
Preprint
Article
Comparative Analysis of the Complete Chloroplast Genomes of Six Endangered Cycas Species: Genomic Features, Comparative Analysis, and Phylogenetic Implications
Altmetrics
Downloads
112
Views
27
Comments
0
A peer-reviewed article of this preprint also exists.

This version is not peer-reviewed
Submitted:
06 September 2023
Posted:
07 September 2023
You are already at the latest version
Alerts
Abstract
Cycas (family Cycadaceae), spread throughout tropical and subtropical regions, is crucial in con-servation biology. Due to the subtle morphological variations between species, a solid spe-cies-level phylogeny for Cycas is lacking. Because of the rapid progress in high-throughput se-quencing technology, it has become feasible to acquire complete chloroplast (cp) genome se-quences, which provide a molecular foundation for phylogenetic research. In the present study, we employed next-generation sequencing technology to assemble and analyze the chloroplast genomes of six Cycas plants, including their genome structure, GC content, and nucleotide di-versity. The Cycas chloroplast genome spans 162,038 to 162,159 bp and contains 131 genes, in-cluding 86 protein-coding genes, 37 transfer RNA (tRNA) genes, and 8 ribosomal RNA (rRNA) genes. Through a comparative analysis, we found that the chloroplast genome of Cycas was highly conserved, as indicated by the contraction and expansion of the inverted repeat (IR) re-gions and sequence polymorphisms. In addition, several non-coding sites (psbK-psbI, petN-psbM, trnE-UUC-psbD, ndhC-trnM-CAU, and rpl32-trnP-GGG) showed significant varia-tion. The utilization of phylogenetic analysis relying on protein-coding genes has substantiated the division of Cycas primarily into four groups. The application of these findings will prove valuable in evaluating genetic diversity and the phylogenetic connections among closely related species. Moreover, it will provide essential support for the advancement of wild germplasm re-sources.
Keywords:
Subject: Biology and Life Sciences - Biochemistry and Molecular Biology
1. Introduction
Cycads are one of the oldest families of living seed plants, dating back to the Late Permian period [1]. More than 360 species of cycads exist, divided into 10 genera and two families, Cycadaceae and Zemitaceae [2]. These plants are found in patches throughout tropical and subtropical regions of Asia, Africa, Oceania, and America [3]. From a morphological perspective, the reproductive systems of cycads are most similar to those of spore plants, making cycads very important in studying the genesis and early evolution of seed plants. From the standpoint of their phylogenetic origin and evolution, cycads have existed and reproduced for at least 280 million years [4], suffered significant environmental changes, and contain a wealth of genetic data. In terms of species protection, cycads have persisted and flourished until the present, although many plant species have perished due to changes in the earth's environment. Therefore, studying the mechanisms underlying the environmental adaptability of cycads is critical in conservation biology.
There is only one genus of Cycadaceae, Cycas, which contains roughly 120 species [4]. This genus contains a large number of species, involves a complicated taxonomy, and covers a wide geographical range. Cycas are thought to have originated in Tertiary East Asia according to recent biogeographic research [5]. In China, there are roughly 20 different species of Cycas, mostly found in the southwest and southeast coasts [6].
There have been some challenges and disagreements in the categorization of Cycas species for the following reasons. First, it is challenging to properly analyze the physical traits of Cycas. Since these species take a long time to reach sexual maturity, there are few flowering plants to be discovered. When combined with variable environments, populations of the same Cycas species can also differ significantly. Additionally, natural hybridization may exist between different species [7]. Hill proposed dividing Cycas into six groups based on long-term observations of cycad plants in the field and integrating reproductive characteristics such as the ovule coat, megaspore leaf shape, and anatomical structure of the seeds: Section Asiorientales, Section Strangeriodes, Section Indosinenses, Section Cycas, Section Panzhihuaenses, and Section Wadeae [8,9]. Recently, Zheng et al. proposed 23 species from four sections in China based on distribution and morphological characteristics [6]. Traditional taxonomy, however, has limited ability to establish species boundaries due to small morphological variations between species induced by either environmental or genetic causes.
The strategy of integrating DNA identification and morphological features was also employed to define Cycas species, and more accurate findings were achieved. Using molecular sequencing techniques, Xiao and Möller performed a phylogenetic study of 31 Cycas species using the nrDNA ITS gene [10]. The chloroplast (cp) genome presents substantial advantages for investigations in the field of plant evolutionary biology due to its genetic stability, well-preserved genome structure, and rate of evolutionary change that outpaces that of mitochondria [11]. Liu et al. used four chloroplast genes and three nuclear genes to conduct a phylogenetic analysis of 104 species and 5 subspecies in the genus Cycas [12]. Nonetheless, the exploration of genomic resources within this genus has remained relatively limited, as evidenced by the small number of studies conducted [13-15]. In GenBank database, the collection of complete cp genome sequences for Cycas species is currently limited to approximately 10 entries.
In this study, the cp genomes of six Cycas species, C. longlingensis, C. longisporophylla, C. guizhouensis, C. ferruginea, C. crassipes, and C. bifiba, were sequenced. Our study encompassed a thorough exploration of the cp genome, encompassing detailed descriptions of its assembly and annotations and the identification of simple sequence repeats (SSRs). Furthermore, we performed phylogenetic analyses of Cycas species based on the coding genes from cp genome sequences, incorporating both our newly sequenced species and those previously published. These findings complement current genetic information on Cycas species and serve as a good reference for Cycas DNA molecular research.
2. Materials and Methods
2.1. Plant Materials and DNA Extraction.
Fresh leaves of six different Cycas species, C. longlingensis, C. longisporophylla, C. guizhouensis, C. ferruginea, C. crassipes, and C. bifiba, were taken from the Guilin Botanical Garden (Guangxi, China; coordinates: N 25°4’14.88’’, E 110°17’57’’) and immediately placed in liquid nitrogen. The total genomic DNA was extracted from fresh leaves (> 1.0 g) using a Magnetic Plant Genomic DNA Kit (TIANGEN Biotech, Beijing, China) following the manufacturer’s instructions. The quality of DNA was evaluated utilizing a TBS-380 Mini-Fluorometer (Invitrogen) and electrophoresis on a 1% agarose gel.
2.2. Chloroplast Genome Sequencing and Assembling.
A total of 1 µg DNA was utilized as an input material for library construction. Using the VAHTS Universal Plus DNA Library Prep Kit for Illumina (Vazyme, Jiangsu, China), we crafted sequencing libraries in accordance with the manufacturer's guidelines while applying index codes to individual sample sequences. Briefly, the DNA sample underwent initial fragmentation into 300-500 bp segments through sonication. Subsequently, the preexisting DNA fragments underwent end-polishing and A-tailing, followed by ligation with full-length adaptors for sequencing. Polymerase chain reaction (PCR) amplification was then performed utilizing a cBot Truseq PE Cluster Kit v3-cBot-HS (Illumina). Lastly, PCR products underwent purification using an AMPure XP system (Beckman Coulter Inc., Brea, CA, USA), their library size distribution was determined using an Agilent 2100 Bioanalyzer, and quantification was carried out via real-time PCR. Using a cBot Cluster Generation System (Illumina Inc.), the indexed samples underwent clustering according to the manufacturer's protocols. Following clustering, the resulting libraries underwent sequencing on an Illumina Novaseq 6000 platform, generating reads with a length of 150 bp in the paired-end configuration.
The raw paired-end reads were subjected to quality assessment using the FastQC v0.11.7 software. Subsequent to quality evaluation, the obtained data were processed through de novo assemblers (Fast-plast, https://github.com/mrmckain/Fast-Plast or GetOrganelle, https://github.com/Kinggerm/GetOrganelle) to generate optimal contigs. The cp sequence of C. szechuanensis (MH341576) was retrieved from GenBank and employed as the reference seed sequence for C. longlingensis, C. longisporophylla, C. ferruginea, C. crassipes, and C. bifiba. Additionally, the cp sequence of C. bifida (MW900434) was utilized as the seed sequence for C. guizhouensis.
Subsequently, the chloroplast (cp) genomes underwent annotation using the PGA software (available at https://github.com/quxiaojian/PGA) and the Geseq software (accessible via https://chlorobox.mpimp-golm.mpg.de/geseq.html) with default settings, followed by manual corrections. The resulting gene maps were visualized using the online tool OGDraw v1.2 [16]. Six newly sequenced complete cp genomes were deposited to GenBank with the following Accession Numbers: Accession Nos. C. bifiba (OQ862764), C. crassipes (OQ862765), C. ferruginea (OQ862766), C. guizhouensis (OQ862767), C. longisporophylla (OQ862768), and C. longlingensis (OQ862769).
2.3. Repeat Sequences and SSRs.
The cp genome sequences of C. szechuanensis (NC_064393.1), C. shiwandashanica (NC_064393.1), and C. segmentifida (NC_064393.1) downloaded from GenBank were coupled with six newly-sequenced cp genomes of Cycas to conduct an analysis of repeat sequences and simple sequence repeats (SSRs). A Perl script called MISA was employed to detect SSRs within the complete cp genome sequences of the nine Cycas species. The thresholds applied for varying lengths of SSRs—mononucleotides, dinucleotides, trinucleotides, tetranucleotides, pentanucleotides, and hexanucleotides—were set to 10, 6, 5, 5, 5, and 5, respectively. Furthermore, the REPuter program was used to identify four categories of repeat sequences: palindromic, forward, reverse, and complement repeats. The recognition of repeat sequences adhered to the following criteria: (1) A Hamming distance of 3; (2) a minimum size of 20 bp; and (3) a sequence identity equal to or greater than 90%.
2.4. Variations and Divergence Hotspot Regions of the cp Genomes.
The mVISTA comparative genomics server was utilized to generate a sequence variation map with annotation of the C. bifiba cp genome as a reference. Evaluation of IR sequence variations, encompassing features such as expansion and contraction, was conducted through the IRscope online program (https://irscope.shinyapps.io/irapp/). To identify intergeneric divergence hotspots, sliding window analysis was carried out using the DnaSP v5.10 software [17]. This analysis involved a window length of 600 bp and a step size of 200 bp.
2.5. Phylogenomic Reconstruction Based on cp Genomes.
We conducted a phylogenomic analysis utilizing six newly sequenced cp genomes of Cycas species. Additionally, we included eight Cycas species sourced from GenBank in our analysis. These sequences were employed to construct a phylogenetic tree using the maximum-likelihood (ML) method, with Encephalartos lehmannii and Bowenia serrulata defined as outgroups. To reconstruct ML trees, we extracted 86 protein-coding genes from the 19 species. Multiple sequence alignment was accomplished using MAFFT [18], and selection of the GTR-GAMMA (GTR + G) model [19] was guided by a model test applying the Bayesian information criterion (BIC) [20]. The MEGA-X software facilitated the execution of maximum likelihood (ML) trees, with 1,000 bootstrap replicates configured to assess the branch support values. Visualization of the resulting phylogenetic tree was carried out using FigTree v1.4.4.
3. Results
3.1. Chloroplast Genome Structure
The chloroplast genome of Cycas, ranging in size from approximately 162,038 to 162,159 bp, is generally similar to the structure of other seed plant chloroplast genomes, with a quadripartite structure consisting of a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeat (IR) regions (Figure 1). The length of the LSC region was distributed between 88,819 and 88,946 bp (Table 1). The GC content of the LSC regions was similar in six species, ranging from 38.70% in C. bifida to 38.73% in C. longisporophylla. The length of the SSC region ranged between 23,102 and 23,124 bp, with GC content of 36.52–36.55%. The length range for the IR regions of six Cycas species was 25,049–25,060 bp, which contained 42.01–42.03% GC content. The total number of genes was 131, including 86 protein-coding genes, 37 tRNAs (transfer RNA), and 8 rRNAs (ribosomal RNA) in the cp genomes of all six Cycas species. The overall organization and structure of the Cycas chloroplast genome were similar to those of other seed plant chloroplast genomes, with a conserved set of genes involved in photosynthesis and other chloroplast functions.
3.2. IR Expansion and Contraction
The IR border regions were compared according to the chloroplast genome sequences and annotation data for the six newly sequenced Cycas species, along with three other Cycas species: C. szechuanensis, C. shiwandashanica, and C. segmentifida (Figure 2). The chloroplast genome organizations were highly conserved across the nine Cycas species with only minor variations. Namely, the sizes of IR ranged between 25,003 bp and 25,060 bp across nine Cycas species. The rpl23 gene was present only in C. shiwandashanica and C. segmentifida, and the rpl2 gene was found only in C. guizhouensis and C. crassipes. With the exception of C. szechuanensis, whose trnI gene was positioned far from 43 bp in the LSC, all studied Cycas species had the junction LSC/IRb (JLB) located inside the trnI gene. The size and location of the ndhF gene was highly conserved among the nine species, with the same sizes (2219 bp) and the same location at the border of the IRb/SSC junction. The chlL gene at the SSCs was far from the border SSC/IRa (JSA) of both 398 bp and 405 bp. In addition, the psbA gene at the LSCs was far from the border IRa/LSC (JLA) in the range of 152–185 bp.
3.3. Variations and Divergence Hotspot Regions
To study the level of sequence polymorphisms, we used both mVISTA and DnaSP6 to calculate the genetic differences between nine Cycas species and compared the whole chloroplast genomes (Figure 3 and 4) with the reference sequence of C. bifiba. Overall, the protein-coding regions in relative species were highly conserved, and highly variable regions were mainly found in in intergenic spacers (IGSs) such as psbK–psbI, petN–psbM, trnE-UUC–psbD, ndhC–trnM-CAU, and rpl32–trnP-GGG. These hot spots could be applied to DNA barcode encoding and phylogenetic analysis of the Cycas genus. The nucleotide variation (Pi) of nine species was negligible, ranging from 0 to 0.0057, with an average value of 0.00104 (Supplementary Table 1). This result agreed with the subtle differences observed in the mVISTA map. The average Pi of the SSC area was 0.00149, that of the LSC area was 0.00126, and that of the IR area was 0.00052.
3.4. SSR and Large Repeats
Simple sequence repeats (SSRs) and large repeats were investigated in nine Cycas cp genomes. The number of SSRs detected was 49–54, and a total of SSR types (mono-/di-/tri-/compound-nucleotide repeats) were detected, with mononucleotide repeats accounting for 77.78–84.91%. The trinucleotide repeat was only found in C. guizhouensis and C. crassipes (Figure 5A). At the same time, the distribution of SSRs in the LSC region (76.32–84.62%) was higher than that in the SSC region (9.09–15.79%) and IR region (5.00–10.53%) (Figure 5B). The number of SSRs in the LSC of C. szechuanensis and C. bifiba (37) was the largest, while that of C. shiwandashanica and C. segmentifida (29) was the lowest.
This study counted all the interspersed repetitive sequences in nine Cycas chloroplast genomes with repeat unit lengths of more than 20 bp. At the same time, we detected four types of repeats, including forward repeats (F), inverted repeats (R), complementary repeats (C), and palindromic repeats (P). Repeat analysis showed that the total repeat number ranged from 47-49, with 10–13 forward repeats, 13–17 reverse repeats, 4–6 complementary repeats, and 15–21 palindromic repeats in nine Cycas species (Figure 5C). Interestingly, although the total number of large repeats was similar between species, the length of the repetitive sequence varied distinctly from species to species (Figure 5D). Namely, the repeat length distribution was the same in C. segmentifida, C. longlingensis, C. longisporophylla, and C.ferruginea, which contained the most abundant short repeats (21-24 bp) and no long repeats (>33 bp). By contrast, C. g and C. c exhibited no short repeats (21-24 bp) and abundant long repeats (15) greater than 33 bp. In addition, C. szechuanensis and C. bifiba presented similar repeat length distribution patterns, with the highest repeat length being 25-28 bp.
3.5. Phylogenomic Analysis
To further understand the phylogenetic status of Cycas plants and their relationship with other closely related species, the shared protein-coding genes of the chloroplast genomes in the 14 Cycas plants were used to construct a phylogenetic tree using the maximum-likelihood (ML) method and bootstrap with 1,000 iterations (Figure 6). The results of the evolutionary tree we constructed can be divided into approximately five parts: C. taiwaniana–C. szechuanensis, C. segmentifida–C. ferruginea, C. debaoensis–C. guizhouensis, C. panzhihuaensis–C. revoluta, and two outgroup species. Among the six newly-sequenced Cycas cp genomes, C. crassipes and C. guizhouensis were sister species. C. ferruginea was found to be a sister to C. longlingensis, and both were further found to be sister species to C. longisporophylla. The cp genes of C. bifiba presented a close relationship with C. szechuanensis.
4. Discussion
The chloroplast genome data provide comprehensive insights for examining plant phylogenetics and analyzing evolutionary relationships [21,22]. The abundant data encapsulated within the chloroplast genome render this genome highly suitable as a DNA barcoding tool for species identification [23]. Cycas, which is critically threatened across the world [3], has been rarely studied to record its cp genome information. The current work presented the whole cp genomes of six Cycas species, four of which (C. longlingensis, C. longisporophylla, C. guizhouensis, and C. crassipes) were reported here for the first time. Then cp genome comparison was conducted with another three species, C. szechuanensis, C. shiwandashanica, and C. segmentifida, to gain insight into the variations between the aforementioned cp genomes. Together with five other Cycas species, a phylogenetic analysis was performed based on complete cp genomes. Studying the cp genome sequences of these species can increase our biological understanding of Cycas species evolution.
In general, plastomes exhibit a high degree of conservation in terms of their genome structures, gene orders, and gene contents [24]. The structural configurations of the entire cp genomes in the six studied Cycas species closely resemble those found in the majority of higher plants [25-27]. The overall structure is characterized by four distinct regions, including an LSC region spanning from 84,839 to 85,598 bp, an SSC region ranging from 17,559 to 17,687 bp, and two IRs from 31,392 to 31,880 bp each. The comparative examination of six intact cp genomes revealed significant similarities in parameters such as genome length (165,607–167,013 bp), structure, IR/SC borders, and GC content (37.8–38.0%). In addition, the equal number of rRNA, tRNA, and coding genes indicated that the analyzed Cycas species are highly conserved. Previous reports indicated that GC content exhibits variation across distinct regions of cp genomes, with the IR regions displaying higher GC content due to the inclusion of rRNAs [25,28], which was in line with our results.
By comparing the variations in cp genome sequences between distinct taxa, it becomes possible not only to efficiently identify DNA fragments rich in information but also to foster the advancement of techniques for species identification and the exploration of population diversity [29]. mVISTA and DnaSP6 were applied to evaluate variations in the cp genomes of different Cycas species, and both methods demonstrated that Cycas cp genomes were highly conserved. The IR regions were less variable than the LSC and SSC regions, which was consistent with the findings of a prior investigation [30]. In addition, a prior report indicated higher susceptibility to mutations in non-coding regions compared to coding regions [31]. In the present study, we observed only high variable regions within IGSs, rather than coding regions, which aligns with this characteristic.
Inheritance of the cp genome is uniparental, and within a given species, there exists a notable degree of variation in SSRs [32]. Consequently, these SSRs serve as valuable molecular indicators for developmental analyses and species identification purposes [33]. Moreover, SSRs frequently find applications as genetic markers in investigations pertaining to community genetics and evolutionary research [34]. Among the repeats, those that showed the greatest enrichment were mononucleotide repeats followed by dinucleotide repeats. Overall, trinucleotide repeats were infrequent across all nine cp genomes. When conducting a comparative assessment of repeat sequences within the cp genomes, the repeat length distribution was the same in C. segmentifida, C. longlingensis, C. longisporophylla, and C. ferruginea, with an average length of 24.146 bp. In contrast, C. guizhouensis and C. crassipes exhibited much longer repeats, with an average of 30.563 bp. Notably, species with similar repeat length distributions of their cp genomes were close in the phylogenetic tree, indicating that large repeats are reliable molecular indicators in evolutionary studies.
Currently, protein-coding genes are commonly used to build phylogenetic trees [35]. The results of this study revealed the genetic relationships between Cycas plants. According to Zheng et al., C. revoluta and C. taitungensis belong to Asiorientales, while C. panzhihuaensis belongs to Panzhihuaenses [6]. Consistent with this classification, the first two species were also grouped into the same clade in our results and further into a clade with C. panzhihuaensis. Additionally, Zheng et al. discovered 18 species of Stangerioides in China [6], but the C. bifiba, C. longisporophylla, and C. longlingensis species analyzed in this study were not included in their research. Here, the phylogenetic tree indicates that these three species should belong to Stangerioides since they are grouped together in the same branch as other species that are part of this section. This categorization is justified for two reasons. Morphologically, the testa coats of this species are yellow to brown, and their microsporangiate cones and microsporophylls are soft to the touch. Geographically, these species are all found in the Guangxi area of China [36,37]. In summary, our phylogenetic analysis of Cycas species relied upon protein-coding genes, which currently constitute the most comprehensive dataset available. This endeavor not only lays theoretical groundwork in this field but also provides the requisite technical details for advancing and effectively utilizing resources derived from Cycas plants.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: The nucleotide variation (Pi) of nine Cycas species
Author Contributions
Conceptualization, J.T.; methodology, R.Z.; validation, T.C.; formal analysis, S.Z. and L.P.; investigation, T.D. (sampling), S.C. (sequencing), R.Z. (data analysis); writing—original draft preparation, J.T.; writing—review and editing, T.C.; visualization, L.P. and S.Z.; supervision, X.W.; funding acquisition, X.W. All co-authors have reviewed and approved the final version of the manuscript for publication. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Key Research and Development Program (No.2022YFF1300700), Guangxi Natural Science Foundation of China (No. 2020GXNSFAA259029),Chinese Academy of Sciences ‘Light of West China’ Program (2022), Guangxi Forestry Science and Technology Promotion Demonstration Project (2023LYKJ03 and [2022]GT23), Guangxi Key Laboratory of Plant Functional Phytochemicals Research and Sustainable Utilization Independent project (No. ZRJJ2022-2), Guilin Innovation Platform and Talent Plan (20210102-3), and Guilin City Technology Application and Promotion Plan (20220134-3).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data cited in the study are publicly available.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhifeng, G.; Thomas, B.A. A review of fossil cycad megasporophylls, with new evidence of Crossozamia Pomel and its associated leaves from the Lower Permian of Taiyuan, China. Review of Palaeobotany and Palynology 1989, 60, 205-223. [CrossRef]
- Calonje, M.; Stevenson, D.; Stanberg, L. The world list of cycads, online edition. Recuperado el 2019, 10.
- Osborne, R. The world cycad census and a proposed revision of the threatened species status for cycad taxa. Biological Conservation 1995, 71, 1-12. [CrossRef]
- Spiekermann, R.; Jasper, A.; Siegloch, A.M.; Guerra-Sommer, M.; Uhl, D. Not a lycopsid but a cycad-like plant: Iratinia australis gen. nov. et sp. nov. from the Irati Formation, Kungurian of the Paraná Basin, Brazil. Review of Palaeobotany and Palynology 2021, 289, 104415. [CrossRef]
- Liu, J.; Lindstrom, A.J.; Chen, Y.S.; Nathan, R.; Gong, X. Congruence between ocean-dispersal modelling and phylogeography explains recent evolutionary history of Cycas species with buoyant seeds. New Phytologist 2021, 232, 1863-1875. [CrossRef]
- Zheng, Y.; Liu, J.; Feng, X.; Gong, X. The distribution, diversity, and conservation status of Cycas in China. Ecology and Evolution 2017, 7, 3212-3224.
- Tao, Y.; Chen, B.; Kang, M.; Liu, Y.; Wang, J. Genome-wide evidence for complex hybridization and demographic history in a group of Cycas from China. Frontiers in Genetics 2021, 12, 717200. [CrossRef]
- Hill, K. Infrageneric relationships, phylogeny, and biogeography of the genus Cycas (Cycadaeceae). In Proceedings of the Proceedings of the Third International Conference on Cycad Biology, 1995; pp. 139-175.
- Hill, K. Character evolution, species recognition and classification concepts in the Cycadaceae. In Cycad classification: concepts and recommendations; CABI Publishing Wallingford UK: 2004; pp. 23-44.
- Xiao, L.-Q.; Möller, M. Nuclear ribosomal ITS functional paralogs resolve the phylogenetic relationships of a late-Miocene radiation cycad Cycas (Cycadaceae). PloS one 2015, 10, e0117971. [CrossRef]
- Saina, J.K.; Li, Z.-Z.; Gichira, A.W.; Liao, Y.-Y. The complete chloroplast genome sequence of tree of heaven (Ailanthus altissima (Mill.)(Sapindales: Simaroubaceae), an important pantropical tree. International journal of molecular sciences 2018, 19, 929.
- Liu, J.; Zhang, S.; Nagalingum, N.S.; Chiang, Y.-C.; Lindstrom, A.J.; Gong, X. Phylogeny of the gymnosperm genus Cycas L.(Cycadaceae) as inferred from plastid and nuclear loci based on a large-scale sampling: evolutionary relationships and taxonomical implications. Molecular Phylogenetics and Evolution 2018, 127, 87-97. [CrossRef]
- Han, J.; Wang, M.; Qiu, Q.; Guo, R. Characterization of the complete chloroplast genome of Cycas panzhihuaensis. Conservation genetics resources 2017, 9, 21-23. [CrossRef]
- Wang, Y.; Wang, Z.; Zhang, L.; Yang, X. The complete chloroplast genome of Cycas Szechuanensis, an extremely endangered species. Mitochondrial DNA Part B 2018, 3, 974-975. [CrossRef]
- Zhang, D.; Cao, Y.; Lu, Z. The complete chloroplast genome of Cycas bifida, an extremely small population protected species. Mitochondrial DNA Part B 2021, 6, 2960-2961.
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Current genetics 2007, 52, 267-274. [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451-1452. [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research 2002, 30, 3059-3066.
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nature methods 2012, 9, 772-772. [CrossRef]
- Posada, D.; Crandall, K.A. MODELTEST: testing the model of DNA substitution. Bioinformatics (Oxford, England) 1998, 14, 817-818. [CrossRef]
- Dong, W.; Xu, C.; Li, W.; Xie, X.; Lu, Y.; Liu, Y.; Jin, X.; Suo, Z. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences. Frontiers in plant science 2017, 8, 1148. [CrossRef]
- Clegg, M.T.; Zurawski, G. Chloroplast DNA and the study of plant phylogeny: present status and future prospects. Molecular systematics of plants 1992, 1-13. [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. The Plant Cell 2001, 13, 645-658.
- Ravi, V.; Khurana, J.; Tyagi, A.; Khurana, P. An update on chloroplast genomes. Plant Systematics and Evolution 2008, 271, 101-122.
- Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): comparative analyses and identification of mutational hotspots. Genomics 2020, 112, 581-591. [CrossRef]
- Tang, J.; Zou, R.; Wei, X.; Li, D. Complete Chloroplast Genome Sequences of Five Ormosia Species: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis. Horticulturae 2023, 9, 796.
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.-M.; Khan, M.A.; Lee, S.-M.; Lee, I.-J. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Frontiers in plant science 2016, 7, 843. [CrossRef]
- Shahzadi, I.; Mehmood, F.; Ali, Z.; Malik, M.S.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 2019, 19, 100199. [CrossRef]
- Song, W.; Ji, C.; Chen, Z.; Cai, H.; Wu, X.; Shi, C.; Wang, S. Comparative analysis the complete chloroplast genomes of nine Musa species: genomic features, comparative analysis, and phylogenetic implications. Frontiers in Plant Science 2022, 13, 832884. [CrossRef]
- Xie, D.-F.; Yu, Y.; Deng, Y.-Q.; Li, J.; Liu, H.-Y.; Zhou, S.-D.; He, X.-J. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. International Journal of Molecular Sciences 2018, 19, 1847. [CrossRef]
- Kelchner, S.A. The evolution of non-coding chloroplast DNA and its application in plant systematics. Annals of the Missouri Botanical Garden 2000, 482-498. [CrossRef]
- Liang, C.; Wang, L.; Lei, J.; Duan, B.; Ma, W.; Xiao, S.; Qi, H.; Wang, Z.; Liu, Y.; Shen, X. A comparative analysis of the chloroplast genomes of four Salvia medicinal plants. Engineering 2019, 5, 907-915. [CrossRef]
- Yaradua, S.S.; Alzahrani, D.A.; Albokhary, E.J.; Abba, A.; Bello, A. Complete chloroplast genome sequence of Justicia flava: genome comparative analysis and phylogenetic relationships among Acanthaceae. BioMed Research International 2019, 2019.
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Frontiers in plant science 2016, 7, 959. [CrossRef]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 2019, 8, 283. [CrossRef]
- Hill, K. The genus Cycas (Cycadaceae) in China. Telopea 2008, 12, 71-118. [CrossRef]
- Whitelock, L.M. The cycads; Timber Press: 2002.
Figure 1.
Gene map of the chloroplast genomes of six Cycas species. Genes shown outside the outer circle are transcribed clockwise, and those shown inside are transcribed counterclockwise. Genes belonging to different functional groups are color-coded. Darker gray shading in the inner circle indicates the GC content of the chloroplast genome; lighter gray corresponds to AT content.
Figure 1.
Gene map of the chloroplast genomes of six Cycas species. Genes shown outside the outer circle are transcribed clockwise, and those shown inside are transcribed counterclockwise. Genes belonging to different functional groups are color-coded. Darker gray shading in the inner circle indicates the GC content of the chloroplast genome; lighter gray corresponds to AT content.
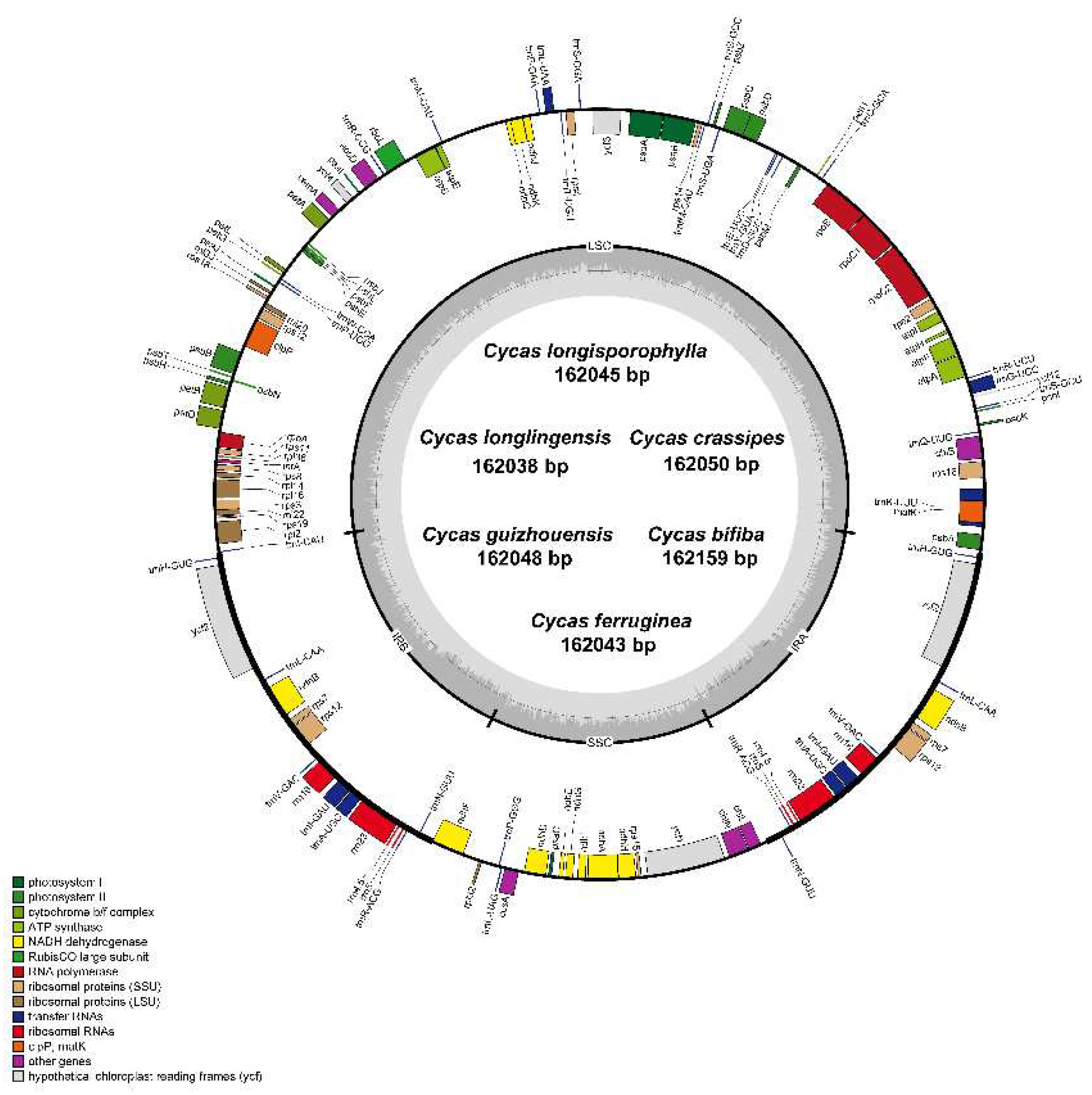
Figure 2.
Comparison of the LSC, SSC, and IR regional boundaries of chloroplast genomes between nine Cycas species. JLB, junction line between LSC and IRb; JSB, junction line between IRb and SSC; JSA, junction line between SSC and IRa; JLA, junction line between IRa and LSC.
Figure 2.
Comparison of the LSC, SSC, and IR regional boundaries of chloroplast genomes between nine Cycas species. JLB, junction line between LSC and IRb; JSB, junction line between IRb and SSC; JSA, junction line between SSC and IRa; JLA, junction line between IRa and LSC.
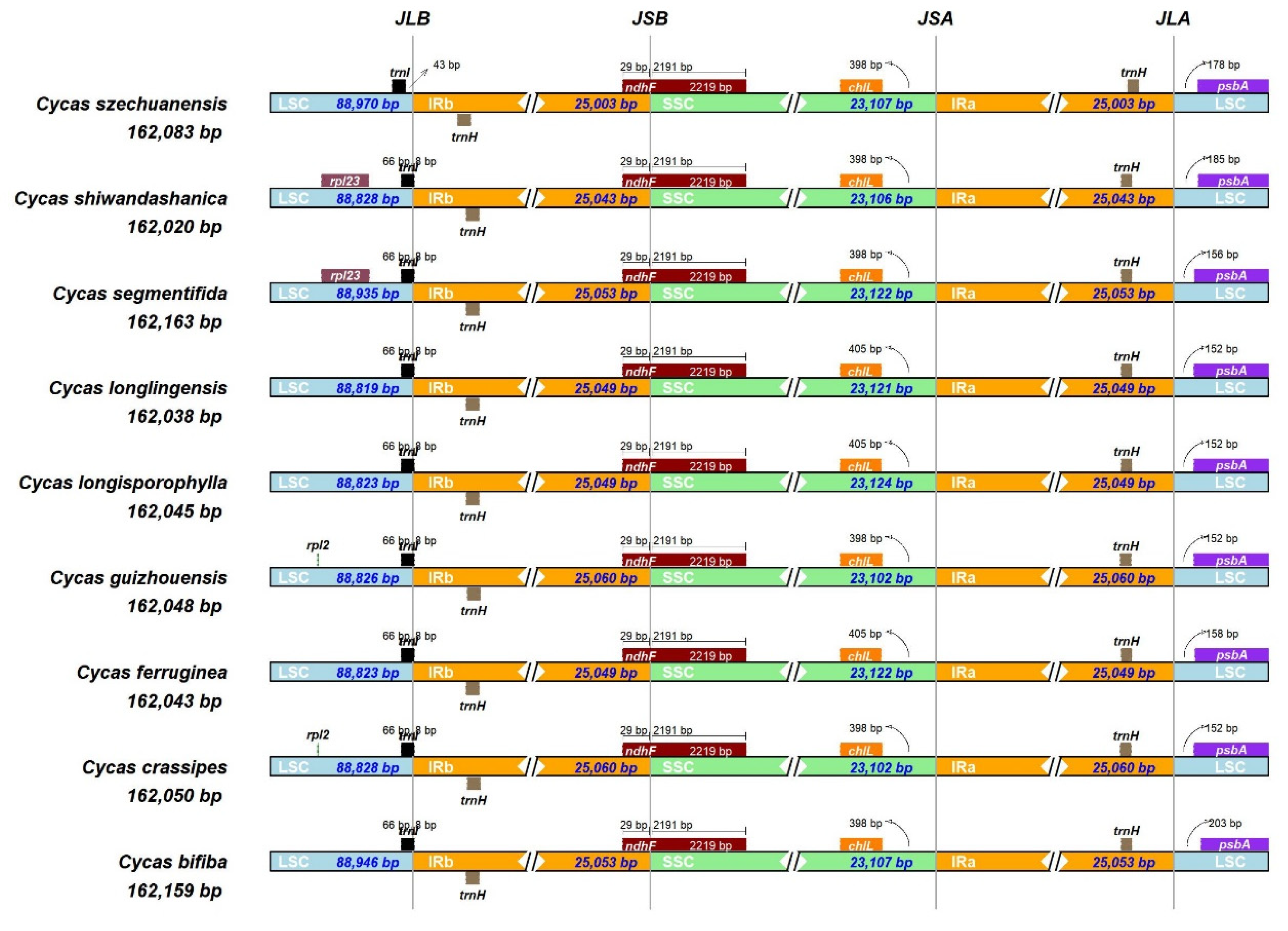
Figure 3.
Comparison of chloroplast genomes via annotation of Cycas bifida as a reference. Vertical scale indicates identify percentages ranging from 50 to 100%; the horizontal scale indicates coordinates within the chloroplast genome. Genome regions are color-coded as exons, introns, untranslated regions (UTRs), and conserved non-coding sequences (CNSs).
Figure 3.
Comparison of chloroplast genomes via annotation of Cycas bifida as a reference. Vertical scale indicates identify percentages ranging from 50 to 100%; the horizontal scale indicates coordinates within the chloroplast genome. Genome regions are color-coded as exons, introns, untranslated regions (UTRs), and conserved non-coding sequences (CNSs).
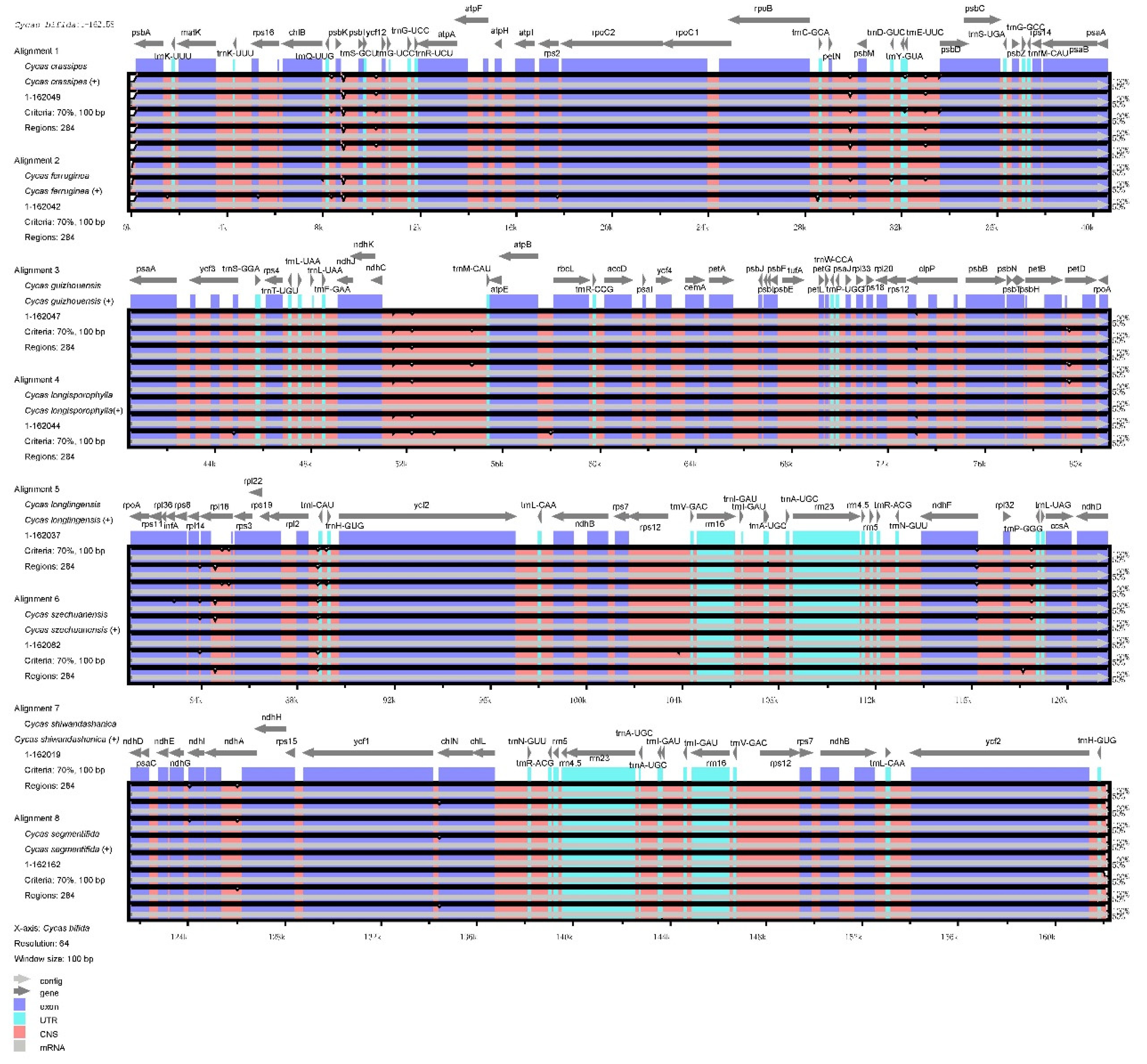
Figure 4.
Sliding window analysis of nine Cycas chloroplast genomes (window length: 600 bp; step size: 200 bp).
Figure 4.
Sliding window analysis of nine Cycas chloroplast genomes (window length: 600 bp; step size: 200 bp).
Figure 5.
Analysis of simple sequence repeats (SSRs) and large repeats in the chloroplast genomes of nine Cycas species: (a) Number of SSRs detected in each species; (b) type and frequency of each identified SSR; (c) four types of repeats; (d) frequency of repeat by length.
Figure 5.
Analysis of simple sequence repeats (SSRs) and large repeats in the chloroplast genomes of nine Cycas species: (a) Number of SSRs detected in each species; (b) type and frequency of each identified SSR; (c) four types of repeats; (d) frequency of repeat by length.
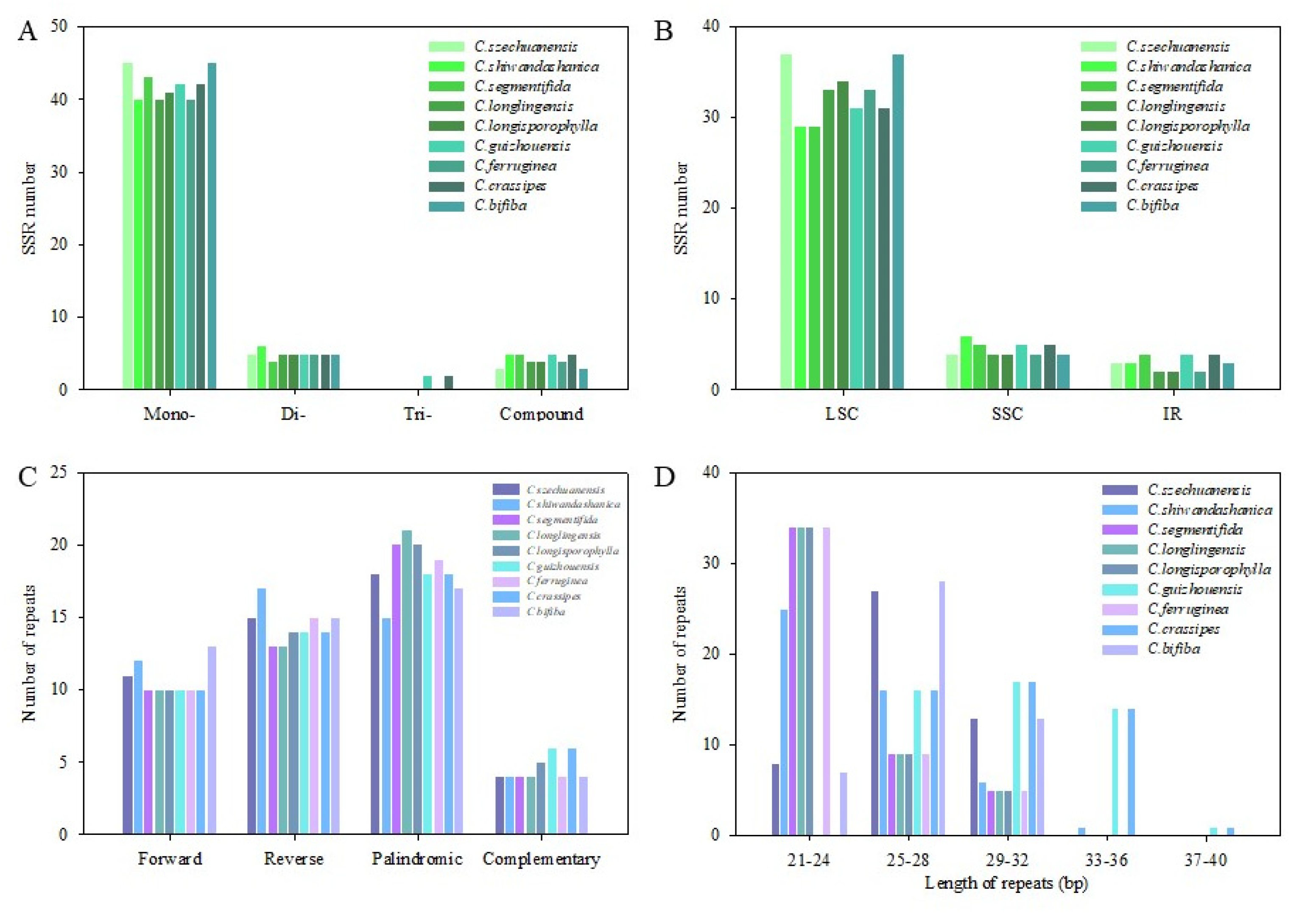
Figure 6.
Phylogenetic tree of six Cycas species and their related species based on complete chloroplast genomes. cp genome sequences were downloaded from GenBank.
Figure 6.
Phylogenetic tree of six Cycas species and their related species based on complete chloroplast genomes. cp genome sequences were downloaded from GenBank.
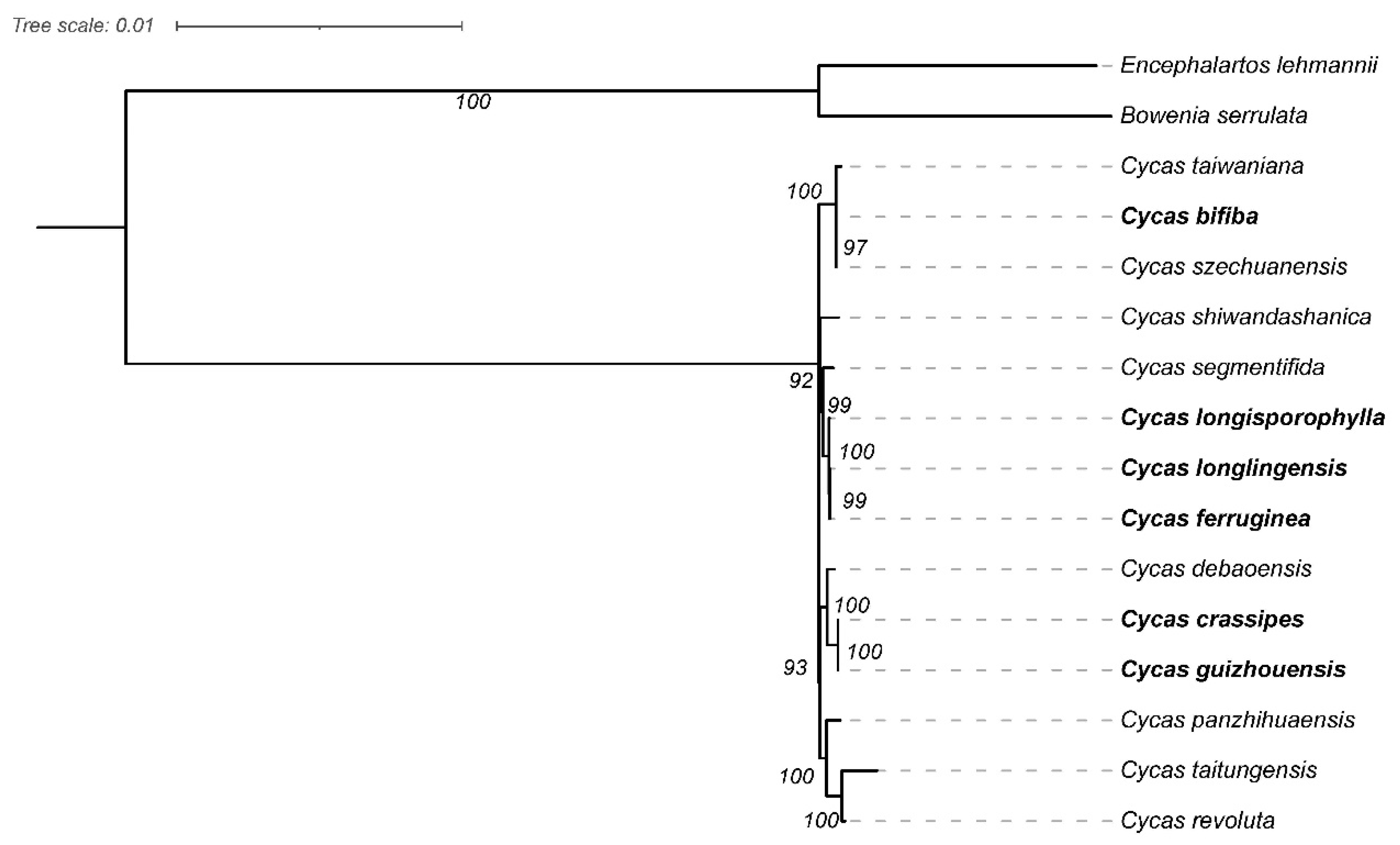

Table 1.
Statistics on the basic features of chloroplast genomes in six Cycas species.
| Sample | Total genome | LSC | IR | SSC | Gene number | |||||||
| Length (bp) | G+C Content (%) |
Length (bp) | G+C Content (%) |
Length (bp) | G+C Content (%) |
Length (bp) | G+C Content (%) | Total | PCGs | tRNA | rRNA | |
| Cycas longisporophylla | 162045 | 39.44 | 88823 | 38.73 | 25049 | 42.03 | 23124 | 36.55 | 131 | 86 | 37 | 8 |
| Cycas bifida | 162159 | 39.42 | 88946 | 38.7 | 25053 | 42.02 | 23107 | 36.52 | 131 | 86 | 37 | 8 |
| Cycas guizhouensis | 162048 | 39.42 | 88826 | 38.71 | 25060 | 42.01 | 23102 | 36.54 | 131 | 86 | 37 | 8 |
| Cycas crassipes | 162050 | 39.42 | 88828 | 38.71 | 25060 | 42.01 | 23102 | 36.54 | 131 | 86 | 37 | 8 |
| Cycas longlingensis | 162038 | 39.44 | 88819 | 38.72 | 25049 | 42.03 | 23121 | 36.55 | 131 | 86 | 37 | 8 |
| Cycas ferruginea | 162043 | 39.43 | 88823 | 38.72 | 25049 | 42.03 | 23122 | 36.55 | 131 | 86 | 37 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated