Submitted:
08 September 2023
Posted:
12 September 2023
You are already at the latest version
Abstract
The combination of wet-lab experimental data on multi-site combinatorial mutations and machine learning is an innovative method in protein engineering. In this study, we present an improved innovative sequence–activity relationship (innov'SAR) methodology based on novel descriptors and digital signal processing (DSP) to construct a predictive model. In this improved approach, 21 experimental (R)-selective amine transaminases from Aspergillus terreus (AT-ATA) were used as an input to predict higher thermostability than that predicted using the existing data. We successfully improved the determination coefficient (R2) of the model from 0.66 to 0.92. In addition, root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were estimated and conformation analysis based on molecular dynamics simulations was performed to verify the enhanced thermal stability of the screened mutants. The improved innov'SAR algorithm enhanced the predictive accuracy, suggesting a method for modifying the stability of AT-ATA, which may help in directed evolutionary screening and open up new avenues for protein engineering.
Keywords:
Artificial Intelligence
; Machine Learning
; Thermostability
; Molecular Dynamics Simulation
; Extended Sequence
; Directed Evolution
1. Introduction
In protein engineering, directed evolution is an important method used for modifying the properties of enzymes. It is widely employed in various industries including chemical or drug synthesis, food production, and waste biodegradation [1,2]. Directed evolution through the natural rule of “mutation selection” remains the most prevalent method for modifying enzymes. Mutant libraries are constructed using procedures such as site-saturation mutation, epPCR, and DNA shuffling. They are screened for target mutants that meet specific requirements by recreating the natural evolutionary process in a laboratory [3,4], thus considerably expanding the range of enzyme applications. To date, directed evolution has been used to generate artificial cysteine lipase with high activity and altered catalytic mechanism [5], a redox-mediated Kemp eliminase [6], modified oxidase for efficient CO2 fixation [7], and modified transaminase for the synthesis of sitagliptin [8]. However, generating and screening mutant libraries are laborious and time-consuming processes, which dramatically impede the progression of the experiment.
With the development of science and computer technology, machine learning has emerged as an efficient technique for designing novel biocatalysts [9,10,11,12,13]. The use of machine learning methods for predicting the secondary structure of proteins was first reported in 1992 [14]. Since then, many new methods based on machine learning algorithms have emerged for rapid and accurate prediction of the stability, activity, and substrate-binding properties of mutant proteins. For example, support vector machine [15,16] and decision tree [17] can be used to predict changes in the stability of enzymes following mutations, random forest can be used to predict protein solubility [18], and the K-nearest neighbor classifier can be used to predict the function and mechanism of enzymes [19]. However, these methods require the modeling of massive experimental data, and the accuracy of prediction requires to be verified through experiments. Innovative sequence–activity relationship (innov'SAR) is a new method for screening mutant libraries that was reported in 2018 [20]. It predicts the effects of mutations on the biological activity of proteins in a small-sample dataset only using sequence information. innov'SAR has been successfully used to improve the enantioselectivity of epoxide hydrolase from Aspergillus niger [21]. However, the method is not suitable for all enzymes. In this study, we modified protein descriptors and used novel strategies to improve the robustness and predictive accuracy of innov'SAR.
As natural biocatalysts, ω-transaminases play a critical role in the asymmetric synthesis of chiral amines and in the racemic splitting of amines, which are used in the manufacture and synthesis of pharmaceutical products, fine chemicals, and agrochemicals [22,23,24]. The amine transaminase from Aspergillus terreus (AT-ATA) has high stereoselectivity and excellent catalytic efficiency [25]; however, its poor thermal stability greatly limits its practical applications. Therefore, improving the thermal stability of transaminases through site-directed mutagenesis is necessary for expanding their industrial applications. We have previously used different protein engineering strategies to improve the thermal stability of transaminases, including the introduction of disulfide bonds, deletion of unstable amino acids on the surface of the loop region, consensus mutagenesis, and the use of B-factor values combined with FoldX energy optimization [26,27,28,29]. In addition, we have used machine learning techniques to improve the stability of AT-ATA [30]. With the increasing demand for chiral amines, further improvement of the thermal stability of AT-ATA can be beneficial for industrial preparation of chiral amines.
In this study, we digitized protein sequences using multiple indices in the AAindex database. Each index represents different biophysical properties, such as hydrophobicity, α-helix and β-turn propensities [31]. The innov'SAR method was optimized by concatenating multiple indices and using an iterative strategy to determine the best protein descriptors. Finally, we successfully used the improved method to predict a more thermally stable AT-ATA, and the predictive accuracy was validated via molecular dynamics simulations [32].
2. Results and Discussion
Protein sequences can be encoded by indexing different physical and chemical properties in the AAindex database to generate different digital sequences. In this study, we connected more indices to obtain more biological information about proteins by coding the indices of different physicochemical properties.
In the innov'SAR method [38], FFT generates protein spectra that significantly contribute to the performance of the model. Therefore, we only considered the coding sequence after connecting FFT. In the encoding phase, each protein variant of the initial experimental dataset was digitized using the indices in the AAindex database. Subsequently, FFT was used to convert the digital sequence into the corresponding protein spectrum as an input for modeling, and this encoded sequence was denoted as the primary sequence (FFT_Seq). A total of 566 indices are available in the AAindex database for FFT-based encoding of protein sequences. Consequently, 566 FFT_Seqs were generated, which were denoted as FFT_Seq1, FFT_Seq2, FFT_Seq3, and so on.
Furthermore, the FFT_Seqs were connected, and the connected sequence of numbers was denoted as an extended sequence (Ext_Seq), which served as the new descriptor after modification. Equation 1 represents the construction of the new descriptor, and the “--” symbol indicates the connection of two FFT_Seqs.
Ext_Seq = FFT_Seqi -- FFT_Seqj -- FFT_Seqk ...
Figure 1 demonstrates the improved innov'SAR method and the transformation of the protein descriptor [38]. Initially, the protein variants were digitally encoded using different indexes in the AAindex database to generate different digital sequences, which were transformed into the corresponding protein spectra in combination with FFT (Figure 1a). Based on the different protein spectra, Ext_Seq was generated via concatenation to obtain more biological information about the protein sequence (Figure 1b). Subsequently, in the encoding phase, the previous FFT_Seq generated using a single index was replaced with the modified Ext_Seq, and the half-lives of the protein variants were integrated and modeled using the partial least squares algorithm. The model with the lowest cvRMSE was identified as optimal using the LOOCV approach. Finally, the predicted half-life was linearly fitted to the actual half-life (Figure 1c), and the predictive power of the model was measured based on R2 in the cross-validation.
More information can be obtained using the abovementioned procedure; however, Ext_Seqs can be combined in thousands of ways owing to the redundancy of indices. The formula used to obtain the number of combinations is mentioned below (Equation 2):
In Equation 2, represents the number of indices and represents the number of FFT_Seqs used to generate Ext_Seq.
Given that the AAindex database has 566 indices, the Ext_Seq set may have 2.4e + 170 possible inputs as modeling codes. Owing to the large number of combinations and considering the performance of the computer, the size of the Ext_Seq set was reduced by filtering a smaller number of FFT_Seqs for combination and identifying the best Ext_Seq by evaluating the modeling performance. We proposed the following two strategies to construct Ext_Seq: (1) using a single index encoding the top five elementary indices for combination and (2) identifying the best Ext_Seq through an iterative connection strategy.
2.1. Combination strategy using multiple index codes
To limit the size of the Ext_Seq set, we restricted N to the top five best indices and defined the extended sequence to have a maximum of five elementary sequence connections, that is, Q ≤ 5. The construction of the extended sequence was independent of the order of elementary sequence connections, that is, FFT_Seq1 -- FFT_Seq2 is equivalent to FFT_Seq2 -- FFT_Seq1. To assess whether Ext_Seq improved the model, we apply the innov'SAR method for Dataset 1. By evaluating the cvRMSE of the model, we select top five indices and use them for combination. Table 1 shows detailed information on the top five indices, including the title, serial number, cvRMSE, and R2 of the indices. According to Equation 2, there are 30 combinations of Ext_Seq. We used each Ext_Seq as a modeling input and ranked the sequences according to R2, the parameter indicating the predictive performance of the model, to identify the best combination of Ext_Seq.
Table 2 shows the top 10 extended sequences based on the top 5 index combinations. Compared with the R2 and cvRMSE of the previous single-index coding model, the R2 of the best extended-index model was increased from 0.81 to 0.86 and its cvRMSE was decreased from 4.56 to 3.64 (Figure 2a, b). The R2 of the previous best single-index AURR980108 model ranked 12th in terms of the combined indices. From the perspective of model performance, the combination of the top-ranked indices resulted in a certain improvement in the performance of the model for the extended sequence.
Furthermore, when the top five indices were concatenated, a longer extended sequence was generated: Ext_Seq = FFT_Seq1 -- FFT_Seq2 -- ...FFT_Seq5. The cvRMSE and R2 of this extended sequence were 4.76 and 0.80, respectively (Figure 2c), ranking 13th in terms of overall ranking, with a reduced performance compared with the best model with single-index coding. Therefore, the length of the extended sequence was not as long as possible. Moreover, the best single-index AURR980108 model was ranked 12th in terms of overall ranking, and the combination of m indices was not better than the combination of n indices (m > n). Based on the abovementioned problems, the combination strategy for selecting the top-ranked indices was not the optimal way to obtain the best Ext_Seq, although the combination strategy improved the predictive performance of the model. Therefore, we used an iterative connection strategy to identify the best Ext_Seq and the best combination of Ext_Seq cases [38].
2.2. Iterative connection strategy
The iterative connection strategy [38] is used to identify the best index for the next connection by continuously expanding the size of Ext_Seq. For each iteration, the current best sequence index (Ext_Seq) was retained through LOOCV, which enabled the construction of Ext_Seq using different index increments. Simultaneously, the best index of modeling performance at the end of the current iteration was determined and prepared for the next iteration.
Figure 3 demonstrates the whole iteration process. In the first iteration, each model was coded according to one of the 566 indices in the AAindex database and was combined with the partial least squares algorithm to construct a total of 566 models. The cvRMSE of each model was evaluated and ranked via cross-validation, and the index with the lowest cvRMSE was selected as the best index. The best index was used for the second iteration, and the retained best index was concatenated with the 566 indices to generate the extended sequence. The extended sequence was used in combination with the partial least squares algorithm to construct the 566 models and determine the best index (extended sequence). For the next iteration, we repeated the same procedure to identify the best index for modeling. A total of 553 models were constructed because of missing values in 13 of the 566 indices in the AAindex database.
Given that the iterative connection strategy can inevitably increase the time complexity of the algorithm, we limited the number of connection indices to 3 (Q ≤ 3): Ext_Seq = FFT_Seq1 -- FFT_Seq2 -- FFT_Seq3. Figure 2a demonstrates the best model obtained through single-index modeling based on the experimental half-life data of 13 AT-ATA with R2 and cvRMSE of 0.81 and 4.56, respectively. Figure 4 shows the results of applying the iterative connection strategy to the abovementioned dataset, with R2 and cvRMSE of 0.96 and 1.93, respectively. Based on the parameters used to evaluate the performance of the model, we concluded that the iterative connection strategy greatly improved the quality and predictive performance of the model with only three indexed connections. Therefore, we increased the sample size by including the new AT-ATA experimental Dataset 2 to assess whether the model could predict variants with higher half-life than the current experimental best half-life. The two datasets were divided to achieve the following two objectives: to compare the robustness of the improved algorithm for the same sample of the model and to observe its effect on the model with different sample sizes.
2.3. Prediction of new improved AT-ATA mutants
The previously described best experimental mutant F115L_L118T harbors two single point mutations in its sequence with a half-life of 65.9 min [27]. To identify better mutants, we constructed a new prediction model using Dataset 2, combined with the iterative connection strategy, which was named model_21. Compared with the R2 of the previous innov'SAR method, the R2 of the new model was successfully improved from 0.66 to 0.92 and all combinations of the 10 single point mutations (210 variants) were computationally generated. The method generated 1003 (210 – 21 = 1003) new variants with multiple point mutations. Figure 5 shows the predicted half-life of all variants in model_21, with 265 mutants having a higher half-life than F115L_L118T, indicating that that model can identify candidates with better thermal stability than F115L_L118T.
2.4. Validation of MD simulation for predicting AT- ATA mutants
To examine the thermal stability of the best predicted mutants, we selected the top two best mutants, named P1 and P2. Details of the mutation sites and predicted half-life of the two mutants are provided in Table 3. We simulated the structures of P1, P2, and the F115L_L118T mutant using YASARA and assessed the stability of the structures based on RMSD. The RMSD values of P1 and P2 were smaller than those of F115L_L118T (Figure 6). Estimation of RMSD revealed that the overall conformational stability of both P1 and P2 was better than that of F115L_L118T. In particular, P2 exhibited lower conformational flexibility and volatility than F115L_L118T, with an average RMSD value of 0.163 nm.
To analyze changes in the RMSF of amino acid residues more clearly, we calculated the average RMSF value for every five residues and used it as the reference value of residues in that region. Estimation of RMSF revealed that the fluctuation level of P1 and P2 residues was lower than that of F115L_L118T. F115L_L118T was more flexible than P1 and P2, indicating that P1 and P2 had a more stable structure (Figure 7a). Specifically, in P2, the site 133 was altered from Glu to Ala, and the RMSF value was decreased from 0.51 nm to 0.15 nm. In addition, the fluctuation of P2 residues was more stable at 130–135 (region 1) and 247–253 (region 2) sites than in the F115L_L118T region. These results indicate that the GLU-ALA mutation at site 133 has a positive effect on the structural stability of the P2 region. To elucidate the reason for the improvement of the stability of mutants, we analyzed protein interactions. A double hydrogen bond was formed among ARG131, ALA133, and ASP134, and a hydrogen bond was formed between ILE135 and ASP134 (Figure 7b). In addition, hydrogen bonds were formed among ALA247, GYL251, and ILE252 with a length of 3.1Å and 2.9Å (Figure 7c). These results suggest that the formation of these hydrogen bonds is closely related to the improvement of the stability of the mutants. Through the analysis of these interactions, we can better understand the structure and stability of the mutants.
3. Materials and Methods
3.1. Aspergillus terreus dataset
The experimental dataset was acquired from a study by Huang et al., and it was divided into two groups. As shown in Table 4, one group was used to test whether the innov'SAR model was improved, and the other group was used to predict the latest experimental dataset 2 [26,27,28,29]. Dataset 1 included a collection of sequences of 12 mutants of AT-ATA and wild-type (WT) AT-ATA and their half-life (T1/2). It was used to test whether the model was improved. Dataset 2 was used to predict more thermally stable AT-ATA. T1/2 was used to measure the thermal stability of enzymes and was defined as the time required for the residual activity of AT-ATA to decline to 50% of its initial activity at 40℃.
3.2. Digital Signal Processing
In the previous innov'SAR method [30], the whole process was divided into three phases: encoding, modeling, and prediction phases. During the encoding phase, the amino acid sequences of proteins are converted into the corresponding digital sequences using the indices of the AAindex database [31], with each standard amino acid letter code being replaced by a numerical value. The AAindex database has more than 500 indices representing various physicochemical properties of the 20 standard amino acids. Digital signal processing, the most critical step, is used for converting the digitized sequences into protein spectra using FFT. Subsequently, FFT converts the digital signals to a representation in energy and frequency domains [33,34] (Equation 3).
In Equation 3, is the input signal of length (amino acid sequence), is the input spectrum (complex number), is the frequency in the spectrum, is the position in the input signal, and is the complex number with i 2 value of -1.
3.3. Evaluation of modeling performance
During the modeling phase, the model was evaluated based on the root mean squared error of cross-validation (cvRMSE) and the coefficient of determination (R2). The leave-one-out cross-validation (LOOCV) approach was used in this study. RMSE was estimated to select the optimal model because it indicated the degree of variation in prediction while using different training sets. R2 was estimated to evaluate the predictive ability of the model, reflecting the degree of concordance between the experimental and predicted half-life. The formulas used for calculating cvRMSE and R2 are mentioned below (Equations 4 and 5):
In the abovementioned equations, is the experimental half-life of the ith sequence, is the half-life of the ith sequence predicted using the ISAR method, is the average of the experimental half-life, and is the number of sequences.
3.4. Molecular dynamics simulation
To verify the validity of the predictive model, we obtained the crystal structure of AT-ATA (PDB ID: 4CE5) from the Protein Data Bank (http://www.rcsb.org) and used molecular dynamics simulation to compare the thermal stability of mutants. Because only the effect of mutation on protein stability was considered, the water of crystallization, impurity ions, and related substrates of AT-ATA were removed during the pretreatment stage. Based on the initial crystal structure, we generated three AT-ATA mutant proteins: the optimal mutant F115L_L118T through experiments and the optimal mutants P1 and P2 through model screening. The three-dimensional (3D) structure of AT-ATA mutants was homology-modeled and optimized using the “BuildModel” command of the FoldX software [35,36].
The simulation process was implemented using the YASARA (version 16.4.6) software [37] (http://www.yasara.org), with the Amber14 force field set for all systems and a constant temperature of 313 K for 100-ns MD simulations. First, the protein was placed in a cube with a density of 0.998 mg/L, and the whole cube was filled with water with a density of 0.998 mg/L. Sodium and chloride ions (0.9%) were added to neutralize the system charge to ensure correct osmotic pressure and electrostatic neutrality, and the ionizable group was protonated according to the PKA value of pH 8.0 in the medium. Subsequently, the gradient descent method was used to minimize the energy of the system. Finally, 100-ns MD simulations with a step size of 2.5 fs were completed under the condition of constant temperature and pressure, and the trajectory file was saved every 25 ps.
During simulation, the cutoff value for van der Waals forces and electrostatic interactions was set to 8.0 Å. At the end of the simulation, YASARA was used to analyze the overall and local changes in the structure of each mutant protein during the simulation. These changes included root mean square deviation (RMSD) of backbone atomic positions and root mean square fluctuation (RMSF) of individual residues.
4. Conclusions
In this study, we successfully used the improved innov'SAR method for the first time for the rational screening and modification of thermally stable AT-ATA. As an efficient combinatorial mutant library screening tool, the half-life of 1024 mutants was predicted using an experimental dataset that accounted for only 2% of the total mutation combinations, reducing the burden of experimental screening. In addition, the improved innov'SAR method was used in combination with the iterative connection strategy to obtain Ext_Seq, and the R2 of the model was improved from 0.66 to 0.92, indicating substantial improvement in the prediction performance of the model. Finally, the screened mutants were validated via molecular dynamics simulations, demonstrating the effectiveness of the improved method in identifying the best mutants.
The greatest advantage of the improved innov'SAR method is that it only requires protein sequence information combined with experimental characteristic values and not the spatial structure information of proteins. The improved method can rapidly predict the characteristic values of combinatorial mutant libraries based on single point mutations and can be used to screen for beneficial mutants. The protein sequences containing non-standard amino acid compositions remains elusive. Moreover, at present, the improved innov'SAR screening tool is only an in silico scripting program. Therefore, the numerical handling of non-standard amino acids should be further elucidated and a publicly accessible web server should be developed so that the improved innov'SAR method can be applied to the directed evolution of various enzymes, thereby opening up new avenues for protein engineering.
Author Contributions
Conceptualization, G.L. and L.J.; methodology, G.L.; software, L.J.; validation, G.L. and K.W.; formal analysis, G.L. and L.J.; investigation, G.L. and L.J.; resources, L.J.; data curation, G.L.; writing—original draft preparation, G.L.; writing—review and editing, L.J.; visualization, G.L.; supervision, T.S. and J.H.; project administration, J.H.; funding acquisition, J.H. and T.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the National Natural Science Foundation of China (Grant nos. 20904047, 21673207, 21873087), the Natural Science Foundation of Zhejiang Province (Grant nos. LY17A040001) .
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors have no conflict of interest for this research article.
Sample Availability
Not applicable.
References
- Romero, P.A.; Arnold, F.H. Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef]
- Packer, M.S.; Liu, D.R. Methods for the Directed Evolution of Proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef]
- Reetz, M.T. Recent Advances in Directed Evolution of Stereoselective Enzymes. In Directed enzyme evolution: Advances and applications; 2017; pp. 69–99. [CrossRef]
- Reetz, M.T. Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef]
- Cen, Y.; Singh, W.; Arkin, M.; Moody, T.S.; Huang, M.; Zhou, J.; Wu, Q.; Reetz, M.T. Artificial Cysteine-Lipases with High Activity and Altered Catalytic Mechanism Created by Laboratory Evolution. Nat. Commun. 2019, 10, 3198–4208. [Google Scholar] [CrossRef]
- Li, A.; Wang, B.; Ilie, A.; Dubey, K.D.; Bange, G.; Korendovych, I.V.; Shaik, S.; Reetz, M.T. A Redox-Mediated Kemp Eliminase. Nat. Commun. 2017, 8, 14876. [Google Scholar] [CrossRef]
- Schwander, T.; von Borzyskowski, L.S.; Burgener, S.; Cortina, N.S.; Erb, T.J. A Synthetic Pathway for the Fixation of Carbon Dioxide in Vitro. Science 2016, 354, 900–904. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef]
- Mazurenko, S.; Prokop, Z.; Damborsky, J. Machine Learning in Enzyme Engineering. ACS Catalysis 2019, 10, 1210–1223. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-Learning-Guided Directed Evolution for Protein Engineering. Nature methods 2019, 16, 687–694. [Google Scholar] [CrossRef]
- Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Machine Learning Applications in Systems Metabolic Engineering. Current opinion in biotechnology 2020, 64, 1–9. [Google Scholar] [CrossRef]
- Woodley, J.M. Accelerating the Implementation of Biocatalysis in Industry. Applied Microbiology and Biotechnology 2019, 103, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Kan, S.J.; Lewis, R.D.; Wittmann, B.J.; Arnold, F.H. Machine Learning-Assisted Directed Protein Evolution with Combinatorial Libraries. Proceedings of the National Academy of Sciences 2019, 116, 8852–8858. [Google Scholar] [CrossRef] [PubMed]
- Muggleton, S.; King, R.D.; Stenberg, M.J. Protein Secondary Structure Prediction Using Logic-Based Machine Learning. Protein Engineering, Design and Selection 1992, 5, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Folkman, L.; Stantic, B.; Sattar, A.; Zhou, Y. EASE-MM: Sequence-Based Prediction of Mutation-Induced Stability Changes with Feature-Based Multiple Models. J. Mol. Biol. 2016, 428, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Srivastava, A.K.; Wang, L. Sequence Feature-Based Prediction of Protein Stability Changes upon Amino Acid Substitutions. BMC Genomics 2010, 11, S5. [Google Scholar] [CrossRef]
- Huang, L.T.; Gromiha, M.M.; Ho, S.Y. IPTREE-STAB: Interpretable Decision Tree Based Method for Predicting Protein Stability Changes upon Mutations. Bioinformatics 2007, 23, 1292–1293. [Google Scholar] [CrossRef]
- Yang, Y.; Niroula, A.; Shen, B.; Vihinen, M. PON-Sol: Prediction of Effects of Amino Acid Substitutions on Protein Solubility. Bioinformatics 2016, 32, 2032–2034. [Google Scholar] [CrossRef]
- Koskinen, P.; Törönen, P.; Nokso-Koivisto, J.; Holm, L. PANNZER: High-Throughput Functional Annotation of Uncharacterized Proteins in an Error-Prone Environment. Bioinformatics 2015, 31, 1544–1552. [Google Scholar] [CrossRef]
- Cadet, F.; Fontaine, N.; Vetrivel, I.; Ng Fuk Chong, M.; Savriama, O.; Cadet, X.; Charton, P. Application of Fourier Transform and Proteochemometrics Principles to Protein Engineering. BMC Bioinformatics 2018, 19, 382. [Google Scholar] [CrossRef]
- Cadet, F.; Fontaine, N.; Li, G.; Sanchis, J.; Ng Fuk Chong, M.; Pandjaitan, R.; Vetrivel, I.; Offmann, B.; Reetz, M.T. A Machine Learning Approach for Reliable Prediction of Amino Acid Interactions and Its Application in the Directed Evolution of Enantioselective Enzymes. Sci Rep 2018, 8, 16757–16772. [Google Scholar] [CrossRef]
- Ferrandi, E.E.; Monti, D. Amine Transaminases in Chiral Amines Synthesis: Recent Advances and Challenges. World Journal of Microbiology and Biotechnology 2018, 34, 1–10. [Google Scholar] [CrossRef]
- Gao, S.; Su, Y.; Zhao, L.; Li, G.; Zheng, G. Characterization of a (R)-Selective Amine Transaminase from Fusarium Oxysporum. Process Biochemistry 2017, 63, 130–136. [Google Scholar] [CrossRef]
- Kelly, S.A.; Mix, S.; Moody, T.S.; Gilmore, B.F. Transaminases for Industrial Biocatalysis: Novel Enzyme Discovery. Applied Microbiology and Biotechnology 2020, 104, 4781–4794. [Google Scholar] [CrossRef] [PubMed]
- Lyskowski, A.; Gruber, C.; Steinkellner, G.; Schürmann, M.; Schwab, H.; Gruber, K.; Steiner, K. Crystal Structure of an (R)-Selective ω-Transaminase from Aspergillus Terreus. PLoS ONE 2014, 9, e87350. [Google Scholar] [CrossRef]
- Xie, D.F.; Fang, H.; Mei, J.Q.; Gong, J.Y.; Wang, H.P.; Shen, X.Y.; Huang, J.; Mei, L.H. Improving Thermostability of (R)-Selective Amine Transaminase from Aspergillus Terreus through Introduction of Disulfide Bonds. Biotechnol Appl Biochem 2018, 65, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Cecylia Severin, L.; Lyu, C.J.; Zhu, W.L.; Wang, H.P.; Jiang, C.J.; Mei, L.H.; Liu, H.G.; Huang, J. Improving Thermostability of (R)-Selective Amine Transaminase from Aspergillus Terreus by Evolutionary Coupling Saturation Mutagenesis. Biochemical Engineering Journal 2021, 167, 107926. [Google Scholar] [CrossRef]
- Xie, D.F.; Yang, J.X.; Lv, C.J.; Mei, J.Q.; Wang, H.P.; Hu, S.; Zhao, W.R.; Cao, J.R.; Tu, J.L.; Huang, J.; et al. Construction of Stabilized (R)-Selective Amine Transaminase from Aspergillus Terreus by Consensus Mutagenesis. Journal of Biotechnology 2019, 293, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xie, D.F.; Feng, Y. Engineering Thermostable (R)-Selective Amine Transaminase from Aspergillus Terreus through in Silico Design Employing B-Factor and Folding Free Energy Calculations. Biochemical and Biophysical Research Communications 2017, 483, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Sun, T.T.; Wang, Y.; Shen, Y. A Machine Learning Study on the Thermostability Prediction of (R)-Omega-Selective Amine Transaminase from Aspergillus Terreus. Biomed Research International 2021, 2021, 2593748. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino Acid Index Database, Progress Report 2008. Nucleic Acids Res 2008, 36, 202–205. [Google Scholar] [CrossRef]
- Cao, J.R.; Fan, F.F.; Lv, C.J.; Wang, H.P.; Li, Y.; Hu, S.; Zhao, W.R.; Chen, H.B.; Huang, J.; Mei, L.H. Improving the Thermostability and Activity of Transaminase From Aspergillus Terreus by Charge-Charge Interaction. Frontiers in Chemistry 2021, 9. [Google Scholar] [CrossRef]
- Veljković, V.; Cosić, I.; Dimitrijević, B.; Lalović, D. Is It Possible to Analyze DNA and Protein Sequences by the Methods of Digital Signal Processing? IEEE Trans Biomed Eng 1985, 32, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.C. Digital Signal Processing Methods for Biosequence Comparison. Nucleic Acids Research 1990, 18, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, Small Molecules and a New Graphical Interface. Bioinformatics 2019, 35, 4168–4169. [Google Scholar] [CrossRef] [PubMed]
- Buss, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches? Comp. Struct. Biotechnol. J.. 2018, 16, 25–33. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View-Molecular Graphics for All Devices-from Smartphones to Workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Fontaine, N.; Cadet, X.; Vetrivel, I. Novel Descriptors and Digital Signal Processing- Based Method for Protein Sequence Activity Relationship Study. IJMS 2019, 20, 5640. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagram of the innov'SAR method for extended sequences. (a) One index was used to generate the corresponding elementary sequence in different index encodings. (b) The elementary sequences were connected after FFT to obtain the extended sequence. (c) The extended sequences and half-life were included in the training dataset to construct regression models. The performance of the model was evaluated by comparing the measured half-life with the predicted half-life.
Figure 1.
Schematic diagram of the innov'SAR method for extended sequences. (a) One index was used to generate the corresponding elementary sequence in different index encodings. (b) The elementary sequences were connected after FFT to obtain the extended sequence. (c) The extended sequences and half-life were included in the training dataset to construct regression models. The performance of the model was evaluated by comparing the measured half-life with the predicted half-life.
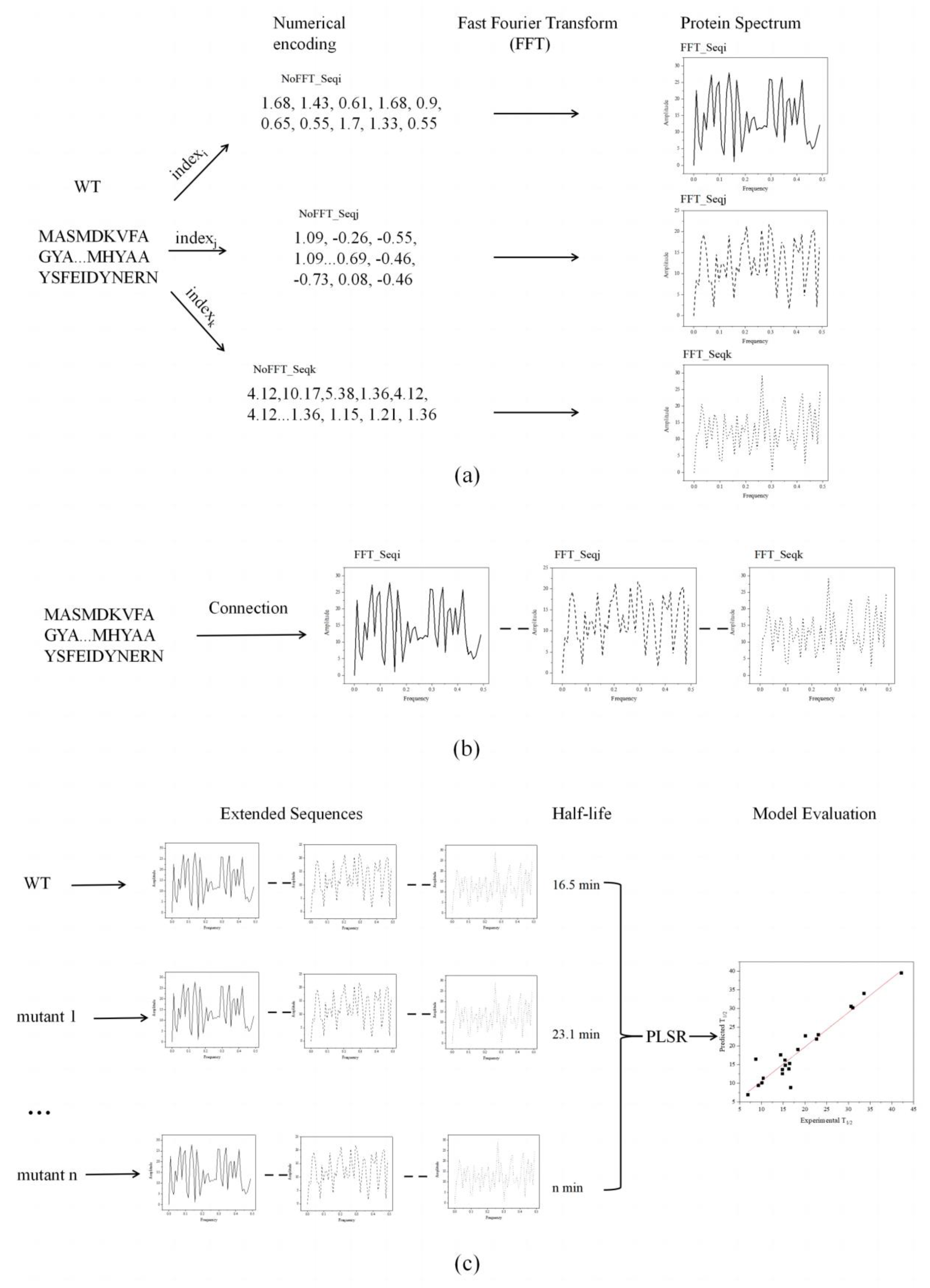
Figure 2.
Thermal stability plots of measured and predicted half-lives of AT-ATA variants. (a) Use of a single index: R2 = 0.81. (b) The optimal combination of indices: R2 = 0.86. (c) Connection of the top five single-index combinations in series: R2 = 0.80.
Figure 2.
Thermal stability plots of measured and predicted half-lives of AT-ATA variants. (a) Use of a single index: R2 = 0.81. (b) The optimal combination of indices: R2 = 0.86. (c) Connection of the top five single-index combinations in series: R2 = 0.80.

Figure 3.
Flow chart of the iterative process of successive concatenation. Each round uses the indices of the previous iteration as the basis for the extended sequence and determines the best index to retain for the current round by evaluating the performance of the model.
Figure 3.
Flow chart of the iterative process of successive concatenation. Each round uses the indices of the previous iteration as the basis for the extended sequence and determines the best index to retain for the current round by evaluating the performance of the model.
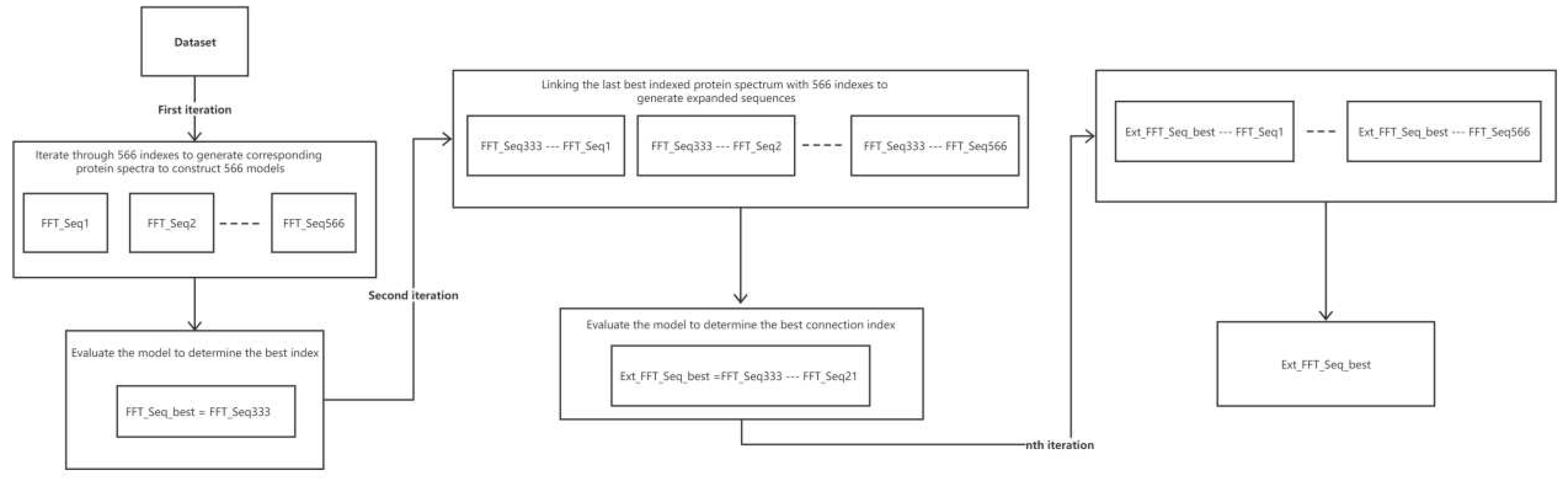
Figure 4.
Plot for experimental versus predicted thermal stability of AT-ATA variants. The graph was plotted using iterative connect indices (AURR980108-MEIH800103-CORJ870104): R2 = 0.96.
Figure 4.
Plot for experimental versus predicted thermal stability of AT-ATA variants. The graph was plotted using iterative connect indices (AURR980108-MEIH800103-CORJ870104): R2 = 0.96.
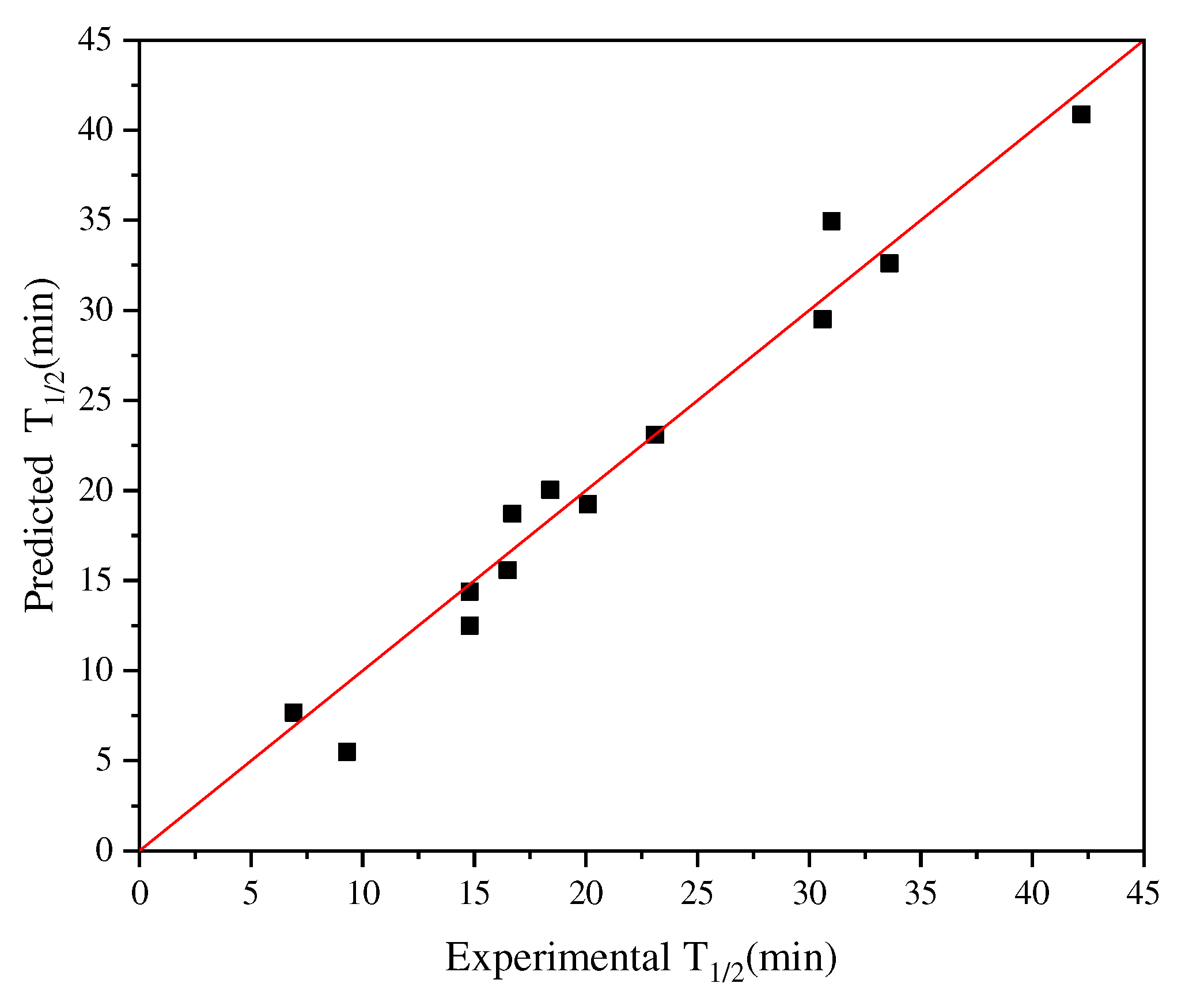
Figure 5.
Half-life of the 1024 possible variants of AT-ATA. (■): half-life measured for WT, (◆): half-life measured for the best experimental mutant F115L_L118T, (▲): half-life measured for the remaining single and multi-site mutants, (●): predicting half-life of all 1024 possible variants.
Figure 5.
Half-life of the 1024 possible variants of AT-ATA. (■): half-life measured for WT, (◆): half-life measured for the best experimental mutant F115L_L118T, (▲): half-life measured for the remaining single and multi-site mutants, (●): predicting half-life of all 1024 possible variants.
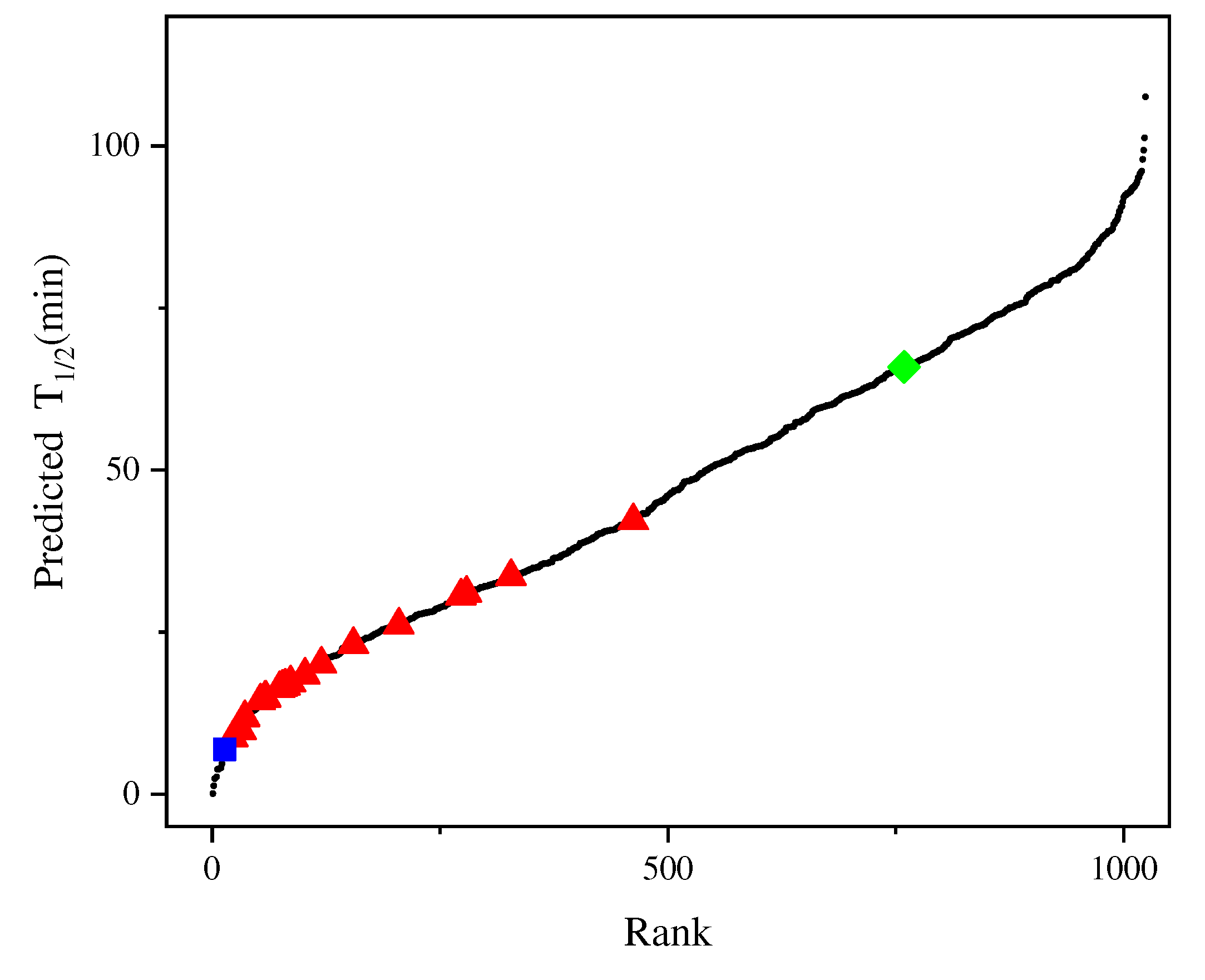
Figure 6.
RMSD values of P1, P2, and F115L_L118T in 100-ns simulations.
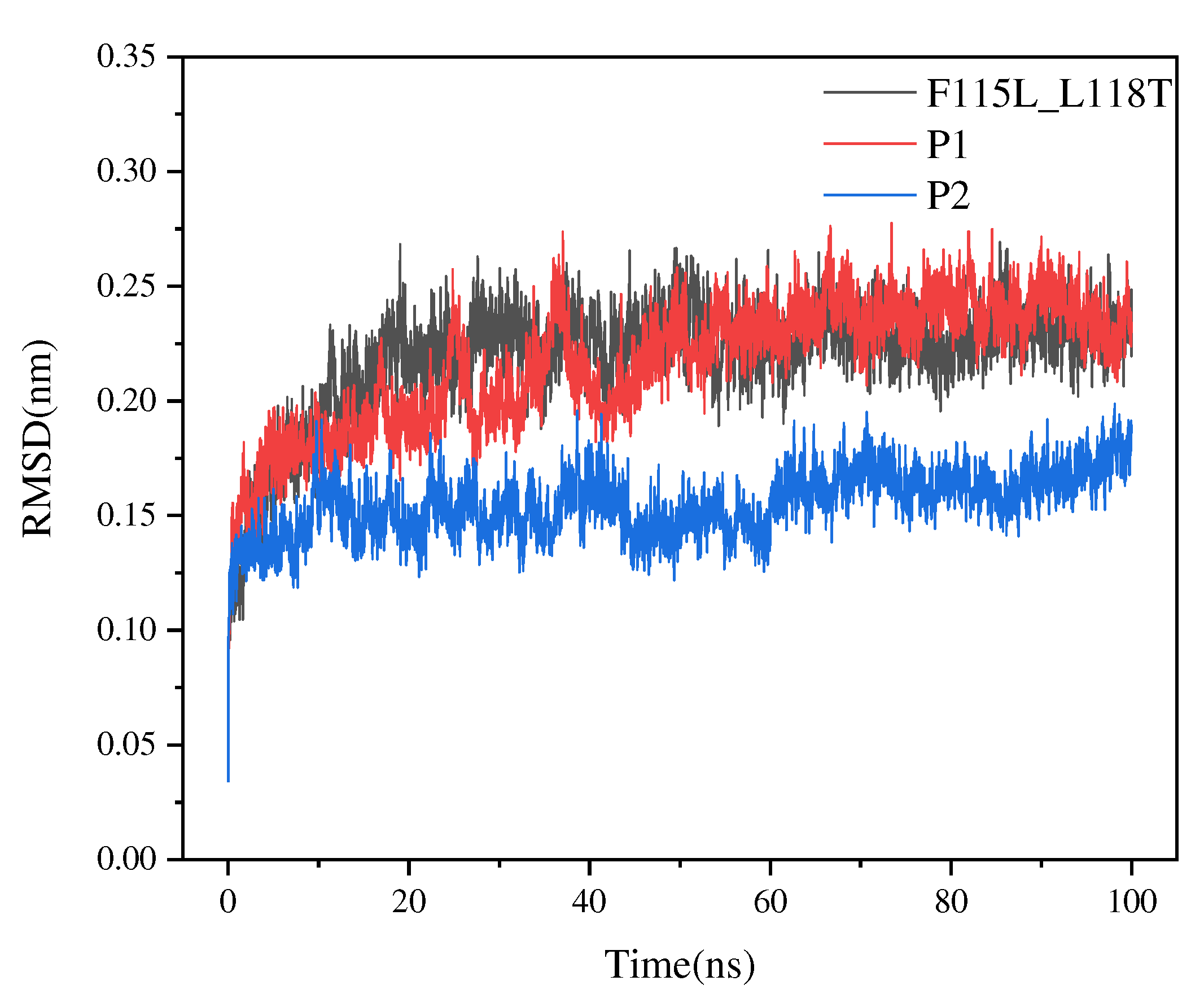
Figure 7.
MD analysis of P1, P2, and F115L_L118T using YASARA at 313 K in the last 20 ns. (a) RMSF of P1, P2, and F115L_L118T. (b, c) 3D structural views of the two protein regions. Hydrogen bonds are indicated by red dashed lines.
Figure 7.
MD analysis of P1, P2, and F115L_L118T using YASARA at 313 K in the last 20 ns. (a) RMSF of P1, P2, and F115L_L118T. (b, c) 3D structural views of the two protein regions. Hydrogen bonds are indicated by red dashed lines.
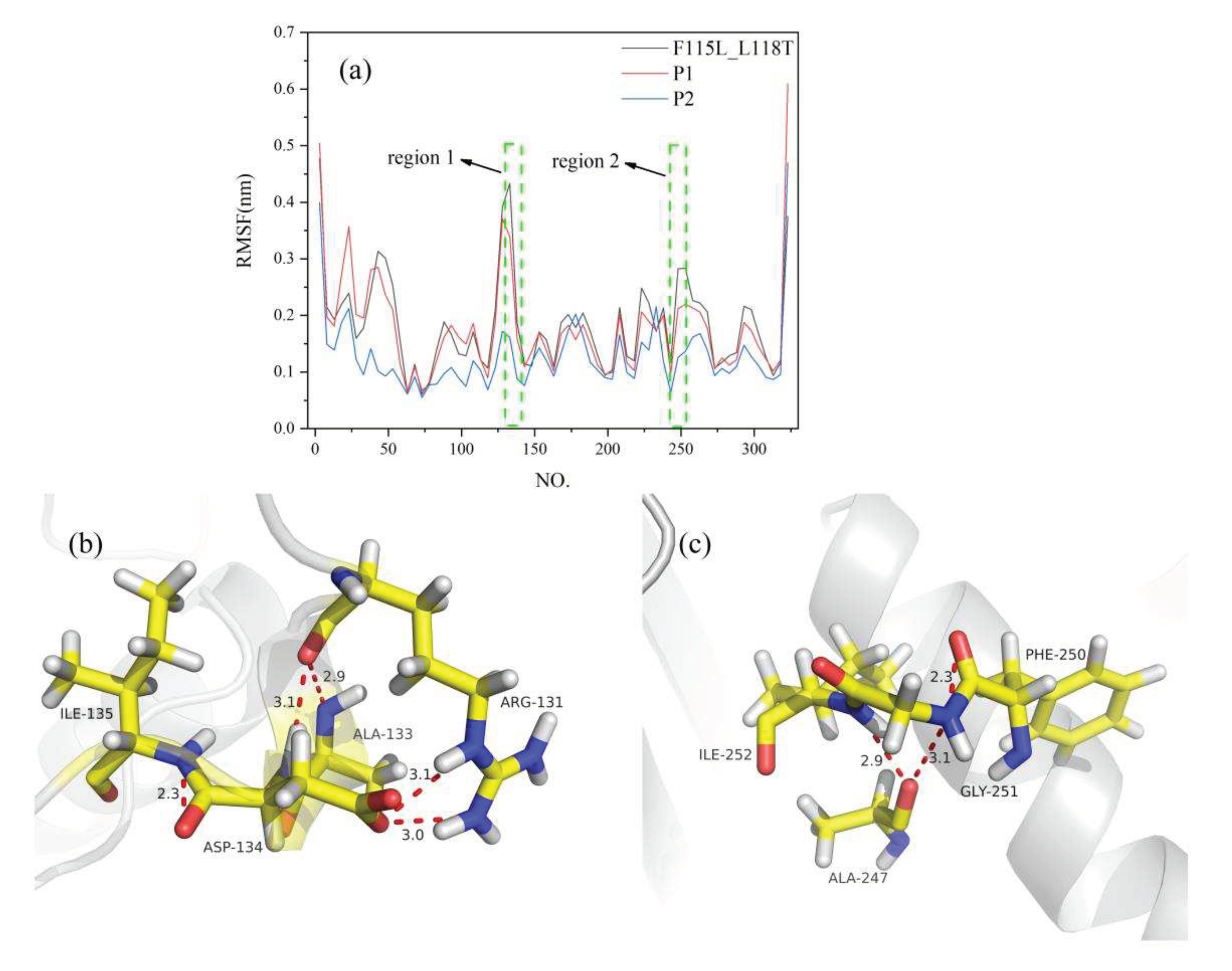

Table 1.
The top five indices encoded by a single index.
| Index | Index Number | cvRMSE | R2 |
|---|---|---|---|
| AURR980108 | 396 | 4.56 | 0.81 |
| OOBM770102 | 201 | 5.33 | 0.76 |
| MUNV940102 | 416 | 6.01 | 0.67 |
| CORJ870102 | 507 | 6.10 | 0.67 |
| GEOR030108 | 484 | 6.58 | 0.59 |
Table 2.
AT-ATA experimental datasetThe top 10 extended sequences selected based on the combination of the top 5 indices.
Table 2.
AT-ATA experimental datasetThe top 10 extended sequences selected based on the combination of the top 5 indices.
| Index number | cvRMSE | R2 |
|---|---|---|
| 396 201 507 484 | 3.64 | 0.86 |
| 396 507 484 | 3.71 | 0.86 |
| 396 201 507 | 3.91 | 0.85 |
| 201 507 | 4.36 | 0.84 |
| 396 416 507 | 4.22 | 0.84 |
| 396 201 416 507 | 4.17 | 0.83 |
| 201 507 484 | 4.20 | 0.83 |
| 396 201 | 4.25 | 0.82 |
| 396 507 | 4.33 | 0.82 |
| 396 201 416 | 4.37 | 0.81 |
Table 3.
The mutation site and predicted half-life of the optimal screened mutants.
| Variant | Mutations | Predicted T1/2 |
|---|---|---|
| P1 | Q97E_F115L_L118T_E133A_H210N_N245D_E253A_G292D | 107.59 |
| P2 | I77L_F115L_L118T_E133A_H210N_N245D_E253A | 101.25 |
Table 4.
AT-ATA experimental dataset.
| Mutations | T1/2 | Note |
|---|---|---|
| WT | 6.9 | Dataset 1/Dataset 2 |
| I77L | 20.1 | Dataset 1/Dataset 2 |
| Q97E | 16.5 | Dataset 1/Dataset 2 |
| F115L | 17.2 | Dataset 2 |
| L118T | 26.1 | Dataset 2 |
| E133A | 9.8 | Dataset 2 |
| H210N | 23.1 | Dataset 1/Dataset 2 |
| N245D | 14.8 | Dataset 1/Dataset 2 |
| E253A | 11.8 | Dataset 2 |
| G292D | 14.8 | Dataset 1/Dataset 2 |
| I295V | 9.3 | Dataset 1/Dataset 2 |
| F115L_L118T | 65.9 | Dataset 2 |
| I77L_H210N | 42.2 | Dataset 1/Dataset 2 |
| Q97E_H210N | 30.6 | Dataset 1/Dataset 2 |
| H210N_N245D | 18.4 | Dataset 1/Dataset 2 |
| H210N_G292D | 33.6 | Dataset 1/Dataset 2 |
| I77L_Q97E_H210N | 31 | Dataset 1/Dataset 2 |
| I77L_H210N_G292D | 16.7 | Dataset 1/Dataset 2 |
| I77L_Q97E_H210N_N245D | 14.4 | Dataset 2 |
| I77L_H210N_N245D_G292D | 16.3 | Dataset 2 |
| I77L_Q97E_H210N_N245D_G292D | 8.7 | Dataset 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



