
Submitted:
11 September 2023
Posted:
13 September 2023
You are already at the latest version
Abstract
Breast Cancer metastasis remains a formidable challenge in cancer research, contributing significantly to patient mortality despite advances in medical research. Lung metastasis, associated with breast cancer, poses an ongoing clinical dilemma with limited curative treatment options. This study delves into the intricate mechanisms underlying Breast Cancer-Lung Metastasis (BC-LM) primarily focusing on the function of SH3PXD2B in maturation of invadopodia, inducing epithelial-to-mesenchymal transition, disruption of proteostasis network, and ultimately leading to metastasis of Breast Cancer (BC) cells. With an extensive analysis of differential gene expression using RNASeq data, comparing normal breast cancer cells to metastatic sub-populations in lung. Employing the New Tuxedo pipeline our investigation notably observes SH3PXD2B as a key regulator in lung metastasis samples. This trend is further substantiated by data from the Cancer Cell Line Encyclopedia (CCLE), and Human Protein Atlas (HPA) which highlights elevated SH3PXD2B expression in MDA-MB-468 cells, underscoring its significance in metastatic adenocarcinoma. Additionally, we checked the overall survival (OS) of metastatic breast cancer (MBC) patients pinpointing SH3PXD2B and its associated partners like SH3PXD2A, MMPs, CTTN, ADAMs, and EMT markers with substantial expression in both BC and lung cancer, prognosticating poorer patient survival. Further, our transcription factor – target gene (TFTG) network essentially elucidated the role of SH3PXD2B as a key node in the network unveiling its roles in regulating cell migration, communication, and developmental processes. Proteomics and Western blotting assays consistently confirm heightened SH3PXD2B expression in BC cell lines, reaffirming our findings. By employing computational structure biology along with cancer systems biology approach, we generated a high-confidence structural model of SH3PXD2B, indicating its SH3_2 and SH3_3 domains crucial for interactions with the drug molecules. Molecular docking simulations identify Eribulin as a promising therapeutic agent capable of targeting these domains. Thus, our multidisciplinary approach seamlessly amalgamates systems medicine principles, aiming to repurpose existing drugs that target SH3PXD2B based on molecular signatures. Targeted therapies have emerged as a promising avenue for addressing MBC and this mechanistic model introduces novel therapeutic avenues for the treatment of BC-LM patients.
Keywords:
Metastatic Breast Cancer
; Breast Cancer Lung Metastasis
; SH3PXD2B
; Eribulin
1. Introduction
Breast cancer (BC) is a pervasive and potentially life-threatening disease among women worldwide characterized by the uncontrolled growth of malignant cells in the breast tissue. It is currently the most widely diagnosed cancer type with an estimation of 2.3 million new cases reported in 2020 and ranking 5th in leading cause of cancer related deaths [1]. Breast cancer is not a homogeneous disease, it is highly diverse and categorized into distinct subtypes, each with discrete characteristics, prognostic factors, and treatment approaches. The primary subtypes of breast cancer are classified based on the presence or absence of specific molecular markers, such as estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). The four classes of breast cancer include – Luminal A tumors (ER/PR+, HER2-), Luminal B tumors (ER+, PR-), HER2-positive tumors (HER2+, ER/PR-) and Triple-negative breast cancer (TNBC) with all ER/PR/HER2-negative. The TNBC subtype is further divided into several groups including basal-like (BL1 and BL2), claudin-low, mesenchymal (MES), luminal androgen receptor (LAR), and immunomodulatory (IM) all of which account to 50-70% of total advanced stage breast cancer cases [2]. The prevalence of breast cancer varies geographically, with higher incidence rates occurring in transitioning countries, such as Melanesia, Western Africa, Polynesia and Caribbean as compared to the transitioned ones – Australia, West Europe, North America and North Europe [3]. Additionally, certain studies suggest the onset of BC in Asian women is earlier than it does in Western women. It affects Asian women at younger age with peak ages of 40-50 years as compared to Western women with peak ages of 60-70 years [4,5]. Research suggests that factors like menstrual cycle and birth cohort may influence these age differences [6,7]. Recent statistic for age-period-cohort indicates that some Asian countries now have later onset ages than the United States, likely due to changing risk factors, screening of BC in women over 50 years and a longer lifespan. Thus, the threat of developing breast cancer rises with age [6]. The risk factors for breast cancer are diverse and can be categorized into non-modifiable and modifiable factors. Non-modifiable factors include race/ethnicity, older age, hormonal imbalance, reproductive history, breast tissue density, genetic mutations, family history, and history of breast diseases. On the other hand, modifiable factors include drug intake, physical activity, body mass index (BMI), alcohol consumption, smoking, insufficient vitamin supplementation, exposure to chemical, etc. [3]. The age-standardized incidence rate (ASIR) was found to be lowest among Asian women (36.8%) as compared to Africa (40.7%), Caribbean (51.9%), Europe (74.3%), Oceania (87.8%), and Northern America (89.4%) in 2020 calculated for per 100,000 females. Similarly, the age-standardized mortality rate (ASMR) was found to be the lowest in Asia (11.6%) as compared to Africa (19.4), Caribbean (13.5), Europe (14.8), Oceania (13.2), and Northern America (16.9) in 2020 calculated for per 100,000 females [8]. Breast cancer mortality rates are higher in low- and middle-income countries as compared to high-income countries. The stage at which breast cancer is detected varies widely across Asian countries, with a higher proportion of localized (Stage I and II) cases in higher-income countries. Low-income countries often have a higher proportion of late-stage diagnoses, leading to increased treatment costs and a greater healthcare burden. Southeast Asia faces over 75% mortalities due to breast cancer within a year owing to financial ruin [8]. Projections for breast cancer incidence and mortality rates for 10years from now in 2035 suggest that there will be approximately 2.97 million new cases reported annually and estimated 0.95 million deaths worldwide [9]. Timely and accurate diagnosis, along with the quality of treatment and care, significantly affect breast cancer survival. According to Cancer Research UK, the five-year survival rates for breast cancer varies by stage: nearly all Stage I patients (about 99.99%) and about 90 in 100 Stage II patients survive five years or more; whereas, this rate drops to 70% for Stage III patients. Moreover, Stage IV patients characterized by the spread of cancer to distant body part indicate poor prognosis with a survival rate near to 25% [10]. Metastasis is a crucial hallmark of advanced stage breast cancer. Targeted therapies aim to inhibit cancer growth by interfering with specific molecules involved in tumor cell proliferation and survival. The choice of targeted treatment for metastatic breast cancer depends on factors like hormone receptor status, Her2 expression, recurrence, metastasis rate, and site of metastasis [11]. Nevertheless, the risk of metastasis persists across all breast cancer types, with no established therapeutic strategy for the most aggressive, invasive and highly metastatic TNBC subtype.
1.1. Treatment approaches, advances and challenges:
Patients diagnosed with early-stage BC undergo primary tumor removal and subsequent therapies like hormone, chemotherapy, or radiation treatments aimed at eradicating residual disease. For patients at risk of metastasis, the dissemination of cancer cells from the primary tumor likely occurred before diagnosis and is halted by radical surgery. Therefore, targeting the metastasis process that has already initiated may not be effective. Since, preventing metastatic lesions from cross-seeding, self-seeding, or re-seeding to other sites may be challenging and poise difficulties in predicting the underlying transient processes. Rather it might be a more meaningful target when dealing with established metastases, where therapies could confine lesions locally and allow for more effective application of cytotoxic treatments [12,13,14]. Cancer treatment generally aims to target a sensitivity window where cancer cells are selectively killed while sparing normal tissues. Metastases are often treated based on the characteristics of their primary tumors, assuming that metastases behave similarly to their tissue of origin. Many primary tumor traits, such as growth dependency and drug sensitivity, are thought to be retained in metastases [15]. The success of hormonal therapy and anti-HER2 therapy for breast cancer subtypes supports this approach [16]. Recently, with FDA approval drugs targeting MBC have been developed and are in use. These include Doxorubicin, Cyclophosphamide, Vorinostat, Zemetostat, Atezolizumab + Paclitaxel, Methotrexate, Tivozanib, Trastuzumab, Lapatinib, Palbociclib, Pertuzumab, and Epirubicin [17,18,19,20,21,22,23,24,25,26,27,28,29]. However, they have many limitations. Emerging targeted therapies against MBC are directed towards oncogenic signaling pathways such as PI3K-Akt-mTOR, MAPK, JAK-STAT, integrin signaling, receptor tyrosine kinases (IGF1R, FGFR, EGFR, ERBB3, MET), cell cycle regulators (CDKs), and epigenetic modifiers (HDAC, DNMT, BET domain chromatin modulators) and are being tested in clinical trials [30]. However, it is important to consider the specific microenvironments of distant organs for development of effective therapies, as relapse and resistance can occur at any point of time. As metastatic tumors may adapt to different microenvironments their growth dependence may considerably shift away from the oncogenic drivers of primary tumors thus, facilitating drug resistance. For example, during breast cancer-bone metastasis various growth factors released from the bone matrix may compensate for the growth stimulus of MBC cells, reducing their reliance on estrogen. This can lead to decreased effectiveness of ER antagonists against ER+ lesions as bone metastasis progresses. Additionally, in some cases the resistance to available therapies may occur due to a phenotypic switch between the subtypes of breast cancer, such as HER2+ to TNBC [12,31,32,33,34]. Hence, these findings suggest that metastatic cancer cells within diverse microenvironments can shift their signaling pathways to ensure survival and growth. The intricate interactions between the cancer cells and various cell types in their surroundings enable them to adjust and switch to alternative growth pathways when one pathway is blocked. Therefore, when developing new therapeutic approaches for metastatic breast cancer, it is crucial to prioritize an understanding of the intricate cellular mechanisms involved, such as alternative activation, feedback mechanisms, and the influence of the microenvironment.
1.2. Breast Cancer Metastasis: Overall survival and Current Findings
Breast Cancer is a disease with high levels of complexities and heterogeneity. One of the most critical factors influencing patient’s prognosis and overall survival with breast cancer is occurrence of metastasis. Metastasis is critical for cancer progression, where an initial tumor gives rise to secondary tumors in distant locations. It is a crucial process for cancer cell survival and progression under stressful conditions. As one of the prominent hallmarks of breast cancer it often leads to treatment failure and patient mortality [35]. According to the survival statistics of metastatic breast cancer patients in Sweden indicates that approximately one-third of early stage (I-III) patients will develop metastatic tumors at distant organs; and the duration of metastasis-free intervals may vary from months to several years. Once distant recurrence occurs, metastatic breast cancer (MBC) is considered incurable, with a median overall survival of 2 to 3 years for metastatic patients. In addition, 3-5% patients diagnosed with advanced stage (IV) breast cancer are likely metastatic. The clinical course of MBC patients varies widely, with some succumbing to the disease shortly after diagnosis, while others survive for a decade or more [36]. TNBC, identified as the most aggressive type of breast cancer is often associated with advanced stages, contributing to low survival rate and high recurrence rate [37,38]. It most commonly metastasizes to bones, brain, lungs and liver with effective targeted therapies [39]. ER+ tumors generally have lower incidence rate within the first five years as compared to TNBC tumors. The different nature of breast cancer subtypes may shed light on their preferred organs. For example, ER+ tumors preferably target bone for metastasizing, as bone is rich in estrogen for maintaining homeostasis and remodeling, it makes easier for ER+ tumor cells to lodge in the bone marrow and hijack the available estrogen. On the other hand, HER2+ tumors driven by ERBB2/ERBB3, may excel at brain metastasis due to their self-sufficient oncogenic signaling and the presence of fewer immune cells in the brain. TNBC tumors more frequently affect visceral organs like the brain and lung [35]. Furthermore, a study conducted by Xiao et. al; showed the distribution and impact of BC subtypes on distant metastatic sites primarily bone, lung, liver and brain. The HR+/HER2− subtype was most common in the entire cohort but gradually decreased in patients with bone (59.9%), lung (47.8%), liver (40.9%), and brain (38.8%) metastases. Whereas, the HER2+ subtypes showed an increased percentage in patients with metastases, particularly in those with liver metastases. In contrast, the triple-negative subtype decreased in patients with bone metastases but increased in those with visceral (lung, liver, and brain) metastases, with the highest increase seen in brain metastases. They further demonstrated the overall survival (OS) based on metastatic sites and breast cancer subtypes. HR+/HER2+ subtype was significantly associated with increased OS in all four types of distant metastases when compared to HR+/HER2− subtype. In the case of HR−/HER2+ subtype, it was significantly associated with worse prognosis in patients with bone and lung metastases but not in patients with liver and brain metastases. The median survival of patients with brain metastasis had worst outcomes (11 months), while bone metastasis had the best outcome (30 months). Conversely, the triple-negative subtype was significantly associated with decreased OS across all metastatic sites. It predicted the worst prognosis with survival ranging from 6 months to 11 months [40].
As discussed earlier, the complexities in therapeutic strategies of metastatic tumors identification of dominant gene patterns in primary tumors and their matched metastases could contribute to novel therapeutic interventions. With powerful high-throughput technologies and computational pipelines research in the past decade attributed to characterizing molecular profiles of breast cancer. Molecular characterization of MBC might help in unraveling the genetic makeup of tumor that influences its response to stress, inducing metastasis. These findings could thus, aid in drug discovery with primary focus on patients with specific genetic abnormalities owing to metastatic setting. The AURORA US metastatic project is aimed at studying the molecular landscape of metastatic breast cancer to enhance our understanding of its molecular evolution and intra-tumor heterogeneity. Additionally, it sought to identify predictive biomarkers for anti-cancer treatment response and generate novel therapeutic hypotheses from genetic findings. The molecular profiles of patients participating in this project were studied based on their biological samples including tissue and metastatic lesion in comparison to primary tumor tissue, additionally, whole blood, plasma and serum samples were also profiled. The profiling of these samples was done using whole genome, whole exome, RNA sequencing and DNA methylation arrays [41]. The most prevalent metastatic sites were the liver, lungs, lymph nodes, and brain. The median age at the time of initial diagnosis was 49 years. The samples had subtype heterogeneity including 34% of the primary lesions with TNBC subtype, 30% with ER+HER2-, 11% with ER+HER2+, and 7% with ER-HER2+. Patients with metastasis received three treatment lines. To conduct a site-specific analysis of metastasis, focusing on the lung, liver, and brain as the most common metastatic sites, the AURORA study integrated datasets from both the RAP101 and GEICAM projects. The multi-omics study by AURORA revealed DNA methylation processes affected the HLA-A and small focal deletions in TNBC/basal-like subtype corresponding to reduced HLA-A expression and decrease in immune cell feature. This finding indicated that immune checkpoint inhibitors (ICIs) may have limited effectiveness against these HLA-A-low tumors, as they cannot be recognized by CD8+ T cells (despite having high neoantigen burdens). Additionally, they suggested a biomarker-driven therapeutic strategy where HLA-A DNA-methylated tumors (acting as the biomarker) could be targeted using DNA demethylating drugs in combination with ICIs. Previously described gene expression subtype discrepancies between primary and metastatic tumors have also been confirmed in the AURORA study. Specifically, gene expression subtype switches in one out of three metastatic breast cancer patients, particularly prevalent in those with luminal/ER+ breast cancer were observed. The results also corroborated with the presence of immune-privileged sites in the brain region making it prone to metastasis. Alongside, it provided insights into the shared immune-privileged characteristics between brain and liver confirming low immune cell features in liver metastasis. The prevalence of BCLM patients within the study is notably substantial; yet, there remains a dearth of knowledge about the comprehensive investigation of lung metastasis occurring in with breast cancer, particularly in relation to elucidating the underlying molecular mechanisms and gene mediators [42].
Despite, lung cancer being one of the deadliest malignancies worldwide, relatively limited researches has been conducted on its clinical relevance in the context of breast cancer lung metastasis (BC-LM). Through this study we aim at unraveling the intricacies of BC-LM, answering vital questions and proposing novel target for therapeutic intervention. In the recent years, mechanisms underlying metastatic tropism have been widely studied, suggesting certain tumors exhibit organ-specific patterns based on the blood flow [39]. However, this explanation does not pertain to breast cancer. According to the seed-soil theory proposed by Paget in 19th century indicated that disseminated cancer cells referred to as “seeds” form a colony when they encounter a microenvironment referred to as “soil” that is conducive for their survival and proliferation [43]. This seed-soil hypothesis thus, raises a series of critical questions: What drives the organ-specific tropism of breast cancer cells to the lungs and why are lungs most prominent organ for MBC? Which crucial gene mediators help TNBC cells in colonizing in lungs? What is the role of the lung microenvironment in supporting or inhibiting the growth of breast cancer metastases, and how can we manipulate these interactions to benefit patient outcomes?
1.3. Breast Cancer – Lung Metastasis: EMT through Invadopodia formation
Approximately 60% of metastatic breast cancer patients experience lung or bone metastasis, with a specific predisposition for lung metastasis in basal-like breast cancer (BLBC) or triple-negative breast cancer [44]. Survival statistics suggest that patients with lung metastasis have a low life expectancy, with a median survival of only 22 months post-treatment [45]. A significant portion, around 60-70%, of metastatic breast cancer patients who ultimately succumb to the disease are initially diagnosed with lung metastasis [46]. Despite various available treatment approaches the patient outcomes for BC-LM remains dismally low. The incidence of lung metastasis is notably higher in triple-negative breast cancer (TNBC), reaching up to 40%, compared to only 20% in non-TNBC cases [47,48,49]. Reports suggest that visceral metastasis, specifically pulmonary metastasis, is more common in TNBC patients, while non-TNBC patients tend to develop bone metastasis [50,51,52,53]. Additionally, luminal A subtype patients tend to avoid lung relapse, while brain metastasis is predominantly observed in those with BLBC and HER2+ breast cancer [45]. Recent SEER database analysis also confirmed that TNBC, particularly BLBC, is primarily linked to lung metastasis [54]. BC-LM carries serious clinical implications and consequences, often resulting in a poor prognosis despite treatment strategies such as chemotherapy, targeted therapy, and endocrine therapy based on molecular receptor profiles [48]. Currently, early diagnosis remains the most effective approach to prevent breast cancer lung metastasis. However, for better patient outcomes with effective therapeutic intervention, when a solitary lung nodule is detected in patients previously treated for breast cancer, it is essential to histologically confirm the diagnosis to differentiate it as recurrent malignancy from primary lung cancer, or benign lung tumors [49,55]. Hence, in order to develop more effective diagnostic and therapeutic approaches for BC-LM patients, it is imperative to gain a comprehensive understanding of the underlying mechanisms. In-depth studies of MBC cells and their interactions with the surrounding microenvironment can offer valuable insights into the factors contributing to the existing challenges. Metastasis is an intricate and multi-step process involving various cellular mechanisms such as detachment from the primary tumor, invasion into surrounding tissues, evading the immune system, and altering the local tissue environment [35,36]. Large amount of evidence suggests that breast cancer stem cells (BCSCs) are recognized as drivers of metastatic growth [56,57]. BCSCs show subtype-specific associations, with studies indicating their enrichment in basal-like breast cancer (BLBC) [58]. CD44v+ BCSCs in primary tumors are linked to distant metastasis promotion, with their expression enhancing lung metastasis by interacting with lung microenvironment factors [59]. However, CD44 alone doesn't identify all BCSCs. Enrichment of BCSCs in BC-LM should be further looked into through intricate signaling network regulating their vital properties facilitating invasiveness and poor prognosis [48]. BCSCs relocating from primary sites to distant microenvironments establish lung niches associated with Notch signaling [60]. Dysregulated activation of the Notch signaling pathway with Notch1 expression can contribute in unchecked proliferation of BCSCs and affect various aspects of BCSC behavior, including self-renewal, proliferation, apoptosis, and epithelial-mesenchymal transition (EMT) [61,62,63,64]. Although the precise role of the Notch pathway in primary tumor cell dissemination to the lung is yet unclear, it likely plays a crucial part in adaptation of MBCs to metastatic niches, possibly interacting with other signaling pathways [48]. The Wnt/β-catenin signaling pathway has a significant role in mammary gland development and breast cancer tumorigenesis; aberrant activation of this pathway through over-expression of β-catenin is associated with worst prognosis in breast cancer, particularly in the triple-negative subtype [65,66]. High Wnt/β-catenin signaling is exhibited by BCSCs and is particularly associated with TNBCs advancing EMT and metastasis in breast cancer patients. However, the action of Wnt family members is multifaceted, governed by canonical and non-canonical activation, and can either hinder or drive breast cancer progression and metastasis depending on the specific signaling context [48]. Additionally, recent research underscores the role of paracrine Hedgehog signaling (Hh) signaling associated with breast cancer growth impacting migration, particularly associated with the poor prognosis of the basal-like phenotype. Studies demonstrate that transcription factors like GLI1 and FOXC1 influence BC-LM through interactions with the CXCL12-CXCR4 axis, controlling angiogenesis, and BCSC properties which are mainly enriched in BLBC [67,68,69]. Dysregulated Hh signaling functions independently as well as through interaction with other signaling pathways in breast cancer. As per a study conducted in hepatocellular carcinoma, the activation of Hh signaling together with TGF- β promoted liver cancer lung metastasis in mouse models, suggesting the same mechanism might be involved in BC-LM which needs to be unveiled [48,70]. Thus, co-activation of Hh, Notch, and Wnt pathways in TNBC samples is linked to shorter survival; however, the precise mechanisms of their coordination in breast cancer metastasis are not yet fully understood and investigating their interaction hold promising therapeutic strategies for BC-LM [71,72,73]. Further, the target tissues for disseminated cancer cells (DCCs) engage in diverse interactions with resident stromal cells, immune cells, tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), extra-cellular matrix (ECM), cytokines, chemokines, and other possible factors that are yet-to-be determined [74,75]. Studies have indicated that even subtle changes in the composition of leukocytes at distant sites from the primary tumor can influence the development of metastases. CXCR2+ neutrophil subtype has been associated with pro-metastatic effects of mesenchymal-stromal cells [76]. Recent findings pertaining to breast cancer mouse models illuminate the role of neutrophils in mediating metastatic initiation by modifying the lung microenvironment before metastases. This suggests that immune cells can influence the creation of metastatic niches in BC-LM [77,78]. TAMs have also been found to be crucial in BC-LM as the induce EMT through the secretion of CCL18 [79]. Additionally, clinical studies have linked pulmonary macrophages to promote the survival of breast cancer cells in lung microenvironment by allowing TAMs to interact with VCAM1 receptor which provide survival benefit for MBCs [79,80,81,82,83]. Next, the CAFs too, play a significant role in BC-LM as they express Tiam1 and osteopontin that regulate the metastatic process [84]. Expression of platelet-derived growth factor receptor β (PDGFRβ), a protein associated with CAFs, has been linked to lung metastasis in breast cancer. Key factors influencing the spread of breast cancer to lungs are associated with epithelial to mesenchymal transition, making it a vital and centric process in BC-LM.
1.3.1. Epithelial to Mesenchymal Transition through Invadopodia Formation
EMT is a biological process by which epithelial cells, which are typically immobile and organized in tightly packed structures, acquire characteristics of mesenchymal cells, which are more motile and invasive having the ability to degrade the ECM. Cells in EMT process thus, demonstrate the gain and loss of certain characteristics while switching between two states. It involves complex molecular changes including the down-regulation of epithelial markers (e.g., E-cadherin) and up-regulation of mesenchymal markers (e.g., N-cadherin, vimentin). Additionally, it describes transcription factors like Snail, Slug, and Twist which play a pivotal role in regulating the process [86]. The regulation of epithelial-mesenchymal transition by TGFβ has been extensively studied and can occur through both SMAD and non-SMAD signaling pathways. Various growth factors, such as EGF, FGF, HGF, and VEGF, stimulate receptor tyrosine kinases (RTKs), initiating multiple signaling cascades that result in the up-regulation of transcription factors associated with EMT. Activation of RTKs or integrins can also activate AKT, leading to increased expression of Snail by inhibiting GSK-3 beta. Furthermore, inflammatory cytokines released by cancer cells itself and also by surrounding immune cells can promote EMT through the induction of Snail and STAT3. Within the tumor microenvironment, the accumulation of HIF-1α drives hypoxic conditions that lead to the expression of TWIST, ultimately inducing EMT [87]. Besides, molecular drivers, development of invadopodia plays a very crucial role in epithelia-to-mesenchymal transition. Invadopodia are defined as actin based subcellular structures that are described as specialized machinery for extracellular matrix degradation [88,89]. Formation of invadopodia is characterized by the development of large protrusions along with branched actin filaments, vesicles and various cytoskeletal components that occupy these protrusions [90]. Invadopodia influence cell motility through various mechanisms, such as coordinating focal adhesion dynamics, lamellipodia formation, and ECM track generation. They can also act as mechanosensors and exert traction forces on the ECM [91]. Many studies have illustrated the structural components of invadopodia. However, currently efforts are being made to understand the governing mechanisms of invadopodia formation.
The maturation formation and maturation process of invadopodia involves the recruitment and simultaneous activation of multiple proteases near the cell's periphery, facilitating ECM degradation and release of cytokine. These proteases include zinc-regulated metalloproteases (e.g., MMP2, MMP9, MT1-MMP, ADAM family), cathepsin cysteine proteases, and serine proteases (e.g., seprase and urokinase plasminogen activator) [92]. Multiple studies have determined that cancer cells stimulated with growth factors such as PDGF, TGFβ, and EGF may trigger the formation of Invadopodia. These stimuli initiate the formation of Invadopodia primarily through their respective signaling cascades with Src and PKC intermediates [93]. The transition of resting cells to migratory cells takes place through the process of focal adhesion degradation (Figure 1). Studies have suggested a reciprocal relationship between focal adhesions and Invadopodia, owing to the role of focal adhesion kinases (FAKs) that acts as a negative regulator of Invadopodia. FAK negatively regulate Invadopodia formation by controlling the spatial activation of Src [94]. Reduction in FAK results in the release of active Src, which enhances the phosphorylation of Invadopodia-related proteins and increase Invadopodia formation. In the Src-transformed cells the initiation of Invadopodia formation occurs near focal adhesions in response to localized production of a lipid called PI3,4-P2. This lipid recruits a protein called Tks5 (hereafter referred as SH3PXD2A), which in turn associates with cortactin (CTTN), a protein crucial for actin regulation. SH3PXD2A is proposed to be the scaffold that recruits cortactin to Invadopodia precursors. SH3PXD2A has the ability to engage with a variety of actin regulatory proteins, including Nck1, Nck2, (N)-WASP, and Grb2. In a similar manner, cortactin also associates with several actin regulatory proteins like (N)-WASP and Arp2/3. It is probable that one or more of these proteins act as intermediaries in the interaction between cortactin and SH3PXD2A. This interaction might be influenced by the phosphorylation state of cortactin, as it can be activated by different kinases such as Src, PAK, and ERK, thereby impacting its interactions with other proteins. Furthermore, cortactin and Tks4 (referred to as SH3PXD2B hereafter) play roles in the advancement of Invadopodia maturation. Cortactin is recognized for its involvement in the secretion of metalloproteases, while SH3PXD2B contributes to the localization or stabilization of MT1-MMPs within Invadopodia, facilitating ECM degradation. Thus, model for Invadopodia maturation demonstrated by Murphy, et.al; states that SH3PXD2A and cortactin cooperate to create invadopodia and release metalloproteases, with SH3PXD2B later aiding in MT1-MMP localization to enable the activation of MMPs and ECM degradation. Therefore, the key players in Invadopodia formation and maturation involve adaptor proteins SH3PXD2B and SH3PXD2A along with cortactin and MMPs [93].
1.3.2. SH3PXD2B in Cancer Metastasis:
In the recent years, SH3PXD2B a closely related protein to SH3PXD2A has been identified as a critical component of invadopodia in Src-transformed fibroblasts and is implicated in metastasis of melanoma [95]. A study carried out by Buschman, et.al; have demonstrated that Src-transformed fibroblast cells lacking SH3PXD2B, developed pre-invadopodia structures with essential proteins properly localized for ECM degradation, however due to the lack of SH3PXD2B the degradation did not occur, thus, suggesting its crucial role in functionality of Invadopodia [96]. This further elucidates that a fully formed and mature invadopodia structure is useless without the presence of SH3PXD2B, and cannot carry out its migratory functions. As discussed, earlier MT1-MMP is recruited by SH3PXD2B and is particularly important for metastasis due to its diverse substrates present in the ECM such as collagens, fibronecting and laminins [97]. The subcellular localization of MT1-MMP aided by SH3PXD2B is crucial for its functionality in transmembrane domain and cytoplasmic tail as this localization regulates its proteolytic and degradation activities [98,99]. This appropriate localization of MT1-MMP thus, contributes to cancer cell growth, studied in 3D ECM environments [100]. In terms of the functionality of SH3PXD2B in cancer metastasis the recruitment of MT1-MMP results in the activation of MMP2 and MMP9 that degrade the extracellular matrix and facilitate the invasion of process by cancer cells [96]. Multiples studies have demonstrated that SH3PXD2B plays a significant role in promoting the invasion and metastasis of various cancer types, including colon cancer, breast cancer, and melanoma [95,101,102]. A recent study carried out in oral squamous cell carcinoma highlights that overexpression of crucial proteins associated with invadopodia including SH3PXD2B elevates metastatic capabilities of cancer cells and facilitate tumor progression [103]. Another recent study investigates the prognostic value of SH3PXD2B in hepatocellular carcinoma (HCC), signifying that high expression of SH3PXD2B is associated with poor overall survival in HCC patients. The findings demonstrate that increased expression of SH3PXD2B in HCC promote tumor growth and metastasis [104]. Even though the role of SH3PXD2B is not yet explored in BC-LM, these findings suggest that SH3PXD2B could be a potential therapeutic target and curbing its expression may significantly inhibit metastasis.
SH3PXD2B is identified as a scaffold protein responsible for regulating intracellular signaling by bringing regulatory proteins, enzymes, or cytoskeletal structures in close proximity [105]. The previous studies directed towards the role of SH3PXD2A in cancer metastasis imply that SH3PXD2B may have comparable functions in cancer progression and metastasis due to their evolutionary conservation and structural resemblance. The structural component of SH3PXD2B is characterized by an N-terminal phox homology (PX) domain, four Src homology 3 (SH3) domains, multiple proline-rich motifs (PRMs), and Src phosphorylation sites [105,106]. The PX domain's primary function is to join the scaffold protein to the cell membrane through phosphoinositide binding [96]. SH3 domains act as docking sites for signaling molecules and facilitate protein-protein interactions. Additionally, the proline-rich motifs serve as contact sites for molecules containing the SH3 domains [105]. SH3PXD2B exhibits two distinct states within cells: a cytoplasmic, inactive state, and a membrane-bound, active state. This transition between states is likely regulated through phosphorylation, although direct evidence for these conformational changes is limited [96,107]. This idea is supported by similar self-regulation activity seen in p47phox, a protein that has a slight structural homology with SH3PXD2B with respect to its N-terminal PX domain [108]. While in cytoplasm, the intramolecular regulatory mechanism in SH3PXD2B includes the binding of SH3 domains to a specific proline-rich motifs within its C-terminal region characterizing its auto-inhibitory state and preventing the PX domain from accessing phosphatidylinositol phosphates, which are important for its function. Consequently, when phosphorylation of C-terminal serine residues that are close to the PRMs takes place this auto-inhibitory state is disrupted and the tandem SH3 domains are exposed, allowing them to bind and interact with their regulatory partners. Simultaneously, the locked PX domain is also freed in order to interact with phospholipids and recruit the MMPs for ECM [105].
Apart from conferring invasive properties to the cell and orchestrating cell motility, Invadopodia may also have a role in cell-cell communication. SH3PXD2B is reported to bind ADAM15 using its fourth SH3 domain [109]. The ADAM family proteins are membrane-localized proteases and have the ability to act as sheddases. They are involved in the activation of growth factors or ligands by cleaving their inactive membrane-anchored forms and release their active forms. This shedding process has been demonstrated for various molecules, including insulin-like growth factor-binding protein (IGF-BP), Delta-like ligand 1 (DLL1), E-cadherin, amphiregulin, heparin-binding EGF-like growth factor (HB-EGF), transforming growth factor alpha (TGFα), EGF, and tumor necrosis factor alpha (TNFα). After cleavage by ADAMs, the released ligands exert their effects on the same, adjacent, or distant cells, facilitating communication between the "signal sender" cell and "receiver" cells [105]. Even though the role of SH3PXD2B has been critically highlighted in cancer metastasis, certain studies suggest its contradictory role. It has been recognized with a novel function in negatively regulating the EMT processes in few cancer models. A study conducted in colon cancer found that SH3PXD2B found to play a part in preventing EMT-like changes in colon cancer cells and when it was knocked down from these cells, they began to display characteristics associated with mesenchymal cells facilitating EMT [110]. However, the exact mechanism and factors associated with the contradictory functions of SH3PXD2B in cancer metastasis is not completely clear and deeper research is needed to elucidate its appropriate functions.
Understanding the contradictory role of SH3PXD2B in cancer metastasis and deciphering its functionality in breast cancer-lung metastasis model requires a multidisciplinary approach that combines cancer systems biology and computational structure biology. Through this approach our study aims at integrating multiomics data from breast cancer patients and breast cancer cell lines with different metastatic outcomes to discern patterns linked to SH3PXD2B expression and its correlation with metastasis. Additionally, it involves construction of comprehensive and inter-regulatory protein-protein networks to unveil potential functional modules and pathways associated with SH3PXD2B. Finally, through the use of computational structure biology platform we seek to dive deeper into the structural aspects SH3PXD2B by eliminating disordered regions and revealing potential target sites for therapeutic interventions.
2. Methodology
2.1. Data acquisition and analysis
To investigate the mechanisms underlying breast cancer lung metastasis (BCLM) progression, we accessed supplementary data (GSE138122) from study conducted by Cai, et.al [111]. This study explored the connection between chromatin landscapes, transcription factors, breast cancer subtypes, and their association with metastatic relapse, particularly to the lung or brain. The study utilized ATACSeq and RNASeq analyses to investigate the mechanisms underlying breast cancer metastasis to the lymph nodes. The transcriptomics data was re-analyzed to determine differentially expressed gene mediators and EMT markers using the New-Tuxedo pipeline described by Pertea et. al. [112]. Next, we corroborated these results with expression data obtained from the Cancer Cell Line Encyclopedia (CCLE) and The Cancer Genome Atlas (TCGA). We conducted a comparative analysis of SH3PXD2B expression levels in metastatic cell lines and BC cell lines indicated by the CCLE. This included the BC-MCF-7 BC epithelial cell line, MDA-MB-231 TNBC-metastatic cell line, and MDA-MB-468 metastatic cell line. Additionally, we also analyzed its expression levels in A549 lung adenocarcinoma (LUAD) cell line and H-1975 metastatic LUAD cell line. Further, to gain insights into translational factors, associated with SH3PXD2B we utilized data from the Human Protein Atlas (HPA). This analysis shed light on the role of SH3PXD2B and its interactions with partners involved in EMT, both in disease development and sub-cellular localization.
2.2. Overall Survival Analysis
The Kaplan-Meier Plot is the simplest way of computing the overall survival of patients periodically. The survival curve determines the probability of surviving in a given length of time (divided in small intervals) while considering the onset of a disease or of a treatment administered. For analyzing the overall survival of patients with the expression of breast cancer to lung cancer metastatic proteins we utilized the user-friendly online tool Kaplan – Meier Plotter (https://kmplot.com/analysis/). It carries out the survival analysis based on three assumptions: 1. It assumes that over the course of disease at any time patients who are recalcitrant and refuse to be a part of the study or patients who may not experience the event of death before the study ends will have the same chance of survival as those who are kept under observation. 2. Secondly it assumes that patients included early or late in the study have the same the survival probabilities. 3. Third it assumes that the event of death of the patient will occur at the specified time (stage) in the disease. Thus, overall survival analysis is the examination of patient data in relation to the expression of mutant genes over the course of time until the patient's death [113,114,115]. To study breast cancer metastasis to the lungs, we began with a list of 31 genes that were uploaded into the database. The samples were then divided into two groups based on their median gene expression levels, creating distinct cohorts with high and low gene expression. We utilized this stratification to create Kaplan-Meier survival curves, which depict the probability of survival over time in small intervals, along with the count of individuals at risk at each time point. The log-rank P test was used to compare the survival curves of the two cohorts (shown in the graph) and test the null hypothesis, which stated that there is no difference regarding survival among the two cohorts.
However, the sheer number of genes under investigation along with the unavailability of sufficient clinical data led to an increase in FDR values for each survival plot. As a result, we had to put the plots through another analysis wherein the z-score was calculated for filtering out most prominent genes. Z-Score defines the standard deviation from the normal estimated variance in order to draw directed conclusion for genes under study in breast cancer to lung cancer metastasis. Z-score was calculated using the formula:
where
2.3. Reconstruction of Transcription Factor Target Gene (TFTG) Network and EMTome network
Based on scientific literature the proteins involved in breast cancer metastasis to lung and published data for their associated transcription factors available in multiple databases was collected to construct an integrated bio-molecular interaction network of Transcription Factor-Target Gene (TFTG). In our study, we used an enlisted 22 metastatic proteins reported through various literatures playing a crucial role in breast cancer lung metastasis [116,117,118,119,120,121,122,123,124,125,126]. The associated transcription factors regulating the expression of these metastatic proteins were fished from the TF2DNA and TF link databases, which offer detailed and extremely reliable information on transcription factor - target gene interactions for numerous organisms, including Homo sapiens. It unifies information from various other transcription factor databases, including JASPAR and TRRUST database, offering accumulative statistics for the organism-specific transcription factors. The integrated TFTG network was built using Cytoscape (v.3.4.0). The network consisted of 22 metastatic proteins with their paired 1036 transcription factors forming a densely interconnected network of 1058 nodes and 2394 edges. This network was analysed using Cytoscape plugin – CytoHubba which runs the network through 12 different algorithms/scoring methods and returns the top 10 nodes, each of which is said to be essential to the network's regulation and connectivity. In order to rank nodes and edges in a network, CytoHubba employs a double screening scheme which aids our understanding about the functionality of an individual node as well as its collaboration with other nodes in a clique. It uses algorithms like BottleNeck, Betweenness Centrality, Radiality, Stress, Edge Percolating Coefficient, and others to present a reliable and more condensed nature of the TFTG network.
In addition to this, to identify the epithelial-to-mesenchymal transition markers associated with the metastatic proteins we constructed another network using the data from EMTome database and StringDB and analysed it employing the same parameters as we used for TFTG network analysis [127]. The network consisted of metastatic proteins with their paired EMT markers forming a densely interconnected network of 3384 nodes and 39551 edges.
2.4. Gene Set Enrichment Analysis (GSEA)
Gene Set Enrichment Analysis (GSEA) is a computational technique used to identify predefined gene sets that exhibit statistically significant differences in gene expression between two specific biological conditions [128]. GSEA method was used to analyse the differential expression of SH3PXD2B in three Breast Cancer cell-lines including MCF7, MDAMB231, MDAMB468 with respect to its expression in non-cancerous cell-line MCF10A. The gene sets are defined based on prior biological knowledge, from published information on biochemical pathways or identified co-expression from previous experiments. The expression data for all the cell-lines was obtained from CCLE.
We took into account the GSEA's capability to evaluate expression of 2725 genes and provide physiologically pertinent details regarding the differential expression of SH3PXD2B amongst the defined phenotype classes –Normal and Tumor, the enrichment analysis was carried out on all BC cell-lines with respect to non-cancerous breast epithelial cell-line. Three such gene set combinations for BC was considered: MCF-7 (metastatic) with MCF10A, MDA-MB-231 (TNBC) with MCF10A, and MDA-MB-468 (TNBC pleural) with MCF10A. To calculate the enrichment score (ES), which indicates how much a particular gene set is over-represented at the top and bottom of the ranking list, the ranking-metric is utilized by GSEA.
2.5. Cell Culture and maintenance
The human Breast Cancer MDA-MB-231, MDA-MB-468, MCF-7, human breast epithelial MCF-10A, human lung cancer A549 and NCI-H1975, and human lung epithelial BEAS-2B cell lines were obtained from the National Centre for Cell Science (NCCS), Pune’s cell repository. MDA-MB-231 and MDA-MB-468 cells were cultured in Leibovitz's L-15 medium supplemented with 2mM glutamine and 15% foetal bovine serum (FBS) without CO2. The MCF-7 cell line was maintained in Dulbecco's Modified Eagle's medium (DMEM) with 10% FBS, along with penicillin and streptomycin (PS), and incubated at 5% CO2. The breast normal epithelial cell line MCF-10A (was gifted by Dr. Shrikant Rapole, NCCS) was cultured using Mammary Epithelial cell growth Basal Medium (MEBM) containing 100 ng/ml cholera toxin and incubated at 5% CO2. In addition to these the H1975 and BEAS-2B cell lines were cultured using Roswell Park Memorial Institute (RPMI) with 10% FBS, PS and 5% CO2. The A549 cell line was maintained using Ham’s F 12 with 10% FBS, PS and 5% CO2.
2.6. Proteomics and Western Blotting
To investigate protein interactions involving SH3PXD2B and its binding partners in MDA-MB-231 and MDA-MB-468 cell lines, a pull-down approach was employed. The procedure involved lysing the cells, immunoprecipitating the protein complex of interest, and eluting the bound proteins for downstream analysis. Mass spectrometry analysis was performed using an Orbitrap Fusion Tribrid LC-MS/MS system. It allowed label-free immunoprecipitation mass spectrometry (IP-MS) analysis, with high sensitivity and accuracy. After immunoprecipitation, elution, and protein digestion, the resulting peptides were desalted and cleaned up. The LC-MS/MS instrument separated the peptides based on their hydrophobicity and solvent gradient. Mass spectrometry was performed using Data-Dependent Acquisition (DDA) or Data-Independent Acquisition (DIA) methods. This allowed the collection of both MS and MS/MS spectra, enabling peptide identification and quantification.
To validate our study, we utilized Western Blotting technique to investigate the protein levels of SH3PXD2B and CTTN in cell lysate samples from breast cancer (MCF7, MDA-MB-231 and MDA-MB-468), lung cancer (A549 and NCI-H1975), breast normal (MCF10A), and lung normal (BEAS-2B) cell lines. β-actin or GAPDH served as control proteins for comparative analysis. Both techniques utilized specific Rb-polyclonal antibody against SH3PXD2B, from ThermoFisher (Catalog No. PA5-57673).
2.7. Structural aspects of SH3PXD2B
We investigated the structural aspects of SH3PXD2B by modelling its structure (NCBI Accession ID: NP_001017995.1). To achieve this, we employed AlphaFold Google Colab [129]. It predicted a high-confidence model of SH3PXD2B that was assessed using the following parameters: 1. Predicted Local Distance Difference Test (pLDDT) used to determine intra-domain confidence, 2. Predicted Aligned Error (PAE) utilized to assess confidence between domains or chains within the protein. However, the coverage of the predicted protein structure was low (covered 362 out of 911 base pairs only) from the native structure of SH3PXD2B (UniProtKB accession ID A1X283). Also, the native structure provided by AlphaFold2 consisted large number of loop structures. Thus, having large numbers of disordered region. In order to develop an understanding of the ordered regions in SH3PXD2B domains we used PrDOS.
2.8. Molecular docking
The drugs or small molecules and drug-antibody conjugates in use for metastatic breast cancer as the “Standard of Care” according to the American Society of Clinical Oncology (ASCO) were retrieved from Cancer.Net (https://www.cancer.net/cancer-types/breast-cancer-metastatic/types-treatment). The structural insights of SH3PXD2B led to the conclusion that the 2nd and 3rd SH3 domains (SH3_2 and SH3_3 respectively) within the structure could serve as potential sites for enhanced drug binding capacity.
The interaction between SH3_2 and SH3_3 domains of SH3PXD2B was determined using molecular docking technique through AutoDock Vina and MGL Tools [130,131]. Molecular docking uses algorithms to stimulate the biding process of protein and small molecule (ligand), thus, providing insights into their binding affinity, binding energy exploring their structural conformations and evaluating their complementary interactions within the provided grid [132,133]. The .sdf files for the ligands were retrieved from PubChem database and the. pdbqt files of protein were generated using Open Babel. Alongside a grid box with dimensions (centre_x = -0.873, centre_y = 26.287, centre_z = 9.705) and sides with (size_x = 92, size_y = 76, size_z = 98) was created for SH3_2 and dimensions with (centre_x = -0.158, centre_y = 15.687, centre_z = -12.823) and sides with (size_x = 94, size_y = 84, size_z = 80) was created for SH3_3 domains of SH3PXD2B to interact with the shortlisted drug candidates. Docking was carried out using a genetic algorithm and the protein-ligand interactions were mapped using PyMOL and Ligplot+ [134]. Docking provided binding positions for each peptide using an empirical scoring method, the best binding position was picked for further analysis depending on their binding energies.
2.9. Pathway Enrichment Analysis
The inclusive analysis of DESeq data and TFTG network provided a list of 56 significant metastatic markers playing a crucial role in giving rise to breast cancer lung metastasis. To find the interacting nodes (proteins) for these metastatic markers we used STRING database (https://string-db.org/). The STRING database is dedicated to constructing protein-protein interacting networks. It mapped the 56 metastatic markers to their interacting proteins displaying a biological network covering all the proteins. Alongside this it also performs automated pathway classification of the metastatic markers listing out their role and functionality in biological process/pathways [135]. In addition to the analysis of occurrence of metastatic markers in classified pathways we performed a thorough pathway enrichment analysis for these markers using Cytoscape plugin – Biological Networks Gene Ontology tool (BiNGO). BiNGO calculates the enrichment score of biological markers or gene ontologies by taking into account both their expression and their occurrence in a given process to determine if they are overrepresented in a particular biological process or network. BiNGO's key benefit over other GO enrichment tools is that it makes use of molecular interaction networks, in this case, the transcription factor-target gene network. The statistical test used for calculating the p-value for enrichment analysis was Hypergeometric test and the false positive values were corrected by using Benjamin Hochberg correction method [13]. Further, to validate our results from proteomics and network biology studies we conducted additional pathway enrichment analysis using StringDB and DICE.
3. Results
3.1. Differentially expressed genes in BC-LM
The data set obtained from Cai, et.al; used ATACSeq and RNASeq techniques to delve into the molecular mechanisms associated with breast cancer metastasis to the lymph nodes [111]. Differential Gene Expression (DGE) analysis was a central component of our research, it enabled us to identify and characterize gene mediators that undergo activation or alteration during metastasis and facilitate migration of cancer cells compared to normal cells. The New Tuxedo pipeline enabled us in obtaining metastatic markers with respect the MDA-MB-231 cell lines. The transcriptome data analysis for BC vs. LM revealed the expression levels of genes such as SH3PXD2B, SH3PXD2A, MMP14, CTTN, ADAMs, and various epithelial-mesenchymal transition (EMT) markers suggesting that the expression of SH3PXD2B was low in BC samples but high in LM samples (Figure 2A).
Further to comprehensively assess mRNA expression of these markers across all cell lines representing both MBC and LUAD we collected data from CCLE. Our analysis, in line with RNA-Seq transcriptomics reports, yielded intriguing results. Notably, SH3PXD2B demonstrated the highest expression level in the MDA-MB-468 cell line, highlighting its pivotal role in metastatic adenocarcinoma (Figure 2B).
Our primary aim was to unravel the underlying mechanisms involved in breast cancer metastasis to the lymph nodes and the results of this analysis provide crucial insights into the molecular processes associated with breast cancer metastasis.
Apart from investigating the transcriptional signatures, we have also looked upon the translational part from the Human Protein Atlas, wherein we found the expression of SH3PXD2B was significantly high in both breast and lung cancer patients as well as cell lines (Figure 2C). This also demonstrated its functionality in disease casualty. Additionally, we also examined the sub-cellular localization of SH3PXD2B and found it to be present in nucleoplasm and neucloli fribrillar center along with NOX4, SNAI1, TWIST1, ZEB1 and ZEB2 its interacting partners co-existed and facilitated EMT during BC-LM.
3.2. Overall Survival Analysis
SH3PXD2B, SH3PXD2A, MMP14, MMP9, CTTN, ADAM12, ADAM15, NOX5, EGF, SRC, GRB2, SNAI1, ZEB1, ZEB2, DUOX1, and CDKN2A, demonstrated significant expression in breast cancer - lung metastasis. The Overall Survival (OS) analysis plots generated predicted poor survival rate of patients in both breast cancer and lung cancer (Figure 3A,B). The log-rank P value for every gene was less than 0.05 showing its significance in cancer metastasis and occurrence of event of death during the specified time intervals. High expression of ADAM15 in breast cancer demonstrated worst OS with log-rank p value 5 times lesser than 0.05. Similarly, patients with high expression of MMP9 in lung cancer showed least OS with a log-rank p value 3 times lesser than 0.05. This could be better understood with the Hazard Ratios (HR). The hazard ratio, often known as the ratio of risk of an event (death) occurring at a certain period of time, measures the magnitude of the variance between the two curves in the survival plot [137]. It can be seen the HR for all the genes was more than 85% of the set cutoff values indicating that the patients with high expression cohorts were at greater risk, supporting worse overall survival. Genes like MMPs, ADAMs, and SH3PXDs may play a significant role in driving the spread of cancer from breast to lung. MMP9, MMP14, ADAM12, ADAM15, SH3PXD2B and SH3PXD2A are all critical components of the metastatic niche. The overall survival analysis provided by Kaplan Meier Plotter is evident and can be used to connect the mRNA expression patterns of these genes to the course of Breast Cancer to Lung metastasis.
Additionally, the utilization of z-scores instead of p-values was of great significance and importance due to their capacity to offer a standardized metric for quantifying the extent to which a data point diverges from the mean. According to the interpretation of Z-scores, the genes that have been identified as crucial in the advanced stage of breast cancer include SH3PXD2B, CYBA, MMP9, CTTN, NOX5, EGF, SRC, GRB2, ZEB1, SNAI1,and CDKN2A. Regarding the examination of overall survival in lung cancer, the genes that have been identified include SH3PXD2A, MMP9, MMP14, ADAM12, GRB2, ZEB1, ZEB2, and CTNNB1 (Figure 3C) (Supplementary Data S1).
3.3. TFTG and EMTome network analysis
Our comprehensive analysis of the GSE138122 DESeq data and TFTG network results revealed 56 essential metastatic proteins and transcription factors that were necessary to the causation of breast cancer to lung metastasis. The significant transcription factors and metastatic proteins in the inter-regulatory TFTG network were determined using Cytoscape plugin-CytoHubba. CytoHubba provided top 10 ranked TFs and TGs critical to the entire network using the 12 scoring methods including Betweenness, BottleNeck, Closeness, Clustering coefficient, Degree, Density of Maximum Neighbourhood Component (DMNC), EcCentricity, Edge Percolating Coefficient (EPC), Maximal Clique Centraity (MCC), Maximum Neighborhood Component (MNC), Radiality and Stress. The top 10 TFs/TGs in the network were determined by their frequency of occurrence in each scoring method respectively. Based on their frequency of occurrence in each of the12 algorithms, top 10 metastatic proteins and transcription factors from the entire network are represented in (Figure 4A–O). The analysis of the TFTG network mainly identified 15 metastatic proteins namely VIM, MMP2, FYN, CTTN, CDH2, CYBA, FGR, NOX5, ADAM15, PIK3CA, DUOX2, SH3PXD2B, MMP9, MMP14 and BLK and 20 transcription factors including CDK2, CEBPA, CNOT3, CPSF3, DEK, EBF3, FOXA3, FOXE3, GATAD1, GFI1, STST3, NFKB1, RELA, SP1, ESR1, KLF1, ARNT, TFAP2A, ZNF589, and ZSCAN9 respectively to be highly enriched in the entire network.
Additionally, the EMTome network analysis revealed key molecular signatures or EMT markers that are closely related to metastatic proteins and involved in the formation of invadopodia during metastasis. Notably, these include FN1, SRC, CTNNB1, EGFR, AKT1, ALB, CDH1, ACTA2, EGF and CCNA2 (Figure 4P) (Supplementary data S2).
3.4. Analysis of differentially expressed SH3PXD2B in BC cell-lines:
The conclusion of the gene set enrichment analysis is the enrichment plot indicating the enrichment score (ES), which determines how much a gene set is over-represented at the top or bottom of a ranked list of genes. In the enrichment plot the magnitude of the decrement in the enrichment score depends on how well the gene set correlates with the defined phenotype (class), the graph here illustrates the negative enrichment of SH3PXD2B in MCF10A (non-cancerous/normal breast cell-line) for all gene set combination showing rapid decrease in the expression of SH3PXD2B in non-cancerous condition (the Normal class) with an enrichment score of -0.8, -0.8 and -0.7 respectively for all the three combinations mentioned above (Figure 5A). The leading-edge subset of a gene in a gene set, or the subset of members contributing more to the ES, has also been seen to adversely correlate more with the negatively enriched normal phenotype. Furthermore, the bottom-most part of the enrichment plot displays the ranking metrics value considered by GSEA to calculate the correlation between each gene in the gene set and the defined phenotype. As we proceed downwards the ranked list, the value of the ranking metrics changes from positive to negative, with a positive value signifying correlation with the diseased phenotype and a negative value signifying correlation with the normal phenotype. According to the ES, the highest ranked genes in the gene sets are found between ranks 0 through 100.
Alongside, the butterfly plot produced by the GSEA analysis shows the positive and negative associations of genes in the ranked list depending on their rank and ranking metric score (Figure 5B). It displays the actual correlation for the top genes within the defined phenotypes in addition to permuted (1%, 5%, 50%) positive and negative correlation. By default, the butterfly plot considers the first and last 100 genes in the ranked list for analysis. In order to support the enrichment plots, the butterfly plot shows that the observed negatively ranked genes which are associated with the normal phenotype are negatively correlated via abnormal expression of SH3PXD2B having more affinity towards the tumor phenotype. Moreover, a heat map is also generated depicting the (clustered) genes in the leading-edge subset of gene set (Figure 5C). For each phenotype, a heat map shows the correlation between the ranked genes and the defined phenotype class. Expression values are depicted on the heat-map as colors; with the hues (red, pink, light blue, dark blue) according to the gene expression value range (high, moderate, low, and lowest). The heat-maps obtained in all three cases demonstrate that the genes targeted by SH3PXD2B have higher expression in the leading-edge subset of genes correlating the tumor phenotype illustrated with red color for MCF7A, MDAMB231 and MDAMB468 respectively as compared to its lowest expression illustrated with dark blue color for MCF10A in the datasets.
3.5. Proteomics and Western Blotting:
For the proteomics study we utilized the SH3PXD2B Rb-polyclonal antibody (ThermoFisher, Catalog No. PA5-57673) to immunoprecipitate SH3PXD2B from MDA-MB-231 and MDA-MB-468 cell lines. Subsequently, the collected samples underwent analysis using the orbitrap mass spectrometry technique, specifically the Orbitrap Fusion Tribrid LC-MS/MS system. The immunoprecipitation experiment required the IgG markers to be negated from the respective cell line markers identified through immunoprecipitation of SH3PXD2B antibody pull-down. Therefore, the markers were converted using DAVID to get the Ensembl GeneID from the UniProt Accession IDs provided by the proteome discoverer analysis tool. Our analysis revealed varying levels of SH3PXD2B expression in both the samples. Notably we observed an increasing trend in the expression of SH3PXD2B associated with MDA-MB-468 (TNBC) cell line that is known to be prevalent in MBC (Figure 6A) (Supplementary data S3 and S4).
Further, we demonstrated the expression of SH3PXD2B in both breast cancer cell lines (MDA-MB-231 and MDA-MB-468) and lung cancer cell lines (BEAS-2B, A549, and NCI-H1975) using Western Blotting techniques. Also, we studied the expression levels of CTTN exclusively in BC cell lines as underscoring its functional relationship with SH3PXD2B in breast cancer metastasis. The results, indicated that SH3PXD2B and was highly expressed in MDA-MB-468 cells as compared to MDA-MB-231 cells corroborating with our proteomics analysis (Figure 6B,E). Additionally, CTTN was also found to be highly expressed in MDA-MB-468 cells as compared to MDA-MB-231 cells (Figure 6C,E). The expression of SH3PXD2B in LC cells (A549 and H1975) was found to b ehigh as compared to normal lung epithelial cells (BEAS-2B) (Figure6 D,F) (Supplementary data S5).
3.6. Structure
The predicted model of SH3PXD2B, generated using AlphaFold, is presented in (Figure 7A). The analysis elucidated high confidence regions using parameters such as predicted LDDT (pLDDT) and predicted aligned error (PAE).The yellow-labeled areas in the ball and stick model correspond to interfaces within SH3PXD2B displaying poor confidence, notably, this includes the 4th SH3 domain. The blue-labeled areas represent interfaces that are confident and very high-confident regions within the modeled structure. This encompasses the SH3_2 and SH3_3 domains.
The conservedness of all domains with specific ordered regions in AlphaFold model of SH3PXD2B was marked to be relatively high, as estimated through PrDOS. The predicted probability score for disordered regions was calculated for all SH3PXD2B domains. The Bokeh plot analysis indicated the disordered probability as per the number of residues in SH3PXD2B. We found that SH3_2 and SH3_3 domains had least disordered probability with 94.92% ordered regions thus, corroborating with our model confidence results. Additionally, we have shown the ordered residues for every domain represented in black and disordered regions represented in red (Figure 7B).
3.7. Docking
Molecular docking of drug candidates retrieved from ASCO Cancer.Net was performed specifically for SH3_2 and SH3_3 domains keeping in mind their structural confidence and high number of ordered regions. The results demonstrated that, Eribulin (PubChem ID 11354606) is found to be efficiently interacting with both the domains with the lowest binding energy as -12.79 kcal/mol for SH3_2 and -10.77 kcal/mol for SH3_3 domain respectively. The results were compared and represented in bar plot with the binding energies (kcal/mol) vs the names of all drugs candidates used for our study (Figure 8A) (Supplementary data S6).
The Eribulin molecule was found to interact with SH3_2 domain at residue number 1D, 2D, 5D and 44D (D represents Chain D) specifically with valine, isoleucine, tyrosine and proline residues at these positions. The interaction was hydrophobic. It also interacted with a hydrogen bond with valine at position 1D. For SH3_3 domain it interacted with 5 amino acid residues positioned at 2E, 3E, 4E, 29E and 42E (E represents Chain E) including glutamic acid, phenylalanine, glutamine, tryptophan and proline respectively. Out of these amino acid residues 2E, 4E and 29E interacted with hydrogen bonding and 3E, 4E, 29E and 42E interacted with hydrophobic interactions (Figure 8B).
3.8. Enriched Pathways
The Cytoscape plugin - BiNGO facilitated the identification of the GO categories or the biological network sub-graph which was noticeably over-represented in association with the expression of metastatic marker. As a result, a graph (Figure 9A) showing the gene ontology terms overrepresented in the network and a table with the data, including the p-value, corrected p-value, and cluster frequency, were generated (Supplementary data S7) where the identified metastatic markers showed involvement in many biological pathways playing a positive role in the regulation of cell migration, cell communication and cellular development.
Additionally, the proteomics-based network analysis was performed using StringDB and DICE to elucidate the enriched pathways associated with the metastatic proteins. The clustering coefficient for MDA-MB-231 and MDA-MB-468 networks of associated partner obtained from StringDB was found to be 0.509 and 0.493 respectively. Thus, indicating that these networks exhibit strong local clustering patterns among nodes, which can be meaningful in the context of understanding protein-protein interactions and functional relationships in biological systems. Further, the associated pathways with these proteins were elucidate by DICE and processes such as regulation of actin-filament, localization of proteins to membrane, anchoring junctions, adhering junction and domain specific binding were observed playing key role in metastasis.
4. Discussion
Metastasis continues to present a formidable challenge in breast cancer patients, maintaining its status as the primary cause of mortality among affected patients, despite significant advances in medical research and therapeutic strategies [17]. Among the various forms of metastasis observed in breast cancer, the emergence of lung metastasis stands as a particularly ominous and life-threatening consequence. Current treatment modalities, including chemotherapy, radiotherapy, and surgical resection, are primarily palliative in nature, offering temporary relief rather than a permanent solution. This underscores the pressing need for innovative approaches to combat breast cancer-lung metastasis effectively with definitive cure [138]. In recent years, the spotlight has shifted toward targeted therapies that are grounded in targeting key molecular players and signaling pathways underlying the intricate processes of metastasis, holding the potential to revolutionize our approach to treat BC-LM.
The disruption of the proteostasis network is emerging as a key factor in breast cancer metastasis. Proteostasis refers to the maintenance of protein homeostasis often through transcriptional and translational changes within cells within cells. It involves the proper folding, assembly, trafficking, and degradation of proteins essential for preserving normal physiological conditions [139]. Recent evidences suggest that disruption of proteostasis network leads to many diseases including cancer at higher risk [140]. The proteostasis network plays a crucial role in cancer cell survival and progression. Cancer cells experience proteotoxic stress due to the rapid synthesis of mutated or misfolded proteins, increased metabolic demands, and a hostile tumor microenvironment. To adapt and thrive, cancer cells often exploit the proteostasis network for their benefit [141]. The disrupted proteostasis network and elevated proteotoxic stress may favor therapeutic resistance, invasion and metastasis of breast cancer cells. Identifying key regulatory nodes and vulnerabilities in this network may lead to the development of novel therapeutic approaches for metastatic breast cancer.
SH3PXD2B plays an instructive role in inducing an EMT like process in cancer cells [142]. In this study, we embarked on a comprehensive exploration of the molecular landscape associated with breast cancer lung metastasis, with a particular focus on the role of SH3PXD2B as a potential key player in the process. The transcriptome data analysis for the comparison between breast cancer and lung metastasis unveiled a number of genes, including SH3PXD2B, SH3PXD2A, MMP14, CTTN, ADAMs, and several epithelial-mesenchymal transition (EMT) markers. Notably, the expression of SH3PXD2B exhibited a stark contrast between BC and LM samples, with low expression in the former and a substantial increase in the latter. This finding thus, emphasized the potential role of SH3PXD2B in driving the metastatic process. To bolster our findings, we conducted a comprehensive analysis of mRNA expression across a spectrum of cell lines, encompassing both MBC and lung adenocarcinoma LUAD from CCLE. The analysis highlighted the prominence of SH3PXD2B expression, particularly in the MDA-MB-468 (TNBC) cell line, reinforcing its significance in metastatic adenocarcinoma. In addition, to this the data drawn from HPA also provided substantial evidence supporting the high expression of SH3PXD2B in both breast and lung cancer patients, as well as cell lines. Further it also revealed the subcellular localization of SH3PXD2B along with its interacting partners NOX4, SNAI1, TWIST1, ZEB1, and ZEB2 all of which have been implicated in EMT processes [105]. Next the OS analysis revealed a bleak prognosis for patients with elevated expression of these genes, with log-rank p-values consistently falling below 0.05, signifying their significance in cancer metastasis and the occurrence of fatal events during specified time intervals. Calculation of z-score reduced the FDR and unearthed genes such as SH3PXD2B, CYBA, MMP9, CTTN, NOX5, EGF, SRC, GRB2, ZEB1, SNAI1, and CDKN2A as key players in advanced breast cancer. The TFTG network and EMTome network analysis corroborated with overall findings indicating these proteins with highest degree of occurrence owing to all algorithmic analysis. Further the top hits obtained from network analysis was subjected to pathway enrichment analysis. This analysis unveiled pathways regulating cell migration, cell communication, protein localization to membranes, anchoring junctions, and adhering junctions all contributing significantly to EMT and metastasis.
These findings highlighted the significant role of SH3PXD2B in MBC, prompting us to focus our analysis specifically on it. The GSEA provided valuable insights into the enrichment of SH3PXD2B expression and its associated partners in BC cell lines. The results indicated a negative correlation of SH3PXD2B expression in normal vs. tumor cell lines. This was supported by the leading-edge subset analysis that affirmed the genes targeted by SH3PXD2B exhibited a stronger correlation with the tumor phenotype, aligning with the downregulation of SH3PXD2B in normal conditions. The combination of Western blot analysis and immunoprecipitation mass spectrometry ensured robust validation of our findings, allowing for the confirmation of SH3PXD2B expression levels in both breast and lung cancer cell lines and the identification of potentially interacting CTTN associated with SH3PXD2B in BC. Proteomics analysis revealed variable levels of SH3PXD2B expression, in BC cell lines itself, with a noteworthy increase associated with the MDA-MB-468 (TNBC) cell line, a hallmark of MBC. Also, the western blot analysis was consistent with the proteomics data for BC and DGE/HPA data for LM. Additionally, the CTTN expression was checked in BC cell lines as its association with SH3PXD2B has been implicated in MBC; the results were in affirmation with SH3PXD2B expression in MDA-MB-468 cell line. Since, SH3PXD2B has been the center of our study and has been validated at multiple steps we were curious to look into its structural aspects to employ effective therapeutic strategy for MBC preventing BC-LM. The structural information aids in understanding the interaction of proteins with other proteins and potential binding sites [143]. Our exploration of SH3PXD2B's structure harnessed the power of AlphaFold, resulting in a predicted model that afforded a high degree of confidence for specific domains. We further planned to eliminate the disordered regions of protein using computational tools. Disordered regions are segments of proteins that lack a stable three-dimensional structure under native conditions. Instead, these regions exist as dynamic ensembles of conformations. When utilizing drugs or therapeutic antibodies, it may be necessary to focus on structured regions of a protein that are more likely to serve as effective binding sites and increase the efficacy of employed drug. Removal of disordered regions highlighted the SH3_2 and SH3_3 domains to be highly ordered with approximately 94.92% of their regions classified as ordered. Next to target SH3PXD2B in MBC at these domains we specifically docked drug molecules that are currently used as “Standard of Care” for BC patients. The molecular docking results revealed that Eribulin emerged as a highly efficient binder, interacting with both domains with low binding energies (-12.79 kcal/mol for SH3_2 and -10.77 kcal/mol for SH3_3).
SH3PXD2B acts as a scaffold protein, facilitating the assembly of signaling complexes. It interacts with various signaling molecules, including Src kinases and focal adhesion proteins. This protein-protein interaction network is essential for intracellular signaling, which regulates cell migration, invasion, and adaptation to the new microenvironment at metastatic sites. Through our study we elucidated SH3PXD2B as a master regulator of metastatic processes and ECM degradation conferring to the disruption of protein homeostasis in MBC cells aggravating tumors to distant organs more specifically lungs due to close proximity of lymph nodes.
Recent years have seen intensive research into small compounds aimed at targeting gene mediators to mitigate disruptions in the proteostasis network, with the ultimate goal of developing anticancer agents. A recent study by Pizzuti et. al; evaluates the existing evidence surrounding Eribulin's efficacy and safety in highly metastatic Triple negative breast cancer subtype. It specifies that Eribulin primarily exerts its effects by inhibiting the polymerization of microtubules. It also, functions in altering tumor hypoxia through vascular remodeling, reversing the epithelial-to-mesenchymal transition and more broadly, diminishing the capacity for invasion, migration and metastatic potential of cancer cells [142]. By employing a multidisciplinary platform encompassing cancer systems biology and computational structural biology, we aimed to elucidate both the functional and structural attributes of SH3PXD2B. In addition to this we integrated systems medicine approach to repurpose the already existing drugs to target SH3PXD2B in BC-LM patients based on their molecular signature (Figure 10). By employing ccomputational tools we screened compound libraries against the predicted structure of SH3PXD2B in order to assess suitability of drugs and guide clinical trials and treatment decisions. Patient-specific therapeutic interventions targeting SH3PXD2B can be designed and monitored for efficacy, leading to more personalized and effective BC-LM therapies. This multidisciplinary approach holds promise for rewiring the proteostasis network and improving outcomes for BC-LM patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
For research articles with several authors, the following statements should be used “Conceptualization, X.X. and Y.Y.; methodology, X.X.; software, X.X.; validation, X.X., Y.Y. and Z.Z.; formal analysis, X.X.; investigation, X.X.; resources, X.X.; data curation, X.X.; writing—original draft preparation, X.X.; writing—review and editing, X.X.; visualization, X.X.; supervision, X.X.; project administration, X.X.; funding acquisition, Y.Y. All authors have read and agreed to the published version of the manuscript.”
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sung, Hyuna, et al. "Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries." CA: a cancer journal for clinicians 71.3 (2021): 209-249. [CrossRef]
- Orrantia-Borunda, Erasmo, et al. "Subtypes of breast cancer." Breast Cancer [Internet] (2022).
- Łukasiewicz, Sergiusz, et al. "Breast cancer—epidemiology, risk factors, classification, prognostic markers, and current treatment strategies—an updated review." Cancers 13.17 (2021): 4287. [CrossRef]
- Leong, Stanley PL, et al. "Is breast cancer the same disease in Asian and Western countries?." World journal of surgery 34 (2010): 2308-2324. [CrossRef]
- Green, Michael, and Vinod Raina. "Epidemiology, screening and diagnosis of breast cancer in the Asia–Pacific region: current perspectives and important considerations." Asia-Pacific Journal of Clinical Oncology 4 (2008): S5-S13. [CrossRef]
- Sung, Hyuna, et al. "Female breast cancer incidence among Asian and Western populations: more similar than expected." Journal of the National Cancer Institute 107.7 (2015): djv107. [CrossRef]
- Lee, Peng-Jhen, et al. "Urban–Rural Disparity in Birth Cohort Effects on Breast Cancer Incidence." Journal of Urban Health (2023): 1-14. [CrossRef]
- Lim, Yu Xian, et al. "Breast cancer in Asia: incidence, mortality, early detection, mammography programs, and risk-based screening initiatives." Cancers 14.17 (2022): 4218. [CrossRef]
- International Agency for Research on Cancer [accessed on September 2, 2023] (https://gco.iarc.fr/tomorrow/en).
- Cancer Research UK [accessed on September 2, 2023] (https://www.cancerresearchuk.org/aboutcancer/breast-cancer/survival).
- Caswell-Jin, Jennifer L., et al. "Change in survival in metastatic breast cancer with treatment advances: meta-analysis and systematic review." JNCI cancer spectrum 2.4 (2018): pky062. [CrossRef]
- Jin, Xin, and Ping Mu. "Targeting breast cancer metastasis." Breast cancer: basic and clinical research 9 (2015): BCBCR-S25460. [CrossRef]
- Kim, Mi-Young, et al. "Tumor self-seeding by circulating cancer cells." Cell 139.7 (2009): 1315-1326. [CrossRef]
- Comen, Elizabeth, Larry Norton, and Joan Massague. "Clinical implications of cancer self-seeding." Nature reviews Clinical oncology 8.6 (2011): 369-377. [CrossRef]
- Chiang, Anne C., and Joan Massagué. "Molecular basis of metastasis." New England Journal of Medicine 359.26 (2008): 2814-2823. [CrossRef]
- Keefe, Dorothy MK, and Emma H. Bateman. "Tumor control versus adverse events with targeted anticancer therapies." Nature reviews Clinical oncology 9.2 (2012): 98-109. [CrossRef]
- Park, Misung, et al. "Breast cancer metastasis: Mechanisms and therapeutic implications." International Journal of Molecular Sciences 23.12 (2022): 6806. [CrossRef]
- Shafei, Ayman, et al. "A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer." Biomedicine & Pharmacotherapy 95 (2017): 1209-1218. [CrossRef]
- Helsby, Nuala, et al. "Cyclophosphamide bioactivation pharmacogenetics in breast cancer patients." Cancer Chemotherapy and Pharmacology 88.3 (2021): 533-542. [CrossRef]
- Miranda Furtado, Cristiana Libardi, et al. "Epidrugs: targeting epigenetic marks in cancer treatment." Epigenetics 14.12 (2019): 1164-1176. [CrossRef]
- Dai, Enyong, et al. "Epigenetic modulation of antitumor immunity for improved cancer immunotherapy." Molecular cancer 20.1 (2021): 1-27. [CrossRef]
- Schmid, Peter, et al. "Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer." New England Journal of Medicine 379.22 (2018): 2108-2121. [CrossRef]
- Corso, Giovanni, et al. "Metaplastic breast cancer: prognostic and therapeutic considerations." Journal of Surgical Oncology 123.1 (2021): 61-70. [CrossRef]
- Mayer, Erica L., et al. "A Phase I dose-escalation study of the VEGFR inhibitor tivozanib hydrochloride with weekly paclitaxel in metastatic breast cancer." Breast cancer research and treatment 140 (2013): 331-339. [CrossRef]
- Cortés, Javier, et al. "Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer." New England Journal of Medicine 386.12 (2022): 1143-1154. [CrossRef]
- Geyer, Charles E., et al. "Lapatinib plus capecitabine for HER2-positive advanced breast cancer." New England journal of medicine 355.26 (2006): 2733-2743. [CrossRef]
- Finn, Richard S., et al. "Palbociclib and letrozole in advanced breast cancer." New England journal of medicine 375.20 (2016): 1925-1936. [CrossRef]
- Von Minckwitz, Gunter, et al. "Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer." New England Journal of Medicine 377.2 (2017): 122-131.
- Plosker, G.L. , Faulds D. Epirubicin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cancer chemotherapy. Drugs. 1993;45:788–856. [CrossRef]
- Zardavas, Dimitrios, José Baselga, and Martine Piccart. "Emerging targeted agents in metastatic breast cancer." Nature reviews Clinical oncology 10.4 (2013): 191-210. [CrossRef]
- Shah, Sohrab P., et al. "Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution." Nature 461.7265 (2009): 809-813. [CrossRef]
- Amir, Eitan, et al. "Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer." Journal of clinical oncology 30.6 (2012): 587. [CrossRef]
- Lindström, Linda Sofie, et al. "Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression." J Clin Oncol 30.21 (2012): 2601-2608. [CrossRef]
- Bedard, Philippe L., et al. "Tumour heterogeneity in the clinic." Nature 501.7467 (2013): 355-364. [CrossRef]
- Van Denderen, Bryce JW, and Erik W. Thompson. "The to and fro of tumour spread." Nature 493.7433 (2013): 487-488.
- Valachis, Antonis, et al. "Overall survival of patients with metastatic breast cancer in Sweden: a nationwide study." British Journal of Cancer 127.4 (2022): 720-725. [CrossRef]
- Garrido-Castro, Ana C., Nancy U. Lin, and Kornelia Polyak. "Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment." Cancer discovery 9.2 (2019): 176-198. [CrossRef]
- Dent, Rebecca, et al. "Triple-negative breast cancer: clinical features and patterns of recurrence." Clinical cancer research 13.15 (2007): 4429-4434. [CrossRef]
- Andre, F. , and C. C. Zielinski. "Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents." Annals of oncology 23 (2012): vi46-vi51. [CrossRef]
- Xiao, Weikai, et al. "Breast cancer subtypes and the risk of distant metastasis at initial diagnosis: a population-based study." Cancer management and research (2018): 5329-5338. [CrossRef]
- Zardavas, Dimitros, et al. "The AURORA initiative for metastatic breast cancer." British journal of cancer 111.10 (2014): 1881-1887. [CrossRef]
- Garcia-Recio, Susana, et al. "Multiomics in primary and metastatic breast tumors from the AURORA US network finds microenvironment and epigenetic drivers of metastasis." Nature Cancer 4.1 (2023): 128-147. [CrossRef]
- Paget, Stephen. "The distribution of secondary growths in cancer of the breast." The Lancet 133.3421 (1889): 571-573. [CrossRef]
- Gennari, Alessandra, et al. "Survival of metastatic breast carcinoma patients over a 20-year period: A retrospective analysis based on individual patient data from six consecutive studies." Cancer 104.8 (2005): 1742-1750.
- Smid, Marcel, et al. "Subtypes of breast cancer show preferential site of relapse." Cancer research 68.9 (2008): 3108-3114. [CrossRef]
- Dan, Zhaoling, et al. "A pH-responsive host-guest nanosystem loading succinobucol suppresses lung metastasis of breast cancer." Theranostics 6.3 (2016): 435.
- Foulkes, William D., Ian E. Smith, and Jorge S. Reis-Filho. "Triple-negative breast cancer." New England journal of medicine 363.20 (2010): 1938-1948.
- Jin, Liting, et al. "Breast cancer lung metastasis: Molecular biology and therapeutic implications." Cancer biology & therapy 19.10 (2018): 858-868. [CrossRef]
- Soh, Junichi, Yoshifumi Komoike, and Tetsuya Mitsudomi. "Surgical therapy for pulmonary metastasis of breast cancer." Translational Cancer Research 9.8 (2020): 5044. [CrossRef]
- Kennecke, Hagen, et al. "Metastatic behavior of breast cancer subtypes." Journal of clinical oncology 28.20 (2010): 3271-3277. [CrossRef]
- Liedtke, Cornelia, et al. "Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer." Journal of clinical oncology 26.8 (2008): 1275-1281. [CrossRef]
- Dent R, Hanna WM, Trudeau M, et al. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res Treat 2009;115:423-8. [CrossRef]
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010;363:1938-48. [CrossRef]
- Wu, Qi, et al. "Breast cancer subtypes predict the preferential site of distant metastases: a SEER based study." Oncotarget 8.17 (2017): 27990. [CrossRef]
- Kong, Fengwei, et al. "Case Report Second primary lung cancer after a first breast cancer: a case report and critical literature review." Int J Clin Exp Med 11.2 (2018): 1049-1054.
- Langerød, Anita, et al. "TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer." Breast cancer research 9 (2007): 1-16. [CrossRef]
- Schmitt, Fernando, et al. "Cancer stem cell markers in breast neoplasias: their relevance and distribution in distinct molecular subtypes." Virchows Archiv 460 (2012): 545-553. [CrossRef]
- Honeth, Gabriella, et al. "The CD44+/CD24-phenotype is enriched in basal-like breast tumors." Breast Cancer Research 10.3 (2008): 1-12.
- Hu, Jing, et al. "A CD44v+ subpopulation of breast cancer stem-like cells with enhanced lung metastasis capacity." Cell death & disease 8.3 (2017): e2679-e2679. [CrossRef]
- McGovern, Marie, et al. "A “latent niche” mechanism for tumor initiation." Proceedings of the National Academy of Sciences 106.28 (2009): 11617-11622.
- Harrison, Hannah, et al. "Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor." Cancer research 70.2 (2010): 709-718. [CrossRef]
- Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M, Santini D, Ceccarelli C, Chieco P, Bonafé M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells. 2007;25(3):807–15. [CrossRef] [PubMed]
- Pal D, Kolluru V, Chandrasekaran B, Baby BV, Aman M, Suman S, Sirimulla S, Sanders MA, Alatassi H, Ankem MK, et al. Targeting aberrant expression of Notch-1 in ALDH+ cancer stem cells in breast cancer. Mol Carcinog. 2017;56(3):1127–1136. [CrossRef]
- Suman S, Das TP, Damodaran C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br J Cancer. 2013;109(10):2587–96. [CrossRef] [PubMed]
- Khramtsov, Andrey I., et al. "Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome." The American journal of pathology 176.6 (2010): 2911-2920. [CrossRef]
- Geyer, Felipe C., et al. "β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation." Modern pathology 24.2 (2011): 209-231. [CrossRef]
- Hayashi, Hisaki, and Tsutomu Kume. "Forkhead transcription factors regulate expression of the chemokine receptor CXCR4 in endothelial cells and CXCL12-induced cell migration." Biochemical and biophysical research communications 367.3 (2008): 584-589. [CrossRef]
- Han, Bingchen, et al. "FOXC1 activates smoothened-independent hedgehog signaling in basal-like breast cancer." Cell reports 13.5 (2015): 1046-1058. [CrossRef]
- Zuo, Hou Dong, and W. Wu Yao. "The role and the potential regulatory pathways of high expression of forkhead box C1 in promoting tumor growth and metastasis of basal-like breast cancer." J BUON 21.4 (2016): 818-25.
- Liu, Jiao, et al. "Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways." Cancer letters 379.1 (2016): 49-59. [CrossRef]
- Pires, Bruno Ricardo Barreto, et al. "Targeting cellular signaling pathways in breast cancer stem cells and its implication for cancer treatment." Anticancer research 36.11 (2016): 5681-5691. [CrossRef]
- Zardawi, Sarah J., et al. "Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer." Histology and histopathology (2009).
- Okuhashi, Yuki, Mai Itoh, and Shuji Tohda. "Hedgehog stimulation suppresses clonogenicity and activates NOTCH signalling in T-lymphoblastic leukaemia Jurkat cells." Anticancer Research 37.9 (2017): 5005-5009. [CrossRef]
- Egeblad, Mikala, Elizabeth S. Nakasone, and Zena Werb. "Tumors as organs: complex tissues that interface with the entire organism." Developmental cell 18.6 (2010): 884-901. [CrossRef]
- Polyak, Kornelia, and Raghu Kalluri. "The role of the microenvironment in mammary gland development and cancer." Cold Spring Harbor perspectives in biology 2.11 (2010): a003244. [CrossRef]
- Yu, P. F. , et al. "TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils." Oncogene 36.4 (2017): 482-490. [CrossRef]
- Acharyya, Swarnali, and Joan Massague. "Arresting supporters: targeting neutrophils in metastasis." Cell research 26.3 (2016): 273-274. [CrossRef]
- Wculek, Stefanie K., and Ilaria Malanchi. "Neutrophils support lung colonization of metastasis-initiating breast cancer cells." Nature 528.7582 (2015): 413-417. [CrossRef]
- Chen, Jingqi, et al. "CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3." Cancer cell 19.4 (2011): 541-555. [CrossRef]
- Li, Xiu Juan, et al. "Role of pulmonary macrophages in initiation of lung metastasis in anaplastic thyroid cancer." International Journal of Cancer 139.11 (2016): 2583-2592. [CrossRef]
- Cao, Haiqiang, et al. "Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer." ACS nano 10.8 (2016): 7738-7748. [CrossRef]
- Vadrevu, Surya Kumari, et al. "Studying the role of alveolar macrophages in breast cancer metastasis." JoVE (Journal of Visualized Experiments) 112 (2016): e54306.
- Chen, Qing, Xiang H-F. Zhang, and Joan Massagué. "Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs." Cancer cell 20.4 (2011): 538-549. [CrossRef]
- Xu, Kun, et al. "The fibroblast Tiam1-osteopontin pathway modulates breast cancer invasion and metastasis." Breast cancer research 18.1 (2016): 1-15. [CrossRef]
- Kim, Hye Min, Woo Hee Jung, and Ja Seung Koo. "Expression of cancer-associated fibroblast related proteins in metastatic breast cancer: an immunohistochemical analysis." Journal of translational medicine 13.1 (2015): 1-11. [CrossRef]
- Nieto, M. Angela, et al. "EMT: 2016." Cell 166.1 (2016): 21-45.
- Gonzalez, David M., and Damian Medici. "Signaling mechanisms of the epithelial-mesenchymal transition." Science signaling 7.344 (2014): re8-re8. [CrossRef]
- Linder, Stefan, Christiane Wiesner, and Mirko Himmel. "Degrading devices: invadosomes in proteolytic cell invasion." Annual review of cell and developmental biology 27 (2011): 185-211.
- Weaver, Alissa M. "Invadopodia: specialized cell structures for cancer invasion." Clinical & experimental metastasis 23 (2006): 97-105. [CrossRef]
- Schoumacher, Marie, et al. "Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia." Journal of Cell Biology 189.3 (2010): 541-556. [CrossRef]
- Petrie, Ryan J., Andrew D. Doyle, and Kenneth M. Yamada. "Random versus directionally persistent cell migration." Nature reviews Molecular cell biology 10.8 (2009): 538-549. [CrossRef]
- Artym, Vira V., et al. "Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function." Cancer research 66.6 (2006): 3034-3043. [CrossRef]
- Murphy, Danielle A., and Sara A. Courtneidge. "The'ins' and'outs' of podosomes and invadopodia: characteristics, formation and function." Nature reviews Molecular cell biology 12.7 (2011): 413-426.
- Chan, Keefe T., Christa L. Cortesio, and Anna Huttenlocher. "FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion." Journal of Cell Biology 185.2 (2009): 357-370. [CrossRef]
- Iizuka, Shinji, et al. "The role of Tks adaptor proteins in invadopodia formation, growth and metastasis of melanoma." Oncotarget 7.48 (2016): 78473.
- Buschman, Matthew D., et al. "The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation." Molecular biology of the cell 20.5 (2009): 1302-1311. [CrossRef]
- Itoh, Yoshifumi, and Motoharu Seiki. "MT1-MMP: a potent modifier of pericellular microenvironment." Journal of cellular physiology 206.1 (2006): 1-8. [CrossRef]
- Uekita, Takamasa, et al. "Cytoplasmic tail–dependent internalization of membrane-type 1 matrix metalloproteinase is important for its invasion-promoting activity." The Journal of cell biology 155.7 (2001): 1345-1356. [CrossRef]
- Lehti, Kaisa, et al. "Regulation of membrane-type-1 matrix metalloproteinase activity by its cytoplasmic domain." Journal of Biological Chemistry 275.20 (2000): 15006-15013. [CrossRef]
- Hotary, Kevin B., et al. "Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix." Cell 114.1 (2003): 33-45. [CrossRef]
- Gianni, Davide, et al. "c-Src–mediated phosphorylation of NoxA1 and Tks4 induces the reactive oxygen species (ROS)–dependent formation of functional invadopodia in human colon cancer cells." Molecular biology of the cell 21.23 (2010): 4287-4298. [CrossRef]
- Leong, Hon S., et al. "Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis." Cell reports 8.5 (2014): 1558-1570. [CrossRef]
- Mitre, Geovanni Pereira, et al. "Key proteins of invadopodia are overexpressed in oral squamous cell carcinoma suggesting an important role of MT1-MMP in the tumoral progression." Diagnostic Pathology 16 (2021): 1-10. [CrossRef]
- Kui, Xiang, et al. "Prognostic value of SH3PXD2B (Tks4) in human hepatocellular carcinoma: a combined multi-omics and experimental study." BMC Medical Genomics 14.1 (2021): 1-10. [CrossRef]
- Kudlik, Gyöngyi, et al. "Advances in understanding TKS4 and TKS5: Molecular scaffolds regulating cellular processes from podosome and invadopodium formation to differentiation and tissue homeostasis." International Journal of Molecular Sciences 21.21 (2020): 8117. [CrossRef]
- Saini, Priyanka, and Sara A. Courtneidge. "Tks adaptor proteins at a glance." Journal of cell science 131.1 (2018): jcs203661. [CrossRef]
- Bögel, Gábor, et al. "Frank-ter Haar syndrome protein Tks4 regulates epidermal growth factor-dependent cell migration." Journal of Biological Chemistry 287.37 (2012): 31321-31329. [CrossRef]
- Teasdale, Rohan D., and Brett M. Collins. "Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: structures, functions and roles in disease." Biochemical Journal 441.1 (2012): 39-59.
- Mao, Mao, et al. "The podosomal-adaptor protein SH3PXD2B is essential for normal postnatal development." Mammalian genome 20 (2009): 462-475. [CrossRef]
- Szeder, Bálint, et al. "Absence of the Tks4 scaffold protein induces epithelial-mesenchymal transition-like changes in human colon cancer cells." Cells 8.11 (2019): 1343. [CrossRef]
- Cai, Wesley L., et al. "Specific chromatin landscapes and transcription factors couple breast cancer subtype with metastatic relapse to lung or brain." BMC medical genomics 13.1 (2020): 1-18. [CrossRef]
- Pertea, Mihaela, et al. "Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown." Nature protocols 11.9 (2016): 1650-1667.
- Lánczky, András, and Balázs Győrffy. "Web-based survival analysis tool tailored for medical research (KMplot): development and implementation." Journal of medical Internet research 23.7 (2021): e27633.
- Goel, Manish Kumar, Pardeep Khanna, and Jugal Kishore. "Understanding survival analysis: Kaplan-Meier estimate." International journal of Ayurveda research 1.4 (2010): 274. [CrossRef]
- Rich, Jason T., et al. "A practical guide to understanding Kaplan-Meier curves." Otolaryngology—Head and Neck Surgery 143.3 (2010): 331-336. [CrossRef]
- Eckert MA, Santiago-Medina M, Lwin TM, Kim J, Courtneidge SA, Yang J. ADAM12 induction by Twist1 promotes tumor invasion and metastasis via regulation of invadopodia and focal adhesions. J Cell Sci. 2017 Jun 15;130(12):2036-2048. Epub 2017 May 3. [CrossRef] [PubMed] [PubMed Central]
- Martner A, Aydin E, Hellstrand K. NOX2 in autoimmunity, tumor growth and metastasis. J Pathol. 2019 Feb;247(2):151-154. Epub 2018 Nov 29. [CrossRef] [PubMed] [PubMed Central]
- Li Y, Zhang H, Gong H, Yuan Y, Li Y, Wang C, Li W, Zhang Z, Liu M, Liu H, Chen J. miR-182 suppresses invadopodia formation and metastasis in non-small cell lung cancer by targeting cortactin gene. J Exp Clin Cancer Res. 2018 Jul 9;37(1):141. [CrossRef] [PubMed] [PubMed Central]
- Li X, Yang J, Wang X, Li X, Liang J, Xing H. Role of TWIST2, E-cadherin and Vimentin in epithelial ovarian carcinogenesis and prognosis and their interaction in cancer progression. Eur J Gynaecol Oncol. 2016;37(1):100-8. [PubMed]
- Mrozik KM, Blaschuk OW, Cheong CM, Zannettino ACW, Vandyke K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer. 2018 Oct 1;18(1):939. [CrossRef] [PubMed] [PubMed Central]
- Liu, K. , Tang, Z., Huang, A., Chen, P., Liu, P., Yang, J., Lu, W., Liao, J., Sun, Y., Wen, S., Hu, Y., Huang, P."Glyceraldehyde-3-phosphate dehydrogenase promotes cancer growth and metastasis through upregulation of SNAIL expression". International Journal of Oncology 50.1 (2017): 252-262. [CrossRef]
- Wang Y, Shi J, Chai K, Ying X, Zhou BP. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets. 2013 Nov;13(9):963-972. [CrossRef] [PubMed] [PubMed Central]
- Fan L, Lei H, Zhang S, Peng Y, Fu C, Shu G, Yin G. Non-canonical signaling pathway of SNAI2 induces EMT in ovarian cancer cells by suppressing miR-222-3p transcription and upregulating PDCD10. Theranostics. 2020 Apr 27;10(13):5895-5913. [CrossRef] [PubMed] [PubMed Central]
- Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. 2015;14(4):481-7. [CrossRef] [PubMed] [PubMed Central]
- Kim, Hye-Ran, et al. “Zinc Finger E-Box Binding Homeobox 2 as a Prognostic Biomarker in Various Cancers and Its Correlation with Infiltrating Immune Cells in Ovarian Cancer.” Current Issues in Molecular Biology, vol. 44, no. 3, Mar. 2022, pp. 1203–14. Crossref. [CrossRef]
- Golubovskaya VM, Ho B, Conroy J, Liu S, Wang D, Cance WG. Gene Expression Profiling Identifies Important Genes Affected by R2 Compound Disrupting FAK and P53 Complex. Cancers (Basel). 2014 Jan 21;6(1):166-78. [CrossRef] [PubMed] [PubMed Central]
- Vasaikar, Suhas V., et al. "EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures." British journal of cancer 124.1 (2021): 259-269. [CrossRef]
- Subramanian, Aravind, et al. "Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles." Proceedings of the National Academy of Sciences 102.43 (2005): 15545-15550. [CrossRef]
- Jumper, John, et al. "Highly accurate protein structure prediction with AlphaFold." Nature 596.7873 (2021): 583-589. [CrossRef]
- Trott, Oleg, and Arthur J. Olson. "AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading." Journal of computational chemistry 31.2 (2010): 455-461.
- Morris, Garrett M., et al. "AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility." Journal of computational chemistry 30.16 (2009): 2785-2791. [CrossRef]
- Rastelli G, et al. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2009;8(11): 879-891. [CrossRef]
- Morris GM, Lim-Wilby M. Molecular docking. Methods Mol Biol. 2008;443:365-382. [CrossRef]
- Laskowski, Roman A., and Mark B. Swindells. "LigPlot+: multiple ligand–protein interaction diagrams for drug discovery." (2011): 2778-2786. [CrossRef]
- Damian Szklarczyk and others, The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets, Nucleic Acids Research, Volume 49, Issue D1, 8 January 2021, Pages D605–D612. [CrossRef]
- Steven Maere and others, BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks, Bioinformatics, Volume 21, Issue 16, August 2005, Pages 3448–3449. [CrossRef]
- Tom Brody,Chapter 9 - Biostatistics—Part I,Editor(s): Tom Brody, Clinical Trials (Second Edition), Academic Press, 2016,Pages 203-226,ISBN 9780128042175. [CrossRef]
- Yousefi, Meysam, et al. "Organ-specific metastasis of breast cancer: molecular and cellular mechanisms underlying lung metastasis." Cellular Oncology 41 (2018): 123-140. [CrossRef]
- Balch, William E., et al. "Adapting proteostasis for disease intervention." science 319.5865 (2008): 916-919. [CrossRef]
- Chen, Xiao-Qiong, et al. "Protein homeostasis in aging and cancer." Frontiers in Cell and Developmental Biology 11 (2023): 1143532.
- Brancolini, Claudio, and Luca Iuliano. "Proteotoxic stress and cell death in cancer cells." Cancers 12.9 (2020): 2385. [CrossRef]
- Merő, Balázs, et al. "Characterization of the intramolecular interactions and regulatory mechanisms of the scaffold protein Tks4." International Journal of Molecular Sciences 22.15 (2021): 8103. [CrossRef]
- Kitchen, Douglas B., et al. "Docking and scoring in virtual screening for drug discovery: methods and applications." Nature reviews Drug discovery 3.11 (2004): 935-949. [CrossRef]
- Pizzuti, Laura, et al. "Eribulin in triple negative metastatic breast cancer: critic interpretation of current evidence and projection for future scenarios." Journal of Cancer 10.24 (2019): 5903. [CrossRef]
Figure 1.
Invadopodia formation facilitating EMT and cancer cell metastasis.
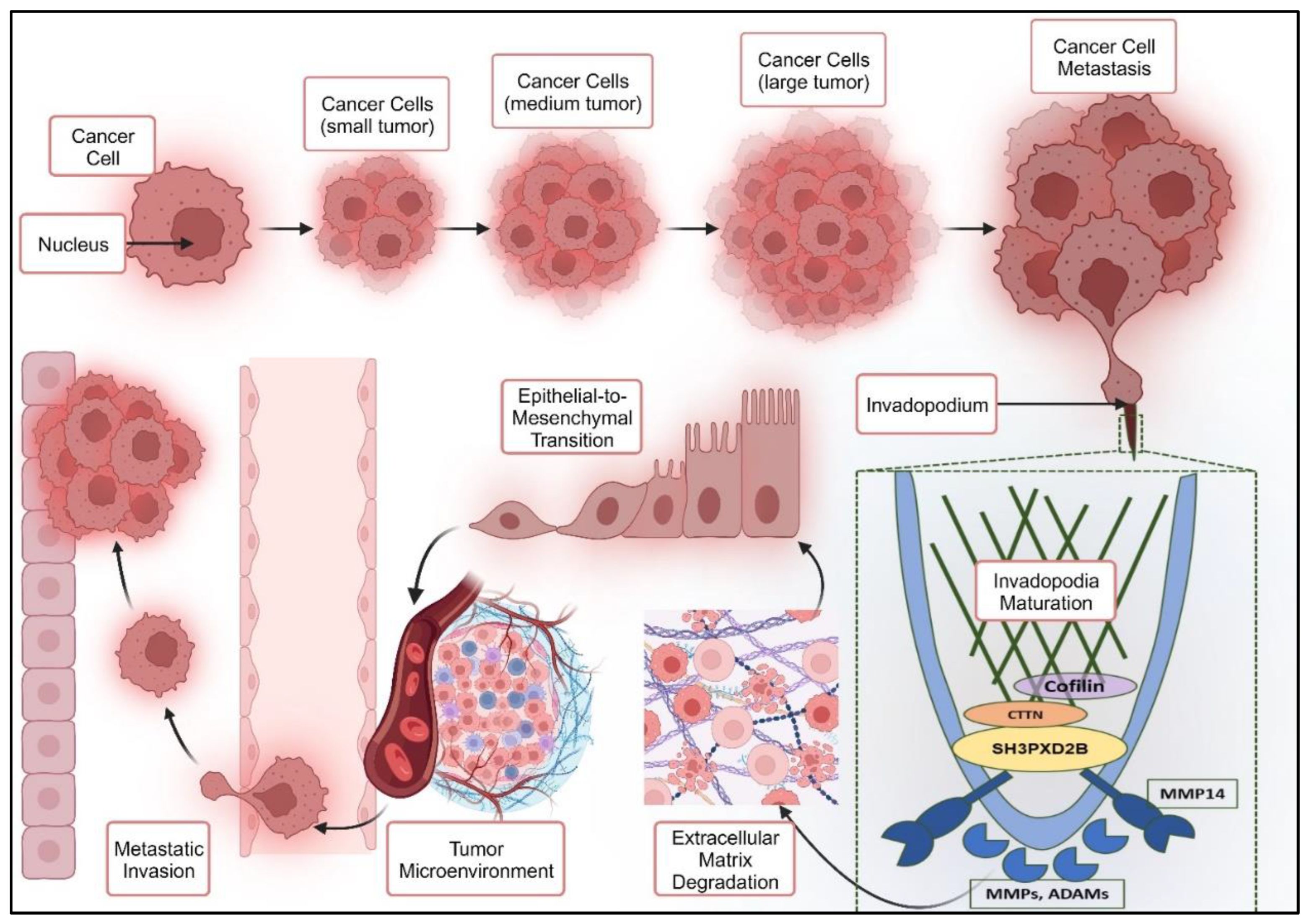
Figure 2.
(A): Differential Gene Expression of metastatic markers. (a) tabulated expression values (b) graphical representation. (B): mRNA expression of metastatic markers from CCLE. (C): Protein Expression data from the Human Protein Atlas database.
Figure 2.
(A): Differential Gene Expression of metastatic markers. (a) tabulated expression values (b) graphical representation. (B): mRNA expression of metastatic markers from CCLE. (C): Protein Expression data from the Human Protein Atlas database.
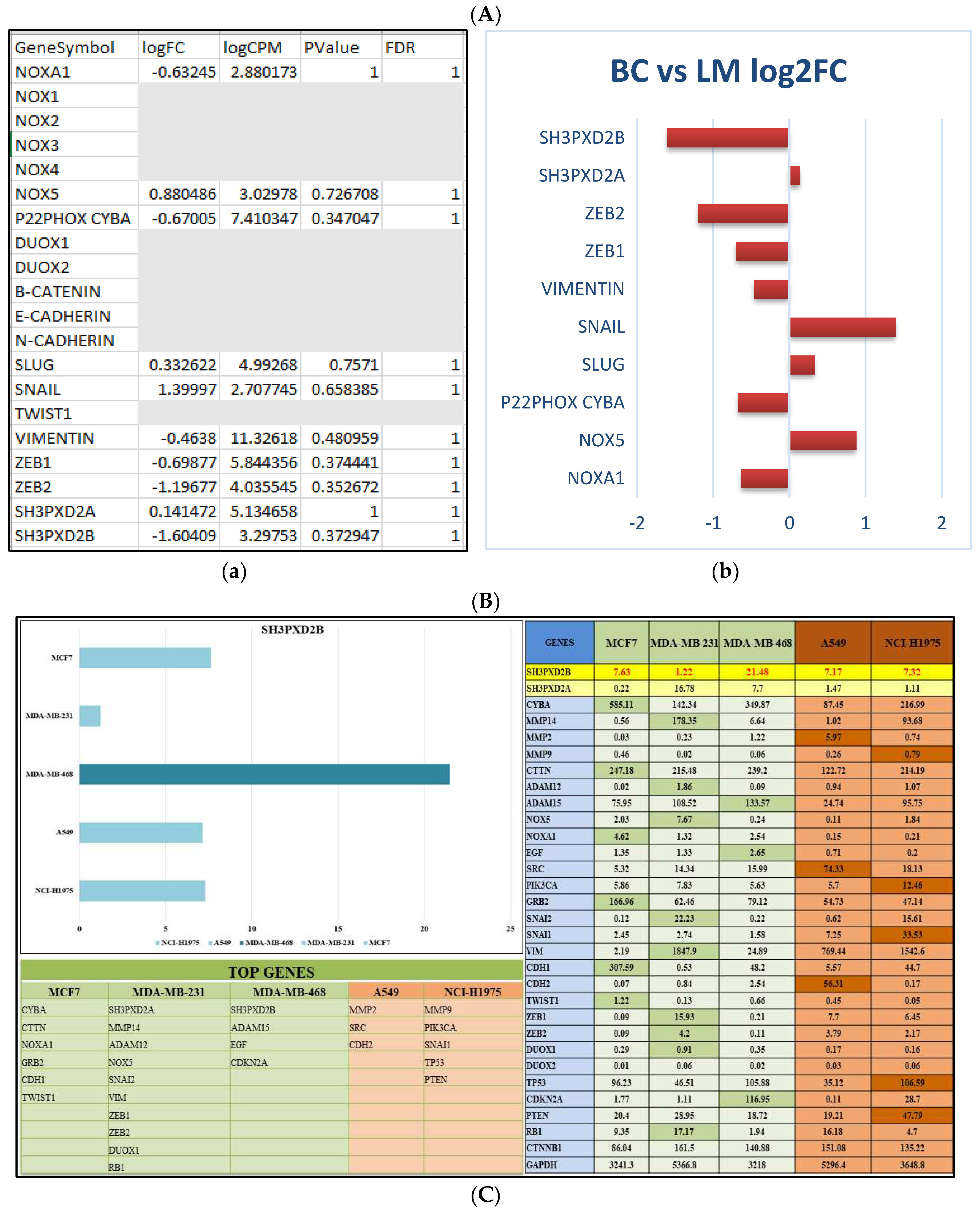
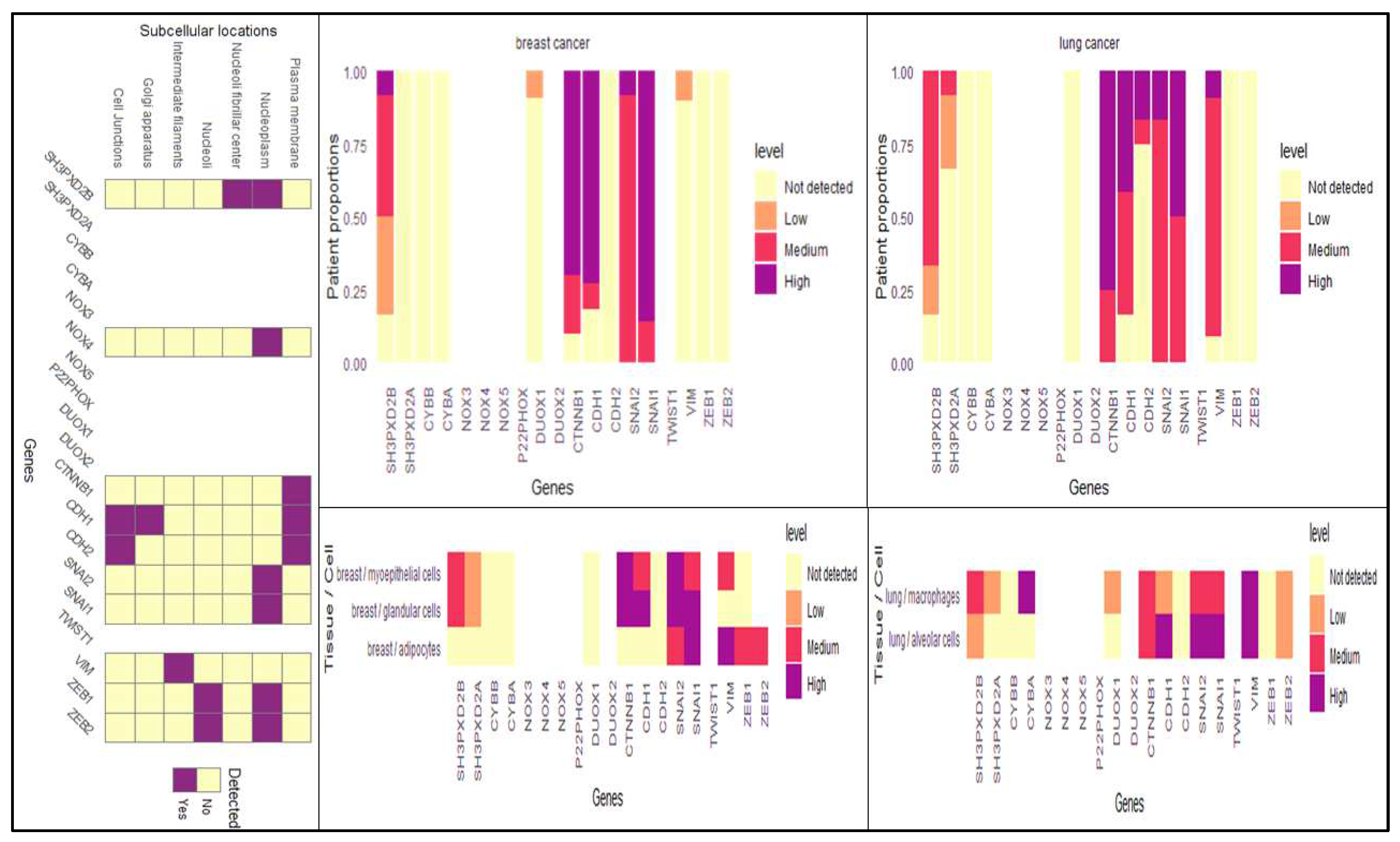
Figure 3.
Expression plots of (A) Overall Survival Analysis for Breast Cancer. (B) Overall Survival Analysis for Lung Cancer. (C) Graphs defining z-score for all metastatic markers involved in BC and LC.
Figure 3.
Expression plots of (A) Overall Survival Analysis for Breast Cancer. (B) Overall Survival Analysis for Lung Cancer. (C) Graphs defining z-score for all metastatic markers involved in BC and LC.
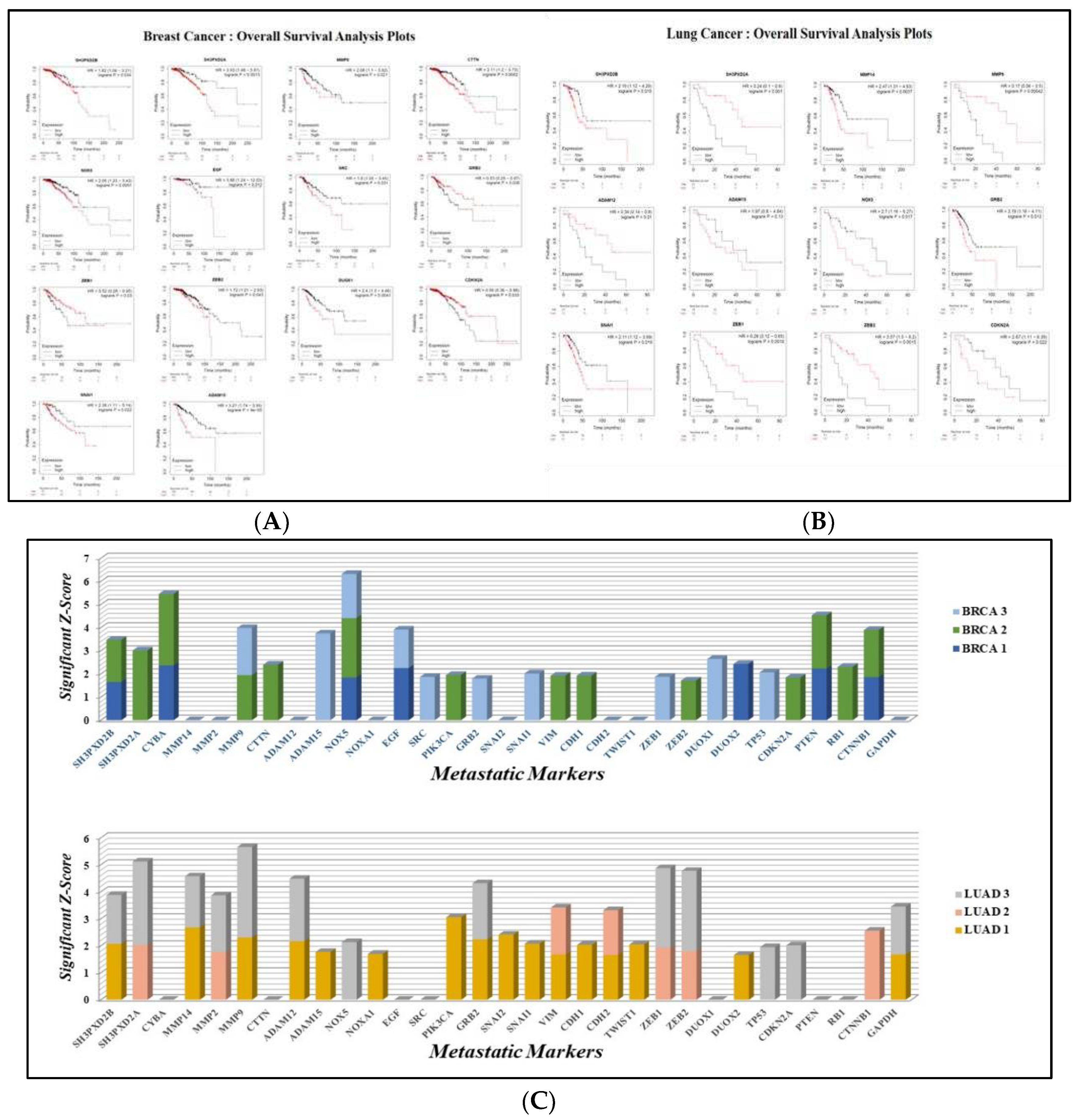
Figure 4.
TF-TG network analysis: (A) Original TFTG Network (B) Betweenness centrality (C) BottleNeck (D) Closeness centrality (E) Clustering Coefficient (F) Degree of Nodes (G) DMNC (H) EcCentricity (I) EPC (J) MCC (K) MNC (L) Radiality (M) Stress (N) Frequency of Occurrence of Transcription factors (O) Frequency of Occurrence of Metastatic Proteins (P) Frequency of Occurrence of EMT markers.
Figure 4.
TF-TG network analysis: (A) Original TFTG Network (B) Betweenness centrality (C) BottleNeck (D) Closeness centrality (E) Clustering Coefficient (F) Degree of Nodes (G) DMNC (H) EcCentricity (I) EPC (J) MCC (K) MNC (L) Radiality (M) Stress (N) Frequency of Occurrence of Transcription factors (O) Frequency of Occurrence of Metastatic Proteins (P) Frequency of Occurrence of EMT markers.
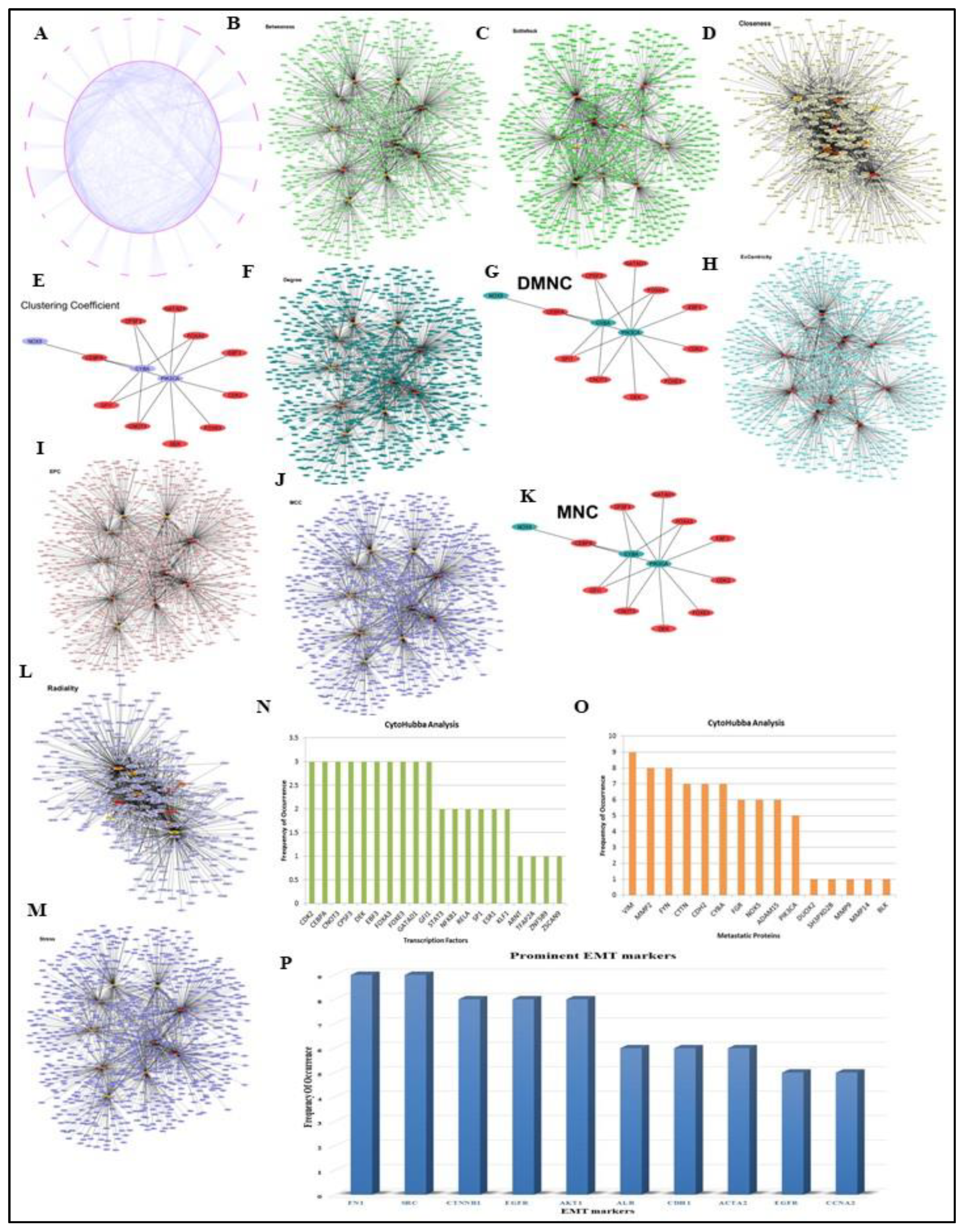
Figure 5.
Gene Set Analysis (GSEA) results for all three combinations respectively: MCF7 vs. MCF10A, MDAMB231 vs. MCF10A and MDAMB468 vs. MCF10A. (A) Enrichment Plots (B) Butterfly Plots (C) Heat-maps.
Figure 5.
Gene Set Analysis (GSEA) results for all three combinations respectively: MCF7 vs. MCF10A, MDAMB231 vs. MCF10A and MDAMB468 vs. MCF10A. (A) Enrichment Plots (B) Butterfly Plots (C) Heat-maps.
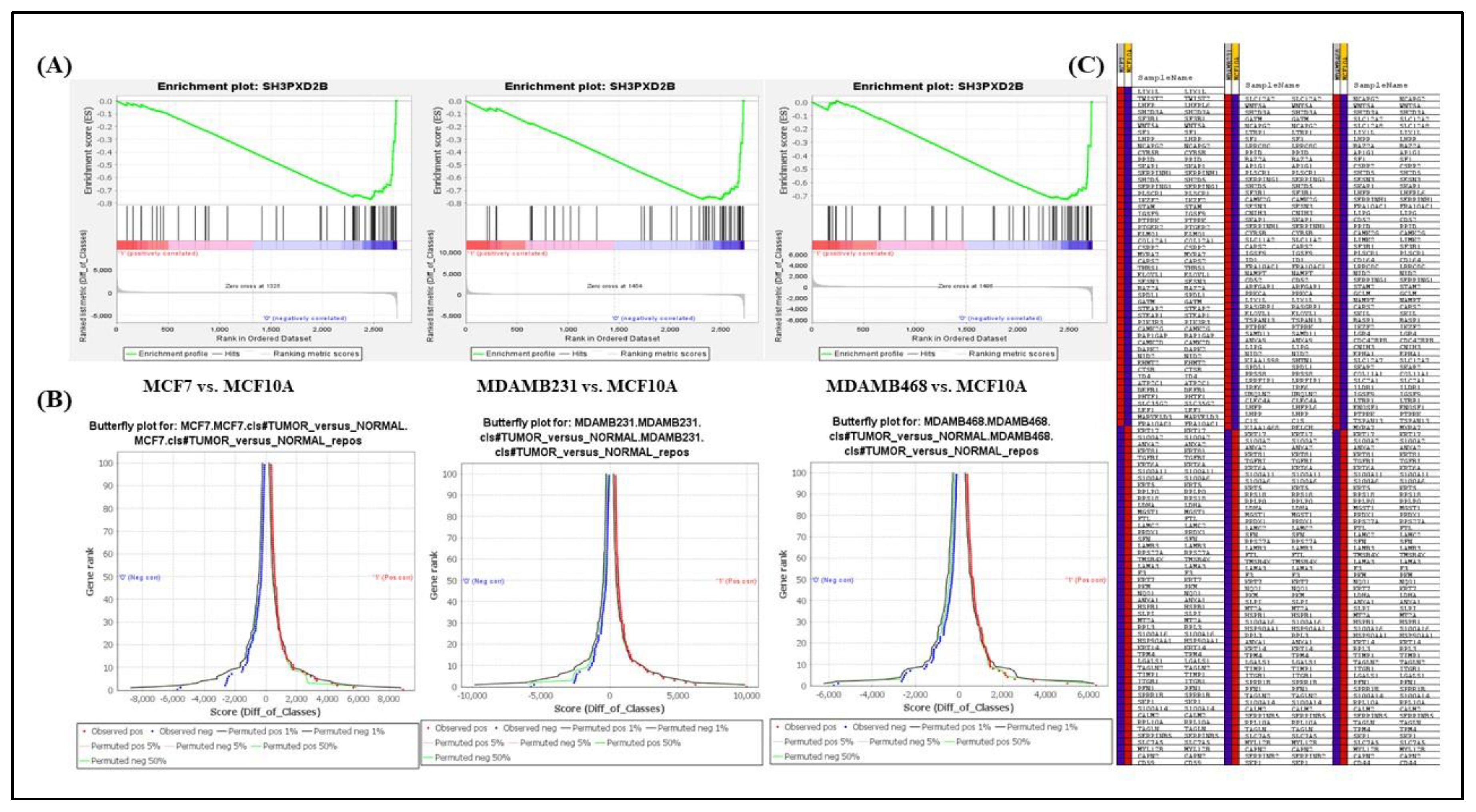
Figure 6.
Proteomics and Western Blot analysis. (A) Proteomic (B) WB of SH3PXD2B for BC cell line (C) WB of CTTN for BC cell line (D) WB of SH3PXD2B for LC and Lung normal cell line (E) & (F) graphical representation of WB analysis in BC and LC cell lines.
Figure 6.
Proteomics and Western Blot analysis. (A) Proteomic (B) WB of SH3PXD2B for BC cell line (C) WB of CTTN for BC cell line (D) WB of SH3PXD2B for LC and Lung normal cell line (E) & (F) graphical representation of WB analysis in BC and LC cell lines.
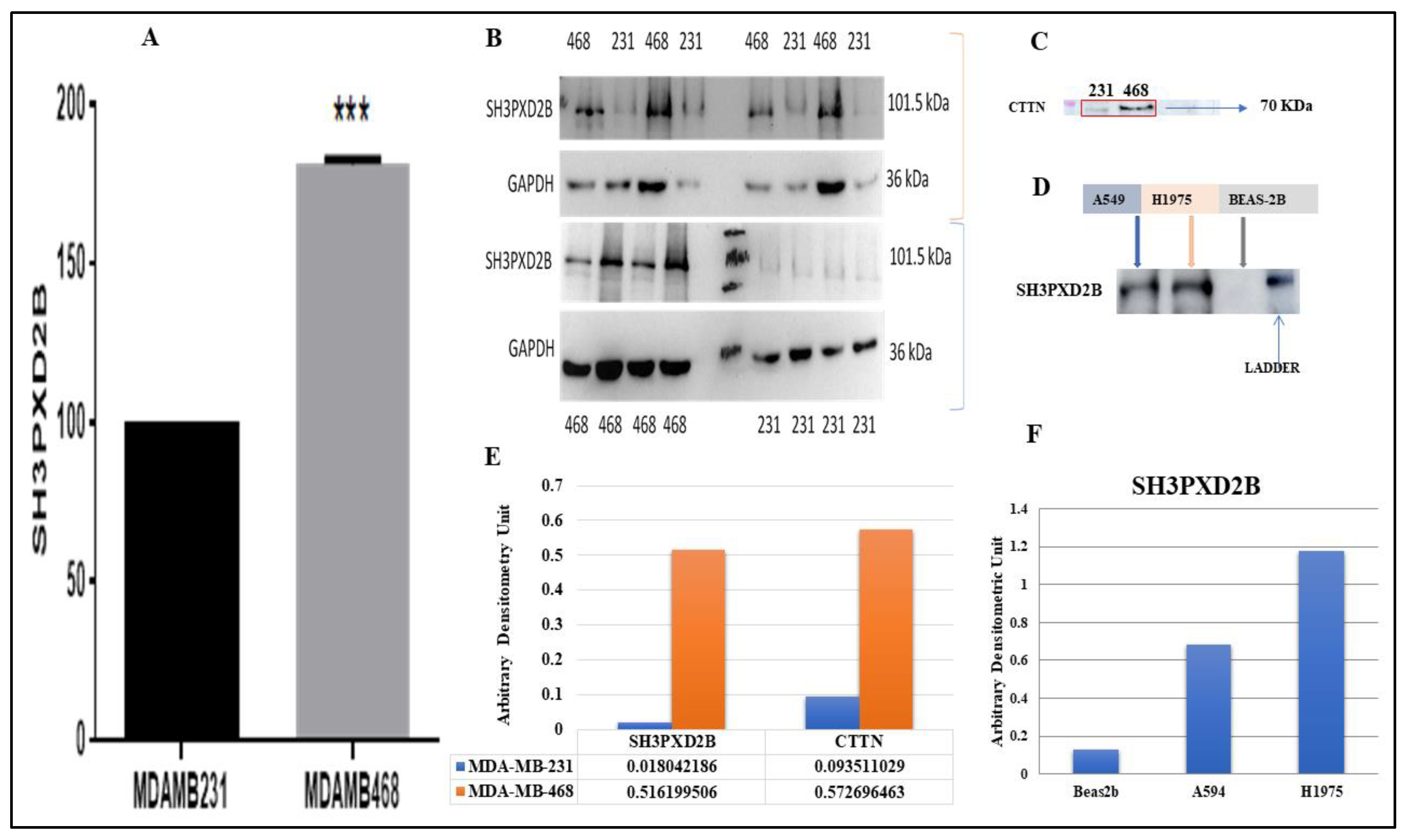
Figure 7.
A. Structural insights into SH3PXD2B. B. Graphical representation of disordered regions in every domain of SH3PXD2B.
Figure 7.
A. Structural insights into SH3PXD2B. B. Graphical representation of disordered regions in every domain of SH3PXD2B.
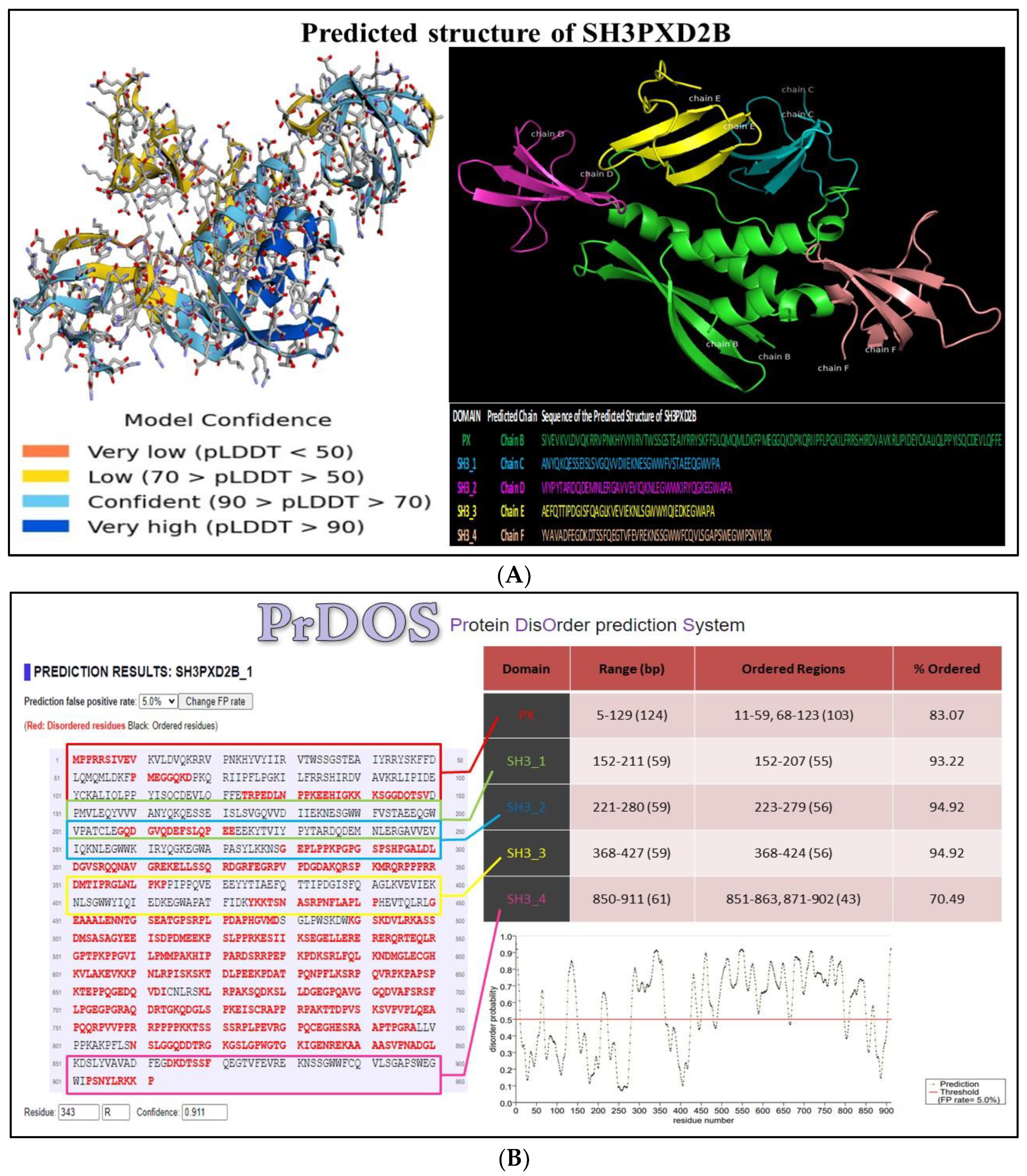
Figure 8.
(A). Graphical representation of binding energies of shortlisted compounds for MBC with SH3_2 and SH3_3 respectively. (B). Eribulin interaction with SH3_2 and SH3_3 domains of SH3PXD2B.
Figure 8.
(A). Graphical representation of binding energies of shortlisted compounds for MBC with SH3_2 and SH3_3 respectively. (B). Eribulin interaction with SH3_2 and SH3_3 domains of SH3PXD2B.
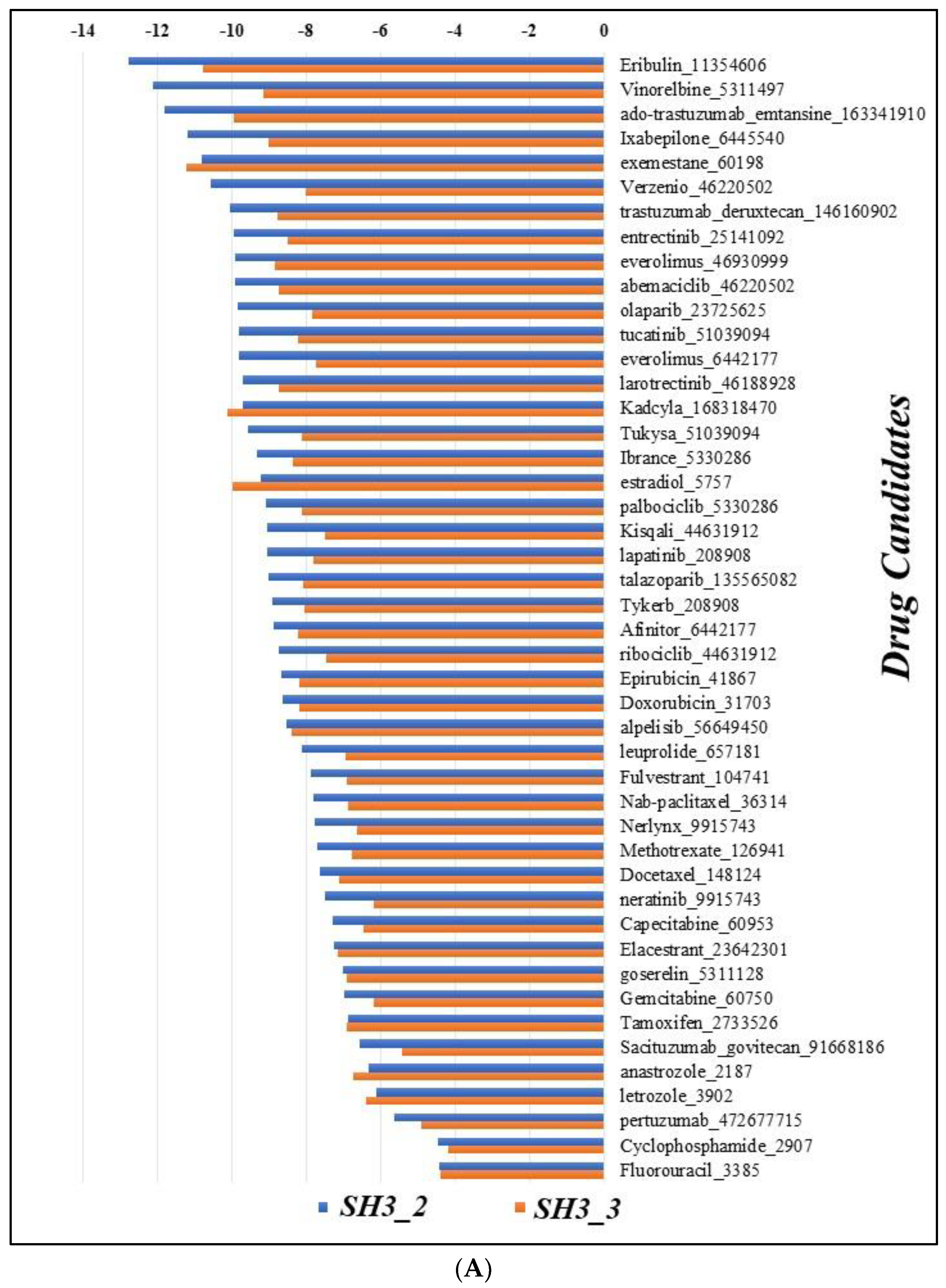
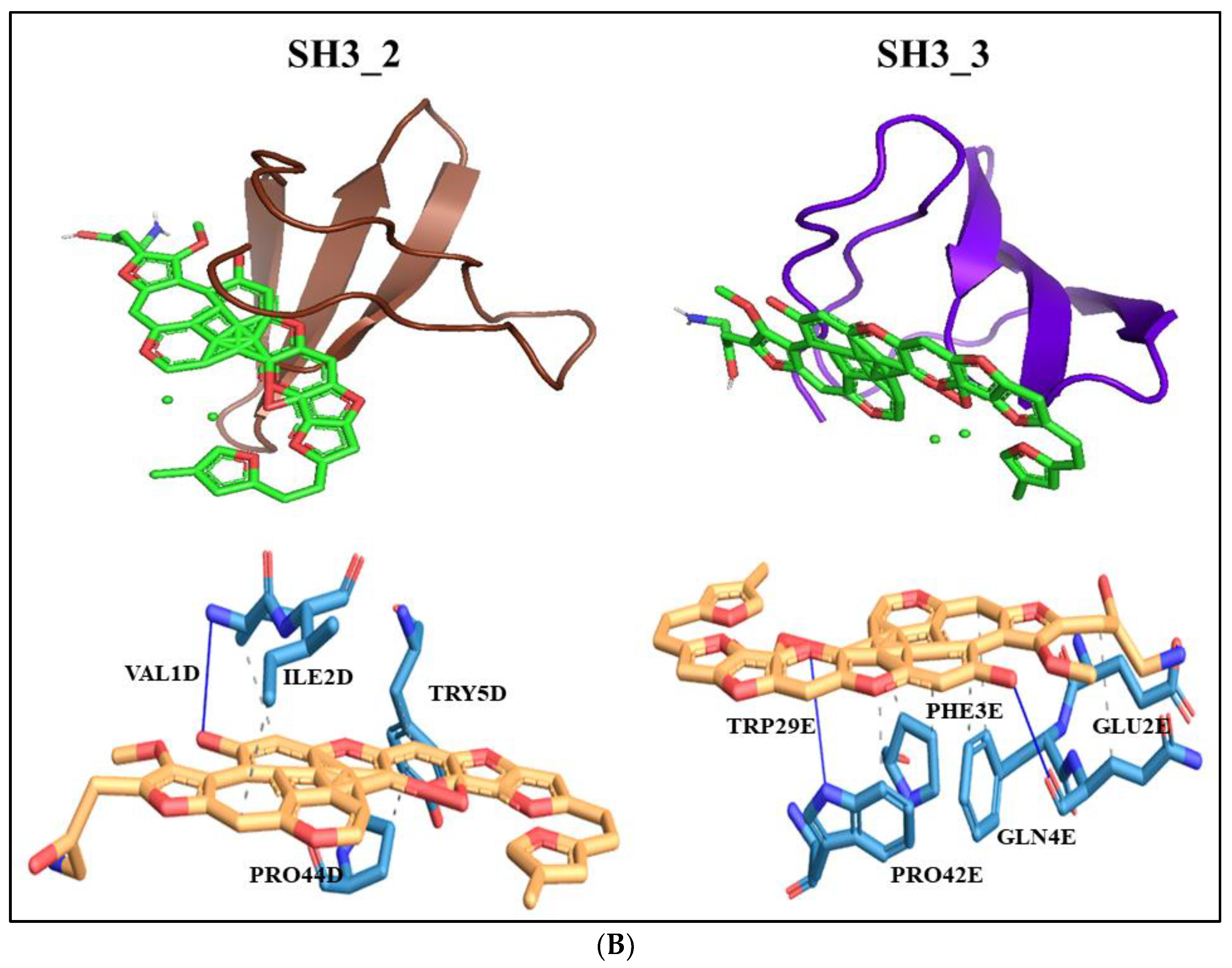
Figure 9.
a: Pathway Enrichment Analysis: (A) BiNGO – biological network sub – graph (B) Top hits of biological processes occurring in breast cancer lung metastasis. b: Proteomic based network analysis and pathway enrichment by DICE.
Figure 9.
a: Pathway Enrichment Analysis: (A) BiNGO – biological network sub – graph (B) Top hits of biological processes occurring in breast cancer lung metastasis. b: Proteomic based network analysis and pathway enrichment by DICE.
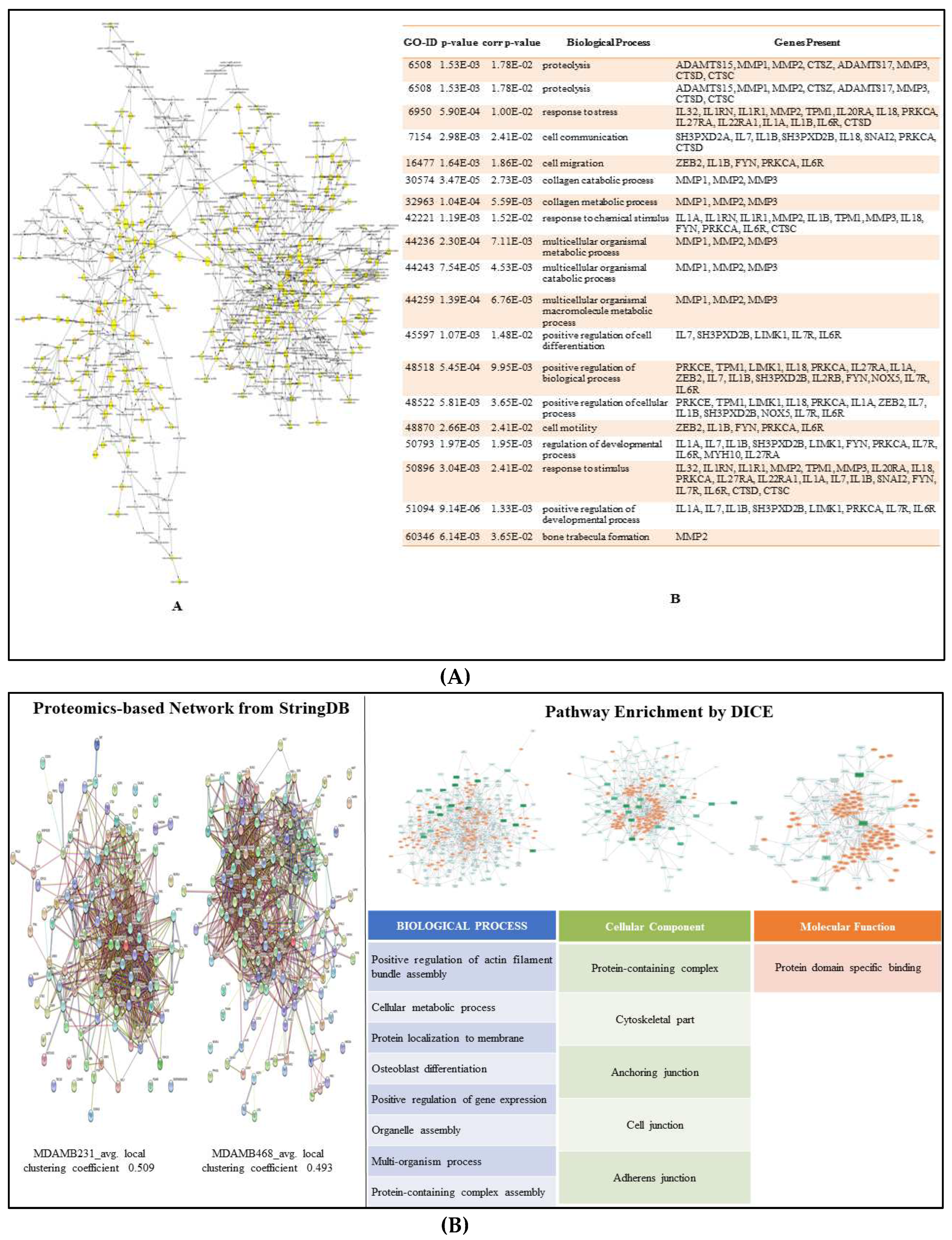
Figure 10.
Mechanistic model illustrating targeting of SH3PXD2B in BC-LM using multidisciplinary approach.
Figure 10.
Mechanistic model illustrating targeting of SH3PXD2B in BC-LM using multidisciplinary approach.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



