
Submitted:
11 September 2023
Posted:
13 September 2023
You are already at the latest version
Abstract
A single paragraph of about 200 words maximum. For research articles, abstracts should give a pertinent overview of the work. We strongly encourage authors to use the following style of structured abstracts, but without headings: (1) Background: Place the question addressed in a broad context and highlight the purpose of the study; (2) Methods: briefly describe the main methods or treatments applied; (3) Results: summarize the article’s main findings; (4) Conclusions: indicate the main conclusions or interpretations. The abstract should be an objective representation of the article and it must not contain results that are not presented and substantiated in the main text and should not exaggerate the main conclusions.
Keywords:
herpesvirus
; ADAR
; RNA editing
; miRNA
; latency
; innate immunity
1. Introduction
Viruses depend on the host cell for replication and constantly evolve in response to host defenses. Various host pattern recognition receptors (PRRs, e.g.., Toll-like receptors (TLRs), retinoic acid-inducible gene 1 (RIG -I)–like receptors, protein kinase R (PKR); interferon-gamma-inducible protein 16 (IFI16), etc.) recognize so-called pathogen-associated molecular patterns (PAMPs), such as viral capsids, surface glycoproteins, viral genomes and transcripts, and trigger signaling cascades that lead to the establishment of the replication nonpermissive state or initiate cell death (reviewed in [1,2,3]). Although viruses respond to host defenses with their countermeasures (i.e., inhibitors of these signaling pathways), the defense mechanisms and activation of downstream effectors must be precisely balanced to prevent overactivation and damage to the host.
Members of the Adenosine Deaminase Acting on RNA (ADAR) protein family catalyze the conversion of adenosines to inosines (A-to-I editing), one of the most common forms of posttranscriptional RNA modification, provide the molecular mark that distinguishes host RNA from foreign RNA (e.g. viral). They suppress the hyperactivation of dsRNA sensors (such as PKR, RIG-I/MDA5, OAS/RNase L and ZBP1) that would lead to autoimmunity (reviewed in [4,5,6]). On the one hand, A-to-I editing of RNA transcripts can affect multiple cellular processes through various mechanisms, including mRNA translation, splicing, RNA structure, and RNA silencing, and on the other hand, it attenuates dsRNA sensing in an editing-dependent and/or -independent manner (reviewed in [7]). Viruses may benefit from all of these mechanisms, and there is ample evidence to support this assumption. This review presents all currently published studies on ADAR -mediated post-transcriptional modifications and the potential role of ADARs in herpesvirus infection, major dsDNA viruses that have been largely neglected in A-to-I studies. The activity of ADAR affects several aspects of gene expression and regulation of replication in various herpesviruses, from modulation of miRNA biogenesis and miRNA targeting to complete alteration of the biological properties of edited transcripts. In addition, there are several lines of evidence that the ADAR proteins are required for efficient reactivation and replication of herpesviruses. Studies of diverse and evolutionarily distant herpesviruses, e.g., from human viruses to viruses of marine snail, suggest a fundamental importance of ADAR for replication of these viruses. The role of the ADAR proteins remains largely unexplored, but it is clear that A-to-I editing adds another level of complexity to the biology of herpesviruses.
2. The ADAR protein family in brief
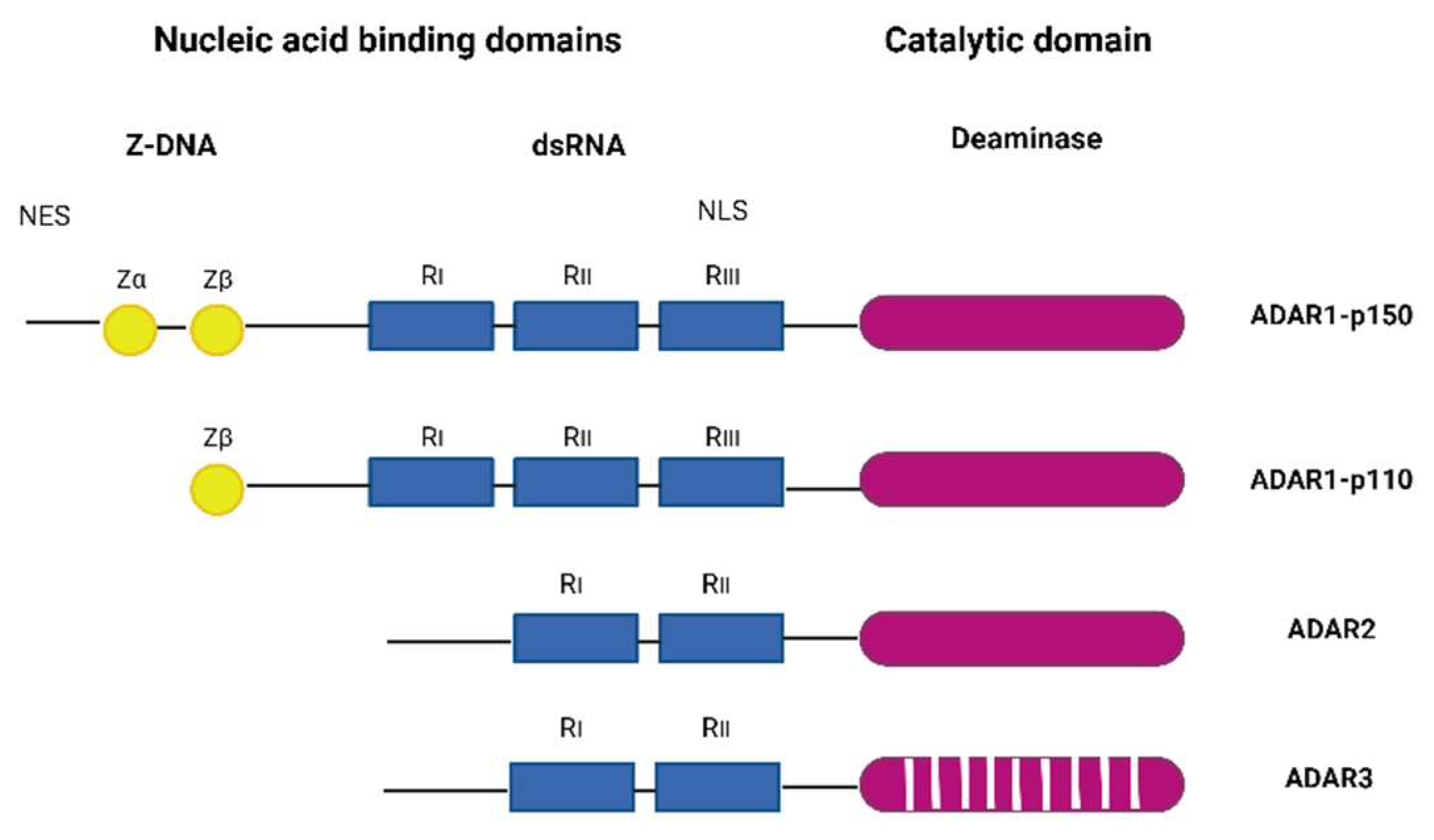
Adenosine deaminases acting on RNA (ADARs) catalyze the C6 deamination of adenosine (A) to inosine (I) in double-stranded RNA (dsRNA), one of the most common post-transcriptional modifications of RNAs, known as A-to-I editing (reviewed in [8]). Inosine forms base pairs with cytosine (C) instead of uracil (U), which alters the structure, stability, biogenesis, and coding ability of transcripts. The ADAR family includes three genes: ADAR1 (ADAR), ADAR2 (ADARB1), and ADAR3 (ADARB2) (Figure 1). ADAR1 encodes the constitutively expressed ADAR1 p110, which is localized in the nucleus, and the interferon-inducible ADAR1 p150, which is localized in the cytoplasm and nucleus. ADAR1 is the major RNA editor in humans, and most of its hundreds of millions of editing sites have been found in Alu elements, many of which are located in introns and 3'-untranslated regions [9,10].
ADAR1 p150 has two Z-DNA binding domains, in contrast to p110 which has only one, and shows a broader range of targets compared to p110 [12]. ADAR1 is ubiquitously expressed and is inducible by many viruses, including human cytomegalovirus (HCMV) [13], Kaposi’s Sarcoma Associated Virus (KSHV)[14], reoviruses [15], etc. In mice, ADAR1 deficiency is embryonically lethal [16], and in humans, dysregulations of ADAR1 activity are associated with a number of diseases, including Aicardi-Goutières syndrome, an autoimmune disorder and interferonopathy [17]; bilateral striatal necrosis dystonia [18]; and dyschromatosis symmetrica hereditaria, a skin pigmentation disorder [19]; and various cancers [20]. The main role of ADAR1 editing is to prevent sensors (PKR; RIG-I/MDA5, LGP2, ZBP1, OAS/RNase L) from recognizing endogenous dsRNA as non-self and to prevent hyperactivation of downstream signaling [21-25]. In addition, ADAR1 forms a complex with a number of different proteins, including Dicer, to promote miRNA processing and RNA-induced gene silencing [26].
In humans, ADAR2 is highly expressed in the brain, arteries, lungs, and bladder and is responsible for site-specific editing (reviewed in [27]). The major physiological function of ADAR2 is attributed to exonic RNA editing of the neuronal glutamate receptor (GluR), a process essential for neuronal homeostasis [28]. In contrast to ADAR1 and ADAR2, the expression of ADAR3 (ADARB2) is mainly restricted to the nervous system, particularly the hippocampus and amygdala [29]. Although ADAR3 has two copies of dsRNA-binding domains it does not exhibit catalytic deaminase activity and has an editing regulatory function [30]. Recently, ADAR3 was shown to also regulate MAVS expression, suggesting that its role extends beyond editing modification [31]. In mice, genetic ablation of Adar3 does not lead to embryonic lethality, but mice lacking ADAR3 show impaired short- and long-term memory [32].
The role of ADAR proteins in viral infections has been studied for a great number of RNA and a few DNA viruses, and proviral and antiviral roles have been demonstrated. These studies are discussed in detail elsewhere (reviewed in [11,33]), and we will focus only on recent discoveries in herpesviruses below.
3. Herpesviruses in brief
Members of the order Herpsesvirales (herpesviruses) are widespread in nature and have been discovered in a variety of animals. The order includes three distinct virus families: a) the Orthoherpesviridae (formerly Herpesviridae; i.e. the herpesviruses of mammals, birds, and reptiles), b) the Alloherpesviridae (the herpesviruses of fish and amphibians), and c) the Malacoherpesviridae (the herpesviruses of mollusks). Herpesviruses are large dsDNA viruses that share several biological properties, including the establishment of latent infection as a mechanism for lifelong persistent infection and transmission [34,35].
The replication cycle of herpesviruses entails two phases, the lytic (i.e., productive) phase and the latent phase. During the lytic phase, viral genes are abundantly expressed in a tightly regulated temporal cascade of gene expression. In the second phase of infection, herpesviruses reach target cells in which they initiate a latent infection program. In general, viral genomes take the form of closed circular and chromatinized molecules, from which only a small subset of specific genes are expressed and no viral progeny are produced. Each herpesvirus establishes latency in a specific group of cells, which may be distinct from the cells in which the virus replicates productively. Importantly, latent genomes retain the ability to reactivate and generate new infectious virions during the lytic phase. The molecular mechanisms of establishment, maintenance, and reactivation from the latency phase vary widely among different herpesviruses but clearly represent an exceptionally successful mechanism for dissemination and persistence in nature [35]. Herpesviruses have co-evolved with their hosts and developed numerous specific adaptations to host defense mechanisms, such that cross-species infections are rare in nature [35,36].
3.1. Orthoherpesviridae
The family Ortoherpesviridae encompasses viruses that infect mammals, birds and reptiles and includes nine clinically important human herpesviruses (human herpesvirus 1 – 5, HHV-6A and -6B, HHV-7 and HHV-8). Based on different biological properties the family is divided into three subfamilies: Alphaherpesvirinae, Betaherpesviriane and Gammaherpesvirinae. Alphaherpesviruses have relatively broad host range, rapid productive phase and establish latent infection in neurons, primarily in sensory ganglia. This subfamily contains viruses isolated from numerus different animals including important human pathogens herpes simplex virus 1 (HSV-1, also HHV-1) and HSV-2 (HHV-2) and varicella zoster virus (VZV, HHV-3). Betaherpesviruses have slow productive cycle and have very restricted host range. Usualy, betaherpesviruses infect various cell types and tissues and establish latency in CD34+ hematopoietic progenitor cells and CD14+ monocytes or T lymphocytes. This subfamily contains numerus viruses including human cytomegalovirus (HCMV, or HHV-5), human herpesvirus 6A and 6B (HHV-6A and HHV-6B), human herpesvirus 7 (HHV-7), murine cytomegalovirus (MCMV), rat cytomegalovirus (RCMV) etc. grouped in several genera. Gammaherpesviruses include human onocoviruses Epstein-Barr virus (EBV, or HHV-4) and Kaposi’s sarcoma-associated virus (KSHV, or HHV-8). Latency is usually established in lymphoid tissues in either B or T lymphocytes. Interestingly, while alpha- and betaherpesviruses primarily enter productive phase in infected cells in culture, gammaherpesviruses predominate in latent phase [34,35].
4. ADAR and herpesviruses
4.1. Evidence for ADAR-mediated RNA editing in Alphaherpesviruses
4.1.1. Herpes simplex virus 1 (HSV-1) – latent miRNA editing
Herpes simplex virus 1 (HSV-1) is an important human pathogen, a widely distributed and extensively studied virus known to cause cold sores. After productive infection in the mucosal epithelium in the oro-labial or genital area, HSV-1 reaches nearby neurons and migrates via axons to cell bodies in sensory ganglia, where it initiates latent infection. During latency, no viral progeny are produced and only transcripts derived from the latency-associated transcript (LAT) locus are abundantly expressed [37]. LATs are multiple non-coding RNAs and precursors of miRNAs and stable introns of 2 kb and 1.5 kb. The exact function of LATs remains unclear, but functions in suppression of gene expression in the productive phase, inhibition of apoptosis, and chromatin remodeling have been demonstrated [38-42]. HSV-1 expresses miRNAs from more than 20 loci (miR-H1 - miR-H18 and miR-H26-H29), and often both strands of the miRNA duplex can be detected, indicating efficient utilization of the coding potential [43-47]. Most HSV-1 miRNAs were detected in productive infections, whereas only a small subset of miRNAs (miR-H2-H8) was detected in latently infected human ganglia [45,48]. Many HSV-1 miRNAs are conserved in HSV-2 and are in antisense to transcripts encoding important viral proteins. For example, miR-H2, -H7, and -H2 are encoded opposite ICP0, an important viral transcriptional coactivator and regulator of innate immunity, suggesting their function in repressing these genes [43,49-52]. Apart from this, the functions of HSV-1 miRNAs are still poorly defined.
The complexity of miRNA regulation of HSV-1 infection was recently further increased by the discovery of posttranscriptional editing of HSV-1 miRNAs in the latency phase. Zubković et al. found that miR-H2-3p, but not other miRNAs encoding HSV-1, are hyperedited in latently infected human and mouse ganglia, suggesting functional significance at this stage of infection [48,53]. miR-H2-3p is edited within the seed sequence that determines biding specificity for transcripts targeted for regulation, and thus editing expands its targeting potential. In an overexpression system, the edited miR-H2-3p was found to have the potential to regulate ICP4 (an essential viral protein) in addition to ICP0, which is encoded in the opposite direction to this miRNA [53]. At present, it is not clear which ADAR protein is responsible for the editing phenomenon or what biological significance the editing has for HSV-1 replication. Nevertheless, these results suggest that other noncoding RNAs abundant in latently infected neurons, such as the LAT intron, may also be edited and that editing may play an important role in the biology of these RNAs.
4.1.2. Varicella zoster virus (VZV) – edited novel viral transcripts
Varicella zoster virus (VZV) is known to cause chickenpox (varicella) and shingles (zoster). VZV is a highly infectious virus transmitted by inhalation of aerosolized virions released from skin lesions. VZV initially infects mucosal cells in the upper respiratory tract, where it is recognized by dendritic cells and transported to lymph nodes, where infection spreads to T lymphocytes, which shed the virus into the skin. Latency is established in ganglia throughout the neuroaxis. The reactivated virus migrates anterograde to the dermatome where it can cause herpes zoster [54]. Latent VZV, similar to HSV-1, expresses only a few restricted transcripts, including ORF63 and the spliced latency-associated VZV transcript (VLT), which is in antisense position to the gene ORF61, an important viral transactivator similar to HSV-1 ICP0 [55]. Despite the obvious similarity in control of latency between HSV-1 and VZV, VZV is the only herpesvirus for which no encoded miRNAs have been identified.
In contrast to HSV-1, the phenomenon of RNA editing of VZV transcripts was studied in its lytic phase. Using long-read nanopore sequencing technology of RNA extracted from lytic VZV infections, Prazsak et al. identified a number of novel VZV transcripts, including several transcripts derived from loci near the origin of replication (oriS), termed nroRNA (near-replication-origin) transcripts [56]. One of these transcripts, NTO3, expressed antisense to ORF62, showed a very high frequency of A-to-G substitutions that was not present in the overlapping other transcripts, suggesting a hyperediting event of this transcript. The authors suggest that editing of NTO3 ensures the thermodynamic stability of the RNA secondary structure and that editing may have an impact on the potential regulation of the antisense transcript ORF62; however, this remains to be investigated.
4.1.2. Gallid herpesvirus 2 (GaHV-2, or Marek’s disease virus 1 (MDV-1)
Gallid herpesvirus 2 (GaHV-2) is an oncogenic virus of chickens causing major losses in poultry production. GaHV-2 induces aggressive T-cell lymphomas in only a few weeks leading to the death of the chicken. The genome of the virus is colinear with HSV-1, including internal repeats that contain a number of important genes (e.g. viral telomerase subunit vTR and meq, the major oncogene), latency associated transcript long non-coding RNA (LAT lncRNA) and three clusters of miRNAs [57-59].
Interestingly, Figueroa et al. recently identified another 5’-caped polyadenylated, alternatively spliced lncRNA, named ERL lncRNA [60]. The transcript is encoded within the repeat flanging the unique long region of the genome overlapping with the transcripts of R-LORF5a, meq and 14D genes and two clusters of miRNAs, mdv1-miR-M9-M4 and -M11-M1. In contrast to LAT lncRNA which is the hallmark of latency, the ERL lncRNA displayed similar levels during primary productive infection, lytic reactivation and latency. Moreover, ERL lncRNA displayed hyperediting (i.e. more than 25% of its transcripts) during primary productive infection, but not during reactivation from latency or in latency [60]. Although the function or ERL lncRNA and biological relevance of its editing is unknown, author have proposed that this transcript play role as natural antisense transcript regulating the two miRNA clusters.
Taken together, current information on roles of ADAR in alphaherpesviruses is very limited. However, the above-mentioned studies clearly show that editing affects transcripts of these viruses, and functional studies are yet to come.
4.2. Evidence of ADAR-mediated RNA editing in Betaherpesviruses – edited host miRNA
Cytomegalovirus (CMV) is a ubiquitously distributed virus that infects up to 60-100% of adults. CMV is the leading cause of congenital disease and an important and life-threatening opportunistic pathogen in immunocompromised individuals, especially transplant recipients. HCMV establishes latency primarily in CD34+ hematopoietic progenitor cells (HPCs), and differentiation of these cells into dendritic cells (DC) or macrophages in response to cytokine and growth factor signaling leads to virus reactivation [61]. The hallmark of HCMV biology is modulation of the host immune response, which includes a large number of viral proteins and gene products that interfere with antigen presentation (e.g., viral miR-UL112 downregulates the activating NK ligand MICB and reduces killing of infected cells) [62,63].
To date, the role of ADAR in CMV infection has been investigated in only one case, yet the study provided several important findings. Nachmani et al. found that ADAR1 p110, but not p150, is significantly upregulated in cells productively infected with HCMV in culture [13]. Interestingly, they did not observe the same upregulation in other dsDNA (HSV-1, HSV-2, adenovirus) or RNA viruses (influenza virus A and human metapneumovirus). It appears that HCMV increases the expression of the ADAR1-p110 isoform by activating one of the four promoters (1B) that regulate ADAR1 gene expression. The increased level of ADAR1 protein was accompanied by an increased rate of the edited form of host miR-376a (miR-376a(e)) compared with mock-infected cells, i.e., from 50% in mock-infected cells to approximately 80% in HCMV-infected cells. Editing of miR-376a occurs in the seed region of this miRNA, resulting in a drastic change in target selection, including loss of regulation of a key NK cell-activating receptor MICB. On the other hand, the edited miRNA gained the ability to downregulate HLA-E, the ligand of two immune receptors, the inhibitory heterodimer CD94/NKG2A and the activating CD94/NKG2C, facilitating NK cell cytotoxicity and immunelimination of HCMV-infected cells. Taken together, on the one hand, the virus uses host and viral miRNAs (miR-UL112 and a number of host miRNAs, including miR-376a) to reduce the elimination of infected cells, and on the other hand, the induced editing miR-376a renders cells susceptible to NK cell toxicity. Although seemingly contradictory, it is quite puzzling which of these activities is dominant and relevant to a particular phase of dynamic viral replication, i.e., from maintenance of latency to reactivation and dissemination [13].
This study did not address the possible editing of viral transcripts or viral miRNAs. However, in a study with a similar virus, murine CMV (MCMV), no evidence of edited MCMV miRNAs was found [64].
4.3. Gammaherpesviruses: Editing dependent and editing independent roles of ADAR proteins
4.3.1. Epstein-Barr virus (EBV, HHV-4) – editing affects miRNA biogenesis
Epstein-Barr virus (EBV, HHV-4) is a ubiquitous human virus that infects more than 90% of the adult population and causes no disease in the vast majority of healthy carriers. However, EBV can induce B lymphocyte proliferation in vivo and has been linked to a number of cancers, including Burkitt's lymphoma, Hodgkin's disease, nasopharyngeal carcinoma, and gastric cancer (reviewed in [65]). Recently, a large longitudinal study of a cohort of more than 10 million young adults found that the risk of developing multiple sclerosis increases 32-fold after infection with EBV [66]. EBV establishes its latency predominantly in B cells, which is divided into five patterns depending on the expression of the viral latency gene expression (latency 0, I, IIa, IIb, and III). In addition, EBV encodes more than 20 miRNAs located in two clusters, the BART and BHRF1 regions of the genome, which are involved in regulating the transition from the lytic to the latent phase and in attenuating the antiviral response [67].
The precursors of several of these miRNAs, including pri-miR-BHRF1-1, pri-miR-BART3, pri-miR-BART6, pri-miR-BART8, pri-miR-BART11, and pri-miR-BART16, have bene found posttranscriptionally edited at specific sites in EBV latently infected cells [68,69]. The editing frequency of one of these pre-miRNAs, pri-miR-BART6, reached as high as 50-70% of all its transcripts. Interestingly, editing of pri-miR-BART6 affected DROSHA processing but not binding of cofactor DGCR8, greatly reducing the amount of mature miR-BART6. Moreover, miR-BART6 targets Dicer, and consequently, processing of its pri-miRNA modulates not only Dicer levels but also global repression by miRNAs [68]. Modulation of Dicer levels may play a role in regulating latency and balancing the on-off switch for reactivation. In a similar study, pri-miR-BART3 was also found to be hyperedited and to affect DROSHA processing and miRNA targeting of Dicer by editing the seed region of miRNA [69]. However, several studies did not find edited mature forms of EBV miRNAs, suggesting that the levels of edited miRNAs are low or exhibit rapid turnover [70]. To date, it remains puzzling which member of the ADAR protein family, ADAR1 or ADAR2, edits EBV pri-miRNAs. However, based on the low expression of ADAR2 in cells harboring latent EBV, it is suggested that ADAR1 p110 may play a predominant role.
In addition to miRNA studies, massive RNA editing was observed on lncRNA transcripts derived from the EBV origin of replication, oriPtL and oriPtR, which have a role in promoting productive gene expression and DNA replication [71]. These newly identified late viral transcripts maintained their localization within the nucleus, and were predicted to form long hairpin structures that provide a configuration for recognition by RNA editing enzymes. Indeed, using immunoprecipitation assays, the authors demonstrated the direct interaction of ADAR1 and oriP transcripts during induced reactivation. Interestingly, oriP transcripts were found in association with the paraspeckle complex assembly factor NONO, suggesting a role for these transcripts in modulating the antiviral stress response [71]. However, it is not known whether RNA editing is required for this interaction.
4.3.2. Kaposi’s sarcoma associated herpesvirus (KSHV, HHV-8)
Kaposi’s sarcoma associated herpesvirus (KSHV, HHV-8) is associated with at least three human malignancies: Kaposi's sarcoma (KS), primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD). KSHV infection in tumors and established PEL cell lines is predominantly latent, and only a limited number of genes are expressed including latency-associated nuclear antigen 1 and 2 (LANA), v-FLIP, K15, K12 and miRNAs [72]. K12 RNA is the most common transcript present during latent phase of viral replication of KSHV and encodes three kaposin proteins (Kaposin A, B, and C) and miR-K10 that retains tumorigenic activity [73-75].
KSHV - RNA editing phenomena
Sequence heterogeneity and potential editing phenomena in herpesviruses were first noted for KSHV [14,76,77], and only recently several studies on KSHV have shed light on the importance and diverse functions of ADAR proteins for herpesvirus infection. In their remarkable study providing the first evidence for herpesvirus-encoded miRNAs, Pfeffer at al., found A-to-G sequence variation within KSHV-encoded miR-K12-10, and suggested posttranscriptional editing [76]. Such editing would also affect the Kaposin A protein, which is encoded from the same locus with the amino acid glycine replaced by a serine. Indeed, the editing of kaposin mRNA was later confirmed in two PEL cell lines using a sensitive PCR and Northern blot assays [78]. Remarkably, editing of K12 transcript, which includes changes within both kaposin A and miR-K10, eliminates entirely its transforming activity, i.e. ability to induce focus formation in transfected cells or produce tumors in nude mice [78]. Moreover, levels of K12 editing increases dramatically with reactivation and lytic progression [14,78], suggesting that its tumorigenic activity is required for maintenance of latency. In addition, authors demonstrated that recombinant ADAR1 highly specifically edits K12 transcript in vitro, suggesting that ADAR1 was responsible for editing of this transcript in PEL cell lines [78]. Nonetheless, the surge in the levels of Kaposin transcript editing is not concomitant with the increase of ADAR1 [79], but it corelates with increased RNA binding during reactivation [79]. Recently Rajendran et al. performed comprehensive analysis of the KSHV editom landscape in different cells and identified numerous additional editing spots in viral transcriptome, some of which are conserved in all cells tested, including editing within, kaposin, LANA and RTA transcript (amino acid change E378G), and pri-miR-K12-4 (two sites within the loop and one within the seed sequence) [79]. Similar to kaposin transcript, editing of pri-miR-K12-4 was abolished in cell devoid of ADAR1 expression. Interestingly, editing of pri-miR-K12-4 had a strong impact on Drosha processivity resulting in reduced levels of mature miRNAs [79]. This result was consistent with the finding that depletion of ADAR1 resulted in increased levels of mature miR-K12-4. Furthermore, authors demonstrated that editing within the seed region not only expands the repertoire of potential RNA that can be targeted but also switches ontological association of potential targets. For example, targets of unedited miR-K12-4-3p are ontologically enriched for processes involved in transcription, metabolic processes and macromolecule biosynthesis, while edited miR-K12-4-3p targets are enriched for cell growth and development [79]. Although biological relevance of miR-K12-4-3p editing remains elusive, it is intriguing that it is required for infectious virion production [79], and that A-to-I editing might contribute to efficient KSHV infection.
KSHV- editing independent roles of ADAR
Herpesvirus infections, including KSHV, are known to trigger innate immune responses by activating host DNA and RNA sensors (e.g., TLRs, RIG -I, MDA5 and cGAS, IFI16), leading to the induction of type I interferons and proinflammatory cytokines [80,81]. On the other hand, the main role of ADAR1 is to mark RNAs as self and prevent overactivation of these signaling pathways. Therefore, it can be predicted that ADAR1 may play a pro-herpesvirus role and that its deficiency may limit efficient herpesvirus infection. Indeed, in their recent work, Zhang et al. have shown that ablation of ADAR1 (both forms p150 and p110) in cells latently infected with KSHV strongly inhibits induced reactivation [82]. Moreover, they show that knockdown of ADAR1 increases IFN production and induces a variety of antiviral genes during KSHV reactivation, suggesting that ADAR1 is required for optimal reactivation of KSHV. Nevertheless, many ADAR1-activated pathways could be responsible for the observed induction of IFN. To address this question, Zhang et al carefully examined potential molecular pathways in cells lacking ADAR1 and showed that depletion of RIG -I and, to a lesser extent, MDA5 rescued KSHV reactivation, abrogated RLR singling and activation of TBK1 and IRF3, ultimately leading to reduced induction of IFN. Depletion of the common downstream adaptor protein MAVS confirmed their conclusions [82]. The exact trigger for activation of RIG -I in reactivating cells remains unknown, but Zhang et al show evidence of enhanced binding of RIG -I to dsRNA in cells lacking ADAR1. To our knowledge, this is the first study to show an editing-independent role for ADAR1 in herpesvirus infection, underscoring the immense importance of this protein. It is reasonable to assume that other herpesviruses also rely on the presence of ADAR proteins in cells and that similar paradigms apply within the same host species. However, different herpesviruses have evolved different adaptations to host defenses, so the need for a specific ADAR-mediated pathway may differ.
4.4. Malacoherpesviridae –herpesviruses of mollusks
Herpesviruses of invertebrates are largely unknown, limiting our understanding of the diversity and evolution of nonmammalian, as well as mammalian, herpesviruses. Osterid herpesvirus-1 (OsHV-1) and Haliotid herpesvirus-1 (HaHV-1), which infect marine bivalves and abalones (sea snails), respectively, are the only two invertebrate herpesviruses isolated to date [83]. However, there are sequence evidence for several new viruses in the family, suggesting a wide diversity of these viruses [84]. They can cause detrimental infections in aquaculture species and significant economic losses in oyster and abalone farming. Unfortunately, studies on malacoherpesviruses are largely limited to infections in vivo and are therefore very challenging .
Nonetheless, using RNA analysis Rosani et al. demonstrated the upregulation of ADAR1 homolog in the bivalve Crossostrea gigas and in the gastropod Haliotis diversicolor supertexta durign OsHV-1 or HaHV-1 infection, respectively, which correlated with the extensive hyperediting of viral and host RNAs [85,86]. The ADAR hyperediting on the OsHV-1 transcripts occurred in genomic hot-spots characterized by the presence of overlapping genes on the opposite strand. Similarly, Bia at al. showed upregulation of ADAR1 homolog in the bivalve Scapharca broughtonii infected with OsHV-1 and in Haliotis diversicolor supertexta infected with HaHV-1 [87]. Although the kinetics of ADAR1 upregulation were somewhat different between these two species, hyperediting increased with time in both infections, reaching maximal levels late in infection. Important to note, in C. gigas ADAR1 transcript was also upregulated after stimulation with poly(I:C), which corelated with hyperediting of a number of genes involved in antiviral response, miRNA maturation and epigenetic regulation [86]. In addition, several lines of evidence led authors to speculate that levels of editing might directly differentiate between oysters resistant and susceptible to virus infection.

Table 1.
Known functions of ADAR proteins in herpesvirus infection.
| Virus taxonomy | Virus | ADAR activity | Ref. | ||
| Herpesvirales | Ortoherpesviridae | Alphaherpesvirinae | HSV-1 (HHV-1) |
ADAR1 expression levels maintained during productive infection. Editing of HSV-1 miR-H2-3p in latency and to lesser extent in productive infection. Function: Increased targeting repertoire of miR-H2-3p. |
[13] [48,53] [53] |
| VZV (HHV-3) |
Dynamics of ADAR expression levels unknown. Editing of lncRNA NTO3 (antisense to ORF63). Function: unknown. |
[56] |
|||
| GaHV-2 |
Dynamics of ADAR expression levels unknown. Editing of ERL lncRNA. Function: unknown. |
[60] |
|||
| Betaherpesvirinae | HCMV (HHV-5) |
ADAR1 p110 is upregulated in productive infection. Editing of host miR-376a. Function: edited miRNA gains specificity to downregulates HLA-E and abolishes targeting of MICB (ligand of activating NKG2D receptor) facilitating elimination of HCMV infected cells. |
[13] |
||
| Gammaherpesvirinae | EBV (HHV-4) |
Dynamics of ADAR expression levels unknown. Editing of pri-BHRF1-1, pri-miR-BART3,-BART6, -BART8, -BART11 and -BART16. Editing of vlncRNA oriPtL and oriPtR. Functions: Affected DROSHA processing of pri-miR-BART6 and -BART3 resulting in lower levels of miRNAs, and loss of posttranscriptional regulation of their targets (Dicer). miR-BART3 seed sequence editing abolished Dicer targeting. Functions of edited oriPtL and oriPtR unknown. |
[68,69] [71] [68,69] |
||
| KSHV (HHV-8) |
ADAR1 expression levels maintained from latent to lytic infection.* ADAR1 (all forms) increased during reactivation.* Editing of K12 transcript, LANA, RTA etc. and pri-miR-K12-10, pri-miR-K12-4 Functions: Editing eliminates K12 transforming activity and reduces pri-miR-K12-4 processing by Drosha. Increased repertoire of miR-K12-4 targets. ADAR1 prevents activation of RIG-I signaling and enables efficient virus reactivation. |
[78,79] [14,82] [76,78,79,82] [82] |
|||
| Malacoherpesviridae |
OsHV-1 HaHV-1 |
ADAR1 upregulated in productively infected host. Editing: Increased global editing of viral and host transcripts during infection. Function: unknown. |
[85,86,87] | ||
* studies show conflicting results.
5. Discussion
Over the years and through intensive studies, much has been learned about the biology and pathogenesis of herpesviruses, which has not only improved understanding but also enabled treatment of disease and improvement of health. The role of ADAR proteins in dsDNA viruses, including herpesviruses, is a rather neglected area of research. This is somewhat understandable considering that ADARs are dsRNA-binding proteins and that most studies have focused on RNA viruses. The goal of this literature review is to provide clues to another level of herpesvirus complexity that has not been adequately explored. We focus exclusively on the potential role of the ADAR proteins, although other RNA editing proteins, such as the activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like’ (AID/APOBEC) protein family also play important roles.
A-to-I editing (ediotome) in herpesvirus infections has been studied in detail in only a few cases. However, current evidence suggests that a substantial amount of editing of viral transcripts occurs during productive and latent infection in all viruses studied. While in RNA viruses editing could affect the genome, contribute to genome diversity and impact fitness (hypermutations) and pathogenesis of the virus (reviewed in [7,33], such scenarios are unlikely in dsDNA viruses, and the focus is on editing of coding and non-coding transcripts. It is important to note that increased editing rate usually correlates with increased expression of ADAR proteins, but differential expression of ADAR proteins has only been studied for some herpesviruses. Nonetheless, some important conclusions can be made. First, productive infection (HCMV, OsHV-1, HaHV-1) [13,85-87], including the reactivation process (KSHV) [82], triggers the expression of ADAR proteins, but not for all herpesviruses (HSV-1 and HSV-2) [13]. Second, HCVM selectively activates only the p110 promoter but not the interferon-inducible p150 [13], whereas KSHV induces both forms [14]. These results strongly suggest that different viruses use different mechanisms and may have different requirements for A-to-I editing at different phases of infection. For example, hyperediting of one of HSV-1 encoded miRNAs (miR-H2-3p), has been found in latently infected human ganglia, and to much lower extent in productively infected cells in culture, clearly indicating a potential importance of this process for latent infection [53]. One may hypothesize that the lower extent of editing in productive infection compared to latent infection is simply a matter of accessibility. On the other hand, KSHV transcripts and miRNAs are edited to a lesser extent during latency and increase during reactivation. Current evidence indicates that ADAR does not randomly edit viral transcripts, but specifically edits multiple transcripts (e.g. kaposin) and a selection of pri-miRNAs to alter the properties of these gene products and generate different cellular environment required by the virus [78]. This is probably only one of the mechanisms, but the dramatic loss of kaposin transformation activity strongly suggests its importance.
In herpesviruses, the functional consequences of editing are best seen in miRNAs. Editing of both KSHV and EBV miRNA precursor transcripts (pri-miRNAs) negatively affects DROSHA processivity, limiting the amounts of these miRNAs and deregulating their direct targets [68,69,79]. Moreover, EBV miR-BART3 and –BART6 directly regulate Dicer, so editing of these miRNAs has a global effect on the RNA-interreference (RNAi) system [68,69]. Whether, the precursor of hyperedited HSV-1 miR-H2 inherits impaired processivity by DROSHA is not known, but 50% rate of miR-H2 editing suggests that it is not [48,53]. Furthermore, there is evidence that edited miR-H2 is loaded onto Ago as efficiently as non-edited miRNA, in contrast to edited miR-BART6 where editing also suppresses RISC loading [53]. It is important to mention that editing- mediated alterations of miRNA biology is not unique to viruses, rather well noted for cellular miRNAs (i.e. editing effects biogenesis, loading, re-targeting and stability of miRNAs).
There are other transcripts with dsRNA structure, but miRNAs, found edited in VZV, Ga-HV-2, KSHV, OsHV-1 and HaHV-1 infections, however clear evidences for relevant biological role are far less obvious [56,71,79,86].
Editing (in)dependent functions of ADAR. The role of ADAR1, primarily ADAR p150, in suppressing antiviral signaling has been demonstrated for several RNA viruses (MeV, VSV, IV, HCV, CoV, etc.)[7]. However, it is not clear whether A-to-I editing is always required for suppressive function or whether it depends only on dsRNA binding and interaction with RNA sensors. There is ample evidence that the absence of ADAR1 p150 leads to overactivation of PKR (e.g., MeV, VSV, EMCV, HIV), but in some viruses other sensing pathways take over (e.g., the OAS pathway in CoV, RIG -I in IV), and sometimes activation of PKR can even be beneficial to the virus (HCV) [7,88]. Therefore, it is very interesting that ADAR1 mediates the immunosuppression required for efficient reactivation via the MDA5 pathway during KSHV reactivation [82]. It is now known whether other sensing pathways are involved in this process. It will be very exciting to learn whether the ADAR1/MDA5 axis is also required for other herpesviruses or whether other pathways are more important.
Some open question. The studies described have given us only a small glimpse of the possible role of ADAR proteins and the complexity they bring to herpesvirus infection. There is still a large gap in basic knowledge about the editome in infected cells. For example, editing of many viruses, including HSV1 and HSV-2, is unknown, and the dynamics of editing during reactivation are unexplored for most viruses. Editing can affect the processing (biogenesis), stability, interactome, and function of RNAs, so uncovering editing will contribute to a better understanding of the basic biological properties of transcripts and their functions. The contribution of the two ADAR proteins ADAR1 and ADAR2 to a variety of replication strategies, latency niches, and hosts remains to be better defined. In addition, how expression of the ADAR proteins is regulated during infection remains to be better understood (e.g., KSHV and HCMV induce ADAR1 expression during productive infection but different protein forms, whereas HSV-1 does not affect ADAR1 levels).
On the other hand, a body of evidence suggests that ADAR p150, the interferon-stimulated form of ADAR, is solely responsible for suppressing cytoplasmic sensors (PKR, RIG -I/MDA5, ZBP1, OAS) and attenuating their antiviral response. However, which signaling pathways are involved (different in different herpesviruses? Do all signaling pathways contribute?); what are the exact triggers of the signaling pathways; how these triggers link ADAR1 and dsRNA sensors; what is a role of various posttranscriptional and posttranslational modifications? and many other questions remain to be addressed.
Finally, no herpesvirus gene product has yet been identified that mimics or directly affects the functions of ADAR, such as the VAI transcript of AdV [89], the NS1 protein of influenza virus [90], and the E3L protein of vaccinia virus [91],.
All in all, the biology of herpes viruses has been studied in great detail, but there is still much to learn, and the complexity of these viruses always offers new surprises.
Author Contributions
Conceptualization, I.J and I.M.; analysis – all authors; writing—all authors; review and editing I.J. and I.M, supervision, I.J.; funding acquisition, I.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Croatian Science Foundation Grant IP-2020-02-2287 and DOK-2012-02-9152, and University of Rijeka support grant prirod-sp-23-502930 to IJ.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 2016, 16, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 2009, 22, 240–273, Table of Contents. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int Rev Immunol 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Gallo, A.; Vukic, D.; Michalik, D.; O'Connell, M.A.; Keegan, L.P. ADAR RNA editing in human disease; more to it than meets the, I. Hum Genet 2017, 136, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Bonamassa, B.; Alisi, A.; Nobili, V.; Locatelli, F.; Gallo, A. ADAR enzyme and miRNA story: a nucleotide that can make the difference. Int J Mol Sci 2013, 14, 22796–22816. [Google Scholar] [CrossRef]
- Tomaselli, S.; Galeano, F.; Locatelli, F.; Gallo, A. ADARs and the Balance Game between Virus Infection and Innate Immune Cell Response. Curr Issues Mol Biol 2015, 17, 37–51. [Google Scholar]
- Samuel, C.E. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J Biol Chem 2019, 294, 1710–1720. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 2014, 24, 365–376. [Google Scholar] [CrossRef]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 2012, 30, 253–260. [Google Scholar] [CrossRef]
- Pfaller, C.K.; George, C.X.; Samuel, C.E. Adenosine Deaminases Acting on RNA (ADARs) and Viral Infections. Annu Rev Virol 2021, 8, 239–264. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Rosenberg, B.R.; Chung, H.; Rice, C.M. Identification of ADAR1 p150 and p110 Associated Edit Sites. Methods Mol Biol 2023, 2651, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Zimmermann, A.; Oiknine Djian, E.; Weisblum, Y.; Livneh, Y.; Khanh Le, V.T.; Galun, E.; Horejsi, V.; Isakov, O.; Shomron, N.; et al. MicroRNA editing facilitates immune elimination of HCMV infected cells. PLoS Pathog 2014, 10, e1003963. [Google Scholar] [CrossRef]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog 2014, 10, e1003847. [Google Scholar] [CrossRef]
- Hood, J.L.; Morabito, M.V.; Martinez, C.R., 3rd; Gilbert, J.A.; Ferrick, E.A.; Ayers, G.D.; Chappell, J.D.; Dermody, T.S.; Emeson, R.B. Reovirus-mediated induction of ADAR1 (p150) minimally alters RNA editing patterns in discrete brain regions. Mol Cell Neurosci 2014, 61, 97–109. [Google Scholar] [CrossRef]
- Wang, Q.; Miyakoda, M.; Yang, W.; Khillan, J.; Stachura, D.L.; Weiss, M.J.; Nishikura, K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 2004, 279, 4952–4961. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 2012, 44, 1243–1248. [Google Scholar] [CrossRef]
- Herbert, A. Mendelian disease caused by variants affecting recognition of Z-DNA and Z-RNA by the Zalpha domain of the double-stranded RNA editing enzyme ADAR. Eur J Hum Genet 2020, 28, 114–117. [Google Scholar] [CrossRef]
- Miyamura, Y.; Suzuki, T.; Kono, M.; Inagaki, K.; Ito, S.; Suzuki, N.; Tomita, Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003, 73, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.R.; Slack, F.J. ADAR1 and its implications in cancer development and treatment. Trends Genet 2022, 38, 821–830. [Google Scholar] [CrossRef]
- Stok, J.E.; Oosenbrug, T.; Ter Haar, L.R.; Gravekamp, D.; Bromley, C.P.; Zelenay, S.; Reis e Sousa, C.; van der Veen, A.G. RNA sensing via the RIG-I-like receptor LGP2 is essential for the induction of a type I IFN response in ADAR1 deficiency. Embo J 2022, 41, e109760. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Deng, P.; Zhu, Z.; Zhu, J.; Wang, G.; Zhang, L.; Chen, A.F.; Wang, T.; Sarkar, S.N.; Billiar, T.R.; et al. Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J Immunol 2014, 193, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Calis, J.J.A.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Dao Thi, V.L.; Shilvock, A.R.; Hoffmann, H.H.; Rosenberg, B.R.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824. [Google Scholar] [CrossRef]
- de Reuver, R.; Verdonck, S.; Dierick, E.; Nemegeer, J.; Hessmann, E.; Ahmad, S.; Jans, M.; Blancke, G.; Van Nieuwerburgh, F.; Botzki, A.; et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature 2022, 607, 784–789. [Google Scholar] [CrossRef]
- Hubbard, N.W.; Ames, J.M.; Maurano, M.; Chu, L.H.; Somfleth, K.Y.; Gokhale, N.S.; Werner, M.; Snyder, J.M.; Lichauco, K.; Savan, R.; et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature 2022, 607, 769–775. [Google Scholar] [CrossRef]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef]
- Hajji, K.; Sedmik, J.; Cherian, A.; Amoruso, D.; Keegan, L.P.; O'Connell, M.A. ADAR2 enzymes: efficient site-specific RNA editors with gene therapy aspirations. RNA 2022, 28, 1281–1297. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. J Biol Chem 2017, 292, 4326–4335. [Google Scholar] [CrossRef]
- Raghava Kurup, R.; Oakes, E.K.; Manning, A.C.; Mukherjee, P.; Vadlamani, P.; Hundley, H.A. RNA binding by ADAR3 inhibits adenosine-to-inosine editing and promotes expression of immune response protein MAVS. J Biol Chem 2022, 298, 102267. [Google Scholar] [CrossRef]
- Mladenova, D.; Barry, G.; Konen, L.M.; Pineda, S.S.; Guennewig, B.; Avesson, L.; Zinn, R.; Schonrock, N.; Bitar, M.; Jonkhout, N.; et al. Adar3 Is Involved in Learning and Memory in Mice. Front Neurosci-Switz 2018, 12. [Google Scholar] [CrossRef]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benko, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J Gen Virol 2021, 102. [Google Scholar] [CrossRef] [PubMed]
- Pellett, E.P.; Roizman, B. Herpesviridae. In Fields of Virology, 6 ed.; D.M., K., P.M., H., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2017; Volume 2.
- Murthy, S.; Couacy-Hymann, E.; Metzger, S.; Nowak, K.; De Nys, H.; Boesch, C.; Wittig, R.; Jarvis, M.A.; Leendertz, F.H.; Ehlers, B. Absence of frequent herpesvirus transmission in a nonhuman primate predator-prey system in the wild. J Virol 2013, 87, 10651–10659. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses,. In Fields Virology, 6th ed.; M., K.D.M.a.H.P., Ed.; Lippincott, Williams & Wilkins: New York, NY, 2013; Volume 2.
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef]
- Thompson, R.L.; Sawtell, N.M. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J. Virol. 2001, 75, 6660–6675. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef]
- Jurak, I.; Kramer, M.F.; Mellor, J.C.; van Lint, A.L.; Roth, F.P.; Knipe, D.M.; Coen, D.M. Numerous Conserved and Divergent MicroRNAs Expressed by Herpes Simplex Viruses 1 and 2. Journal of Virology 2010, 84, 4659–4672. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef]
- Umbach, J.L.; Wang, K.; Tang, S.; Krause, P.R.; Mont, E.K.; Cohen, J.I.; Cullen, B.R. Identification of viral microRNAs expressed in human sacral ganglia latently infected with herpes simplex virus 2. J. Virol. 2010, 84, 1189–1192. [Google Scholar] [CrossRef]
- Han, Z.Y.; Liu, X.J.; Chen, X.Q.; Zhou, X.S.; Du, T.; Roizman, B.; Zhou, G.Y. miR-H28 and miR-H29 expressed late in productive infection are exported and restrict HSV-1 replication and spread in recipient cells. P Natl Acad Sci USA 2016, 113, E894–E901. [Google Scholar] [CrossRef] [PubMed]
- Cokaric Brdovcak, M.; Zubkovic, A.; Ferencic, A.; Sosa, I.; Stemberga, V.; Cuculic, D.; Rokic, F.; Vugrek, O.; Hackenberg, M.; Jurak, I. Herpes simplex virus 1 miRNA sequence variations in latently infected human trigeminal ganglia. Virus Res 2018, 256, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pesola, J.M.; Li, G.; McCarron, S.; Coen, D.M. Mutations Inactivating Herpes Simplex Virus 1 MicroRNA miR-H2 Do Not Detectably Increase ICP0 Gene Expression in Infected Cultured Cells or Mouse Trigeminal Ganglia. J Virol 2017, 91. [Google Scholar] [CrossRef]
- Flores, O.; Nakayama, S.; Whisnant, A.W.; Javanbakht, H.; Cullen, B.R.; Bloom, D.C. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J Virol 2013, 87, 6589–6603. [Google Scholar] [CrossRef]
- Barrozo, E.R.; Nakayama, S.; Singh, P.; Neumann, D.M.; Bloom, D.C. Herpes Simplex Virus 1 MicroRNA miR-H8 Is Dispensable for Latency and Reactivation In Vivo. Journal of Virology 2021, 95. [Google Scholar] [CrossRef]
- Jurak, I.; Silverstein, L.B.; Sharma, M.; Coen, D.M. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J Virol 2012, 86, 10093–10102. [Google Scholar] [CrossRef]
- Zubkovic, A. ; al., e. HSV-1 miRNAs are posttranscriptionaly edited in latently infected human ganglia. https://www.biorxiv.org/content/10.1101/2023.05.26.542484v1; ACCEPTED, 2023. [Google Scholar]
- Arvin, A.; Gilden, D. Varicela-Zoster Virus. In Fileds of Virology, 6th ed.; D.M., K., P.M., H., Eds.; Lippincott, Williams & Wilkins: New York, NY, : 2013; Volume 2.
- Depledge, D.P.; Sadaoka, T.; Ouwendijk, W.J.D. Molecular Aspects of Varicella-Zoster Virus Latency. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Prazsak, I.; Moldovan, N.; Balazs, Z.; Tombacz, D.; Megyeri, K.; Szucs, A.; Csabai, Z.; Boldogkoi, Z. Long-read sequencing uncovers a complex transcriptome topology in varicella zoster virus. Bmc Genomics 2018, 19, 873. [Google Scholar] [CrossRef] [PubMed]
- Osterrieder, N.; Wallaschek, N.; Kaufer, B.B. Herpesvirus Genome Integration into Telomeric Repeats of Host Cell Chromosomes. Annual Review of Virology, Vol 1 2014, 1, 215–235. [Google Scholar] [CrossRef]
- Osterrieder, N.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Trapp, S. Marek's disease virus: from miasma to model. Nat Rev Microbiol 2006, 4, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Cantello, J.L.; Anderson, A.S.; Morgan, R.W. Identification of latency-associated transcripts that map antisense to the ICP4 homolog gene of Marek's disease virus. J Virol 1994, 68, 6280–6290. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, T.; Boumart, I.; Coupeau, D.; Rasschaert, D. Hyperediting by ADAR1 of a new herpesvirus lncRNA during the lytic phase of the oncogenic Marek's disease virus. J Gen Virol 2016, 97, 2973–2988. [Google Scholar] [CrossRef]
- Goodrum, F. The complex biology of human cytomegalovirus latency. Adv Virus Res 2022, 112, 31–85. [Google Scholar] [CrossRef]
- Slavuljica, I.; Krmpotic, A.; Jonjic, S. Manipulation of NKG2D ligands by cytomegaloviruses: impact on innate and adaptive immune response. Front Immunol 2011, 2, 85. [Google Scholar] [CrossRef]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat Immunol 2010, 11, 806–813. [Google Scholar] [CrossRef]
- Dolken, L.; Perot, J.; Cognat, V.; Alioua, A.; John, M.; Soutschek, J.; Ruzsics, Z.; Koszinowski, U.; Voinnet, O.; Pfeffer, S. Mouse cytomegalovirus microRNAs dominate the cellular small RNA profile during lytic infection and show features of posttranscriptional regulation. J Virol 2007, 81, 13771–13782. [Google Scholar] [CrossRef]
- Frappier, L. Epstein-Barr virus: Current questions and challenges. Tumour Virus Res 2021, 12, 200218. [Google Scholar] [CrossRef]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Cullen, B.R. EBV Noncoding RNAs. Curr Top Microbiol Immunol 2015, 391, 181–217. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Wulff, B.E.; Alla, N.R.; Maragkakis, M.; Megraw, M.; Hatzigeorgiou, A.; Iwakiri, D.; Takada, K.; Wiedmer, A.; Showe, L.; et al. Editing of Epstein-Barr Virus-encoded BART6 MicroRNAs Controls Their Dicer Targeting and Consequently Affects Viral Latency. J Biol Chem 2010, 285, 33358–33370. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Yuen, K.S.; Tsao, S.W.; Chen, H.; Kok, K.H.; Jin, D.Y. Perturbation of biogenesis and targeting of Epstein-Barr virus-encoded miR-BART3 microRNA by adenosine-to-inosine editing. J Gen Virol 2013, 94, 2739–2744. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog 2012, 8, e1002484. [Google Scholar] [CrossRef]
- Cao, S.; Moss, W.; O'Grady, T.; Concha, M.; Strong, M.J.; Wang, X.; Yu, Y.; Baddoo, M.; Zhang, K.; Fewell, C.; et al. New Noncoding Lytic Transcripts Derived from the Epstein-Barr Virus Latency Origin of Replication, oriP, Are Hyperedited, Bind the Paraspeckle Protein, NONO/p54nrb, and Support Viral Lytic Transcription. J Virol 2015, 89, 7120–7132. [Google Scholar] [CrossRef]
- Dissinger, N.J.; Damania, B. Recent advances in understanding Kaposi's sarcoma-associated herpesvirus. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Zhong, W.; Wang, H.; Herndier, B.; Ganem, D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc Natl Acad Sci U S A 1996, 93, 6641–6646. [Google Scholar] [CrossRef]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi's sarcoma-associated herpesvirus. J Virol 1999, 73, 5722–5730. [Google Scholar] [CrossRef]
- Forte, E.; Raja, A.N.; Shamulailatpam, P.; Manzano, M.; Schipma, M.J.; Casey, J.L.; Gottwein, E. MicroRNA-mediated transformation by the Kaposi's sarcoma-associated herpesvirus Kaposin locus. J Virol 2015, 89, 2333–2341. [Google Scholar] [CrossRef]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef]
- Gottwein, E.; Cai, X.; Cullen, B.R. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J Virol 2006, 80, 5321–5326. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.Z.; Linnstaedt, S.D.; Muralidhar, S.; Cashman, K.A.; Rosenthal, L.J.; Casey, J.L. RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. Journal of Virology 2007, 81, 13544–13551. [Google Scholar] [CrossRef]
- Rajendren, S.; Ye, X.; Dunker, W.; Richardson, A.; Karijolich, J. The cellular and KSHV A-to-I RNA editome in primary effusion lymphoma and its role in the viral lifecycle. Nat Commun 2023, 14, 1367. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Rao, Y.; Tian, M.; Zhang, S.; Feng, P. Modulation of Innate Immune Signaling Pathways by Herpesviruses. Viruses 2019, 11. [Google Scholar] [CrossRef]
- O'Connor, C.M.; Sen, G.C. Innate Immune Responses to Herpesvirus Infection. Cells 2021, 10. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, G.; Damania, B. ADAR1 Facilitates KSHV Lytic Reactivation by Modulating the RLR-Dependent Signaling Pathway. Cell Rep 2020, 31, 107564. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.; Karin, E.L.; Pupko, T. Sequence analysis of malacoherpesvirus proteins: Pan-herpesvirus capsid module and replication enzymes with an ancient connection to "Megavirales". Virology 2018, 513, 114–128. [Google Scholar] [CrossRef]
- Rosani, U.; Shapiro, M.; Venier, P.; Allam, B. A Needle in A Haystack: Tracing Bivalve-Associated Viruses in High-Throughput Transcriptomic Data. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Rosani, U.; Bai, C.M.; Maso, L.; Shapiro, M.; Abbadi, M.; Domeneghetti, S.; Wang, C.M.; Cendron, L.; MacCarthy, T.; Venier, P. A-to-I editing of Malacoherpesviridae RNAs supports the antiviral role of ADAR1 in mollusks. BMC Evol Biol 2019, 19, 149. [Google Scholar] [CrossRef]
- Rosani, U.; Bortoletto, E.; Montagnani, C.; Venier, P. ADAR-Editing during Ostreid Herpesvirus 1 Infection in Crassostrea gigas: Facts and Limitations. mSphere 2022, 7, e0001122. [Google Scholar] [CrossRef]
- Bai, C.M.; Rosani, U.; Zhang, X.; Xin, L.S.; Bortoletto, E.; Wegner, K.M.; Wang, C.M. Viral Decoys: The Only Two Herpesviruses Infecting Invertebrates Evolved Different Transcriptional Strategies to Deflect Post-Transcriptional Editing. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Garaigorta, U.; Chisari, F.V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009, 6, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Liu, Y.; Samuel, C.E. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology 1998, 245, 188–196. [Google Scholar] [CrossRef] [PubMed]
- de Chassey, B.; Aublin-Gex, A.; Ruggieri, A.; Meyniel-Schicklin, L.; Pradezynski, F.; Davoust, N.; Chantier, T.; Tafforeau, L.; Mangeot, P.E.; Ciancia, C.; et al. The interactomes of influenza virus NS1 and NS2 proteins identify new host factors and provide insights for ADAR1 playing a supportive role in virus replication. PLoS Pathog 2013, 9, e1003440. [Google Scholar] [CrossRef]
- Liu, Y.; Wolff, K.C.; Jacobs, B.L.; Samuel, C.E. Vaccinia virus E3L interferon resistance protein inhibits the interferon-induced adenosine deaminase A-to-I editing activity. Virology 2001, 289, 378–387. [Google Scholar] [CrossRef]
Figure 1.
The ADAR protein family. ADAR1, interferon inducible p150 and constitutively expressed p110 form, ADAR2 and ADAR3. ADAR1 and ADAR2 have deaminase activity. ADAR3 lacks deaminase activity and has a role in regulation of editing. Z-DNA binding domains (Za and Zb) are, shown as yellow circles, the dsRNA binding domains (RI, RII and RIII) are shown as blue rectangles, and deaminase catalytic domain are shown are purple ovals. Striped purple oval indicated lack of deaminase activity. Figure adapted from [11] and generated in BioRender.
Figure 1.
The ADAR protein family. ADAR1, interferon inducible p150 and constitutively expressed p110 form, ADAR2 and ADAR3. ADAR1 and ADAR2 have deaminase activity. ADAR3 lacks deaminase activity and has a role in regulation of editing. Z-DNA binding domains (Za and Zb) are, shown as yellow circles, the dsRNA binding domains (RI, RII and RIII) are shown as blue rectangles, and deaminase catalytic domain are shown are purple ovals. Striped purple oval indicated lack of deaminase activity. Figure adapted from [11] and generated in BioRender.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



