Submitted:
10 September 2023
Posted:
12 September 2023
You are already at the latest version
Abstract
Lung adenocarcinoma (LUAD) is the most common lung cancers, which accounts for about 35%-40% of all lung cancer patients. Despite therapeutic advancements in recent years, the overall survival time of the LUAD patients still remain poor, especially KRAS mutant LUAD. Therefore, it is necessary to further explore novel targets and drugs to improve the prognosis of LUAD. Ferroptosis, an iron-dependent regulated cell death (RCD)caused by lipid peroxidation, has attracted much attention recently as an alternative target for apoptosis in LUAD therapy. Ferroptosis has been found closely related with LUAD, at its every stage, including initiation, proliferation and progression. In this review, we will provide a comprehensive overview on ferroptosis mechanisms, it’s regulation in LUAD, and application of targeting ferroptosis for LUAD therapy.
Keywords:
ferroptosis
; regulation
; lung adenocarcinoma
; therapy
1. Introduction
Lung cancer is the malignant tumor with the highest morbidity and mortality rate in China and even in the world, of which lung adenocarcinoma (LUAD) is the most common pathological type, accounting for about 40% of lung cancer[1]. LUAD requires individualized treatment based on tumor progression: early-stage LUAD is mainly treated surgically[2], whereas advanced-stage and metastatic patients require systemic therapy[3]. These treatments can significantly improve patients' symptoms and prolong their overall survival to some extent. However, anti-tumor efficiency in LUAD patients is usually limited by chemotherapy resistance and apoptotic escape, leading to tumor recurrence and poor prognosis[4,5]. In addition, about 25-30% of lung adenocarcinoma patients obtain KRAS mutations[6]. The small size and smooth surface of the KRAS protein makes it hard to be targeted for small molecular drugs[7]. Therefore, the search for specific molecular targets and the development of non-apoptosis-inducing drugs remains a high priority in treatment of LUAD, especially KRAS-mutant LUAD.
Ferroptosis is a novel type of regulated cell death (RCD) with unique molecular alterations and morphological features identified by Dixon et al in 2012 [8]. The essence of ferroptosis is a membrane rupture cell death caused by the accumulation of iron-dependent lipid peroxides[9]. There is increasing evidence that application of ferroptosis inducers (FINs) or modulation of ferroptosis-regulated genes (FRGs) can inhibit tumor cell growth and overcome drug resistance due to escape from apoptosis, including LUAD[10]. However, a comprehensive review of the regulation and targeted-therapy of ferroptosis in LUAD has yet not been performed. Here, we examine the core mechanisms of ferroptosis and discuss its regulation and therapeutic implications in the context of LUAD.
2. Molecular mechanisms of ferroptosis
2.1. Iron metabolism
Iron is one of the essential trace elements in the human body and plays important physiological and biochemical functions[11]. Iron in the body generally exists in ferrous (Fe2+) or ferric (Fe3+) forms, of which Fe2+ is essential in inducing ferroptosis. Excess Fe2+ will undergo a Fenton reaction with hydrogen peroxide, generating free hydroxide (OH-) and hydroxyl (OH.) radicals that can rapidly promote peroxidation of membrane lipids, leading inito ferroptosis[11]. Iron in normal tissues is in a dynamic equilibrium: circulating Fe3+ enters the cell via transferrin (TF) and transferrin receptor (TFR), and intracellular Fe3+ is stored as ferritin (including two subtypes, ferritin heavy chain [FHL] and ferritin light chain [FTL]) or reduced to Fe2+ and transferred out of the cell to participate in iron recirculation to maintain intracellular iron homeostasis[12,13]. Increasing TF-mediated iron uptake or NCOA4-mediated ferritinophagy leads to an increase in intracellular labile iron. Ferritinophagy refers to the specific recognition of and binding to ferritin by nuclear receptor coactivator 4 (NCOA4), which mediates the autophagic degradation of ferritin in the lysosome thereby promoting the release of Fe2+[14]. The expression of NCOA4 can promote erastin-induced ferroptosis in LUAD cells by promoting FHL autophagy[15].
2.2. Lipid metabolism
Another important factor for inducing ferroptosis is the peroxidation of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs), especially arachidonic acid (AA) and adrenic acid (AdA)[16]. The formation of lipid peroxides needs three key enzymes, acyl-CoA synthetase long-chain family member (ACSL4), lys phosphatidylcholine acyltransferase 3 (LPCAT3) and lipoxygenases (ALOXs)[16,17]. Among them, ACSL4 is responsible for catalyzing the acylation between AA/AdA and CoA to generate AA-CoA/AdA-CoA; while LPCAT3 is responsible for catalyzing the re-esterification of AA/AdA-CoA and membrane phosphatidylethanolamines (PLs) to generate AA/AdA-PE; and the ALOXs, a class of enzymes with iron as a cofactor, is responsible for oxidizing PUFAs to produce hydroperoxyl derivatives such as 4-malondialdehyde(MDA) and Hydrodynamical(HNE). Common ALOXs are ALOXE3, ALOX5, ALOX12, ALOX12B, ALOX15, and ALOX15B[17]. Besides, Cytochrome P450 oxidoreductase (POR) can also mediate lipid peroxidation and promote ferroptosis[18]. Notably, ACSL4 and LPCAT3 were expressed at elevated levels in highly malignant LUAD patients[19], whereas 4-HNE was lower in highly staged tumors relative to low-staged tumors and normal tissues[20], all of which associated with high ferroptosis sensitivity, suggesting that lipid metabolism modulates the ferroptosis sensitivity in LUAD in a multifaceted and multi-mechanistic manner. On the other hand, monounsaturated fatty acids (MUFAs), synthesized by stearoyl-CoA desaturase1 (SCD1), can competitively bind to cell membranes thereby inhibiting ferroptosis[19].
2.3. Antioxidant defenses
Cell defenses against ferroptosis by multiple antioxidant systems, such as the solute carrier family 7 member 11 (SLC7A11)-reduced glutathione (GSH)-glutathione peroxidase 4 (GPX4) pathway[21], apoptosis inducing factor mitochondria-associated 2 (AIFM2, the newly renamed as ferroptosis suppressor protein 1 (FSP1))-coenzyme Q (CoQ10) axis[22], and the GTP cyclohydrolase-1(GCH1)-tetrahydrobiopterin /dihydrobiopterin (BH4/BH2) pathway[23].
Among them, the SLC7A11-GSH-GPX4 pathway is the most important and earliest discovered antioxidant system[21]. SLC7A11 is a cystine/glutamate transporter, which can uptake extracellular cystine into the cell, and transport glutamate out of the cell simultaneously[24]. And the cystine that enters the cell can be used for the synthesis of GSH, which is not only an important antioxidant, but also a preferred substrate for GPX4. GPX4, a selenoprotease, can act as a detoxifier by converting highly toxic lipid hydroperoxides into non-toxic lipiodols and hydroperoxides into water. Erastin, a ferroptosis inducer, directly inhibits the activity of the XC system, leading to the accumulation of intracellular glutamate, a decrease in GSH synthesis, and a reduction in the activity of GPX4, thereby leading to cell ferroptosis[8]. Thus, SLC7A11 and GPX4 are important ferroptosis regulators as well as potential targets for cancer therapy.
Next, the FSP1-CoQ10 pathway was recently found to be able to protect cells from ferroptosis by reducing CoQ10 to producing ubiquinol, a lipid radical trapping agent[22]. Notably, FSP1-CoQ10 functions in a subcellular organelle-specific manner, with pro-apoptotic effects in mitochondria and anti-ferroptosis in the plasma membrane. In non-small cell lung cancer (NSCLC), colorectal cancer (CRC) and pancreatic ductal adenocarcinoma (PDAC), FSP1 is expressed at higher levels in KRAS-mutant tumor cells compared to normal cells[25]. But considering the diversity of FSP1 functions, it is reasonable to believe that FSP1 may also promote tumor progression through mechanisms other than ferroptosis. Finally, the GCH1-BH4/BH2 pathway can function as antioxidants protecting from ferroptosis in the absence of GPX4[23].
3. Ferroptosis in LUAD
3.1. Ferroptosis in LUAD tumorigenesis and progression
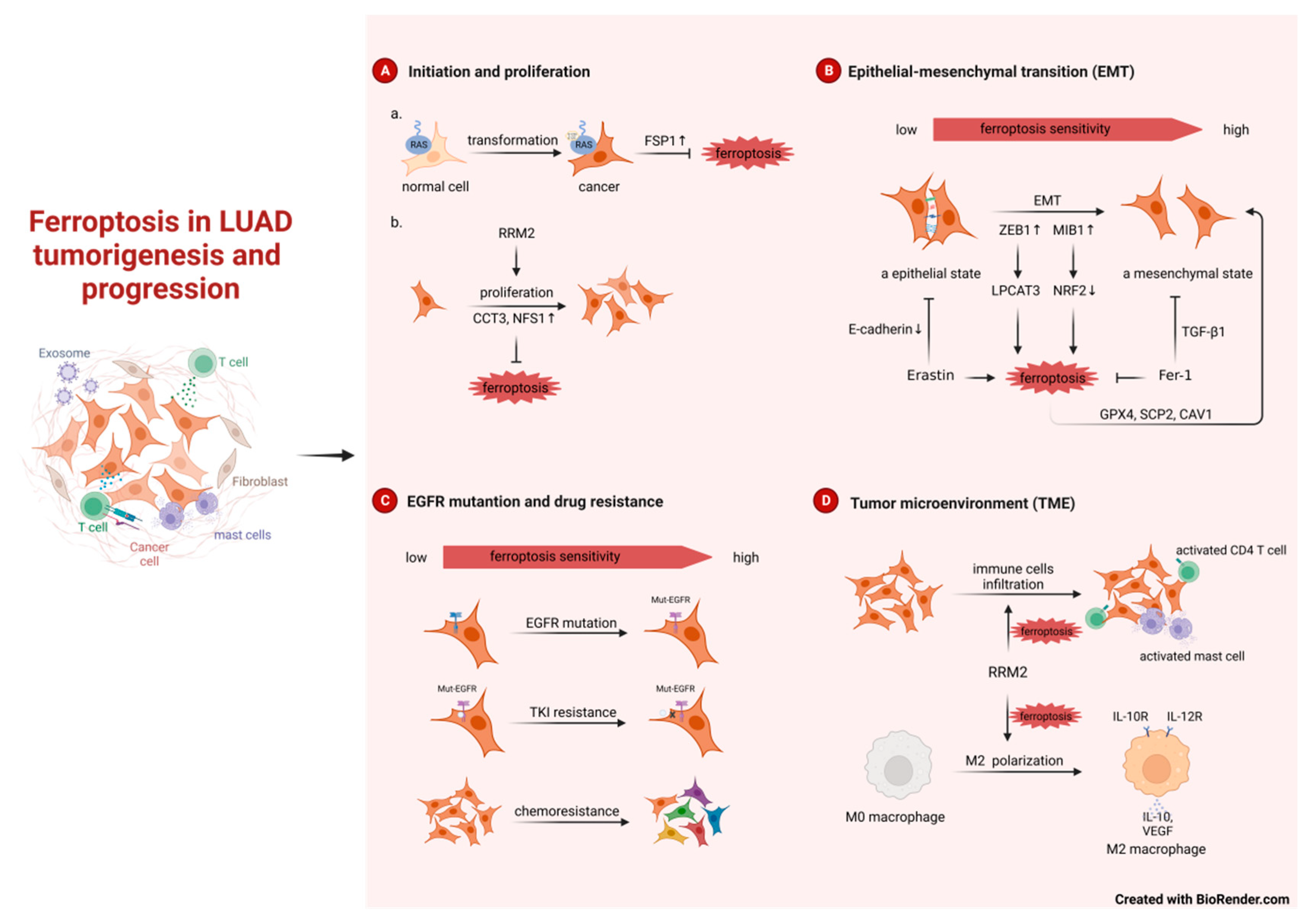
3.1.1. Ferroptosis in LUAD tumorigenesis
About 25-30% of lung adenocarcinomas develop KRAS mutations, with the KRAS-G12C point mutation being the most common[6,26]. When G12C mutation occurs in KRAS, its GTP hydrolase activity will be lost, which will activate several signaling pathways such as RAF-MEK-ERK, PI3K-AKT-mTOR and Ral-GDS, and then the cells will have malignant potential and gradually become cancerous, which will eventually lead to the development of LUAD[27]. It is established that KRAS-mediated cellular transformation requires the generation of reactive oxygen species (ROS) due to elevated expression of NADPH oxidase 1(Nox1)[28]. So how do cells expressing mutant KRAS alleviate ROS-induced cell death? That is, FSP1 expression was sufficient in KRAS-mutated LUAD to significantly promote 3D spheroid growth in vitro and accelerate tumor onset in vivo by protecting from ferroptosis[25]. Consistent with this finding, Zang et al showed that KRAS-driven LUAD has a greater resistance to ferroptosis owing to a reprogrammed lipid metabolism by a higher level of acyl-coenzyme A synthetase long-chain family member 3 (ACSL3) expression[29]. Due to the small size and smooth surface of the KRAS protein, there is no suitable binding pocket for other small molecules except the one for GTP/GDP, and the affinity of RAS for GTP is very high, which makes it difficult for drugs to compete with the substrate[7]. The treatment of KRAS-mutated LUAD has been a challenge. Intervention of FSP1 and expression in the early stage of atypical adenomatoid hyperplasia (AAH) to adenocarcinoma in situ (AIS) or to micro infiltrating adenocarcinoma (MIA) and thus intervention in ferroptosis tumor initiation, may be a novel therapeutic direction for KRAS-mutated LUAD.
3.1.2. Ferroptosis in LUAD progression
Ferroptosis is also linked to the growth and development of LUAD. Researches found many ferroptosis inhibitors act as indicators of poor prognosis, promotes tumor cells growth in LUAD, such as CCT3 and NFS1 [30,31]. In addition, bioinformatics analyses have revealed a potential clinical connection between the ferroptosis-related genes (FRGs) and LUAD patients. A prognostic model integrating several FRGs was used to predict the prognosis, mutation burden, TME cell infiltration characteristics and immunotherapy effects, chemotherapy sensitivities in patients with LUAD[32,33,34,35,36,37].
Epithelial-mesenchymal transition (EMT) and ferroptosis are two important processes in tumor progression and recent studies reported that they may form a positive feedback loop to a certain extent in LUAD. On the one hand, it has been known that highly mesenchymal-like tumor cells are indeed more sensitive to ferroptosis inducers. The E3 Ligase MIB1 can promote EMT, while at the same time, promote ferroptosis by NRF2 degradation[38]. ZEB1 is one of the major transcription factors that regulate EMT by binding to the E-box in E-cadherin[39]. In LUAD, ZEB1 correlates with the transcription of LPCAT3 and thus increases the susceptibility to ferroptosis[19]. On the other hand, ferroptosis tendency also enhances the development of EMT. Erastin, which is an inducer of ferroptosis, can reduces the expression of E-cadherin and causes de-epithelialization. While Ferrostatin-1(Fer-1), an inhibitor of ferroptosis, can partially inhibit EMT induced by TGF-β1[40]. Not coincidentally, some ferroptosis markers (GPX4, SCP2, CAV1) again suggest that ferroptosis can positively regulate the occurrence of EMT in vivo [19]. However, there is also negative feedback loop between EMT and ferroptosis. High ARNTL2 expression was associated with EMT and lymph node metastasis in patients with LUAD, while it plays an inhibiting role in ferroptosis[41].
Epidermal growth factor receptor (EGFR) is the most common mutation in LUAD, with a prevalence of 15% of Caucasians and 50% of Asians[42,43]. EGFR tyrosine kinase inhibitors (EGFR-TKIs) are used to treat EGFR mutant LUAD, first- to third-generation EGFR-TKIs have been approved both domestically and internationally, and fourth-generation EGFR-TKIs such as BLU-945 have entered clinical studies[44,45]. EGFR-mutant LUAD cells also increase cellular sensitivity to ferroptosis. Low-dose selenite synergized with osimertinib in EGFR-mutant H1975[46]. Acquired resistance is still inevitable with the use of EGFR-TKIs, and about 20%-30% of EGFR mutant LUADs are intrinsically resistant to EGFR-TKIs[47,48,49]. In addition to mesenchymal state and EGFR-mutant tumor cells mentioned above, EGFR-TKI resistant LUAD cells also increase cellular sensitivity to ferroptosis, and the histone deacetylase inhibitor Vorinostat can further downregulate the expression of xCT in EGFR mutant LUAD cells and enhance the effect of ferroptosis induction therapy[50]. Further work on the clinical effect of these drugs and their combination with TKI on EGFR mutation-LUAD are warranted. Besides, chemoresistance also makes LUAD cells more sensitive to ferroptosis, and promoting ferroptosis can overcome or reverse the resistance of tumor cells to cisplatin, pemetrexed and Lapatinib[51,52,53].
The tumor microenvironment constitutes the balance between tumor cells and immune cells and can have both adverse and beneficial consequences during tumor progression. Ribonucleotide reductase subunit M2 (RRM2) not only affects tumor cells proliferation but also regulates immune cells infiltration, thereby influence lung cancer progression in a ferroptosis-dependent manner. For one thing, depletion or silencing RRM2 inhibited the proliferation and induce ferroptosis in H1975 and H358 LUAD cells[54,55]. For another, RRM2 effectively promoted M2 macrophage polarization, facilitating tissue repair and LUAD development in vitro and in vivo [56]. RRM2 also regulated the infiltration levels of activated mast cells and activated CD4 memory T cells, again suggesting that RRM2 may be engaged in immune infiltration[54]. Not coincidentally, Bioinformatics has demonstrated that others FRGs can modify the behavior of TME cells, and that these subtypes of TME cells exhibited distinct biological features and communicate extensively with tumor epithelial cells. Patients with a higher abundance of these ferroptosis-related TME cell subtypes have a better clinical outcome[53]. In addition, Ferroptosis-related prognostic signatures, such as prognostic ferroptosis-related lncRNA signature and GPX4-related prognostic signature, are not only correlated with multiple tumor-infiltrating immune cells and immune-associated processes and pathways in TME, but also with the response to immunotherapy, chemotherapy, and targeted therapy[57,58].
Figure 1.
Ferroptosis in LUAD tumorigenesis and progression. RRM2, ribonucleotide reductase subunit M2;CCT3, chaperonin containing TCP1 subunit 3;NFS1, nitrogen fixation 1;ZEB1, zinc finger E-box binding homeobox 1;Fer-1, ferrostatin-1;TKI, tyrosine kinase inhibitor.
Figure 1.
Ferroptosis in LUAD tumorigenesis and progression. RRM2, ribonucleotide reductase subunit M2;CCT3, chaperonin containing TCP1 subunit 3;NFS1, nitrogen fixation 1;ZEB1, zinc finger E-box binding homeobox 1;Fer-1, ferrostatin-1;TKI, tyrosine kinase inhibitor.
3.2. Regulation of ferroptosis in LUAD
Ferroptosis is essentially a form of cell death caused by oxidative damage. Like other RCDs (e.g., apoptosis, pyroptosis, and necrosis), it is modifiable. In LUAD, the core regulators of ferroptosis are mainly regulated at the transcriptional and epigenetic levels.
3.2.1. Transcriptional regulation
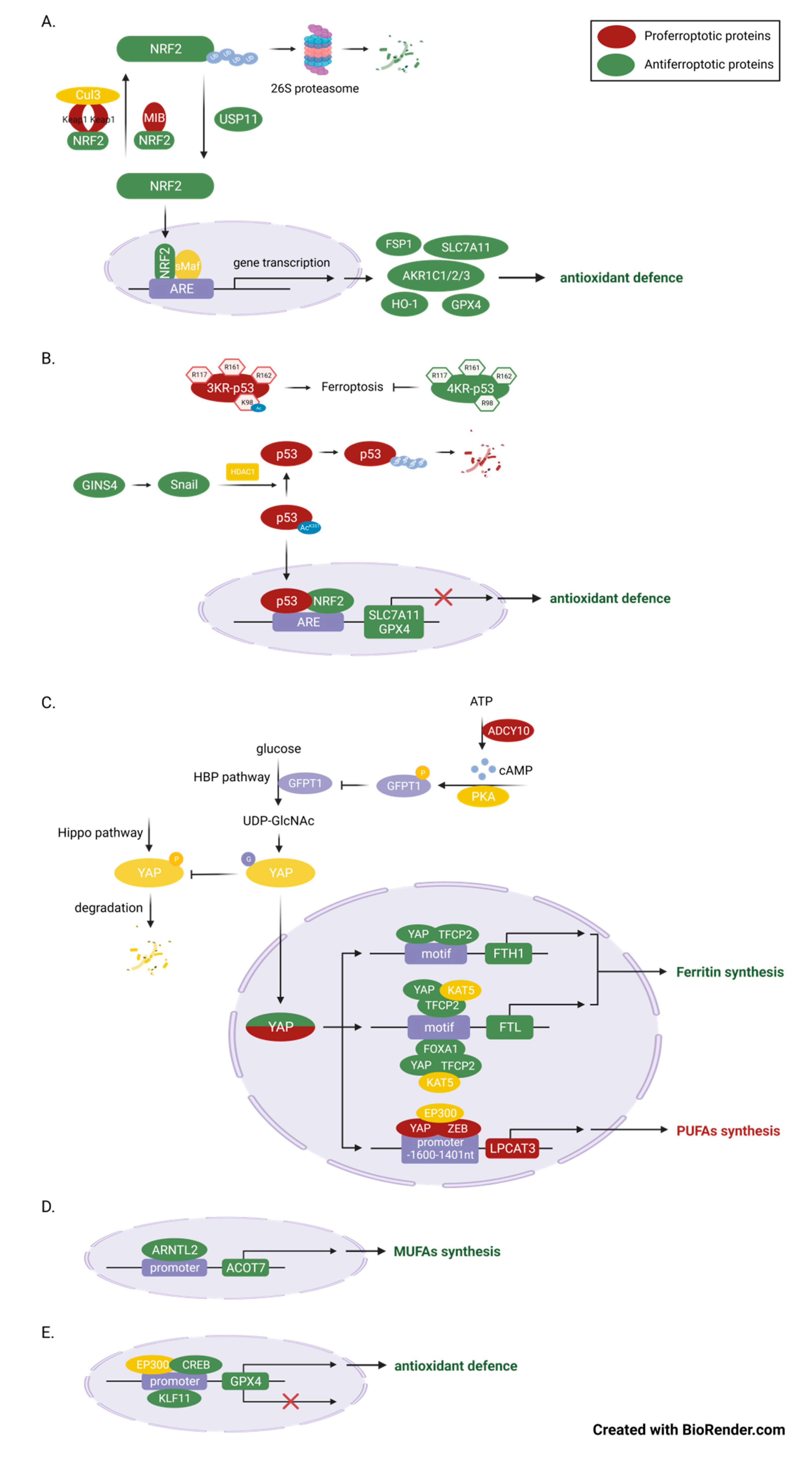
In LUAD, transcriptional factors and cofactors such as NRF2, YAP, ARNTL2, CREB and KLF11, etc., are key regulators in various step of ferroptosis and thus in tumor pathogenesis and progression.
NRF2
The master transcriptional factor nuclear factor erythroid 2-related factor 2 (NFE2L2, also known as NRF2), plays an important antioxidant role in maintaining redox homeostasis. The role of NRF2 in ferroptosis is a popular subject of ongoing research,with a major of studies suggest that NRF2 plays an anti-ferroptotic role in LUAD cells. As we know, under the induction of oxidative stress, NRF2 can transfers into nucleus, where it binds to partner proteins such as Maf and subsequently initiates the transcription of a series of antioxidant genes, including NAD(P)H quinone dehydrogenase 1 (NQO1)[59], GPX4, SLC7A11, the aldo-keto-reductase-1C (AKR1C) family[60] and heme oxygenase-1 (HO-1), as well as FSP1 [25]. In KRAS-mutant cells, the Aifm2 mRNA, encoding the protein FSP1, is upregulated because of MAPK-NRF2 pathway activation[25]. The expression of FSP1 can protect cells from ferroptosis-associated lipid ROS accumulation[22,61]. Although this is contrary to the previous finding that overexpression of oncogenic KRAS in fibroblasts resulted in sensitization to erastin-induced ferroptosis[62]. It coincides with the fact that GPX4 deletion in pancreatic intraepithelial neoplasia (PanINs) is not sufficient to trigger ferroptosis in genetically engineered mouse models, supporting the idea that KRAS-mutant cells have evolved additional antioxidant mechanisms to counteract ferroptosis[63]. Among, NRF2-mediated transcriptional activation may be critical in determining whether oncogenic KRAS expression pro-ferroptosis or anti-ferroptosis. Up to half of patients present with activating KRAS mutations[25], it is therefore tempting to speculate whether combined induction of ferroptosis and inhibition of FSP1 can increase the therapeutic effectiveness of KRAS mutated LUAD in a clinical setting.
Ubiquitination and proteasome-mediated degradation are the most important mechanisms regulating the expression and activity of NRF2, which in turn controls the ferroptotic resistance to LUAD. First and most important is the Keap1-cullin 3 (Cul3)-E3-mediated Ubiquitinated protein degradation[64]. Kelch-like ECH-associated protein 1 (KEAP1), which maintains low levels of NRF2 in homeostasis, mediates 26S proteasome degradation by recruiting a Cul3-containing E3 ubiquitin ligase complex to NRF2[64,65]. KEAP1-mutant lung adenocarcinoma account for about 17% of LUAD[66], inactivation of KRAP1 leads to constituent activation of NRF2 signaling which favors tumorigenesis, so that even with the intervention of chemotherapy and immunotherapy, the survival of such LUAD patients is shorter [67,68]. For example, activation of the KRAP1-NRF2-AKRs pathway can maintain the proliferation rate of LUAD cells, where AKR1C1/2/3 antagonizes ferroptosis through promoting detoxification of active intermediates of aldehydes and ketones. Among others, this effect is greatly enhanced by the cooperativeness of STK11 and KEAP1 loss-of-function, independent of KRAS mutation status[60,67]. It was found that, in addition to KEAP1, NRF2 is also capable of being degraded by MIB1, another E3 ubiquitin ligase[38]. High expression of MIB1 in A549 cells affects HMOX1 transcription and subsequent lipid ROS clearance by downregulating NRF2. Lung squamous and adenocarcinoma patients with high MIB1 expression are also associated with lower survival rates. Vice versa, ubiquitination of NRF2 can be removed by de-ubiquitinating enzymes(DUBs) because ubiquitination is a dynamic and reversible process, and this process in non-small cell lung cancer(NSCLC) is mediated by ubiquitin-specific processing protease 11 (USP11) [64]. Our study revealed that RSL3 promotes ferroptosis in A549 and H2122 cells by directly inhibiting USP1-mediated de-ubiquitination and promoting degradation of NRF2, thereby repressing transcription of SLC7A11 and GCL[69].
p53
Wild-type TP53 is an suppressor that negatively regulates the growth of LUAD,while mutant TP53 has oncogenic activity and is one of the critical causes of LUAD transformation. Approximately 33% of LUAD patients are associated with mutations in the TP53 gene, and Thompson et al reported that LUAD NCI-H1299 cells expressing mutant p53 had increased sensitivity to the ferroptosis inducer, Erastin, compared to WT p53 cells[70]. This may be due to it suppression on SLC7A11 expression, through entrapping the transcription factor NRF2[71]. In addition, p53 R273H cells were more vulnerable to AF-induced ferroptotic cell death due to downregulation of GPX4 and lipid peroxidation[72]. Acetylation is an important layer of p53 functional regulation mechanism in ferroptosis and tumor inhibition. In fact, mouse p53 3KR mutations at K117, K161, and K162 activated ferroptosis and inhibited tumor growth, but the additional mutation of p53 at K98 would abolish this ferroptosis-promoting process[73]. Additionally, GINS4 suppressed p53-mediated ferroptosis through activating Snail that antagonized the acetylation of p53 lysine residue 351 (K351 for human p53), thereby resulted in an upregulation of the ubiquitination level and a decrease in the stability of p53[74].
YAP
YAP, a cotranscription factor acting downstream of the Hippo pathway, not only functions as a proto-oncoprotein, but is also closely related to ferroptosis sensitivity in LUAD[75]. Inhibition of system Xc activity, which reduces cystine entry into the cell, leads to intracellular glutamate accumulation simultaneously. As previously mentioned, ferroptosis sensitivity varies widely in cells and tissues and may be regulated by the release of liable iron and lipid peroxides[14]. Zhang et al firstly found that erastin inhibition of system Xc-induced endogenous glutamate accumulation was able to determine ferroptosis sensitivity of LUAD cells via inhibiting the ADCY/PKA/HBP/YAP axis[15]. Glutamate accumulation activates ADCY10 in a Ca2+-dependent manner, ADCY10 confers varies impacts of glutamate by produce cAMP and cAMP and PKA-dependent phosphorylation and suppression of GFPT1[15]. Thereby, the O-GlcNAcylation and stability of YAP are reduced. YAP is a coactivator that does not function as a direct transcription factor and requires the formation of transcription-promoting complex with other transcription factors[76]. Next, they also found that YAP regulates iron-dependent ferroptosis sensitivity through the formation of the YAP-TFCP2-complex[15,20]. Inhibition of YAP promotes NCOA4-mediated ferritinophagy in different LUAD cell lines[15]. And inhibits FTL transcription via YAP-TFCP2-FOXA1 complex in PC9 and H1299 cells lines, thus doubly promotes labile iron production to enhance the ferroptotic effect[20]. Finally, Zhang et al also found that YAP regulates lipid-dependent ferroptosis sensitivity through the formation of YAP-ZEB-complex. As previously stated, the lipid metabolism-related enzymes, LPCAT3, is a ferroptosis inducer [77]. YAP-ZEB-complex binds to LPCAT3 promoter, promoting its transcription, lipid peroxidation and ferroptosis[19]. These studies have not only identified therapeutic targets of ferroptosis (such as YAP, FTL and FTH), but have also identified several ferroptotic sensitivity biomarkers (such as CODY10 and LPCAT3) in LUAD, all of which may provide support for ferroptosis use in clinical oncology therapy.
Others
Ferroptosis is, in fact, an iron-dependent, unchecked lipid peroxidation form of cell death [8]. In addition to the regulation on ferroptosis by NRF2 and YAP mentioned above, any other transcription factors capable of influencing the transcription of lipid oxidation and anti-lipid oxidation genes would be able to intervene in ferroptosis. As a first example, the aryl hydrocarbon receptor nuclear translocator like 2 (ARNTL2), as a circadian transcription factor, increase the expression of acyl-CoA thioesterase 7(ACOT7) via binding to the ACOT7 promoter, which in turn promotes the synthesis of MUFAs such as oleic acid and palmitoleic acid[78]. Given that ARNTL2 promotes EMT and cell proliferation and migration in addition to inhibiting lipid peroxidation and iron death via ACOT7, ARNTL2 may be a potential poor biomarker for LUAD[41]. As a second example, cAMP response element-binding protein (CREB) and kruppel-like factor 11 (KLF11) can promote or repress GPX4 expression by binding to the GPX4 promoter, respectively. The high expression of CREB in LUAD patient’s tumor tissues and KLF11 in RSL3/IKE-treat A549 and PC9 cells further supports the ideas that CREB or KLF11 is a positive or negative regulators of antioxidant defense[79,80].
Figure 2.
Master transcriptional regulators of ferroptosis in LUAD. (A) NFE2L2 is a crucial anti-ferroptotic transcription factor in LUAD cells, as it regulates multiple genes involved in antioxidant defense. The expression of NFE2L2 is primarily controlled by ubiquitination and proteasome-mediated degradation. Multiple E3 ubiquitin ligases or de-ubiquitinating enzymes regulate the sensitivity to ferroptosis by controlling the expression of NFE2L2 in LUAD cells. (B) TP53 inhibits ferroptosis and LUAD progression by suppressing the expression of SLC7A11 and GPX4. Acetylation is very important in the regulation of its function in ferroptosis because it increases the stability of p53 protein. (C) YAP1 plays a dual role in ferroptosis, depending on the expression of its target genes. The O-GlcNAcylation of YAP1, which is mediated by O-GlcNAc, enhances the activity of YAP1 and its transcriptional ability. In LUAD cells, the sensitivity to ferroptosis can be regulate by controlling its upstream ADCY/PKA/HBP axis. (D) ARNTL2 promotes ferroptosis by promoting the expression of ACOT7, a MUFA synthesis-related enzymes. (E) CREB1 inhibits while KLF11 promotes ferroptosis by binding to the promoter of GPX4.
Figure 2.
Master transcriptional regulators of ferroptosis in LUAD. (A) NFE2L2 is a crucial anti-ferroptotic transcription factor in LUAD cells, as it regulates multiple genes involved in antioxidant defense. The expression of NFE2L2 is primarily controlled by ubiquitination and proteasome-mediated degradation. Multiple E3 ubiquitin ligases or de-ubiquitinating enzymes regulate the sensitivity to ferroptosis by controlling the expression of NFE2L2 in LUAD cells. (B) TP53 inhibits ferroptosis and LUAD progression by suppressing the expression of SLC7A11 and GPX4. Acetylation is very important in the regulation of its function in ferroptosis because it increases the stability of p53 protein. (C) YAP1 plays a dual role in ferroptosis, depending on the expression of its target genes. The O-GlcNAcylation of YAP1, which is mediated by O-GlcNAc, enhances the activity of YAP1 and its transcriptional ability. In LUAD cells, the sensitivity to ferroptosis can be regulate by controlling its upstream ADCY/PKA/HBP axis. (D) ARNTL2 promotes ferroptosis by promoting the expression of ACOT7, a MUFA synthesis-related enzymes. (E) CREB1 inhibits while KLF11 promotes ferroptosis by binding to the promoter of GPX4.
3.2.2. Epigenetic regulation
In addition to ferroptosis regulation at the transcriptional level, the susceptibility of LUAD to ferroptosis can also be regulated at several epigenetic levels, including DNA methylation, histone modification and various non-coding RNA-mediated processes. Despite recent emerging evidence supporting a role for DNA methylation in regulating GPX4 expression and the widespread use of ferroptosis-related DNA methylation signature as predictive and prognostic biomarkers for a wide range of cancers[81,82,83], DNA methylation in ferroptosis sensitivity of LUAD remains relatively understudied. For example, a recent study mentions that selenite is a potent LUAD FIN along with the activation of DNA methylation. However, selenite induction of ROS and possible inhibition of xCT expression, as well as downregulation of DNMT1 mediated TET1 DNA methylation are two parallel antineoplastic mechanisms in EGFR- and potentially KRAS-mutant lung cancer cells (H1975 and H358 cell lines), and there is no mention of an interconnection between the two[46]. So, we next focus on the role of two other epigenetic mechanisms in the regulation of LUAD ferroptosis in the following.
Histone modification
As we all know, histone modifications such as methylation and acetylation can regulate transcriptional activity[84]. The acetyltransferase E1A binding protein P300(EP300), for example, promotes the binding of transcription factors to target genes by increasing H3K27ac[85]. Most typically EP300 binds to the bZIP domain of CREB via its CBP/p300-HAT domain to form a transcription-promoting complex[86]. This transcriptional complex greatly enhances the binding of CREB to the Gpx4 promoter in the previously mentioned A549 and H1299 cells[79]. Even, EP300 can form a ternary complex with YAP and ZEB, wherein the YAP WW domain interacts with the ZEB ZF domain, and YAP also binds to the Bromo domain of EP300, while ZEB binds to the CBP/p300-HAT domain of EP300[19]. Given that the ternary complex formed by two-by-two binding of the three proteins is much more stable than the binary complex, targeting this EP300-YAP-ZEB transcription complex may be a new strategy for treating LUAD.
Non-coding RNA-mediated processes
- miRNAs
An increasing number of miRNAs, small (17-24 nucleotides) non-coding RNAs, have been recognized as important players in ferroptosis regulation. They can bind to the 3'-UTR of messenger RNAs (mRNAs) and mediate the post-transcriptional silencing of FRGs[87]. For example, miR-27a-3p regulates SLC7A11 expression to alter the sensitivity of A549 and Calu-3 LUAD cells to erastin-trigged ferroptosis [88]. miR-324-3p regulates GPX4 to alter the desensitization of A549 DDP cells to cisplatin-induced ferroptosis [51], and bioinformatics analyses predicted that SLC7A11 and GPX4 are targets of miRNAs such as has-mir-37 and miRNA-126-3p/5p[89]. But whether and how these miRNAs regulate them deserves further investigation.
- CircRNAs
Speaking of miRNAs, it's important to mention competing endogenous RNAs (ceRNAs), generally circRNAs and lncRNAs, which act as "miRNA sponges" to mitigate the inhibitory effects of miRNAs on their target genes[90,91]. Pan et al added another example of this in LUAD. They found that circRNA P4HB acts as a sponge for miRNA-1184 and competitively binds miRNA-1184 to SLC7A11, thereby inhibiting its inhibitory effect on Slc7a11 and promoting Slc7a11 expression and subsequent GSH synthesis[92]. This circP4HB /miR-1184/ SLC7A11 axis protects LUAD from erastin-induced ferroptosis and promotes tumor growth in vivo and in vitro. In addition to functioning as ceRNAs, circRNAs also directly interact with proteins to inhibit ferroptosis in LUAD[93]. The exosome-derived circRNA, circ_1010093, acts as a novel ferroptosis suppressor in LUAD. Autocrine cir93 secreted by tumor cells inhibits AA incorporation into the plasma membrane and reduce lipid peroxidation in a FABP3-dependent manner. Mechanistically, the exosomal cir93 maintains intracellular levels of cir93 and promotes its binding to the AA transporter protein fatty acid-binding protein 3 (FABP3) thereby reducing AA global levels via promotes the reaction of AA with taurine. Moreover, N-arachidonoyl taurine (NAT), the product of AA and taurine, inhibit ACSL4, LPCAT3 and PLTP expression by competitive binding to transcription factor TAX2, thereby further reducing AA incorporation into the plasma membrane[94]. This shows that cellular communication (autocrine secretion of cir93) in the tumor microenvironment is also capable of activating the antioxidant system and inhibiting ferroptosis. Both circP4HB in tumor cells and cir93 in patient plasma are biomarkers of LUAD and deserve further investigation in the early diagnosis and treatment of LUAD.
- LncRNAs
Another typical ceRNA is long non-coding RNAs (lncRNAs), a large number of which act as oncogenes and against cellular ferroptosis in LUAD, including the LINC00324/miR-200c-3p/TFAP2A pathway[32], the lncRNA GSEC/miR-101-3p/CISD1 pathway[95], the LncRNA T-UCR Uc.339/miR-339/SLC7A11 pathway[96] and the LINC00336/miR-6852/CBS pathway[97]. Of these, LINC00336 is more specific. Not only is LINC00336 able to regulate miR-6852, but also miR-6852 is also able to inhibit LINC00336 in turn. Furthermore, this ferroptosis resistance is not solely dependent on a simple lncRNA-miRNA interaction. There is also a lncRNA-protein interaction between LINC00336 and ELAV-like RNA-binding protein 1 (ELAVL1). Binding of ELAVL1 to LINC00336 prolongs its half-life, thereby further enhancing its resistance to ferroptosis[97]. There are also some lowly expressed lncRNAs in LUAD are ferroptosis promotor and tumor suppressor. As the first example, the GMDS-AS1 and LINC01128 specifically reduce miR-6077 and sensitize LUAD cells to cell-cycle arrest and ferroptosis[59,98]. A possible explanation is that miR-6077 interacts with and suppresses CDKN1A and KEAP1, thereby inhibiting CDKN1A-mediated G2/M arrest and KEAP1-NRF2-SLC7A11/NQO1-mediated ferroptosis. Thus, the GMDS-AS1 and LINC01128 can restore the sensitivity of LUAD to cisplatin/pemetrexed combination chemotherapy[59]. As the second example, the LINC00551 promotes ferroptosis of LUAD cells in an autophagy-dependent manner by upregulating DDIT4 via competitively binding miR-4328. DNA damage-inducible transcript 4(DDIT4) can inhibit mTOC1 complex, thereby promoting autophagy and ferroptosis[98]. This is consistent with previous findings that DDIT4 is a driver of autophagy-dependent ferroptosis in pancreatic cancer cells[99].
Collectively, the above non-coding RNAs associated with ferroptosis may be novel targets or biomarkers for LUAD prevention and treatment.
- m6A
- modification
N6-methyladenosine (m6A) is a WER system consisted by the writers (W), erasers (E) and readers (R), and writers are methyltransferases while erasers are enzymes catalyze demethylation[100]. Unlike writers and erasers, readers function as RNA-binding proteins that recognize m6A modifications and regulate mRNA splicing, export, translation, and stability[101]. Emerged research show that m6A modification and its associated proteins is significant regulators of ferroptosis LUAD tumorigenesis[102,103]. For example, methyltransferase-like 3 (METTL3) desensitizes ferroptosis by enhancing the stability and translation of SLC7A11 mRNA via recruiting TYHDF1 to SLC7A11 m6A modification[104]. The m6A reader insulin-like growth factor 2 mRNA binding protein 3 (IGF2BP3) is identified as an ferroptosis inhibitor via sustaining m6A-methylated mRNAs encoding anti-ferroptosis factors, including GPX4, SLC3A2, ACSL3, and FTH1[105]. Both METTL3 and IGF2BP3 are highly expressed in LUAD patients and associated with poor prognosis[104,105]. Moreover, the m6A reader YT521-B homology containing 2 (YTHDC2) was identified as an endogenous ferroptosis inducer and can inhibit LUAD tumorigenesis[106]. Mechanically, the two subunit of system XC, both SLC7A11 and SLC3A2, can be suppressed in a m6A-dependently manner. YTHDC2 was able to accelerate the degradation of SLC7A11 mRNA by directing binding to the 3'UTR of SLC7A11 mRNA. YTHDC2 was also able to indirectly inhibit SLC3A5 expression by destabilizing the SLC3A5 transcription factor HOXA13 mRNA. Thereby increasing YTHDC2 in LUAD patients may be an attractive and alternative ferroptosis-targeted therapy[106].

Table 1.
Nonding RNAs that regulate ferroptosis in LUAD.
| ncRNAs | Epigenetic mechanism | Functions in ferroptosis | Expression in LUAD patients | References |
|---|---|---|---|---|
| EP300 | histone modification | ①EP300/CREB complex: suppressor; increase GPX4 expression② EP300/YAP/ZEB complex: promoter; increase LPCAT3 expression | N/A | [19,79] |
| miR-27a-3p | miRNA | promoter; Inhibit SLC7A11 expression | downregulation | [88] |
| miR-324-3p | miRNA | promoter; Inhibit GPX4 expression | Downregulation in A549/DDP cells | [51] |
| P4HB | circRNA | Suppressor; sponge miRNA-1184 and increase SLC7A11 expression | Upregulation | [92] |
| circ_1010093 | circRNA | Suppressor; inhibit lipid (ACSL4, LPCAT3 and PLTP) | Upregulation | [94] |
| LINC00324 | lncRNA | Suppressor; sponge miR-200c-3p and promote TFAP2A-NRF2 axis | Upregulation | [37] |
| GSEC | lncRNA | Suppressor; sponge miR-101-3p and increase CISD1(a mitochondrial iron-sulfur protein) | Upregulation | [95] |
| Uc.339 | lncRNA | Suppressor; sponge pri-miR-339, inhibits the production of mature miR-339 and increase SLC7A11 expression | Upregulation | [96] |
| LINC00336 | lncRNA | Suppressor; sponge miR-6852 and increases cystathionine-β-synthase (CBS, involves in transsulfuration pathway and synthesizes cysteine ) expression | Upregulation | [97] |
| GMDS-AS1 and LINC01128 | lncRNA | Suppressor; sponge miR-6077 and promote KEAP1-NRF2-SLC7A11/NQO1 pathway | downregulation | [59] |
| LINC00551 | lncRNA | Promotor; sponge miR-4328 and increase DDIT4 expression,DDIT4 inhibits mTOR activity and promote autophagy-dependent ferroptosis | downregulation | [98] |
| METTL3 | m6A | Suppressor; enhance the stability and translation of SLC7A11 | upregulation | [104] |
| IGF2BP3 | m6A | Suppressor; enhance the stability and translation of anti-ferroptotic factors (GPX4, SLC3A2, ACSL3, and FTH1) | upregulation | [105] |
| YTHDC2 | m6A | Promotor; inhibit system Xc (directly inhibit SLC7A11 and indirectly inhibit SLC3A5 expression) | downregulation | [106] |
3.3. Ferroptosis in LUAD therapy
Targeting ferroptosis has the potential to be a prospective strategy for combating LUAD, especially in chemotherapy-resistance LUAD patients. Combinatory therapy with ferroptosis inducers and chemotherapeutic agents may produce improved therapeutic effects in LUAD patients with Cisplatin/sorafenib/Osimertinib resistance[51,59]. In addition to small molecular compounds like sorafenib[107,108], Chinese medicines[109,110], natural products[111] and nanoparticles[112] are showing as potential ferroptosis inducers in LUAD.
The multi-targeting properties Chinese medicine have attracted research into whether it can modulate ferroptosis in LUAD. Tetrandrine (Tet) is isolated from the plant Stephania tetrandra S. Moore and has been widely used in pulmonary fibrosis and anti-tumor therapy[113]. It has also been identified as a potential FIN that can inhibit GPX4 by suppressing the expression of SLC7A11[109]. Mixing Tet with citric acid in ddH2O at a ratio of 4:1 produces Tetrandrine Citrate (TetC), which also overcomes the hydrophobicity of Tet that makes it difficult to form drugs. Moreover, Hedyotis diffusa, another antitumor effector, is also able to induce ferroptosis by inhibiting Bcl2 expression, promoting Bax expression, and thus promoting the activation of VDAC2/3[110]. Voltage-dependent anion channel 2/3(VDAC2/3) is a mitochondrial channel protein whose sustained activation promotes mitochondrial depolarization, facilitates ROS release, [114,115]and induces ferroptosis. Previous studies have shown that Erastin can also induce ferroptosis through activation of VDAC2/3[116]. Both TetC and VDAC2/3 have the advantage of low toxicity, with little drug damage to the heart, liver, and kidneys. Combining natural products likes Ginkgetin with cisplatin can enhance anticancer effect in NSCLC. Ginkgetin derived from Ginkgo biloba leaves induces ferroptosis by disrupting the Nrf2/HO-1 antioxidant system, increasing ROS release and mitochondrial membrane potential loss[111]. Moreover, an inhalable nanoreactor was proposed to enhance LUAD ferroptosis therapy[112].
4. Conclusion and Perspective
The discovery of ferroptosis has provided imaginative insights into the treatment of LUAD and overcoming drug resistance. In recent years, the study of ferroptosis in LUAD has grown considerably and many other signaling pathways, such as FSP1-CoQ10 and (DHODH)-CoQ10, which have been found to regulate ferroptosis in addition to the classical SLC7A11-GPX4 antioxidant pathway. These signalings are potential anti-tumor targets that could be used to complement FIN therapies based on small molecule compounds such as Erastin and RSL3. However, there are many challenges to the pharmacisation of these inhibitors. Firstly, most of the inhibitors act via one-point to one-point. As we know, excessive lipid peroxidation, unbalanced redox hemostasis, and labile iron overload are three major hallmarks of ferroptosis. The ideal cost-effective strategy to induce ferroptosis is to simultaneously inhibit these factors. Whether a single target is sufficient to induce ferroptosis and exert clinical efficacy is worth investigating, as the present study demonstrated that the oncogenic effect of MPA, a pan-inhibitor targeting AKR1Cs (a family of ferroptosis-protective gene), was not significant enough to be used as a therapeutic strategy[67]. In addition, the pharmacological properties of many FINs are not well understood, and the possible side effects of their clinical application have yet to be investigated. Finally, the specificity of FINs is also an issue, and FINs that attack tumor cells and normal cells indiscriminately has no clinical value.
Research on the regulation and application of ferroptosis in LUAD is still in its infancy. There are still many aspects worth exploring or deepening, such as the link between ferroptosis and epithelial–mesenchymal transition (EMT). Next, other levels of regulation, such as the regulation of ferroptosis at the protein level, are also worth investigating. Current studies have focused on transcriptional regulation and epigenetic modifications, with only a few studies describing the molecular mechanisms by which protein autophagy regulates ferroptosis[15], while other molecular biochemical alterations, such as protein post-translational modifications (PTMs), have been less well studied on ferroptosis.
Author Contributions
X.W., X.L., S.H. and R.C. figured out the idea of writing this review. X.W., X.L. and S.H. summarized the published results and drafted the manuscript. R.C. and J.C. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The review was funded by the Innovative Research Team of High-level Local Universities in Shanghai and by the National Science Foundation of China 82173352, 81872230.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Herbst, R.S., D. Morgensztern, and C. Boshoff, The biology and management of non-small cell lung cancer. Nature. 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Hirsch, F.R. , et al., Lung cancer: current therapies and new targeted treatments. Lancet. 2017, 389, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G. , et al., Adjuvant Systemic Therapy and Adjuvant Radiation Therapy for Stage I to IIIA Completely Resected Non-Small-Cell Lung Cancers: American Society of Clinical Oncology/Cancer Care Ontario Clinical Practice Guideline Update. J Clin Oncol. 2017, 35, 2960–2974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. , et al., Elevated TRIM23 expression predicts cisplatin resistance in lung adenocarcinoma. Cancer Sci. 2020, 111, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C. and G.J. Riely, Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. JAMA. 2019, 322, 764–774. [Google Scholar] [CrossRef]
- Liu, S.-Y. , et al., Clinical characteristics and prognostic value of the KRAS G12C mutation in Chinese non-small cell lung cancer patients. Biomarker Research. 2020. 8(1). 2020. [Google Scholar]
- Huang, L. , et al., KRAS mutation: from undruggable to druggable in cancer. Signal Transduction and Targeted Therapy.2021. 6(1).
- Dixon, S.J. , et al., Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. , et al., Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Stockwell, B.R. , et al., Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- van Swelm, R.P.L., J. F.M. Wetzels, and D.W. Swinkels, The multifaceted role of iron in renal health and disease. Nat Rev Nephrol 2020, 16, 77–98. [Google Scholar] [CrossRef]
- Wang, Y. , et al., NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun 2020, 531, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Song, N. , et al., Ferritin: A Multifunctional Nanoplatform for Biological Detection, Imaging Diagnosis, and Drug Delivery. Acc Chem Res 2021, 54, 3313–3325. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D. , et al., Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Zhang, X. , et al., Endogenous glutamate determines ferroptosis sensitivity via ADCY10-dependent YAP suppression in lung adenocarcinoma. Theranostics 2021, 11, 5650–5674. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E. , et al., Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S. , et al., Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Zou, Y. , et al., Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Cui, J. , et al., LPCAT3 is transcriptionally regulated by YAP/ZEB/EP300 and collaborates with ACSL4 and YAP to determine ferroptosis sensitivity. Antioxid Redox Signal 2023.
- Wang, Y., et al., Transcriptional Repression of Ferritin Light Chain Increases Ferroptosis Sensitivity in Lung Adenocarcinoma. Front Cell Dev Biol 2021. 9: p. 719187.
- Yang, W.S. , et al., Regulation of ferroptotic cancer cell death by GPX4. Cell 2014. 156(1-2): p. 317-331.
- Doll, S. , et al., FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Kraft, V.A.N. , et al., GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Bridges, R.J., N. R. Natale, and S.A. Patel, System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol 2012, 165, 20–34. [Google Scholar] [CrossRef]
- Muller, F. , et al., Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ 2023, 30, 442–456. [Google Scholar] [CrossRef]
- Engelman, J.A. , et al., Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature Medicine 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. , Targeting RAS signalling pathways in cancer therapy. Nature Reviews Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- <56-6.pdf>.
- Zhang, Y. , et al., BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nature Cell Biology 2018, 20, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Wang, K., et al., Upregulation of CCT3 predicts poor prognosis and promotes cell proliferation via inhibition of ferroptosis and activation of AKT signaling in lung adenocarcinoma. BMC Molecular and Cell Biology 2022. 23(1).
- Alvarez, S.W. , et al., NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef]
- <14_2023 Aging (Albany NY)..pdf>.
- Gao, C., et al., Risk stratification of lung adenocarcinoma using a nomogram combined with ferroptosis-related LncRNAs and subgroup analysis with immune and N6-methyladenosine modification. BMC Medical Genomics 2022. 15(1).
- Shen, Y., et al., Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Frontiers in Immunology 2023. 13.
- Wang, Y., et al., A Novel Predictive Model Incorporating Ferroptosis-Related Gene Signatures for Overall Survival in Patients with Lung Adenocarcinoma. Medical Science Monitor 2021. 27.
- Zhang, W., et al., Molecular subtypes based on ferroptosis-related genes and tumor microenvironment infiltration characterization in lung adenocarcinoma. OncoImmunology 2021. 10(1).
- Zhu, G. , et al., Prognostic value of ferroptosis-related genes in patients with lung adenocarcinoma. Thoracic Cancer 2021, 12, 1890–1899. [Google Scholar] [CrossRef]
- Wang, H. , et al., The E3 Ligase MIB1 Promotes Proteasomal Degradation of NRF2 and Sensitizes Lung Cancer Cells to Ferroptosis. Mol Cancer Res 2022, 20, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E. , et al., ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. Journal of Clinical Investigation 2016, 126, 3219–3235. [Google Scholar] [CrossRef]
- Sun, L. , et al., Lipid Peroxidation, GSH Depletion, and SLC7A11 Inhibition Are Common Causes of EMT and Ferroptosis in A549 Cells, but Different in Specific Mechanisms. DNA and Cell Biology 2021, 40, 172–183. [Google Scholar] [CrossRef]
- Zhang, H., et al., ARNTL2 is an indicator of poor prognosis, promotes epithelial-to-mesenchymal transition and inhibits ferroptosis in lung adenocarcinoma. Transl Oncol 2022. 26: p. 101562.
- Yatabe, Y. , et al., EGFR Mutation Testing Practices within the Asia Pacific Region: Results of a Multicenter Diagnostic Survey. Journal of Thoracic Oncology 2015, 10, 438–445. [Google Scholar] [CrossRef]
- Han, B., et al., <em>EGFR</em> mutation prevalence in Asia-Pacific and Russian patients with advanced NSCLC of adenocarcinoma and non-adenocarcinoma histology: The IGNITE study. Lung Cancer 2017. 113: p. 37-44.
- Recondo, G. , et al. , Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI? Nature Reviews Clinical Oncology 2018, 15, 694–708. [Google Scholar] [PubMed]
- Tan, C.-S., et al., Third generation EGFR TKIs: current data and future directions. Molecular Cancer 2018. 17(1).
- Chan, L.S. , et al., Selenite as a dual apoptotic and ferroptotic agent synergizes with EGFR and KRAS inhibitors with epigenetic interference. Clin Epigenetics 2023, 15, 36. [Google Scholar] [CrossRef]
- Wang, J. , et al., Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small-cell lung cancer with activating EGFR mutations. OncoTargets and Therapy 2016.
- Tumbrink, H.L., A. Heimsoeth, and M.L. Sos, The next tier of EGFR resistance mutations in lung cancer. Oncogene 2020, 40, 1–11. [Google Scholar] [CrossRef]
- Reita, D., et al., Molecular Mechanism of EGFR-TKI Resistance in EGFR-Mutated Non-Small Cell Lung Cancer: Application to Biological Diagnostic and Monitoring. Cancers 2021. 13(19).
- Zhang, T. , et al., Targeting histone deacetylase enhances the therapeutic effect of Erastin-induced ferroptosis in EGFR-activating mutant lung adenocarcinoma. Transl Lung Cancer Res 2021, 10, 1857–1872. [Google Scholar] [CrossRef]
- Deng, S.H. , et al., miR-324-3p reverses cisplatin resistance by inducing GPX4-mediated ferroptosis in lung adenocarcinoma cell line A549. Biochem Biophys Res Commun 2021. 549: p. 54-60.
- Bi, G. , et al., miR-6077 promotes cisplatin/pemetrexed resistance in lung adenocarcinoma via CDKN1A/cell cycle arrest and KEAP1/ferroptosis pathways. Molecular Therapy - Nucleic Acids, 2022; 28, 366–386. [Google Scholar]
- Wang, P. Wang, and Y. Wang, Ferroptosis patterns modulate immunocyte communication in tumor microenvironments: clinical value and therapeutic guidance of lung adenocarcinoma. Functional & Integrative Genomics, 2023; 23(2). [Google Scholar]
- Deng, B. , et al., Identification and validation of a ferroptosis-related gene to predict survival outcomes and the immune microenvironment in lung adenocarcinoma. Cancer Cell International, 2022; 22(1). [Google Scholar]
- Luo, L. Chen, and F. Huang, Machine learning revealed ferroptosis features and ferroptosis-related gene-based immune microenvironment in lung adenocarcinoma. Chemico-Biological Interactions, 2023; 378, 110471. [Google Scholar]
- Tang, B. , et al., Identification of critical ferroptosis regulators in lung adenocarcinoma that RRM2 facilitates tumor immune infiltration by inhibiting ferroptotic death. Clinical Immunology 2021, 232, 108872. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y. , et al., Identification of a prognostic ferroptosis-related lncRNA signature in the tumor microenvironment of lung adenocarcinoma. Cell Death Discovery, 2021; 7(1). [Google Scholar]
- Feng, Z. , et al., Identification and Validation of a GPX4-Related Immune Prognostic Signature for Lung Adenocarcinoma. Journal of Oncology 2022, 2022, 1–24. [Google Scholar]
- Bi, G. , et al., miR-6077 promotes cisplatin/pemetrexed resistance in lung adenocarcinoma via CDKN1A/cell cycle arrest and KEAP1/ferroptosis pathways. Mol Ther Nucleic Acids 2022, 28, 366–386. [Google Scholar] [CrossRef]
- Jung, K.A. , et al., Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol Lett 2013, 218, 39–49. [Google Scholar] [CrossRef]
- Bersuker, K. , et al., The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Yagoda, N. , et al., RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Dai, E. , et al., Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun 2020, 11, 6339. [Google Scholar] [CrossRef]
- Zhang, D.D. , et al., Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Molecular and Cellular Biology 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. , Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013, 53, 401–26. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. , Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Wohlhieter, C.A. , et al., Concurrent Mutations in STK11 and KEAP1 Promote Ferroptosis Protection and SCD1 Dependence in Lung Cancer. Cell Rep 2020, 33, 108444. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C. , et al., Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res 2018, 24, 334–340. [Google Scholar] [CrossRef]
- Zhang, W. , et al., The RSL3 Induction of KLK Lung Adenocarcinoma Cell Ferroptosis by Inhibition of USP11 Activity and the NRF2-GSH Axis. Cancers (Basel) 2022. 14(21).
- Thompson, L.R. , et al., Distinct TP53 Mutation Types Exhibit Increased Sensitivity to Ferroptosis Independently of Changes in Iron Regulatory Protein Activity. International Journal of Molecular Sciences 2020. 21(18).
- Liu, D.S. , et al., Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nature Communications 2017. 8(1).
- Freire Boullosa, L. , et al., Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biology 2021. 42.
- Wang, S.-J. , et al., Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Reports 2016, 17, 366–373. [Google Scholar] [CrossRef]
- Proceedings of the National Academy of Sciences.
- Zhao, B. , et al., Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Zhu, C. , et al., A Non-canonical Role of YAP/TEAD Is Required for Activation of Estrogen-Regulated Enhancers in Breast Cancer. Mol Cell 2019, 75, 791–806. [Google Scholar] [CrossRef]
- Thomas, C. , et al., LPCAT3 deficiency in hematopoietic cells alters cholesterol and phospholipid homeostasis and promotes atherosclerosis. Atherosclerosis 2018, 275, 409–418. [Google Scholar] [CrossRef]
- Wang, T. , et al., ARNTL2 upregulation of ACOT7 promotes NSCLC cell proliferation through inhibition of apoptosis and ferroptosis. BMC Mol Cell Biol 2023, 24, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. , et al., CREB stimulates GPX4 transcription to inhibit ferroptosis in lung adenocarcinoma. Oncol Rep 2021. 45(6).
- Zhao, G. , et al. , KLF11 regulates lung adenocarcinoma ferroptosis and chemosensitivity by suppressing GPX4. Commun Biol 2023, 6, 570. [Google Scholar]
- Logie, E. , et al., Ferroptosis Induction in Multiple Myeloma Cells Triggers DNA Methylation and Histone Modification Changes Associated with Cellular Senescence. Int J Mol Sci 2021. 22(22).
- Xu, Y. , et al., Ferroptosis-associated DNA methylation signature predicts overall survival in patients with head and neck squamous cell carcinoma. BMC Genomics 2022, 23, 63. [Google Scholar] [CrossRef]
- Zhang, X. , et al. , Homocysteine induces oxidative stress and ferroptosis of nucleus pulposus via enhancing methylation of GPX4. Free Radic Biol Med 2020, 160, 552–565. [Google Scholar] [PubMed]
- Tessarz, P. and T. Kouzarides, Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A. , et al., Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nat Chem Biol 2019, 15, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Tong, Q. , et al., MnTE-2-PyP reduces prostate cancer growth and metastasis by suppressing p300 activity and p300/HIF-1/CREB binding to the promoter region of the PAI-1 gene. Free Radic Biol Med 2016, 94, 185–94. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y. , et al., Correction: miR-1260b, mediated by YY1, activates KIT signaling by targeting SOCS6 to regulate cell proliferation and apoptosis in NSCLC. Cell Death Dis 2020, 11, 261. [Google Scholar] [CrossRef] [PubMed]
- Lu, X. , et al. , MiR-27a-3p Promotes Non-Small Cell Lung Cancer Through SLC7A11-Mediated-Ferroptosis. Front Oncol 2021, 11, 759346. [Google Scholar]
- Wei, B. , et al., Comprehensive analysis of tumor immune infiltration associated with endogenous competitive RNA networks in lung adenocarcinoma. Pathol Res Pract 2019, 215, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Li, X. , et al., NUDT21 regulates circRNA cyclization and ceRNA crosstalk in hepatocellular carcinoma. Oncogene 2020, 39, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Kong, X. , et al. , LncRNA-CDC6 promotes breast cancer progression and function as ceRNA to target CDC6 by sponging microRNA-215. J Cell Physiol 2019, 234, 9105–9117. [Google Scholar] [PubMed]
- Pan, C.F. , et al., CircP4HB regulates ferroptosis via SLC7A11-mediated glutathione synthesis in lung adenocarcinoma. Transl Lung Cancer Res 2022, 11, 366–380. [Google Scholar] [CrossRef]
- Zhou, W.Y. , et al., Circular RNA: metabolism, functions and interactions with proteins. Mol Cancer 2020, 19, 172. [Google Scholar] [CrossRef]
- Zhang, X. , et al., Essential roles of exosome and circRNA_101093 on ferroptosis desensitization in lung adenocarcinoma. Cancer Commun (Lond) 2022, 42, 287–313. [Google Scholar] [CrossRef]
- Jiang, X., et al., Systematic Analysis and Validation of the Prognosis, Immunological Role and Biology Function of the Ferroptosis-Related lncRNA GSEC/miRNA-101-3p/CISD1 Axis in Lung Adenocarcinoma. Front Mol Biosci 2021. 8: p. 793732.
- Zhang, N. , et al., LncRNA T-UCR Uc.339/miR-339/SLC7A11 Axis Regulates the Metastasis of Ferroptosis-Induced Lung Adenocarcinoma. J Cancer 2022, 13, 1945–1957. [Google Scholar] [CrossRef]
- Wang, M. , et al., Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ 2019, 26, 2329–2343. [Google Scholar] [CrossRef]
- Peng, X. , et al. , Overexpression of LINC00551 promotes autophagy-dependent ferroptosis of lung adenocarcinoma via upregulating DDIT4 by sponging miR-4328. PeerJ 2022, 10, e14180. [Google Scholar]
- Liu, J. , et al., Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem Biol 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Shi, H., J. Wei, and C. He, Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Fu, Y. , et al., Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Lan, Q. , et al., The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res 2019, 79, 1285–1292. [Google Scholar] [CrossRef]
- Song, Z., et al., Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci 2021. 276: p. 119399.
- Xu, Y. , et al., METTL3 promotes lung adenocarcinoma tumor growth and inhibits ferroptosis by stabilizing SLC7A11 m(6)A modification. Cancer Cell Int 2022, 22, 11. [Google Scholar] [CrossRef]
- Xu, X. , et al., IGF2BP3 is an essential N(6)-methyladenosine biotarget for suppressing ferroptosis in lung adenocarcinoma cells. Mater Today Bio, 2022; 17, 100503. [Google Scholar]
- Ma, L. , et al., Targeting SLC3A2 subunit of system X(C)(-) is essential for m(6)A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic Biol Med. 2021; 168, 25–43. [Google Scholar]
- <82-Lachaier.pdf>.
- He, H. , et al., KIF20A is associated with clinical prognosis and synergistic effect of gemcitabine combined with ferroptosis inducer in lung adenocarcinoma. Front Pharmacol. 2022; 13, 1007429. [Google Scholar]
- Mo, X. , et al., Tetrandrine citrate suppresses lung adenocarcinoma growth via SLC7A11/GPX4-mediated ferroptosis. Discov Oncol. 2023, 14, 85. [Google Scholar] [CrossRef]
- Huang, F., et al., Hedyotis diffusa injection induces ferroptosis via the Bax/Bcl2/VDAC2/3 axis in lung adenocarcinoma. Phytomedicine. 2022. 104: p. 154319.
- Lou, J.S., et al., Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine. 2021. 80: p. 153370.
- Wang, W. , et al., Inhalable Biomimetic Protein Corona-Mediated Nanoreactor for Self-Amplified Lung Adenocarcinoma Ferroptosis Therapy. ACS Nano. 2022, 16, 8370–8387. [Google Scholar] [CrossRef] [PubMed]
- Luan, F., X. He, and N. Zeng, Tetrandrine: a review of its anticancer potentials, clinical settings, pharmacokinetics and drug delivery systems. J Pharm Pharmacol. 2020, 72, 1491–1512. [Google Scholar] [CrossRef]
- DeHart, D.N., et al., Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem Pharmacol. 2018. 148: p. 155-162.
- Lipper, C.H. , et al., Redox-dependent gating of VDAC by mitoNEET. Proc Natl Acad Sci U S A. 2019, 116, 19924–19929. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.N. , et al., Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



