
Submitted:
06 September 2023
Posted:
07 September 2023
You are already at the latest version
Abstract
Cyanobacteria, microorganisms performing oxygenic photosynthesis, must adapt their metabolic processes to environmental challenges such as day and night changes. PipX, a unique regulatory protein from cyanobacteria provides a mechanistic link between signalling protein PII, a widely conserved (in bacteria and plants) transducer of carbon/nitrogen/energy richness, and the transcriptional regulator NtcA, which controls a large regulon involved in nitrogen assimilation. PipX is also involved in translational regulation by interaction with the ribosome-assembly GTPase EngA. However, increases of the PipX/PII ratio are toxic, presumably by abnormally increased binding of PipX to other partner(s). Here we present mutational and structural analyses of reported PipX-PII and PipX-NtcA complexes, leading to identification of single amino acid changes that decrease or abolish PipX toxicity. Four out of 11 mutations decreasing toxicity did not decrease PipX levels, suggesting that the targeted residues (F12, D23, L36, R54) provide toxicity determinants. In addition, one of those four mutations (D23A) argued against the over-activation of NtcA as the cause of PipX toxicity. Since most mutations at residues contacting PII decreased PipX levels, PipX stability would depend on its ability to bind to PII, a conclusion supported by the light-induced decrease in PipX levels observed in Synechococcus elongatus PCC7942.
Keywords:
NtcA
; EngA
; Synechococcus elongatus
; nitrogen regulation network
; light and dark conditions
; PipX toxicity
; energy sensing
; mutational analysis
1. Introduction
Cyanobacteria, phototrophic prokaryotes that perform oxygenic photosynthesis, are the main contributors to marine primary production [1,2] and have a very important ecological impact on global carbon, nitrogen and oxygen cycles. They have evolved sophisticated systems to maintain the homeostasis of carbon/nitrogen assimilation (reviewed by [3,4]), the two most abundant elements in all living forms. Cyanobacteria can use different nitrogen sources that are first converted into ammonium and then incorporated into amino acids and other N-containing compounds via the glutamine synthetase-glutamate synthase (GS-GOGAT) pathway using 2-oxoglutarate (2-OG) as a carbon skeleton [5]. The metabolite 2-OG, a universal indicator of the intracellular carbon-to-nitrogen balance [6,7], appears to be particularly suitable for this role in cyanobacteria [4].
In bacteria and plants, 2-OG is sensed by the widely distributed and highly conserved PII family of homotrimeric signal transduction proteins, encoded by glnB in cyanobacteria. PII regulates the activity of proteins involved in nitrogen and carbon metabolism by direct protein-protein interactions [8], perceiving metabolic information through the competitive binding of ATP or ADP and the synergistic binding of ATP and 2-OG [9,10]. The PII trimer has three binding sites for ATP/ADP (in some species AMP) and 2-OG. Despite the remarkable structural conservation of PII proteins, unique PII targets are found in cyanobacteria.
The first PII targets identified in cyanobacteria were detected in a search for proteins of the unicellular strain Synechococcus elongatus PCC7942 (hereafter S. elongatus) that interact with PII (a protein 112 amino acids long), namely NAGK (N-Acetyl Glutamate Kinase) and PipX (PII interacting protein X), a small protein of 89 amino acids restricted to cyanobacteria [11,12,13,14,15,16]. PipX is composed of two domains. The N-terminal domain (residues 1-53) is a Tudor-Like Domain [17] that is assimilated to the KOW domain [18]. The C-terminal domain (residues 54-89) is composed of two alpha-helices. In the PipX-PII complexes (Figure 1A), one PII trimer sequesters three PipX molecules [17]. PipX-PII complexes from Anabaena sp. PCC 7120 are virtually identical to those of S. elongatus [19].
Cyanobacterial genomes always contain at least as many copies of the essential gene glnB (encoding PII) as of the non-essential gene pipX [20]. In addition, loss-of-function mutations at pipX allow inactivation of the glnB gene in S. elongatus [21,22,23,24,25], suggesting that a relatively high PII/PipX ratio is needed to sequester PipX to prevent uncontrolled binding to additional partners, resulting in “PipX toxicity”.
PipX also binds to the global transcriptional regulator NtcA, which is involved in nitrogen assimilation in cyanobacteria [12,26,27]. PipX provides a mechanistic link between PII signalling and gene expression in response to nitrogen limitation of the global NtcA regulon [28,29]. The PipX–NtcA complex (Figure 1B; [17]) consists of one active (2-OG-bound) NtcA dimer and two PipX molecules. Each NtcA subunit binds one PipX molecule. PipX stabilizes the conformation of NtcA that is transcriptionally active and probably helps local recruitment of RNA polymerase. Binding of PipX to PII or NtcA is antagonistically tuned by 2-OG levels: whereas high levels of 2-OG favour the interaction of PipX with NtcA, they prevent the PipX-PII interaction [12,17,22,28].
PipX uses the same surface from its TLD/KOW domain to bind to either 2-OG-bound NtcA, stimulating DNA binding and transcriptional activity, or to 2-OG-free PII. PII sequestration of PipX at low 2-OG renders PipX unavailable for NtcA binding and activation, reducing the expression of NtcA-dependent gene targets [16,17,19,21,22,23,30]. In addition, the interaction between PII and PipX is highly sensitive to fluctuations in the ATP/ADP ratio, and thus the energy state of the cells [31,32].
In S. elongatus, pipX forms a bicistronic operon with the downstream gene pipY [33], encoding a member of the widely distributed and highly conserved family of pyridoxal phosphate (PLP)-binding proteins (COG0325/PLPBP) that are involved in vitamin B6 and amino acid homeostasis [34,35]. Previous work indicates functional interactions in S. elongatus and further suggests tight co-regulation and translational coupling between PipX and PipY in cyanobacteria [33,36,37].
While the identification of PipX as a binding partner for PII or NtcA lead to the characterisation of PipX as a player in the context of transcriptional regulation linked to carbon/nitrogen metabolism, more recent studies suggest the influence of PipX in additional processes and at additional levels. Yeast three-hybrid searches with PipX–PII as bait identified the transcriptional regulator PlmA, a protein found exclusively in cyanobacteria, as an additional PipX target [13], suggesting the involvement in more extensive transcription control. Transcriptomic analyses of null and gain-of-function pipX mutants revealed a strong connection between PipX and translation [28]. PipX acts in cis to upregulate PipY levels post transcriptionally [33,36], presumably to maintain appropriate PipX-PipY stoichiometry or regulated control of PipY protein levels. Gradient profiling by sequencing (Grad-seq) showed that PipX co-localizes with either metabolic regulators PII, NtcA and PlmA or with RNA-protein complexes involved in transcription, RNA metabolism and translation initiation [38]. Last but not least, cyanobacterial synteny [14] led to the identification of the ribosome-assembly GTPase EngA as a PipX regulatory target [39,40], thus providing additional evidence of the regulatory connections between PipX and ribosome function in S. elongatus.
The aim of this work was to gain additional insights into the determinants involved in the putative interaction of PipX with its “toxic” partner(s) by expanding previous mutational analyses of PipX. However, most of the tested mutations decreased protein levels, and only four single point mutations that decrease PipX toxicity without impairing protein levels were identified and thus discussed in this work. Since most of the mutations target residues involved in binding to PII or NtcA the results further suggested that the in vivo stability of PipX is affected by its ability to bind to PII, its most abundant partner. Last but not least, we show that disruption of PipX-PII complexes in S. elongatus cultures under environmentally relevant conditions such the transition from darkness to light transiently decrease PipX levels. The implications of these results for the PipX interaction and regulatory network are discussed.
2. Materials and Methods
2.1. Cyanobacterial Growth Conditions
S. elongatus cultures were routinely grown at 30°C in BG11 media (BG110 plus 17.5 mM sodium nitrate (NaNO3) and 10 mM HEPES/NaOH pH 7.8; [41]), under constant illumination provided by cool white-fluorescent lights in baffled flasks (shaking: 150 rpm, 70 μmol photons m–2s–1) or on plates (50 μmol photons m–2s–1). For solid media, 1.5% (w/v) agar and 0.5 mM sodium thiosulfate (Na2S2O3; after autoclaving) were added. The transformations were performed essentially as described by [42]. To select genetically modified strains, solid media were supplemented with the antibiotics: chloramphenicol (Cm; 3.5 μg mL–1), streptomycin (Sm; 15 μg mL–1), or kanamycin (Km; 12 μg mL–1).
For growth in liquid, 25–30 mL of cultures were adjusted to an initial optical density at 750 nm (OD750nm) of 0.1, measured using an Ultrospec 2,100 pro UV–Vis Spectrophotometer (Amersham), and grown until they reached 0.5–0.7. For the transition from darkness to light, cultures at 0.7 OD750nm were subjected to 12 hours of darkness followed by exposure to light.
2.2. Plasmid and Strains Construction
Strains and plasmids used in this work are listed in Table 1 and Table 2, respectively, and oligonucleotides in Table S1. Cloning procedures were carried out in Escherichia coli DH5α using standard techniques [43]. All constructs were analyzed by automated dideoxy DNA sequencing.
To obtain plasmids pUAGC948, pUAGC945, pUAGC937 and pUAGC618 carrying fusion Φ(C.K1-pipXH9ApipY), Φ(C.K1-pipXY16ApipY), Φ(C.K1-pipXR70ApipY) and Φ(C.K1-pipXL80QpipY) respectively, QuickChange mutagenesis using pUAGC410 as template was performed with the following pairs of primers PipX-H9A-1F/1R, PipX-Y16A-1F/1R, PipX-R70A-1F/1R and PipX-L80Q-1F/1R.
To generate the C.K1X*Y S. elongatus derivative strain, ∆pipX strain was transformed with the appropriate plasmids, and the transformants were selected on plates with kanamycin. Completed segregation and correct construction insertion were analysed by PCR with primers PipX-126F and PipX-5R.
CK1XH9AY, CK1XY16AY, CK1XR70AY and CK1XL80QY strains were transformed with the construction to inactivate glnB. Sm resistant clones were analysed with primers Glnb-1F and Glnb-1R to check the segregation of glnB null allele.
2.3. Protein Extraction and Immunodetection Assays
For immunodetection assays, 10 mL of cultures were sampled at different times and quickly harvested by centrifugation at 7300g for 6 min (4ºC), flash frozen in liquid nitrogen and stored at 20 ºC until use. The pellets were resuspended in 60 μl of lysis buffer (25 mM Tris/HCl pH 7.5, 0.4 mM EDTA, 1 mM DTT, 0.8 mg/mL protease inhibitor, 50 mM NaCl) and cells were disrupted using a spoon of 0.1 µm glass beads, as described in [13]. Mixtures were subjected to three cycles of 60 s at a speed of 5 m/s in a high-speed homogenizer Minibeadbeater always followed by a 60 s of repose at 4 ºC after each cycle. Samples were centrifuged (5500g for 5 min) and the supernatant fractions (crude protein extracts) were transferred to a new tube and stored at -20ºC until needed.
Protein concentrations were estimated by the Bradford method using the PierceTM detergent compatible Bradford assay kit (ThermoScientific) in a VICTOR3TM 1420 Multilabel Plate Reader (PerkinElmer). Immunodetection was performed by charging 60 µg of total protein extract in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 15% polyacrylamide), followed by wet immunoblotting in 0.1 μm polyvinylidene fluoride membranes (from GE Healthcare). The membranes were blocked with Tris-Buffered Saline (TBS; 20 mM Tris/HCl pH 7.5, 500 mM NaCl) solution containing 5% non-fat dried milk for 30 min at room temperature and then incubated overnight in TBS solution containing 2% non-fat dried milk and the primary antibody. Then, the membranes were incubated at room temperature for 1.5 hours with a 1:150000 dilution of ECL rabbit IgG, HRP-linked F(ab’)2 fragment (from donkey; GE Healthcare). The signal was detected with the addition of the SuperSignal WestFemto reagent (ThermoScientific) in a Biorad ChemiDoc Imager using the automatic exposure mode and avoiding pixel saturation or using X-ray and scanning the films. All the membranes were immunodetected first with a 1:5000 dilution of primary anti-PipX antibody and then with a 1:5000 anti-PlmA. At least two independent western-blot assays of one to three independent clones of each strain were performed for each of the mutant strains. Antisera against PipX (Pineda Antikörper Service, Berlin, Germany) and PlmA (Genosphere Biotechnologies, Paris) were produced in rabbits.
2.4. Computational Methods
Graphical representations of the protein structures were generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.7.1.7, Schrödinger, LLC.). Atom-atom contacts were automatically calculated using the default range defined in LigPlot+ Version v.2.2.8 [45].
Protein intensity levels were quantified from the western blot images using the ImageJ software. Bands were picked up using the “rectangle” function and the area plot corresponding to the intensity was measured with the “wand” tool. Each area from the PipX immunodetection was normalized using the corresponding area of PlmA and referred to the control strain. Statistical analysis of the results was performed in the RStudio program [46]. Detailed quantifications of protein expressions can be found in Table S2.
3. Results and Discussion
3.1. PipX Toxicity is Altered by Point Mutations Targeting Residues at Each of Its Two Domains
The identification, after unsuccessful attempts to inactivate glnB in S. elongatus, of spontaneous suppressor mutations at the promoter or at coding sequences of the pipX gene [22] led to the realization that decreasing PipX levels and/or activity makes PII dispensable. However, the mechanism involved in PipX toxicity remains to be addressed. Amongst the spontaneous suppressor mutations obtained in the different studies only two were single substitutions (R54C and L65Q) and could thus give clues on PipX residues that may play a role in toxicity in the absence of PII. Their location at the C-terminal domain of PipX (Figure 2A), which is not involved in the well characterized interactions with PII and NtcA partners, suggests a rather important role for this domain in PipX toxicity.
R54 is the first of the 6 arginine residues of a basic R-rich patch located at alpha helix 1 and, although it does not interact with PII and it only makes one direct and one water-mediated hydrogen bond with NtcA in the available structures of the corresponding complexes, the R54C substitution impaired Y2H interactions with NtcA [17,22]. L65 interacts with L80 of alpha helix 2 in the “flexed” conformation of the two-helix C-terminal domain of PipX and both residues are exposed in the “extended” conformation. The flexed form is found in all PipX-NtcA structures, in some of the PipX-PII molecules [17], and when PipX is not bound to its partners [47]. While L65Q did not impair Y2H interactions with PII or NtcA, it disrupted three-hybrid interactions of PipX-PII with PlmA [13]. Similar to L65Q, and in contrast to R54C, the L80Q substitution also disrupted three-hybrid interactions of PipX-PII with PlmA, supporting the involvement of L65 and L80 in common functions.
The ability of PipX mutant derivatives to alter PipX toxicity in the absence or at lower doses of PII has previously been addressed by genetic approaches and allowed classification of mutations into three simple categories regarding toxicity: gain-of-function, loss-of-function or wild type function. Analysis of PipX toxicity initially involves replacing the pipX gene with the marker fusion Φ(C.K1-pipX) in S. elongatus [22,23]. In the resulting strain (CK1XY), reproducibly higher levels of the PipX protein are produced [33], allowing identification of gain-of-function mutations that increase toxicity. On the other hand, distinction between neutral and loss-of-function mutations requires that mutant strains (collectively called CK1X*Y, where * symbolizes each of the PipX mutations), are subsequently used as recipients for the inactivation allele glnB::C.S3.
The role of residues L65 and L80 in promoting conformational changes in PipX prompted us to examine the toxicity of the L80Q variant. Along similar lines we also examined the toxicity of the R70A variant. R70, located in the R-rich patch of the C-terminal domain, is one of the PipX residues with the highest mobility and it has been proposed to act as a hinge for the opening of the second helix [47].
We next introduced R70A and L80Q variants in the appropriate plasmids and strains and tested them for PipX toxicity alongside a wild type control and other mutations of interest. The latter included two substitutions at the TLD/KOW that were not previously analysed for toxicity, H9A, known to impair Y2H signals with both PII and NtcA, and Y16A which only impaired Y2H signal with PII [17].
As shown in Figure 2B the glnB::C.S3 allele segregated in strains CK1XH9AY and CK1XL80QY but not in CK1XY16AY, CK1XR70QY or the wild type control, indicating that H9A and L80Q, but not Y16A or R70A, suppressed toxicity. Since neither R69A or R70A suppress toxicity, it appears that the R-rich patch is not directly involved in toxicity, although we cannot exclude the possibility of functional redundancy between R70 and its neighbour R69, which also does not suppress toxicity. On the other hand, suppression of toxicity by the L80Q variant supported the importance of the flexed conformation for the toxic function of PipX.
The location on PipX structure and toxicity phenotypes conferred by these and all previously studied mutations are illustrated in Figure 2C. It should be noted that, except for the two spontaneous mutations (R54C, L65Q), the remaining ones target residues chosen in the context of studies on PipX interactions with PII, NtcA or to a lesser extent with PlmA, and thus their distribution along PipX sequence is non-random.
3.2. Most but Not All PipX Point Mutations alter Toxicity and NtcA Coactivation in a Similar Manner
Because toxicity is also triggered by PipX overexpression [23,37,39], a very simplistic explanation for the loss- or gain-of-function mutations would be that they correlate with, respectively, lower or higher levels of PipX. However, previous studies concerning the two spontaneous mutations suppressing PipX toxicity (R54C and L65Q; [22]) and the two gain-of-function mutations contradicted the idea, since that correlation stands only for L65Q and, remarkably, the gain-of-function proteins PipXE4A or PipXY32A are detected at lower levels than PipX in the corresponding CS3XY derivative strains [23]. The implication is that toxicity must depend on the increased binding of these gain-of-function proteins to additional partner(s).
Increasing productive interactions with NtcA to upregulate transcription of target genes was one of the first working hypotheses to explain PipX toxicity, prompting studies in which we analysed the effect of mutations altering toxicity on co-activation of NtcA-dependent genes [22,23,28]. Altogether, there is a good but incomplete correlation between the effects of mutations in NtcA coactivation and toxicity. All three tested mutations that were wild type for PipX toxicity were also wild type for NtcA coactivation (R69A, Q82A, Q86A), while the two mutations that increased toxicity (E4A and Y32A) and most mutations that decreased PipX toxicity (Y6A, Q34A, R35A, F38A, R54C, L65Q) were, respectively, gain-of-function and loss-of-function for NtcA coactivation. However, three loss-of-function mutations for toxicity (F12A, D23A and L36A) did not decrease NtcA coactivation, indicating separation of functions.
An additional indication that there is not a clear cause-effect relationship between PipX toxicity and NtcA activity was provided by the finding that PII also counteracted PipX toxicity under conditions in which NtcA is known to be mainly inactive, as it is the case in cultures with ammonium as a nitrogen source [32]. Therefore, it appears that up regulation of NtcA gene targets might contribute to but would not be the only factor involved in PipX toxicity.
3.3. Not All Loss-of-Function Mutations for Toxicity Decrease PipX Levels in S. elongatus
We wondered to which extent loss-of-function mutations for toxicity corresponded to decreased PipX levels in S. elongatus and thus they would not be informative on the molecular mechanism of toxicity. To investigate this issue, we obtained CK1X*Y strains corresponding to each of the loss-of-function or neutral mutations available and subsequently performed western blots to compare PipX levels. It is worth noting that the effect on PipX* levels of mutations E4A and Y32A could not be tested in parallel since they are lethal in strain CK1XY. However, they were previously shown to decrease PipX* levels in strain CS3XY [23].
As shown in Figure 3A, significantly lower levels of PipX* were found amongst most of the strains carrying loss-of-function mutations for toxicity (Y6A, H9A, Q34E, R35A, F38A, L65Q, L80Q) while not amongst any of the strains classified as wild type for toxicity (carrying substitutions Y16A, R69A, R70Q, Q82A or Q86A). In addition, we identified four mutations that decreased toxicity without significantly influencing the levels of PipX (F12A, D23A, L36A, and R54C). On the other hand, the variant Y16A, while not altering toxicity, conferred slightly higher levels of PipX*.
Taken together, the results shown in Figure 3A reveal a good correlation between loss-of-function for toxicity and impaired levels of PipX. Importantly, it shows that most of the mutations discussed here, particularly those located at the TLD/KOW domain, have negative impact on PipX levels, possibly because of destabilization via misfolding or decreased stability of the properly folded protein.
Next, we focused on the toxicity-suppressing mutations that did not alter PipX levels. Three of them replace residues of the TLD/KOW domain (F12, D23, L36), the first two mapping at loops on opposite edges of the β-sheet (loops β1-β2 and β2-β3, respectively) and L36 at the beginning of β4, on the same edge of the sheet than F12 (Figure 3B). The fourth one (R54C) is at the beginning of C-terminal domain and in close proximity to L36. Mutations F12A, L36A and R54C impair interaction of PipX with at least NtcA and, although mutations F12A, L36A did not impair NtcA coactivation of reporter genes [23], we cannot exclude the possibility that suppression of PipX toxicity by these mutations might derive from a negative effect on NtcA coactivation, affecting particular NtcA-dependent genes. That mechanism would be more likely in the case of R54C, specifically altering yeast-two hybrid interaction with NtcA and impairing coactivation [22]. Importantly, residue D23 makes no direct contact with PII or NtcA (see PDB files 2XG8 and 2XKO), and thus the same mechanism for suppression of PipX toxicity would not apply here.
3.4. Most Mutations Impairing Binding to PII and NtcA Reduce PipX Levels in S. elongatus
The finding that several mutations targeting residues involved in binding to PII or NtcA, including the two known gain-of-function mutations (E4A and Y32A), impaired PipX levels in S. elongatus suggested to us that the stability of PipX in vivo may depend on its ability to form complexes with its more abundant partners, particularly with PII. Interestingly, proteins PipXE4A, PipXY6A or PipXY32A, detected at low levels in S. elongatus ([23]; Figure 3A) yielded purification levels similar to wild-type PipX when they were expressed in E. coli [17]. Thus, the finding that these PipX* derivatives were not particularly unstable in E. coli, suggests that PipX may be more susceptible to proteases in cyanobacteria.
To further explore the idea that the stability of PipX in vivo may depend on its ability to form complexes with PII or NtcA, we represented PipX complexes in Figure 3C and determined PipX-PII and PipX-NtcA contacts using LigPlot+. PipX chains E and C were respectively used from PDBs 2XG8 and 2XKO complexes.
The most drastic effects on PipX* protein levels were produced by the substitution F38A, targeting an invariant residue at beta sheet 4 not contacting PII or NtcA that has a key structural role in filling the hydrophobic nucleus that stabilizes the curved conformation of the TLD/KOW domain [17,23]. Thus, this substitution has misfolding effects that are independent of the interaction of PipX with its partners. The three substitutions in the TLD/KOW domain that decreased PipX levels (Y6A, Q34A and R35A) involve residues with extensive interactions with PII and NtcA, while the substitutions that do not decrease PipX levels (Y16A and D23A), target residues having no direct interaction with PII or NtcA.
Two mutations suggested that binding to PII may be more critical for PipX levels than binding to NtcA: E4A, that specifically impairs binding to PII and decreases PipX levels and R54C, that specifically impair binding to NtcA and do not decrease PipX levels. Since under standard culture conditions PipX monomers are an order of magnitude more abundant than NtcA monomers, while PII trimers are in 40-fold excess over NtcA dimers [48], a role as PipX chaperon would make physiological sense in the case of PII but not so much for NtcA.
While the results available so far support the notion that the stability of PipX in vivo was affected by its ability to form complexes, particularly with PII (summarized in Figure 3D), two mutations were apparently at odds with this idea. F12A and L36A target residues at the PII and NtcA interaction surfaces of the TLD/KOW domain, they abolish the signal in Y2H assays with PII and with NtcA [17] but did not reduce PipX* levels or impair NtcA co-activation [23]. Thus, it appears that cellular factors in S. elongatus may specifically ameliorate the impact of these substitutions on interactions with PII and NtcA or that the mutations stabilise PipX, perhaps by increasing its affinity for alternative partners.
The L65Q and L80Q variants which confer lower levels of PipX* represent residues outside the TDL/KOW domain. Since L65 and L80 are involved in the maintenance of the flexed conformation, and this is the conformation adopted by PipX when not bound to its targets [47] it is tempting to propose that the flexed form contributes to PipX stability.
Altogether, the results of mutational analyses in S. elongatus suggest that complex formation regulate the stability of PipX, raising questions on the physiological significance and molecular details involved.
3.5. PipX Levels are Down Regulated by Switching from Dark to Light Conditions
It is worth noting that studies in which S. elongatus cultures have been subjected to different conditions of nitrogen, temperature or light regimes indicated that PipX levels do not fluctuate between the corresponding experimental conditions compared [23,40,48]. However, this constancy of PipX levels under different culture conditions is still compatible with transient decreases on PipX levels in response to environmental changes that trigger intracellular increases of 2-OG levels and/or of the ATP/ADP ratio and therefore disrupt PipX-PII complexes.
Since the intracellular ATP/ADP ratio can easily be increased by transferring to light S. elongatus cultures that have been previously maintained in darkness [32,49], we next investigated the effect on PipX levels of changes from darkness to light using culture conditions previously shown to simultaneously affect the ATP/ADP ratio and PipX-PII complex formation [32].
S. elongatus cultures were subjected to a 12-hour period of darkness before being transferred to light, and timepoints were taken at different intervals up to 120 minutes of the light exposure. As shown in Figure 4, a decrease of circa 30 % of the PipX levels was already observed 5 min after the light switch, followed by a slow recovery that was complete at the end of the experiment. Therefore, the results confirm that disrupting PipX-PII complexes results in a transient decrease of PipX levels.
3.6. Regulatory Complexities of the PipX-PII Interaction Network, the Dual Role of PII
Sequestration into PipX-PII complexes have been regarded as a molecular mechanism to negatively control NtcA transcriptional activity [12,17] or, considering PipX binding partners subsequently identified, to interfere in general with formation of other PipX complexes that would thus be negatively regulated by PII in response to intracellular signals of carbon/nitrogen or energy status. This appears to be the case with PipX coactivation of the large NtcA-dependent regulon [28] or with the less well-known regulatory interactions involving PipX, including modulation of the ribosome-assembly GTPase EngA [39,40] or the putative interaction with the “toxic” partner. However, we show here that PII-PipX complex formation also provides a mechanism to maintain a relatively large pool of PipX, implying that PII, acting as a PipX chaperon, is also an activator of PipX functions.
It is worth noting that, before we reported the identification of PipX [11,12], genetic analysis suggested that PII was a positive regulator of NtcA-activated genes [50]. Since we subsequently found that inactivation of PII requires the obtention of suppressor mutations that often map at pipX [21,24], PipX deficiency provided a straightforward explanation for the impaired NtcA activity in the glnB null mutant. The present study provides a molecular mechanism for the positive regulatory role of PII on NtcA activity, and presumably also in other PipX controlled processes. PII would thus have dual regulatory roles over different PipX complexes. Interference with formation of other PipX complexes would be favored by the great excess of PII, a very abundant protein, over other PipX partners [32,39], while the stimulation of formation of those other PipX complexes, in response to lowering the intracellular levels of 2-OG or the ATP/ADP ratio, would depend on the amount of PipX previously sequestered by PII.
The positive role of PII on PipX levels raises questions on the extent to which the levels of other PII binding proteins, particularly small regulatory ones, are modulated by complex formation with PII. Small proteins appear to play critical roles in key cyanobacterial processes such photosynthesis or regulation of the carbon to nitrogen balance [51,52,53,54,55,56,57,58,59], with paradigmatic examples such as the inhibition by nitrogen abundance of glutamine synthetase, executed by two small proteins instead of by covalent modification, which is the main mechanism in proteobacteria [60,61,62,63]. Recent work in Synecocystis sp. PCC 6803 [64,65,66] has shown that PII binds other small proteins, collectively called Pirs (PII-interacting regulators) that, in contrast to PipX, are not widely conserved in cyanobacteria. Two of them PirA and PirC/CfrA were highly enriched in the PII-target searches [64] and have been shown to be involved in regulation of the carbon to nitrogen balance. PirA antagonises the PII-dependent activation of NAGK, the key enzyme in arginine biosynthesis, and can be considered a PII-sequestrator while PirC allosterically inhibits the glycolytic enzyme 2,3-phosphoglycerate-independent phosphoglycerate mutase (PGAM) and it is thus sequestered by PII, as it is the case with PipX. However, the levels of PirA and PirC (in response to signals of the carbon/nitrogen status) are highly (and inversely) up regulated [64,65,66], a common feature shared by small regulatory proteins. In contrast, PipX, whose intracellular levels appear to be subjected just to relatively small and transient changes is so far the only small cyanobacterial protein whose levels appear to be down regulated essentially by the same signals that trigger disruption of PII-PipX complexes.
The finding that PipX levels are down regulated by disruption of complexes with PII emphasizes the importance of the later as a dual regulator of the multiple interactions mediated by PipX. In the case of PipX-EngA complexes, in addition to PII effectors, EngA effectors GTP and GDP [67,68] need to be also considered and may be particularly relevant under transition from darkness to light, when GTP/GDP ratios are expected to increase prior to reinitiating culture growth [69]. Since in vitro pull-down assays suggested that binding of PipX to EngA is decreased by GDP [39], after transfer of S. elongatus cultures to light, disruption of PipX-PII complexes combined with an increased GTP/GDP ratio should favor formation of PipX-EngA complexes, a response that in turn would be attenuated by the subsequent and transient decrease in PipX levels. Last but not least, the transient decrease in PipX levels would provide a protective mechanism against PipX toxicity. Further work is needed to address the possible involvement of EngA in this phenomenon.
4. Conclusions
To gain further insights into the regulatory complexities of the PipX interaction network, we have performed mutational analyses to investigate the connections between PipX toxicity and protein levels or interactions with binding partners PII or NtcA. The impact of 16 independent substitutions on the levels of PipX in S. elongatus have been analysed alongside control strains. Toxicity phenotypes were also determined here for mutant strains. As a result of these analyses, we have identified four mutations that suppress toxicity without impairing the levels of PipX in S. elongatus. The location of the targeted residues suggests that the yet uncharacterized “toxic” partner(s) might bind to PipX regions overlapping only partially with the surface binding to PII or NtcA, a scenario incompatible with formation of ternary complexes.
As a result of the mutational analysis presented here, we have been able to infer, and subsequently demonstrate, that PII regulates PipX levels in S. elongatus. This introduces a previously ignored additional level of control in the context of the PipX interaction network.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Table S1: Oligonucleotides, Table S2: Western blot quantification data.
Author Contributions
AC designed research. AL, LT, and RC performed research. AC wrote the paper. All authors analyzed data, contributed to manuscript revision, and read and approved the submitted version.
Funding
This work was supported by grant PID220-118816GB-I00 from the Spanish Government (MICINN) and grants VIGROB22-126, VIGROB23-126, and GRE20-04-C from the University of Alicante.
Acknowledgments
The authors thank C. V. Racovac, T. Mata, J. Espinosa, J. I. Labella, S. Bibak and L. Fuertes for technical contributions, and V. Rubio and R. Dixon for constructive discussions and critical reading of a previous version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blank, C.E.; Sanchez-Baracaldo, P. Timing of morphological and ecological innovations in the cyanobacteria--a key to understanding the rise in atmospheric oxygen. Geobiology 2010, 8, 1–23. [Google Scholar] [CrossRef]
- Lee, H.-W.; Noh, J.-H.; Choi, D.-H.; Yun, M.; Bhavya, P.S.; Kang, J.-J.; Lee, J.-H.; Kim, K.-W.; Jang, H.-K.; Lee, S.-H. Picocyanobacterial Contribution to the Total Primary Production in the Northwestern Pacific Ocean. Water 2021, 13, 1610. [Google Scholar] [CrossRef]
- Zhang, Y.; Burkhardt, D.H.; Rouskin, S.; Li, G.W.; Weissman, J.S.; Gross, C.A. A Stress Response that Monitors and Regulates mRNA Structure Is Central to Cold Shock Adaptation. Mol. Cell 2018, 70, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Forchhammer, K.; Selim, K.A. Carbon/nitrogen homeostasis control in cyanobacteria. FEMS Microbiol. Rev. 2020, 44, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Muro-Pastor, M.I.; Reyes, J.C.; Florencio, F.J. Ammonium assimilation in cyanobacteria. Photosynth. Res. 2005, 83, 135–150. [Google Scholar] [CrossRef]
- Senior, P.J. Regulation of nitrogen metabolism in Escherichia coli and Klebsiella aerogenes: Studies with the continuous-culture technique. J. Bacteriol. 1975, 123, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Huergo, L.F.; Dixon, R. The Emergence of 2-Oxoglutarate as a Master Regulator Metabolite. Microbiol. Mol. Biol. Rev. 2015, 79, 419–435. [Google Scholar] [CrossRef]
- Forchhammer, K.; Selim, K.A.; Huergo, L.F. New views on PII signaling: From nitrogen sensing to global metabolic control. Trends Microbiol. 2022, 30, 722–735. [Google Scholar] [CrossRef]
- Kamberov, E.S.; Atkinson, M.R.; Ninfa, A.J. The Escherichia coli PII signal transduction protein is activated upon binding 2-ketoglutarate and ATP. J. Biol. Chem. 1995, 270, 17797–17807. [Google Scholar] [CrossRef]
- Forchhammer, K.; Hedler, A. Phosphoprotein PII from cyanobacteria--analysis of functional conservation with the PII signal-transduction protein from Escherichia coli. Eur. J. Biochem. 1997, 244, 869–875. [Google Scholar] [CrossRef]
- Burillo, S.; Luque, I.; Fuentes, I.; Contreras, A. Interactions between the nitrogen signal transduction protein PII and N-acetyl glutamate kinase in organisms that perform oxygenic photosynthesis. J. Bacteriol. 2004, 186, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.; Forchhammer, K.; Burillo, S.; Contreras, A. Interaction network in cyanobacterial nitrogen regulation: PipX, a protein that interacts in a 2-oxoglutarate dependent manner with PII and NtcA. Mol. Microbiol. 2006, 61, 457–469. [Google Scholar] [CrossRef]
- Labella, J.I.; Obrebska, A.; Espinosa, J.; Salinas, P.; Forcada-Nadal, A.; Tremino, L.; Rubio, V.; Contreras, A. Expanding the Cyanobacterial Nitrogen Regulatory Network: The GntR-Like Regulator PlmA Interacts with the PII-PipX Complex. Front. Microbiol. 2016, 7, 1677. [Google Scholar] [CrossRef]
- Labella, J.I.; Llop, A.; Contreras, A. The default cyanobacterial linked genome: An interactive platform based on cyanobacterial linkage networks to assist functional genomics. FEBS Lett. 2020, 594, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Labella, J.I.; Cantos, R.; Salinas, P.; Espinosa, J.; Contreras, A. Distinctive Features of PipX, a Unique Signaling Protein of Cyanobacteria. Life (Basel) 2020, 10, 79. [Google Scholar] [CrossRef]
- Forcada-Nadal, A.; Llacer, J.L.; Contreras, A.; Marco-Marin, C.; Rubio, V. The P(II)-NAGK-PipX-NtcA Regulatory Axis of Cyanobacteria: A Tale of Changing Partners, Allosteric Effectors and Non-covalent Interactions. Front. Mol. Biosci. 2018, 5, 91. [Google Scholar] [CrossRef]
- Llacer, J.L.; Espinosa, J.; Castells, M.A.; Contreras, A.; Forchhammer, K.; Rubio, V. Structural basis for the regulation of NtcA-dependent transcription by proteins PipX and PII. Proc. Natl. Acad. Sci. USA 2010, 107, 15397–15402. [Google Scholar] [CrossRef]
- Kyrpides, N.C.; Woese, C.R.; Ouzounis, C.A. KOW: A novel motif linking a bacterial transcription factor with ribosomal proteins. Trends Biochem. Sci. 1996, 21, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.X.; Jiang, Y.L.; Xu, B.Y.; Chen, Y.; Zhang, C.C.; Zhou, C.Z. Crystal structure of the cyanobacterial signal transduction protein PII in complex with PipX. J. Mol. Biol. 2010, 402, 552–559. [Google Scholar] [CrossRef] [PubMed]
- (20) Laichoubi, K.B.; Beez, S.; Espinosa, J.; Forchhammer, K.; Contreras, A. The nitrogen interaction network in Synechococcus WH5701, a cyanobacterium with two PipX and two P(II)-like proteins. Microbiology (Reading) 2011, 157 Pt 4, 1220–1228. [Google Scholar] [CrossRef]
- Espinosa, J.; Castells, M.A.; Laichoubi, K.B.; Contreras, A. Mutations at pipX suppress lethality of PII-deficient mutants of Synechococcus elongatus PCC 7942. J Bacteriol 2009, 191, 4863–4869. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, J.; Castells, M.A.; Laichoubi, K.B.; Forchhammer, K.; Contreras, A. Effects of spontaneous mutations in PipX functions and regulatory complexes on the cyanobacterium Synechococcus elongatus strain PCC 7942. Microbiology (Reading) 2010, 156 Pt 5, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Laichoubi, K.B.; Espinosa, J.; Castells, M.A.; Contreras, A. Mutational analysis of the cyanobacterial nitrogen regulator PipX. PLoS ONE 2012, 7, e35845. [Google Scholar] [CrossRef]
- Chang, Y.; Takatani, N.; Aichi, M.; Maeda, S.; Omata, T. Evaluation of the effects of P(II) deficiency and the toxicity of PipX on growth characteristics of the P(II)-Less mutant of the cyanobacterium Synechococcus elongatus. Plant Cell Physiol. 2013, 54, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Takatani, N.; Sonoike, K.; Jimbo, H.; Nishiyama, Y.; Omata, T. Dissection of the Mechanisms of Growth Inhibition Resulting from Loss of the PII Protein in the Cyanobacterium Synechococcus elongatus PCC 7942. Plant Cell Physiol. 2021, 62, 721–731. [Google Scholar] [CrossRef]
- Herrero, A.; Muro-Pastor, A.M.; Flores, E. Nitrogen control in cyanobacteria. J. Bacteriol. 2001, 183, 411–425. [Google Scholar] [CrossRef]
- Esteves-Ferreira, A.A.; Inaba, M.; Fort, A.; Araujo, W.L.; Sulpice, R. Nitrogen metabolism in cyanobacteria: Metabolic and molecular control, growth consequences and biotechnological applications. Crit. Rev. Microbiol. 2018, 44, 541–560. [Google Scholar] [CrossRef]
- Espinosa, J.; Rodriguez-Mateos, F.; Salinas, P.; Lanza, V.F.; Dixon, R.; de la Cruz, F.; Contreras, A. PipX, the coactivator of NtcA, is a global regulator in cyanobacteria. Proc. Natl. Acad. Sci. USA 2014, 111, E2423–E2430. [Google Scholar] [CrossRef]
- Giner-Lamia, J.; Robles-Rengel, R.; Hernandez-Prieto, M.A.; Muro-Pastor, M.I.; Florencio, F.J.; Futschik, M.E. Identification of the direct regulon of NtcA during early acclimation to nitrogen starvation in the cyanobacterium Synechocystis sp. PCC 6803. Nucleic Acids Res 2017, 45, 11800–11820. [Google Scholar] [CrossRef]
- Espinosa, J.; Forchhammer, K.; Contreras, A. Role of the Synechococcus PCC 7942 nitrogen regulator protein PipX in NtcA-controlled processes. Microbiology (Reading) 2007, 153 Pt 3, 711–718. [Google Scholar] [CrossRef]
- Zeth, K.; Fokina, O.; Forchhammer, K. Structural basis and target-specific modulation of ADP sensing by the Synechococcus elongatus PII signaling protein. J. Biol. Chem. 2014, 289, 8960–8972. [Google Scholar] [CrossRef]
- Espinosa, J.; Labella, J.I.; Cantos, R.; Contreras, A. Energy drives the dynamic localization of cyanobacterial nitrogen regulators during diurnal cycles. Environ. Microbiol. 2018, 20, 1240–1252. [Google Scholar] [CrossRef]
- Labella, J.I.; Cantos, R.; Espinosa, J.; Forcada-Nadal, A.; Rubio, V.; Contreras, A. PipY, a Member of the Conserved COG0325 Family of PLP-Binding Proteins, Expands the Cyanobacterial Nitrogen Regulatory Network. Front. Microbiol. 2017, 8, 1244. [Google Scholar] [CrossRef] [PubMed]
- Ito, T. Role of the conserved pyridoxal 5’-phosphate-binding protein YggS/PLPBP in vitamin B6 and amino acid homeostasis. Biosci. Biotechnol. Biochem. 2022, 86, 1183–1191. [Google Scholar] [CrossRef]
- Tremino, L.; Llop, A.; Rubio, V.; Contreras, A. The Conserved Family of the Pyridoxal Phosphate-Binding Protein (PLPBP) and Its Cyanobacterial Paradigm PipY. Life (Basel) 2022, 12, 1622. [Google Scholar] [CrossRef]
- Cantos, R.; Labella, J.I.; Espinosa, J.; Contreras, A. The nitrogen regulator PipX acts in cis to prevent operon polarity. Environ. Microbiol. Rep. 2019, 11, 495–507. [Google Scholar] [CrossRef]
- Llop, A.; Labella, J.I.; Borisova, M.; Forchhammer, K.; Selim, K.A.; Contreras, A. Pleiotropic effects of PipX, PipY, or RelQ overexpression on growth, cell size, photosynthesis, and polyphosphate accumulation in the cyanobacterium Synechococcus elongatus PCC7942. Front. Microbiol. 2023, 14, 1141775. [Google Scholar] [CrossRef]
- Riediger, M.; Spat, P.; Bilger, R.; Voigt, K.; Macek, B.; Hess, W.R. Analysis of a photosynthetic cyanobacterium rich in internal membrane systems via gradient profiling by sequencing (Grad-seq). Plant Cell 2021, 33, 248–269. [Google Scholar] [CrossRef]
- Jerez, C.; Salinas, P.; Llop, A.; Cantos, R.; Espinosa, J.; Labella, J.I.; Contreras, A. Regulatory Connections Between the Cyanobacterial Factor PipX and the Ribosome Assembly GTPase EngA. Front. Microbiol. 2021, 12, 781760. [Google Scholar] [CrossRef] [PubMed]
- Llop, A.; Bibak, S.; Cantos, R.; Salinas, P.; Contreras, A. The ribosome assembly GTPase EngA is involved in redox signaling in cyanobacteria. Front. Microbiol. 2023, 14, 1242616. [Google Scholar] [CrossRef] [PubMed]
- Rippka, R.; Deruelles, J.; Waterbury, J.B.; Hermand, M.; Stanier, R.Y. Generic Assignments, Strain Histories and Properties of Pure Cultures of Cyanobacteria. J. General. Microbiol. 1979, 111, 1–61. [Google Scholar] [CrossRef]
- Golden, S.S.; Sherman, L.A. Optimal conditions for genetic transformation of the cyanobacterium Anacystis nidulans R2. J. Bacteriol. 1984, 158, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.R.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor: Berlin/Heidelberg, Germany, 1989. [Google Scholar]
- Glover, D.M. DNA Cloning: A Practical Approach; Glover: 1985; p. 109.
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- RStudio: Integrated Development for, R. RStudio; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Forcada-Nadal, A.; Palomino-Schatzlein, M.; Neira, J.L.; Pineda-Lucena, A.; Rubio, V. The PipX Protein, When Not Bound to Its Targets, Has Its Signaling C-Terminal Helix in a Flexed Conformation. Biochemistry 2017, 56, 3211–3224. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, A.C.; Benevento, M.; Lehmann, R.; van Breukelen, B.; Post, H.; Giansanti, P.; Maarten Altelaar, A.F.; Axmann, I.M.; Heck, A.J. Daily rhythms in the cyanobacterium synechococcus elongatus probed by high-resolution mass spectrometry-based proteomics reveals a small defined set of cyclic proteins. Mol. Cell Proteomics 2014, 13, 2042–2055. [Google Scholar] [CrossRef]
- Rust, M.J.; Golden, S.S.; O’Shea, E.K. Light-driven changes in energy metabolism directly entrain the cyanobacterial circadian oscillator. Science 2011, 331, 220–223. [Google Scholar] [CrossRef]
- Fadi Aldehni, M.; Sauer, J.; Spielhaupter, C.; Schmid, R.; Forchhammer, K. Signal transduction protein P(II) is required for NtcA-regulated gene expression during nitrogen deprivation in the cyanobacterium Synechococcus elongatus strain PCC 7942. J Bacteriol 2003, 185, 2582–2591. [Google Scholar] [CrossRef]
- Collier, J.L.; Grossman, A.R. A small polypeptide triggers complete degradation of light-harvesting phycobiliproteins in nutrient-deprived cyanobacteria. EMBO J. 1994, 13, 1039–1047. [Google Scholar] [CrossRef]
- Salinas, P.; Ruiz, D.; Cantos, R.; Lopez-Redondo, M.L.; Marina, A.; Contreras, A. The regulatory factor SipA provides a link between NblS and NblR signal transduction pathways in the cyanobacterium Synechococcus sp. PCC 7942. Mol. Microbiol. 2007, 66, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Groben, R.; Kaloudas, D.; Raines, C.A.; Offmann, B.; Maberly, S.C.; Gontero, B. Comparative sequence analysis of CP12, a small protein involved in the formation of a Calvin cycle complex in photosynthetic organisms. Photosynth. Res. 2010, 103, 183–194. [Google Scholar] [CrossRef]
- Storz, G.; Wolf, Y.I.; Ramamurthi, K.S. Small proteins can no longer be ignored. Annu. Rev. Biochem. 2014, 83, 753–777. [Google Scholar] [CrossRef] [PubMed]
- Klahn, S.; Schaal, C.; Georg, J.; Baumgartner, D.; Knippen, G.; Hagemann, M.; Muro-Pastor, A.M.; Hess, W.R. The sRNA NsiR4 is involved in nitrogen assimilation control in cyanobacteria by targeting glutamine synthetase inactivating factor IF7. Proc. Natl. Acad. Sci. USA 2015, 112, E6243–E6252. [Google Scholar] [CrossRef] [PubMed]
- Klahn, S.; Bolay, P.; Wright, P.R.; Atilho, R.M.; Brewer, K.I.; Hagemann, M.; Breaker, R.R.; Hess, W.R. A glutamine riboswitch is a key element for the regulation of glutamine synthetase in cyanobacteria. Nucleic Acids Res. 2018, 46, 10082–10094. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, D.; Kopf, M.; Klahn, S.; Steglich, C.; Hess, W.R. Small proteins in cyanobacteria provide a paradigm for the functional analysis of the bacterial micro-proteome. BMC Microbiol. 2016, 16, 285. [Google Scholar] [CrossRef]
- Brandenburg, F.; Klahn, S. Small but Smart: On the Diverse Role of Small Proteins in the Regulation of Cyanobacterial Metabolism. Life (Basel) 2020, 10, 322. [Google Scholar] [CrossRef]
- Krauspe, V.; Fahrner, M.; Spat, P.; Steglich, C.; Frankenberg-Dinkel, N.; Macek, B.; Schilling, O.; Hess, W.R. Discovery of a small protein factor involved in the coordinated degradation of phycobilisomes in cyanobacteria. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Merida, A.; Candau, P.; Florencio, F.J. Regulation of glutamine synthetase activity in the unicellular cyanobacterium Synechocystis sp. strain PCC 6803 by the nitrogen source: Effect of ammonium. J Bacteriol 1991, 173, 4095–4100. [Google Scholar] [CrossRef]
- Reyes, J.C.; Florencio, F.J. A novel mechanism of glutamine synthetase inactivation by ammonium in the cyanobacterium Synechocystis sp. PCC 6803. Involvement of an inactivating protein. FEBS Lett 1995, 367, 45–48. [Google Scholar] [CrossRef]
- Reyes, J.C.; Muro-Pastor, M.I.; Florencio, F.J. Transcription of glutamine synthetase genes (glnA and glnN) from the cyanobacterium Synechocystis sp. strain PCC 6803 is differently regulated in response to nitrogen availability. J Bacteriol 1997, 179, 2678–2689. [Google Scholar] [CrossRef]
- Garcia-Dominguez, M.; Reyes, J.C.; Florencio, F.J. Glutamine synthetase inactivation by protein-protein interaction. Proc. Natl. Acad. Sci. USA 1999, 96, 7161–7166. [Google Scholar] [CrossRef]
- Watzer, B.; Spat, P.; Neumann, N.; Koch, M.; Sobotka, R.; Macek, B.; Hennrich, O.; Forchhammer, K. The Signal Transduction Protein P(II) Controls Ammonium, Nitrate and Urea Uptake in Cyanobacteria. Front. Microbiol. 2019, 10, 1428. [Google Scholar] [CrossRef] [PubMed]
- Bolay, P.; Rozbeh, R.; Muro-Pastor, M.I.; Timm, S.; Hagemann, M.; Florencio, F.J.; Forchhammer, K.; Klahn, S. The Novel P(II)-Interacting Protein PirA Controls Flux into the Cyanobacterial Ornithine-Ammonia Cycle. mBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, T.; Scholl, J.; Spat, P.; Lucius, S.; Koch, M.; Macek, B.; Hagemann, M.; Forchhammer, K. The novel P(II)-interactor PirC identifies phosphoglycerate mutase as key control point of carbon storage metabolism in cyanobacteria. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Brown, E.D. Phenotypic investigations of the depletion of EngA in Escherichia coli are consistent with a role in ribosome biogenesis. FEMS Microbiol. Lett. 2014, 353, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Acharya, A.; Tomar, S.K.; Prakash, B. Disrupting domain-domain interactions is indispensable for EngA-ribosome interactions. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Gaal, T.; Bartlett, M.S.; Ross, W.; Turnbough, C.L., Jr.; Gourse, R.L. Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science 1997, 278, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PipX complexes with PII and NtcA. A. PipX-PII (PDB:2XG8), B. PipX-NtcA (PDB:2XKO). PipX structures are shown in ribbon representation and are colored in different hues of grey. PII and NtcA are shown in surface representation with each subunit in a different hue of blue (PII) or green (NtcA). In B) NtcA-bound 2-OG molecules are shown in spheres representation and are colored brown.
Figure 1.
PipX complexes with PII and NtcA. A. PipX-PII (PDB:2XG8), B. PipX-NtcA (PDB:2XKO). PipX structures are shown in ribbon representation and are colored in different hues of grey. PII and NtcA are shown in surface representation with each subunit in a different hue of blue (PII) or green (NtcA). In B) NtcA-bound 2-OG molecules are shown in spheres representation and are colored brown.
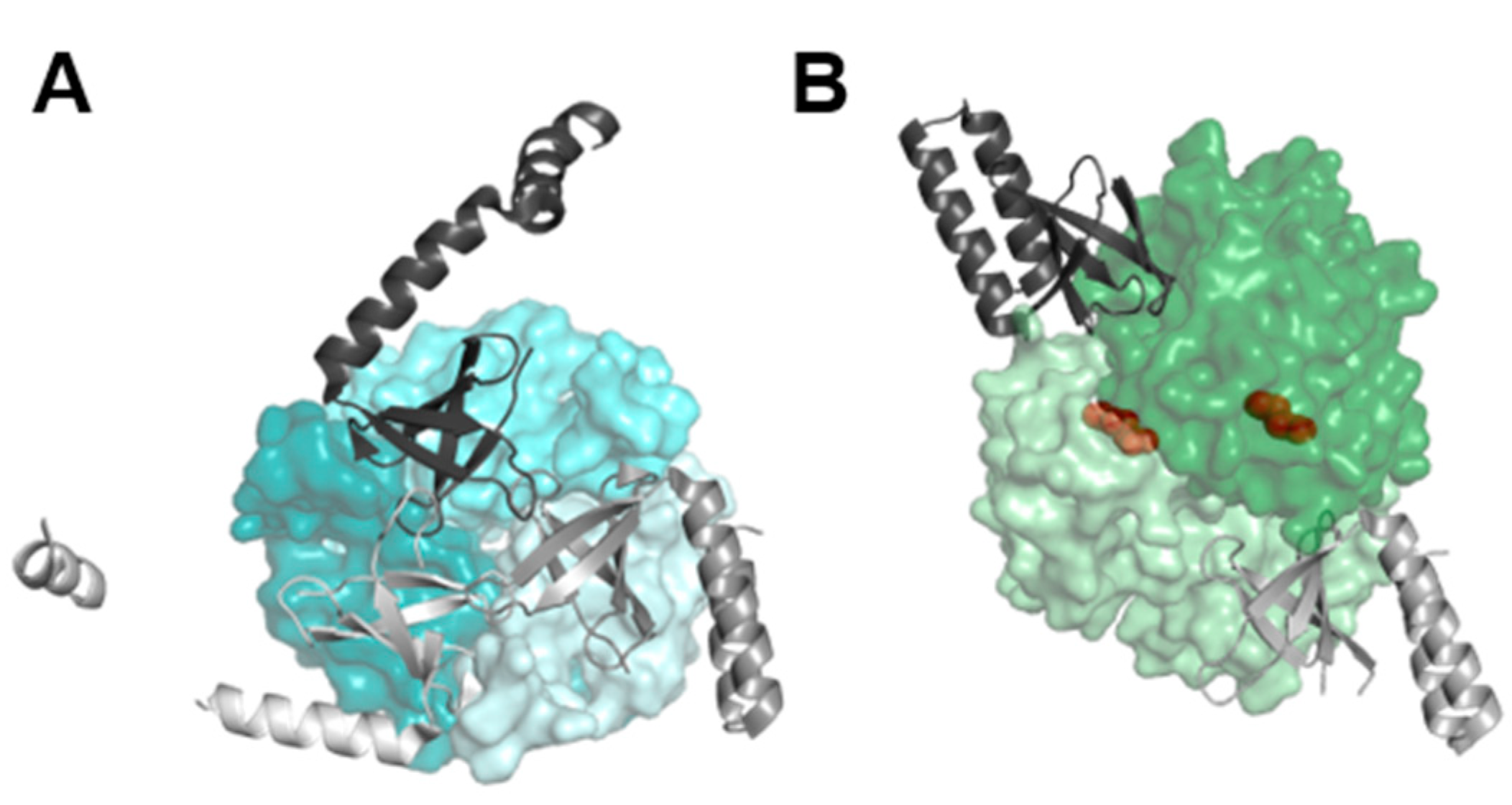
Figure 2.
PipX point mutations and toxicity. A. Ribbon representation of C-terminal helices of PipX in “flexed” (light grey; taken from chain D of 2XKO) and “extended” (dark grey, taken from chain D of 2XG8) forms. Discussed residues are indicated, with arginines in purple. Selected side chains are shown (sticks representation), with N atoms colored blue. B. Schematic representation of the wild type (glnB) and mutant (glnB::C.S3) glnB alleles whose segregation in CK1X*Y strains is shown below by PCR analysis. Positions of primers and expected sizes of PCR products are indicated. C. Two views as indicated of the PipX subunit structure (chain D of PDB file 2XG8) in the extended conformation of its two C-terminal helices. The surface of the subunit is represented in semi-transparent form, rendering visible the fold of the polypeptide chain in cartoon representation. The figure illustrates the location of the residues for which mutations are discussed here, indicating the PipX toxicity phenotype confered by each mutation according to the following color code: grey, neutral; blue, loss-of-function; red, gain-of-function.
Figure 2.
PipX point mutations and toxicity. A. Ribbon representation of C-terminal helices of PipX in “flexed” (light grey; taken from chain D of 2XKO) and “extended” (dark grey, taken from chain D of 2XG8) forms. Discussed residues are indicated, with arginines in purple. Selected side chains are shown (sticks representation), with N atoms colored blue. B. Schematic representation of the wild type (glnB) and mutant (glnB::C.S3) glnB alleles whose segregation in CK1X*Y strains is shown below by PCR analysis. Positions of primers and expected sizes of PCR products are indicated. C. Two views as indicated of the PipX subunit structure (chain D of PDB file 2XG8) in the extended conformation of its two C-terminal helices. The surface of the subunit is represented in semi-transparent form, rendering visible the fold of the polypeptide chain in cartoon representation. The figure illustrates the location of the residues for which mutations are discussed here, indicating the PipX toxicity phenotype confered by each mutation according to the following color code: grey, neutral; blue, loss-of-function; red, gain-of-function.
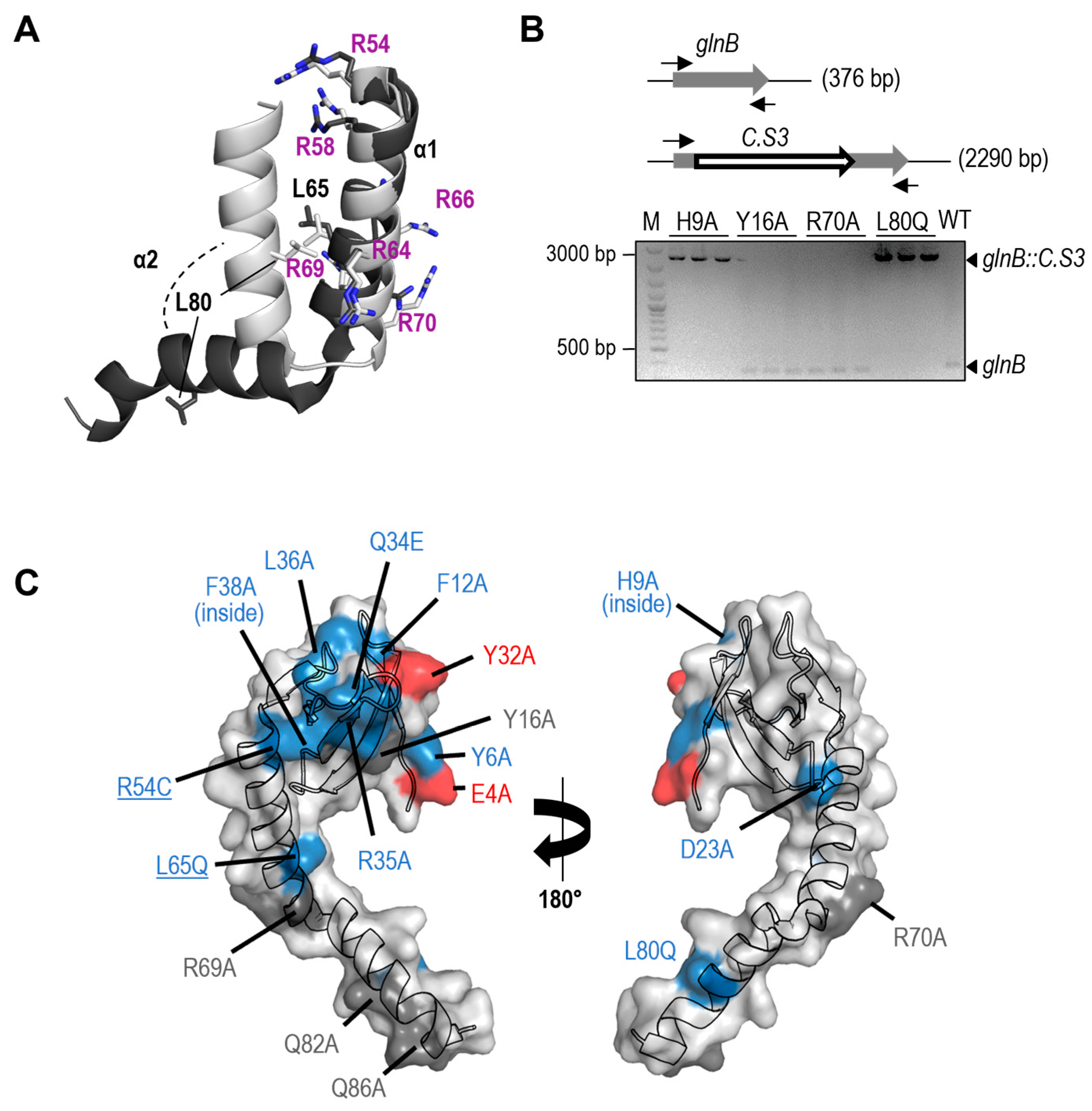
Figure 3.
Effects of PipX point mutations on protein levels and location of residues in complexes. A. Top, representative illustrations of PipX bands from Western-blots of the different control or mutant S. elongatus strains. Main panel, quantification (means and SD) of PipX band intensities for the indicated number of biological replicates (figures above the bars) normalized to the intensity in the same blot of endogenous PlmA. Wilcoxon rank sum test with Holm-Bonferroni correction of the mutants versus control strain (CK1XY) produced p-values <0.05(∗), <0.01(**) or <0.001 (***). Blue bars indicate loss of toxicity. Bottom, schematic linear representation of PipX polypeptide to indicate the position of tested mutations. B. Localization in the PipX structure (chain E taken from PDB 2XG8; secondary structure elements are labelled) of residues whose studied mutations decreased toxicity without significantly decreasing PipX levels. Residues side chains are shown in sticks representation, with C, O and N atoms colored orange, red and blue, respectively. The inter-residue distances indicated by the dashed red lines are also given. C. Stereoviews of PipX-PII (top) or PipX-NtcA (bottom) zooming on PipX (in cartoon representation) and the region of PII or NtcA (in surface representation) that accommodates PipX. Side chains of PipX residues discussed here are shown in sticks representation (O and N atoms in red and blue, respectively) and are identified in black or red coloring depending on whether they do or do not make direct interactions with PII or NtcA D. The same residues are represented as spheres on the isolated PipX structure (from PDB:2XG8 chain E) and residues interacting or not with PII in cyan or pink, respectively. Pale and dark tones correspond to low or WT protein levels, respectively.
Figure 3.
Effects of PipX point mutations on protein levels and location of residues in complexes. A. Top, representative illustrations of PipX bands from Western-blots of the different control or mutant S. elongatus strains. Main panel, quantification (means and SD) of PipX band intensities for the indicated number of biological replicates (figures above the bars) normalized to the intensity in the same blot of endogenous PlmA. Wilcoxon rank sum test with Holm-Bonferroni correction of the mutants versus control strain (CK1XY) produced p-values <0.05(∗), <0.01(**) or <0.001 (***). Blue bars indicate loss of toxicity. Bottom, schematic linear representation of PipX polypeptide to indicate the position of tested mutations. B. Localization in the PipX structure (chain E taken from PDB 2XG8; secondary structure elements are labelled) of residues whose studied mutations decreased toxicity without significantly decreasing PipX levels. Residues side chains are shown in sticks representation, with C, O and N atoms colored orange, red and blue, respectively. The inter-residue distances indicated by the dashed red lines are also given. C. Stereoviews of PipX-PII (top) or PipX-NtcA (bottom) zooming on PipX (in cartoon representation) and the region of PII or NtcA (in surface representation) that accommodates PipX. Side chains of PipX residues discussed here are shown in sticks representation (O and N atoms in red and blue, respectively) and are identified in black or red coloring depending on whether they do or do not make direct interactions with PII or NtcA D. The same residues are represented as spheres on the isolated PipX structure (from PDB:2XG8 chain E) and residues interacting or not with PII in cyan or pink, respectively. Pale and dark tones correspond to low or WT protein levels, respectively.
Figure 4.
PipX levels after the transition from darkness to light. Representative immunodetection pictures of PipX and PlmA detected in the WT strain, and relative PipX levels, normalized by PlmA signal and referred to the point 0. Means and error bars (SD) from the number of biological replicates (two independent experiments) are indicated in brackets below the points. Wilcoxon rank sum test with Holm-Bonferroni correction versus 120 min point produced p-values < 0.05 (*).
Figure 4.
PipX levels after the transition from darkness to light. Representative immunodetection pictures of PipX and PlmA detected in the WT strain, and relative PipX levels, normalized by PlmA signal and referred to the point 0. Means and error bars (SD) from the number of biological replicates (two independent experiments) are indicated in brackets below the points. Wilcoxon rank sum test with Holm-Bonferroni correction versus 120 min point produced p-values < 0.05 (*).
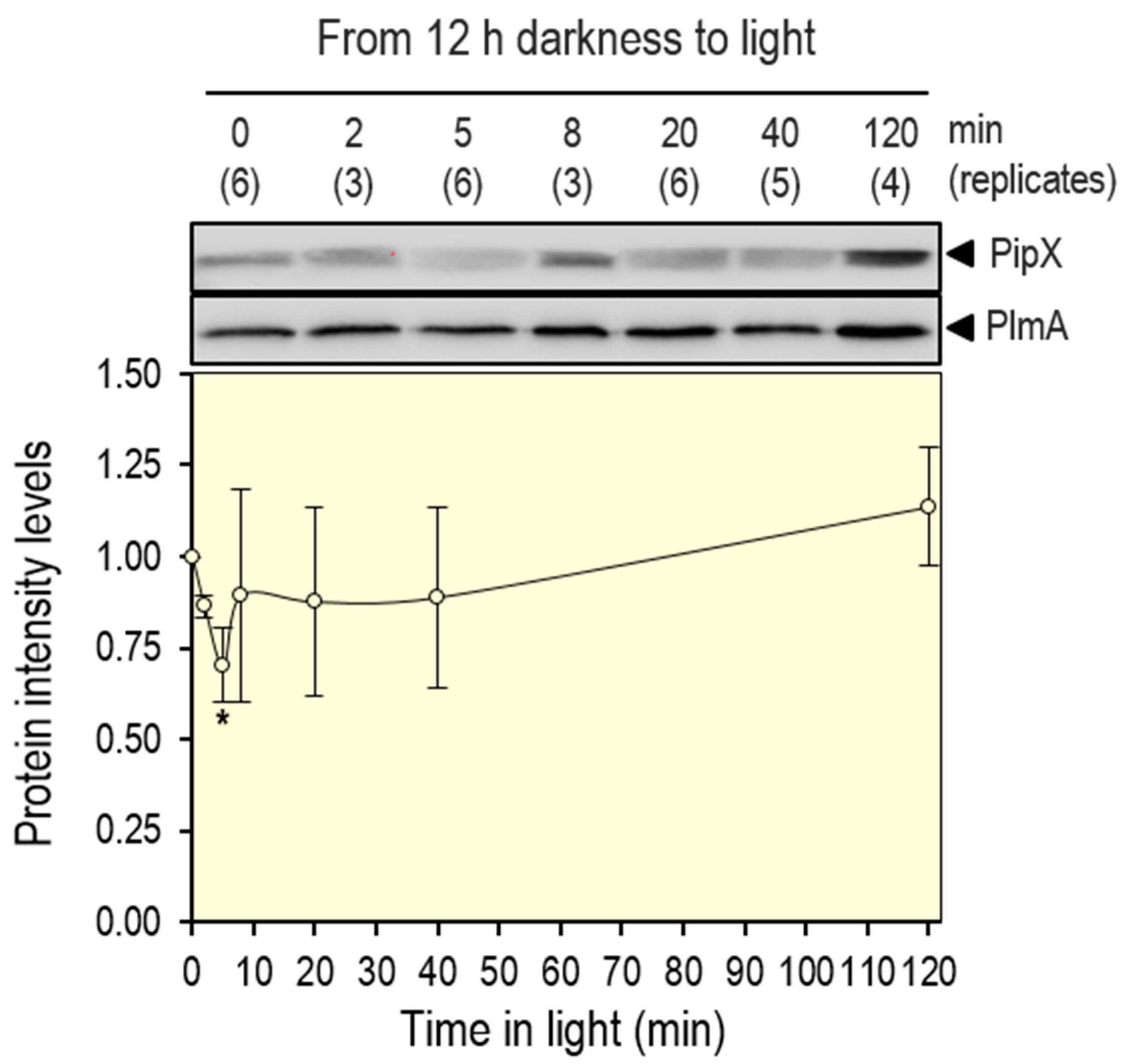

Table 1.
Strains.
| Strain | Genotype or relevant characteristics | Source or reference |
|---|---|---|
| E. coli DH5α | F-φ80 dlacZΔM15Δ(lacZYA-argF)U169 endA1 recA1 hsdR17(rK- mK+) deoR thi-1 supE44 gyrA96 relA1λ- | [44] |
| WT | Wild type S. elongatus PCC7942 | Pasteur Culture Collection |
| ∆pipX | pipX::cat, CmR | [33] |
| CK1XY | Φ(C.K1-pipXpipY), KmR | [12] |
| CK1XY6AY | Φ(C.K1-pipXY6ApipY), KmR | [23] |
| CK1XH9AY | Φ(C.K1-pipXH9ApipY), KmR | This work |
| CK1XH9AY/glnB | Φ(C.K1-pipXH9ApipY)/glnB::CS3, KmR SmR | This work |
| CK1XF12AY | Φ(C.K1-pipXF12ApipY), KmR | [23] |
| CK1XY16AY | Φ(C.K1-pipXY16ApipY), KmR | This work |
| CK1XD23AY | Φ(C.K1-pipXD23ApipY), KmR | [23] |
| CK1XQ34EY | Φ(C.K1-pipXQ34EpipY), KmR | [23] |
| CK1XR35AY | Φ(C.K1-pipXR35ApipY), KmR | [23] |
| CK1XL36AY | Φ(C.K1-pipXL36 ApipY), KmR | [23] |
| CK1XF38AY | Φ(C.K1-pipXF38ApipY), KmR | [23] |
| CK1XR54CY | Φ(C.K1-pipXR54CpipY), KmR | [22] |
| CK1XL65QY | Φ(C.K1-pipXL65QpipY), KmR | [22] |
| CK1XR69AY | Φ(C.K1-pipXR69ApipY), KmR | [23] |
| CK1XR70AY | Φ(C.K1-pipXR70ApipY), KmR | This work |
| CK1XL80QY | Φ(C.K1-pipXL80QpipY), KmR | This work |
| CK1XL80QY/glnB | Φ(C.K1-pipXL80QpipY)/glnB::CS3, KmR SmR | This work |
| CK1XQ82AY | Φ(C.K1-pipXQ82ApipY), KmR | [23] |
| CK1XQ86AY | Φ(C.K1-pipXQ86ApipY), KmR | [23] |
Table 2.
Plasmids.
| Plasmid | Genotype or relevant characteristics | Source or reference |
|---|---|---|
| pUAGC126 | pipX replaced with cat, ApR CmR | [33] |
| pUAGC701 | CS3(-) into glnB, ApR SmR | [21] |
| pUAGC410 | [Φ(C.K1-pipXpipY)], ApR KmR | [12] |
| pUAGC685 | [Φ(C.K1-pipXY6ApipY)], ApR KmR | [23] |
| pUAGC948 | [Φ(C.K1-pipXH9ApipY)], ApR KmR | This work |
| pUAGC939 | [Φ(C.K1-pipXF12ApipY)], ApR KmR | [23] |
| pUAGC945 | [Φ(C.K1-pipXY16ApipY)], ApR KmR | This wok |
| pUAGC680 | [Φ(C.K1-pipXD23ApipY)], ApR KmR | [23] |
| pUAGC687 | [Φ(C.K1-pipXQ34EpipY)], ApR KmR | [23] |
| pUAGC688 | [Φ(C.K1-pipXR35ApipY)], ApR KmR | [23] |
| pUAGC976 | [Φ(C.K1-pipXL36ApipY)], ApR KmR | [23] |
| pUAGC940 | [Φ(C.K1-pipXF38ApipY)], ApR KmR | [23] |
| pUAGC681 | [Φ(C.K1-pipXR54CpipY)], ApR KmR | [22] |
| pUAGC682 | [Φ(C.K1-pipXL65QpipY)], ApR KmR | [22] |
| pUAGC689 | [Φ(C.K1-pipXR69ApipY)], ApR KmR | [23] |
| pUAGC937 | [Φ(C.K1-pipXR70ApipY)], ApR KmR | This work |
| pUAGC618 | [Φ(C.K1-pipXL80QpipY)], ApR KmR | This work |
| pUAGC619 | [Φ(C.K1-pipXQ82ApipY)], ApR KmR | [23] |
| pUAGC683 | [Φ(C.K1-pipXQ86ApipY)], ApR KmR | [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



