
Submitted:
08 September 2023
Posted:
12 September 2023
You are already at the latest version
Abstract
Concatemeric viral DNA is packaged into Bacteriophage P22 procapsids via a headful packaging mechanism mediated by a molecular machine consisting of small (gp3) and large (gp2) terminase subunits. Although a negative stain reconstruction exists for the terminase holoenzyme, it is not clear how this complex binds dodecameric portal protein located at a 5-fold mismatch vertex. Herein, we describe new assemblies for the holoenzyme. Both native mass spectrometry and transmission electron microscopy reveal that the P22 terminase complex adopts three main assemblies, which include a nonameric S-terminase bound to two L-terminase 1(gp3)9:2(gp2), two nonameric S-terminase bound to five L-terminase 2(gp3)9:5(gp2), and three nonameric S-terminase bound to seven L-terminase 3(gp3)9:7(gp2). Native agarose gel electrophoresis shows that the terminase complex interacts with procapsid with mild crosslinking. These results herein illustrate the P22 terminase complex can adopt a variety of confirmations and assembly states.
Keywords:
Salmonella virus
; viral genome-packaging motor
; small terminase
; large terminase
; Bacteriophage P22
; electron microscopy
1. Introduction
The Salmonella-phage P22 packages its genome from concatemeric dsDNA using a powerful molecular motor that consists of a large (gp2, 499 amino acids, 57.6 kDa [1]) and small (gp3, 162 amino acids, 18.6 kDa [2]) terminase subunits. In vitro studies have been performed to show that gp2 and gp3 form an oligomeric complex [3]. The recognition subunit, gp3, binds to packaging initiation (pac) sites [4] in the P22 genome and positions viral dsDNA to the large terminase subunit gp2, which uses ATP hydrolysis to translocate a single genome copy into empty procapsids. This reaction is very efficient and in similar phages proceeds at rates as high as 2,000 bp/sec [5]. The nuclease domain of gp2 also cleaves the concatameric dsDNA once the head is filled [4]. Upon cleavage, the gp3:gp2 complex quickly disassociates from the capsid enabling binding of the tail factors gp4, gp10 and gp26 [6,7] that seal the portal protein and stabilize the genome inside the capsid. This is followed by the attachment of six copies of the trimeric tailspike gp9 [8].
Gp3 spontaneously assembles into nine radially positioned protomers [9]. Initially, an 18 Å resolution structure obtained by negative stain electron microscopy (EM) revealed a C-terminally cleaved gp3 nonamer [10]. Subsequently, a crystal structure of 1.75 Å resolution along with the full-length EM structure of 18 Å resolution revealed an outer diameter of 95 Å, and an inner diameter of 23 Å; wide enough to compensate for hydrated dsDNA [11].
Gp2 consists of an N-terminal ATPase fold (residues 38-284), a flexible linker, a C-terminal nuclease domain (313-482), and a C-terminal basic tail (residues 483-499) [12,13,14]. The ATPase domain is conserved in phages and large-terminase containing viruses. It contains a typical nucleotide-binding fold [15] with two subdomains comprised of ATP/GTP-binding Walker A and B motifs. The nuclease domain of gp2 has been solved via x-ray crystallography [16]. It has an RNAse H-fold and is a member of the ribonuclease H/resolvase/integrase superfamily [17]. The nuclease domain of gp2 has a mixed α/β fold and is globular in structure. The catalytic site has two Mg2+ ions. One Mg2+ is octahedrally coordinated with four Asp and four waters, while the other Mg2+ is tetrahedrally coordinated with Asn, His, and two waters. Catalytic site Asp321 is conserved in related phages and critical to nuclease activity [16].
The structure of the gp2 α-helical hairpin residues 1-33 bound to Fab4 has been determined through crystallography [18], however, the 3-D structure of the P22 gp2 ATPase domain has not been determined to date. Since the fold is conserved, it is likely the 3D structure resembles that of Sf6 [19] and T4 [20]. Monomeric gp2 assembles into a pentamer bound to dodecameric portal (gp1) [21,22]. Bacteriophage P22 has a symmetry mismatch at one of its 5-fold vertices. This vertex contains the 12-fold portal protein through which genome packaging and ejection occur.
There is a full-length crystal structure for the Sf6 large terminase. The sequence identity of P22 to Sf6 is 15% and 12% for the corresponding large and small terminases respectively [23]. Although sequence identity is low for large terminases, 3D homology remains relatively high. Vis-à-vis other large terminases, we expect the 3-D structure of monomeric gp2 to resemble that of other published large terminases.
A negative stain transmission electron microscopy (TEM) reconstruction has been determined for the first P22 terminase holoenzyme complex [24] which illustrates a stoichiometry of 1(gp3)9:2(gp2). Not only has a high-resolution cryo-EM structure not been determined since the negative stain reconstruction was reported, but the stoichiometry of the complex of the large terminase complex that associates with dodecameric portal has not been determined. The large terminase from T4 has been shown to form a pentamer and associate with the portal [25]. In this paper, we use a combination of electron microscopy coupled to mass spectrometry to show that the terminase complex is capable of adopting variable stoichiometric assemblies, which include 5 copies of gp2. These results provide new insights into the biological relevance of heterogeneity observed in this dynamic molecular machine.
2. Materials and Methods
Biochemical techniques
The S:L-terminase complex was expressed in E. coli strain BL21-AI (Life Technologies) by inducing at 18 °C for 12-16 hours with a final concentration 0.2% L-arabinose and 0.1 mM IPTG. Cell pellets were resuspended in Lysis buffer containing 20 mM Tris-Cl pH 8.0, 300 mM NaCl, 1 mM MgCl2, 1% glycerol, 0.1% n-Dodecyl-β-D-maltoside, 3 mM β-mercaptoethanol, 1.0 mM phenylmethylsulfonyl fluoride and cells were disrupted by sonication. The S:L-terminase complex was purified on amylose beads (New England Biolabs) and after washing with 500 ml of lysis buffer the complex was incubated with 1 mM AMP-PNP (Sigma) and PreScission Protease to cleave off MBP or GST. The day after, cleaved species coming off beads were further purified on a Superdex 200 16/60 gel filtration column (GE Healthcare) in GF-buffer (20 mM Tris-Cl pH 8.0, 250 mM NaCl, 1 mM MgCl2, 1% glycerol. The gel filtration column was calibrated with molecular weight markers as previously described [26]. Isolated S:L-terminase and S-LBD:L-terminase complexes were concentrated to ~10 mg/ml using a 30 MWCO ultrafiltration spin column (Vivaspin 20, Sartorius Stedim Biotech GmbH).
EM specimen prep
To determine the structure and organization of the gp2:gp3 complex, Peak 2 (Figure 1) was analyzed with negative stain EM. To enhance contrast of the sample, approximately 2-5 nm carbon was floated off mica support film and placed over C-flat grids containing 3x3 hole patterns with 2 μm hole diameter and 0.5 μm hole spacing (Protochips (EMS) CF-2/0.5-4C-50). The grids were then glow discharged. Upon screening the appropriate sample concentration, 3 ml sample was applied to the grid for 1 minute. The grid was gently blotted and passed through four 50 ml volumes of 2% uranyl formate. Subsequently, the grid was blotted, air-dried, and stored under desiccation.
EM Data collection and processing
Because of sample heterogeneity, it was necessary to use the Random Conical Tilt (RCT) data collection strategy to obtain initial 3-D models [27]. Image tilt pairs, untilted and -55°, were acquired for 1100 images using a Tecnai 12 electron microscope operating at 120 keV, with a dose near 20 e-/Å2 (Figure S1) sourced from a Denka M-3 LaB6 Phillips Cathode (product No. 1451). The C-Flat grid 3x3 hole patterns were targeted for RCT using Leginon [28]. We applied a focus node at the center of the 3x3 holes, and subsequently captured images of the remaining eight holes. Data processing was conducted using the APPION suite [29]. DoG Picker was used to pick particles from all images [30]. Particles within image pairs were correlated using TiltAutoAligner [31]. Upon making stacks of the zero tilt image particles (22,744) and corresponding tilted image particles, the zero tilt images were iteratively classified using the CL2D algorithm [32]. This resulted in 18,767 zero tilt particles. Class averages of zero tilt particles matched the negative tilt particle reconstruction (data not shown).
Focused Classification and 2D Alignment
After classification with CL2D produced class averages where side views of gp3 were discernable, representative class averages were used for referenced-based alignment using SPIDER [33]. Following referenced-based alignment, a mask was generated to select each lobe from an image of each class average (Figure 2A). A low pass Gaussian filter of 0.0125 was applied to the mask. The mask was then used to crop out corresponding areas each aligned class average. All the masked particles from each class average were combined into stacks with the class they originated from, centered, and subsequently submitted to 2D classification in both RELION [34] and SPARX [35], separately.
2D Alignment
Unmasked images were aligned and clustered iteratively, using ISAC [36]. Irreproducible clusters were used as a seed for subsequent rounds of alignment and clustering, up to twelve generations. ISAC produced the best class averages for this heterogenous dataset.
Native MALDI mass spectrometry
Prior to native mass spec measurement, the purified S:L-terminase complex was buffer exchanged into 150 mM aqueous ammonium acetate (AmAc) (pH 8.0), by ultrafiltration (Vivaspin 500, Sartorius Stedim Biotech, Germany) with a 10 kDa cut off. 1-2 µL of sample, at a final concentration of 2 µM, was loaded into a nanoflow gold-plated borosilicate electrospray capillary (made in house). The higher order oligomers of S:L-terminase complex were analyzed on a modified QTOF-2 (Waters/MS Visions) operated on positive ion mode. Xenon was used as collision gas. MS parameters: backing pressure 10 mbar; capillary 1300 V; cone 60 V; extracted cone 0 V; pressure in the collision cell 2 × 10 -2 mbar; collision energy 30 V. The sample was analyzed on a modified Exactive Plus EMR Orbitrap instrument (Thermo Fisher Scientific, Bremen) over m/z range 500-20000 [37,38]. Manual tuning of the voltage offset on the flatapole, transport multipole, ion lenses was used for mass filtering of the incoming protein ions, as previously described [39]. Nitrogen was used for the HCD cell at a gas pressure of 6-8 × 10-10 bar. Mass spec parameters: spray voltage 1.3-1.4 V, source fragmentation 30 V, source temperature 250 °C, collision energy 40-50 V, 10000 resolution at m/z 200. Maxent was used to deconvolute the peaks and determine molecular weights.
Procapsid production and purification
P22 phage and Salmonella were provided by Peter Prevelige (University of Alabama at Birmingham). The Salmonella strains used were derivatives of Salmonella typhimurium LT2, DB700 suppressor minus (leuA- 414(am) supo) and supE derivative supressor plus MS1363. The supplied phage contains an amber mutation in gp2 and gp3. This phage was used to infect MS1363 suppressor plus Salmonella, which lead to intact phage and enables high titer production. Upon reaching the desired titer of 1012 phage/ml, DB7000 suppressor minus Salmonella was infected with a multiplicity of infection (MOI) of 7. This enabled production of procapsids without packaged double stranded DNA. Cell pellets were lysed and clarified before loading onto a linear 10-40% sucrose gradient. Procapsids were extracted from the side of the tube using an 18-gauge needle and subsequently loaded onto a CsCl step gradient where they migrate to the d=1.2/1.4 CsCl interface (Figure S2) as previously described [40].
Native agarose gel electrophoresis
Native agarose gel electrophoresis was conducted as previously published [40]. Briefly, native agarose gels were prepared in Tris-borate-magnesium (TBM) buffer (45 mM Tris, 45 mM borate, 1 mM MgCl2). Reaction conditions utilized 0.5 mg procapsid and 62 mg terminase complex on ice for 30 minutes in the presence of 0.01% or 0.1% glutaraldehyde. Crosslinking was quenched using 1 M Tris-HCl pH 7.0. The samples were then mixed with 4X loading buffer (1.5 x TBM, 12.5% v/v) Ficoll, 0.01% (w/v) bromophenol blue, 0.02% NaN3), and electrophoresed at 4 V/cm for three hours at 4 °C. After electrophoresis, the gel was gently rocked in 50 mM EDTA for 20 to 30 minutes. The gel was then stained for one to two hours with Coomassie blue (10% (v/v) methanol, 10% (v/v) acetic acid, 0.01% (w/v) Coomassie blue R-250) prior to dehydration with a gel drier. The gel was destained overnight in 10% acetic acid.
3. Results
To examine the structure and stoichiometry of the gp2-gp3 complex, Peak 2 from gel filtration (Figure 1) was visualized by negative stain EM. Approximately 35,534 particles were picked from 1,101 images using Difference of Gaussians (DoG) Picker. These particles were initially sorted to remove poor particles using reference-free techniques resulting in 18,767 particles. Subsequently, representative class averages were used as templates for reference-based alignment to separate the different assembly and conformational states.
3.1. Multiple (gp3)9 in gp2-gp3 complex
The individual gp2 and gp3 components identified in the class averages were based on first recognizing gp3 (Figure 2A). Gp3 was easily recognized with striking side views of its nonamer β-hairpin and α-helical core based on projections of the crystal and EM structures [11]. The centralized ellipsoid density was featureless but was assumed to be gp2 (Figure 2A). The poor resolution in the gp2 region could be explained due to flexibility of gp2 domains. At this early stage, the class averages contained one, two or three (gp3)9.
3.2. Focused classification reveals gp2-gp3 domain interaction
To obtain domain-level resolution in the putative gp2 ellipsoid density region, a focused classification strategy was utilized. Briefly, particles were aligned to major class averages consisting of two or three (gp3)9 bound to gp2 ellipsoid density (Figure 2A). Individual stacks were made for each type of (gp3)9 organization, and the particles were again aligned to corresponding 2D class averages using SPIDER [33]. After verifying via a visual analysis that the particles were properly aligned, a mask was drawn around the gp3 density and a small portion of the ellipsoid density for each class average. The mask was then used to crop the density of the aligned particles using the corresponding class average. The cropped particles containing gp3 and a small portion of ellipsoid density were next subjected to 2D classification using RELION [34]. This focused 2D classification strategy produced a class average where gp2 density interacting with gp3 could be resolved. Beneath the gp3 mushroom-like density, two spherical densities could be resolved (Fig. 2B). Each density is the size of an individual gp2 domain and agree with the (gp3)9:2(gp2) terminase holoenzyme reconstruction previously published [24]. These results support that the ellipsoid density with multiple gp3 attached corresponds to a gp2 core (Figure 2C).
3.3. Variable (gp3)9 attach to large terminase core
Initial class averages were low in resolution in the (gp3)9 region because programs could not manage alignment with concurrent conformational heterogeneity and variable stoichiometry in the dataset. This is likely because they focus on aligning particles to their overall shape. To obtain better detail and define gp3 localization in the heterogeneous terminase holoenzyme populations, 2D classification of entire raw particles was performed using iterative alignment and classification (ISAC) algorithm. ISAC clearly showed holoenzyme complexes consisting of one (Figure 3A), two or three gp3′s with different relative positions (Figure 3B). The (gp3)9 predominantly showed up with a side view preferred orientation on the grid (Figure 3B and C).
In order to resolve the number of gp2 associated with each terminase holoenzyme assembly, native matrix-assisted laser desorption/ionization (MALDI) mass spectrometric analysis of purified samples was performed on initial Peak 2 shown in Figure 1. The analysis revealed mass measurements consistent with 1(gp3)9:1(gp2) *(2AMP-PNP), 1(gp3)9:2(gp2) *(2AMP-PNP), 2(gp3)9:5(gp2) *(10AMP-PNP), and 3(gp3)9:7(gp2) *(14AMP-PNP) (Figure 4A). There are at least two populations for the holoenzyme complex. One population is approximately 290 kDa consisting of 1(gp3)9:2(gp2-AMP-PNP). A second peak is around 642 kDa consisting of 2(gp3)9:5(gp2-AMP-PNP). A third peak consisting of 3(gp3)9:7(gp2-AMP-PNP) is likely formed by a combination of peaks 1 and 2. The mass spectroscopy results support the main populations initially identified with transmission electron microscopy (Figure 4B).
3.4. Binding of Terminase complex to P22 procapsid
To examine if the terminase complex sample discussed herein could interact with P22 portal protein in procapsids, procapsids were incubated with terminase complex and analyzed using native agarose electrophoresis. In this native gel which separates by size and charge, to our surprise the terminase migrates slower than procapsid (Figure 5). Incubation of procapsid and terminase only produced a new species in the presence of glutaraldehyde (Figure 5). In fact, a discrete procapsid-terminase complex band appeared with 0.01% glutaraldehyde, while incubation with 0.1% glutaraldehyde produced a smear.
3.5. Figures
Figure 1.
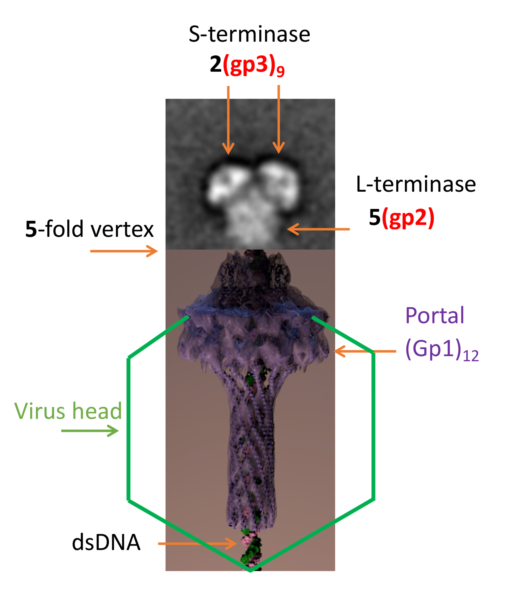
Co-expression and purification of the gp3:gp2 complex. (A) Domain organization of P22 S- and L-terminase subunits. (B) MBP-tagged gp3 is co-expressed with gp2 in E. coli. The complex is isolated on amylose beads, MBP tag is cleaved and the complex subsequently purified by size exclusion chromatography using a Superdex 200 column, which yields two peaks. Peak 2 is consistent with ~300-350 kDa, whereas Peak 1 is larger (1 MDa-700 kDa). Peak 2 was run three times over gel filtration to obtain a single peak (red trace). Two fractions from each peak were analyzed on SDS-Page gels. Peaks 1 and 2 are indistinguishable in SDS gel analysis.
Figure 1.
Co-expression and purification of the gp3:gp2 complex. (A) Domain organization of P22 S- and L-terminase subunits. (B) MBP-tagged gp3 is co-expressed with gp2 in E. coli. The complex is isolated on amylose beads, MBP tag is cleaved and the complex subsequently purified by size exclusion chromatography using a Superdex 200 column, which yields two peaks. Peak 2 is consistent with ~300-350 kDa, whereas Peak 1 is larger (1 MDa-700 kDa). Peak 2 was run three times over gel filtration to obtain a single peak (red trace). Two fractions from each peak were analyzed on SDS-Page gels. Peaks 1 and 2 are indistinguishable in SDS gel analysis.
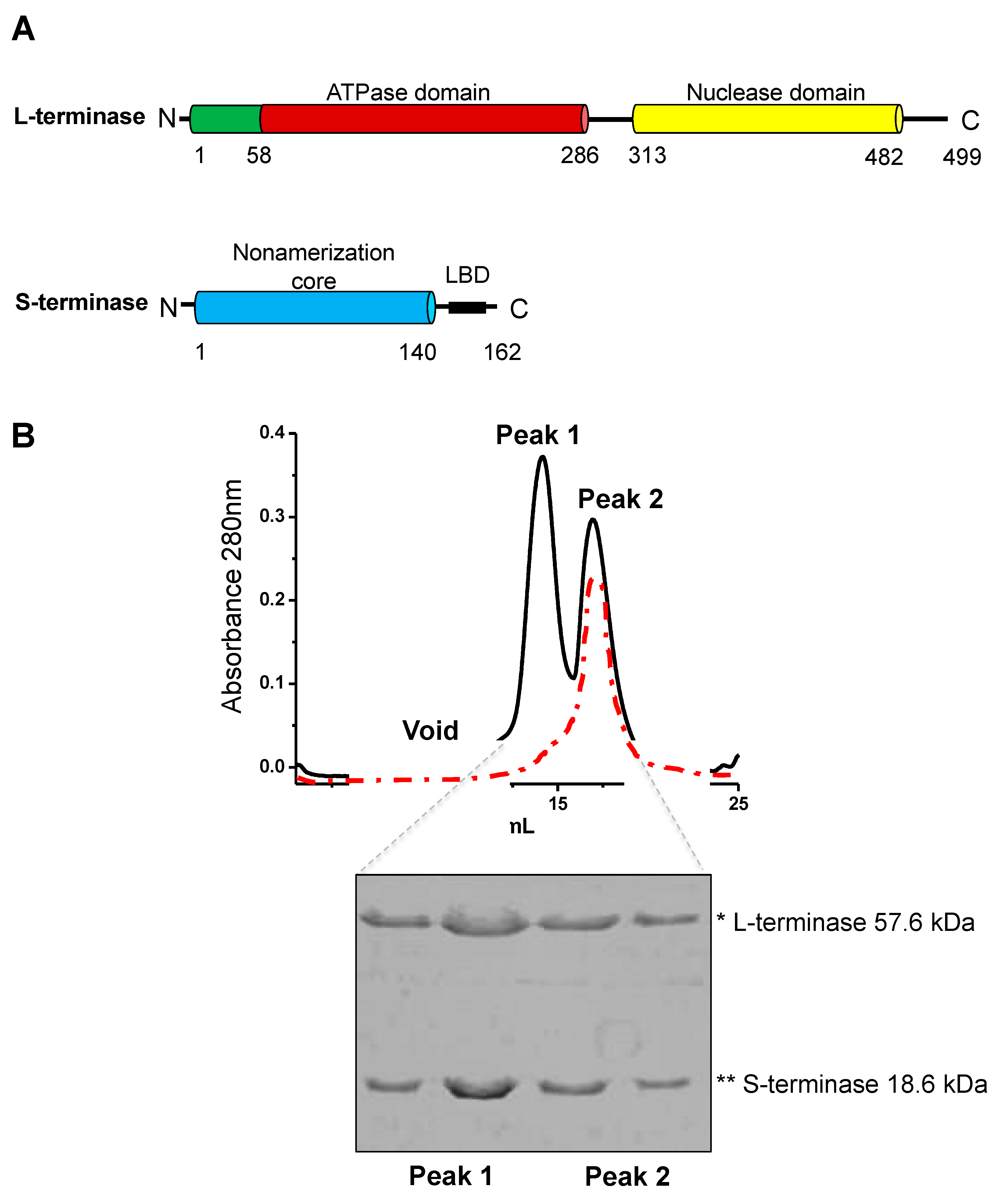
Figure 2.
Figure 2. Focused classification in 2D. (A) Class averages representing various assembly states after reference-based alignment with Spider [33]. Masked regions for focused classification are circled red. Putative gp3 and gp2 regions are denoted with blue and green arrows, respectively. Ellipsoid gp2 region denoted with blue arrow. (B) Resultant 2D class averages with RELION after masking. (C) The 9-mer S-terminase density strongly resembles the crystal structure, giving confidence in focused classification results. The 2D density shows S-terminase (blue) with two gp2′s (purple and green) attached at S-terminase C-terminus. The full-length gp2 is from Sf6 (PDB 4IEEE) [19].
Figure 2.
Figure 2. Focused classification in 2D. (A) Class averages representing various assembly states after reference-based alignment with Spider [33]. Masked regions for focused classification are circled red. Putative gp3 and gp2 regions are denoted with blue and green arrows, respectively. Ellipsoid gp2 region denoted with blue arrow. (B) Resultant 2D class averages with RELION after masking. (C) The 9-mer S-terminase density strongly resembles the crystal structure, giving confidence in focused classification results. The 2D density shows S-terminase (blue) with two gp2′s (purple and green) attached at S-terminase C-terminus. The full-length gp2 is from Sf6 (PDB 4IEEE) [19].
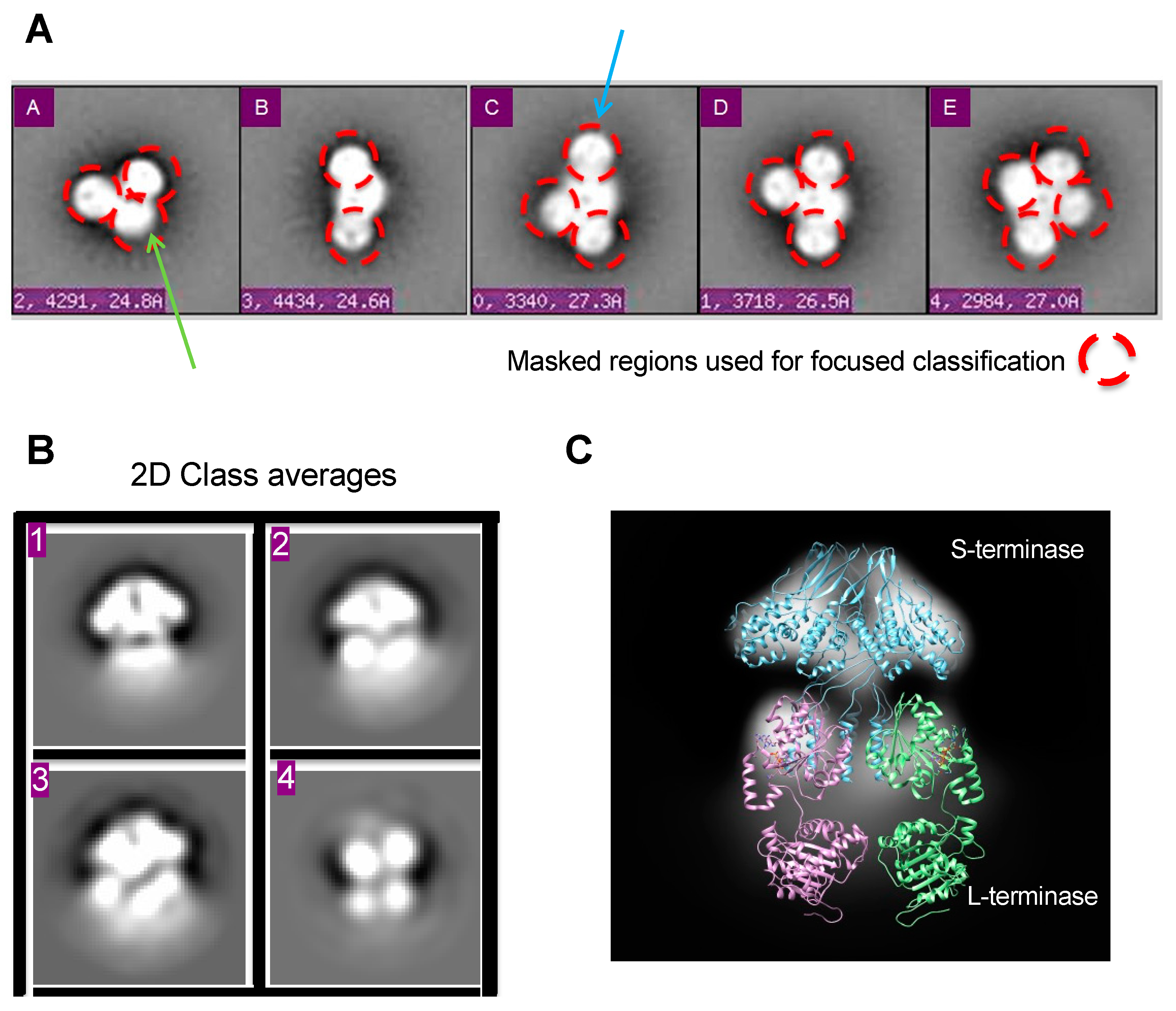
Figure 3.
S-terminase (gp3)9 attached to a L-terminase (gp2) core. (A) Class average matches P22 terminase holoenzyme complex containing 1(gp3)9 (cyan) and 2 gp2 (ATPase – red and nuclease – yellow). (B) Iterative Stable Alignment and Classification (ISAC) 2D class averages. Class averages contained one, two, or three (gp3)9 (blue arrow) attached to a gp2 core (red arrow). Scale bar is 100 Å. (C) Class averages from (B). Gp3 residues 1-139 (purple) manually docked into representative gp3 density.
Figure 3.
S-terminase (gp3)9 attached to a L-terminase (gp2) core. (A) Class average matches P22 terminase holoenzyme complex containing 1(gp3)9 (cyan) and 2 gp2 (ATPase – red and nuclease – yellow). (B) Iterative Stable Alignment and Classification (ISAC) 2D class averages. Class averages contained one, two, or three (gp3)9 (blue arrow) attached to a gp2 core (red arrow). Scale bar is 100 Å. (C) Class averages from (B). Gp3 residues 1-139 (purple) manually docked into representative gp3 density.
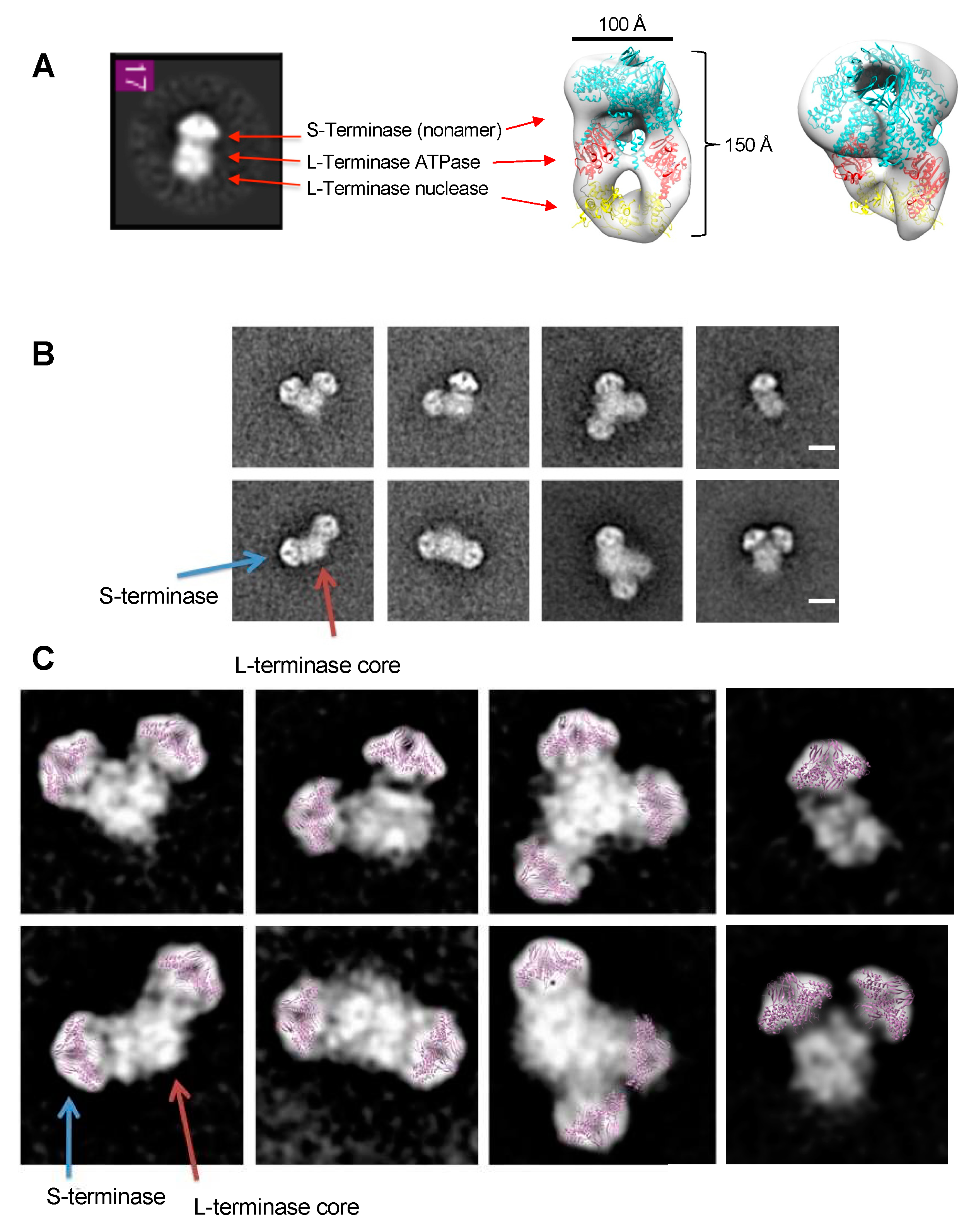
Figure 4.
Mass Spectrometric analysis of purified gp3:gp2 complex. (A) The differential assembly is consistent with at least two distinct gp3:gp2 populations of ~290 kDa (stoichiometry (1 x gp3) + (2 x gp2:AMP-PNP)) and ~642 kDa (stoichiometry (2 x gp39) + (5 x gp2:AMP-PNP)) and a ~925 kDa oligomer of the first two complexes combined (stoichiometry (3 x gp39) + (7 x gp2:AMP-PNP)). (B) Representative class averages of the three major assemblies identified by mass spec.
Figure 4.
Mass Spectrometric analysis of purified gp3:gp2 complex. (A) The differential assembly is consistent with at least two distinct gp3:gp2 populations of ~290 kDa (stoichiometry (1 x gp3) + (2 x gp2:AMP-PNP)) and ~642 kDa (stoichiometry (2 x gp39) + (5 x gp2:AMP-PNP)) and a ~925 kDa oligomer of the first two complexes combined (stoichiometry (3 x gp39) + (7 x gp2:AMP-PNP)). (B) Representative class averages of the three major assemblies identified by mass spec.
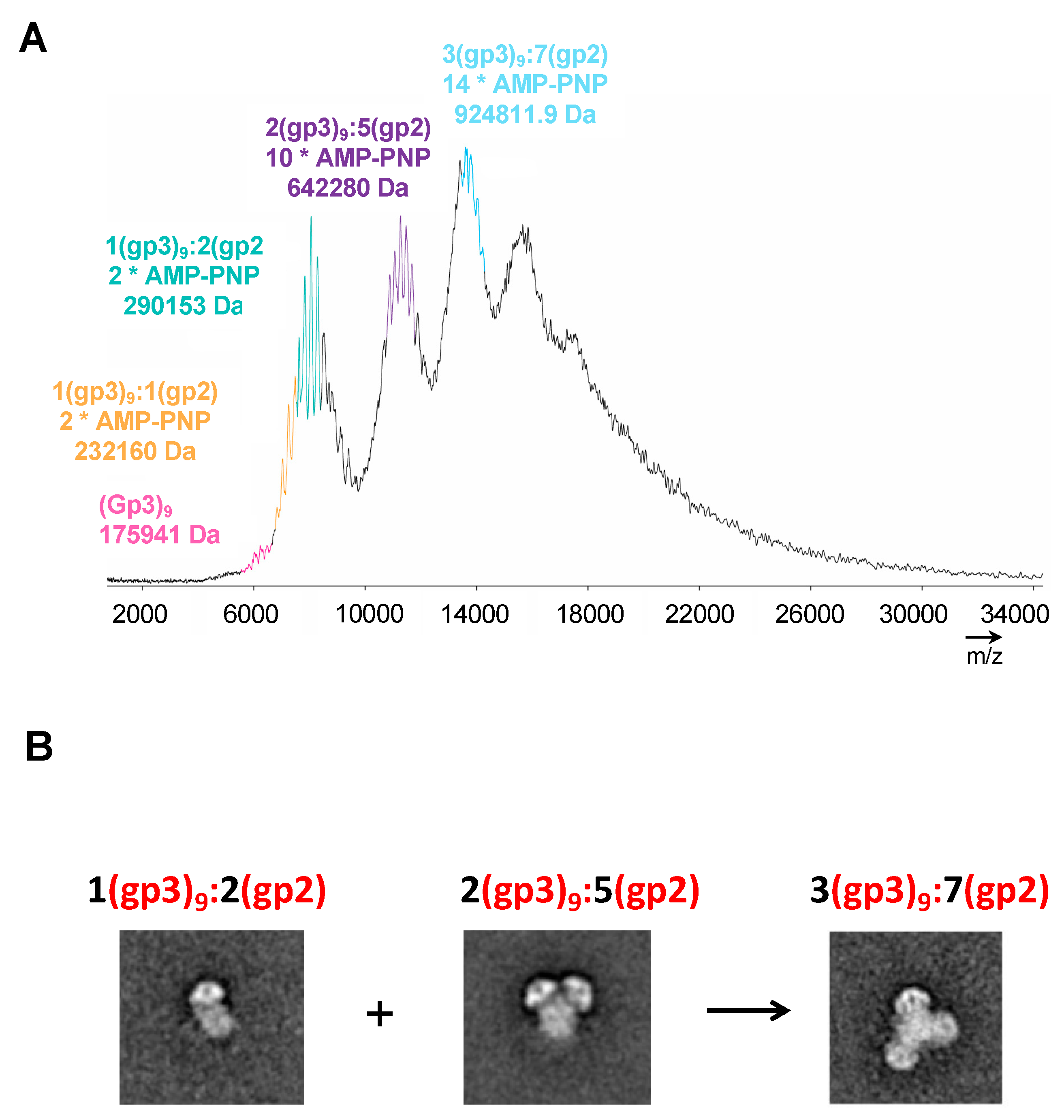
Figure 5.
Generation of procapsid-terminase complex. Native agarose gel with procapsid (P) incubated with terminase complex (T) forms a new complex (P/T) in the presence of glutaraldehyde crosslinking. Lanes 1-3 contain 0.01% glutaraldehyde and lanes 6-8 contain 0.1% glutaraldehyde. The crosslinked procapsid:terminase complex appears in lane 3 denoted as P/T complex (dashed yellow rectangle).
Figure 5.
Generation of procapsid-terminase complex. Native agarose gel with procapsid (P) incubated with terminase complex (T) forms a new complex (P/T) in the presence of glutaraldehyde crosslinking. Lanes 1-3 contain 0.01% glutaraldehyde and lanes 6-8 contain 0.1% glutaraldehyde. The crosslinked procapsid:terminase complex appears in lane 3 denoted as P/T complex (dashed yellow rectangle).

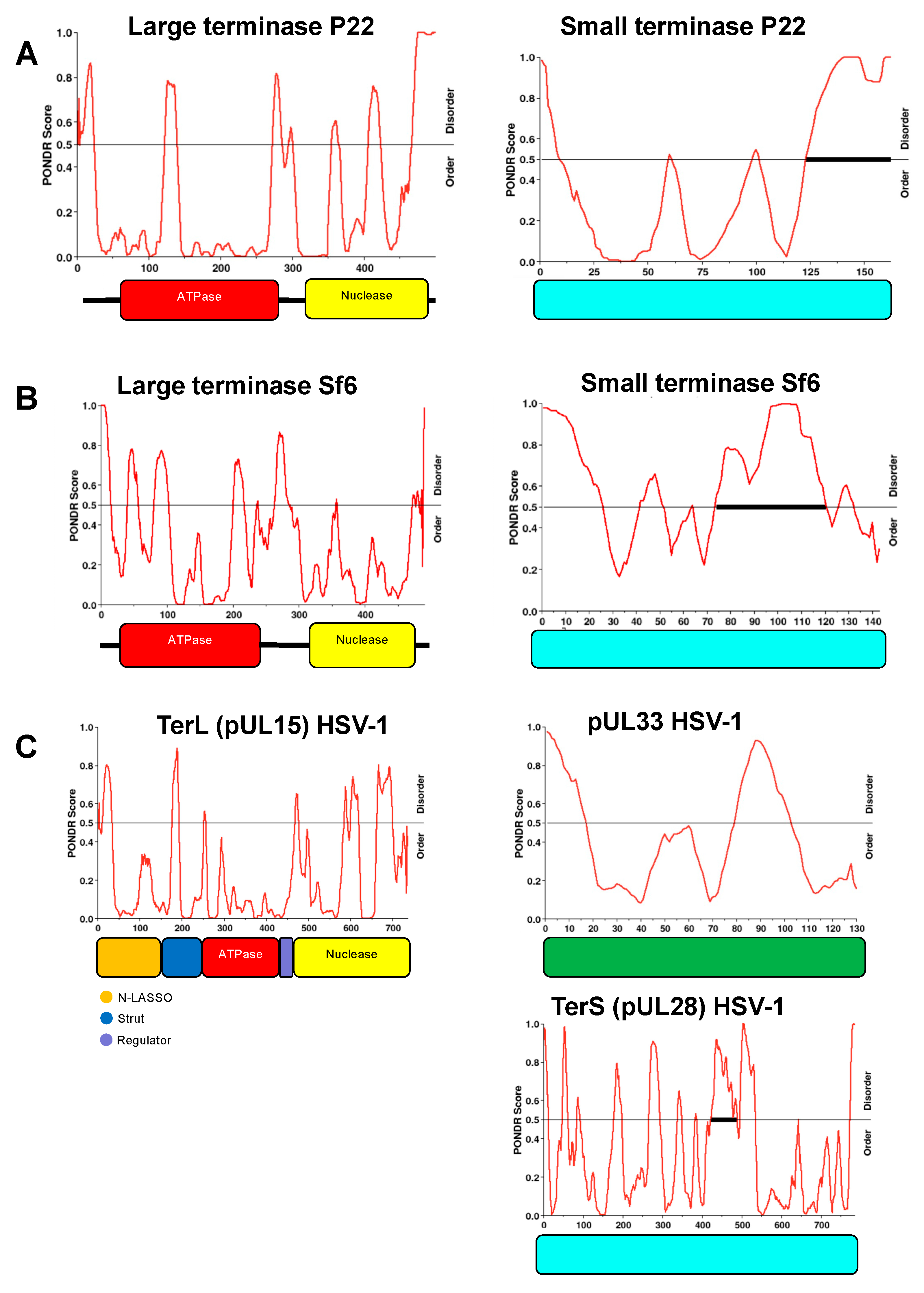
Figure 6.
Figure 6. Intrinsic disorder region (IDR) distribution for P22, Sf6, and HSV-1 terminases. PONDR analysis indicates similar IDR’s for L-terminases (left) across different species. Similar IDR’s are also observed for small terminases (right). The ratio of order to disorder is shown for: (A) P22– gp2 and gp3; (B) Sf6– gp2 and gp1; (C) HSV-1– pUL15 (TerL), pUL33, and pUL28 (TerS).
Figure 6.
Figure 6. Intrinsic disorder region (IDR) distribution for P22, Sf6, and HSV-1 terminases. PONDR analysis indicates similar IDR’s for L-terminases (left) across different species. Similar IDR’s are also observed for small terminases (right). The ratio of order to disorder is shown for: (A) P22– gp2 and gp3; (B) Sf6– gp2 and gp1; (C) HSV-1– pUL15 (TerL), pUL33, and pUL28 (TerS).
4. Discussion
Terminases are transient complexes that package viral genomes into preformed procapsids. After nearly a decade since the first moderate resolution P22 terminase holoenzyme was determined, a high-resolution cryo-EM structure is still lacking. This is likely due to biological heterogeneity observed with this complex illustrated herein. Unlike bacteriophages whose terminase complex is made of 2 proteins (L-terminase and S-terminase), herpesviruses have 3 proteins that comprise the terminase complex (pUL15, pUL28, pUL33). Recently the terminase complex for herpesvirus has been determined and express as monomeric, hexameric, or dodecameric in form [41]. Thus, the heterogeneity observed for herpesvirus terminases, which express in 3 main stoichiometries, are consistent with that observed herein for P22 terminase complexes.
We found that P22 terminase complex could assemble into 3 main classes: 1(gp3)9:2(gp2), 2(gp3)9:5(gp2), 3(gp3)9:7(gp2). The variability in subunit assembly is supported by native mass spectroscopy, indicating that the observed assemblies are not an artifact of negative stain sample preparation. We propose the differential assembly of these complexes is due to S-terminase C-terminal helices that are free to associate with additional L-terminase ATPase subunits. Subsequent binding of an additional S-terminase nonamer enables additional L-terminase subunits to bind. The C-terminal helices of S-terminase fit the canonical definition of an intrinsically disordered region (IDR) according to several IDP software programs including PONDR (Figure 6). Moreover, L-terminase also contains several IDR’s that would contribute to conformational and structural heterogeneity observed herein. The intrinsic disorder distribution within the protein sequence of small and large terminase is similar across species (Figure 6). Previous structural studies show that L-terminase is monomeric in solution but forms a pentamer at the portal vertex [21,22]. Based on this, it is likely the holoenzyme with 2(gp3)9:5(gp2) can directly bind and headful-package concatemeric genomic DNA inside P22 procapsids. Data shows that one or more of the complexes are relevant because they bind procapsid in the presence of gentle glutaraldehyde crosslinking.
A nearly complete structural characterization exists for Bacteriophage P22 ranging from mature viral assembly [7,42] to genomic injection conduit formation [43], with most protein structures and their locations known for the virus. Future experiments for the cryo-EM structure of P22 terminase complex bound to procapsid may be facilitated by supplementing with P22 genomic DNA or gentle crosslinking as suggested herein.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Julia Elise Cabral and Yanfei Qiu analyzed data; Albert J.R. Heck performed mass spectroscopy; Reginald McNulty performed electron microscopy and image analysis. Reginald McNulty wrote the original draft; Julia Elise Cabral, Yanfei Qiu, and Albert J.R. Heck, and Reginald McNulty reviewed and edited the manuscript.
Funding
This research was funded by National Institutes of Health grants K22AI139444 to R.M. Native MS measurement was performed by A.J.R.H., who have been supported by the ManiFold project, grant number 317371, and in part by the PRIME-XS project, grant number 262067, both funded by the European Union Seventh Framework Program. The APC was funded by University of California Irvine.
Data Availability Statement
All relevant data are contained within this publication.
Acknowledgments
We thank Prof. Gino Cingolani (Thomas Jefferson University) for providing purified terminase complex. We thank Peter Prevelige Jr. (University of Alabama at Birmingham) for helpful instructions on producing P22 procapsids. We thank Prof. John (Jack) Johnson (Scripps Research) for initiating the project and guidance. We thank Yang Yang (Utrecht University) for some acquisition of native MS data. We thank T.M.M. and K.L.M for graphical abstract recommendations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eppler, K.; Wyckoff, E.; Goates, J.; Parr, R.; Casjens, S. , Nucleotide sequence of the bacteriophage P22 genes required for DNA packaging. Virology 1991, 183, 519–38. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.; Sampson, L.; Randall, S.; Eppler, K.; Wu, H.; Petri, J. B.; Schmieger, H. , Molecular genetic analysis of bacteriophage P22 gene 3 product, a protein involved in the initiation of headful DNA packaging. Journal of molecular biology 1992, 227, 1086–99. [Google Scholar] [CrossRef] [PubMed]
- Poteete, A. R.; Botstein, D. , Purification and properties of proteins essential to DNA encapsulation by phage P22. Virology 1979, 95, 565–73. [Google Scholar] [CrossRef]
- Wu, H.; Sampson, L.; Parr, R.; Casjens, S. , The DNA site utilized by bacteriophage P22 for initiation of DNA packaging. Molecular microbiology 2002, 45, 1631–46. [Google Scholar] [CrossRef] [PubMed]
- Fuller, D. N.; Raymer, D. M.; Kottadiel, V. I.; Rao, V. B.; Smith, D. E. , Single phage T4 DNA packaging motors exhibit large force generation, high velocity, and dynamic variability. Proceedings of the National Academy of Sciences of the United States of America 2007, 104, 16868–73. [Google Scholar] [CrossRef]
- Chang, J.; Weigele, P.; King, J.; Chiu, W.; Jiang, W. , Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure 2006, 14, 1073–82. [Google Scholar] [CrossRef]
- Lander, G. C.; Tang, L.; Casjens, S. R.; Gilcrease, E. B.; Prevelige, P.; Poliakov, A.; Potter, C. S.; Carragher, B.; Johnson, J. E. , The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science 2006, 312, 1791–5. [Google Scholar] [CrossRef]
- King, J.; Lenk, E. V.; Botstein, D. , Mechanism of head assembly and DNA encapsulation in Salmonella phage P22. II. Morphogenetic pathway. Journal of molecular biology 1973, 80, 697–731. [Google Scholar] [CrossRef]
- Nemecek, D.; Gilcrease, E. B.; Kang, S.; Prevelige, P. E., Jr.; Casjens, S.; Thomas, G. J., Jr. , Subunit conformations and assembly states of a DNA-translocating motor: the terminase of bacteriophage P22. Journal of molecular biology 2007, 374, 817–36. [Google Scholar] [CrossRef]
- Nemecek, D.; Lander, G. C.; Johnson, J. E.; Casjens, S. R.; Thomas, G. J., Jr. , Assembly architecture and DNA binding of the bacteriophage P22 terminase small subunit. Journal of molecular biology 2008, 383, 494–501. [Google Scholar] [CrossRef]
- Roy, A.; Bhardwaj, A.; Datta, P.; Lander, G. C.; Cingolani, G. , Small terminase couples viral DNA binding to genome-packaging ATPase activity. Structure 2012, 20, 1403–13. [Google Scholar] [CrossRef] [PubMed]
- Duffy, C.; Feiss, M. , The large subunit of bacteriophage lambda’s terminase plays a role in DNA translocation and packaging termination. Journal of molecular biology 2002, 316, 547–61. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, S.; Kondabagil, K.; Rossmann, M. G.; Rao, V. B. , The functional domains of bacteriophage t4 terminase. The Journal of biological chemistry 2004, 279, 40795–801. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M. E.; Gaussier, H.; Catalano, C. E. , The DNA maturation domain of gpA, the DNA packaging motor protein of bacteriophage lambda, contains an ATPase site associated with endonuclease activity. Journal of molecular biology 2007, 373, 851–65. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M. G.; Moras, D.; Olsen, K. W. , Chemical and biological evolution of nucleotide-binding protein. Nature 1974, 250, 194–9. [Google Scholar] [CrossRef]
- Roy, A.; Cingolani, G. , Structure of p22 headful packaging nuclease. The Journal of biological chemistry 2012, 287, 28196–205. [Google Scholar] [CrossRef]
- Ponchon, L.; Boulanger, P.; Labesse, G.; Letellier, L. , The endonuclease domain of bacteriophage terminases belongs to the resolvase/integrase/ribonuclease H superfamily: a bioinformatics analysis validated by a functional study on bacteriophage T5. The Journal of biological chemistry 2006, 281, 5829–36. [Google Scholar] [CrossRef]
- Lokareddy, R. K.; Ko, Y. H.; Hong, N.; Doll, S. G.; Paduch, M.; Niederweis, M.; Kossiakoff, A. A.; Cingolani, G. , Recognition of an alpha-helical hairpin in P22 large terminase by a synthetic antibody fragment. Acta Crystallogr D Struct Biol 2020, (Pt 9) Pt 9, 876–888. [Google Scholar] [CrossRef]
- Zhao, H.; Christensen, T. E.; Kamau, Y. N.; Tang, L. , Structures of the phage Sf6 large terminase provide new insights into DNA translocation and cleavage. Proceedings of the National Academy of Sciences of the United States of America 2013, 110, 8075–80. [Google Scholar] [CrossRef]
- Sun, S.; Kondabagil, K.; Gentz, P. M.; Rossmann, M. G.; Rao, V. B. , The structure of the ATPase that powers DNA packaging into bacteriophage T4 procapsids. Molecular cell 2007, 25, 943–9. [Google Scholar] [CrossRef]
- Dauden, M. I.; Martin-Benito, J.; Sanchez-Ferrero, J. C.; Pulido-Cid, M.; Valpuesta, J. M.; Carrascosa, J. L. , Large terminase conformational change induced by connector binding in bacteriophage T7. The Journal of biological chemistry 2013, 288, 16998–17007. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Kondabagil, K.; Draper, B.; Alam, T. I.; Bowman, V. D.; Zhang, Z.; Hegde, S.; Fokine, A.; Rossmann, M. G.; Rao, V. B. , The structure of the phage T4 DNA packaging motor suggests a mechanism dependent on electrostatic forces. Cell 2008, 135, 1251–62. [Google Scholar] [CrossRef] [PubMed]
- Parent, K. N.; Gilcrease, E. B.; Casjens, S. R.; Baker, T. S. , Structural evolution of the P22-like phages: comparison of Sf6 and P22 procapsid and virion architectures. Virology 2012, 427, 177–88. [Google Scholar] [CrossRef]
- McNulty, R.; Lokareddy, R. K.; Roy, A.; Yang, Y.; Lander, G. C.; Heck, A. J. R.; Johnson, J. E.; Cingolani, G. , Architecture of the Complex Formed by Large and Small Terminase Subunits from Bacteriophage P22. Journal of molecular biology 2015, 427, 3285–3299. [Google Scholar] [CrossRef]
- Hegde, S.; Padilla-Sanchez, V.; Draper, B.; Rao, V. B. , Portal-large terminase interactions of the bacteriophage T4 DNA packaging machine implicate a molecular lever mechanism for coupling ATPase to DNA translocation. J Virol 2012, 86, 4046–57. [Google Scholar] [CrossRef] [PubMed]
- Lokareddy, R. K.; Bhardwaj, A.; Cingolani, G. , Atomic structure of dual-specificity phosphatase 26, a novel p53 phosphatase. Biochemistry 2013, 52, 938–48. [Google Scholar] [CrossRef]
- Yoshioka, C.; Pulokas, J.; Fellmann, D.; Potter, C. S.; Milligan, R. A.; Carragher, B. , Automation of random conical tilt and orthogonal tilt data collection using feature-based correlation. J Struct Biol 2007, 159, 335–46. [Google Scholar] [CrossRef]
- Suloway, C.; Pulokas, J.; Fellmann, D.; Cheng, A.; Guerra, F.; Quispe, J.; Stagg, S.; Potter, C. S.; Carragher, B. , Automated molecular microscopy: the new Leginon system. J Struct Biol 2005, 151, 41–60. [Google Scholar] [CrossRef]
- Lander, G. C.; Stagg, S. M.; Voss, N. R.; Cheng, A.; Fellmann, D.; Pulokas, J.; Yoshioka, C.; Irving, C.; Mulder, A.; Lau, P. W.; Lyumkis, D.; Potter, C. S.; Carragher, B. , Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 2009, 166, 95–102. [Google Scholar] [CrossRef]
- Voss, N. R.; Yoshioka, C. K.; Radermacher, M.; Potter, C. S.; Carragher, B. , DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol 2009, 166, 205–13. [Google Scholar] [CrossRef]
- Hauer, F.; Gerle, C.; Kirves, J. M.; Stark, H. , Automated correlation of single particle tilt pairs for Random Conical Tilt and Orthogonal Tilt Reconstructions. J Struct Biol 2013, 181, 149–54. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C. O.; Bilbao-Castro, J. R.; Shkolnisky, Y.; Alcorlo, M.; Melero, R.; Caffarena-Fernandez, G.; Li, M.; Xu, G.; Marabini, R.; Carazo, J. M. , A clustering approach to multireference alignment of single-particle projections in electron microscopy. J Struct Biol 2010, 171, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T. R.; Gao, H.; Baxter, W. T.; Asturias, F. J.; Boisset, N.; Leith, A.; Frank, J. , SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat Protoc 2008, 3, 1941–74. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S. H. , RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 2012, 180, 519–30. [Google Scholar] [CrossRef]
- Hohn, M.; Tang, G.; Goodyear, G.; Baldwin, P. R.; Huang, Z.; Penczek, P. A.; Yang, C.; Glaeser, R. M.; Adams, P. D.; Ludtke, S. J. , SPARX, a new environment for Cryo-EM image processing. J Struct Biol 2007, 157, 47–55. [Google Scholar] [CrossRef]
- Yang, Z.; Fang, J.; Chittuluru, J.; Asturias, F. J.; Penczek, P. A. , Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure 2012, 20, 237–47. [Google Scholar] [CrossRef]
- Rose, R. J.; Damoc, E.; Denisov, E.; Makarov, A.; Heck, A. J. , High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nature methods 2012, 9, 1084–6. [Google Scholar] [CrossRef]
- Rosati, S.; Rose, R. J.; Thompson, N. J.; van Duijn, E.; Damoc, E.; Denisov, E.; Makarov, A.; Heck, A. J. , Exploring an orbitrap analyzer for the characterization of intact antibodies by native mass spectrometry. Angewandte Chemie 2012, 51, 12992–6. [Google Scholar] [CrossRef]
- Snijder, J.; van de Waterbeemd, M.; Damoc, E.; Denisov, E.; Grinfeld, D.; Bennett, A.; Agbandje-McKenna, M.; Makarov, A.; Heck, A. J. , Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by Orbitrap mass spectrometry. Journal of the American Chemical Society 2014, 136, 7295–9. [Google Scholar] [CrossRef]
- Moore, S. D.; Prevelige, P. E., Jr. , Bacteriophage p22 portal vertex formation in vivo. Journal of molecular biology 2002, 315, 975–94. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, P.; Wang, N.; Chen, Z.; Su, D.; Zhou, Z. H.; Rao, Z.; Wang, X. , Architecture of the herpesvirus genome-packaging complex and implications for DNA translocation. Protein Cell 2020, 11, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Lander, G. C.; Olia, A. S.; Li, R.; Casjens, S.; Prevelige, P., Jr.; Cingolani, G.; Baker, T. S.; Johnson, J. E. , Peering down the barrel of a bacteriophage portal: the genome packaging and release valve in p22. Structure 2011, 19, 496–502. [Google Scholar] [CrossRef] [PubMed]
- McNulty, R.; Cardone, G.; Gilcrease, E. B.; Baker, T. S.; Casjens, S. R.; Johnson, J. E. , Cryo-EM Elucidation of the Structure of Bacteriophage P22 Virions after Genome Release. Biophys J 2018, 114, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



