
Submitted:
16 August 2023
Posted:
06 September 2023
You are already at the latest version
Abstract
Preterm infants often experience frequent intermittent hypoxia (IH) episodes which is associated with neuroinflammation. We tested the hypotheses that early caffeine and/or non-steroidal in-flammatory drugs (NSAIDs) confer superior therapeutic benefits for protection against IH-induced neuroinflammation than late treatment. Newborn rats were exposed to IH or hy-peroxia (50% O2) from birth (P0) to P14. For early treatment, the pups were administered: 1) dai-ly caffeine citrate (Cafcit, 20 mg/kg IP loading on P0, followed by 5 mg/kg from P1-P14); 2) ke-torolac topical ocular solution in both eyes from P0 to P14; 3) ibuprofen (Neoprofen, 10 mg/kg loading dose on P0 followed by 5 mg/kg/day on P1 and P2); 4) caffeine+ketorolac co-treatment; 5) caffeine+ibuprofen co-treatment; or 6) equivalent volume saline. On P14, animals were placed in room air (RA) with no further treatment. For late treatment, pups received similar treatments from P15-P21 (caffeine and/or ketorolac), or P15-P17 (ibuprofen). RA control were similarly treated. At P21, whole brains were assessed for histopathology, apoptosis, mye-lination, and biomarkers of inflammation. IH caused significant inflammation, reduced mye-lination, and apoptosis in preterm rat brains, an effect that was improved predominantly with combined caffeine/NSAID early treatment. Early caffeine/NSAID co-treatment confers syner-gistic neuroprotection against IH-induced damage.
Keywords:
Brain
; Caffeine
; Cytokines
; Ibuprofen
; Intermittent Hypoxia
; Myelination
; Neuroinflammation
; Non-steroidal Anti-inflammatory Drugs
1. Introduction
Every year in the United States, about 63,000 infants are born with a very low birth weight (VLBW; ≤1500 g) [1]. This group represents 1·5% of all live births and has increased gradually over the past decade due to increased survival (50-70%) [2,3]. Disability in this subset exceeds 50% in most studies [1,3], with neurocognitive delays in the later preterm population unaccounted for. In terms of neurologic impairments, preterm births account for cognitive, behavioral, attention or socialization deficits in 25–50% and major motor deficits (e.g., cerebral palsy) in 5–10 % [3,4]. The epiCURE study described structural brain disease in 17% of the extremely low birth weight (ELBW) babies, with parenchymal cysts and/or hydrocephalus [5]. These impairments lead to reduced quality of life and productivity, increased healthcare services utilization, higher dependency and need for assisted living [6,7]. Insults to the central nervous system (CNS) account for over 40% of the disability adjusted life years (DALYs) in neonates, mainly because of prematurity and hypoxic ischemic encephalopathy (HIE) [5,6,7].
Several animal studies have investigated mechanisms of injury that lead to increased neurologic morbidity [8,9,10]. The effect from hypoxia/hypoxemia appears to be an important and dominant mechanism of injury [11]. Current continuous recordings of oxygen saturation reveal a much higher frequency of intermittent hypoxia (IH) events that were previously un-documented in medical charts and provide insight to high-risk patterns associated with both short-term and long-term morbidity [12,13]. Although interpretation of absolute values of cerebral tissue oxygenation have yet to be determined, recent data have shown a greater adverse impact from IH than bradycardic events [14]. Regarding hypoxia exposure, chronic IH (CIH) has been observed to cause greater extent and degree of damage to the premature brain than chronic continuous hypoxia (CCH) [15,16] due to increased inflammation as CCH is unlikely to stimulate adaptive responses in neurons [17,18]. Various regions in the CNS, such as the sub cortex and especially the hippocampus have been shown to be more vulnerable than others to hypoxic exposure [18]. CIH can result in considerable damage to the hippocampal formation and IH exposure can lead to apoptosis in the hippocampal CA1 region [18,19,20]. The atlas of brain maps has proven useful in several publications for illustrating and comparing the distribution patterns of neuroanatomical data on templates and providing detailed mapping results [21]. As the pathology of brain injury becomes clearer, there is increased interest in developing methods to ameliorate it. The excessive production and release of proinflammatory cytokines are hallmarks of neuroinflammation and these cytokines serve as neurotoxic factors involved in the pathogenesis of white matter injury after IH in the immature brain [22,23]. Therapeutic approaches to protect the premature brain from inflammatory damage is of utmost importance.
Caffeine (Caff) and non-steroidal anti-inflammatory drugs (NSAIDs), such as Ibuprofen (Ibu) are often used concomitantly in ELBW babies for apnea of prematurity (AOP) and patent ductus arteriosus (PDA) closure, respectively [24]. Caff is a respiratory stimulant that reduces the incidence of AOP, and ameliorates clinical symptoms associated with AOP [25,26] and has become the drug of choice in neonatal care for improvement of respiratory outcomes [25]. Its use for neuroprotection in neonates and prevention of hypoxemic episodes has been investigated and proven effective [27,28]. Ibuprofen use has been shown to reduce inflammation in the brain of neonatal rats [29] and growth restricted piglets [23,30] suggesting a potential therapeutic intervention to prevent white matter damage. However, its chronic use appears to worsen the outcomes [31]. Ketorolac (Keto), another NSAID, is used to reduce the need for more traditional pain medication such as opioid analgesics [32,33,34]. A systematic review involving preterm and term infants showed that Keto in appropriate doses is a viable analgesic and does not increase the risk of bleeding or adverse renal effects [35]. Keto has been used in preterm infants to prevent retinopathy of prematurity (ROP), but the results were not consistent [36,37]. Both Ibu and Keto act by decreasing prostaglandin (PG) synthesis through non-selective inhibition of cyclooxygenase (COX)-1 and -2. However, there are no studies investigating whether Keto confers neuroprotection by reduction of inflammation, or the synergistic effects of Caff and NSAIDs for reduction of IH-induced neuroinflammation.
Using our neonatal IH model, which consistently produces oxidative stress in neonatal rats [38,39] we tested the hypothesis that early caffeine and/or NSAIDs confer superior therapeutic benefits for protection against IH-induced neuroinflammation than late treatment. Our hypothesis was tested with the following objectives: 1) To examine the effects of neonatal IH on inflammation in the neonatal rat brain; 2) To compare the effects of caffeine, ketorolac or ibuprofen given individually during neonatal IH on brain inflammation; 3) To establish whether Caff and/or NSAID co-treatment has synergistic effects for reducing brain inflammation and apoptosis; 4) To examine whether topical ocular Keto has effects in the brain; and 5) To investigate whether early treatment during IH exposure is more beneficial than late treatment during recovery/reoxygenation. The primary outcome was brain histopathology and morphometry, and the secondary outcomes were biomarkers of oxidative stress, inflammation, myelination, and apoptosis.
2. Materials and Methods
2.1. Animals
All experiments were approved by the State University of New York, Downstate Medical Center Institutional Animal Care and Use Committee, Brooklyn, NY (Protocol #11-10255). Certified infection-free, timed-pregnant Sprague Dawley rats were purchased from Charles River Laboratories (Wilmington, MA) at 18 days gestation. The animals were housed in an animal facility with a 12-hour-day/12-hour-night cycle and provided standard laboratory diet and water ad libitum until delivery of their pups.
2.2. Experimental Design
Within 2-4 hours of birth, newborn rat pups delivering on the same day were pooled and randomly assigned to expanded litters of 18 pups/litter (9 males and 9 females). Gender was identified by the anogenital distance. The expanded litter size was used to simulate poor nutrition and relative postnatal malnutrition of ELBW infants who are at increased risk for severe oxidative stress and need for caff and/or NSAIDs. Two phases of treatment were conducted: 1) Early Treatment: Animals were exposed to IH from P0 to P14 and treated with either intraperitoneal (IP) and topical ocular saline (Sal/Sal), IP Caff, topical ocular Keto, IP Ibu, combined IP Caff and topical ocular Keto (Caff/Keto), or combined IP Caff and Ibu (Caff/Ibu). Treatment with Caff consisted of 20 mg/kg (diluted in sterile normal Sal) loading dose on P0 followed by 5 mg/kg/day from P1 to P14 (20 µL volume). Treatment of Keto consisted of a single drop of Acuvail (4% ocular solution), into both eyes from P0 to P14. Treatment with Ibu consisted of 10 mg/kg loading dose on P0 followed by 5 mg/kg/day on P1 and P2. Treatment with Caff/Keto consisted of equivalent doses and volumes of IP Caff and topical ocular Acuvail, and treatment with Caff/Ibu consisted of equivalent doses and volumes of Caff and Ibu. Treatment with Sal/Sal consisted of equivalent IP and topical ocular volumes from P0 to P14. Rats were allowed to recover in room air (RA) until P21 with no further treatment; and 2) Late Treatment: Animals were exposed to IH from P0 to P14 after which they were removed from IH conditions and placed in RA until P21. The pups were administered doses as described above, of Sal, Caff, or Keto from P14 to P21, Ibu from P15 to P17, Caff/Keto from P15-P21; and Caff (P15-P17)/Ibu (P15-P17). A flow chart representing the early and late phases is presented in Figure 1.
2.3. Neonatal Intermittent Hypoxia (IH) Profiles
Animals randomized to IH were placed with the dams in specialized oxygen chambers attached to an oxycycler (BioSpherix, NY, USA). The IH profile consisted of an initial exposure of hyperoxia (50% O2) for 30 minutes followed by three brief, 1-minute, clustered hypoxic events (12% O2), with a 10-minute re-oxygenation in 50% O2 between each hypoxic event. Recovery from IH occurred in 50% O2 following each clustered IH event for 2.5 hours for a total of 8 clustering IH episodes per day for 14 days, as previously described [38,39]. Oxygen saturation was confirmed on a sentinel un-anesthetized rat pup from each group using the MouseOx Pulse Oximeter and WinDaq Waveform Browser software (STARR Life Sciences Corp., Oakmont PA) before and after IH exposure.
2.4. Sample Collection & Processing
At euthanasia (P21), whole brains were removed, weighed and placed in 10% neutral buffered formalin (NBF). Fixed coronal sections were placed in cassettes and sent to the Histowiz, Inc. (Long Island City, NY USA) for processing, paraffin embedding, sectioning and hematoxylin and eosin (H&E) staining using standard laboratory techniques. Unstained sections were used for TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling), Luxol fast blue, and immunohistochemistry (IHC). Fresh brain samples excised from the cerebral cortex (n=6 samples/group) were placed in sterile Lysing Matrix D 2.0 mL tubes containing 1.4 mm ceramic spheres (MP Biomedicals, Santa Ana, CA, USA) and 1.0 mL PBS then snap-frozen in liquid nitrogen. Samples were stored at -80oC until analysis of cytokines using enzyme-linked immunosorbent assay (ELISA).
2.5. Brain histopathology and Morphometry
H&E stained images were captured at 20X magnification (scale bar=20 µm) using an Olympus BX53 microscope, DP72 digital camera, and CellSens imaging software (Olympus, Center Valley, PA), attached to a Dell Precision T3500 computer (Dell, Round Rock, TX). Morphometric analyses included total brain width (frontal-occipital and inter-parietal); and width of layers I-VI of the cerebral cortex [40]. Twelve sections were analyzed for each group. Measurements were made using the count and measure tool of the Olympus CellSens imaging software
2.6. Apoptosis (TUNEL Stain)
Apoptosis was determined in the unstained brain sections using the TUNEL assay kit purchased from Abcam (Waltham, MA, USA), according to the manufacturer’s protocol. Sections were counterstained with Methyl Green.
2.7. Myelin Stain
Myelination was determined using the Luxol fast blue (LFB) stain (VitroVivo Biotech, Rockville, MD, USA). Briefly, unstained cross-sections were de-paraffinized with xylenes and alcohols prior to overnight incubation with Luxol fast blue solution at 56oC. Slides were washed and differentiated in lithium carbonite solution and 70% ethanol, then counter stained with Cresyl Violet solution, washed and mounted with Permount. Images were captured at 40X magnification using an Olympus BX53 microscope, DP72 digital camera, and CellSens imaging software (Olympus, Center Valley, PA, USA), attached to a Dell Precision T3500 computer (Dell, Round Rock, TX, USA). Stain intensity was determined using the count and measure on region of interest tool of the CellSens imaging software (12 measurements/group).
2.8. IHC Assays
Unstained sections were used for IHC of phosphorylated (p) nuclear factor kappa B (NFkB), IkB, and pro-inflammatory cytokines, including interleukin-1(IL-1)β, IL-6, tumor necrosis factor (TNF)α, and IL-10. Unstained sections (2 sections per slide) were de-paraffinized with xylenes and alcohols prior to unmasking of antigens. Sections were washed several times in PBS containing triton X-100 (PBS-T) and incubated in blocking solution (Cell Signaling Technologies, Danvers, MA) for 1 hour. The blocking solution was removed and primary antibodies for cytokines (Santa Cruz Biotechnology, Inc., Dallas, TX) were added and sections were incubated overnight. The sections were washed and incubated with Signal Boost antibody serum secondary antibodies (Cell Signaling) for 1 hour. After washing, sections were counterstained with hematoxylin Images were captured at 20X magnification (scale bar=20 µm) using an Olympus BX53 microscope, DP72 digital camera, and CellSens imaging software (Olympus, Center Valley, PA), attached to a Dell Precision T3500 computer (Dell, Round Rock, TX). Twelve sections were analyzed for each group. Measurements were made using the count and measure tool of the Olympus CellSens imaging software.
2.9. ELISA Assays
All samples were analyzed on the same day. On the day of analyses, the tubes were allowed to defrost on ice and placed in a high-speed FastPrep-24 instrument (MP Biomedicals, Santa Ana, CA, USA), which utilizes a unique, optimized motion to efficiently homogenize biological samples within 40 seconds via multidirectional simultaneous beating of the Lysing Matrix ceramic beads on the tissue. The homogenates were then centrifuged at 4oC at 10,000 rpm for 20 minutes. The supernatant was filtered, and the filtrate was used for the assay of total cellular proteins, IL-1β, TNFα, IL-6, and IL-10 using commercially available kits purchased from Millipore Sigma Millipore-Sigma (St. Louis, MO, USA) and MyBioSource (San Diego, CA). All samples were processed and assayed according to the manufacturer’s protocol. A total of 6 samples per experimental group were analyzed. All data were standardized using total cellular protein levels.
2.10. Total Cellular Protein Levels
On the day of assays, an aliquot (10 µL) of the cerebral cortex homogenates was used for total cellular protein levels using the Bradford method (Bio-Rad, Hercules, CA USA) with bovine serum albumin as a standard.
2.11. Statistical Analysis
To determine differences among the treatment groups, a test for normality was first conducted using Bartlett’s test. Normally distributed data were analyzed using two-way analysis of variance (ANOVA) with Dunnett’s post-hoc tests. Non-normally distributed data were analyzed using Kruskall Wallis test with Dunn’s multiple comparison test. Data are presented as mean±standard error of the mean (SEM) and a p-value of <0.05 was considered as statistically significant, using SPSS version 26.0 (SPSS Inc., Chicago, IL, USA). Graphs were prepared using GraphPad Prizm version 7.03 (GraphPad, San Diego, CA, USA).
3. Results
3.1. Brain Weight
Whole brain weight and brain to body weight ratios are presented in Figure 2. Panels A and C represent body weight and brain/body weight ratios for the early treatment groups, respectively; and panels B and D represent the corresponding late treatment groups. The solid white bar represents the groups exposed to room air (RA), the checked bar represents the groups exposed to 50% O2 (hyperoxia); and the solid black bar represents the groups exposed to neonatal IH. In the early treatment groups, animals exposed to neonatal IH and treated with Sal/Sal or Sal/Ibu had significantly higher brain/body weight ratios compared to their RA or hyperoxia littermates (Figure 2C). In the late treatment groups, animals exposed to hyperoxia and neonatal IH and treated with ketorolac, ibuprofen with and without caffeine had persistently higher brain to body weight ratios (Figure 2D).
3.2. Histopathology
Representations of the H&E-stained cerebral cortex sections for the early treatment groups are presented in Figure 3. The upper panels represent the groups exposed to RA; the middle panels represent the groups exposed to hyperoxia; and the lower panels represent the groups exposed to neonatal IH. Treatment groups are labeled in the top panels. Microscopically, the following horizontal organizations of cellular layers (Laminae) were seen as previously described [40] in the cortex: Molecular (Plexiform) layer I consists of apical dendritic tufts of the pyramidal neurons, horizontally organized axons, Cajal-Retzius cells and glial cells. External granular layer II consists of mainly granular cells and small pyramidal cells. External pyramidal layer III consists of small and medium-sized pyramidal cells. Internal granular layer IV has small round granular cells (spiny stellate cells, spiny interneurons) and small pyramidal cells. Internal pyramidal layer V has the largest pyramidal cells. Axons of the pyramidal cells leave the cortex and reach subcortical areas. Multiform (Polymorphous) layer VI: Excitatory cells like spiny stellate cells, pyramidal cells, inverted pyramidal neurons, bipolar/fusiform cells, and odd-shaped cells are present in layer VI. It also holds inhibitory interneurons.
In the RA-exposed group, cellular layers were wider and showed increased cellularity in groups treated with Ketorolac, both alone and in conjunction with Caff. Caff, Ibu and the Caff/Ibu groups all displayed increased brain dimensions on gross specimen analysis. However, cellular layers V and VI with Caff/Ibu co-treatment were reduced in thickness to a significant degree. In the hyperoxia early treatment group, saline treatment resulted in showed significantly reduced frontal-occipital diameter compared to RA controls, but on cellular morphometric analysis, increased cortical thickness in all 6 layers was found. Increased brain dimensions were grossly appreciated in NSAID treated groups; however cellular layer analysis showed layer III was significantly increased in thickness in all treatment subgroups. In the neonatal IH treatment groups, Caff and Keto individual treatment showed marked increase in size of the specimen as well as increased width of layers III, IV and VI, and this effect was maintained in the groups subjected to co-treatment of the 2 agents also. Ibuprofen treatment also conferred increased brain size as well as increase in cell layer width of layers III and V, both alone and in conjunction with Caff. Overall, layers IV and VI were narrower in all treatment groups compared to the control IH group. All histological findings were corroborated with morphometric analyses presented in Table 1.
Representations of the H&E-stained cerebral cortex sections for the late treatment groups are presented in Figure 4. The upper panels represent the groups exposed to RA; the middle panels represent the groups exposed to hyperoxia; and the lower panels represent the groups exposed to neonatal IH. Treatment groups are labeled in the top panels. In groups exposed to RA, Caff treatment conferred significant increased biparietal length and increased width of layers I and II as compared to controls. Keto treatment increased width of cell layer II, holding pyramidal cells, to a greater extent as compared to layer III. Ibu treatment showed increased frontal-occipital (FO) diameter, however only cell layer II showed increase in width. Caff/Keto co-treatment showed an increase in biparietal diameter of the gross brain specimen, while Caff/Ibu increased FO diameter as well increased width of cell layers II, V and VI. In the hyperoxia groups, control animals, as well as all treatment groups were found to have increased brain volumes as shown by increased cross sectional diameters compared to RA counterparts, perhaps due to increased cellular proliferation attributable to higher oxygenation. Caff alone showed increased layer I, and Ibu showed increased layer III cells, together their co-treated groups showed increased width in layers I, II and V. In the neonatal IH groups, all treatment groups were found to have increased brain volumes as shown by increased cross sectional diameters compared to RA counterparts. Cellular layer morphometric analysis showed increased cell I layer width in treatment with Caff or Ibu separately but not in the co-treatment group, which instead had wide layers III and V. Caff/Keto co-treatment had significantly wider layers V and VI compared to the control IH group. All histological findings were corroborated with morphometric analyses presented in Table 2.
3.3. Apoptosis
Apoptosis, as evidenced by the TUNEL stain for early and late treatment groups are presented in Figure 5 and Figure 6, respectively. Apoptosis is indicated by the brown stain. Sections were counter stained with methyl green which indicates negativity. In the early treated groups, apoptosis was less evident in the Sal, Keto, and Caff/Keto groups exposed to RA. Ibu treatment caused the highest TUNEL positivity. In hyperoxia, TUNEL positivity was highest in the Sal and Ibu groups. Although there was some positive staining with Keto and Caff/Keto, there was also a high presence of methyl green. Caff appeared to have the most effect for reducing apoptosis in hyperoxia. In neonatal IH, all groups showed TUNEL positivity, but treatment with Keto and Caff/Keto were most effective for reducing apoptosis (Figure 5). The late treatment groups apoptosis was highest with Caff/Keto and lowest with Sal and Keto in RA. In hyperoxia, Keto and Caff/Keto reduced apoptosis compared to all other groups. In neonatal IH, treatment with Caff, Keto, and Caff/Keto reduced apoptosis compared to Sal, Ibu and Caff/Ibu (Figure 6). Quantitative analysis of the stain intensity is presented in Table 3 and Table 4 for early and late treatment, respectively.
3.4. Myelination
Representative cerebral cortex sections stained for myelination as indicated by the LFB stain for early and late treatment groups are presented in Figure 7 and Figure 8, respectively. In the early RA groups, there was increased myelination with Ibu and Caff/Ibu treatment. In early hyperoxia, myelination was significantly increased in the Sal group compared to the Sal RA group, and declined with the other treatments in hyperoxia, although treatment with Caff and Caff/Ibu produced the highest myelination compared to the Keto, Ibu, and Caff/Keto groups. Myelination was reduced in all groups exposed to early IH (Figure 7). In the late treatment groups, myelination was highest with Caff, Keto, Ibu, and Caff/Ibu treatment in RA. However, exposure to hyperoxia and neonatal IH decreased myelination in all groups (Figure 8). Quantitative analysis of the stain intensity is presented in Table 3 and Table 4 for early and late treatment, respectively.
3.5. Cytokines Levels
Cytokine levels in the brain homogenates for the early and late treatment groups are presented in Figure 9 and Figure 10, respectively. All cytokines were elevated in the Sal-treated groups exposed to neonatal IH. This response was reversed with Caff, Keto, and Caff/Keto. However, IL-1β and IL-10 were also reduced with Ibu and Caff/Ibu, but not TNFα and IL-6. For the late treatment groups, all cytokines were lower with all treatments and exposures compared to saline controls in RA. Overall, cytokine levels were lower in the hyperoxia and neonatal IH groups. Of all groups Caff/Keto most effectively reduced cytokines in the neonatal IH exposed groups.
3.6. IHC
Immunoreactivity of IkB, IL-1β, and TNFα (brown stain) are presented in Figure 11, Figure 12, Figure 13, Figure 14, Figure 15 and Figure 16. No differences were noted among the groups for NFkB, IL-6 and IL-10 (data not shown). IHC of IkB in the early and late treatment groups is presented in Figure 11 and Figure 12, respectively. In early RA, IkB was induced with Keto and to a lesser degree Caff/Keto. Hyperoxia decreased IkB in all groups, but treatment with Keto and Caff/Keto showed mild immunostaining. This effect was also evident in neonatal IH, but to a greater degree. In the late treatment groups, IkB was also induced with Keto and Caff/Keto in all oxygen environments. Overall, Keto and Caff/Keto treatment appears to have a positive influence on IkB induction regardless of early or late treatment. Quantitative analysis of the stain intensity is presented in Table 3 and Table 4 for early and late treatment, respectively. IHC of IL-1β in the early and late treatment groups is presented in Figure 13 and Figure 14, respectively. In the RA groups, only Keto treatment increased IL-1β, and to a lesser degree, Caff/Ibu. There was no staining in any of the groups exposed to hyperoxia and exposure to neonatal IH show minor staining in the cerebral cortex layer I with Caff, and punctate staining with Ibu. Late treatment also showed no appreciative IHC staining for IL-1β, except for minor staining in the cerebral cortex layer I with Caff as shown with early treatment. TNFα IHC in the early and late treatment groups are presented in Figure 15 and Figure 16, respectively. In the early RA groups, treatment with Sal, Ibu and Caff/Ibu showed highest staining in RA, while in hyperoxia, the highest staining was noted with Sal, Caff, Caff/Keto and Caff/Ibu. In IH, minor staining was noted with Caff and Caff/Keto late treatment. Quantitative analyses of the cytokine staining intensities are presented in Table 3 and Table 4 for early and late treatment, respectively.
4. Discussion
The present study was conducted to address specific questions regarding: 1) the effects of early exposure of hyperoxia and neonatal IH on inflammation in the neonatal rat brain; 2) the comparative efficacy of Caff, Keto, or Ibu on inflammation in the neonatal rat brain; 3) synergistic effects of Caff and/or NSAID co-treatment for reducing hyperoxia- and IH-induced brain inflammation; 4) topical ocular Keto effects in the brain; and 5) whether early treatment during hyperoxia or IH exposure is more beneficial than late treatment during recovery/reoxygenation. We utilized a model of neonatal IH developed in our laboratory that consistently produces oxidative stress, as previously reported [38,39]. Using rodent models of brain injury, studies have shown that rodents at postnatal day 7-10 is developmentally similar to a term human infant [41,42,43,44]. For our experiments, we used newborn rats on the first day of life which corresponds to a very preterm infant brain. Early treatment correlated with the preterm infant brain and late treatment with the term infant brain. The major findings of these experiments were: 1) early exposure of the neonatal brain to either hyperoxia or neonatal IH impairs brain weight and development, and causes induced inflammation; 2) Given individually, Caff, and Keto, but not Ibu were most beneficial for reducing apoptosis and inflammation; 3) Caff/Keto demonstrated better synergistic effects for inducing IkB and reducing apoptosis and inflammation than Caff/Ibu; 4) topical ocular Keto enters the brain and confers beneficial effects with or without Caff; and 5) early treatment during exposure is more beneficial for inducing myelination, reducing apoptosis and overall improvement in neuro inflammation and cortical organization than late treatment during recovery/reoxygenation. These findings support our hypothesis and demonstrate that not only the choice of treatment also the timing of its administration has significant effects in the premature brain.
Exposure to hyperoxia and IH showed significant elevations in brain/body weight ratios, in all early groups except Caff and in late Ibu, Caff/Keto and Caff/Ibu groups. This may be due to the lower body weights in response to abnormal oxygenation [45]. Organ weight is one of the most sensitive indicators of drug toxicity and organ weight relative to body weight accounts for changes in overall body weight [46], and may suggest an overall physiological response to the drug. Regarding cerebral cortex organization, the RA groups showed an overall increase in cell differentiation and cortical thickness that was relatively preserved in the earlier cohort, however in the late treatment groups, the effects of increased thickness of layers I-V became less pronounced and only layer II in NSAID treatment groups remained prominent. Literature shows that layer 3 consists of pyramidal cells mainly and serves as association and commissural corticocortical fibers [42]. With an ever-increasing body of evidence in favor of dysmaturation of white matter as the underlying mechanism of neurodysregulation of the ELGAN’s brain [47,48,49]. Our findings shed further light on the effects of hyperoxia and neonatal IH on cortical cell organization and deserving further research. In the hyperoxia groups, all treatment groups were found to have increased brain volumes as shown by increased cross sectional diameters compared to RA counterparts, perhaps due to increased cellular proliferation attributable to higher oxygenation that persisted, albeit to a lesser extent, in the late treatment littermates. In cellular layer analysis, however, effects on layer 3 were largely positive in the early treatment cohort but less so in the later one. It remains to be seen whether this increase in cell mass was due to hypertrophy or hyperplasia, as this study did not aim to find the cytopathologic pathologic nature of hyperoxia-induced cell changes, but studies by Kato et. al [9,10] seem to suggest this.
In the IH exposed groups, gross size of the specimen was enlarged in both early and late groups even in controls, which seems to suggest that this effect was conferred by IH and not by the treatment. Further examination of the cell layers revealed that layer III remained mostly unchanged, however layers IV and VI became narrower in the early treatment groups but cell layers in late treatment cohort did not show appreciable changes from controls. This may suggest that early treatment, at a time when the brain is still developing, is detrimental to the internal granular layer. This layer, also known as the main input cortical station, receives sensory input from periphery, and layer VI or the fusiform/multiform layer comprised of axons of the fusiform cells which distribute commissural fibers and corticothalamic projections ending in the thalamus [40]. As the early treatment coincides with cell migration stage in the preterm population, perhaps reduced cell migration and axonal projection lead to suppression of abnormal circuitry that contribute to sensory and emotional disturbances that are a hallmark of neonatal brain injury. Furthermore, late treatment did not produce similar cell layer findings suggesting that after migration, cells are less susceptible to the adverse effects of hyperoxia and/or IH.
The t½ of Caff is about 4 days, while Keto and Ibu are eliminated much faster, having a half-life of 4-6 hours. This would suggest that treatment with Ibu, as happens with PDA closure, would confer short term anti-inflammatory effects while Caff would provide more sustained and pervasive effects in the tissue. The findings from our study suggest however that the effect of the NSAID’s on brain differentiation is related more to the dosing being timed with stages of neural tissue development than their duration of action. The effects of topical ocular Keto alone and in conjunction with Caff indicate that the drug crosses the blood-ocular barrier and enters into the brain, and provides further evidence of a connection between the eye and the brain [50].
Both early and late treatment with Keto and Caff/Keto reduced apoptosis or programmed cell death in all exposures, while Caff, Ibu or their combination was not effective. This seems to suggest that Caff may have diminished neuroprotective in chronic hyperoxia and IH even with Ibu co-treatment. This must also be seen in the context of apoptosis that occurs in later stages of preterm brain maturation as part of normal neuronal development once cells have reached their “programmed” destination in the brain [49]. Many studies have shown that Caff alone [51] and with morphine induces apoptosis [52]. The mechanism of Caff induction of apoptosis even in RA conditions, remains to be determined.
Myelination is considered a proxy for white matter (WM) development and has been positively correlated with brain development [53,54]. In our study, early Ibu and Caff/Ibu in RA as well as hyperoxia seemed to favor, while late treatment caused reduced myelination. This confirms that late treatment may be more injurious. Overall, IH seems to negatively affect the process of myelination regardless of early or late treatment. This finding confirms previous reports which shows irreversible dysmyelination with chronic hypoxia and intermittent hypoxia [55]. Although several of the treatment groups showed higher myelination, it is debatable whether function is correlated with these quantities. WM dysmaturation studies have shown that increased activity of pre-OL's is responsible for abnormal corticofugal tracks, helping the authors form the theory that perhaps better markers of “meaningful myelination” need to be found as predictors of brain development [49].
Studies have shown that separate IkB molecules preferentially inhibit distinct Rel/NF-kB protein dimers [56]. This interaction enables the IkB proteins to mask the nuclear localization signal and prevent nuclear translocation of Rel/NFkB protein dimers. Many aspects of the inflammatory responses are also controlled by NFkB including the production of acute-phase proteins such as serum amyloid A, angiotensinogen and factors of the complement cascade and the induction of pro- inflammatory cytokines such as IL-1 (both α and β) and TNFα [57]. IkB is a the most studied inhibitor of NFkB. During activation of NFkB, numerous stimuli, including IL-1 and TNFα, activate a complex of IkB kinases to degrade IkB [58]. While there were no appreciable differences in NFkB, we showed that Ibu alone and in combination with Caff induces IkB, although early treatment was more effective that late treatment. Since Ibu is a potent anti-inflammatory drug, it is likely that those effects may be in part due to induction of IkB. To our knowledge, this is the first report of Ibu effect on IkB in the brain, in the setting of hyperoxia or IH. However, studies have shown a suppressive effect of Ibu on NFkB in respiratory cells [59].
The other cytokines namely Il-1β, IL-6, and TNFα, seem to vary in their relationship as predictors of response to drug treatment in different oxygen environments. In the CNS, cytokines exert their function through both traditional engagement of their receptors, which are expressed by both glial and neuronal cells and through less traditional means such as modulation of neurotransmitter receptor function. For example, IL-1 modulates γ-aminobutyric acid-responsive neurotransmitter receptors to enhance inhibitory responses, and both IL-1 and IL-6 modulate synaptic plasticity through inhibiting formation of long-term potentiation [60]. Excitotoxicity is a major pathway of neuronal cell death that is associated with ischemia, trauma, and neurodegenerative diseases and results from an uncontrolled elevation in intracellular calcium that enters the cell through chronically activated N-methyl-D-aspartatic acid receptors. Agents such as antioxidants, growth factors, and certain cytokines protect against excitotoxicity [61]. In RA, IL-1β, IL-6, and TNF-α seem to show similar trends, with a reduction in both early and late NSAID treatment groups. In the presence of hyperoxia, Il-1β, and TNFα behave similarly and are reduced in response to early NSAID treatment, but IL-6 seems to rise, suggesting a mechanism of action different than the other 2. IL-6 has often been proposed as the “exercise factor” as it rises in response to strenuous exercise [62]. Perhaps hyperoxia seems to affect IL-6 differently by virtue of its response to increased oxygen as seen in exercise and our hyperoxia model. Response to Caff is interesting in that all cytokines show an increase in early group, suggesting perhaps that Caff alone can work through cyclic AMP and adrenaline to increase their levels. In later treatment, this effect is seen in the Caff/Ibu group, suggesting decreased effect of Ibu and confirming beneficial synergism with Caff. In IH, all cytokines seem to decrease with treatment, suggesting a role for use these neuroprotective agents in IH.
Although the present study has clinical significance, there are limitations. We did not examine the responses at P14. This would have provided further information regarding immediate effects of the exposures with or without treatment. We also did not differentiate between the outcomes of male and female as gender may be confounding factor in response to neuroprotective therapies. Nevertheless, our study shows that the use of Caff and/or NSAIDs during early postnatal life appears to confer better neurologic outcomes in terms of cortical development and neuroinflammation, when given in the setting of hyperoxia or neonatal IH. In contrast, later treatment has reduced efficacy for promoting myelination and normal cortical development. These findings suggest that achieving better oxygenation to ensure desired effects of neonatal management with Caff and/or NSAIDs should remain a priority. We also showed that the developing brain responds to hypoxia by upregulating programmed cell death, confirming previous studies [63,64].
5. Conclusions
In conclusion, the effects of medication in the neonate are a challenge for pharmacists and pharmacologists due to their unique physiologic attributes. Ascertaining safety takes precedence over efficacy, yet pursuit of both is desirable for the achievement of the ultimate goal, a healthy infant. While neonatal medicine has made significant improvements in care and ventilation of preterm infants, the incidence of apnea of prematurity and frequent intermittent hypoxia remain high. Further studies are needed to determine whether our findings can be extrapolated to the human situation.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, M.B., J.V.A. and K.D.B.; methodology, M.B., C.L.C. and K.D.B.; validation, J.V.A and K.D.B.; formal analysis, M.B., C.L.C, K.D.B., investigation, M.B., C.L.C. and K.D.B.; resources, J.V.A. and K.D.B.; data curation, M.B. and K.D.B.; writing—original draft preparation, M.B.; writing—review and editing, M.B., C.L.C., J.V.A., and K.D.B.; visualization, M.B. and K.D.B.; supervision, J.V.A. and K.D.B; project administration, J.V.A. and K.D.B.; funding acquisition, J.V.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (Grant #U54HD071594) and the Alicia and Madu Rao Family Research Foundation (Grant # EIN 262145011).
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board of State University of New York, Downstate Health Sciences University Institutional Animal Care and Use Committee, Brooklyn, NY (Protocol #11-10255).
Data Availability Statement
Data are available upon a reasonable request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martin, J.A.; Kung, H.C.; Mathews, T.J.; Hoyert, D.L.; Strobino, D.M.; Guyer, B.; Sutton, S.R. Annual summary of vital statistics: 2006. Pediatrics 2008, 121, 788-801. [CrossRef]
- Woodward, L.J.; Edgin, J.O.; Thompson, D.; Inder, T.E. Object working memory deficits predicted by early brain injury and development in the preterm infant. Brain 2005, 128, 2578-2587. [CrossRef]
- Bayless, S.; Stevenson, J. Executive functions in school-age children born very prematurely. Early Hum Dev 2007, 83, 247–254. [CrossRef]
- Platt, M.J.; Cans, C.; Johnson, A.; Surman, G.; Topp, M.; Torrioli, M.G.; Krageloh-Mann, I. Trends in cerebral palsy among infants of very low birthweight (<1500 g) or born prematurely (<32 weeks) in 16 European centres: a database study. Lancet 2007, 369, 43-50.
- Wood, N.S.; Costeloe, K.; Gibson, A.T.; Hennessy, E.M.; Marlow, N.; Wilkinson, A.R. The EPICure study: associations and antecedents of neurological and developmental disability at 30 months of age following extremely preterm birth. Arch Dis Child 2005, 90, F134–F140. [CrossRef]
- Newton, C.R. Global Burden of Pediatric Neurological Disorders. Semin Pediatr Neurol 2018, 27, 10-15. [CrossRef]
- Marlow, N.; Wolke, D.; Bracewell, M.A.; Samara, M. Neurologic and developmental disability at six years of age after extremely preterm birth. N Engl J Med 2005, 352, 9-19. [CrossRef]
- Kato, H.; Liu, Y.; Araki, T.; Kogure, K. Temporal profile of the effects of pretreatment with brief cerebral ischemia on the neuronal damage following secondary ischemic insult in the gerbil: cumulative damage and protective effects. Brain Res 1991; 553:238±242. [CrossRef]
- Kato, H.; Kogure, K. Neuronal damage following non-lethal but repeated cerebral ischemia in the gerbil. Acta Neuropathol (Berl) 1990, 79, 494-500. [CrossRef]
- Kato, H.; Kogure, K.; Nakano, S. Neuronal damage following repeated brief ischemia in the gerbil. Brain Res 1989, 479, 366-370. [CrossRef]
- Volpe, J.J. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol 2009, 8, 110–124. [CrossRef]
- Zhang, Q.; Ding, Y.; Yao, Y.; Yu, Y.; Yang, L.; Cui, H. Creating rat model for hypoxic brain damage in neonates by oxygen deprivation. PLoS One 2013, 8, e83589. [CrossRef]
- Nagata N, Saji M, Ito T, Ikeno S, Takahashi H, Terakawa N. Repetitive intermittent hypoxia-ischemia and brain damage in neonatal rats. Brain Dev 2000, 22, 315-20. [CrossRef]
- Schmid, M.B.; Hopfner, R.J.; Lenhof, S.; Hummler, H.D.; Fuchs, H. Cerebral oxygenation during intermittent hypoxemia and bradycardia in preterm infants. Neonatology 2015, 107, 137-46. [CrossRef]
- Bunn, H.F.; Poyton, R.O. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev 1996, 76, 839-885. [CrossRef]
- Hochachka, P.W.; Buck, L.T.; Doll, C.J.; Land, S.C. Unifying theory of Hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 1996, 93, 9493-9498. [CrossRef]
- Gozal, E.; Sachleben, L.R. Jr.; Rane, M.J.; Vega, C.; Gozal, D. Mild sustained, and intermittent hypoxia induce apoptosis in PC-12 cells via different mechanisms. Am J Physiol Cell Physiol 2005, 288, C535-C542.
- Rice, J.E. 3rd.; Vannucci, R.C.; Brierley. J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981, 9, 131-141.
- Gozal, D.; Daniel, J.M.; Dohanich, G.P. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci 2001, 21, 2442-2450. [CrossRef]
- Gozal, E.; Row, B.W.; Schurr, A.; Gozal, D. Developmental differences in cortical and hippocampal vulnerability to intermittent hypoxia in the rat. Neurosci Lett 2001, 305, 197-201. [CrossRef]
- Swanson, L.W. Brain maps 4.0—Structure of the rat brain: An open access atlas with global nervous system nomenclature ontology and flatmaps. J Comp Neurol 2018, 526, 935–943. [CrossRef]
- Rezaie, P.; Dean, A. Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology 2002, 22, 106-32. [CrossRef]
- Wixey, J.A.; Sukumar, K.R.; Pretorius, R.; Lee, K.M.; Colditz, P.B.; Bjorkman, S.T.; Chand, K.K. Ibuprofen Treatment reduces the neuroinflammatory response and associated neuronal and white matter impairment in the growth restricted newborn. Front Physiol 2019, 10, 541. [CrossRef]
- Aranda, J.V.; Salomone, F.; Valencia, G.B.; Beharry, K.D. Non-steroidal anti-inflammatory drugs in newborns and infants. Pediatr Clin North Am 2017, 64, 1327-1340. [CrossRef]
- Schmidt, B.; Roberts, R.S.; Anderson, P.J.; Asztalos, E.V.; Costantini, L.; Davis, P.G.; Dewey, D.; D'Ilario, J.; Doyle, L.W.; Grunau, R.E.; Moddemann, D.; Nelson, H.; Ohlsson, A.; Solimano, A.; Tin, W.; Caffeine for Apnea of Prematurity (CAP) Trial Group. Academic performance, motor function, and behavior 11 years after neonatal caffeine citrate therapy for apnea of prematurity: An 11 year follow-up of the CAP randomized clinical trial. JAMA Pediatr 2017, 171, 564-572.
- Abu-Shaweesh, J.M.; Martin, R.J. Caffeine use in the neonatal intensive care unit. Semin Fetal Neonatal Med 2017, 22, 342-347. [CrossRef]
- Bucher, H.U.; Duc, G. Does caffeine prevent hypoxaemic episodes in premature infants? A randomized controlled trial. Eur J Pediatr 1988, 147, 288-91.
- Rhein, L.M.; Dobson, N.R.; Darnall, R.A.; Corwin, M.J.; Heeren, T.C.; Poets, C.F.; McEntire, B.L.; Hunt, C.E.; Caffeine Pilot Study Group. Effects of caffeine on intermittent hypoxia in infants born prematurely: a randomized clinical trial. JAMA Pediatr 2014, 168, 250-7.
- Carty, M.L.; Wixey, J.A.; Reinebrant, H.E.; Gobe, G.; Colditz, P.B.; Buller, K.M. Ibuprofen inhibits neuroinflammation and attenuates white matter damage following hypoxia-ischemia in the immature rodent brain. Brain Res 2011, 1402, 9-19. [CrossRef]
- Chand, K.K.; Miller, S.M.; Cowin, G.J.; Mohanty, L.; Pienaar, J.; Colditz, P.B.; Bjorkman, S.T.; Wixey, J.A. Neurovascular Unit Alterations in the Growth-Restricted Newborn Are Improved Following Ibuprofen Treatment. Mol Neurobiol 2022, 59, 1018-1040. [CrossRef]
- Browne, K.D.; Iwata, A.; Putt, M.E.; Smith, D.H. Chronic ibuprofen administration worsens cognitive outcome following traumatic brain injury in rats. Exp Neurol 2006, 201, 301-7. [CrossRef]
- Papacci, P.; De Francisci, G.; Iacobucci, T.; Giannantonio, C.; De Carolis, M.P.; Zecca, E.; Romagnoli, C. Use of intravenous ketorolac in neonate and premature babies. Paediatr Anaesth 2004, 14, 487-92. [CrossRef]
- Lynn, A.M.; Bradford, H.; Kantor, E.D.; Seng, K.Y.; Salinger, D.H.; Chen, J.; Ellenbogen, R.G.; Vicini, P.; Anderson, G.D. Postoperative ketorolac tromethamine use in infants aged 6-18 months: the effect on morphine usage, safety assessment, and stereo-specific pharmacokinetics. Anesth Analg 2007, 104, 1040-51.
- Stone, S.B. Ketorolac in postoperative neonates and infants: A systematic review. J Pediatr Pharmacol Ther 2021, 26, 240-247. [CrossRef]
- Zuppa, A.F.; Mondick, J.T.; Davis, L.; Cohen, D. Population pharmacokinetics of ketorolac in neonates and young infants. Am J Ther 2009, 16, 143-146. [CrossRef]
- Avila-Vazquez, M.; Maffrand, R.; Sosa, M.; Franco, M.; De Alvarez, B.V.; Cafferata, M.L.; Bergel, E. Treatment of retinopathy of prematurity with topical ketorolac tromethamine: a preliminary study. BMC Pediatr 2004, 4, 15. [CrossRef]
- Giannantonio, C.; Papacci, P.; Purcaro, V.; Cota, F.; Tesfagabir, M.G.; Molle, F.; Lepore, D.; Baldascino, A.; Romagnoli, C. Effectiveness of ketorolac tromethamine in prevention of severe retinopathy of prematurity. J Pediatr Ophthalmol Strabismus 2011, 48, 247-51. [CrossRef]
- Manlapaz-Mann, A.; Cai, C.L.; Bodkin, D.; Mustafa, G.; Aranda, J.V.; Beharry, K.D. Effects of omega 3 polyunsaturated fatty acids, antioxidants, and/or non-steroidal inflammatory drugs in the brain of neonatal rats exposed to intermittent hypoxia. Int J Dev Neurosci 2021, 81, 448-460.
- Soontarapornchai, K.; Cai, C.L.; Ahmad, T.; Aranda, J.V.; Hand, I.; Beharry, K.D. Pharmacodynamic effects of standard versus high caffeine doses in the developing brain of neonatal rats exposed to intermittent hypoxia. Int J Mol Sci 2021, 22, 3473. [CrossRef]
- Chauhan, P.; Rathawa, A.; Jethwa, K.; Mehra, S. The anatomy of the cerebral cortex. In: Pluta R, editor. Cerebral Ischemia. Brisbane (AU): Exon Publications; 2021, Chapter 1.
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 2013, 106-107, 1-16. [CrossRef]
- Craig, A.; Ling Luo, N.; Beardsley, D.J.; Wingate-Pearse, N.; Walker, D.W.; Hohimer, A.R.; Back, S.A. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Experimental Neurology 2003, 181, 231-240. [CrossRef]
- Vannucci, R.C.; Connor, J.R.; Mauger, D.T.; Palmer, C.; Smith, M.B.; Towfighi, J.; Vannucci, S.J. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res 1999, 55, 158-63.
- Brochu, M.E.; Girard, S.; Lavoie, K.; Sébire, G. Developmental regulation of the neuroinflammatory responses to LPS and/or hypoxia-ischemia between preterm and term neonates: An experimental study. J Neuroinflammation 2011, 8, 55. [CrossRef]
- García-Fuentes, E.; Santiago-Fernández, C.; Gutiérrez-Repiso, C.; Mayas, M.D.; Oliva-Olivera, W.; Coín-Aragüez, L.; Alcaide, J.; Ocaña-Wilhelmi, L.; Vendrell, J.; Tinahones, F.J.; Garrido-Sánchez, L. Hypoxia is associated with a lower expression of genes involved in lipogenesis in visceral adipose tissue. J Transl Med 2015, 13, 373. [CrossRef]
- Lazic, S.E.; Semenova, E.; Williams, D.P. Determining organ weight toxicity with Bayesian causal models: Improving on the analysis of relative organ weights. Sci Rep 2020, 10, 6625. [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 2001, 21, 1302-12. [CrossRef]
- Volpe, J.J. Dysmaturation of Premature Brain: Importance, Cellular Mechanisms, and Potential Interventions. Pediatr Neurol 2019, 95, 42-66. [CrossRef]
- Back, S.A.; Miller, S.P. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann Neurol 2014, 75, 469-486.
- Jindal, V. Interconnection between brain and retinal neurodegenerations. Mol Neurobiol 2015, 51, 885-92. [CrossRef]
- Saiki, S.; Sasazawa, Y.; Imamichi, Y.; Kawajiri, S.; Fujimaki, T.; Tanida, I.; Kobayashi, H.; Sato, F,; Sato, S.; Ishikawa, K.; Imoto, M.; Hattori, N. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011, 7, 176-87. [CrossRef]
- Kasala, S.; Briyal, S.; Prazad, P.; Ranjan, A.K.; Stefanov, G.; Donovan, R.; Gulati, A. Exposure to morphine and caffeine induces apoptosis and mitochondrial dysfunction in a neonatal rat brain. Front Pediatr 2020, 8, 593. [CrossRef]
- Dubois, J.; Dehaene-Lambertz, G.; Kulikova, S.; Poupon, C.; Hüppi, P.S.; Hertz-Pannier, L. The early development of brain white matter: a review of imaging studies in fetuses, newborns and infants. Neuroscience 2014, 276, 48-71. [CrossRef]
- Back, S.A. White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol 2017, 134, 331-349. [CrossRef]
- Kanaan, A.; Farahani, R.; Douglas, R.M.; Lamanna, J.C.; Haddad, G.G. Effect of chronic continuous or intermittent hypoxia and reoxygenation on cerebral capillary density and myelination. Am J Physiol Regul Integr Comp Physiol 2006, 290, R1105-14. [CrossRef]
- Li, N.; Karin, M. Is NF-kappaB the sensor of oxidative stress? Faseb J 1999, 13, 1137-1143.
- Carlson, N.G.; Wieggel, W.A.; Chen, J.; Bacchi, A.; Rogers, S.W.; Gahring, L.C. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol 1999, 163, 3963-8.
- Jobin, C.; Sartor, R.B. The I kappa B/NF-kappa B system: a key determinant of mucosalinflammation and protection. Am J Physiol Cell Physiol 2000, 278, C451-62.
- Dauletbaev, N.; Lam, J.; Eklove, D.; Iskandar, M.; Lands, L.C. Ibuprofen modulates NF-kB activity but not IL-8 production in cystic fibrosis respiratory epithelial cells. Respiration 2010, 79, 234-42.
- May, M.J.; Ghosh, S. Rel/NF-κB and IκB proteins: an overview. Semin Cancer Biol 1997, 8, 63-73.
- Johnston, M.V. Excitotoxicity in perinatal brain injury. Brain Pathol 2005, 15, 234-40. [CrossRef]
- Pedersen, B.K.; Steensberg, A.; Fischer, C.; Keller, C.; Keller, P.; Plomgaard, P.; Wolsk-Petersen, E.; Febbraio, M. The metabolic role of IL-6 produced during exercise: is IL-6 an exercise factor? Proc Nutr Soc 2004, 63, 263-7.
- Dewitte, K.; Claeys, M.; Van Craenenbroeck, E.; Monsieurs, K.; Heidbuchel, H.; Hoymans, V.; Stoop, T. Role of oxidative stress, angiogenesis and chemo-attractant cytokines in the pathogenesis of ischaemic protection induced by remote ischaemic conditioning: Study of a human model of ischaemia-reperfusion induced vascular injury. Pathophysiology 2019, 26, 53-59. [CrossRef]
- Hess, D.C.; Blauenfeldt, R.A.; Andersen, G.; Hougaard, K.D.; Hoda, M.N.; Ding, Y.; Ji, X. Remote ischaemic conditioning-a new paradigm of self-protection in the brain. Nat Rev Neurol 2015, 11, 698-710. [CrossRef]
Figure 1.
Experimental Design of Early and Late Treatment. RA (room air); 50% O2 (50% oxygen); IH (intermittent hypoxia); IP (intraperitoneal), OC (topical ocular); P0-P2 (postnatal day 0 to postnatal day 2); P0-P14 (postnatal day 0 to postnatal day 14); P15-P17 (postnatal day 15 to postnatal day 17); P15-P21 (postnatal day 15 to postnatal day 21).
Figure 1.
Experimental Design of Early and Late Treatment. RA (room air); 50% O2 (50% oxygen); IH (intermittent hypoxia); IP (intraperitoneal), OC (topical ocular); P0-P2 (postnatal day 0 to postnatal day 2); P0-P14 (postnatal day 0 to postnatal day 14); P15-P17 (postnatal day 15 to postnatal day 17); P15-P21 (postnatal day 15 to postnatal day 21).
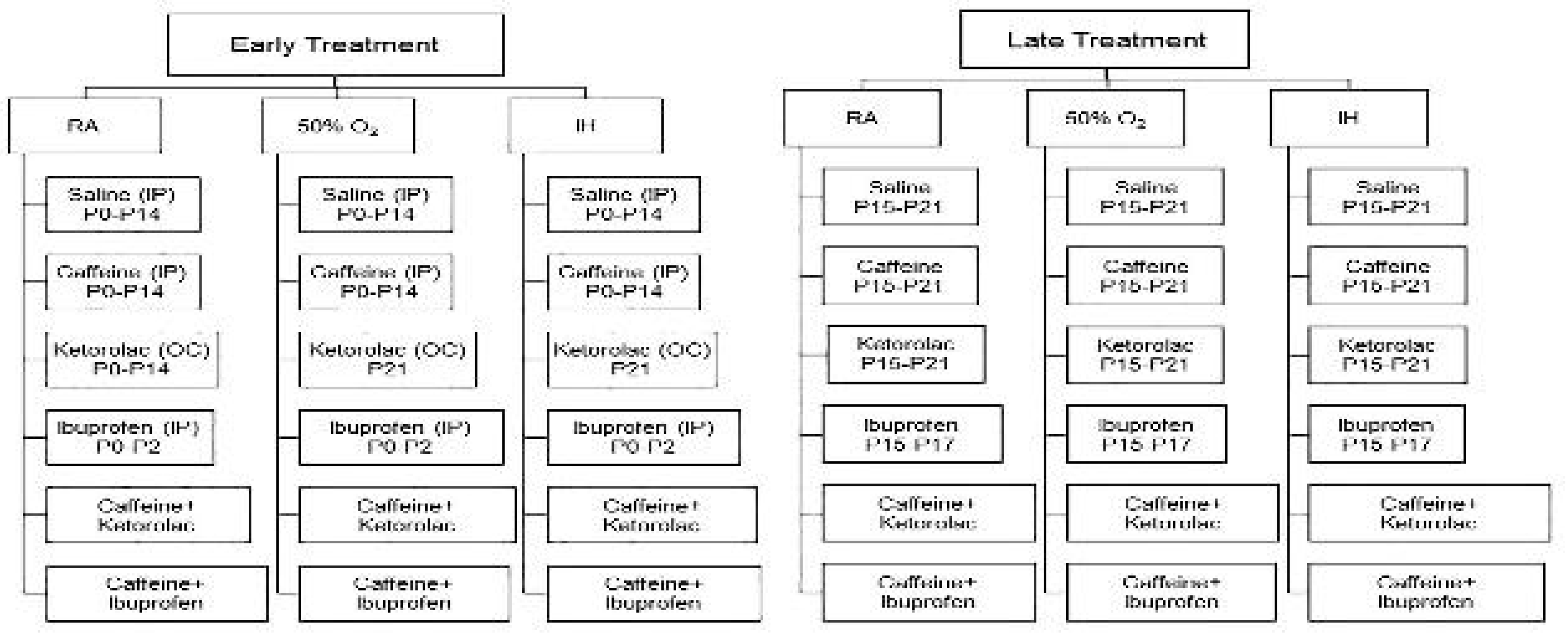
Figure 2.
Effect of neonatal intermittent hypoxia (IH) and early or late caffeine/NSAIDs treatment on brain weight and brain/body weight ratios. Panels A and C represent the early treatment, respectively; and panels B and C represent the late treatment, respectively. The solid white bar represents the groups exposed to room air (RA), the checked bar represents the groups exposed to 50% O2; and the solid black bar represents the groups exposed to neonatal IH. Groups are labeled as follows: Sal/Sal (ocular saline/IP saline); Sal/Caff (ocular saline/IP caffeine); Sal/Keto (IP saline/ocular ketorolac); Sal/Ibu (ocular saline/IP ibuprofen); Caff/Keto (IP caffeine/ocular ketorolac); and Caff/Ibu (IP caffeine/IP ibuprofen). Data are mean±SEM (n=18 samples per group).
Figure 2.
Effect of neonatal intermittent hypoxia (IH) and early or late caffeine/NSAIDs treatment on brain weight and brain/body weight ratios. Panels A and C represent the early treatment, respectively; and panels B and C represent the late treatment, respectively. The solid white bar represents the groups exposed to room air (RA), the checked bar represents the groups exposed to 50% O2; and the solid black bar represents the groups exposed to neonatal IH. Groups are labeled as follows: Sal/Sal (ocular saline/IP saline); Sal/Caff (ocular saline/IP caffeine); Sal/Keto (IP saline/ocular ketorolac); Sal/Ibu (ocular saline/IP ibuprofen); Caff/Keto (IP caffeine/ocular ketorolac); and Caff/Ibu (IP caffeine/IP ibuprofen). Data are mean±SEM (n=18 samples per group).
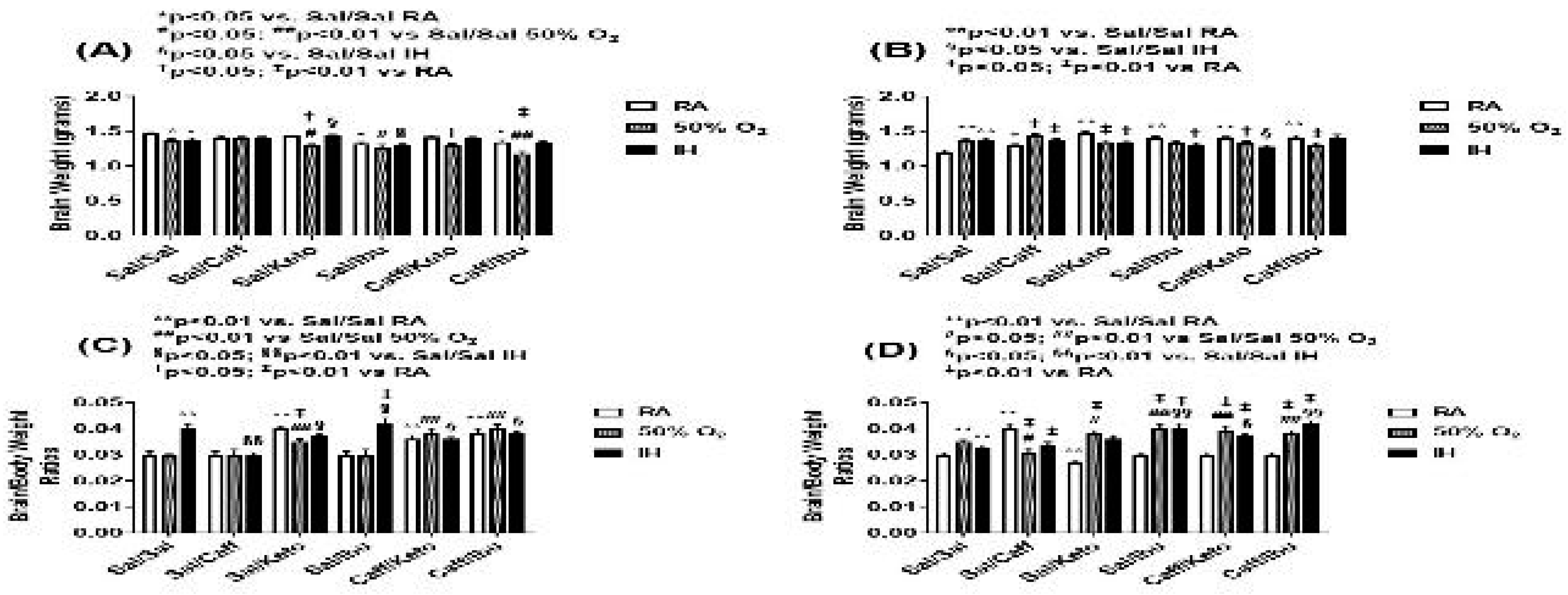
Figure 3.
Representations of the H&E stained cerebral cortex sections for the early treatment groups. The upper panels represent the groups exposed to RA; the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH. Images are 20X magnification, scale bar is 50 µm.
Figure 3.
Representations of the H&E stained cerebral cortex sections for the early treatment groups. The upper panels represent the groups exposed to RA; the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH. Images are 20X magnification, scale bar is 50 µm.
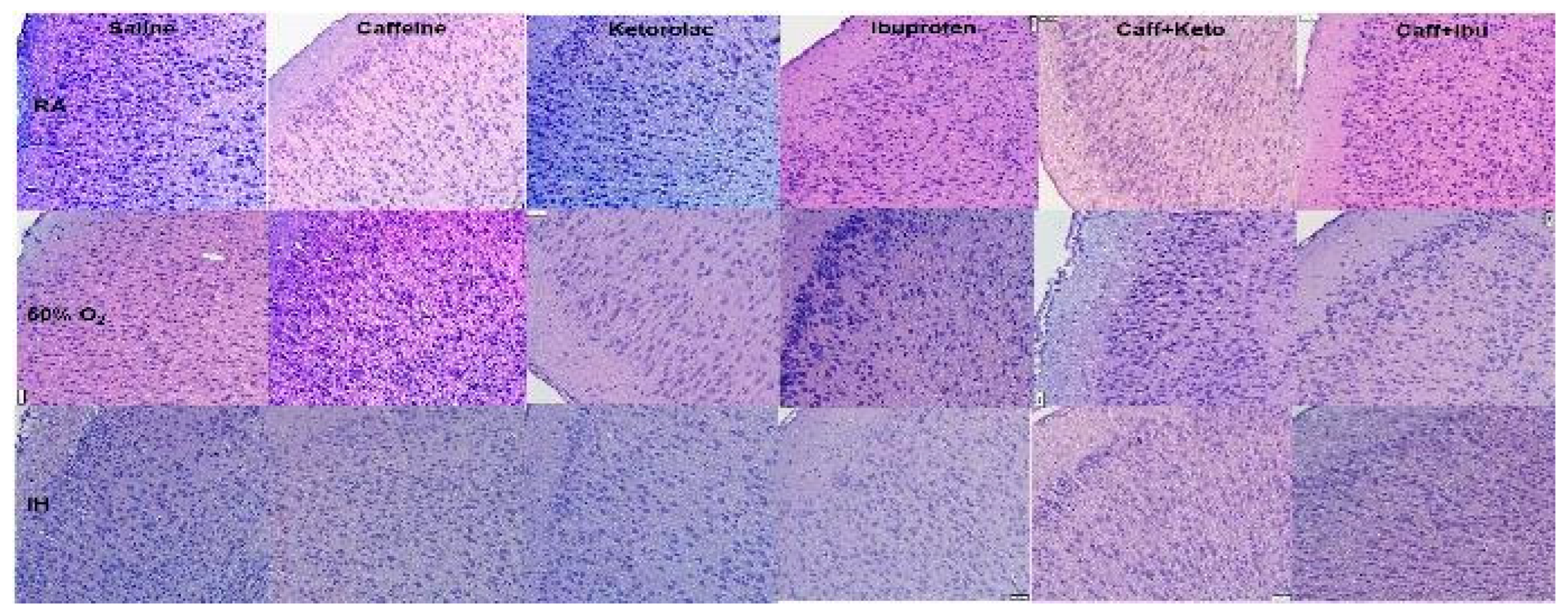
Figure 4.
Representations of the H&E stained cerebral cortex sections for the late treatment groups. The upper panels represent the groups exposed to RA; the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH. Images are 20X magnification, scale bar is 50 µm.
Figure 4.
Representations of the H&E stained cerebral cortex sections for the late treatment groups. The upper panels represent the groups exposed to RA; the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH. Images are 20X magnification, scale bar is 50 µm.
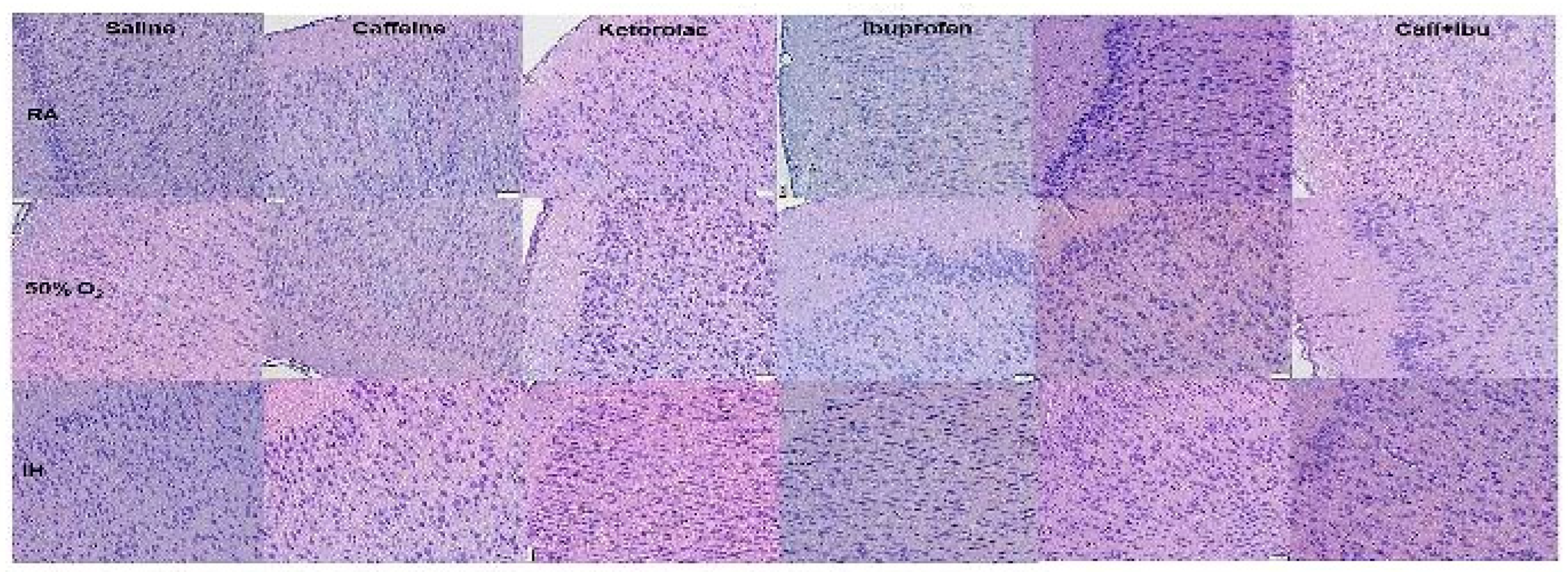
Figure 5.
Representations of the TUNEL stained cerebral cortex sections for the early treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Caff (caffeine); Keto (ketorolac); Ibu (ibuprofen). Apoptosis is indicated by the brown stain. Sections were counter stained with methyl green which indicates negativity. Images are 20X magnification, scale bar is 50 µm.
Figure 5.
Representations of the TUNEL stained cerebral cortex sections for the early treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Caff (caffeine); Keto (ketorolac); Ibu (ibuprofen). Apoptosis is indicated by the brown stain. Sections were counter stained with methyl green which indicates negativity. Images are 20X magnification, scale bar is 50 µm.

Figure 6.
Representations of the TUNEL stained cerebral cortex sections for the late treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Apoptosis is indicated by the brown stain. Sections were counter stained with methyl green which indicates negativity. Images are 20X magnification, scale bar is 50 µm.
Figure 6.
Representations of the TUNEL stained cerebral cortex sections for the late treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Apoptosis is indicated by the brown stain. Sections were counter stained with methyl green which indicates negativity. Images are 20X magnification, scale bar is 50 µm.
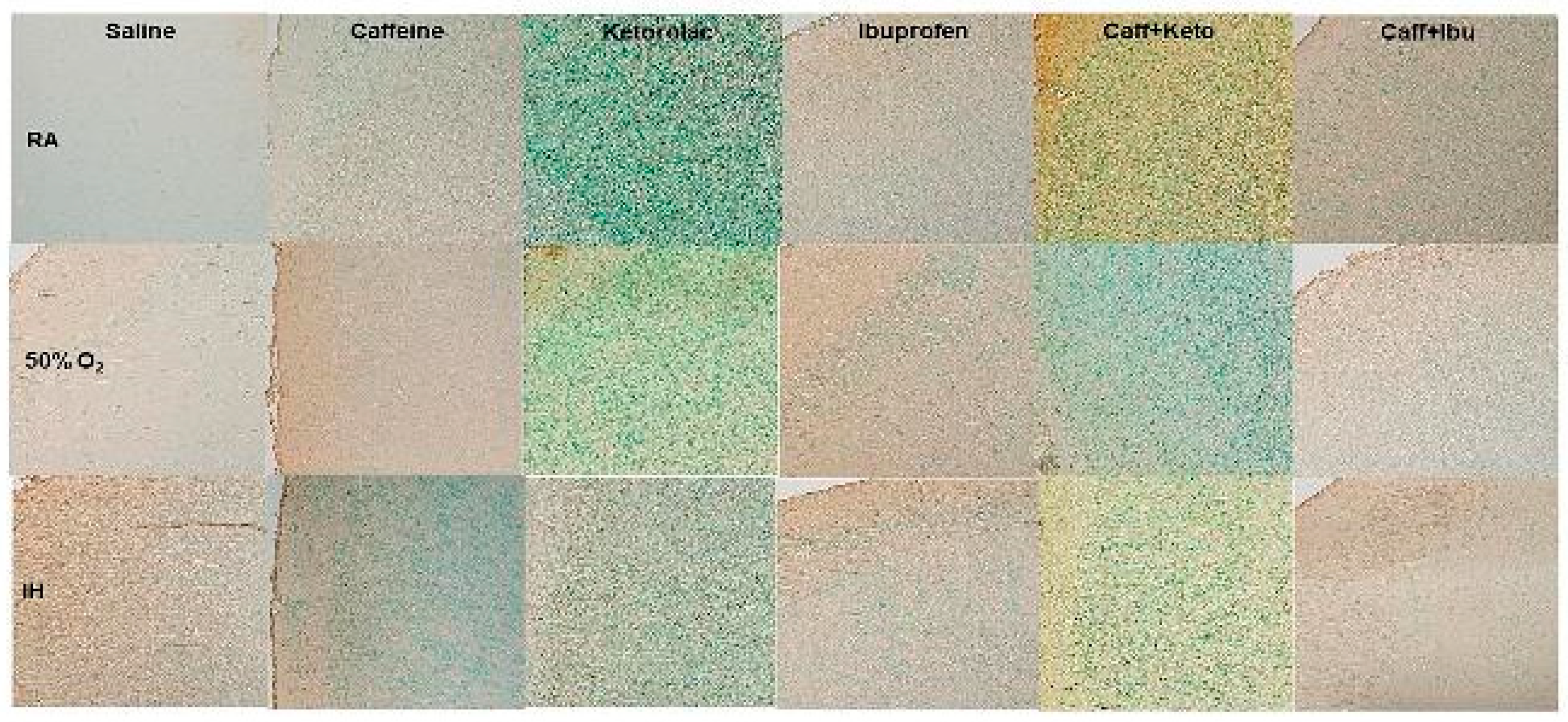
Figure 7.
Representations of the Luxol fast blue (LFB) stained cerebral cortex sections indicating myelination, for the early treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Caff (caffeine); Keto (ketorolac); Ibu (ibuprofen). Myelination is indicated by the blue stain. Images are 20X magnification, scale bar is 50 µm.
Figure 7.
Representations of the Luxol fast blue (LFB) stained cerebral cortex sections indicating myelination, for the early treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Caff (caffeine); Keto (ketorolac); Ibu (ibuprofen). Myelination is indicated by the blue stain. Images are 20X magnification, scale bar is 50 µm.

Figure 8.
Representations of the Luxol fast blue (LFB) stained cerebral cortex sections indicating myelination, for the late treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Caff (caffeine); Keto (ketorolac); Ibu (ibuprofen). Myelination is indicated by the blue stain. Images are 20X magnification, scale bar is 50 µm.
Figure 8.
Representations of the Luxol fast blue (LFB) stained cerebral cortex sections indicating myelination, for the late treatment groups. The upper panels represent the groups exposed to RA (room air); the middle panels represent the groups exposed to hyperoxia (50% O2); and the lower panels represent the groups exposed to neonatal IH (intermittent hypoxia). Caff (caffeine); Keto (ketorolac); Ibu (ibuprofen). Myelination is indicated by the blue stain. Images are 20X magnification, scale bar is 50 µm.

Figure 9.
Effect of neonatal intermittent hypoxia (IH) and early caffeine/NSAIDs treatment on cytokines in the cerebral cortex homogenates. The groups are as described in Figure 2. Data are mean±SEM (n=6 samples per group).
Figure 9.
Effect of neonatal intermittent hypoxia (IH) and early caffeine/NSAIDs treatment on cytokines in the cerebral cortex homogenates. The groups are as described in Figure 2. Data are mean±SEM (n=6 samples per group).
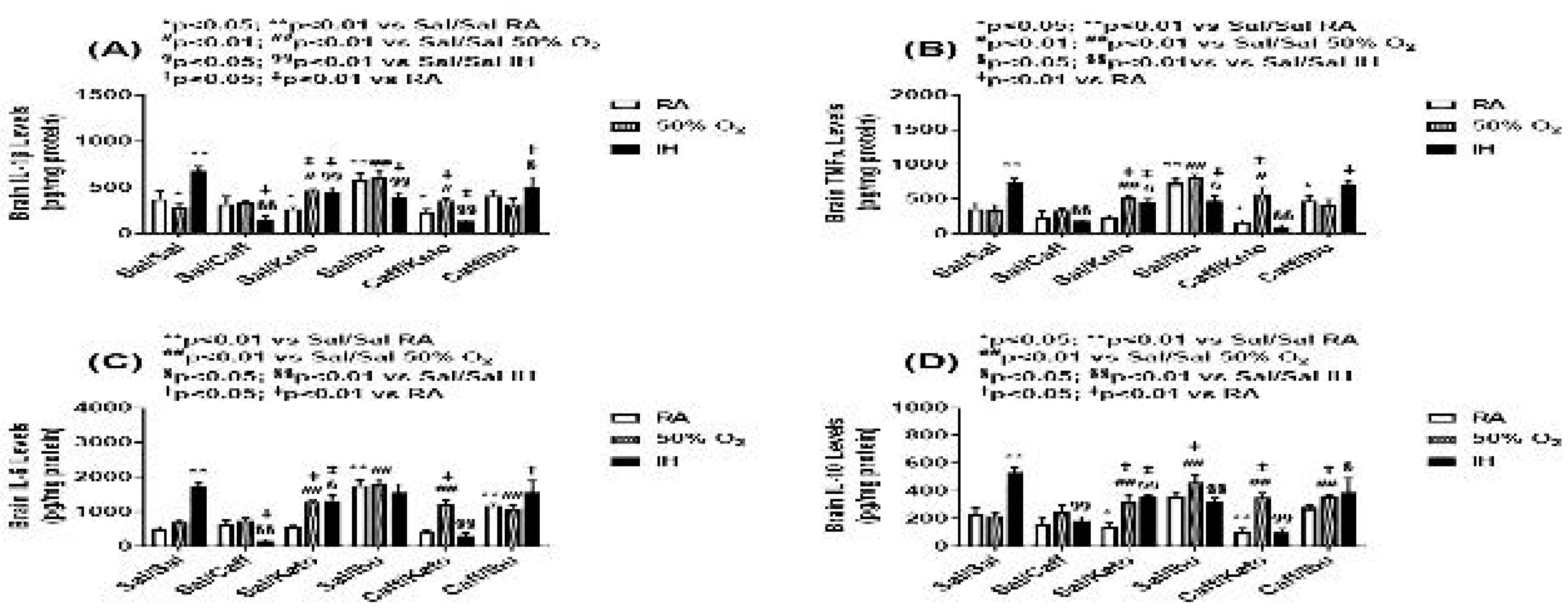
Figure 10.
Effect of neonatal intermittent hypoxia (IH) and late caffeine/NSAIDs treatment on cytokines in the cerebral cortex homogenates. The groups are as described in Figure 2. Data are mean±SEM (n=6 samples per group).
Figure 10.
Effect of neonatal intermittent hypoxia (IH) and late caffeine/NSAIDs treatment on cytokines in the cerebral cortex homogenates. The groups are as described in Figure 2. Data are mean±SEM (n=6 samples per group).
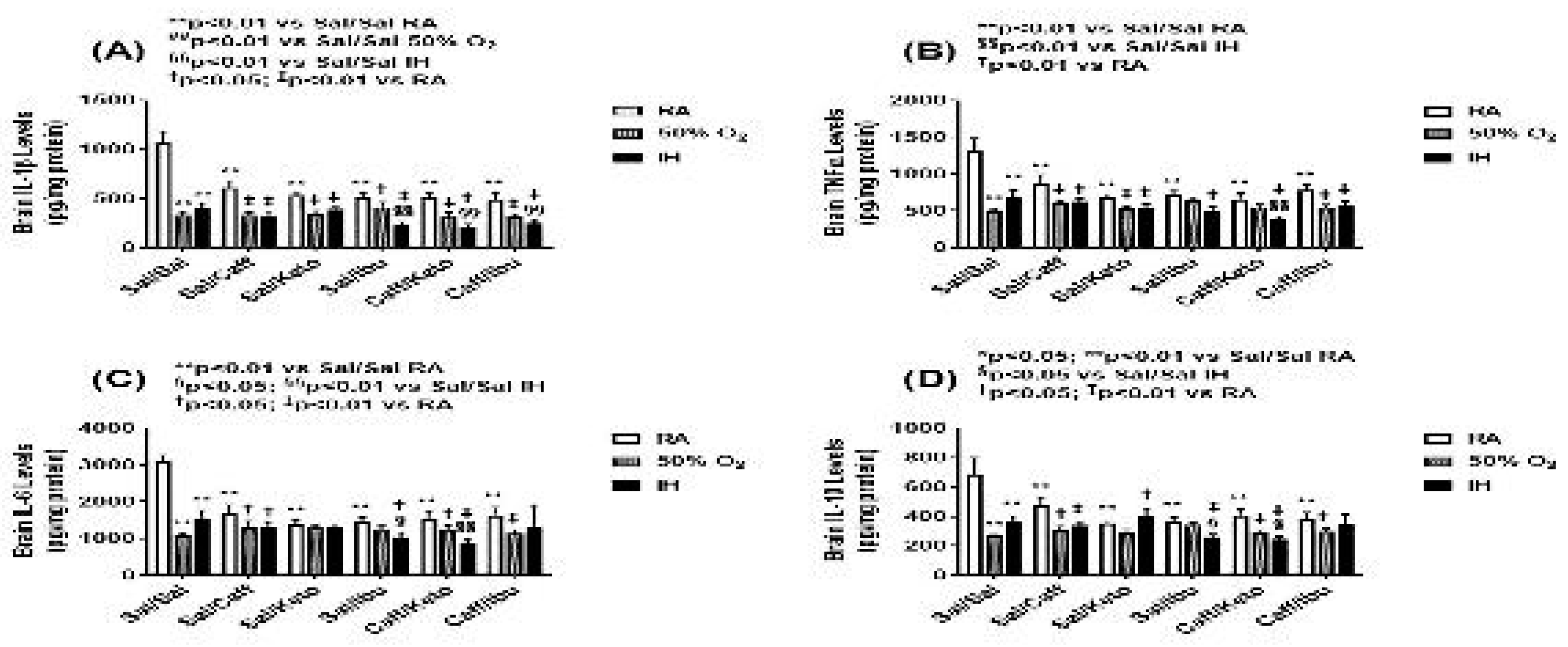
Figure 11.
Representative image of immunoreactivity of IkB in the early treatment groups. IkB is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
Figure 11.
Representative image of immunoreactivity of IkB in the early treatment groups. IkB is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.

Figure 12.
Representative image of immunoreactivity of IkB in the late treatment groups. IkB is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
Figure 12.
Representative image of immunoreactivity of IkB in the late treatment groups. IkB is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
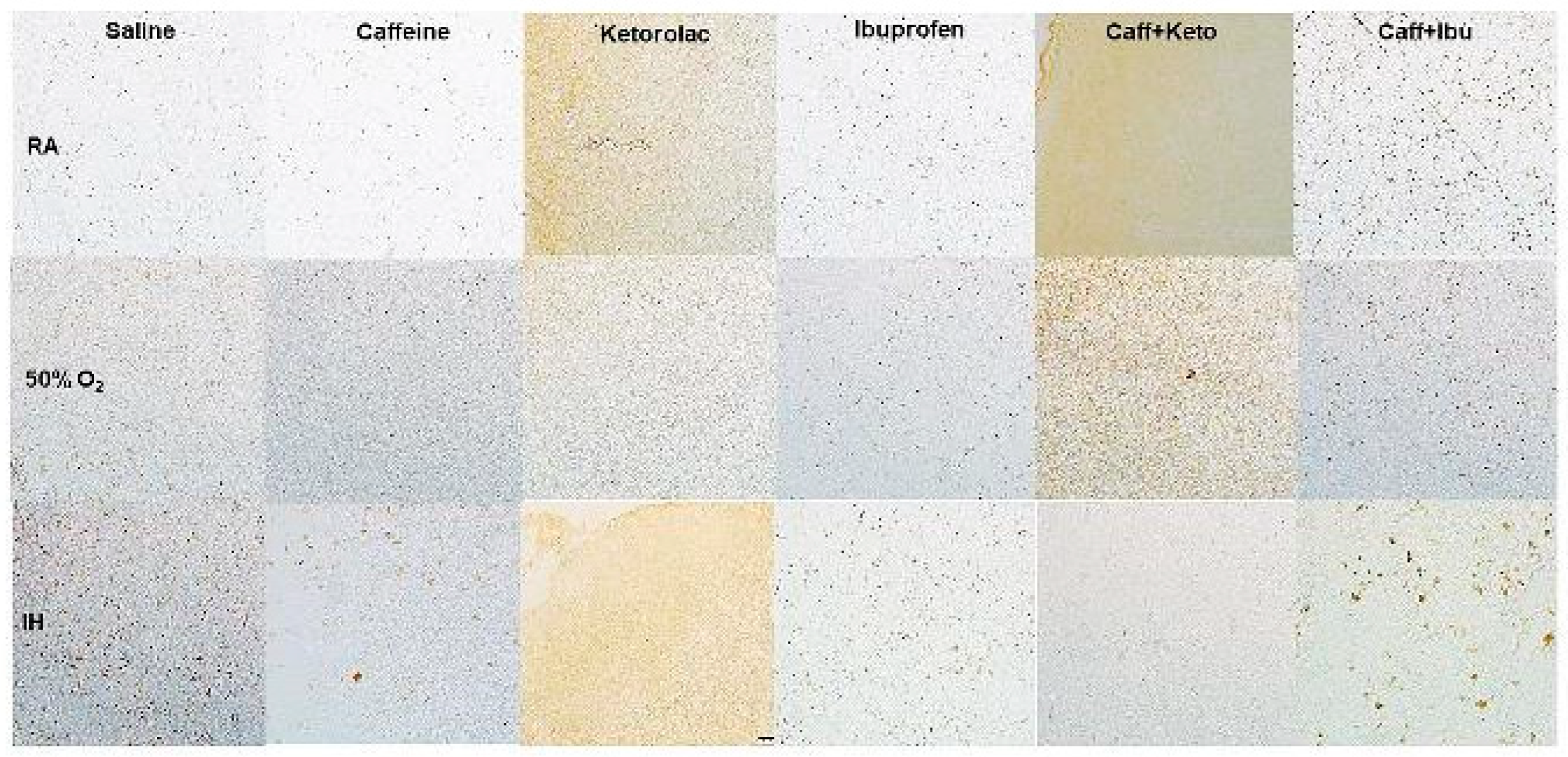
Figure 13.
Representative image of immunoreactivity of IL-1β in the early treatment groups. IL-1β is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
Figure 13.
Representative image of immunoreactivity of IL-1β in the early treatment groups. IL-1β is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
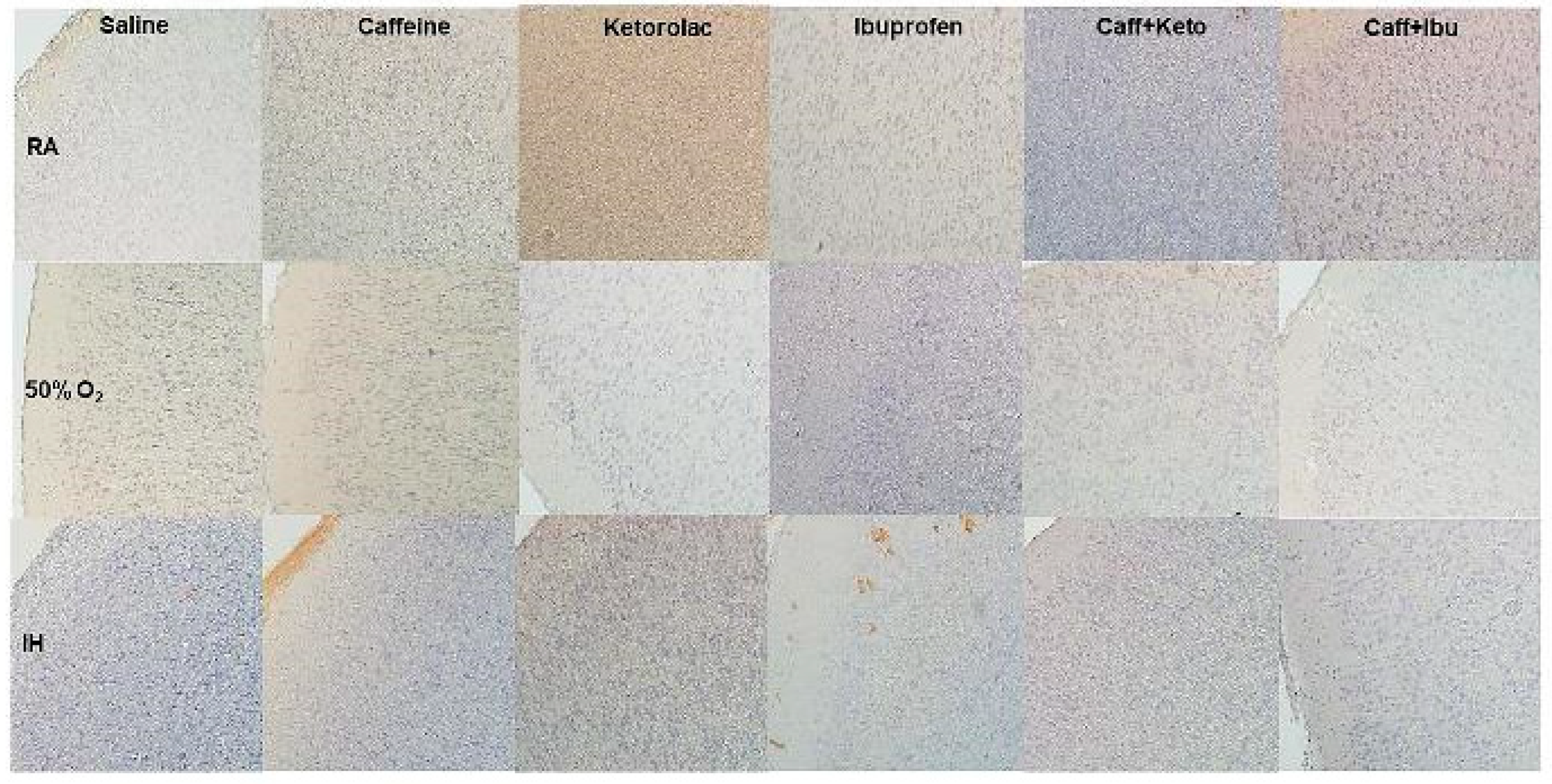
Figure 14.
Representative image of immunoreactivity of IL-1β in the late treatment groups. IL-1β is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
Figure 14.
Representative image of immunoreactivity of IL-1β in the late treatment groups. IL-1β is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
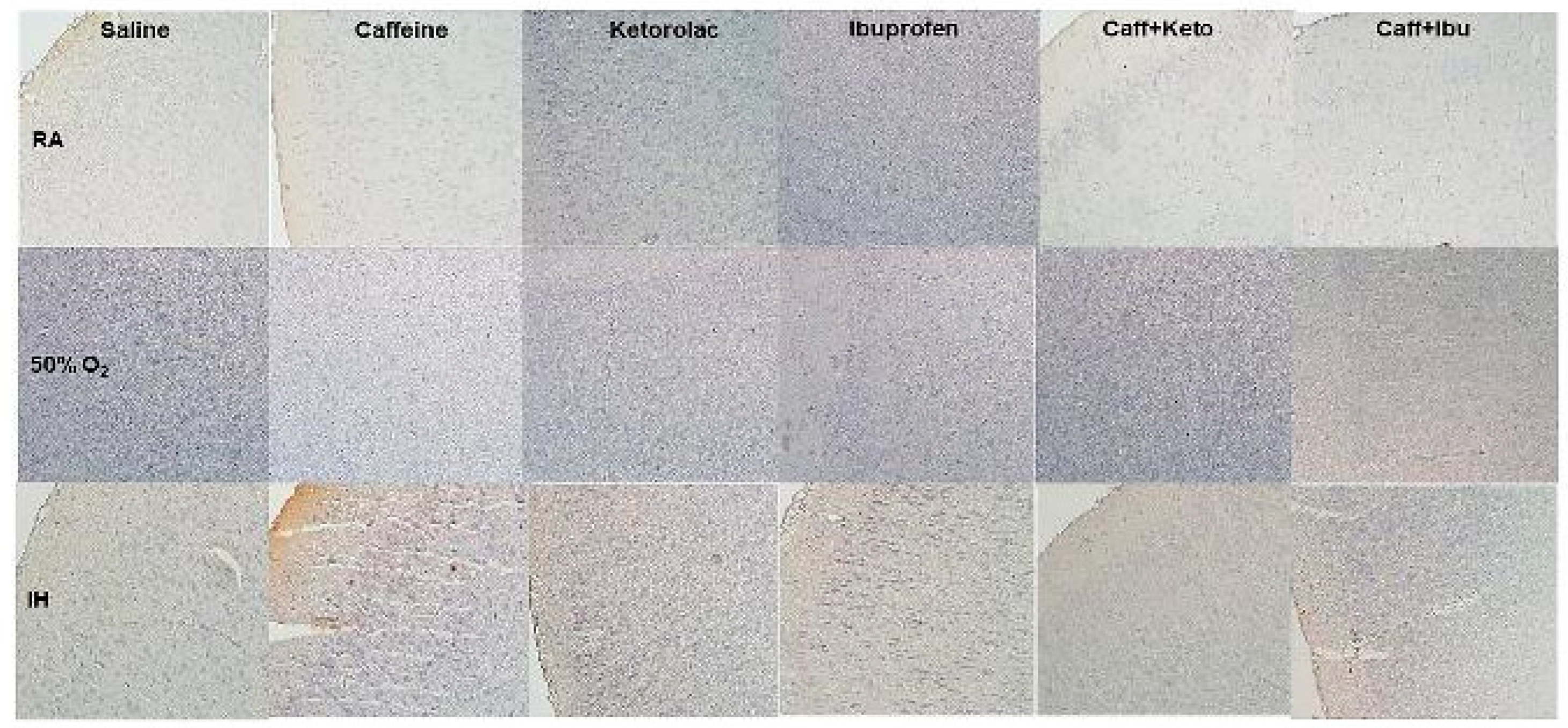
Figure 15.
Representative image of immunoreactivity of TNFα in the early treatment groups. TNFα is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
Figure 15.
Representative image of immunoreactivity of TNFα in the early treatment groups. TNFα is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.

Figure 16.
Representative image of immunoreactivity of TNFα in the late treatment groups. TNFα is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.
Figure 16.
Representative image of immunoreactivity of TNFα in the late treatment groups. TNFα is indicated by the brown stain. The sections were counterstain with hematoxylin (blue). Images are 20X magnification, scale bar is 50 µm.


Table 1.
Morphometric Analyses (Early).
| G | Total Brain Width (frontal-occipital) |
Total Brain Width (inter-parietal) |
Layer I | Layer II | Layer III | Layer IV | Layer V | Layer VI |
|---|---|---|---|---|---|---|---|---|
| RA: | ||||||||
| Sal | 7836.6±88.0 | 12337.9±87.7 | 109.1±8.0 | 80.5±7.8 | 101.7±7.8 | 146.2±21.4 | 236.7±12.6 | 185.4±17.3 |
| Caff | 5591.1±7.1** | 10406.4±17.8 ** |
134.9±7.4 | 63.4±6.9 | 109.3±9.0 | 130.3±9.2 | 120.1±7.9** | 137.7±12.2 |
| Keto | 8543.5±22.3** | 14631.0±289.0** | 182.6±15.4 ** |
96.6±7.1** | 294.1±22.8** | 239.9±18.5 ** |
133.2±8.4** | 155.4±15.7 |
| Ibu | 6794.7±45.4** | 10041.3±14.2 ** |
136.9±10.4 | 101.0±7.1 | 142.4±26.3 | 120.8±20.1 | 223.3±20.4 | 143.6±10.8 |
| Caff+Keto | 8439.3±35.0** | 14048.7±397.4** | 150.8±10.1* | 76.9±5.2** | 228.8±23.3** | 242.4±13.1** | 125.9±10.5** | 155.1±18.0 |
| Caff+Ibu | 6310.7±57.3** | 10917.3±36.9 ** |
123.5±3.4 | 82.6±9.8 | 133.0±20.3 | 142.6±13.9 | 150.3±17.0** | 113.3±12.9** |
|
50% O2: | ||||||||
| Sal | 5890.2±33.0** | 9471.5±159.4 | 109.0±11.3 | 71.5±1.8 | 174.0±18.7 | 241.5±17.0 | 198.0±28.9 | 137.3±11.1 |
| Caff | 5851.9±20.5† | 9312.3±23.7 | 1174.8±27.0##‡ | 2726.4±144.2 ##‡ |
2000.5±214.6 ##‡ |
1973.7±46.9##‡ | 1749.2±302.3 ##‡ |
1327.3±118.4 **‡ |
| Keto | 8387.7±256.4## | 13652.5±915.8## | 214.9±13.9 ## |
99.3±13.0 | 235.5±15.4 | 260.2±34.3 | 276.9±90.6 | 122.0±16.7 |
| Ibu | 7667.6±55.7 ##‡ |
8820.2±1820.7‡ | 122.0±3.0 | 79.0±5.4 | 154.6±13.2† | 244.5±25.3‡ | 258.9±62.4 | 149.2±20.9 |
| Caff+Keto | 6894.5±92.7##‡ | 12334.0±145.3‡ | 153.4±8.4 | 82.5±7.5 | 238.8±0.87‡ | 323.8±9.6 | 122.5±8.0 | 153.4±8.4 |
| Caff+Ibu | 7313.6±33.0##‡ | 11509.6±70.7‡ | 121.5±5.40 | 88.9±4.7 | 170.4±13.0† | 244.0±25.5‡ | 165.3±15.0 | 144.6±10.6 |
|
Neonatal IH: | ||||||||
| Sal | 8270.1±25.2 | 11218.5±21.6 | 312.1±115.9 | 299.3±135.6* | 567.9±265.9 | 575.4±185.0 | 290.8±89.2 | 416.1±107.4* |
| Caff | 8063.0±116.2‡ | 12813.3±17.7‡ | 153.2±34.0 | 130.4±17.7 | 158.2±26.4‡ | 310.5±44.7 §§‡ |
228.3±55.8 | 147.6±36.3 §§ |
| Keto | 9106.0±136.5§§‡ | 13711.7±56.2 | 156.3±12.2 | 140.5±12.0 | 303.1±46.3 | 294.2±47.8 §§ |
210.5±41.5 | 150.0±36.0 §§ |
| Ibu | 8840.0±116.4§§‡ | 12924.0±79.5‡ | 144.4±8.6 | 122.3±13.3 | 186.3±9.9‡ | 273.4±47.8 §§‡ |
415.5±179.5 | 179.5±17.3 §§ |
| Caff+Keto | 6935.5±76.4 §§‡ |
12410.0±133.3‡ | 156.0±7.5 | 88.4±8.9 | 173.0±23.4 §§‡ |
182.9±17.6 §§ |
201.4±37.9 | 182.6±27.9 §§ |
| Caff+Ibu | 8711.1±116.6§‡ | 12730.0±42.4‡ | 158.9±14.6 §† |
87.2±24.9 | 160.3±26.8§ | 221.5±36.8 §§ |
314.3±53.5‡ | 188.7±15.1 §‡ |
RA (room air); 50% O2 (hyperoxia); IH (intermittent hypoxia); Caff (Caffeine); Keto (Ketorolac); Ibu (Ibuprofen). Animals were treated with caffeine citrate from birth (P0) to P14; ketorolac eye drops from P0 to P14; or ibuprofen from P0 to P3. Data were analyzed using two-way ANOVA (n=6/group). *p<0.05, **p<0.01 vs. Saline RA; #p<0.05##p<0.01 vs. Saline 50% O2; §p<0.05, §§p<0.01 vs Saline IH; †p<0.05, ‡p<0.01 vs RA.
Table 2.
Morphometric Analyses (Late).
| G | Total Brain Width (frontal-occipital) |
Total Brain Width (inter-parietal) |
Layer I | Layer II | Layer III | Layer IV | Layer V | Layer VI |
|---|---|---|---|---|---|---|---|---|
| RA: | ||||||||
| Sal | 7705.2±67.0 | 13309.8±276.3 | 154.5±6.1 | 56.1±3.6 | 147.8±33.9 | 202.9±19.1 | 241.0±9.2 | 127.0±16.2 |
| Caff | 7651.1±109.7 | 11572.0±113.5** | 104.0±5.1** | 85.9±5.6** | 241.5±47.9 | 259.8±34.0 | 292.5±30.5 | 140.4±19.5 |
| Keto | 7595.6±219.0 | 12598.4±250.0 | 149.6±10.4 | 101.4±7.6** | 280.1±52.3* | 211.2±5.1 | 247.0±16.4 | 165.7±13.8 |
| Ibu | 8404.0±81.8** | 13263.0±530.0 | 166.6±12.9 | 87.2±5.1** | 180.9±15.6 | 258.3±25.0 | 234.4±25.9 | 145.3±12.2 |
| Caff+Keto | 7902.7±30.3 | 11506.7±88.2** | 133.7±7.5 | 70.7±5.1 | 205.0±26.8 | 181.5±30.2 | 186.5±25.0 | 158.4±7.3 |
| Caff+Ibu | 8281.1±207.2* | 13327.4±280.4 | 136.6±8.3 | 86.5±5.6** | 191.1±23.9 | 270.6±18.9 | 405.0±29.7** | 198.9±9.1** |
| 50% O2: | ||||||||
| Sal | 8947.5±21.2** | 13534.8±255.2 | 179.8±15.2 | 102.4±10.4 | 243.0±23.4 | 237.8±33.6 | 192.5±18.5 | 151.3±7.0 |
| Caff | 9335.5±122.5##‡ | 14038.0±174.4‡ | 130.8±6.5#§ | 83.5±8.4 | 211.0±20.2 | 298.0±31.0 | 305.2±49.2 | 155.5±13.8 |
| Keto | 8773.5±195.7‡ | 11511.0±165.7##‡ | 141.8±2.4† | 100.0±6.8 | 222.0±12.2 | 277.3±16.9† | 190.8±39.0 | 164.0±17.5 |
| Ibu | 9093.5±76.8‡ | 13285.5±288.6 | 182.6±8.0 | 111.6±8.4 | 240.3±16.4† | 238.5±18.1 | 313.6±23.1 | 162.3±12.7 |
| Caff+Keto | 7656.5±7.1##† | 13158.5±297.0‡ | 161.1±6.6 | 126.9±8.0‡ | 240.3±23.4 | 253.4±18.5 | 218.5±24.9 | 132.6±10.0 |
| Caff+Ibu | 9272.0±36.4‡ | 13344.7±194.4 | 171.0±8.7‡ | 107.4±4.3‡ | 201.5±15.0 | 235.8±22.4 | 299.8±37.6† | 154.0±16.4 |
| Neonatal IH: | ||||||||
| Sal | 8270.1±29.1** | 11218.5±217.6** | 187.7±57.9 | 163.8±65.6 | 367.6±118.3* | 369.8±98.4 | 284.9±41.8 | 260.9±65.0* |
| Caff | 8817.0±18.3 §§‡ |
13775.3±280.9 §§‡ |
178.6±7.7‡ | 114.5±19.6 | 324.5±42.2 | 243.4±56.9 | 242.3±32.1 | 178.9±8.5 |
| Keto | 7501.7±39.8§§ | 11466.0±160.1‡ | 141.4±10.8 | 90.3±5.8 | 275.5±47.9 | 254.3±24.0 | 240.6±27.1 | 171.8±25.9 |
| Ibu | 9481.3±68.4 §§‡ |
15107.3±280.5 §§‡ |
226.6±9.6‡ | 116.5±24.2 | 227.8±17.7 | 285.6±30.1 | 229.7±42.6 | 178.0±9.4 |
| Caff+Keto | 7461.7±93.7 §§‡ |
698.0±91.1§§‡ | 143.0±3.9 | 97.1±15.9 | 250.3±29.8 | 252.6±17.5 | 127.3±6.1§§ | 125.0±11.9§ |
| Caff+Ibu | 9465.5±206.6§§‡ | 14610.0±242.4 §§‡ |
124.4±2.2 | 93.4±3.5 | 325.0±32.1‡ | 256.0±17.1 | 169.7±20.8 §‡ |
180.4±23.1 |
RA (room air); 50% O2 (hyperoxia); IH (intermittent hypoxia); Caff (Caffeine); Keto (Ketorolac); Ibu (Ibuprofen). Animals were treated with caffeine citrate from birth (P0) to P14; ketorolac eye drops from P0 to P14; or ibuprofen from P0 to P3. Data were analyzed using two-way ANOVA (n=6/group). *p<0.05, **p<0.01 vs. Saline RA; #p<0.05##p<0.01 vs. Saline 50% O2; §p<0.05, §§p<0.01 vs Saline IH; †p<0.05, ‡p<0.01 vs RA.
Table 3.
IHC Quantification (Early).
| Group | Apoptosis (TUNEL) |
Myelin (LFB) | pNFkB | IkB | IL-1β | IL-6 | TNFα |
|---|---|---|---|---|---|---|---|
| RA: | |||||||
| Sal | 489.5±178.2 | 1369.5±265.7 | 0.8±0.36 | 181.3±19.8 | 391.6±37.3 | 381.7±29.1 | 3134.4±193.9 |
| Caff | 4387.7±724.1** | 1038.7±106.5 | 2.3±1.3 | 51.1±5.2 | 61.6±15.5** | 1.5±0.3** | 88.2±21.6** |
| Keto | 225.3±17.5 | 2797.6±124.0** | 18.5±12.3 | 3277.7±470.2** | 29.6±7.5** | 20.3±5.6** | 1460.8±551.0** |
| Ibu | 6809.3±283.7** | 3374.5±203.0** | 3444.3±475.3** | 70.5±4.6 | 46.1±5.8** | 214.6±42.5 | 298.1±39.0** |
| Caff+Keto | 4462.7±594.3** | 27.8±2.0** | 196.5±61.3 | 25477.3±1440.6** | 56.8±20.4** | 63.3±10.3* | 795.7±131.0** |
| Caff+Ibu | 8517.8±182.8** | 2653.0±441.5** | 29.4±7.2 | 331.3±14.3 | 8.4±3.0** | 453.4±165.8 | 737.3±133.7** |
| 50% O2: | |||||||
| Sal | 2917.6±390.8** | 18.4±2.9** | 2049.9±952.0* | 195.2±19.8 | 72.4±11.4** | 19.0±6.1** | 131.7±43.8** |
| Caff | 6915.8±292.0##† | 4.9±1.5‡ | 144.8±31.0 | 51.7±8.2 | 29.7±3.8 | 34.5±7.2‡ | 2258.9±368.0##‡ |
| Keto | 1297.2±75.7##‡ | 26.0±14.4‡ | 711.9±77.9‡ | 467.0±92.1##‡ | 23.2±3.7 | 0.31±0.19 | 184.6±67.9† |
| Ibu | 3940.6±593.7‡ | 0±0##‡ | 2275.3±274.7† | 51.7±4.6 | 77.8±6.5 | 8.2±1.8‡ | 198.9±62.4 |
| Caff+Keto | 365.0±41.7##‡ | 0±0#‡ | 3068.9±320.3‡ | 1701.1±137.0‡ | 59.9±6.4 | 14.7±3.8‡ | 338.1±97.1 |
| Caff+Ibu | 5658.0±310.9##† | 0±0##‡ | 3225.3±699.0‡ | 60.9±7.2† | 330.8±40.7##‡ | 163.3±42.6## | 2556.8±300.0##‡ |
| Neonatal IH: | |||||||
| Sal | 306.4±63.1** | 0±0** | 1302.6±162.2 | 182.3±11.2 | 66.0±8.9** | 50.6±11.3** | 1688.9±186.6** |
| Caff | 2400.0±329.1‡ | 0±0‡ | 1026.7±111.0‡ | 85.8±13.8† | 77.7±8.9 | 5.5±1.6 | 10.6±3.4§§ |
| Keto | 777.3±73.7§‡ | 0±0‡ | 1379.3±202.6‡ | 885.8±89.4§§‡ | 61.4±22.5 | 209.4±49.2§§‡ | 805.1±289.4§§ |
| Ibu | 3670.5±582.7‡ | 0±0‡ | 149.2±20.6§§‡ | 204.8±29.6‡ | 189.7±40.3§§‡ | 41.4±25.3‡ | 3.2±0.57§§‡ |
| Caff+Keto | 1851.3±396.9‡ | 0±0‡ | 971.7±179.1 | 93.9±6.9‡ | 53.3±10.8 | 67.6±11.7 | 919.5±6.9§§ |
| Caff+Ibu | 8004.7±1326.2§§ | 0±0‡ | 369.0±53.1§§ | 905.1±134.4§§‡ | 22.5±3.6 | 56.7±8.8† | 382.8±66.8§§ |
RA (room air); 50% O2 (hyperoxia); IH (intermittent hypoxia); Caff (Caffeine); Keto (Ketorolac); Ibu (Ibuprofen); pNFkB (phosphorylated nuclear factor kappa B); IL (interleukin); TNF (tumor necrosis factor). . Animals were treated with caffeine citrate from birth (P0) to P14; ketorolac eye drops from P0 to P14; or ibuprofen from P0 to P3. Data were analyzed using two-way ANOVA. *p<0.05, **p<0.01 vs. Saline RA; #p<0.05##p<0.01 vs. Saline 50% O2; §p<0.05, §§p<0.01 vs Saline IH; †p<0.05, ‡p<0.01 vs RA.
Table 4.
IHC Quantification (Late).
| G | Apoptosis (TUNEL) |
Myelin (LFB) | pNFkB | IkB | IL-1β | IL-6 | TNFα |
|---|---|---|---|---|---|---|---|
| RA: | |||||||
| Sal | 65.4±17.5 | 916.2±165.5 | 0.69±0.41 | 38.3±3.6 | 1699.9±230.6 | 43.5±9.8 | 4268.9±681.2 |
| Caff | 16.0±2.5 | 5027.7±723.4** | 253.8±66.4 | 93.9±5.6 | 2155.9±598.8** | 1691.8±502.1** | 1992.7±379.7** |
| Keto | 2315.2±462** | 1600.0±306.4 | 4425.4±990** | 1765.7±223.7** | 9113.9±419.7** | 13.2±1.8 | 256.2±116.8** |
| Ibu | 262.0±24.1 | 5576.5±470.0** | 24.3±4.4 | 174.5±15.0 | 52.0±12.6** | 423.4±203.1 | 524.2±235.1** |
| Caff+Keto | 1526.1±251.9** | 563.3±47.8 | 4751.7±607.3** | 653.9±79.9** | 4.9±0.87** | 50.7±7.0 | 281.5±43.7** |
| Caff+Ibu | 5.1±1.5 | 5495.0±693.9** | 29.4±4.3 | 34.8±6.3 | 205.8±32.2** | 41.4±5.8 | 2479.9±368.4** |
| 50% O2: | |||||||
| Sal | 876.2±108.4** | 13042.9±438.2** | 3239.5±800.3** | 30.0±2.8 | 1151.5±175.2* | 4.1±0.9 | 2742.2±746.7 |
| Caff | 33.5±8.7## | 4083.8±764.4## | 12668.9±2030 | 89.2±5.2 | 5365.3±531.0##‡ | 86.5±27.8#‡ | 5411.4±735.4##‡ |
| Keto | 106.8±23.7##‡ | 2348.2±579.3## | 2924.9±1037.3 | 869.2±202.6## | 1875.3±690.3‡ | 61.2±27.2 | 2522.4±202.6‡ |
| Ibu | 4.7±1.4##‡ | 1710.2±346.9##‡ | 2275.3±274.7‡ | 186.9±17.0 | 135.7±56.0 | 29.7±14.2† | 169.0±48.2## |
| Caff+Keto | 1116.6±54.7† | 1172.7±118.9##† | 22.1±3.6##‡ | 645.6±77.9## | 213.6±86.9 | 61.4±7.7# | 630.3±120.1## |
| Caff+Ibu | 44.2±15.6##† | 1692.1±370.8##‡ | 14.5±2.6## | 31.1±5.9 | 3.7±1.5‡ | 99.4±16.9## | 559.1±186.3##‡ |
| Neonatal IH: | |||||||
| Sal | 270.0±39.3 | 41.3±4.5 | 852.3±140.0 | 81.9±7.5** | 32.4±5.5** | 545.4±71.3* | 168.0±36.9* |
| Caff | 21.0±11.7 | 1279.1±229.4§§‡ | 1454.9±395.0 | 59.3±4.0‡ | 3731.0±481.4§§ | 312.6±51.9§§‡ | 643.4±234.3 |
| Keto | 543.3±135.5‡ | 1364.7±100.8§§ | 735.1±109.3‡ | 2139.4±398.3§§ | 123.3±15.4‡ | 4.1±1.1§§ | 17.3±6.9 |
| Ibu | 194.3±46.2 | 1744.7±153.3§§‡ | 343.1±54.4 | 71.0±6.1‡ | 18.1±4.7 | 5.5±2.2§§† | 1031.7±184.7§§ |
| Caff+Keto | 441.3±110.9‡ | 998.8±60.0§§ | 3363.9±234.0§§‡ | 3227.0±320.6§§‡ | 40.9±9.6 | 56.9±18.8§§ | 720.3±310.0 |
| Caff+Ibu | 75.4±5.8‡ | 993.0±87.2§§‡ | 579.9±53.5‡ | 210.6±12.2‡ | 50.6±38.0‡ | 319.4±50.1§§‡ | 254.3±83.3‡ |
RA (room air); 50% O2 (hyperoxia); IH (intermittent hypoxia); Caff (Caffeine); Keto (Ketorolac); Ibu (Ibuprofen); pNFkB (phosphorylated nuclear factor kappa B); IL (interleukin); TNF (tumor necrosis factor). Animals were treated with caffeine citrate from birth (P0) to P14; ketorolac eye drops from P0 to P14; or ibuprofen from P0 to P3. Data were analyzed using two-way ANOVA (n=6/group). *p<0.05, **p<0.01 vs. Saline RA; #p<0.05##p<0.01 vs. Saline 50% O2; §p<0.05, §§p<0.01 vs Saline IH; †p<0.05, ‡p<0.01 vs RA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



