Submitted:
06 September 2023
Posted:
07 September 2023
You are already at the latest version
Abstract
Consumption of high amounts of ethanol is a risk factor for development of cardiovascular diseases such as arterial hypertension. The hypertensive state induced by ethanol is a complex multi-factorial event, and oxidative stress is a pathophysiological hallmark of vascular dysfunction associated with ethanol consumption. Increasing levels of reactive oxygen species (ROS) in the vasculature triggers important processes underlying vascular injury, including accumulation of intracellular Ca2+ ions, reduced bioavailability of nitric oxide (NO), activation of mitogen-activated protein kinases (MAPKs), endothelial dysfunction, and loss of the anticontractile effect of perivascular adipose tissue (PVAT). The enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase plays a central role in vascular ROS generation in response to ethanol. Activation of the renin-angiotensin-aldosterone system (RAAS) is an upstream mechanism which contributes to NADPH oxidase stimulation, overproduction of ROS and vascular dysfunction. This review discusses the mechanisms of vascular dysfunction induced by ethanol, detailing the contribution of ROS to these processes. Data examining the association between neuroendocrine changes and vascular oxidative stress induced by ethanol are also reviewed and discussed. These issues are of paramount interest to public health as ethanol contributes to blood pressure elevation in the general population, and it is linked to cardiovascular conditions and diseases.
Keywords:
Oxidative stress
; NADPH oxidase
; Ethanol
; Blood pressure
; Endothelium
1. Introduction
The causal relation between heavy ethanol consumption and arterial hypertension was first established in 1915 by French army physician Camille Lian [1]. Although his findings were largely ignored for decades, Lian’s findings were eventually corroborated by several epidemiologic studies carried out in the late 1960s and in subsequent years in variable-sized populations [2,3,4,5,6]. Attempts to evaluate a possible mechanism for ethanol-induced hypertension in humans were hindered by several limitations, including differences in type and frequency of ethanol consumption and variability in age, ethnicity, gender, body mass index, salt use, comorbidities (e.g., obesity, hypertension), and the use of medications. Studies conducted in the late 1970s and in the early 1980s provided evidence that heavy ethanol drinkers showed increased circulating levels of noradrenaline and renin, which suggested that ethanol promoted activation of both the sympathetic nervous system and the renin-angiotensin-aldosterone system (RAAS) [7,8,9,10]. However, these studies did not establish a causal relation between neuroendocrine changes and the hypertensive state induced by ethanol consumption. In the absence of a well-defined causality, experimental models of alcoholism became crucial to the understanding of the mechanisms underlying ethanol-induced hypertension.
Studies in animals substantiated the initial epidemiological findings in humans, confirming that ethanol consumption promoted neuroendocrine changes and led to increased blood pressures [11,12]. Moreover, those studies established a relationship between ethanol consumption and vascular dysfunction, suggesting the existence of a myogenic mechanism that might alter the contractile/relaxant properties of the vasculature, thereby contributing to the elevation of blood pressure. Current knowledge shows that chronic ethanol intake leads to functional and structural changes of the vasculature including endothelial dysfunction, inflammation, remodeling, and functional alterations, which are characterized by hypercontractility and impaired vasorelaxation [13]. Data support the idea that oxidative stress is a central mechanism whereby ethanol promotes its deleterious effects in the vasculature [14]. Distinctive vascular cell types can produce reactive oxygen species (ROS), and those include endothelial, smooth muscle and adventitial cells. In addition, immune system cells (e.g., neutrophils and macrophages) may also contribute to ROS generation in response to ethanol [15,16].
Described as the major source of ROS in the vasculature, the enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase plays a central role in the redox unbalance induced by ethanol in the vascular system [14]. This multi-subunit transmembrane enzyme consists of membrane and cytosolic subunits that relay specific information intracellularly in response to a wide variety of stimuli. In the vasculature, RAAS promotes activation and overexpression of NADPH oxidase through the action of angiotensin II and aldosterone, which further increases ROS production, leading to vascular dysfunction [17]. Ethanol increases both the activity and expression of some components of the NADPH oxidase, favoring overproduction of ROS. The redox imbalance induced by ethanol is further aggravated by decreased antioxidant responses [14].
This review discusses the mechanisms of vascular dysfunction induced by consumption of high amounts of ethanol detailing the contribution of ROS to the pathobiology of ethanol in the vasculature. Data illustrating the association among neuroendocrine changes, ROS generation and ethanol-induced vascular dysfunction are also addressed. A MEDLINE-based search was conducted using the keywords that follow: ethanol, alcohol, alcoholism, hypertension, blood pressure, vascular dysfunction, redox imbalance, reactive oxygen species, endothelial dysfunction, oxidative stress, NADPH oxidase, and antioxidant. Articles were limited to those containing abstracts that were published in English. Information analysis started with the title, followed by the abstract and, finally, the complete report. Reasons for the exclusion of articles include unclear ethanol dose or ingestion period. Amount, duration and pattern of ethanol consumption influence the effects of this compoundin the cardiovascular system. Here, we focus on the effects of chronic comsumption of high amounts of ethanol.
2. Ethanol Consumption: A Risk Factor for Arterial Hypertension
In the 1970s, the Kaiser-Permanente Multiphasic Health Examination Data, a landmark observational study, showed an increase of 11 mmHg or more in systolic blood pressure in individuals consuming more than 5 drinks per day (one standard drink contains 14 g of ethanol) in comparison to non-drinkers [2]. The study established a relationship between the amount of ethanol consumed and an increase in blood pressure, an observation that was subsequently corroborated by other reports [4,6], including the second Kaiser-Permanente study, published in 1986 [18]. Analysis of six prospective studies and 29 cross-sectional studies in North America, Australia, Japan, Europe, and New Zealand, among other countries, showed that the association between arterial hypertension and ethanol consumption is independent of gender, age, body mass index, and smoking status. Prevalence of hypertension in individuals consuming 3–4 drinks/day is approximately 50% greater than that in non-drinkers, while in individuals that consumed 6–7 drinks/day the prevalence is 100% greater [1]. However, a specific population may itself be considered an important modifier of the prevalence of arterial hypertension induced by ethanol consumption. For example, it has been estimated that around 30% of the cases of arterial hypertension are attributed to ethanol in the United States and England [19], while in Australia the prevalence of hypertension attributed to ethanol consumption is 7% [20] in comparison, prevalence was shown to be 24% in France [21].
Establishing a standard threshold for ethanol effects on blood pressure has been difficult and continues to be controversial. Whilst 1–2 drinks/day have been described to produce slight or no effect on blood pressure [2,3,4,22], consumption of 3–5 drinks/day has been associated with increases in blood pressure, with more than 5 drinks/day promoting substantial increases in systolic blood pressure [2]. However, other studies have shown the threshold to occur at lower drinking levels [2,6]. In addition, to the variations in the effect threshold, the extent of blood pressure increase due to ethanol consumption is also controversial. The Kaiser-Permanente Multiphasic Health Examination Data showed that more than 5 drinks/day promoted an increase of at least 11 mmHg in systolic blood pressure [2]. Other studies have shown that an acute intake of 5 drinks leads to increased values of systolic and diastolic blood pressures by 4–7 mmHg and 4–6 mmHg, respectively [23,24,25]. For chronic, albeit a comparatively more moderate consumption of ethanol of 2–3 drinks/day for 4 weeks, average increases of 2.3 mmHg and 1.3 mmHg have been reported for systolic and diastolic blood pressures, respectively [26]. A separate study conducted with chronic drinkers (≈4 drinks/day) for an average period of 21 years has shown that consumption of the same intake levels of 4 drinks/day of wine or beer leads to increases in systolic blood pressure of 2.9 and 1.9 mmHg, respectively [27].
Another important modifier of the ethanol-risk relationship for arterial hypertension is gender. It is well established that prevalence of arterial hypertension induced by ethanol consumption is lower in women than in men [2,18]. As described in the Risk Factor Prevalence Study [20], no more than 1% of hypertension cases in women were attributed to ethanol consumption. A systematic review and meta-analysis on the effects of ethanol consumption on blood pressure has revealed the existence of a J-shaped relationship for women, showing that consumption of 1 drink/day is associated with a reduced relative risk for hypertension, a somewhat protective effect, whereas 2 drinks/day significantly increases the risk [28]. Similar findings have been described in another meta-analysis study that has also reported a J-shaped relationship for women [29]. More recently, it has been described consumption of 1–2 drinks/day leads to increased risk of hypertension in male chronic drinkers in comparison to abstainers whereas no significant difference is observed between female chronic drinkers and corresponding abstainers [30].
Collectively, these studies have provided evidence that chronic consumption of high amounts of ethanol is an important risk factor for arterial hypertension development. However, available data for humans have not been sufficient to establish a substantial mechanism for blood pressure elevation as a result of ethanol consumption. The inherent limitations of epidemiologic studies in humans have been compensated by development of experimental models, which then corroborate the epidemiologic findings and confirm the pressor effects of ethanol. Additionally, the experimental studies showed a positive relationship between increases in blood pressure and the extent of ethanol consumption, describing the period of exposure to ethanol as an important factor in the development of arterial hypertension [31,32]. Blood ethanol concentration is another modifier of the relationship between ethanol consumption and hypertension. Based on experimental findings, it was proposed that higher levels of blood ethanol accounts for earlier development of arterial hypertension [31,33,34].
In 1983, in a pioneer study of the hypertensive effects of ethanol, Sutter et al. described a 25% increase in mean blood pressure (from 98 to 122 mmHg) in male Wistar rats treated with ethanol (20%v) for 12 weeks [11,35]. Subsequently, they showed that the ethanol-induced hypertensive state was accompanied by augmented levels of circulating noradrenaline [36], as it had been observed in humans. Increased sympathetic activity and hypertensive states were also described in rats treated with ethanol (20%v) [31]. In ethanol-fed Sprague-Dawley rats, blood pressures were higher at week 6 (from 106 to 147 mmHg), while an increase from 117 to 149 mmHg in blood pressure was observed in ethanol-fed Wistar rats only after 8 weeks of treatment [31]. It is important to note that such changes in blood pressure were not the result of an upsurge but they rather had been gradually observed since the early stages of ethanol consumption. In some studies, for instance, blood pressure elevation was observed after 4 weeks of treatment with ethanol [32,37], whilst others evidenced this same response after only 2 weeks [34,38,39]. Differences in ethanol concentration in the blood may help explain the disparity among studies, since higher levels of blood ethanol were associated with earlier increases in blood pressure [37,40].
Taking into consideration that elevated blood pressure is a strong predictor of impending/imminent cardiovascular diseases, hypertension associated with ethanol consumption may have important long-term clinical consequences. Some mechanisms have been postulated to explain the hypertensive response to chronic ethanol consumption: while human epidemiologic studies have proposed the involvement of a neuroendocrine mechanism, experimental studies have suggested the existence of a myogenic mechanism in which ethanol consumption promotes alterations in the vascular tonus. The latter is based on results obtained from studies describing that ethanol-induced hypertension occurs in parallel to changes in vascular responsiveness [32,37,40]. These mechanisms are discussed in the following sections.
3. Mechanisms Underlying Ethanol-Induced Hypertension: The Neuroendocrine and Myogenic Theories
3.1. Neuroendocrine Changes
A clear relationship between ethanol consumption and arterial hypertension had already been established by epidemiological studies by the end of the 1970s. It became important then to understand the mechanisms by which ethanol affected blood pressure. Studies in humans have attempted to address this question by evaluating the participation of the sympathetic nervous system and RAAS, two important regulators of blood pressure.
The contribution of the catecholamines noradrenaline and adrenaline to the pressor effects of ethanol was investigated in humans by measuring the circulating levels of these hormones, whose corresponding plasma concentrations were found to be higher after consumption of ethanol [10,41,42]. Based on those findings, it was proposed that the sympathetic nervous system would be mediating the pro-hypertensive effects of ethanol. However, other investigators showed that, despite blood pressure being higher among ethanol drinkers, no changes were detected in plasma levels of adrenaline or noradrenaline [6,23]. The reasons for such discrepancies are still uncertain, but they may be due to dose, frequency and pattern of ethanol consumption. Experimental studies in animals corroborated the findings in humans, showing that consumption of ethanol increased both blood pressure and the circulating levels of the catecholamines [11,38]. However, neither the human studies nor the animal models managed to establish a causal relation between the activation of the sympathetic nervous system and the ethanol-induced increase in blood pressure. More recently, it was shown that ethanol promotes increases in both blood pressure and in the circulating levels of adrenaline and noradrenaline in male Wistar rats. Nebivolol (a selective β1-adrenergic receptor antagonist) prevented these responses, implicating a role of the sympathetic nervous system in the hypertensive state associated with ethanol consumption [43].
Regarding the participation of RAAS in ethanol-induced hypertension, pioneering works by many researchers revealed increases in the circulating levels of renin and aldosterone in heavy drinkers [7,8,44,45] and experimental studies strengthened the proposal that ethanol consumption activates RAAS. According to these data, ethanol increases plasma activity of the renin and angiotensin converting enzyme (ACE) and also increases the circulating levels of angiotensin I and II, and aldosterone [38,46,47]. RAAS plays a critical role in mediating ethanol-induced hypertension since initial findings suggest that the increase in blood pressure induced by ethanol is mediated by angiotensin II [39]. Moreover, recent findings show the involvement of aldosterone in the hypertensive state induced by ethanol consumption [48]. The increase in blood pressure promoted by RAAS results from its effects in the vasculature and does not seem to be the result of changes in fluid and electrolyte balance [39,48].
Both the sympathetic nervous system and RAAS play a role in the pressor effects of ethanol and a relationship between these two systems to promote this response does exist. However, it is unknown if over activation of those systems caused by ethanol intake are independent and work synergically, or if they communicate to each other in a positive feedback fashion. Ethanol-induced hypertension is accompanied by increases in the circulating levels of the catecholamines adrenaline and noradrenaline as well as by increased plasma levels of renin, angiotensin I, angiotensin II, and ACE activities. These responses are prevented by nebivolol, a selective β1-adrenergic receptor antagonist, showing that β1 receptors mediate RAAS activation [43]. Importantly, AT1 receptors blockade with losartan prevent the increase in blood pressure induced by ethanol, implicating RAAS in such response [39].
Results from clinical and experimental studies led to the conclusion that the two major systems controlling blood pressure under physiological conditions, the sympathetic nervous system and RAAS, are implicated in the hypertensive state induced by ethanol, and that there is an interplay between their corresponding responses. We discuss these findings further in Section 4.
3.2. The Myogenic Theory
Much of the experimental studies that investigated the effects of ethanol consumption on blood pressure also addressed the impact of the former on vascular responsiveness. Altogether, data from such reports revealed that ethanol both promote vascular hypercontractility and impair vascular relaxation in distinctive vascular territories.
Increased vascular reactivity to adrenoreceptor agonists has been described in the rat aorta, and the carotid and mesenteric arteries [12,49,50,51,52,53]. The hypertensive state induced by ethanol is accompanied by increases in the pressor effects of intravenous phenylephrine (a selective α1-adrenoreceptor agonist) and endothelin-1 [34,54], supporting the concept that increased vascular responsiveness in vivo may be involved in the elevation of blood pressure induced by ethanol. Vascular hypercontractility induced by ethanol in the aorta is endothelium-independent and maintained by two mechanisms: an augmented production and release of thromboxane A2 (TXA2), a smooth muscle-derived vasoconstrictor prostanoid; and increased extracellular Ca2+ influx [51]. In the aorta, increased TXA2 production is mediated by the cyclooxygenase (COX)-2 enzyme [48], but in resistance arteries, the mechanism whereby ethanol increases vasoconstriction in the presence of adrenergic agonists differs from that described in the aorta. In the mesenteric arterial bed, ethanol consumption induces an endothelium-dependent increase in phenylephrine-induced contraction. This response is the result of an augmented release of endothelial-derived vasoconstrictor prostanoids and an impaired modulatory action of endothelial nitric oxide (NO); the latter is likely associated with downregulation of the endothelial NO synthase enzyme (eNOS) [55].
The pro-contractile effect of ethanol is a complex and multi-mechanistic process. In the carotid artery, downregulation of ETB receptors explains the hyper-contractile effect to endothelin-1induced by ethanol. These receptors counteract the contraction induced by endothelin-1 by releasing NO [52,56]. In the microcirculation, ethanol consumption potentiates the vasocontractile effect of endothelin-1 by promoting upregulation of ETA receptors, which mediate the contractile response induced by the peptide [57]. Overproduction of ROS is another mechanism by which ethanol promotes its vasocontractile effects. Drugs that display antioxidant effects are capable of preventing the pro-contractile effects of ethanol [43,58,59]. In fact, increased oxidative stress is a central mechanism whereby ethanol promotes vascular dysfunction, being responsible not only for the contractile effect but also for the impaired relaxation [14]. A more detailed overview on the contribution of oxidative stress to the vascular dysfunction induced by ethanol is provided in Section 5.
Impairment of vascular relaxation also accounts for the deleterious effects of ethanol in the vasculature. In the aorta, ethanol decreased the endothelium-dependent relaxation induced by acetylcholine and adrenomedullin [47,60,61]. Similar findings were described in resistance arteries in which the relaxation induced by these vasoactive agents was impaired after consumption of ethanol [55,62]. The NO-cyclic guanosine monophosphate (cGMP) pathway plays a prominent role in the vascular relaxation induced by adrenomedullin and acetylcholine [63], suggesting that the endothelial dysfunction is trigged by decreased NO bioavailability. In fact, the vascular expression of the enzyme eNOS as well as the levels of NO are reduced in the vasculature of ethanol-treated rats. These responses have a negative impact in the NO-cGMP pathway and are described to be mediated by ROS [64]. The participation of oxidative stress in the endothelial dysfunction induced by ethanol will be further discussed in Section 5.5.
Downregulation of endothelial ETB receptors is also proposed as a mechanism whereby ethanol impairs vascular relaxation [56]. Activation of these receptors leads to the production of NO, and, consequently, ETB receptors mediate vascular relaxation and counteract vascular contraction [65]. Relaxation induced by IRL1620 (a selective endothelin ETB receptor agonist), has been shown to be reduced in carotid arteries from ethanol-treated rats, while, as mentioned before, ethanol favored the contractile effect of endothelin-1 in carotid arteries [56]. In view of these observations, it is possible to conclude that downregulation of ETB receptors figures as a mechanism impairing vascular relaxation and favoring vascular contraction.
Neuroendocrine changes followed by vascular hypercontractility are proposed as a sequence of events that explains the pathophysiological effects of ethanol in the cardiovascular system.
4. Interplay of Neuroendocrine and Myogenic Changes in Mediating the Pressor Effects of Ethanol
RAAS is a major regulator of blood pressure under physiological conditions. Most of its actions, including sodium retention, aldosterone secretion (by the adrenal glands) and vasoconstriction, are mediated by angiotensin II, which exerts its biological actions through G-protein-coupled receptors, termed AT1 and AT2. Ethanol-induced hypertension correlates with elevated plasma angiotensin II levels, endothelial dysfunction and impaired vascular relaxation [38,47]. Blockade of AT1 receptors prevents both the hypertensive state and the vascular hypercontractility induced by ethanol [39,59], showing the existence of a relationship between ethanol-induced RAAS activation and vascular changes. More recently, evidences on the participation of aldosterone on ethanol-induced vascular hypercontractility were provided [48]. Ethanol consumption increases the circulating levels of both angiotensin II and aldosterone [39,48]. The latter is a steroid hormone synthesized mainly by the adrenal glands, and whose secretion occurs primarily in response to activation of AT1 receptors by angiotensin II [66]. Blockade of AT1 receptors prevents ethanol-induced aldosterone release, showing that this response is mediated by angiotensin II [39]. The increase in blood pressure as well as vascular hypercontractility promoted by ethanol is abrogated by blockade of mineralocorticoid receptors [48], suggesting that aldosterone acts as the final mediator of RAAS to promote the pressor and pro-contractile effects of ethanol. Thus, the positive relationship between RAAS activation and vascular hypercontractility in response to ethanol consumption might help to explain the hypertensive effect of ethanol.
Production of angiotensin II also occurs locally within the vascular wall. Local RAAS may generate angiotensin II even when systemic RAAS is suppressed or normal. Under pathological conditions, vascular RAAS may amplify the effects of systemic RAAS, contributing to vascular oxidative stress and hypertension [67]. Ethanol consumption does not change the concentrations of angiotensin I or angiotensin II, neither does it alterthe expression and activity of ACE in the vasculature. In addition, vascular expression of both AT1 and AT2 receptors is not affected by ethanol consumption, suggesting that, in spite of activating the systemic RAAS, ethanol does not activate the vascular angiotensin II-generating system [39].
Ethanol-induced RAAS activation is proposed to be mediated by the sympathetic nervous system, since the hypertensive and pro-contractile effects of ethanol occur in parallel to increases in circulating levels of catecholamines [36,38] (Figure 1). The cardiovascular effects of ethanol involve a direct action of the sympathetic nervous system through activation of α- and β-adrenoreceptors [34,43]. Moreover, the sympathetic nervous system may trigger RAAS activation via β1-adrenergic receptors located in the juxtaglomerular cells, which synthesize, store, and secrete the enzyme renin. Blockade of β1-adrenergic receptors prevents an ethanol-promoted increase in the circulating levels of renin, angiotensin I, angiotensin II, and ACE activities, showing that the sympathetic nervous system modulates RAAS activation in response to the alcohol [43]. The central nervous system plays an important role in regulating sympathetic outflow and arterial pressure in response to ethanol consumption. It is proposed that ethanol-induced increase in sympathetic outflow involves activation of NMDA receptors in the central nucleus of the amygdala neurons that projects to the rostral ventrolateral medulla [68].
Pro-contractile and hypertensive effects of ethanol result from the interplay between neuroendocrine and vascular changes. Ethanol centrally stimulates the sympathetic nervous system, which is responsible for triggering RAAS activation. The latter is critically involved in the vascular hypercontractility induced by ethanol consumption (Figure 1). Neuroendocrine changes promoted by ethanol triggers overproduction of ROS in the vasculature leading to disruption of the NO-cGMP pathway in the endothelium, thus favoring the accumulation of intracellular Ca2+. These responses culminate in vascular dysfunction and hypertension.
5. Oxidative Stress: The Major Mediator of Vascular Dysfunction Induced by Ethanol
Ethanol alters vascular tonus by disrupting the mechanisms that control and maintain the balance between contraction and relaxation. A number of possible mechanisms have been postulated to explain the pathogenesis of ethanol toxicity in the vasculature, which highlights the importance of identifying the biochemical/molecular basis of the ethanol effects (Figure 2). Overproduction of ROS is pointed out as a central mechanism whereby ethanol promotes vascular dysfunction and hypertension through increased generation of ROS and activation of redox-sensitive pathways, thereby reducing NO bioavailability and increasing intracellular Ca2+ ion levels, actions that mediate the pro-contractile effects of ethanol.
5.1. NADPH Oxidase is a Major Mediator of ROS Generation in Response to Ethanol
ROS stands for radical oxygen species produced either as intermediates or as a final reaction product. The vasculature produces superoxide anions (O2•-) and hydrogen peroxide (H2O2). These two compounds have important functions in the maintenance of vascular integrity and control of vascular tone by interacting with redox-sensitive target proteins under physiological conditions and activating redox-signaling pathways [69].
Oxidative stress is often described as a disturbance in the equilibrium established between ROS production and antioxidant defenses, which results in an increased bioavailability of ROS. Oxidative stress triggers pathophysiological processes including inflammation (by promoting platelet aggregation and monocytes migration), tissue hypertrophy, cellular proliferation, fibrosis, hypercontractility, and endothelial dysfunction, processes that are all involved in vascular dysfunction [70]. Thus, vascular dysfunction promoted by redox imbalance not only induces direct oxidative damage to macromolecules, but also triggers redox-signaling pathways in the vasculature that leads to changes in gene transcription and in oxidative modifications of proteins causing [71].
A number of enzymatic systems may generate O2•- using oxygen as substrate, including xanthine oxidase, lipoxygenase, COX, uncoupled NO synthase (NOS), cytochrome P450 reductase and some enzymes of the mitochondrial electron transport chain [72]. In these cases, ROS are generated as secondary products of the main chemical reactions catalyzed by those enzymes; however, for the enzyme NADPH oxidase, ROS consist of the main reaction product, with this enzyme being the major source of ROS in the vasculature, utilizing either NADPH or NADH as electron donors to promote the reduction of molecular oxygen into O2•- [73]. The so-called “NOX family” is composed of seven isoforms of NADPH oxidase, which differ in their catalytic subunit (Nox1–5, Duox1, Duox2) and regulation. Whilst Nox1-4 are regulated by cytosolic proteins, Nox5 and Duox1 and Duox2 are activated by Ca2+ ions, which binds to specific domains located in these proteins [74]. In human vascular cells, the isoforms Nox1, Nox2, Nox4, and Nox5 are expressed and functionally active. NADPH oxidase-derived ROS plays a role in both physiological and pathophysiological processes in the vasculature [73].
Overproduction of ROS by ethanol is crucial to its vascular pathophysiology. Similar to physiological conditions, O2•- and H2O2 are also central to the deleterious effects of ethanol in the vasculature, as well as in a variety of tissues, with enzymes of the NOX family playing a key role in the process [75,76,77,78]. Ethanol consumption has been described to promote increases in vascular NADPH oxidase activity, ROS generation and lipoperoxidation. These responses are concomitant to changes in vascular tonus, which include hypercontractility and impaired vasorelaxation [47]. The augmented activity of NADPH oxidase promoted by ethanol may be associated to increased expression and phosphorylation of p47phox, a cytosolic protein that regulates the activation of Nox2 [79,80,81]. The effects of ethanol on NADPH oxidase also include increases in the expression of the catalytic subunits Nox1 and Nox2. This effect may help explain the augmented production of ROS in the vasculature promoted by ethanol consumption. In fact, NADPH oxidase is involved in other actions of ethanol including lipoperoxidation, vascular hypercontractility and impaired vasorelaxation [58,64,82].
In the microcirculation, ethanol impairs vascular relaxation, increases NADPH oxidase activity, expression of Nox2 and translocation of p47phox, which is a crucial step for Nox2 activation [81]. Blockade of NADPH oxidase prevents the deleterious effects of ethanol in the microcirculation and attenuates the resulting increase in blood pressure, suggesting that the hypertensive state associated with ethanol consumption involves formation of ROS [64,81,83]. It is noteworthy that antioxidants are capable of preventing ethanol-induced overexpression of Nox1 and Nox2, implicating ROS in such response [43,58,81]. In this scenario, NADPH oxidase-derived ROS favor a positive feedback loop that amplify the starting signal. Thus, the causal relationship between ethanol, ROS and hypertension most likely occurs at the vascular level, where ethanol promotes activation/overexpression of NADPH oxidase, subsequently generating ROS, which are then implicated in vascular hypercontractility and impaired vasorelaxation. Altogether, these responses contribute to increases in vascular resistance and blood pressure.
NADPH oxidase activation is a multimediated and complex process since this enzyme responds to a wide range of stimuli. In the vasculature, mediators that control vascular tonus, such as endothelin-1, aldosterone and angiotensin II, may promote NADPH activation [73]. The physiological actions of angiotensin II in the vasculature are predominantly mediated by AT1 receptors, which are also implicated in the pathophysiological actions of this peptide. Angiotensin II regulates the onset and progression of cardiovascular diseases by increasing NADPH oxidase activity and leading to upregulation of vascular Nox1 and Nox2, which are important in redox-mediated hypertension in various cardiovascular diseases [84]. The actions of angiotensin II on NADPH oxidase favors overproduction of O2•-, which, in turn, influence downstream signaling pathways. Angiotensin II figures as an important mediator of NADPH oxidase activation in response to ethanol. The actions of angiotensin II occur via AT1 receptors and include increased activity of NADPH oxidase and overproduction of ROS, responses that are directly implicated in the vascular hypercontractility and hypertension induced by ethanol [39,59].
There is also evidence showing the involvement of aldosterone in NADPH oxidase activation in response to ethanol. Aldosterone induces upregulation and increases in NADPH oxidase activity in the vasculature through activation of mineralocorticoid receptors [85,86]. The latter are implicated in the upregulation of Nox1 and in the increase in NADPH oxidase activity promoted by ethanol consumption in the vasculature. Activation of mineralocorticoid receptors leads to overproduction of ROS, which then mediate upregulation of COX2 and overproduction of the vasocontractile prostanoid TXA2, which ultimately induces vascular contraction [48].
NADPH oxidase is the major source of ROS in the vasculature and, consequently, this enzyme is implicated in the pathophysiology of multiple cardiovascular diseases [69]. Increased expression and activity of this oxidase is accepted as a central mechanism of the vascular effects promoted by ethanol [14]. NADPH oxidase-derived ROS are considered as key factors in the development of endothelial dysfunction and hypercontractility induced by ethanol. Additionally, ethanol may lead to overproduction of ROS by activating targets other than NADPH oxidase. Other sources of ROS production in response to ethanol consumption are discussed in the next section.
5.2. Other Sources of Ethanol-Induced ROS Generation in the Vasculature
Ethanol-induced ROS generation may also occur through uncoupled eNOS and xanthine oxidase. In addition, ethanol metabolism is also associated with ROS production. However, the contribution of these sources to the deleterious effects of ethanol in the vasculature is not well determined.
The enzyme eNOS is a constitutive isoform of NOS present in the vasculature, where it promotes the generation of NO and L-citrulline using the amino acid L-arginine as substrate. The synthesis of NO occurs when eNOS is in its dimeric form, being L-arginine and the cofactor tetrahydrobiopterin (BH4) crucial to its dimerization and activity. In its uncoupled form, eNOS may produce both O2•- and NO, contributing to vascular dysfunction [87]. The reaction of these two molecules generates peroxynitrite (ONOO-), a very reactive free radical that oxidizes BH4, leading to eNOS uncoupling and subsequent generation of O2•-. Moreover, O2•- is capable of oxidizing BH4, a process that also favors eNOS uncoupling [87]. Few studies assessed the role of uncoupled eNOS to ROS generation in response to ethanol. In this regard, ethanol consumption augments hepatic and renal concentrations of ONOO-. In the vasculature, ethanol consumption induced increased staining for nitrotyrosine, suggesting the production of high levels of ONOO- [43]. Ethanol consumption promotes BH4 deficiency, which occurs in parallel to an impaired eNOS-dependent vasodilatation [88,89]. In the microcirculation, ethanol consumption compromises endothelium-dependent vasorelaxation and this response is reversed by administration of BH4, suggesting a role for uncoupled eNOS in such response [90]. Those are indirect evidence that suggest a possible contribution of uncoupled eNOS to ROS generation induced by ethanol.
Xanthine oxidase contributes to O2•- generation in conditions such as hypertension and coronary arterial disease [91]. By promoting the reduction of xanthine or hypoxanthine to uric acid, xanthine oxidase also reduces one or two electrons of molecular oxygen leading to the generation of O2•- or H2O2 as intermediaries [92]. During the reduction of xanthine or hypoxanthine, one atom of hydrogen is transferred from these substrates to NAD generating NADH. Ethanol may influence xanthine oxidase activity by promoting an imbalance of the NAD/NADH ratio during its oxidation [93]. In the liver, ethanol was described to promote an increase in lipid peroxidation that was mediated by xanthine oxidase [94]. Despite the playing a role in the hepatic effects of ethanol, the participation of xanthine oxidase in the vascular effects of ethanol remain elusive.
The central route whereby ethanol is metabolized in the liver takes place in the cytosol where the enzyme alcohol dehydrogenase converts ethanol in acetaldehyde, which is then oxidized to acetate in the mitochondria by the enzyme acetaldehyde dehydrogenase. The conversion of ethanol in acetaldehyde may also occur in the peroxisome and microsome by the enzymes catalase and cytochrome P450 2E1 (CYP2E1), respectively. Ethanol metabolism by the CYP2E1 pathway produces ROS. This response may be amplified when ethanol is chronically consumed since in this case it induces the expression of CYP2E1. In addition, ethanol is metabolized by alcohol dehydrogenase and CYP2E1 in extra-hepatic tissues the sympathetic nervous system and RAAS [95]. ROS-derived from ethanol metabolism react with macromolecules (e.g., nucleophilic proteins, phospholipids and nucleic acids), and also activate intracellular pathways that leads to tissue inflammation and apoptosis.
Both, alcohol dehydrogenase and CYP2E1 are functionally active in the vasculature [96]. Ethanol promotes direct actions in the vasculature where it induces both catalytic activity and expression of ethanol-metabolizing enzymes. These responses occur in parallel with overproduction of ROS. The increase in oxidative stress induced by ethanol leads to activation of the myosin light chain (MLC) kinase, subsequent phosphorylation of MLC and tight-junction proteins, decreased blood-brain barrier integrity, and increased monocyte migration across blood-brain barrier [96,97]. Thus, vascular metabolism of ethanol leads to generation of ROS that will, in turn, affect vascular integrity. In addition, inhibition of the ethanol-metabolizing enzymes dehydrogenase and CYP2E1, reduces the direct vasomotor effects exerted by ethanol, showing that ethanol metabolism in the vasculature influences vascular tonus [62].
5.3. Impairment of Antioxidant Systems May Contribute to Ethanol-Induced ROS Accumulation
Cellular levels of ROS are regulated by enzymatic and non-enzymatic antioxidant systems. While the enzymes superoxide dismutase (SOD), catalase and glutathione peroxidase comprise the most important enzymatic systems, ascorbate, tocopherols, glutathione, bilirubin, and uric acid are pointed out as the major non-enzymatic antioxidants [98]. Enzymes of the enzymatic antioxidant system are expressed on blood vessels, where they play an important role in the control of redox balance.
There are three isoforms of SOD: the cytoplasmic Cu/ZnSOD or SOD1, the mitochondrial MnSOD or SOD2 and the extracellular Cu/ZnSOD or SOD3); all of which are expressed in the vasculature. These enzymes promote the dismutation of O2•- in H2O2 and oxygen in distinctive intracellular compartments [99]. Since O2•- is a highly instable and reactive molecule, enzymes of the SOD family are considered as the first line of defense against free radicals. The reaction of these antioxidant enzymes with O2•-maintains the physiological levels of O2•-, preventing cellular damage. Ethanol can diminish SOD activity. For example, heavy drinkers (6 or more drinks/day) show decreased plasma SOD activity, when compared to abstainers [100]. Similar findings are described in an animal model of ethanol consumption [43]. Experimental findings showed that ethanol decreased total SOD activity in the vasculature [101]. Activity of both Cu/ZnSOD and Mn-SOD were depressed in the vasculature of ethanol-treated rats, a response that related to decrease NO bioavailability and endothelial dysfunction. The exact mechanism whereby ethanol diminishes SOD activity is unknown, but this response is proposed to be mediated by ONOO- [47]. In the microcirculation, ethanol consumption reduces SOD activity and SOD1 expression, being these effects attributed to ROS [81].
H2O2 is a stable and membrane permeable ROS that is involved in the activation of distinctive redox-signaling pathways. H2O2 is displays mild oxidant properties and for this reason it is inert to most biomolecules. In fact, H2O2 contributes to the physiological regulation of the vascular tone by promoting activation of potassium channels and increasing the generation of NO production [102]. However, H2O2 promotes alterations of amino acid residues (e.g., cysteine residues) located in active or allosteric sites of some proteins leading to modifications of their activity and function. Phosphatases, transcription factors, ion channels, antioxidant enzymes, structural proteins and protein kinases are examples of proteins that may be modified by H2O2 [103].
Among those proteins, a group of protein kinases named mitogen-activated protein kinases (MAPKs) is of special interest. MAPKs belong to four families of proteins [the extracellular signal regulated kinase (ERK1/2), p38MAPK, jun N-terminal kinase (JNK) and the extracellular signal-regulated kinase 5 (ERK5)] that are key components of signaling pathways. While proteins of the ERK cascade play a role in proliferation, differentiation, growth and cell survival, JNK and p38MAPK are involved in apoptosis/inflammation and inflammatory responses, respectively [103]. Since all MAPKs are targets of H2O2, cellular consequences derived from their redox regulation by H2O2 are ample.
The concentration of H2O2 is regulated by intracellular and extracellular enzymes, such as catalase, which converts H2O2 into H2O and O2. Since catalase is a key enzyme in the metabolism of H2O2, decreases in its expression and/or activity may result in increased H2O2 bioavailability [104]. Ethanol consumption decreases vascular catalase activity, but the consequences of this response are unknown [47,101]. Diminished catalase activity may favor an increase in H2O2 concentration in vascular cells leading to redox regulation of signaling pathways. In fact, ethanol consumption is linked to MAPKs activation in the vasculature. Inhibitors of p38MAPK and ERK1/2 attenuate ethanol-induced contraction and increase in intracellular Ca2+, showing the involvement of these proteins in the vasocontractile response to ethanol [105,106]. Ethanol consumption increases vascular p38MAPK and SAPK/JNK phosphorylation as well as expression of SAPK/JNK, responses that occur in parallel to increased NADPH oxidase-derived ROS and vascular hypercontractility [39,58,64,80,107]. Thus, MAPKs contribute to the vascular pathobiology of ethanol, but the involvement of H2O2 in their activation needs further clarification.
The effects of ethanol on catalase activity are dependent on the period of consumption and amount of ethanol consumed. Some reports have shown that ethanol may increase catalase activity in the vasculature, leading to a decreased bioavailability of H2O2 [81,82,83]. The increase in catalase activity in response to ethanol was also described in other tissues as a compensatory mechanism to protect against the deleterious effects displayed by H2O2 [81,108]. However, at low concentrations in the in the vasculature, H2O2 exerts vasocontractile actions [102]. In the microvasculature, ethanol-induced increase in catalase activity leads to a decrease in H2O2 concentration and these responses are associated with impaired vasodilatation [81]. Thus, the increase in catalase activity in the vasculature may favor H2O2-mediated vascular contraction.
Glutathione peroxidase promotes the reduction H2O2to H2O and catalyzes the conversion of lipid hydroperoxides to their corresponding alcohols. Eight isoforms of glutathione peroxidase are currently described (glutathione peroxidase 1-8). These enzymes vary in cellular location and substrate specificity. They work with SOD and catalase, forming an enzymatic antioxidant system that promotes ROS reduction, limiting their cellular toxicity. In order to reduce H2O2 and alkyl hydroperoxides, all members of the glutathione peroxidase family use glutathione (GSH) as substrate, which is a reducing agent that is converted to glutathione disulfide (GSSG), its oxidized form, during the reduction process [109]. Clinical and experimental studies revealed that ethanol consumption promotes reduction in the activity of glutathione peroxidase in serum [100,110,111]. In the vasculature, ethanol induces decreases of glutathione peroxidase activity, GSH levels and of the GSH/GSSG ratio [47,82,101]. The possible contribution of these responses to the deleterious effects of ethanol in the vasculature is unknown. As described for catalase, a decrease in glutathione peroxidase activity may favor an increase of H2O2, which acts as a signaling second messenger in the vasculature triggering multiple signaling pathways involved in vascular dysfunction.
Increased levels of ROS may be the result of an imbalance between their generation and elimination by antioxidant systems. Ethanol promotes a negative regulation of antioxidant enzymes and this response may contribute to vascular increases in ROS levels induced by ethanol.
5.4. Ethanol-Induced Oxidative Stress Leads to Ca2+ ion Accumulation in the Vasculature
Ca2+ is essential for smooth muscle contraction. Increases in intracellular Ca2+ ion concentration during excitation may occur due to Ca2+ ion release from intracellular stores (sarcoplasmic reticulum or mitochondria) or extracellular Ca2+ ion influx through voltage- or ligand-gated ion channels located in the cell membrane. The increase in intracellular Ca2+ in smooth muscle cells is one mechanism by which ethanol consumption promotes vascular hypercontractility. In vivo and in vitro studies provided evidence that ethanol augments vascular concentration of Ca2+ ions by promoting increases in Ca2+ ions uptake in smooth muscle cells [112,113]. Of importance, ethanol-induced intracellular Ca2+ accumulation is associated with vascular hypercontractility [51,114].
Redox-sensitive signaling pathways mediate the contraction and increase in intracellular Ca2+ induced by ethanol in vascular smooth muscle cells. In vitro, antioxidants attenuate both the elevation in intracellular Ca2+and the vasocontractile effect induced by ethanol, implicating ROS in such responses [115,116]. Ethanol effects on Ca2+ ion accumulation are mediated by H2O2 and O2•-, which trigger production of COX-derived vasoconstrictor prostanoids (prostaglandin F2α and TXA2) that ultimately increases intracellular Ca2+ ion concentration in vascular smooth muscle cells and their contraction [117] (Figure 2).
In vivo, ethanol consumption promoted elevations in vascular Ca2+ influx and this response occurred in parallel to hypertension [112,118,119]. The increase in Ca2+ influx mediated by ethanol occurs through voltage-sensitive channels and is linked to the pro-contractile effect exerted by ethanol [120]. As seem in studies in vitro, ROS are also implicated in the ability of ethanol to promote Ca2+ accumulation in vivo. The vascular hypercontractility associated with ethanol consumption is mediated by TXA2, a vasoconstrictor prostanoid that stimulates Ca2+ influx through the cell membrane [51]. The increase in TXA2 production is mediated by the pro-inflammatory enzyme COX2, whose vascular expression is induced by ethanol. ROS are implicated in the upregulation of COX2, overproduction of TXA2 and vascular hypercontractility being their production regulated by aldosterone [48]. Thus, the RAAS, through the action of aldosterone, triggers vascular hypercontractility to ethanol. By activating mineralocorticoid receptors, aldosterone induces ROS generation, which will in turn induce upregulation of COX2 and overproduction of TXA2. The latter will ultimately stimulates extracellular Ca2+ influx leading to vascular contraction (Figure 2).
A role for ROS in Ca2+ mobilization and vasoconstriction induced by ethanol is well established. Overproduction of ROS, Ca2+ influx and vascular contraction are interrelated and might contribute to ethanol-induced hypertension.
5.5. Role of ROS in Endothelial Dysfunction Induced by Ethanol
The vascular endothelium produces NO, a relaxing factor that plays a key role inthe control of vascular tone. NOis a free radical generated from the amino acid L-arginine by the action of the three isoforms of the enzyme NOS. While neuronal NOS (nNOS or NOS1) and endothelial NOS (eNOS/NOS3) are the isoforms constitutivelly expressed in the vasculture, the inducible NOS (iNOS/NOS2) isoform is expressed in response to inflammatory stimuli. In the vascular endothelium, eNOS is the main isoform reponsible for NO production.Once synthesized in the endothelium, NO diffuses to vascular smooth muscle cells where it stimulates the soluble isoform of guanylyl cyclase (sGC), an enzyme that catalyzes the synthesis of cGMP from guanosine 5′-triphosphate (GTP). cGMP activates protein kinase G (PKG) that, acting by multiple mechanisms, will induce smmoth muscle relaxation [121].
Endothelial dysfunction is a systemic pathological state of the endothelium characterized by a decrease in NO bioavailability and activation of the NO-cGMP pathway. In general, reduced NO bioavalilability may be the result of a decreased production of NO by endothelial eNOS, or, more frequently, an increased breakdown by O2•-, which reacts with NO leading to the genertaion of ONOO-. Endothelial dysfunction is triggered by different cardiovascular risk factors such as hypertension, obesity and diabetes [122]. Ethanol consumption induces overproduction of ROS in the vasculature and for this reason is considered a risk factor for endotheial dysfunction.
Endothelial dysfunction is assessed in vitro by evaluating endothelium-dependent vasorelaxation induced by vasoactive substances (e.g., acetylcholine) that stimulates endothelial release of NO. This pharmacological approach has been widely used to evaluate the impacts of ethanol consumption in endothelial function. Ethanol consumption decreases the endothelium-dependent relaxation induced by acetylcholine, a response that is mediated by endothelial-derived NO [123]. The impairment in endothelium-dependent vasorelaxation promoted by ethanol is related to a decreased production of NO by eNOS [124,125]. In fact, ethanol consumption reduces eNOS expression and this response occurs in parallel to a decrease in acetylcholine-induced relaxation [55,62]. Antioxidants prevented the downregulation of eNOS as well as the decrease in NO bioavailability and impairment in acetylcholine-dependent relaxation, implicating ROS in such responses [64,89,126]. Thus, ROS mediate endothelial dysfunction in response to ethanol by downregulating eNOS and by inducing NO inactivation. NADPH oxidase is implicated in the production of endothelial ROS that will further promote endothelial dysfunction induced by ethanol. Inhibition of this enzyme restored the impairment of endothelium-dependent relaxation induced by ethanol consumption. Endothelial dysfunction mediated by NADPH oxidase-derived ROS is linked to both impaired vascular relaxation and hypertension induced by ethanol consumption. As discussed in Section 5.1, ethanol-induced activation and expression of NADPH oxidase in the vasculature is mediated by the RAAS [39,48].
Ethanol consumption also compromises the systhesis of NO by interfering with BH4, a cofactor that is necessary for eNOS during the sysnthesis of NO. By decreasing BH4, ethanol consumption impairs endothelium-dependent dilatation [89]. Reduced metabolism of BH4 leads to eNOS uncoupling resulting in an increased generation of O2•- and a reducedproduction of NO [127]. Ethanol consumption promotes a decreased in arteriolar flow-induced vasodilation. The microvascular dysfunction is restored by BH4 administration, showing that reduction of the eNOS cofactor has a negative impact in NO production. In additon, it may be concluded that uncoupled eNOS may contribute to the impared vasorelaxation of the microcirculation induced by ethanol [128]. Ethanol-induced decreased availability or utilization of BH4 favors O2•- generation resulting in an imbalance between O2•- and NO, thereby contributing to endothelial dysfunction, presumably by NO inactivation [89](Figure 2).
Ethanol metabolism in the vasculature may play a role in the endothelial dysfunction induced by ethanol consumption. ADH and CYP2E1 are ethanol-metabolizing enzymes that are constitutively expressed and functionally active in the vasculature. CYP2E1 promotes the conversion of ethanol into acetaldehyde, but this process leads to the generation of O2•- [96]. The latter reacts with NO decreasing its bioavailability, a response that can account for the impaired endothelium-dependent relaxation promoted by ethanol consumption [80,117].
Endothelial dysfunction induced by ethanol may be aggravated by overexpression of iNOS, the inducible isoform of NOS [82,120]. This enzyme induces a substantial and sustained release of NO that readily reacts with O2•-, forming ONOO-, an oxidizing molecule that is linked to endothelial dysfunction. In the microcirculation, induction of iNOS expression by ethanol is associated with a decrease in NO bioavailability and impaired endothelium-dependent relaxation. These responses occurred in parallel to overproduction of O2•- and were prevented by the antioxidant apocynin, showing that ethanol-induced iNOS upregulation is mediated by ROS [81].
Under physiological conditions, endothelium-derived NO counteracts vascular contraction. In this sense, endothelial dysfunction not only compromises vascular relaxation, but it also favors vascular contraction. Decreased endothelial modulation of the vascular contraction was described after ethanol consumption. In all cases, this response was a consequence of reduced NO bioavailability and occurred in parallel to impaired endothelium-dependent vascular relaxation [12,55,56].
Endothelial function may also be assed in vivo using the non-invasive method of brachial artery flow-mediated dilation (FMD) [129]. This method is widely used to determine endothelium- and NO-mediated vasodilatation in vivo in experimental and clinical studies. Lower values of brachial artery FMD are linked to a higher risk of future cardiovascular events. Individuals with a history of chronic alcoholism (≥6 drinks/day for ≥2 years) or with a history of repeated binge drinking show decreased brachial artery FMD, when compared to nondrinkers [130,131,132,133]. Reduced endothelial function induced by heavy ethanolconsumption may predispose individuals to future cardiovascular diseases, including hypertension [128].
Decrease of NO bioavailability is a central mechanism whereby ethanol promotes endothelial dysfunction (Figure 2). This response results fromprejudiced activation or downregulation of eNOS, responses that are mediated by ROS. In addition, O2•- generated by NADPH oxidase, uncoupled eNOS or ethanol metabolism reacts with NO reducing its bioavailability and resulting in impaired vasorelaxation. Endothelial dysfunction impairs vasorelaxation and represents an important mechanism underlying the effects of ethanol on blood pressure.
5.6. Perivascular Adipose Tissue (PVAT) and Its Role in Ethanol-Induced ROS Production
PVAT is a complex tissue composed predominantly by adipocytes, but other cell types including mesenchymal stem cells and immune cells are also found in PVAT. It surrounds most blood vessels and displays phenotypic heterogeneity depending on the vascular territory. While PVAT surrounding thoracic aorta has a brown-like adipose tissue phenotype, PVAT that surrounds the abdominal aorta and coronary arteries is a mixture of white-like and brown-like adipose tissues. Conversely, mesenteric, femoral, and carotid arteries are surrounded by PVAT that is predominantly composed by white-like adipose tissue [134,135].
PVAT displays an anticontractile effect by releasing a wide range of vasoactive substances, such as NO, H2S, H2O2, prostacyclin, palmitic acid methyl ester and angiotensin 1-7. The contribution of each one of these substances to the regulation of vascular tone is dependent on PVAT composition (brown-like or white-like adipocytes) and, for this reason, varies according to the vascular bed. The anticontractile phenotype of PVAT is seemed under physiological conditions, but it may shift to a pro-contractile one under certain pathophysiological circumstances, such as hypertension and obesity [136]. The pro-contractile effects of PVAT are mediated by decreased production/release of anticontractile substances and increased generation of pro-contractile factors, such as O2•-, angiotensin II, noradrenaline, prostaglandins and chemerin [135].
There are few reports describing the impact exerted by ethanol exposure in PVAT. Current data show that the effects of ethanol in PVAT vary according to the vascular territory and pattern of ethanol consumption. PVAT counteracts the pro-contractile effect induced by a single dose of ethanol. In this scenario, the vascular protective effect of PVAT is the result of a decreased activity of catalase that favors an increase in H2O2 concentration. In this case, the anticontractile effect displayed by PVAT-derived H2O2 is partially mediated by NO [137].
In periaortic PVAT, long term ethanol consumption increases the production of ROS via NADPH oxidase activation. As a consequence, there is a reduction in NO bioavailability in PVAT. Despite inducing molecular changes, ethanol does not favor a pro-contractile phenotype of periaortic PVAT or induces loss of its anticontractile effect [16]. However, chronic ethanol consumption favors pro-contractile phenotype of PVAT that surrounds mesenteric arteries. This response is mediated by the pro-inflammatory cytokine IL-6, whose concentration is augmented in plasma and PVAT after ethanol consumption. The pro-contractile phenotype induced by IL-6 involves activation of NADPH oxidase in PVAT, with further increase in O2•- generation. IL-6 derived from PVAT also mediates intravascular recruitment of neutrophils in response to ethanol, showing that PVAT may shift to a pro-inflammatory phenotype in response to ethanol [16].
So far, studies support the notion that PVAT may be a target of the effects of ethanol, while it also contributes to the deleterious effects displayed by ethanol in the vasculature (Figure 3). PVAT is a metabolically active organ that under non-physiological conditions contributes critically to cardiovascular disease onset and progression. In this scenario, dissecting the precise role of PVAT in the vascular effects of ethanol is of paramount interest.
6. Conclusion
Ethanol consumption is associated with a robust oxidative response in the vasculature that correlates to hypertension and neuroendocrine changes. The latter are characterized by increased symphtatetic activiy and activation of RAAS, which is an upstream mechanism that contributes to NADPH oxidase activation, overproduction of ROS and vascular dysfunction. NADPH oxidase-derived ROS triggers important processes underlying vascular injury including intracellular Ca2+ ion accumulation, reduced NO bioavailability, MAPKs activation, endothelial dysfunction and loss of the anticontractile effect of PVAT. Redox imbalance induced by ethanol in the vasculature leads to impared vasodilatation and hypercontractily, which are recoganized as central events in the hypertensive state induced by ethanol consumption. Thus, it is of great importance to invest in implementing strategies that help to prevent alcoholism, thus reducing the risk of ethanol-associated cardiovascular diseases.
Author’s Contributions: Júlio C. Padovan: Conceptualization, Writing—original draft, Writing—review & editing. Thales M.H. Dourado and Gustavo F. Pimenta: Writing—review & editing. Thiago Bruder-Nascimento: Supervision, Writing—original draft, Writing—review & editing. Carlos R. Tirapelli: Conceptualization, Funding acquisition, Supervision, Writing—original draft, Writing—review & editing.
Funding:This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; grant #2019/25189-5) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (CAPES, Finance Code 001.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- MacMahon, S. Alcohol consumption and hypertension. Hypertension 1987, 9, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Klatsky, A.L.; Friedman, G.D.; Siegelaub, A.B.; Gérard, M.J. Alcohol Consumption and Blood Pressure. New Engl. J. Med. 1977, 296, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.A.; Chapman, J.M.; Coulson, A.H. Effects of various factors on systolic and diastolic blood pressure in the Los Angeles heart study. J. Chronic Dis. 1967, 20, 571–581. [Google Scholar] [CrossRef]
- Gyntelberg, F.; Meyer, J. Relationship between blood pressure and physical fitness, smoking and alcohol consumption in copenhagen males aged 40–59. Acta Medica Scand. 1974, 195, 375–380. [Google Scholar] [CrossRef]
- Dyer, A.R.; Stamler, J.; Paul, O.; Berkson, D.M.; Lepper, M.H.; McKean, H.; Shekelle, R.B.; A Lindberg, H.; Garside, D. Alcohol consumption, cardiovascular risk factors, and mortality in two Chicago epidemiologic studies. Circulation 1977, 56, 1067–1074. [Google Scholar] [CrossRef]
- Arkwright, P.D.; Beilin, L.J.; Rouse, I.; Armstrong, B.K.; Vandongen, R. Effects of alcohol use and other aspects of lifestyle on blood pressure levels and prevalence of hypertension in a working population. Circulation 1982, 66, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Linkola, J.; Fyhrquist, F.; Ylikahri, R. Renin, aldosterone and Cortisol during ethanol intoxication and hangover. Acta Physiol. Scand. 1979, 106, 75–82. [Google Scholar] [CrossRef]
- Ibsen, H.; Christensen, N.J.; Rasmussen, S.; Hollnagel, H.; Nielsen, M.D.; Giese, J. The Influence of Chronic High Alcohol Intake on Blood Pressure, Plasma Noradrenaline Concentration and Plasma Renin Concentration. Clin. Sci. 1981, 61, 377s–379s. [Google Scholar] [CrossRef]
- Potter, J.; Beevers, D. Pressor effect of alcohol in hypertension. Lancet 1984, 323, 119–122. [Google Scholar] [CrossRef]
- Howes, L.G.; Reid, J.L. Changes in plasma free 3,4-dihydroxyphenylethylene glycol and noradrenaline levels after acute alcohol administration. Clin. Sci. 1985, 69, 423–428. [Google Scholar] [CrossRef]
- Chan, T.C.; Sutter, M.C. Ethanol consumption and blood pressure. Life Sci. 1983, 33, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Pinardi, G.; Brieva, C.; Vinet, R.; Penna, M. Effects of chronic ethanol consumption on α-adrenergic-induced contractions in rat thoracic aorta. Gen. Pharmacol. Vasc. Syst. 1992, 23, 245–248. [Google Scholar] [CrossRef]
- Phillips, S.A.; Osborn, K.; Hwang, C.-L.; Sabbahi, A.; Piano, M.R. Ethanol Induced Oxidative Stress in the Vasculature: Friend or Foe. Curr. Hypertens. Rev. 2020, 16, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ceron, C.S.; Marchi, K.C.; Muniz, J.J.; Tirapelli, C.R. Vascular oxidative stress: a key factor in the development of hypertension associated with ethanol consumption. Curr. Hypertens. Rev. 2015, 10, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Leite, L.N.; Gonzaga, N.A.; Simplicio, J.A.; Vale, G.T.D.; Carballido, J.M.; Alves-Filho, J.C.; Tirapelli, C.R. Pharmacological characterization of the mechanisms underlying the vascular effects of succinate. Eur. J. Pharmacol. 2016, 789, 334–343. [Google Scholar] [CrossRef]
- Simplicio, J.A.; Dourado, T.M.; Awata, W.M.; Vale, G.T.D.; Dias, V.R.; Barros, P.R.; de Martinis, B.S.; Tostes, R.C.; Tirapelli, C.R. Ethanol consumption favors pro-contractile phenotype of perivascular adipose tissue: A role for interleukin-6. Life Sci. 2023, 319, 121526. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. , Angiotensin II, NADPH Oxidase, and Redox Signaling in the Vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef]
- Klatsky, A.L.; Friedman, G.D.; A Armstrong, M.; Y, O.; T, S.; F, I.; K, Y.; H, I.; H, W.; T, M.; et al. The relationships between alcoholic beverage use and other traits to blood pressure: a new Kaiser Permanente study. Circulation 1986, 73, 628–636. [Google Scholar] [CrossRef]
- Mathews, J.D. Alcohol Usage as a Possible Explanation for Socio-economic and Occupational Differentials in Mortality from Hypertension and Coronary Heart Disease in England and Wales*. Aust. New Zealand J. Med. 1976, 6, 393–397. [Google Scholar] [CrossRef]
- MacMahon, S.W.; Blacket, R.B.; Macdonald, G.J.; Hall, W. Obesity, Alcohol Consumption and Blood Pressure in Australian Men and Women The National Heart Foundation of Australia Risk Factor Prevalence Study. J. Hypertens. 1984, 2, 85–91. [Google Scholar] [CrossRef]
- Milon, H.; Froment, A.; Gaspard, P.; Guidollet, J.; Ripoll, J.P. Alcohol consumption and blood pressure in a French epidemiological study. Eur. Hear. J. 1982, 3, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Harburg, E.; Ozgoren, F.; Hawthorne, V.M.; A Schork, M. Community norms of alcohol usage and blood pressure: Tecumseh, Michigan. Am. J. Public Heal. 1980, 70, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.F.; Watson, R.D.; Skan, W.; Beevers, D.G. The pressor and metabolic effects of alcohol in normotensive subjects. Hypertension 1986, 8, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Seppä, K.; Sillanaukee, P. Binge Drinking and Ambulatory Blood Pressure. Hypertension 1999, 33, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Rosito, G.A.; Fuchs, F.D.; Duncan, B.B. Dose-dependent biphasic effect of ethanol on 24-h blood pressure in normotensive subjects. Am. J. Hypertens. 1999, 12, 236–240. [Google Scholar] [CrossRef]
- Mori, T.A.; Burke, V.; Beilin, L.J.; Puddey, I.B.; Stranges, S.; Wu, T.; Dorn, J.M.; Freudenheim, J.L.; Muti, P.; Farinaro, E.; et al. Randomized Controlled Intervention of the Effects of Alcohol on Blood Pressure in Premenopausal Women. Hypertension 2015, 66, 517–523. [Google Scholar] [CrossRef]
- Zilkens, R.R.; Burke, V.; Hodgson, J.M.; Barden, A.; Beilin, L.J.; Puddey, I.B. Red Wine and Beer Elevate Blood Pressure in Normotensive Men. Hypertension 2005, 45, 874–879. [Google Scholar] [CrossRef]
- Briasoulis, A.; Agarwal, V.; Messerli, F.H. Alcohol Consumption and the Risk of Hypertension in Men and Women: A Systematic Review and Meta-Analysis. J. Clin. Hypertens. 2012, 14, 792–798. [Google Scholar] [CrossRef]
- Taylor, B.; Irving, H.M.; Baliunas, D.; Roerecke, M.; Patra, J.; Mohapatra, S.; Rehm, J. Alcohol and hypertension: gender differences in dose-response relationships determined through systematic review and meta-analysis. Addiction 2009, 104, 1981–1990. [Google Scholar] [CrossRef]
- Roerecke, M.; Tobe, S.W.; Kaczorowski, J.; Bacon, S.L.; Vafaei, A.; Hasan, O.S.M.; Krishnan, R.J.; Raifu, A.O.; Rehm, J. Sex-Specific Associations Between Alcohol Consumption and Incidence of Hypertension: A Systematic Review and Meta-Analysis of Cohort Studies. J. Am. Hear. Assoc. 2018, 7, e008202. [Google Scholar] [CrossRef]
- A Abdel-Rahman, A.; Wooles, W.R. Ethanol-induced hypertension involves impairment of baroreceptors. Hypertension 1987, 10, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Strickland, J.A.; Wooles, W.R. Effect of acute and chronic ethanol on the agonist responses of vascular smooth muscle. Eur. J. Pharmacol. 1988, 152, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, A.R.; Dar, M.S.; Wooles, W.R. Effect of chronic ethanol administration on arterial baroreceptor function and pressor and depressor responsiveness in rats. J. Pharmacol. Exp. Ther. 1985, 232. [Google Scholar]
- Resstel, L.B.; Tirapelli, C.R.; Lanchote, V.L.; Uyemura, S.A.; de Oliveira, A.M.; Corrêa, F.M. Chronic ethanol consumption alters cardiovascular functions in conscious rats. Life Sci. 2006, 78, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.C.; Godin, D.V.; Sutter, M.C. Erythrocyte membrane properties of the chronic alcoholic rat. Drug Alcohol Depend. 1983, 12, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.C.; A Wall, R.; Sutter, M.C.; M, E.-M.; A, A.-R.; S, V.; C, S.; V, P.; P, H.; P, R.; et al. Chronic ethanol consumption, stress, and hypertension. Hypertension 1985, 7, 519–524. [Google Scholar] [CrossRef]
- Utkan, T.; Yildiz, F.; Ilbay, G.; Ozdemirci, S.; Erden, B.F.; Gacar, N.; Ulak, G. Blood pressure and vascular reactivity to endothelin-1, phenylephrine, serotonin, KCl and acetylcholine following chronic alcohol consumption in vitro. Fundam. Clin. Pharmacol. 2001, 15, 157–165. [Google Scholar] [CrossRef]
- da Silva, A.L.; Ruginsk, S.G.; Uchoa, E.T.; Crestani, C.C.; Scopinho, A.A.; Correa, F.M.A.; De Martinis, B.S.; Elias, L.L.K.; Resstel, L.B.; Antunes-Rodrigues, J. Time-Course of Neuroendocrine Changes and Its Correlation with Hypertension Induced by Ethanol Consumption. Alcohol Alcohol. 2013, 48, 495–504. [Google Scholar] [CrossRef]
- Passaglia, P.; Ceron, C.S.; Mecawi, A.S.; Antunes-Rodrigues, J.; Coelho, E.B.; Tirapelli, C.R. Angiotensin type 1 receptor mediates chronic ethanol consumption-induced hypertension and vascular oxidative stress. Vasc. Pharmacol. 2015, 74, 49–59. [Google Scholar] [CrossRef]
- A Brown, R.; Ilg, K.J.; Chen, A.F.; Ren, J. Dietary Mg2+ supplementation restores impaired vasoactive responses in isolated rat aorta induced by chronic ethanol consumption. Eur. J. Pharmacol. 2002, 442, 241–250. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Lambie, D.G.; Johnson, R.H. Effects of ethanol on plasma catecholamines and norepinephrine clearance. Clin. Pharmacol. Ther. 1983, 34, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Ireland, M.A.; Vandongen, R.; Davidson, L.; Beilin, L.J.; Rouse, I.L. Acute effects of moderate alcohol consumption on blood pressure and plasma catecholamines. Clin. Sci. 1984, 66, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Vale, G.T.D.; Simplicio, J.A.; Gonzaga, N.A.; Yokota, R.; Ribeiro, A.A.; Casarini, D.E.; de Martinis, B.S.; Tirapelli, C.R. Nebivolol prevents vascular oxidative stress and hypertension in rats chronically treated with ethanol. Atherosclerosis 2018, 274, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Linkola, J.; Fyhrquist, F.; Nieminen, M.M.; Weber, T.H.; Tontti, K. Renin-Aldosterone Axis in Ethanol Intoxication and Hangover. Eur. J. Clin. Investig. 1976, 6, 191–194. [Google Scholar] [CrossRef]
- Puddey, I.B.; Vandongen, R.; Beilin, L.J.; Rouse, I.L. Alcohol Stimulation of Renin Release in Man: Its Relation to the Hemodynamic, Electrolyte, and Sympatho-Adrenal Responses to Drinking*. J. Clin. Endocrinol. Metab. 1985, 61, 37–42. [Google Scholar] [CrossRef]
- Wright, J.W.; Morseth, S.L.; Abhold, R.H.; Harding, J.W. Elevations in plasma angiotensin II with prolonged ethanol treatment in rats. Pharmacol. Biochem. Behav. 1986, 24, 813–818. [Google Scholar] [CrossRef]
- Husain, K.; Vazquez, M.; Ansari, R.A.; Malafa, M.P.; Lalla, J. Chronic alcohol-induced oxidative endothelial injury relates to angiotensin II levels in the rat. Mol. Cell. Biochem. 2007, 307, 51–58. [Google Scholar] [CrossRef]
- Dourado, T.M.; Assis, V.O.; Awata, W.M.; de Mello, M.M.; Cárnio, E.C.; Castro, M.M.; Tirapelli, C.R. Mineralocorticoid receptors contribute to ethanol-induced vascular hypercontractility through reactive oxygen species generation and up-regulation of cyclooxygenase 2. Eur. J. Pharmacol. 2023, 949, 175723. [Google Scholar] [CrossRef]
- Hatton, D.C.; Bukoski, R.D.; Edgar, S.; McCarron, D.A. Chronic alcohol consumption lowers blood pressure but enhances vascular contractility in Wistar rats. J. Hypertens. 1992, 10, 529–537. [Google Scholar] [CrossRef]
- Stewart, C.W.; Kennedy, R.H. Effects of chronic ethanol consumption on aortic constriction in male and female rats. Eur. J. Pharmacol. 1999, 366, 55–60. [Google Scholar] [CrossRef]
- Tirapelli, C.R.; Al-Khoury, J.; Bkaily, G.; D'Orléans-Juste, P.; Lanchote, V.L.; Uyemura, S.A.; de Oliveira, A.M. Chronic Ethanol Consumption Enhances Phenylephrine-Induced Contraction in the Isolated Rat Aorta. Experiment 2005, 316, 233–241. [Google Scholar] [CrossRef]
- Tirapelli, C.R.; Leone, A.F.C.; Coelho, E.B.; Resstel, L.B.M.; A Corrêa, F.M.; Lanchote, V.L.; A Uyemura, S.; Padovan, C.M.; de Oliveira, A.M. Effect of ethanol consumption on blood pressure and rat mesenteric arterial bed, aorta and carotid responsiveness. J. Pharm. Pharmacol. 2007, 59, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Ladipo, C.; Adigun, S.; Nwaigwe, C.; Adegunloye, B. Chronic Ethanol Consumption Alters Vascular Smooth Muscle Responses In Rats. Clin. Exp. Pharmacol. Physiol. 2002, 29, 707–709. [Google Scholar] [CrossRef]
- Tirapelli, C.R.; Legros, E.; Brochu, I.; Honoré, J.-C.; Lanchote, V.L.; A Uyemura, S.; De Oliveira, A.M.; D'Orléans-Juste, P. Chronic ethanol intake modulates vascular levels of endothelin-1 receptor and enhances the pressor response to endothelin-1 in anaesthetized rats. Br. J. Pharmacol. 2008, 154, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Tirapelli, C.R.; Leone, A.F.C.; Yogi, A.; Tostes, R.C.; Lanchote, V.L.; A Uyemura, S.; Resstel, L.B.M.; A Corrêa, F.M.; Padovan, C.M.; de Oliveira, A.M.; et al. Ethanol consumption increases blood pressure and alters the responsiveness of the mesenteric vasculature in rats. J. Pharm. Pharmacol. 2008, 60, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Tirapelli, C.R.; Casolari, D.A.; Montezano, A.C.; Yogi, A.; Tostes, R.C.; Legros, E.; D'Orléans-Juste, P.; Lanchote, V.L.; Uyemura, S.A.; de Oliveira, A.M. Ethanol Consumption Enhances Endothelin-1-Induced Contraction in the Isolated Rat Carotid. Experiment 2006, 318, 819–827. [Google Scholar] [CrossRef]
- Leite, L.N.; Lacchini, R.; Carnio, E.C.; Queiroz, R.H.; Tanus-Santos, J.E.; de Oliveira, A.M.; Tirapelli, C.R. Ethanol Consumption Increases Endothelin-1 Expression and Reactivity in the Rat Cavernosal Smooth Muscle. Alcohol Alcohol. 2013, 48, 657–666. [Google Scholar] [CrossRef]
- Marchi, K.C.; Ceron, C.S.; Muniz, J.J.; De Martinis, B.S.; Tanus-Santos, J.E.; Tirapelli, C.R. NADPH Oxidase Plays a Role on Ethanol-Induced Hypertension and Reactive Oxygen Species Generation in the Vasculature. Alcohol Alcohol. 2016, 51, 522–534. [Google Scholar] [CrossRef]
- Ceron, C.S.; Vale, G.T.D.; Simplicio, J.A.; Passaglia, P.; Ricci, S.T.; Tirapelli, C.R. Data on the effects of losartan on protein expression, vascular reactivity and antioxidant capacity in the aorta of ethanol-treated rats. Data Brief 2017, 11, 111–116. [Google Scholar] [CrossRef]
- Abou-Agag, L.H.; Khoo, N.K.; Binsack, R.; White, C.R.; Darley-Usmar, V.; Grenett, H.E.; Booyse, F.M.; Digerness, S.B.; Zhou, F.; Parks, D.A. Evidence of cardiovascular protection by moderate alcohol: Role of nitric oxide. Free. Radic. Biol. Med. 2005, 39, 540–548. [Google Scholar] [CrossRef]
- Hipólito, U.V.; Rocha, J.T.; Martins-Oliveira, A.; Tirapelli, D.P.; Jacob-Ferreira, A.; Batalhão, M.E.; Tanus-Santos, J.E.; Carnio, E.C.; Cunha, T.M.; Queiroz, R.H.; et al. Chronic ethanol consumption reduces adrenomedullin-induced relaxation in the isolated rat aorta. Alcohol 2011, 45, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.T.; Hipólito, U.V.; Martins-Oliveira, A.; Tirapelli, D.P.; Batalhão, M.E.; Carnio, E.C.; Queiroz, R.H.; Coelho, E.B.; Cunha, T.M.; Tanus-Santos, J.E.; et al. Ethanol Consumption Alters the Expression and Reactivity of Adrenomedullin in the Rat Mesenteric Arterial Bed. Alcohol Alcohol. 2011, 47, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Champion, H.C.; Pierce, R.L.; Bivalacqua, T.J.; Murphy, W.A.; Coy, D.H.; Kadowitz, P.J. Analysis of responses to hAmylin, hCGRP, and hADM in isolated resistance arteries from the mesenteric vascular bed of the rat. Peptides 2001, 22, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Simplicio, J.A.; Vale, G.T.D.; Gonzaga, N.A.; Leite, L.N.; Hipólito, U.V.; Pereira, C.A.; Tostes, R.C.; Tirapelli, C.R. Reactive oxygen species derived from NAD(P)H oxidase play a role on ethanol-induced hypertension and endothelial dysfunction in rat resistance arteries. J. Physiol. Biochem. 2016, 73, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Tirapelli, C.R.; A Casolari, D.; Yogi, A.; Montezano, A.C.; Tostes, R.C.; Legros, E.; D'Orléans-Juste, P.; De Oliveira, A.M. Functional characterization and expression of endothelin receptors in rat carotid artery: involvement of nitric oxide, a vasodilator prostanoid and the opening of K+channels in ETB-induced relaxation. Br. J. Pharmacol. 2005, 146, 903–912. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Tostes, R.C.; Paradis, P.; Schiffrin, E.L. Aldosterone, Inflammation, Immune System, and Hypertension. Am. J. Hypertens. 2020, 34, 15–27. [Google Scholar] [CrossRef]
- Mascolo, A.; Scavone, C.; Rafaniello, C.; De Angelis, A.; Urbanek, K.; di Mauro, G.; Cappetta, D.; Berrino, L.; Rossi, F.; Capuano, A. The Role of Renin-Angiotensin-Aldosterone System in the Heart and Lung: Focus on COVID-19. Front. Pharmacol. 2021, 12, 667254. [Google Scholar] [CrossRef]
- Chapp, A.D.; Gui, L.; Huber, M.J.; Liu, J.; Larson, R.A.; Zhu, J.; Carter, J.R.; Chen, Q.-H. Sympathoexcitation and pressor responses induced by ethanol in the central nucleus of amygdala involves activation of NMDA receptors in rats. Am. J. Physiol. Circ. Physiol. 2014, 307, H701–H709. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: key regulators in vascular health and diseases. Br. J. Pharmacol. 2017, 175, 1279–1292. [Google Scholar] [CrossRef]
- Cheng, X.-M.; Hu, Y.-Y.; Yang, T.; Wu, N.; Wang, X.-N. Reactive Oxygen Species and Oxidative Stress in Vascular-Related Diseases. Oxidative Med. Cell. Longev. 2022, 2022, 1–11. [Google Scholar] [CrossRef]
- Zhao, S.; Cheng, C.K.; Zhang, C.-L.; Huang, Y. Interplay Between Oxidative Stress, Cyclooxygenases, and Prostanoids in Cardiovascular Diseases. Antioxidants Redox Signal. 2021, 34, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Anagnostopoulou, A.; Camargo, L.L.; Rios, F.J.; Montezano, A.C. Vascular Biology of Superoxide-Generating NADPH Oxidase 5—Implications in Hypertension and Cardiovascular Disease. Antioxidants Redox Signal. 2019, 30, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; E Nagy, L.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-α production. J. Leukoc. Biol. 2006, 79, 1348–1356. [Google Scholar] [CrossRef]
- De Minicis, S.; A Brenner, D. Oxidative stress in alcoholic liver disease: Role of NADPH oxidase complex. J. Gastroenterol. Hepatol. 2008, 23, S98–S103. [Google Scholar] [CrossRef]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflammation 2012, 9, 5–5. [Google Scholar] [CrossRef]
- Yeligar, S.M.; Harris, F.L.; Hart, C.M.; Brown, L.A.S. Ethanol Induces Oxidative Stress in Alveolar Macrophages via Upregulation of NADPH Oxidases. J. Immunol. 2012, 188, 3648–3657. [Google Scholar] [CrossRef]
- Husain, K.; Ferder, L.; A Ansari, R.; Lalla, J. Chronic ethanol ingestion induces aortic inflammation/oxidative endothelial injury and hypertension in rats. Hum. Exp. Toxicol. 2010, 30, 930–939. [Google Scholar] [CrossRef]
- Yogi, A.; Callera, G.E.; Mecawi, A.S.; Batalhão, M.E.; Carnio, E.C.; Antunes-Rodrigues, J.; Queiroz, R.H.; Touyz, R.M.; Tirapelli, C.R. Acute ethanol intake induces superoxide anion generation and mitogen-activated protein kinase phosphorylation in rat aorta: A role for angiotensin type 1 receptor. Toxicol. Appl. Pharmacol. 2012, 264, 470–478. [Google Scholar] [CrossRef]
- Leite, L.N.; Vale, G.T.D.; Simplicio, J.A.; De Martinis, B.S.; Carneiro, F.S.; Tirapelli, C.R. Ethanol-induced erectile dysfunction and increased expression of pro-inflammatory proteins in the rat cavernosal smooth muscle are mediated by NADPH oxidase-derived reactive oxygen species. Eur. J. Pharmacol. 2017, 804, 82–93. [Google Scholar] [CrossRef]
- Ceron, C.S.; Vale, G.T.D.; Simplicio, J.A.; Ricci, S.T.; De Martinis, B.S.; de Freitas, A.; Tirapelli, C.R. Chronic ethanol consumption increases vascular oxidative stress and the mortality induced by sub-lethal sepsis: Potential role of iNOS. Eur. J. Pharmacol. 2018, 825, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Simplicio, J.A.; Gonzaga, N.A.; Nakashima, M.A.; De Martinis, B.S.; Cunha, T.M.; Tirapelli, L.F.; Tirapelli, C.R. Tumor necrosis factor-α receptor 1 contributes to ethanol-induced vascular reactive oxygen species generation and hypertension. J. Am. Soc. Hypertens. 2017, 11, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.M.; Griendling, K.K. NADPH oxidases and angiotensin II receptor signaling. Mol. Cell. Endocrinol. 2009, 302, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Callera, G.E.; Touyz, R.M.; Tostes, R.C.; Yogi, A.; He, Y.; Malkinson, S.; Schiffrin, E.L.; L, C.; A, H.; F, R.; et al. Aldosterone Activates Vascular p38MAP Kinase and NADPH Oxidase Via c-Src. Hypertension 2005, 45, 773–779. [Google Scholar] [CrossRef]
- Ohmine, T.; Miwa, Y.; Takahashi-Yanaga, F.; Morimoto, S.; Maehara, Y.; Sasaguri, T. The involvement of aldosterone in cyclic stretch-mediated activation of NADPH oxidase in vascular smooth muscle cells. Hypertens. Res. 2009, 32, 690–699. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Sun, H.; Patel, K.P.; Mayhan, W.G. Impairment of neuronal nitric oxide synthase-dependent dilation of cerebral arterioles during chronic alcohol consumption. Alcohol. Clin. Exp. Res. 2002, 26. [Google Scholar]
- Sun, H.; Mayhan, W.G. Sex Difference in Nitric Oxide Synthase???Dependent Dilatation of Cerebral Arterioles During Long-Term Alcohol Consumption. Alcohol. Clin. Exp. Res. 2005, 29, 430–436. [Google Scholar] [CrossRef]
- Hwang, C.; Bian, J.; Thur, L.A.; Peters, T.A.; Piano, M.R.; Phillips, S.A. Tetrahydrobiopterin Restores Microvascular Dysfunction in Young Adult Binge Drinkers. Alcohol. Clin. Exp. Res. 2019, 44, 407–414. [Google Scholar] [CrossRef]
- Schuchardt, M.; Herrmann, J.; Tolle, M.; Van Der Giet, M. Xanthine Oxidase and its Role as Target in Cardiovascular Disease: Cardiovascular Protection by Enzyme Inhibition? Curr. Pharm. Des. 2017, 23, 3391–3404. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol. 2013, 1, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Aberle, N.S.; Ren, J. Experimental assessment of the role of acetaldehyde in alcoholic cardiomyopathy. Biol. Proced. Online 2003, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kawase, T.; Alderman, J.; Inatomi, N.; Lieber, C.S. Role of xanthine oxidase in ethanol-induced lipid peroxidation in rats. Gastroenterology 1990, 98, 203–210. [Google Scholar] [CrossRef]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef]
- Haorah, J.; Knipe, B.; Gorantla, S.; Zheng, J.; Persidsky, Y. Alcohol-induced blood?brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J. Neurochem. 2007, 100, 324–336. [Google Scholar] [CrossRef]
- Kotha, R.R.; Tareq, F.S.; Yildiz, E.; Luthria, D.L. Oxidative Stress and Antioxidants—A Critical Review on In Vitro Antioxidant Assays. Antioxidants 2022, 11, 2388. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M.; Kim, J.H.; Lee, M.-R.; Hong, Y.-C.; Choquet, H.; Trapani, E.; Goitre, L.; Trabalzini, L.; Akers, A.; et al. Superoxide Dismutases: Role in Redox Signaling, Vascular Function, and Diseases. Antioxidants Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Rua, R.M.; Ojeda, M.L.; Nogales, F.; Rubio, J.M.; Romero-Gómez, M.; Funuyet, J.; Murillo, M.L.; Carreras, O. Serum selenium levels and oxidative balance as differential markers in hepatic damage caused by alcohol. Life Sci. 2014, 94, 158–163. [Google Scholar] [CrossRef]
- Singh, K.; Ahluwalia, P.; Kaur, P.; Sharma, J. Effect of monosodium glutamate on various lipid fractions and certain antioxidant enzymes in arterial tissue of chronic alcoholic adult male mice. Toxicol. Int. 2012, 19, 9–14. [Google Scholar] [CrossRef]
- Bretón-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef]
- Byon, C.H.; Heath, J.M.; Chen, Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016, 9, 244–253. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-W.; Wang, J.; Zheng, T.; Altura, B.T.; Altura, B.M. Ethanol-Induced Contractions in Cerebral Arteries. Stroke 2001, 32, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-W.; Wang, J.; Zheng, T.; Altura, B.T.; Altura, B.M. Roles of tyrosine kinase–, 1-phosphatidylinositol 3-kinase–, and mitogen-activated protein kinase–signaling pathways in ethanol-induced contractions of rat aortic smooth muscle: possible relation to alcohol-induced hypertension. Alcohol 2002, 28, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga, N.A.; Callera, G.E.; Yogi, A.; Mecawi, A.S.; Antunes-Rodrigues, J.; Queiroz, R.H.; Touyz, R.M.; Tirapelli, C.R. Acute ethanol intake induces mitogen-activated protein kinase activation, platelet-derived growth factor receptor phosphorylation, and oxidative stress in resistance arteries. J. Physiol. Biochem. 2014, 70, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Orellana, M.; Valdés, E.; Fernández, J.; Rodrigo, R. Effects of Chronic Ethanol Consumption on Extramitochondrial Fatty Acid Oxidation and Ethanol Metabolism by Rat Kidney. Gen. Pharmacol. Vasc. Syst. 1998, 30, 719–723. [Google Scholar] [CrossRef]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef]
- Husain, K.; Mejia, J.; Lalla, J.; Kazim, S. Time response of alcohol-induced alterations in blood pressure, nitric oxide and oxidant to antioxidant balance in the plasma of rats. Exp. Clin. Cardiol. 2004, 9, 229–34. [Google Scholar]
- Rendón-Ramírez, A.; Cortés-Couto, M.; Martínez-Rizo, A.B.; Muñiz-Hernández, S.; Velázquez-Fernández, J.B. Oxidative damage in young alcohol drinkers: A preliminary study. Alcohol 2013, 47, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, S.; A Sampson, C.; Prabhakaran, V.M. Platelet-free calcium and vascular calcium uptake in ethanol-induced hypertensive rats. Hypertension 1991, 18, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Altura, B.M.; Zhang, A.; Cheng, T.P.-O.; Altura, B.T. Exposure of piglet coronary arterial muscle cells to low alcohol results in elevation of intracellular free Ca2+: relevance to fetal alcohol syndrome. Eur. J. Pharmacol. 1996, 314, R9–R11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Cheng, T.P.-O.; Altura, B.T.; Altura, B.M. Chronic treatment of cultured cerebral vascular smooth cells with low concentration of ethanol elevates intracellular calcium and potentiates prostanoid-induced rises in [Ca2+]i: Relation to etiology of alcohol-induced stroke. Alcohol 1997, 14, 367–371. [Google Scholar] [CrossRef]
- Zheng, T.; Li, W.; Zhang, A.; Altura, B.T.; Altura, B.M. α-Tocopherol prevents ethanol-induced elevation of [Ca2+]i in cultured canine cerebral vascular smooth muscle cells. Neurosci. Lett. 1998, 245, 17–20. [Google Scholar] [CrossRef]
- Li, W.; Liu, W.; Altura, B.T.; Altura, B.M. Catalase prevents elevation of [Ca2+]i induced by alcohol in cultured canine cerebral vascular smooth muscle cells: Possible relationship to alcohol-induced stroke and brain pathology. Brain Res. Bull. 2003, 59, 315–318. [Google Scholar] [CrossRef]
- Yogi, A.; Callera, G.E.; Hipólito, U.V.; Silva, C.R.; Touyz, R.M.; Tirapelli, C.R. Ethanol-induced vasoconstriction is mediated via redox-sensitive cyclo-oxygenase-dependent mechanisms. Clin. Sci. 2010, 118, 657–668. [Google Scholar] [CrossRef]
- Hsieh, S.T.; Sano, H.; Saito, K.; Kubota, Y.; Yokoyama, M.; L, R.; M, B.; L, D.; J, V.; J, N.; et al. Magnesium supplementation prevents the development of alcohol-induced hypertension. Hypertension 1992, 19, 175–182. [Google Scholar] [CrossRef]
- Vasdev, S.; Gupta, I.P.; A Sampson, C.; Longerich, L.; Parai, S. Ethanol induced hypertension in rats: reversibility and role of intracellular cytosolic calcium. Artery 1993, 20. [Google Scholar]
- Tirapelli, C.R.; Fukada, S.Y.; Yogi, A.; Chignalia, A.Z.; Tostes, R.C.; Bonaventura, D.; Lanchote, V.L.; Cunha, F.Q.; de Oliveira, A.M. Gender-specific vascular effects elicited by chronic ethanol consumption in rats: a role for inducible nitric oxide synthase. Br. J. Pharmacol. 2008, 153, 468–479. [Google Scholar] [CrossRef]
- Król, M.; Kepinska, M. Human Nitric Oxide Synthase—Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef] [PubMed]
- Brizzolara, A.L.; Morris, D.G.; Burnstock, G. Ethanol affects sympathetic cotransmission and endothelium-dependent relaxation in the rat. Eur. J. Pharmacol. 1994, 254, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Mayhan, W.G.; Didion, S.P. Effect of Chronic Alcohol Consumption on Responses of Cerebral Arterioles. Alcohol. Clin. Exp. Res. 1996, 20, 538–542. [Google Scholar] [CrossRef]
- Mayock, D.E.; Ness, D.; Mondares, R.L.; Gleason, C.A.; Sun, H.; Patel, K.P.; Mayhan, W.G. Responses of cerebral arterioles during chronic ethanol exposure. Am. J. Physiol. Circ. Physiol. 1992, 262, H787–H791. [Google Scholar] [CrossRef]
- Sun, H.; Mayhan, W.G.; Mayock, D.E.; Ness, D.; Mondares, R.L.; Gleason, C.A.; Faraci, F.M.; Patel, K.P. Temporal effect of alcohol consumption on reactivity of pial arterioles: role of oxygen radicals. Am. J. Physiol. Circ. Physiol. 2001, 280, H992–H1001. [Google Scholar] [CrossRef]
- Cosentino, F.; Katusic, Z.S. Tetrahydrobiopterin and Dysfunction of Endothelial Nitric Oxide Synthase in Coronary Arteries. Circulation 1995, 91, 139–144. [Google Scholar] [CrossRef]
- Piano, M.R.; Phillips, S.A.; Hwang, C.-L. The effects of alcohol consumption on flow-mediated dilation in humans: A systematic review. Physiol. Rep. 2021, 9, e14872. [Google Scholar] [CrossRef]
- Thijssen, D.H.J.; Bruno, R.M.; Van Mil, A.C.C.M.; Holder, S.M.; Faita, F.; Greyling, A.; Zock, P.L.; Taddei, S.; Deanfield, J.E.; Luscher, T.; et al. Expert consensus and evidence-based recommendations for the assessment of flow-mediated dilation in humans. Eur. Hear. J. 2019, 40, 2534–2547. [Google Scholar] [CrossRef]
- Di Gennaro, C.; Saccani-Jotti, G.; Pinelli, S.; Venturi, N.; Palombi, F.; Manfredi, G.; Pellegrino, A.; Bicchieri, L.; Sansoni, P.; Montanari, A. Endothelial Dysfunction and High Cardiovascular Risk Profile in Severe Alcoholics Improve Only Partially Following a Medium-Term Alcohol Withdrawal. Alcohol. Clin. Exp. Res. 2011, 36, 242–250. [Google Scholar] [CrossRef]
- Goslawski, M.; Piano, M.R.; Bian, J.-T.; Church, E.C.; Szczurek, M.; Phillips, S.A. Binge Drinking Impairs Vascular Function in Young Adults. J. Am. Coll. Cardiol. 2013, 62, 201–207. [Google Scholar] [CrossRef]
- Luo, R.; Shen, J.; Zhou, Q.; Liu, Y.; Li, G. Evaluation of the brachial artery endothelial function in chronic alcohol consumption among males by high-frequency ultrasonography. Echocardiography 2016, 34, 226–231. [Google Scholar] [CrossRef]
- Oda, N.; Kajikawa, M.; Maruhashi, T.; Kishimoto, S.; Yusoff, F.M.; Goto, C.; Nakashima, A.; Tomiyama, H.; Takase, B.; Yamashina, A.; et al. Endothelial function is preserved in light to moderate alcohol drinkers but is impaired in heavy drinkers in women: Flow-mediated Dilation Japan (FMD-J) study. PLOS ONE 2020, 15, e0243216. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, Z.; Zhu, Y.Z. Regional Heterogeneity of Perivascular Adipose Tissue: Morphology, Origin, and Secretome. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Bibi, A.; Valoti, M.; Fusi, F. Perivascular Adipose Tissue and Vascular Smooth Muscle Tone: Friends or Foes? Cells 2023, 12, 1196. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Ding, H.; Jiang, M.; Yin, H.; Gollasch, M.; Huang, Y. Perivascular adipose tissue: Fine-tuner of vascular redox status and inflammation. Redox Biol. 2023, 62, 102683. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga, N.A.; Awata, W.M.; Vale, G.T.D.; Marchi, K.C.; Muniz, J.J.; Tanus-Santos, J.E.; Tirapelli, C.R. Perivascular adipose tissue protects against the vascular dysfunction induced by acute ethanol intake: Role of hydrogen peroxide. Vasc. Pharmacol. 2018, 111, 44–53. [Google Scholar] [CrossRef]
Figure 1.
Proposal of the mechanisms underlying ethanol-induced hypertension. Neuroendocrine changes promoted by ethanol triggers overproduction of ROS in the vasculature leading to vascular dysfunction. These responses culminate in hypercontractility and hypertension. RAAS: renin-angiotensin-aldosterone system; ROS: reactive oxygen species.
Figure 1.
Proposal of the mechanisms underlying ethanol-induced hypertension. Neuroendocrine changes promoted by ethanol triggers overproduction of ROS in the vasculature leading to vascular dysfunction. These responses culminate in hypercontractility and hypertension. RAAS: renin-angiotensin-aldosterone system; ROS: reactive oxygen species.
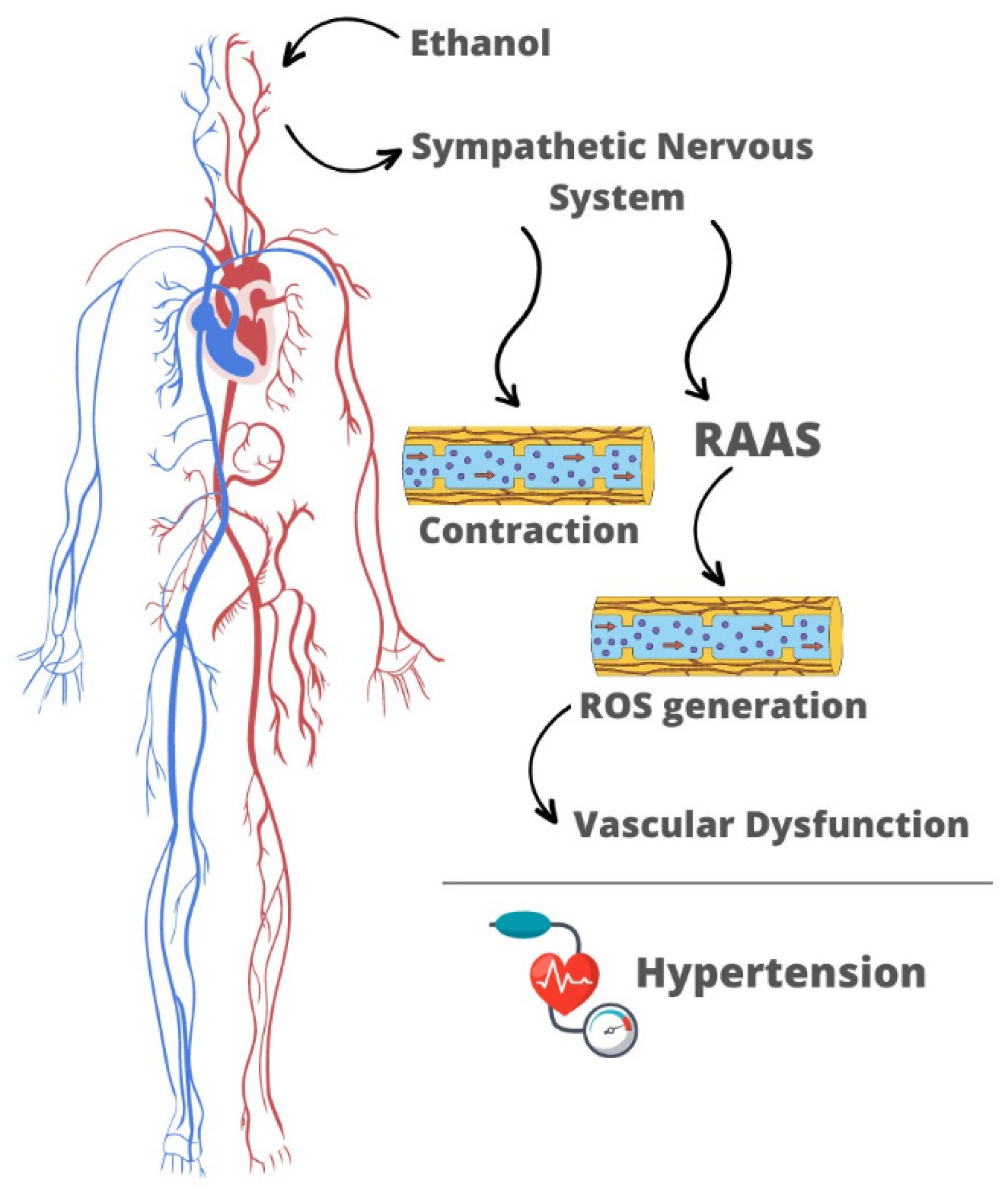
Figure 2.
Summary of the molecular mechanisms underlying ethanol-induced vascular dysfunction. RAAS is an upstream mechanism that leads to NADPH oxidase activation, an enzyme that plays a central role in vascular ROS generation in response to ethanol consumption. Augmented levels of ROS triggers important processes underlying vascular injury including intracellular Ca2+ accumulation, reduced NO bioavailability, MAPKs activation and endothelial dysfunction. VSMC: vascular smooth muscle cells; RAAS: renin-angiotensin-aldosterone system; ROS: reactive oxygen species; CYP2E1: cytochrome P450 2E1; COX2: cyclooxygenase 2; TXA2: thromboxane A2; MAPKs: mitogen-activated protein kinases; NO: nitric oxide; eNOS: endothelial NO synthase; BH4: tetrahydrobiopterin; BH2: dihydrobiopterin; TP: thromboxane receptor; SOD: superoxide dismutase; GPx: glutathione peroxidase.
Figure 2.
Summary of the molecular mechanisms underlying ethanol-induced vascular dysfunction. RAAS is an upstream mechanism that leads to NADPH oxidase activation, an enzyme that plays a central role in vascular ROS generation in response to ethanol consumption. Augmented levels of ROS triggers important processes underlying vascular injury including intracellular Ca2+ accumulation, reduced NO bioavailability, MAPKs activation and endothelial dysfunction. VSMC: vascular smooth muscle cells; RAAS: renin-angiotensin-aldosterone system; ROS: reactive oxygen species; CYP2E1: cytochrome P450 2E1; COX2: cyclooxygenase 2; TXA2: thromboxane A2; MAPKs: mitogen-activated protein kinases; NO: nitric oxide; eNOS: endothelial NO synthase; BH4: tetrahydrobiopterin; BH2: dihydrobiopterin; TP: thromboxane receptor; SOD: superoxide dismutase; GPx: glutathione peroxidase.
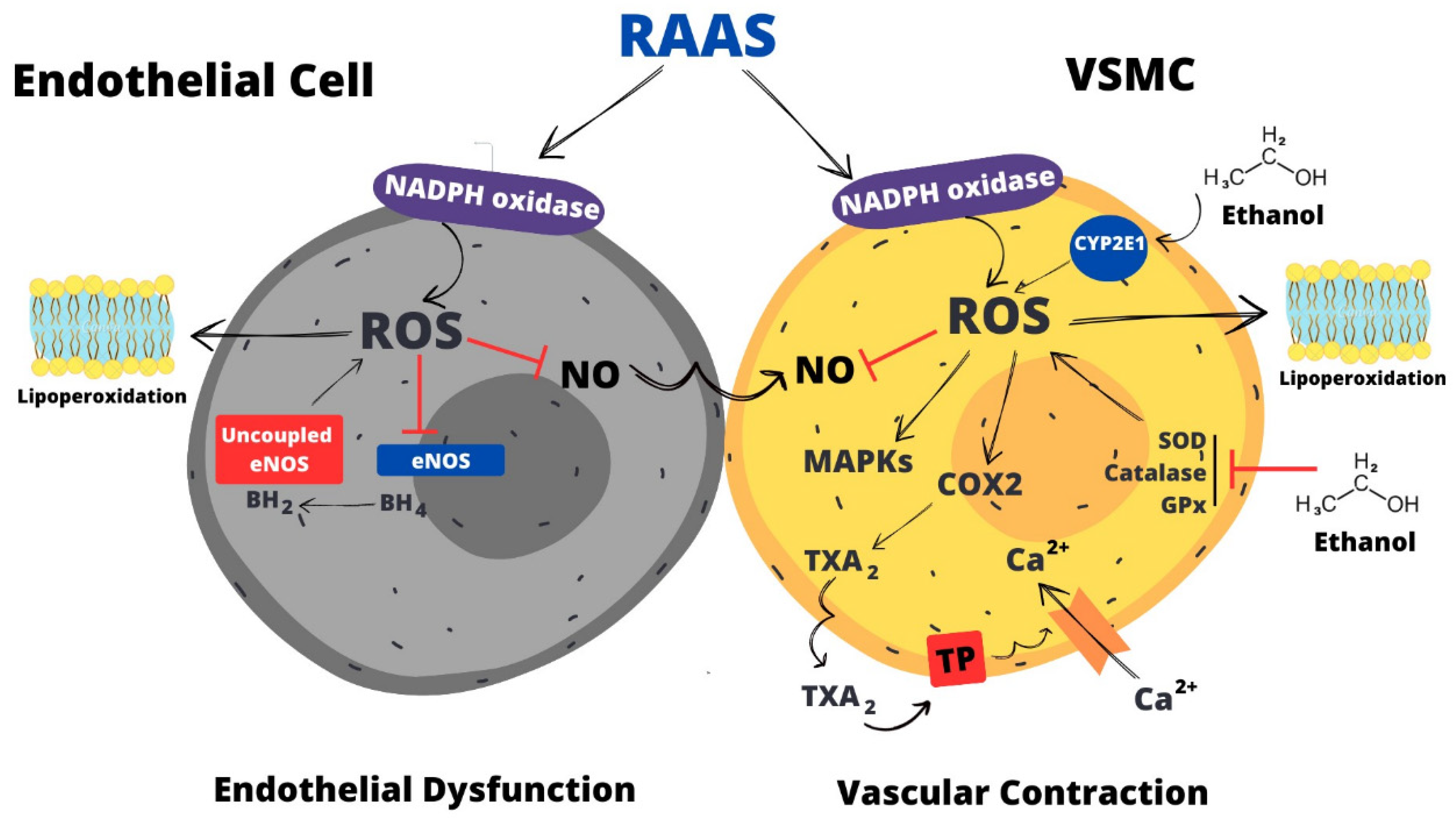
Figure 3.
Ethanol consumption promotes dysfunction of PVAT. Ethanol consumption favors a pro-inflammatory phenotype of PVAT by promoting increases in the levels of IL-6 and TNF-α. These cytokines are linked to activation of NADPH oxidase, ROS generation and decreased bioavailability of NO. PVAT: perivascular adipose tissue; O2•-: superoxide; NO: nitric oxide.
Figure 3.
Ethanol consumption promotes dysfunction of PVAT. Ethanol consumption favors a pro-inflammatory phenotype of PVAT by promoting increases in the levels of IL-6 and TNF-α. These cytokines are linked to activation of NADPH oxidase, ROS generation and decreased bioavailability of NO. PVAT: perivascular adipose tissue; O2•-: superoxide; NO: nitric oxide.
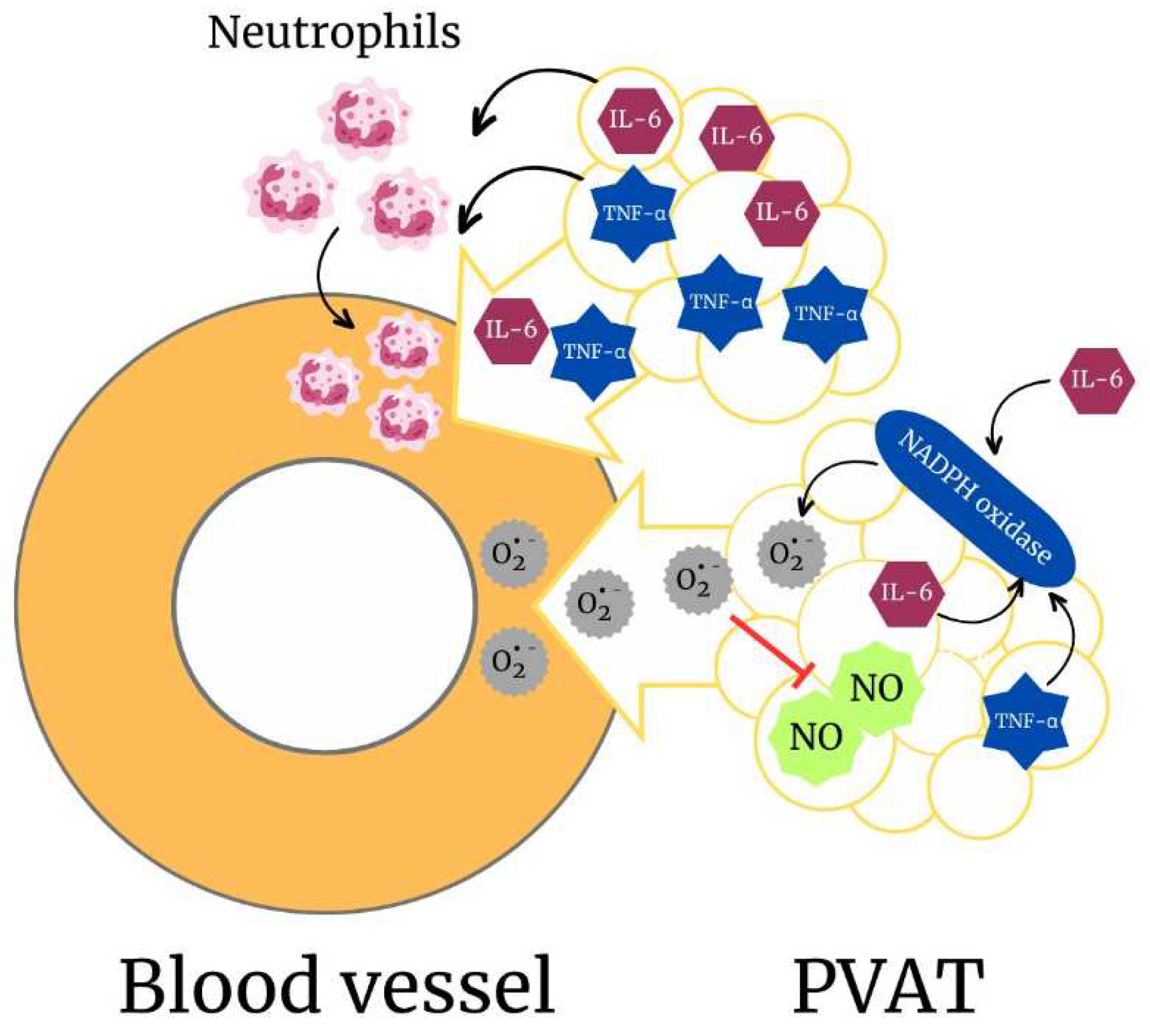
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



