neighboring residues. The black numbers represent the total number of sequences retrieved by MEME for the PNV, FERLW, LHS, and PIX motifs, while the red numbers represent the number of occurrences of each motif in the three classes of enzymes.
Figure 5.
The positions of validated functional motifs in protein structures, along with the number of motifs and specific residues near the motifs in the three classes of PdiTPSs. (a) The crystal structure of Taxadiene synthase (diTPS I); (b) The crystal structure of ent-copalyl diphosphate synthase (diTPS II); (c) The crystal structure of Abietadiene cyclase (diTPS I/II); (d) The crystal structure of Casbene synthase (diTPS I). (e) Seqlogo of the PNV and FERLW motifs and their neighboring residues; (f) Seqlogo of the LHS motif and its neighboring residues; (g) Seqlogo of the PIX motif and its neighboring residues. The black numbers represent the total number of sequences retrieved by MEME for the PNV, FERLW, LHS, and PIX motifs, while the red numbers represent the number of occurrences of each motif in the three classes of enzymes.
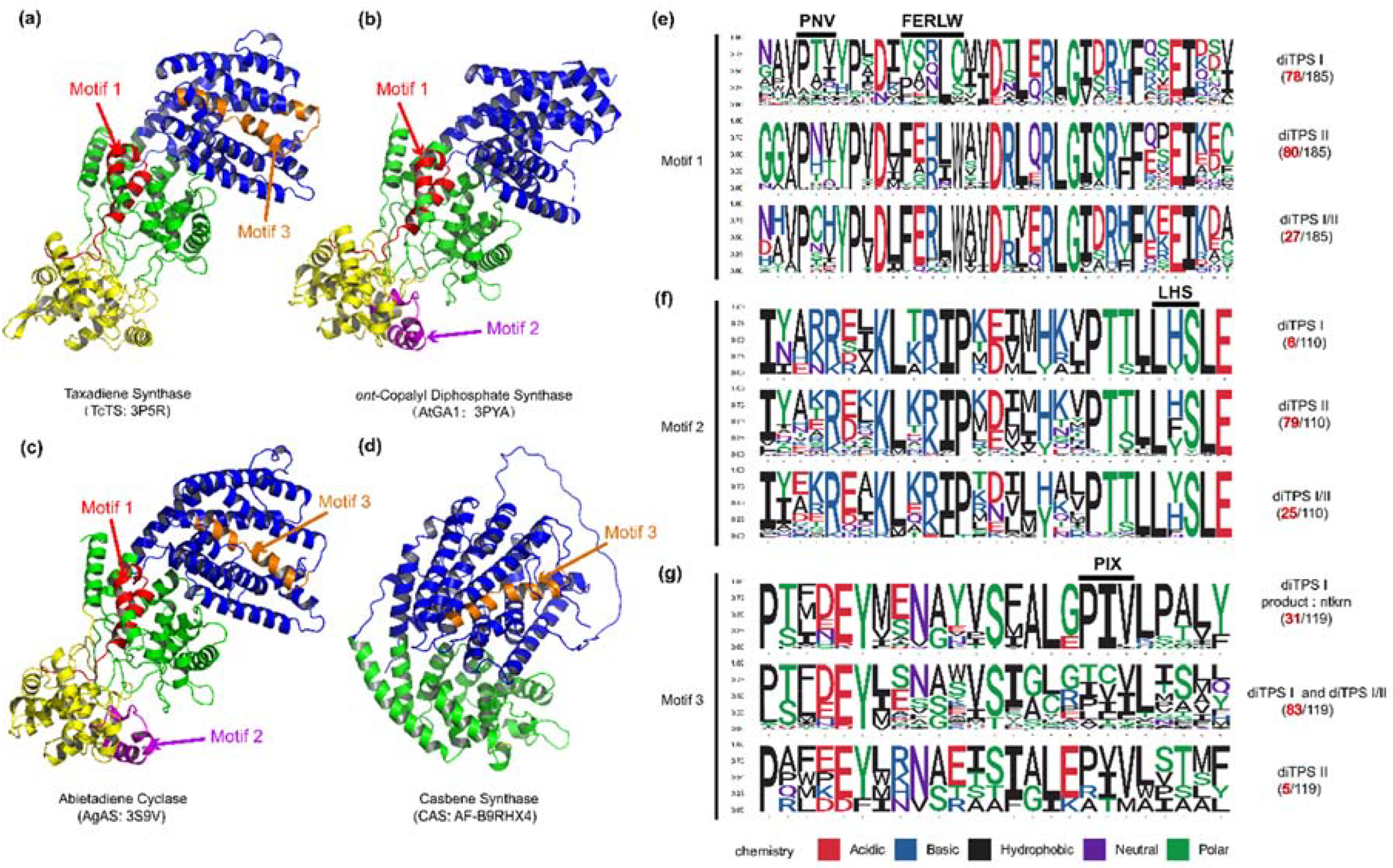
Figure 6.
Preference of 5 types of amino acids in the residues around the substrate and their relative spatial position in the structure. (a) Frequency distribution of five types of amino acids (aliphatic, aromatic, negatively charged polar, positively charged polar, and uncharged) within 4 Å, 6 Å, 8 Å, and 10 Å from the substrate compared to their overall frequency within the protein structure. (b). Comparison of the frequency of 20 amino acids within the protein structure to their frequency within 4 Å, 6 Å, 8 Å, and 10 Å from the substrate. (c) Superimposition of substrate-proximal residues (within 6 Å) of 15 diterpene synthases producing different skeletons, as well as those of diTPS I and diTPS I/II that have undergone mutagenesis studies. (d) Superimposition of substrate-proximal residues (within 8 Å) of diterpene synthases producing different intermediates, as well as those of diTPS II and diTPS I/II that have undergone mutagenesis studies. In c and d, aromatic residues and residues that have been shown to affect product outcome in mutagenesis studies are highlighted. Green fonts indicate the residue with the highest frequency in that position, and red fonts indicate mutated residues (also with the highest frequency). Asterisks indicate statistical significance (*: p < 0.05, **: p < 0.01, ***: p < 0.001), and "NS" indicates no statistical difference.
Figure 6.
Preference of 5 types of amino acids in the residues around the substrate and their relative spatial position in the structure. (a) Frequency distribution of five types of amino acids (aliphatic, aromatic, negatively charged polar, positively charged polar, and uncharged) within 4 Å, 6 Å, 8 Å, and 10 Å from the substrate compared to their overall frequency within the protein structure. (b). Comparison of the frequency of 20 amino acids within the protein structure to their frequency within 4 Å, 6 Å, 8 Å, and 10 Å from the substrate. (c) Superimposition of substrate-proximal residues (within 6 Å) of 15 diterpene synthases producing different skeletons, as well as those of diTPS I and diTPS I/II that have undergone mutagenesis studies. (d) Superimposition of substrate-proximal residues (within 8 Å) of diterpene synthases producing different intermediates, as well as those of diTPS II and diTPS I/II that have undergone mutagenesis studies. In c and d, aromatic residues and residues that have been shown to affect product outcome in mutagenesis studies are highlighted. Green fonts indicate the residue with the highest frequency in that position, and red fonts indicate mutated residues (also with the highest frequency). Asterisks indicate statistical significance (*: p < 0.05, **: p < 0.01, ***: p < 0.001), and "NS" indicates no statistical difference.
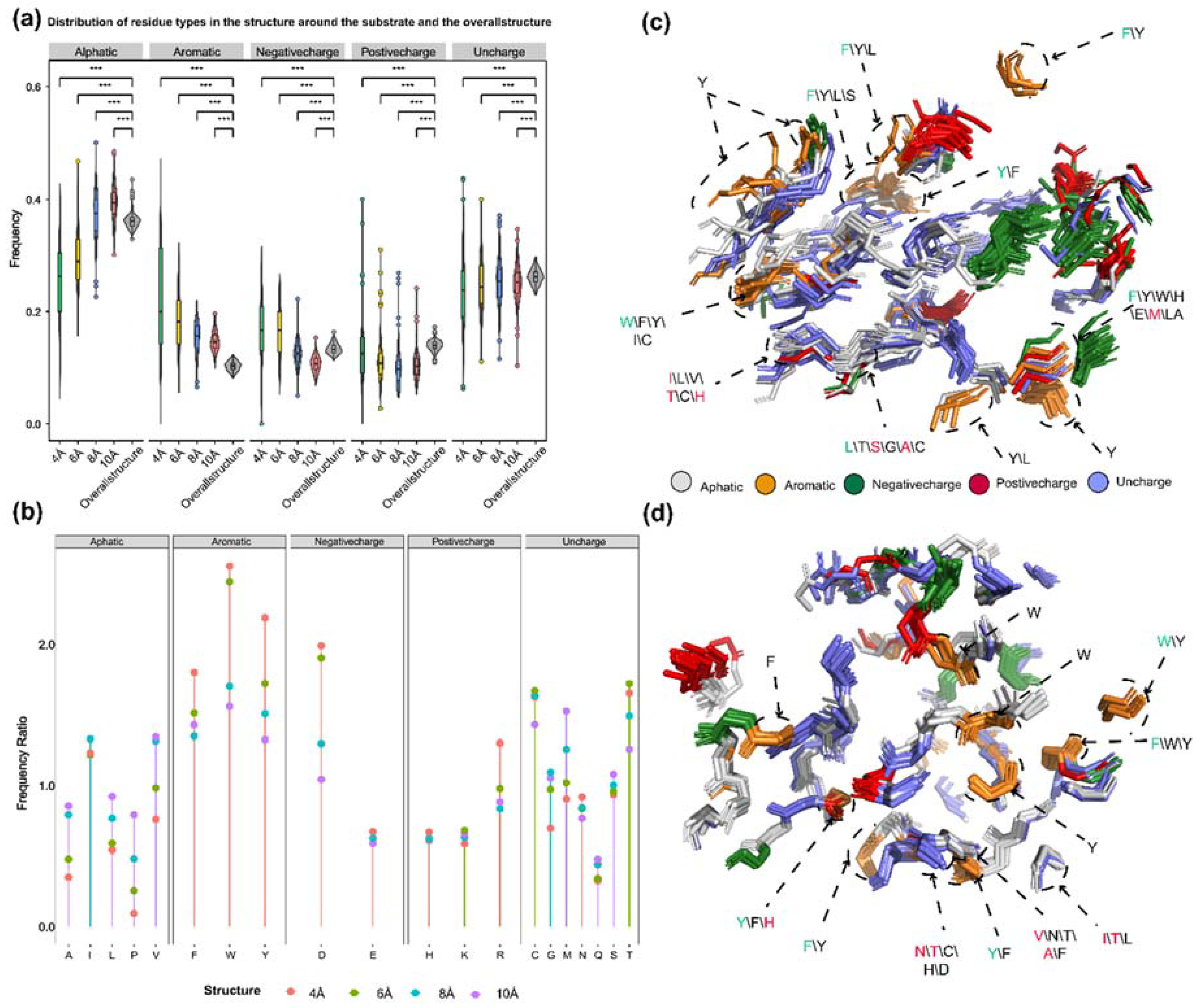

Table 1.
PCC for PdiTPSs sequences, structures, and products
| Content | Pearson correlation coefficient |
| 4Å : Products | 0.38 |
| 6Å : Products | 0.4 |
| 8Å : Products | 0.39 |
| 10Å : Products | 0.4 |
| overallstructure : Products | 0.36 |
| Csequence : Products | 0.35 |
| Nsequence : Products | 0.54 |
| NCsequence : Products | 0.54 |
| overallstructure : Products | 0.55 |
Note: All p values < 0.001 in the table’s statistical results.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



