
Submitted:
06 September 2023
Posted:
07 September 2023
You are already at the latest version
Abstract
Over the years, network analysis became a promising strategy to analyze complex system, i.e. systems composed by a large number of interacting elements. In particular, multilayer networks have emerged as a powerful framework for modelling and analysing complex systems with multiple types of interactions. Network analysis can be applied to pharmacogenomics to gain insights into the interactions between genes, drugs, and diseases. By integrating network analysis techniques with pharmacogenomic data, the goal consists to uncover complex relationships and identify key genes to use in pathway enrichment analysis to figure out biological pathways involved in drug response and adverse reactions. In this study, we model omics, diseases and drugs data together through the multilayer network representation. Then, we mined the multilayer network with a community detection algorithm by obtaining top communities. After that, we used the identified list of genes from the communities to perform pathway enrichment analysis (PEA) to figure out the biological function affected form the selected genes. The results show that the genes forming the top community have the multiple roles through different pathways.
Keywords:
pharmacogenomics
; network analysis
; multilayer networks
; community detection
; pathway enrichment analysis
1. Introduction
Network analysis is a branch of network science that deals with the study of complex networks. In order to investigate complex relationships, network analysis adopts theories and methods typical of several research areas [1]. Networks and network analysis methods are a keystone in computational biology and bioinformatics and are increasingly used to study biological and clinical data in an integrated way. In detail, network analysis consists of a collection of techniques with a shared methodological perspective, which allows to depict relations among entities and to analyze the structures that emerge from the recurrence of these relations. The basic assumption is that better explanations of different phenomena are yielded by the analysis of the relations among entities. A classical network analysis method is represented by community detection [2]. Community detection is one of the most popular research areas in various complex systems, such as biology, sociology, medicine, and transportation systems [3,4]. The reason is that the community structures, defined as groups of nodes that are more densely connected than the rest of the network, represent significant characteristics for understanding the functionalities and organisations of complex systems modelled as network [2]. It is expected that the communities play significant roles in the structure-function relationship. For example, in biological networks such as Protein-Protein Interaction (PPI) networks, the communities represent proteins involved in a similar function; in neuroscience, the communities detected on brain networks mean regions of interest (ROI) that are active during tasks, in social networks, communities can be groups of friends or colleagues, in World Wild Web, the communities represent the web pages sharing the same topic [5]. Thus, the discovery of communities in these systems has become an interesting approach to figuring out how network structure relates to system behaviours. In the last years, network analysis has become an essential tool in pharmacogenomics [6,7]. By providing a powerfull framework to model the data, network analysis allowed researchers to analyze and interpret complex interactions between genes, proteins, and drugs in pharmacogenomics field. This enable to uncover underlying biological mechanisms, identify potential drug targets and biomarkers, facilitate drug repurposing efforts, and enable personalized medicine approaches. In particular, network analysis can identify potential drug targets by constructing biological networks that integrate various data sources, such as protein-protein interactions, gene expression data, and pathway information. Furthermore, by analyzing the network topology and identifying key nodes or modules, it is possible to pinpoint genes or proteins that play crucial roles in disease pathways or drug response [8,9,10]. This information can guide the development of targeted therapies. Also, the network analysis can aid in the discovery of genetic biomarkers that predict drug response or adverse reactions. By integrating genomic and clinical data, researchers can construct networks that capture the relationships between genetic variations, clinical phenotypes, and drug response [11]. Network-based approaches can identify modules or subnetworks that are highly associated with specific drug responses, enabling the discovery of potential biomarkers for personalized medicine. Also, network analysis can uncover the interconnected pathways and biological processes affected by genetic variations or drug treatments. By mapping genetic variants onto biological networks, pathways that are significantly enriched for these variants can be identify [12]. This knowledge helps in understanding the molecular mechanisms underlying drug response and identifying potential targets for intervention.
By analyzing the interactions between drugs, genes, and diseases in a network context, potential off-target effects or repurpose drugs for different indications can be revealed [13]. Finally, network analysis can contribute to personalized medicine approaches by integrating patient-specific genetic and clinical data into networks.
Recently, the need to investigate more complicate frameworks than the classical networks leads to introduction of multilayer approach as extension of graph theory. The reason is that many real networks can not be exhaustively explained with a classical network approach, but need more complex structures [14,15,16]. The introduction of multilayer networks provides a more comprehensive and realistic representation of complex systems where multiple types of relationships coexist. It allows to analyze and understand the dynamics and behavior of interconnected entities in a more nuanced manner [17].
Multilayer network analysis enables the study of various properties and phenomena that are not easily captured by traditional network analysis approaches. It allows for the examination of interdependencies, correlations, and patterns that emerge across different layers. This can provide insights into how different layers influence each other, the resilience of the system, the spread of information or diseases, and the identification of key nodes or communities in the network.
The work aims to model omics, diseases and drugs data together through the multilayer network representation. Multilayer network representation enables the identification of essential genes from genes-diseases-drug communities. The identified genes are detached from their biological context, making impossible to know in which biological mechanisms and functions they are involved. To understand which biological mechanisms are affected from these communities of essential genes, it is mandatory to link each gene to the opportune biological reference context by means of the pathway enrichment analysis (PEA).
For this aim, we built a biological multilayer network comprising genes, drugs, diseases and their associations. Then, we analyzed the network by applying a community detection algorithm and we selected the top communities. After that, we used the identified list of genes from the communities to perform pathway enrichment analysis (PEA) to figure out the biological function affected form the selected genes. PEA links genes and groups of gene to the influenced biological pathways responsible to the disease development, the adverse drugs reactions as well as the different overall survival of patients treated with the same drugs. Thus, the new knowledge allow to develop new treatments more effective than the drug repositioning strategies, as well as to realize more adequate drugs reducing or even better eliminating the onset of possible adverse drug reactions.
2. Background on Multilayer Networks
Multilayer networks have emerged as a powerful framework for modelling and analysing complex systems with multiple types of interactions. Unlike classical networks, which only consider one type of relationship between nodes, multilayer networks present the interdependencies between the entities of a system and the interacting layers, see Figure 1 for complete example [18]. Formally, a multilayer network can be traced back to a set of nodes, edges and layers that take into account the physical and functional relationships between them [17,19].
In particular, each layer in a multilayer network represents a specific aspect or type of relationship between nodes. For example, in a social network, different layers can represent friendships, professional connections, or family relationships. Each layer can have its own set of nodes and edges, and there can be connections between nodes across different layers.
These networks provide a realistic representation of complex real-world systems, finding their way into practical applications in various domains, such as social network analysis, transport organisation, biological systems and technological networks [16,20]. Formally, a multilayer network graph may be described as a tuple
=, where is a set of layer. For each layer k we have a graph (intra layer edges), and for each pair of layer, k, h we have a set of edges (interlayer edges) connecting nodes of the layers v, and k [21].
Examples of multilayer networks come from many different fields, from social network analysis to biological networks. For instance, Figure 1 represents an example of biological multilayer network representing the interplay among diseases, genes and drugs. Multilayer networks provide a powerful tool for analyzing complex genetic and clinical data in pharmacogenomics, enabling better predictions of drug response, identification of drug targets, and acceleration of drug discovery and development processes. Multilayer networks can be used for general data analysis in pharmacogenomics. They can integrate and analyze large-scale genomic, transcriptomic, and proteomic data to uncover hidden relationships between genes, proteins, and drug responses. This can lead to a better understanding of the underlying mechanisms of drug response and aid in the development of personalized treatment strategies.
Furthermore, multilayer networks can be used to predict how individuals will respond to specific drugs based on their genetic information. By training the network on a dataset of patients’ genetic profiles and corresponding drug responses, it can learn complex patterns and make predictions for new patients. This can help identify individuals who are likely to experience adverse drug reactions or those who are more likely to respond positively to a particular medication. Multilayer networks can help identify genetic markers associated with the risk of adverse drug reactions (ADRs). By integrating genetic and clinical data, the network can identify patterns that link specific genetic variations to ADRs. This information can be used to develop personalized medicine approaches, where patients at higher risk of ADRs can be identified and alternative treatment options can be explored. Multilayer networks can aid in the identification of potential drug targets by analyzing genetic data and can assist in drug discovery and repurposing efforts by analyzing genetic data and identifying potential drug candidates. By integrating information from various sources such as gene expression, protein-protein interactions, and biological pathways, the network can identify key genes or proteins that play a crucial role in disease development or drug response.
3. Result and Discussion
3.1. Case Study
We considered the following datasets of the Stanford Biomedical Network Dataset Collection (BioSNAP) [22]:
- Drug-Drug Interaction (DrDrI) network of interactions between drugs, approved by the U.S. Food and Drug Administration (FDA): 1514 nodes and 48514 edges.
- Disease-Disease (DD) network of interaction between inherited: 6878 nodes and 6877 edges.
- Gene-Gene (GG) network of interaction between inherited: 25825 nodes and 208836746 edges.
- Disease-Drug Association (DDrA) network, a set of curated relationships between diseases and drugs: 5535 disease nodes, 1662 drug nodes, and 466656 edges.
- Gene-Disease (GDA) Association network, a set of relationships between genes and disease: 7294 genes nodes, 519 disease nodes, and 21357 edges.
- Gene-Drug Interaction network (GDrI) network, a set of relationships between genes and drugs: 3648 genes nodes, 284 drug nodes, and 18690 edges.
We build a multilayer network with three layers obtained from the DDI, DDr and GG databases. Then, we add interlayer edges by considering the DDrA, GDA, GDrI databases. Finally, the resulting multilayer network, that, for convenience, we called GDD multilayer network, consisted of 52640 nodes and 208892137 interaction, of which 506703 inter-edges.
3.2. Community Detection on GDD multilayer network
Once built, we analyze GDD multilayer network by applying one of most useful exploratory technique, for the analysis networks, i.e. community detection. Community detection is considered a first step in understanding network analysis and community structures, defined as groups of nodes that are more densely connected than the rest of the network, represent significant characteristics for understanding the functionalities and organisations of complex systems modelled as networks. Thus, Community extraction provides the identification of densely connected nodes within multilayer networks, that play significant roles in the structure–function relationship. For this study, we selected Infomap [23] because according to literature, it outperform other community detection methods for multilayer networks. [24].
Then, we applied Infomap on GDD multilayer network obtaining 153 communities. Infomap extracted three typologies: i) communities containing genes, diseases and drugs, ii) communities containing diseases and drugs, iii) communities containing genes. For our aim, we focus on the first typology of communities containing genes, diseases and drugs. Then, we selected the top 10 communities, i.e. the communities in comprising interlayer relations, for example gene-drug and gene-disease relations.
3.3. Pathway enrichment analysis
Pathway enrichment analysis (PEA) helps researchers to comprehend the biological meaning of gene lists obtained from high-throughput experiments, such as RNA sequencing, genome-wide association studies, or proteomics. These experiments identify genes, including proteins and metabolites, that differ between the condition of interest. However, this gene list alone is insufficient for understanding the biological differences between these conditions. Therefore, PEA assists researchers in interpreting large gene lists and developing hypotheses about the underlying biology [25]. To identify the biological mechanisms and/or functions affected from the identified communities of genes. In particular, we describe the enrichment performed using the communities with identifier 10 by means of the software tool BiopaxParser (BiP) [26].
Table 1 reports the enriched pathways using the list of proteins belonging to community 10.
Next, we used BiP to know for each input gene which are the influenced pathways. Table 2 presents the relation between gene and affected pathways. Inside the community 10 a total of 36 genes are member of layer 5, e.g., disease gene layer, and layer 6, e.g., drug gene layer, revealing the multiple role of a gene through different pathways.
Analysing Table 2, it is worthy to note that the activity of metabolism of proteins pathway a well known pathway related to the adverse or normal drugs response as well as to the disease progression or decline, is regulated from the interactions of more multilayer genes P43088, P15170, P18509, P05546, Q9Y277, Q02817, Q13285, O75976 and P15328, contributing to reinforce the benefits of the multilayer formalism to represent complex networks.
3.4. Discussion
Pharmacogenomics is a complex field where the drug response of the living organism is due to the interactions of several different biological entities like genes, enzymes, and small and large molecules that cooperate in a synchronized fashion to accomplish the task. Multilayer network representation allows for more comprehensive and realistic modeling of these heterogeneous interactions than traditional ones. In addition, multilayer networks enable the identification of multilayer communities that are a bunch of genes more densely connected among them and the correlations through the different layers, information that can be used to perform PEA to comprehend the affected underlying biological mechanisms.
Performing PEA using the detected gene communities from layers 10 enriches several biological pathways as reported in Table 1. Analyzing Table 1 content is worth noting that the enriched pathways present multiple intertwinements among them, some of which are more explicit than others. The link between olfactory and leishmania is reported in several scientific publications and in particular in [27], where the authors describe the spatial and/or olfactory memory in sandflies in an endemic area for American cutaneous leishmaniasis.
Table 2 clearly displays the association between genes and pathways, emphasizing that a single gene can be involved in multiple pathways. The enrichment was calculated by implicitly incorporating topological and structural network properties, resulting in improved enrichment outcomes, as opposed to using more general genes as described in [28].
In Figure 3, community1 is represented as a network. The green nodes with red labels in the network correspond to the genes listed in Table 2. These genes play a crucial role in the network’s connectivity and are known as hub genes in literature. For instance, if we remove O95182, P15328, and P09012, the network loses its complete connectivity. This highlights the importance of multilayer modeling and enables the selection of more relevant genes from the network for performing PEA.
Figure 2.
The figure displays a network diagram depicting the interactions among the genes that belong to community1. In the network, green nodes with red labels indicate the genes listed in Table 2.
Figure 2.
The figure displays a network diagram depicting the interactions among the genes that belong to community1. In the network, green nodes with red labels indicate the genes listed in Table 2.
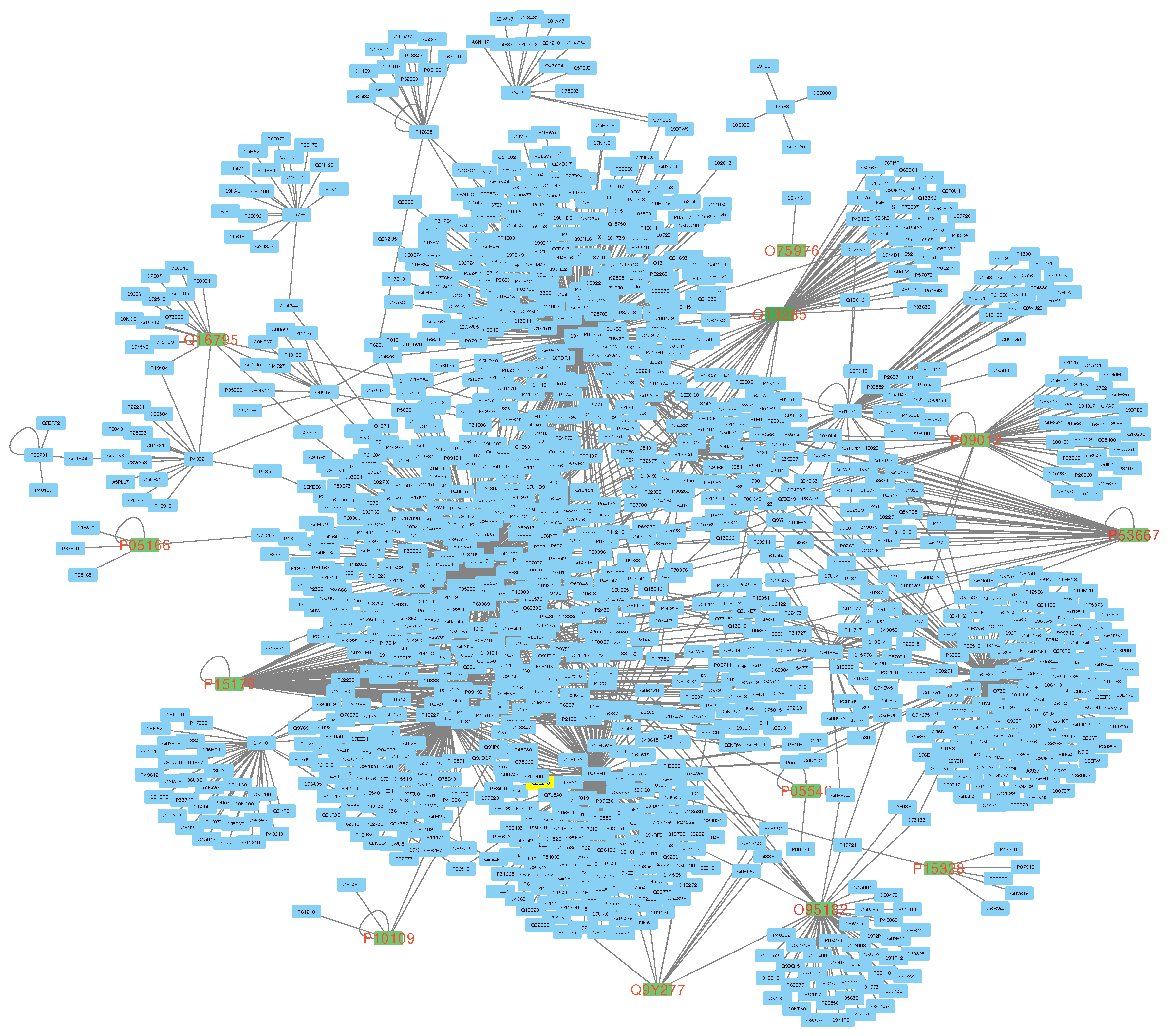
If we rely solely on a traditional network representation, we may overlook crucial information, such as the fact that gene P18509 does not belong to community1. However, through a multilayer representation, we can observe that gene P18509 interacts with P10109 as illustrated in Figure 3.
Figure 3.
The figure displays a network diagram depicting the interactions among the genes that belong to community2.
Figure 3.
The figure displays a network diagram depicting the interactions among the genes that belong to community2.
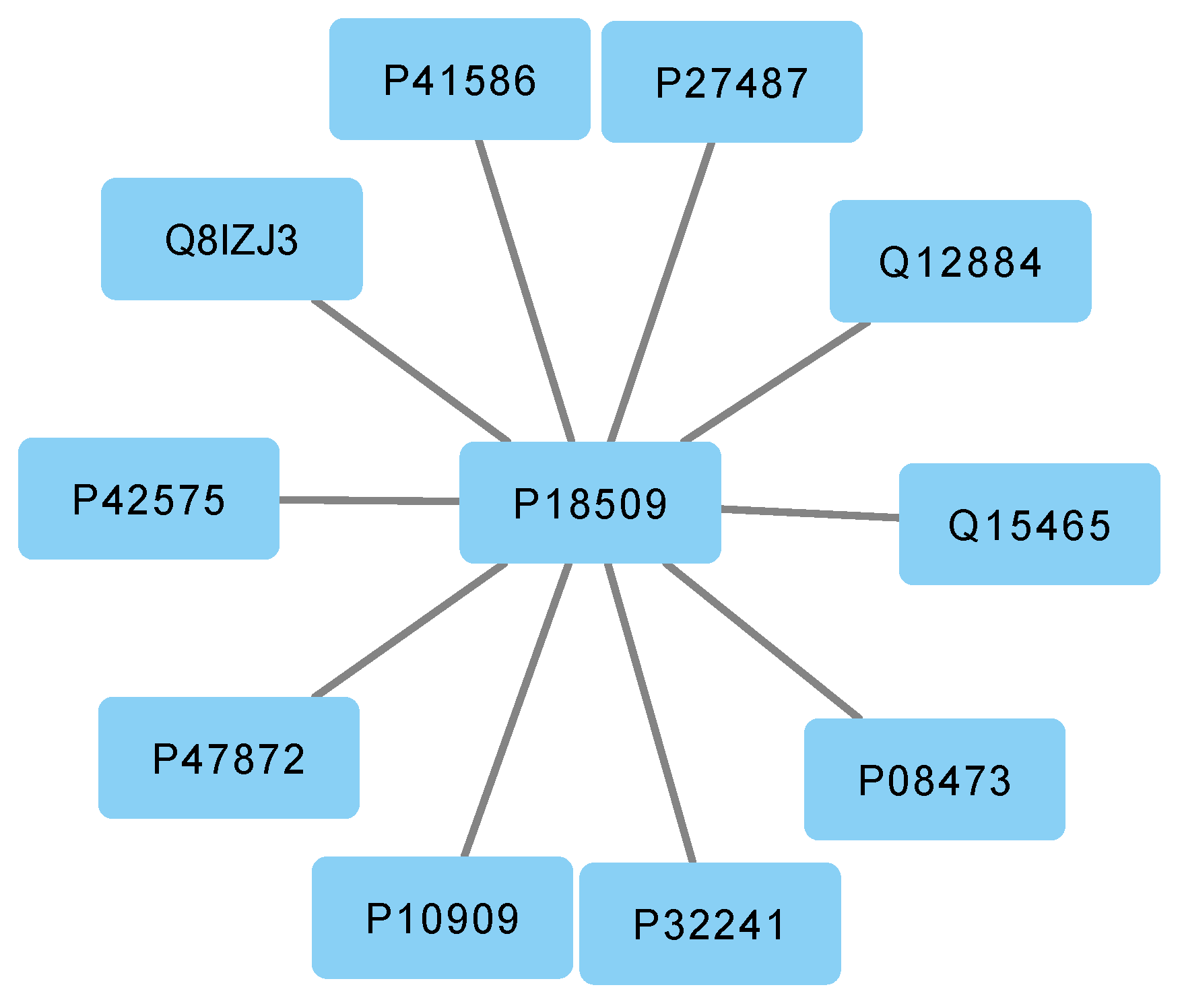
Reason for what both genes affect the same category of pathway related to the metabolism. In conclusion, a multilayer approach can help researchers capture more information and obtain a more accurate understanding of gene interactions.
4. Conclusions
In this work we explore the application of network analysis on pharmacogenomics field. In particular, we used multilayer network representation to model the interaction among genes, drugs, diseases and their associations. Then, we analyzed the network by applying a community detection algorithm in order to discover the top communities. Finally, we used the identified list of genes from the communities to perform pathway enrichment analysis (PEA) to figure out the biological function affected form the selected genes. The results demonstrate that the genes forming the communities extracted from the multilayer network regulate the activity of the protein metabolism pathway related to adverse or normal drug response as well as progression or decline of disease, demonstrating the advantages of the multilayer formalism to represent pharmacogenomics domains.
Author Contributions
Conceptualization, MM and GA.; methodology, MM and GA.; software, MM and GA. ; data curation, MM and GA.; writing—original draft preparation, MM, GA and MC.; writing—review and editing, MM, GA. and MC.; funding acquisition, MC. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Next Generation EU - Italian NRRP, Mission 4, Component 2, Investment 1.5, call for the creation and strengthening of ’Innovation Ecosystems’, building ’Territorial R&D Leaders’ (Directorial Decree n. 2021/3277) - project Tech4You - Technologies for climate change adaptation and quality of life improvement, n. ECS0000009. This work reflects only the authors’ views and opinions, neither the Ministry for University and Research nor the European Commission can be considered responsible for them.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PPI | Protein-Protein Interaction |
| ROI | Regions Of Interest |
| ADRs | Adverse Drug Reactions |
| PEA | Pathway Enrichment Analysis |
| GDD | Gene-Drug-Disease |
| BiP | BiopaxParser |
References
- Milano, M.; Cannataro, M. Network models in bioinformatics: modeling and analysis for complex diseases, 2023. [CrossRef]
- Fortunato, S.; Hric, D. Community detection in networks: A user guide. Physics reports 2016, 659, 1–44. [CrossRef]
- Lancichinetti, A.; Fortunato, S. Consensus clustering in complex networks. Scientific reports 2012, 2, 1–7. [CrossRef]
- Gligorijević, V.; Panagakis, Y.; Zafeiriou, S. Fusion and community detection in multi-layer graphs. 2016 23rd International Conference on Pattern Recognition (ICPR). IEEE, 2016, pp. 1327–1332. [CrossRef]
- Wang, Z.; Li, Z.; Yuan, G.; Sun, Y.; Rui, X.; Xiang, X. Tracking the evolution of overlapping communities in dynamic social networks. Knowledge-Based Systems 2018, 157, 81–97. [CrossRef]
- Zhou, Y.; Lauschke, V.M. Pharmacogenomic network analysis of the gene-drug interaction landscape underlying drug disposition. Computational and Structural Biotechnology Journal 2020, 18, 52–58. [CrossRef]
- Shiota, M.; Kusakabe, H.; Hikita, Y.; Nakao, T.; Izumi, Y.; Iwao, H. Pharmacogenomics of cardiovascular pharmacology: molecular network analysis in pleiotropic effects of statin—an experimental elucidation of the pharmacologic action from protein-protein interaction analysis. Journal of pharmacological sciences 2008, 107, 15–19. [CrossRef]
- Kushwaha, S.K.; Shakya, M. Protein interaction network analysis—approach for potential drug target identification in Mycobacterium tuberculosis. Journal of theoretical biology 2010, 262, 284–294. [CrossRef]
- Hasan, S.; Bonde, B.K.; Buchan, N.S.; Hall, M.D. Network analysis has diverse roles in drug discovery. Drug discovery today 2012, 17, 869–874. [CrossRef]
- Iorio, F.; Isacchi, A.; di Bernardo, D.; Brunetti-Pierri, N. Identification of small molecules enhancing autophagic function from drug network analysis. Autophagy 2010, 6, 1204–1205. [CrossRef]
- Giacomini, K.M.; Brett, C.M.; Altman, R.B.; Benowitz, N.; Dolan, M.; Flockhart, D.; Johnson, J.; Hayes, D.; Klein, T.; Krauss, R.; others. The pharmacogenetics research network: from SNP discovery to clinical drug response. Clinical Pharmacology & Therapeutics 2007, 81, 328–345. [CrossRef]
- Milano, M.; Agapito, G.; Cannataro, M. Challenges and limitations of biological network analysis. BioTech 2022, 11, 24. [CrossRef]
- Lotfi Shahreza, M.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J.R. A review of network-based approaches to drug repositioning. Briefings in bioinformatics 2018, 19, 878–892. [CrossRef]
- Boccaletti, S.; Bianconi, G.; Criado, R.; Del Genio, C.I.; Gómez-Gardenes, J.; Romance, M.; Sendina-Nadal, I.; Wang, Z.; Zanin, M. The structure and dynamics of multilayer networks. Physics reports 2014, 544, 1–122. [CrossRef]
- Milano, M.; Milenković, T.; Cannataro, M.; Guzzi, P.H. L-HetNetAligner: A novel algorithm for Local Alignment of Heterogeneous Biological Networks. Scientific Reports 2020, 10. [CrossRef]
- Milano, M.; Guzzi, P.H.; Cannataro, M. Design and Implementation of a New Local Alignment Algorithm for Multilayer Networks. Entropy 2022, 24. [CrossRef]
- Kinsley, A.C.; Rossi, G.; Silk, M.J.; VanderWaal, K. Multilayer and Multiplex Networks: An Introduction to Their Use in Veterinary Epidemiology. Front Vet Sci 2020, 7, 596. [CrossRef]
- Hammoud, Z.; Kramer, F. Multilayer networks: aspects, implementations, and application in biomedicine. Big Data Analytics 2020, 5. [CrossRef]
- Finn, K.R.; Silk, M.J.; Porter, M.A.; Pinter-Wollman, N. The use of multilayer network analysis in animal behaviour. Animal behaviour 2019, 149, 7–22. [CrossRef]
- Kumar, T.; Sethuraman, R.; Mitra, S.; Ravindran, B.; Narayanan, M. MultiCens: Multilayer network centrality measures to uncover molecular mediators of tissue-tissue communication. PLOS Computational Biology 2023, 19, e1011022. [CrossRef]
- Kivelä, M.; Arenas, A.; Barthelemy, M.; Gleeson, J.P.; Moreno, Y.; Porter, M.A. Multilayer networks. Journal of complex networks 2014, 2, 203–271. [CrossRef]
- Zitnik, M.; Sosic, R.; Leskovec, J. BioSNAP Datasets: Stanford biomedical network dataset collection. Note: http://snap. stanford. edu/biodata Cited by 2018, 5.
- De Domenico, M.; Lancichinetti, A.; Arenas, A.; Rosvall, M. Identifying modular flows on multilayer networks reveals highly overlapping organization in interconnected systems. Physical Review X 2015, 5, 011027. [CrossRef]
- Magnani, M.; Hanteer, O.; Interdonato, R.; Rossi, L.; Tagarelli, A. Community detection in multiplex networks. ACM Computing Surveys (CSUR) 2021, 54, 1–35. [CrossRef]
- Chicco, D.; Agapito, G. Nine quick tips for pathway enrichment analysis. PLoS computational biology 2022, 18, e1010348. [CrossRef]
- Agapito, G.; Pastrello, C.; Guzzi, P.H.; Jurisica, I.; Cannataro, M. BioPAX-Parser: parsing and enrichment analysis of BioPAX pathways. Bioinformatics 2020, 36, 4377–4378. [CrossRef]
- de Freitas, J.S.; Reinhold-Castro, K.R.; Casanova, C.; da Silva, J.P.; Previdelli, I.; Teodoro, U. Spatial and/or olfactory memory in sandflies in an endemic area for American cutaneous leishmaniasis, southern Brazil. Revista da Sociedade Brasileira de Medicina Tropical 2009, 42. [CrossRef]
- Agapito, G.; Cannataro, M. Using BioPAX-Parser (BiP) to enrich lists of genes or proteins with pathway data. BMC bioinformatics 2021, 22, 1–35. [CrossRef]
Figure 1.
The figure shows two toy examples of a classical biological networks (a) and a multilayer network (b). The first example (a) reports three different networks, i.e. gene-gene interaction network, disease-disease interaction network and drug-drug interaction network. In the second example each of those networks represent a distinct layer in the multilayer network. The nodes of multilayer network are the genesm the diseases and the drugs, both discriminated by belonging to the respective layer. The intra-edges represent the gene-gene, drug-drug and the disease-disease associations, while the inter-edges are the gene-disease, gene-drug and disease-drug associations.
Figure 1.
The figure shows two toy examples of a classical biological networks (a) and a multilayer network (b). The first example (a) reports three different networks, i.e. gene-gene interaction network, disease-disease interaction network and drug-drug interaction network. In the second example each of those networks represent a distinct layer in the multilayer network. The nodes of multilayer network are the genesm the diseases and the drugs, both discriminated by belonging to the respective layer. The intra-edges represent the gene-gene, drug-drug and the disease-disease associations, while the inter-edges are the gene-disease, gene-drug and disease-drug associations.
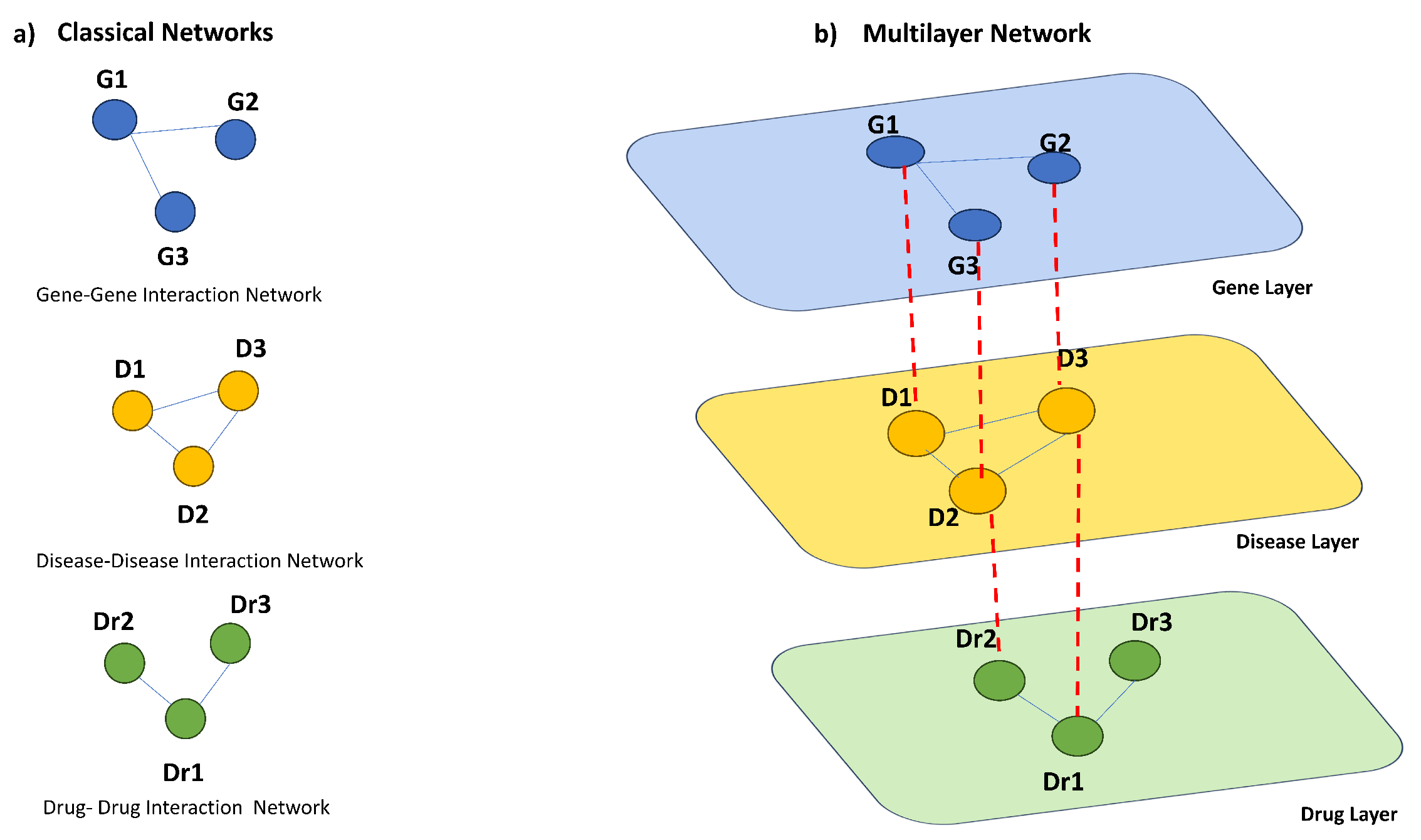

Table 1.
The first 10 enriched pathways, in order of statistical relevance, obtained using the gene list of community 10 as input data.
Table 1.
The first 10 enriched pathways, in order of statistical relevance, obtained using the gene list of community 10 as input data.
| PathwayName | Pvalue | FDRCorrection | BonferroniCorrection | |
| 1) | Olfactory Signaling Pathway | 1.25E-07 | 1.54E-04 | 1.54E-04 |
| 2) | Metabolism of proteins | 1.06E-05 | 0.007 | 0.013 |
| 3) | Post-translational protein modification | 1.16E-05 | 0.005 | 0.014 |
| 4) | Leishmania parasite growth and survival | 6.71E-05 | 0.021 | 0.082 |
| 5) | Anti-inflammatory response favouring Leishmania parasite infection | 6.71E-05 | 0.016 | 0.082 |
| 6) | Signaling by Rho GTPases, Miro GTPases and RHOBTB3 | 6.83E-05 | 0.014 | 0.084 |
| 7) | Signaling by Rho GTPases | 7.20E-05 | 0.013 | 0.089 |
| 8) | TCF dependent signaling in response to WNT | 9.41E-05 | 0.014 | 0.116 |
| 9) | Signaling by WNT | 1.17E-04 | 0.016 | 0.143 |
| 10) | Transcriptional regulation by RUNX1 | 1.20E-04 | 0.015 | 0.148 |
Table 2.
The 20 multi layer genes and the affected biological pathways.
| Gene | Pathways |
| O43739 | Vesicle-mediated transport; Membrane Trafficking; Intra-Golgi and retrograde Golgi-to-ER traffic; |
| P15170 | Translation; Metabolism of RNA; Metabolism of proteins; |
| Q16795 | Respiratory electron transport; Respiratory electron transport, ATP synthesis by chemiosmotic coupling, and heat production by uncoupling proteins; Complex I biogenesis; |
| P18509 | Signaling Pathways; Metabolism of proteins; |
| P05546 | Hemostasis; Metabolism of proteins; Post-translational protein modification |
| P09012 | mRNA Splicing; pre-mRNA splicing; Metabolism of RNA; Processing of Capped Intron-Containing Pre-mRNA |
| Q9Y277 | Metabolism of proteins; Post-translational protein modification. |
| P10109 | Diseases of metabolism; Disease |
| Q02817 | Disease; Diseases of metabolism; Defective C1GALT1C1 causes TNPS; Termination of O-glycan biosynthesis; O-linked glycosylation; O-linked glycosylation of mucins; Metabolism of proteins; Post-translational protein modification |
| Q13285 | Post-translational protein modification; Gene expression (Transcription); Metabolism of proteins; |
| O95182 | Respiratory electron transport; Respiratory electron transport, ATP synthesis by chemiosmotic coupling, and heat production by uncoupling proteins; Complex I biogenesis; |
| P53667 | Thrombin signalling through proteinase activated receptors (PARs); GPVI-mediated activation cascade; G alpha (12/13) signalling events; Signaling Pathways; Disease; Signaling by Rho GTPases, Miro GTPases and RHOBTB3 |
| O43677 | Respiratory electron transport; Respiratory electron transport, ATP synthesis by chemiosmotic coupling, and heat production by uncoupling proteins; Complex I biogenesis; |
| O75976 | Signaling Pathways; Metabolism of proteins; Signaling by Rho GTPases, Miro GTPases and RHOBTB3; Signaling by Rho GTPases; RHO GTPase cycle; |
| P05166 | Metabolism of vitamins and cofactors; Biotin transport and metabolism; |
| P15328 | Metabolism of proteins; Post-translational protein modification; Vesicle-mediated transport; Membrane Trafficking; Asparagine N-linked glycosylation; Transport to the Golgi and subsequent modification; ER to Golgi Anterograde Transport; COPI-mediated anterograde transport; |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



