
Submitted:
13 September 2023
Posted:
14 September 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Coronavirus Disease 2019 (COVID-19) is an infectious respiratory illness caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The disease, first identified in the Chinese city of Wuhan in November 2019 and has since spread worldwide, is the latest human pandemic, and has officially infected over 800 million people and has caused nearly seven million deaths to date. Although SARS-CoV-2 belongs to the large family of coronaviruses, it has some unique biological characteristics in its interplay with the human host. Therefore, this narrative review aims to provide an up-to-date overview of the structure of the virus, incubation and shedding in the human host, infectivity and biological evolution over time, as well as the main mechanisms for invading human host cells and replicating within. We also proffer that ongoing epidemiological surveillance of newly emerged variants must always be accompanied by biological studies aimed at deciphering new advantageous traits that may contribute to increasing virulence and pathogenicity, such that the most appropriate strategies for establishing a (relatively) safe coexistence with the human host can be implemented.
Keywords:
COVID-19
; SARS-CoV-2
; Coronavirus
; Biology
; Genetics
1. Introduction
Coronaviruses are not new viruses to mankind, as coronavirus disease (COVID-19) is the third coronavirus outbreak to occur in the last 20 years, following severe acute respiratory syndrome (SARS) in 2002-2003 and Middle East respiratory syndrome (MERS) in 2012 [1].
The current epidemiology of COVID-19 is challenging to define, as it no longer seems possible to obtain reliable epidemiological figures at this time due to the significant burden of undertesting and underreporting [2], especially after the World Health Organization (WHO) finally decided to no longer define COVID-19 as a public health emergency of international concern [3]. Nonetheless, the COVID-19 dashboard from WHO paints an epidemiological picture of nearly 800 million official infection cases with up to 7 million COVID-related deaths [4]. These numbers are far below the more than 50 million deaths during the Spanish flu pandemic in 1918 and 1919 [1]. However, we can classify COVID-19 as one of the deadliest infectious diseases in human history.
An interesting analysis conducted through September 2022 and published in June 2023 by Jones et al. [5] found that over 96% of persons aged 16 years or older in the United States have SARS-CoV-2 antibodies derived from previous infection and vaccination. About 23% of these antibodies could be from infection alone, around 26% from vaccination alone, and nearly 48% from hybrid immunity. This means that over 70% of the entire U.S. population may have already been infected with SARS-CoV-2 at that time.
2. The pathogen
According to the International Committee on Taxonomy of Viruses [6], the responsible pathogen causing COVID-19 was recognized in a beta coronavirus belonging to the same family of viruses that can cause influenza-like syndromes and colds, which was eventually named Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). It is an enveloped virus with a single-stranded positive sense RNA genome composed of 13 to 15 open reading frames (ORFs), 12 of which are functional. Also, it contains four major structural proteins: spike (S, which includes the receptor-binding domain (RBD)), envelope (E), membrane (M), and nucleocapsid (N), along with additional genes encoding accessory proteins, including the RNA-dependent RNA polymerase (RdRp) (Figure 1) [7].
The total length of the SARS-CoV-2 genome is approximately 30,000 bases, making it one of the longest among the different virus types and having important consequences for proofreading during transcription, particularly with respect to the risk of introducing errors [8].
The key enzyme of all RNA viruses, both negative and positive-sense, is represented by the RdRp, which is essential for replicating viral RNA and transcription. In positive-sense single strand RNA viruses, such as SARS-CoV-2, the enzyme directly transcribes the positive-sense RNA, which acts exactly like a messenger RNA, but also double convert positive-sense RNA in negative-sense RNA and then again in positive-sense RNA, to be assembled in the final viral particle [9].
The spike protein of SARS-CoV-2 is composed of 1273 amino acids and contains two major subunits called S1 (between amino acids 14 and 685) and S2 (between amino acids 686 and 1273) [10]. The first subunit binds to the host cell receptors, while the second subunit contains the machinery responsible for fusion with the host membrane. Subunit S1 has three main domains: the N-terminal domain (between amino acids 14 and 305), the receptor binding domain (between amino acids 319 and 541), and a carboxy-terminal domain (between amino acids 542 and 681). Subunit S2 instead consists of the hydrophobic fusion peptide (between amino acids 788 and 806), the two heptapeptides repeat 1 (between amino acids 912 and 984) and repeat 2 (between amino acids 1163 and 1213), as well as the so-called transmembrane domain (between amino acids 1213 and 1237) and the cytoplasmic domain (between amino acids 1237 and 1273) (Figure 2).
In the native form, the spike protein forms a trimer on the viral surface, with the three protomers, each consisting of an S1 and S2 domain, determining the typical extracellular stalk and bulbous “crown.” After cleavage of the S1 subunit, the S2 subunit is prepared for fusion with the host cell membrane by its fusion peptide.
Overall, SARS-CoV-2 has a mean size of 91 nm and has a mean number of 24 spike proteins on its surface, approximately 10 nm apart [11]. Regarding the possible origin of this coronavirus, phylogenetic analysis with structural modeling of the spike protein of SARS-CoV-2 revealed that this protein shares a cumulative 97% sequence identity with that of another bat coronavirus and shares 97% sequence identity with a pangolin coronavirus in the receptor binding domain [12]. This may suggest that SARS-CoV-2 likely arose from the recombination of these two animal coronaviruses in an intermediate host and then infected humans as a result of a spill-over. SARS-CoV-2 also has 76% structural homology with SARS-CoV-1, the virus that caused the SARS outbreak nearly 20 years ago. However, the structural homology between these two viruses in the receptor binding domain is relatively low, less than 50%, which may contribute to explaining the same biological and clinical features between SARS and COVID-19.
Besides unsubstantiated hypotheses or other conspiracy theories that SARS-CoV-2 escaped or was created or replicated in a research laboratory, the animal spillover theory is the most widely accepted and is also consistent with what has happened in the past with other coronaviruses [13]. One possible theory is that SARS-CoV-2 likely originated from a bat coronavirus, perhaps BatCoV-RaTG13, that was transmitted to another intermediate animal, perhaps a pangolin, where the precursor virus underwent initial recombination within the animal. This new virus may then have been transmitted to the first human index case (where it became similar to the ancestral strain of SARS-CoV-2), which then caused an initial local outbreak. Since the initial spread of the virus in a likely limited human niche could not be detected in time, it is conceivable that further intra-human recombination would have occurred, eventually producing the highly virulent pathogen that caused the pandemic. This theory is accredited by phylogenetic studies, which revealed that the ancestral BatCoV-RaTG13 coronavirus, which probably appeared in 2013, already possessed a latent capacity to bind to human cellular targets. However, this potency has been completely unmasked only after the acquisition of successive mutations in the intermediate host and the human niche.
3. Biological characteristics of SARS-CoV-2 in humans
3.1. Incubation
Regarding incubation time, the different SARS-CoV-2 lineages that have gradually emerged since 2019 have heterogeneous biological characteristics, including potentially different incubation times. In particular, it seems evident that the incubation period has gradually shortened over time, from about 5 days for the ancestral strain to around 3 days for the Omicron lineages [14]. An important aspect that has emerged in several studies [15,16] is that the Omicron sublineages may be characterized by a higher viral load in the nasopharynx compared to the earlier strains, which may support the epidemiological evidence of higher transmissibility since SARAS-CoV-2 is a mainly airborne pathogen. The apparent consequence of the higher viral load of Omicron particles compared to other variants in the upper respiratory tract is that people infected with these lineages exhale a greater amount of viral particles during normal breathing, as shown in the study by Tan et al. [17], thus supporting the higher infectivity of Omicron.
3.2. Viral shedding
A comprehensive meta-analysis on the duration of viral shedding of ancestral SARS-CoV-2 in different categories of patients found that the pooled value was approximately 17 days and was longer in women, adults, patients with chronic diseases, persons with symptomatic infection, as well as in those undergoing steroid therapy. This duration was significantly shorter in recipients of the COVID-19 vaccine [18]. However, the situation has changed over time as the later emergence of different variants has been associated with a progressive decrease in the time infected patients remain positive, with the lowest duration of positivity reported for the Omicron variant (i.e., ~10 days) compared to Delta (i.e., ~12 days), Alpha (i.e., ~13 days), and Ancestral (i.e., ~17 days) [19].
It is important to recall that there is a very close relationship between viral load and infectivity in patients with SARS-CoV-2 infection, with both parameters following a different pattern than in patients infected with SARS-CoV-1 or MERS-CoV, in whom the peak of viral load and infectivity coincided with the onset of symptoms [20]. This is not true for SARS-CoV-2 because viral load increases substantially 3 to 5 days before the onset of symptoms, such that infected patients in the presymptomatic phase could be infectious, although to a lesser extent, because they would not cough or sneeze and thus would contribute less to the spread of airborne viral particles. However, suppose these individuals increase their exhaled air by loud talking, shouting, or singing. In that case, the number of viral particles emitted by these presymptomatic individuals becomes relevant to infectivity [20], considering that the range of the minimum infectious dose of SARS-CoV-2 is estimated to be between 1.26-7 ×106.25 plaque-forming units according to current evidence [21]. Paradigmatic is the case of a single, probably super-spreader singer who was able to infect nearly 86.7% of 61 choir rehearsal attendees after 2.5 hours of singing [22]. However, an important aspect to emphasize is that the positivity of molecular tests does not always equate to infectivity. As shown in a meta-analysis [23], the positivity of a molecular test can be as long as 25 to 29 days, whereas the duration of infectivity determined with viral culture is much shorter, ranging from 10 to 13 days. This is due to the fact that molecular tests amplify RNA and nonviable viral particles, i.e., cell-free RNA or nucleic material of the virus contained in degenerated endothelial cells, dysmorphic syncytial elements, pneumocytes, or dead neutrophil plugs [24]. A recent study has shown that viral RNA shedding in individuals infected with the Omicron lineage can last up to 8-12 days, with the probability of a positive viral culture becoming zero after day 10 [25].
3.3. Infectivity
The reproduction number is a mathematical term that indicates how contagious an infectious disease is, either at baseline (R0) or after changes in population immunity, prevention strategies, and other factors have affected transmission (Rt). Thus, R0 essentially expresses the average number of people likely to be infected by an already infected person. Hence, a value >1 means the virus will continue spreading among susceptible hosts [26]. Accurately determining the reproduction number of SARS-CoV-2 can be challenging, not only because of the variable means of virus transmission but also because it can vary widely depending on various factors such as individual susceptibility to infection, the emergence of new variants, and many environmental factors such as climate, geography, and lifestyle. Nevertheless, a meta-analysis estimated that the initial reproductive number of the SARS-CoV-2 prototype strain ranged from 2.8 to 3.8, with a mean value of around 3.3 [26].
As is well known and will be discussed in more detail later in this article, SARS-CoV-2 has undergone a whole series of mutations that have affected its biological properties, including its infectious potential and the consequent replication number. Each of the new variants of interest has been characterized by higher infectivity. As for the different variants that emerged, Delta was characterized by an R0 value of about 5, but the first Omicron lineages exhibited an almost threefold higher infectivity, raising the potential R0 value to around 15 [27], apparently more elevated than any other infectious virus that has challenged humanity so far [28]. Predictably, a slight increase in infectious potential was observed as new Omicron lineages emerged, with strains BA.2, 4, and 5 showing further increases. Notably, the recombinant sublines BQ.1 and XBB showed an additional 20-25% increase in reproductive numbers, translating to an impressive potential R0 of around 20-25 [29].
Besides structural changes that have enabled a higher affinity with its host cell receptors, the enhanced stability of the Omicron lineages compared to ancestral variants is a possible explanation for its increased infectivity. Stability has especially increased on common materials, especially paper [30], and at higher temperatures, leading to enhanced infectivity even during warmer periods [31].
3.4. The role of super-spreaders
The so-called superspreading events are a unique feature of the COVID-19 pandemic, i.e., circumstances in which certain individuals infect a disproportionate number of secondary cases compared with an “average” infectious person. Several aspects support the concept that these events play a noticeable role in the spread of the virus [32], e.g., the much higher number of infections compared with those expected from simple transmission by droplets or fomites, evidence of long-distance transmission, infection sustained by asymptomatic or presymptomatic persons who do not actively cough or sneeze, higher risk of transmission indoors than outdoors, reports of nosocomial infections occurred despite using strict contact- and-droplet containing measures, evidence of viable SARS-CoV-2 particles in the air, as well as in air filters and building ducts. Other evidence includes the fact that separately confined uninfected animals infected caged animals via air ducts, but also that no reliable study to date has disproved the hypothesis of SARS-CoV-2 transmission via air. Overall, it has been estimated that nearly 2% of cases infected with SARS-CoV-2 may be directly responsible for 20% of all infections [33]. This is supported by a recent study that showed that 11% of subjects emitted 86% of the total volume of airborne virus, most within three days of symptom onset, and that viral emissions correlated strongly with viral load in nasal swabs [34].
4. SARS-CoV-2 evolution
There is little doubt that SARS-COV-2 has undergone many mutations since its earlier appearance in Wuhan and will continue to do so while it remains among us. This is not surprising since viruses that encode their genomes directly in RNA, including HIV and influenza, rapidly insert mutations into their RNA because these microorganisms replicate within their hosts where enzymes that copy RNA are prone to error [35]. Before specifically addressing the important issue of SARS-CoV-2 variants, it is important to recall some basic concepts of natural selection [36]. First, variants of interest are defined as those that exhibit particular characteristics that warrant further evaluation. Variants of concern are defined as those with more specific and worrisome characteristics, such as increased transmissibility, association with more severe disease, or potential to affect diagnosis, therapy, or vaccine efficacy.
Regarding the origin of mutations, they are subject to two main pressures. Adaptive evolution is the process by which some advantageous non-synonymous substitutions become dominant in the viral sequence by positive selection. Viral strains carrying these mutations may have selection advantages, replacing earlier variants and becoming dominant over time [37]. Conversely, convergent evolution is the process by which favorable nonsynonymous substitutions in the viral sequence arise independently but simultaneously in different organisms, even in distant locations, due to positive selection because they provide a substantial advantage [37]. Therefore, these mutations will characterize independent and even phylogenetically distant viral lineages. A paradigmatic example is the E484K mutation within the spike protein, which occurs in lineages B.1.351, P.1, and B.1.1.7. These two aspects are not infrequently found in nature.
Thus, SARS-CoV-2 has mutated and will continue to do so for many important reasons, most notably to increase its affinity for host cell receptors or to evade the immune response, both of which may act synergistically to increase the likelihood of host cell infection. Most mutations are also triggered by natural or vaccine-induced immunity, as well as by antiviral drugs that contribute to the enrichment of the viral genome with mutations. This pressure leads to the emergence and disappearance of new SARS-CoV-2 variants over time [38]. At this point, it is worth recalling that in acute infection, viral load increases rapidly until the host immune response begins to clear the virus, whereupon viral load suddenly decreases. During this period, which is thus a combination of increasing and decreasing viral load, new mutations can evolve in the viral genome that can be incorporated and transmitted to other individuals [39]. Although recent data support the concept that the mutation rate of SARS-CoV-2 is many times (up to 20) lower than that of influenza virus [40], phylogenetic analyses have allowed the estimation that the SARS-CoV-2 genome undergoes nearly 8 mutations per month, mainly caused by random episodic increases in the substitution rate [41].
Numerous studies have helped to clarify the important intra-host evolution of SARS-CoV-2. In their analysis, Shen and colleagues found that the median number of intra-host variants of SARS-CoV-2 in infected patients ranged from 1 to 4, with a very wide range spanning between 0 to 51 in different patients [42]. In a subsequent study involving a large number of COVID-19 patients with persistent viral shedding [43], the authors found that nearly 93% of the samples collected from infected individuals contained at least one intra-host single nucleotide variant. Each sample contained a mean of approximately 20 intra-host single nucleotide variants, and the risk of developing intra-host single nucleotide variants was higher in older adults, male subjects, and individuals with prolonged viral shedding. In a subsequent investigation, the authors found the presence of intra-host single nucleotide variants in 68% of COVID-19 patients, with a median frequency of 1 and a range between 0 and 45. Up to 24% of these intra-host single nucleotide variants were synonymous, whereas 76% were instead non-synonymous. The S gene was that with the highest frequency of non-synonymous intra-host single nucleotide variants, followed by ORF1a, ORF1b, and N [44]. An even higher rate of intra-host single nucleotide variation was found in a large multicenter study conducted by Pathak et al. [45], with such variations occurring in up to 82% of all samples tested. Importantly, this analysis confirmed that the Delta and Kappa lineage-defining variations first developed as intra-host single nucleotide variations before becoming fixed in the population, thus confirming that this is the most likely pathway supporting the emergence of novel SARS-CoV-2 variants. Other interesting aspects have emerged from the study published by Laskar and colleagues [46]. Despite the large number of intra-host variants detected, consistent with previous data, the authors also found that the number of intra-host variations was higher in deceased patients (i.e., almost twice compared to asymptomatic patients). Not unexpectedly, the highest number of mutations involved the spike protein, followed by the nucleocapsid and NSP12 protein.
An important aspect of the emergence of new variants is the fact that the probability of occurrence of non-synonymous mutations and new variants seems to be influenced by the duration of active infection, thus suggesting that higher variability could be observed with longer viral persistence and replication in the human body [43]. Nevertheless, one myth that has since been dispelled is that vaccination can increase the likelihood of new variants evolving within the host. According to data published by Al-Khatib et al. [47], the number of quasispecies detected in vaccinated patients with breakthrough infections was almost identical-if not lower–than in unvaccinated patients with homologous infection with the same SARS-CoV-2 strain. Similarly, analysis of the number of intra-host variants in patients with infections by different SARS-CoV-2 strains did not reveal significant differences between unvaccinated and vaccinated individuals.
Another mechanism worth highlighting that leads to the emergence of new SARS-CoV-2 variants is intra-host recombination between two different SARS-CoV-2 variants [48]. The recombination events between the two lineages can thus generate a third viral lineage that may contain selective mutations of each original strain. This mechanism has likely become very common recently, as many individuals have become infected with multiple variants. It is likely the basis for the recent emergence of the so-called “recombinant strains” [49], which now include the XBB Omicron sublineages (probably derived by recombination of BA.2 with BA.2.75.2).
BA.2.86 is the most recent SARS-CoV-2 variant that WHO is intensively monitoring because it has a substantial number of mutations in the spike protein that may contribute to its immune escape potential. Four of these mutations (K417N, S477N, N501Y, and P681R, which exist only at BA.2.86) have raised serious concerns [50]. Interestingly, P681R is located at the furin cleavage site and is thought to enhance cleavage of the S1 and S2 subunits of the spike protein, leading to better penetration into host cells. This mutation was already present in the Delta variant and likely contributed to the wide distribution and the high pathogenetic potential of this former SARS-CoV-2 strain [50].
What is concerning now, especially with the emergence of XBB sublineages and BA.2.86, is that the level of antigenic difference compared to the original Wuhan strain is equivalent to the difference between SARS-CoV-1 and SARS-CoV-2, which has persuaded some authors to suggest the paradox to re-classify these novel variants as “SARS-CoV-3” [51,52].
However, from a clinical perspective, a comprehensive assessment of the clinical impact of the most prevalent SARS-CoV-2 variants over time has shown that the burden of hospitalizations and ICU admissions has decreased dramatically over time. This change can be attributed to a kaleidoscope of factors, such as more efficient and timely diagnosis of SARS-CoV-2 infection, better availability of human and technical resources in the healthcare system, improved prevention and therapeutic treatment, the spread of vaccine-triggered or natural immunity, but certainly also to an attenuation of the aggressiveness and pathogenicity of SARS-CoV-2 [53]. Very similar evidence has emerged from an England-based nationwide study, where it was found that the worst variant in terms of hospitalizations and deaths was the Alpha, followed by Delta, ancestral, and Omicron [54].
Post-mortem studies clearly demonstrate that the clinical burden of COVID-19 has varied widely throughout the pandemic. In a recent study, Schwab et al. reported that the rate of patients with SARS-CoV-2 infection who died directly from COVID-19 was higher during the predominance of the Alpha variant, intermediate during the waves dominated by the Delta and ancestral strains, and became the lowest (i.e., around 10%) after the emergence of the Omicron sublineages [55]. Accordingly, cumulative lung injury was the highest in patients who died from the Alpha and Delta variants compared with those who died from infection with the Omicron sublineages.
5. Human host cell penetration
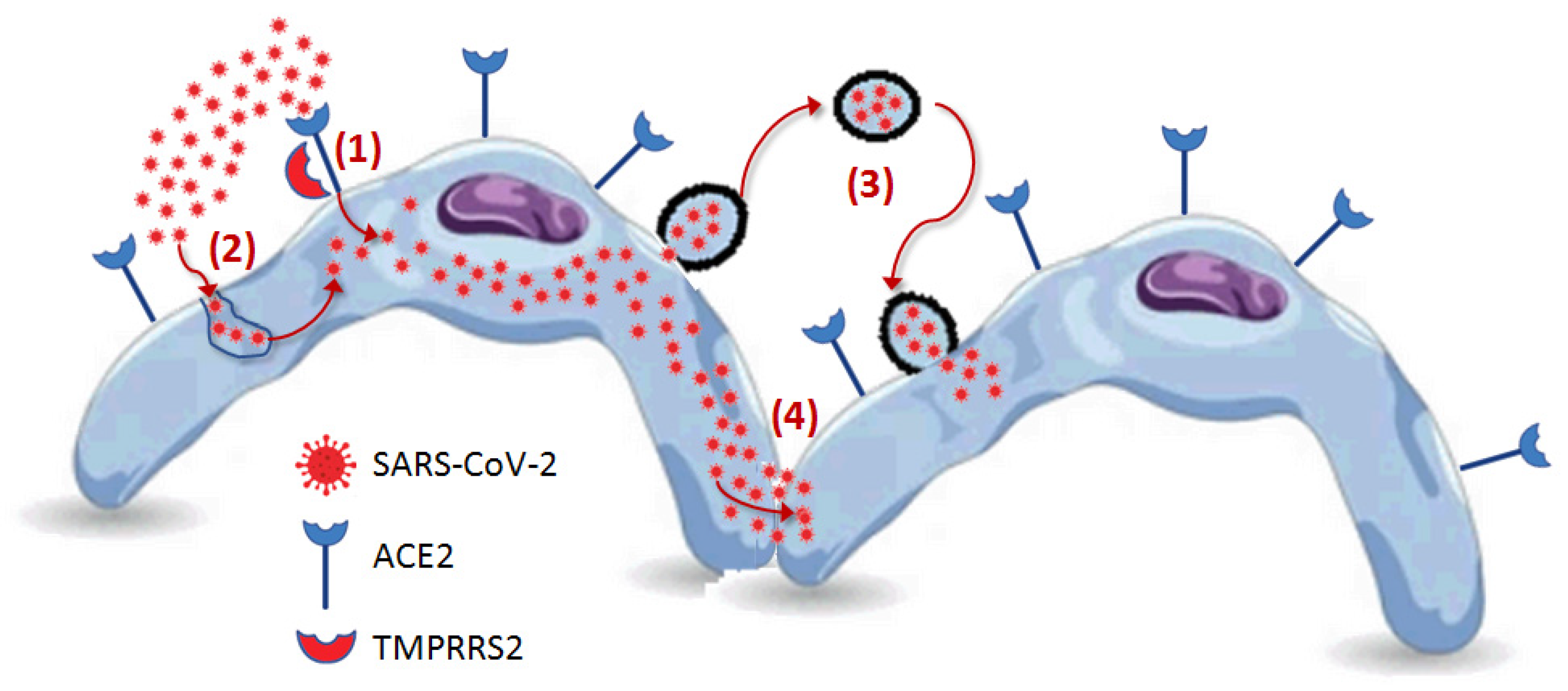
To date, at least four potential mechanisms for SARS-CoV-2 entry into host cells have been identified: direct attachment to receptors expressed on the surface of host cells, cathepsin-dependent endocytosis, fusion of SARS-CoV-2-bearing extracellular particles with the host cell membrane and cell-to-cell transmission [56] (Figure 3).
5.1. Receptor-mediated penetration
As for the mechanism of SARS-CoV-2 entry into host cells, it has now been clearly established that the major cellular receptor is angiotensin-converting enzyme 2 (ACE2), which is expressed on the surface of numerous human cells, such as respiratory epithelia, pneumocytes, and cells of the heart, kidney, gastrointestinal tract, and even testis. Binding between ACE2 and SARS-CoV-2 involves the spike protein, which must be “primed” by another protein on the cell surface, which may be transmembrane serine protease 2 (TMPRSS2) or other proteases may be involved. Once the virus is bound to ACE2, it becomes internalized within the cell and starts its replication process [57].
The essential role of TMPRSS2 is now undisputed, as this enzyme can increase the entry of SARS-CoV-2 into host cells by over 100-fold [57,58]. Interestingly, transmembrane serine protease 11 and 13 also have similar, albeit less pronounced, activity in promoting virus entry [58]. Molecular mimicry may be one of the main reasons for the high virulence and pathogenicity of SARS-CoV-2. Molecular mimicry is usually defined as an evolutionary strategy employed by viruses to exploit the host cellular machinery [59]. As for SARS-CoV-2, this coronavirus has evolved a unique S1/S2 cleavage site in its spike protein characterized by a four-amino acid insert not present in any previous sequenced coronavirus, resulting in a striking mimicry of an identical furin-cleavable peptide on the human epithelial sodium channel-α subunit. On the one hand, this explains the need for proteolytic activation by the host, but on the other, it also justifies the facilitated cell entry enabled by this sequence variation. Furin-mediated proteolysis of the spike protein of SARS-CoV-2 increases binding affinity with ACE2 by cleaving the spike protein into subunits S1 and S2, which remain non-covalently linked in the developing viral particle.
The importance of the furin cleavage site on the spike protein was confirmed by the study of Johnson et al. [60], in which it was shown that a mutant SARS-CoV-2 variant with deletion of the four unique amino acid insertions corresponding to the furin cleavage site was characterized by overall lower pathogenicity but identical immunogenicity compared to wild type, as evidenced by reduced infection of respiratory cells, attenuated disease in a hamster model of COVID-19, and generation of robust immunity to reinfection with the wild-type strain. This demonstrates that the unique insert at the S1 and S2 junction cleaved by furin likely enhances viral tropism in cells expressing active proteases and appropriate receptors.
According to recent findings, the interaction of SARS-CoV-2 with host cells seems to be further facilitated by a number of so-called “attachment cofactors," which essentially include heparan sulphate proteoglycans, phosphatidylserine receptor, Neuropilin-1, CD147, and even C-type lectins. In addition, several potential ACE2-independent SARS-CoV-2 receptors have also been identified. In particular, these include AXL (a tyrosine-protein kinase cellular receptor), KREMEN1 (a negative regulator of Wnt signaling involved in controlling cell survival), Asialoglycoprotein receptor-1 (a calcium-dependent C-type lectin receptor), LDLRAD3 (which promotes the activity of E3 ubiquitin ligases), and TMEM30A (also known as CDC50A, the beta subunit of phospholipid flippase that modulates the translocation of phospholipids on the plasma membrane) [61,62].
Neuropilin-1, in particular, is capable of binding other furin-cleaved substrates such as vascular endothelial growth factor A and class 3 semaphorins. Preliminary data suggest that co-expression of TMPRSS2 with either ACE2 or neuropilin-1 enhances infection, with ACE2 together with TMPRSS2 being twice as efficient as neuropilin-1 with TMPRSS2 [63]. As for leptins, an elegant study has recently demonstrated the existence of a lectin-assisted pathway of infection of host cells by SARS-CoV-2, which is particularly evident in those cellular elements with low surface expression of ACE2, supporting the theory that the virus may use lectins more or less as attachment receptors to facilitate further direct interaction with the ACE2 receptor, even on the surface of cells with low expression of this receptor [64]. This conclusion is supported by evidence that ectopic leptin expression was not associated with SARS-CoV-2 infection of ACE2-negative cells but also that anti-leptin antibodies to ACE2-negative cells were not effective in preventing infection of ACE2-expressing cells.
In summary, the main mechanism of cell penetration by SARS-CoV-2 is as follows [65]. SARS-CoV-2 first binds with the S1 subunits of its spike protein to ACE2 on the surface of host cells. This binding triggers conformational changes in the spike protein, which is converted to a prefusion state. The S2 subunit is then cleaved by TMPRSS2 or other host proteases, resulting in irreversible refolding into a post-fusion conformation that allows subsequent fusion between cell and viral membranes, allowing entry of the viral genome into the host cell. During the entry steps, the S1/S2 site of the spike protein is also cleaved by the intracellular enzyme furin for enhancing viral genome delivery into the infected cell.
5.2. Cathepsin L-mediated endocytosis
An intriguing aspect related to TMPRSS2 is that although its role appears to be essential for enhancing SARS-CoV-2 infection in cells actively expressing this enzyme, virus entry into enzyme-lacking cells appears to occur via a different pathway involving mainly viral endocytosis and sorting into endolysosomes, from where SARS-CoV-2 can enter the host cell via an acid-activated cathepsin L mechanism [66]. This mechanism has become particularly important after emergence of the Omicron lineages, as confirmed by the study of Iwata-Yoshikawa et al. [67], which showed that mice knockout for the gene encoding TMPRSS2 exhibited lower pathogenicity of the SARS-CoV-2 Gamma variant. At the same time, no similar evidence has emerged for infection Omicron lineages, thus implicitly implying that these variants also use alternative mechanisms to penetrate the host cells [67].
5.3. SARS-CoV-2 bearing extracellular particles
A third possible mechanism for host cell entry is extracellular SARS-CoV-2-bearing particles. As shown in the study by Kongsomros et al. [68], extracellular vesicles containing SARS-CoV-2 are regularly formed upon infection of human cells (mean particle diameter: 118±2 nm; concentration: 8.6±1.5×107 particles/ml), which could thus represent an important vehicle for virus propagation. In another study, Xia et al. showed that larger extracellular SARS-CoV-2-bearing microparticles (i.e., with a mean diameter between 1600 and 9500 nm) can be actively released from infected cells by a mechanism of infection-induced pyroptosis and are capable of actively infecting nearby naïve tissues by direct fusion with the plasma membrane of the cell [69]. There is clear evidence that SARS-CoV-2-containing microparticles are present in the bloodstream of infected individuals as early as one day before the onset of symptoms and can persist for up to four weeks, as shown by Liu et al. [70]. Notably, the presence of microparticles in the bloodstream between 20 and 60 days has been observed in individuals with persistent infection.
5.4. Cell to cell propagation
A fourth mechanism of infection has been postulated, involving cell-to-cell spread through endosomal membrane fusion [71]. This pathway of cell invasion appears to be independent of direct binding of the virus to cellular receptors. It is understandably more resistant to neutralizing antibodies, as it does not require extracellular spread of viral particles [71]. This specific mechanism of host cell infection was further explored in the work of Li et al. [72], who introduced the concept that cell-to-cell transmission of SARS-CoV-2 is facilitated by the expression of spike proteins on the surface of infected host cells, which would then bind to the ACE2 receptors on the plasma membrane of neighboring host cells and promote fusion with the cell membrane and the eventual formation of multinucleated cells or syncytia.
6. Intracellular processing
There are a number of multifaceted mechanisms for the intracellular processing of SARS-CoV-2, described in detail in the article by Martin-Sancho et al. [73] and will therefore only be briefly summarized here. These events can be divided into a series of sequential steps, starting with virus-cell interaction, followed by membrane fusion and release of viral RNA into the host cell, which is translated and replicated by the RdRp, as well as viral particle assembly, budding, and extracellular release. A single viral particle originating in the host cell is estimated to contain approximately 35-40 viral RNA–protein complexes consisting of approximately 12 nucleocapsid copies and 800 nucleotides of genomic RNA [74].
The lack of a timely and appropriate antiviral response is another critical mechanism related to the intracellular processing of SARS-CoV-2. In particular, the protein ORF6 and possibly other viral proteins such as NSP1, NSP3, NSP12, NSP13, NSP14, ORF3, and M appear to play a role in limiting early interferon production and associated downstream signaling [75]. Because the type I interferon system is an essential component of the innate immune response, neutralizing this pathway may be responsible for some of the virulence of SARS-CoV-2. This concept has been confirmed by other studies, such as that of Xia et al. [76], which showed that all three of the more lethal coronaviruses, SARS-CoV-1, SARS-CoV-2, and MERS-CoV, are capable of inhibiting the interferon I pathway with varying degrees of efficacy. Although some viral proteins effectively inhibit interferon I signaling, the NSP1 and NSP6 proteins of SARS-CoV-2 were characterized by higher efficiency than the homologous proteins of the other two coronaviruses.
7. Conclusions
Although the WHO has recently stated that COVID-19 should no longer be considered a global health emergency [77], it is predicted that SARS-CoV-2 will remain among the human species for many years to come [78], causing frequent, less lethal waves consisting of high-volume typically mild infections, and fostered by the incessant spread of new variants [79]. Therefore, a continued and comprehensive understanding of the evolving biology of SARS-CoV-2, particularly the multiple features that characterize its interplay with the human host, will be the cornerstone of future strategies to prevent or mitigate the adverse clinical, social, and economic consequences of COVID-19. Ongoing epidemiological surveillance of newly emerged variants must, therefore, be accompanied by biological studies aimed at deciphering new advantageous traits that may contribute to increasing virulence and pathogenicity so that the most appropriate strategies for establishing (relatively) safe coexistence with the human host can be implemented, similar to what has happened with other respiratory pathogens, particularly the influenza virus.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, G.L.; writing—original draft preparation, G.L.; writing—review and editing, F.S.G., C.M, B.M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sampath S, Khedr A, Qamar S, Tekin A, Singh R, Green R, Kashyap R. Pandemics Throughout the History. Cureus. 2021 Sep 20;13(9):e18136. [CrossRef] [PubMed] [PubMed Central]
- Lippi G, Mattiuzzi C, Henry BM. Uncontrolled confounding in COVID-19 epidemiology. Diagnosis (Berl). 2022 Dec 7;10(2):200-202. [CrossRef] [PubMed]
- Wise, J. Covid-19: WHO declares end of global health emergency. BMJ. 2023 May 9;381:1041. [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available at: https://covid19.who.int/. Last accessed: September 10, 2023.
- Jones JM, Manrique IM, Stone MS, Grebe E, Saa P, Germanio CD, Spencer BR, Notari E, Bravo M, Lanteri MC, Green V, Briggs-Hagen M, Coughlin MM, Stramer SL, Opsomer J, Busch MP. Estimates of SARS-CoV-2 Seroprevalence and Incidence of Primary SARS-CoV-2 Infections Among Blood Donors, by COVID-19 Vaccination Status - United States, April 2021-September 2022. MMWR Morb Mortal Wkly Rep. 2023 Jun 2;72(22):601-605. [CrossRef] [PubMed] [PubMed Central]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020 Apr;5(4):536-544. Epub 2020 Mar 2. [CrossRef] [PubMed] [PubMed Central]
- Lippi G, Mattiuzzi C, Bovo C, Plebani M. Current laboratory diagnostics of coronavirus disease 2019 (COVID-19). Acta Biomed. 2020 May 11;91(2):137-145. [CrossRef] [PubMed] [PubMed Central]
- Mariano G, Farthing RJ, Lale-Farjat SLM, Bergeron JRC. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front Mol Biosci. 2020 Dec 17;7:605236. [CrossRef] [PubMed] [PubMed Central]
- Machitani M, Yasukawa M, Nakashima J, Furuichi Y, Masutomi K. RNA-dependent RNA polymerase, RdRP, a promising therapeutic target for cancer and potentially COVID-19. Cancer Sci. 2020 Nov;111(11):3976-3984. Epub 2020 Sep 12. [CrossRef] [PubMed] [PubMed Central]
- Sakkiah S, Guo W, Pan B, Ji Z, Yavas G, Azevedo M, Hawes J, Patterson TA, Hong H. Elucidating Interactions Between SARS-CoV-2 Trimeric Spike Protein and ACE2 Using Homology Modeling and Molecular Dynamics Simulations. Front Chem. 2021 Jan 5;8:622632. [CrossRef] [PubMed] [PubMed Central]
- Ke Z, Oton J, Qu K, Cortese M, Zila V, McKeane L, Nakane T, Zivanov J, Neufeldt CJ, Cerikan B, Lu JM, Peukes J, Xiong X, Kräusslich HG, Scheres SHW, Bartenschlager R, Briggs JAG. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020 Dec;588(7838):498-502. Epub 2020 Aug 17. [CrossRef] [PubMed] [PubMed Central]
- Jaimes JA, André NM, Chappie JS, Millet JK, Whittaker GR. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J Mol Biol. 2020 May 1;432(10):3309-3325. Epub 2020 Apr 19. [CrossRef] [PubMed] [PubMed Central]
- Lippi G, Henry BM, Sanchis-Gomar F. The real origin of SARS-CoV-2: does it really matter? J Lab Precis Med 2021;6:9. [CrossRef]
- Wu Y, Kang L, Guo Z, Liu J, Liu M, Liang W. Incubation Period of COVID-19 Caused by Unique SARS-CoV-2 Strains: A Systematic Review and Meta-analysis. JAMA Netw Open. 2022 Aug 1;5(8):e2228008. Erratum in: JAMA Netw Open. 2022 Sep 1;5(9):e2235424. [CrossRef] [PubMed] [PubMed Central]
- Salvagno GL, Henry BM, Pighi L, De Nitto S, Montagnana M, Lippi G. SARS-CoV-2 Omicron infection is associated with high nasopharyngeal viral load. J Infect. 2022 Jun;84(6):834-872. Epub 2022 Feb 26. [CrossRef] [PubMed] [PubMed Central]
- Woodbridge Y, Amit S, Huppert A, Kopelman NM. Viral load dynamics of SARS-CoV-2 Delta and Omicron variants following multiple vaccine doses and previous infection. Nat Commun. 2022 Nov 7;13(1):6706. [CrossRef] [PubMed] [PubMed Central]
- Tan KS, Ong SWX, Koh MH, Tay DJW, Aw DZH, Nah YW, Abdullah MRB, Coleman KK, Milton DK, Chu JJH, Chow VTK, Tambyah PA, Tham KW. SARS-CoV-2 Omicron variant shedding during respiratory activities. Int J Infect Dis. 2023 Jun;131:19-25. Epub 2023 Mar 21. [CrossRef] [PubMed] [PubMed Central]
- Yan D, Zhang X, Chen C, Jiang D, Liu X, Zhou Y, Huang C, Zhou Y, Guan Z, Ding C, Chen L, Lan L, Fu X, Wu J, Li L, Yang S. Characteristics of Viral Shedding Time in SARS-CoV-2 Infections: A Systematic Review and Meta-Analysis. Front Public Health. 2021 Mar 19;9:652842. [CrossRef] [PubMed] [PubMed Central]
- Owens K, Esmaeili-Wellman S, Schiffer JT. Heterogeneous SARS-CoV-2 kinetics due to variable timing and intensity of immune responses. medRxiv [Preprint]. 2023 Aug 29:2023.08.20.23294350. [CrossRef] [PubMed] [PubMed Central]
- Marc A, Kerioui M, Blanquart F, Bertrand J, Mitjà O, Corbacho-Monné M, Marks M, Guedj J. Quantifying the relationship between SARS-CoV-2 viral load and infectiousness. Elife. 2021 Sep 27;10:e69302. [CrossRef] [PubMed] [PubMed Central]
- SeyedAlinaghi S, Karimi A, Mojdeganlou H, Pashaei Z, Mirzapour P, Shamsabadi A, Barzegary A, Afroughi F, Dehghani S, Janfaza N, Fakhfouri A, Khodaei S, Mehraeen E, Dadras O. Minimum infective dose of severe acute respiratory syndrome coronavirus 2 based on the current evidence: A systematic review. SAGE Open Med. 2022 Aug 11;10:20503121221115053. [CrossRef] [PubMed] [PubMed Central]
- Hamner L, Dubbel P, Capron I, Ross A, Jordan A, Lee J, Lynn J, Ball A, Narwal S, Russell S, Patrick D, Leibrand H. High SARS-CoV-2 Attack Rate Following Exposure at a Choir Practice - Skagit County, Washington, March 2020. MMWR Morb Mortal Wkly Rep. 2020 May 15;69(19):606-610. [CrossRef] [PubMed]
- Park M, Pawliuk C, Nguyen T, Griffitt A, Dix-Cooper L, Fourik N, Dawes M. Determining the communicable period of SARS-CoV-2: A rapid review of the literature, March to September 2020. Euro Surveill. 2021 Apr;26(14):2001506. [CrossRef] [PubMed] [PubMed Central]
- Ceulemans LJ, Khan M, Yoo SJ, Zapiec B, Van Gerven L, Van Slambrouck J, Vanstapel A, Van Raemdonck D, Vos R, Wauters E, Wauters J, Carmeliet P, Mombaerts P. Persistence of SARS-CoV-2 RNA in lung tissue after mild COVID-19. Lancet Respir Med. 2021 Aug;9(8):e78-e79. Epub 2021 Jun 9. [CrossRef] [PubMed] [PubMed Central]
- Takahashi K, Ishikane M, Ujiie M, Iwamoto N, Okumura N, Sato T, Nagashima M, Moriya A, Suzuki M, Hojo M, Kanno T, Saito S, Miyamoto S, Ainai A, Tobiume M, Arashiro T, Fujimoto T, Saito T, Yamato M, Suzuki T, Ohmagari N. Duration of Infectious Virus Shedding by SARS-CoV-2 Omicron Variant-Infected Vaccinees. Emerg Infect Dis. 2022 May;28(5):998-1001. Epub 2022 Mar 15. [CrossRef] [PubMed] [PubMed Central]
- Alimohamadi Y, Taghdir M, Sepandi M. Estimate of the Basic Reproduction Number for COVID-19: A Systematic Review and Meta-analysis. J Prev Med Public Health. 2020 May;53(3):151-157. Epub 2020 Mar 20. [CrossRef] [PubMed] [PubMed Central]
- Nishiura H, Ito K, Anzai A, Kobayashi T, Piantham C, Rodríguez-Morales AJ. Relative Reproduction Number of SARS-CoV-2 Omicron (B.1.1.529) Compared with Delta Variant in South Africa. J Clin Med. 2021 Dec 23;11(1):30. [CrossRef] [PubMed] [PubMed Central]
- Omer SB, Yildirim I, Forman HP. Herd Immunity and Implications for SARS-CoV-2 Control. JAMA. 2020 Nov 24;324(20):2095-2096. [CrossRef] [PubMed]
- Tamura T, Ito J, Uriu K, Zahradnik J, Kida I, Anraku Y, Nasser H, Shofa M, Oda Y, Lytras S, Nao N, Itakura Y, Deguchi S, Suzuki R, Wang L, Begum MM, Kita S, Yajima H, Sasaki J, Sasaki-Tabata K, Shimizu R, Tsuda M, Kosugi Y, Fujita S, Pan L, Sauter D, Yoshimatsu K, Suzuki S, Asakura H, Nagashima M, Sadamasu K, Yoshimura K, Yamamoto Y, Nagamoto T, Schreiber G, Maenaka K; Genotype to Phenotype Japan (G2P-Japan) Consortium; Hashiguchi T, Ikeda T, Fukuhara T, Saito A, Tanaka S, Matsuno K, Takayama K, Sato K. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat Commun. 2023 May 16;14(1):2800. [CrossRef] [PubMed] [PubMed Central]
- Hosseini M, Poon LLM, Chin AWH, Ducker WA. Effect of Surface Porosity on SARS-CoV-2 Fomite Infectivity. ACS Omega. 2022 May 23;7(22):18238-18246. [CrossRef] [PubMed] [PubMed Central]
- Mattiuzzi C, Henry BM, Lippi G. Regional Association between Mean Air Temperature and Case Numbers of Multiple SARS-CoV-2 Lineages throughout the Pandemic. Viruses. 2022 Aug 30;14(9):1913. [CrossRef] [PubMed] [PubMed Central]
- Greenhalgh T, Jimenez JL, Prather KA, Tufekci Z, Fisman D, Schooley R. Ten scientific reasons in support of airborne transmission of SARS-CoV-2. Lancet. 2021 May 1;397(10285):1603-1605. Epub 2021 Apr 15. Erratum in: Lancet. 2021 May 15;397(10287):1808. [CrossRef] [PubMed] [PubMed Central]
- Lau MSY, Grenfell B, Thomas M, Bryan M, Nelson K, Lopman B. Characterizing superspreading events and age-specific infectiousness of SARS-CoV-2 transmission in Georgia, USA. Proc Natl Acad Sci U S A. 2020 Sep 8;117(36):22430-22435. Epub 2020 Aug 20. [CrossRef] [PubMed] [PubMed Central]
- Zhou J, Singanayagam A, Goonawardane N, Moshe M, Sweeney FP, Sukhova K, Killingley B, Kalinova M, Mann AJ, Catchpole AP, Barer MR, Ferguson NM, Chiu C, Barclay WS. Viral emissions into the air and environment after SARS-CoV-2 human challenge: a phase 1, open label, first-in-human study. Lancet Microbe. 2023 Aug;4(8):e579-e590. Epub 2023 Jun 9. Erratum in: Lancet Microbe. 2023 Aug;4(8):e576. [CrossRef] [PubMed] [PubMed Central]
- Callaway, E. The coronavirus is mutating - does it matter? Nature. 2020 Sep;585(7824):174-177. [CrossRef] [PubMed]
- Corey L, Beyrer C, Cohen MS, Michael NL, Bedford T, Rolland M. SARS-CoV-2 Variants in Patients with Immunosuppression. N Engl J Med. 2021 Aug 5;385(6):562-566. [CrossRef] [PubMed] [PubMed Central]
- Losos, JB. Convergence, adaptation, and constraint. Evolution. 2011 Jul;65(7):1827-40. Epub 2011 Apr 7. [CrossRef] [PubMed]
- Boyle L, Hletko S, Huang J, Lee J, Pallod G, Tung HR, Durrett R. Selective sweeps in SARS-CoV-2 variant competition. Proc Natl Acad Sci U S A. 2022 Nov 22;119(47):e2213879119. Epub 2022 Nov 3. [CrossRef] [PubMed] [PubMed Central]
- Markov PV, Ghafari M, Beer M, Lythgoe K, Simmonds P, Stilianakis NI, Katzourakis A. The evolution of SARS-CoV-2. Nat Rev Microbiol. 2023 Jun;21(6):361-379. Epub 2023 Apr 5. [CrossRef] [PubMed]
- Kawasaki Y, Abe H, Yasuda J. Comparison of genome replication fidelity between SARS-CoV-2 and influenza A virus in cell culture. Sci Rep. 2023 Aug 11;13(1):13105. [CrossRef] [PubMed] [PubMed Central]
- Tay JH, Porter AF, Wirth W, Duchene S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol Biol Evol. 2022 Feb 3;39(2):msac013. [CrossRef] [PubMed] [PubMed Central]
- Shen Z, Xiao Y, Kang L, Ma W, Shi L, Zhang L, Zhou Z, Yang J, Zhong J, Yang D, Guo L, Zhang G, Li H, Xu Y, Chen M, Gao Z, Wang J, Ren L, Li M. Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin Infect Dis. 2020 Jul 28;71(15):713-720. Erratum in: Clin Infect Dis. 2021 Dec 16;73(12):2374. [CrossRef] [PubMed] [PubMed Central]
- Li J, Du P, Yang L, Zhang J, Song C, Chen D, Song Y, Ding N, Hua M, Han K, Song R, Xie W, Chen Z, Wang X, Liu J, Xu Y, Gao G, Wang Q, Pu L, Di L, Li J, Yue J, Han J, Zhao X, Yan Y, Yu F, Wu AR, Zhang F, Gao YQ, Huang Y, Wang J, Zeng H, Chen C. Two-step fitness selection for intra-host variations in SARS-CoV-2. Cell Rep. 2022 Jan 11;38(2):110205. Epub 2021 Dec 16. [CrossRef] [PubMed] [PubMed Central]
- Armero A, Berthet N, Avarre JC. Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses. 2021 Jan 19;13(1):133. [CrossRef] [PubMed] [PubMed Central]
- Pathak AK, Mishra GP, Uppili B, Walia S, Fatihi S, Abbas T, Banu S, Ghosh A, Kanampalliwar A, Jha A, Fatma S, Aggarwal S, Dhar MS, Marwal R, Radhakrishnan VS, Ponnusamy K, Kabra S, Rakshit P, Bhoyar RC, Jain A, Divakar MK, Imran M, Faruq M, Sowpati DT, Thukral L, Raghav SK, Mukerji M. Spatio-temporal dynamics of intra-host variability in SARS-CoV-2 genomes. Nucleic Acids Res. 2022 Feb 22;50(3):1551-1561. [CrossRef] [PubMed] [PubMed Central]
- Laskar R, Ali S. Differential mutation profile of SARS-CoV-2 proteins across deceased and asymptomatic patients. Chem Biol Interact. 2021 Sep 25;347:109598. Epub 2021 Jul 23. [CrossRef] [PubMed] [PubMed Central]
- Al-Khatib HA, Smatti MK, Ali FH, Zedan HT, Thomas S, Ahmed MN, El-Kahlout RA, Al Bader MA, Elgakhlab D, Coyle PV, Abu-Raddad LJ, Al Thani AA, Yassine HM. Comparative analysis of within-host diversity among vaccinated COVID-19 patients infected with different SARS-CoV-2 variants. iScience. 2022 Nov 18;25(11):105438. Epub 2022 Oct 25. [CrossRef] [PubMed] [PubMed Central]
- He Y, Ma W, Dang S, Chen L, Zhang R, Mei S, Wei X, Lv Q, Peng B, Chen J, Kong D, Sun Y, Tang X, Wu W, Chen Z, Li S, Wan J, Zou X, Li M, Feng T, Ren L, Wang J. Possible recombination between two variants of concern in a COVID-19 patient. Emerg Microbes Infect. 2022 Dec;11(1):552-555. [CrossRef] [PubMed] [PubMed Central]
- Mohapatra RK, Kandi V, Tuli HS, Chakraborty C, Dhama K. The recombinant variants of SARS-CoV-2: Concerns continues amid COVID-19 pandemic. J Med Virol. 2022 Aug;94(8):3506-3508. Epub 2022 Apr 27. [CrossRef] [PubMed] [PubMed Central]
- Scarpa F, Ciccozzi M. On the SARS-CoV-2 BA.2.86 lineage: a mutation point of view. J Med Virol. 2023 Sep;95(9):e29079. [CrossRef] [PubMed]
- Wang Q, Iketani S, Li Z, Liu L, Guo Y, Huang Y, Bowen AD, Liu M, Wang M, Yu J, Valdez R, Lauring AS, Sheng Z, Wang HH, Gordon A, Liu L, Ho DD. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell. 2023 Jan 19;186(2):279-286.e8. Epub 2022 Dec 14. [CrossRef] [PubMed] [PubMed Central]
- Mykytyn AZ, Fouchier RA, Haagmans BL. Antigenic evolution of SARS coronavirus 2. Curr Opin Virol. 2023 Aug 28;62:101349. Epub ahead of print. [CrossRef] [PubMed]
- Mattiuzzi C, Lippi G. Timeline analysis of clinical severity of COVID-19 in the general population. Eur J Intern Med. 2023 Apr;110:97-98. Epub 2022 Dec 16. [CrossRef] [PubMed] [PubMed Central]
- Perez-Guzman PN, Knock E, Imai N, Rawson T, Elmaci Y, Alcada J, Whittles LK, Thekke Kanapram D, Sonabend R, Gaythorpe KAM, Hinsley W, FitzJohn RG, Volz E, Verity R, Ferguson NM, Cori A, Baguelin M. Epidemiological drivers of transmissibility and severity of SARS-CoV-2 in England. Nat Commun. 2023 Jul 17;14(1):4279. [CrossRef] [PubMed] [PubMed Central]
- Schwab C, Merle U, Schirmacher P, Longerich T. Lethality of SARS-CoV-2 infection-a comparative autopsy study focusing on COVID-19 development and virus variants. Histopathology. 2023 Aug;83(2):242-251. Epub 2023 May 5. [CrossRef] [PubMed]
- Nocini R, Henry BM, Mattiuzzi C, Lippi G. Improving Nasal Protection for Preventing SARS-CoV-2 Infection. Biomedicines. 2022 Nov 17;10(11):2966. [CrossRef] [PubMed] [PubMed Central]
- Lippi G, Lavie CJ, Henry BM, Sanchis-Gomar F. Do genetic polymorphisms in angiotensin converting enzyme 2 (ACE2) gene play a role in coronavirus disease 2019 (COVID-19)? Clin Chem Lab Med. 2020 Jun 29;58(9):1415-1422. [CrossRef] [PubMed]
- Kishimoto M, Uemura K, Sanaki T, Sato A, Hall WW, Kariwa H, Orba Y, Sawa H, Sasaki M. TMPRSS11D and TMPRSS13 Activate the SARS-CoV-2 Spike Protein. Viruses. 2021 Feb 28;13(3):384. [CrossRef] [PubMed] [PubMed Central]
- Anand P, Puranik A, Aravamudan M, Venkatakrishnan AJ, Soundararajan V. SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife. 2020 May 26;9:e58603. [CrossRef] [PubMed] [PubMed Central]
- Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, Zou J, Zhang X, Juelich T, Smith JK, Zhang L, Bopp N, Schindewolf C, Vu M, Vanderheiden A, Winkler ES, Swetnam D, Plante JA, Aguilar P, Plante KS, Popov V, Lee B, Weaver SC, Suthar MS, Routh AL, Ren P, Ku Z, An Z, Debbink K, Diamond MS, Shi PY, Freiberg AN, Menachery VD. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. 2021 Mar;591(7849):293-299. Epub 2021 Jan 25. [CrossRef] [PubMed] [PubMed Central]
- Evans JP, Liu SL. Role of host factors in SARS-CoV-2 entry. J Biol Chem. 2021 Jul;297(1):100847. Epub 2021 May 28. [CrossRef] [PubMed] [PubMed Central]
- Lim S, Zhang M, Chang TL. ACE2-Independent Alternative Receptors for SARS-CoV-2. Viruses. 2022 Nov 16;14(11):2535. [CrossRef] [PubMed] [PubMed Central]
- Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T, Anastasina M, Smura T, Levanov L, Szirovicza L, Tobi A, Kallio-Kokko H, Österlund P, Joensuu M, Meunier FA, Butcher SJ, Winkler MS, Mollenhauer B, Helenius A, Gokce O, Teesalu T, Hepojoki J, Vapalahti O, Stadelmann C, Balistreri G, Simons M. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020 Nov 13;370(6518):856-860. Epub 2020 Oct 20. [CrossRef] [PubMed] [PubMed Central]
- Lempp FA, Soriaga LB, Montiel-Ruiz M, Benigni F, Noack J, Park YJ, Bianchi S, Walls AC, Bowen JE, Zhou J, Kaiser H, Joshi A, Agostini M, Meury M, Dellota E Jr, Jaconi S, Cameroni E, Martinez-Picado J, Vergara-Alert J, Izquierdo-Useros N, Virgin HW, Lanzavecchia A, Veesler D, Purcell LA, Telenti A, Corti D. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature. 2021 Oct;598(7880):342-347. Epub 2021 Aug 31. [CrossRef] [PubMed]
- Cai Y, Zhang J, Xiao T, Peng H, Sterling SM, Walsh RM Jr, Rawson S, Rits-Volloch S, Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020 Sep 25;369(6511):1586-1592. Epub 2020 Jul 21. [CrossRef] [PubMed] [PubMed Central]
- Koch J, Uckeley ZM, Doldan P, Stanifer M, Boulant S, Lozach PY. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021 Aug 16;40(16):e107821. Epub 2021 Jul 13. [CrossRef] [PubMed] [PubMed Central]
- Iwata-Yoshikawa N, Kakizaki M, Shiwa-Sudo N, Okura T, Tahara M, Fukushi S, Maeda K, Kawase M, Asanuma H, Tomita Y, Takayama I, Matsuyama S, Shirato K, Suzuki T, Nagata N, Takeda M. Essential role of TMPRSS2 in SARS-CoV-2 infection in murine airways. Nat Commun. 2022 Oct 15;13(1):6100. [CrossRef] [PubMed] [PubMed Central]
- Kongsomros S, Pongsakul N, Panachan J, Khowawisetsut L, Somkird J, Sangma C, Kanjanapruthipong T, Wongtrakoongate P, Chairoungdua A, Pattanapanyasat K, Newburg DS, Morrow AL, Hongeng S, Thitithanyanont A, Chutipongtanate S. Comparison of viral inactivation methods on the characteristics of extracellular vesicles from SARS-CoV-2 infected human lung epithelial cells. J Extracell Vesicles. 2022 Dec;11(12):e12291. [CrossRef] [PubMed] [PubMed Central]
- Xia B, Pan X, Luo RH, Shen X, Li S, Wang Y, Zuo X, Wu Y, Guo Y, Xiao G, Li Q, Long XY, He XY, Zheng HY, Lu Y, Pang W, Zheng YT, Li J, Zhang LK, Gao Z. Extracellular vesicles mediate antibody-resistant transmission of SARS-CoV-2. Cell Discov. 2023 Jan 6;9(1):2. [CrossRef] [PubMed] [PubMed Central]
- Ning B, Huang Z, Youngquist BM, Scott JW, Niu A, Bojanowski CM, Zwezdaryk KJ, Saba NS, Fan J, Yin XM, Cao J, Lyon CJ, Li CZ, Roy CJ, Hu TY. Liposome-mediated detection of SARS-CoV-2 RNA-positive extracellular vesicles in plasma. Nat Nanotechnol. 2021 Sep;16(9):1039-1044. Epub 2021 Jul 22. [CrossRef] [PubMed] [PubMed Central]
- Zeng C, Evans JP, King T, Zheng YM, Oltz EM, Whelan SPJ, Saif LJ, Peeples ME, Liu SL. SARS-CoV-2 spreads through cell-to-cell transmission. Proc Natl Acad Sci U S A. 2022 Jan 4;119(1):e2111400119. [CrossRef] [PubMed] [PubMed Central]
- Li X, Yuan H, Li X, Wang H. Spike protein mediated membrane fusion during SARS-CoV-2 infection. J Med Virol. 2023 Jan;95(1):e28212. Epub 2022 Oct 25. [CrossRef] [PubMed] [PubMed Central]
- Martin-Sancho L, Lewinski MK, Pache L, Stoneham CA, Yin X, Becker ME, Pratt D, Churas C, Rosenthal SB, Liu S, Weston S, De Jesus PD, O'Neill AM, Gounder AP, Nguyen C, Pu Y, Curry HM, Oom AL, Miorin L, Rodriguez-Frandsen A, Zheng F, Wu C, Xiong Y, Urbanowski M, Shaw ML, Chang MW, Benner C, Hope TJ, Frieman MB, García-Sastre A, Ideker T, Hultquist JF, Guatelli J, Chanda SK. Functional landscape of SARS-CoV-2 cellular restriction. Mol Cell. 2021 Jun 17;81(12):2656-2668.e8. Epub 2021 Apr 13. [CrossRef] [PubMed] [PubMed Central]
- Lu S, Ye Q, Singh D, Cao Y, Diedrich JK, Yates JR 3rd, Villa E, Cleveland DW, Corbett KD. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat Commun. 2021 Jan 21;12(1):502. [CrossRef] [PubMed] [PubMed Central]
- Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020 Jul 30;11(1):3810. [CrossRef] [PubMed] [PubMed Central]
- Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, Menachery VD, Rajsbaum R, Shi PY. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020 Oct 6;33(1):108234. Epub 2020 Sep 19. [CrossRef] [PubMed] [PubMed Central]
- Wise, J. Covid-19: WHO declares end of global health emergency. BMJ. 2023 May 9;381:1041. [CrossRef] [PubMed]
- Lippi G, Plebani M. COVID-19: the global health emergency is over for the WHO, but not yet for laboratory medicine. J Lab Precis Med 2023;8:17. [CrossRef]
- Callaway, E. COVID's future: mini-waves rather than seasonal surges. Nature. 2023 May;617(7960):229-230. [CrossRef] [PubMed]
- Ruaño G, Ha T. Living with respiratory viruses: The next saga in human/viral coexistence? Bioessays. 2021 Apr;43(4):e2000321. Epub 2021 Jan 6. [CrossRef] [PubMed] [PubMed Central]
Figure 1.
Genetic structure of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
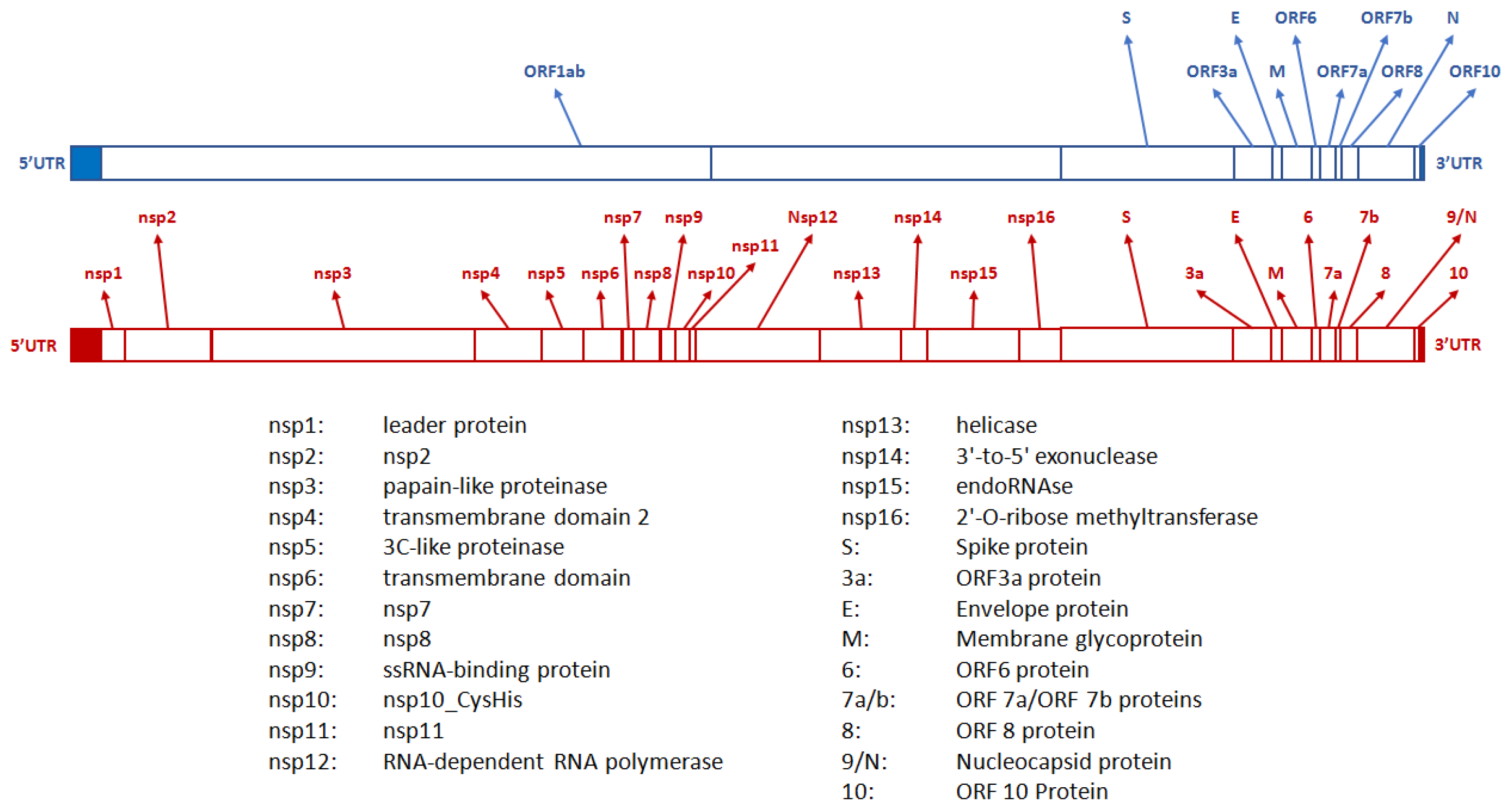
Figure 2.
Genetic structure of the gene encoding for the spike protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Figure 2.
Genetic structure of the gene encoding for the spike protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
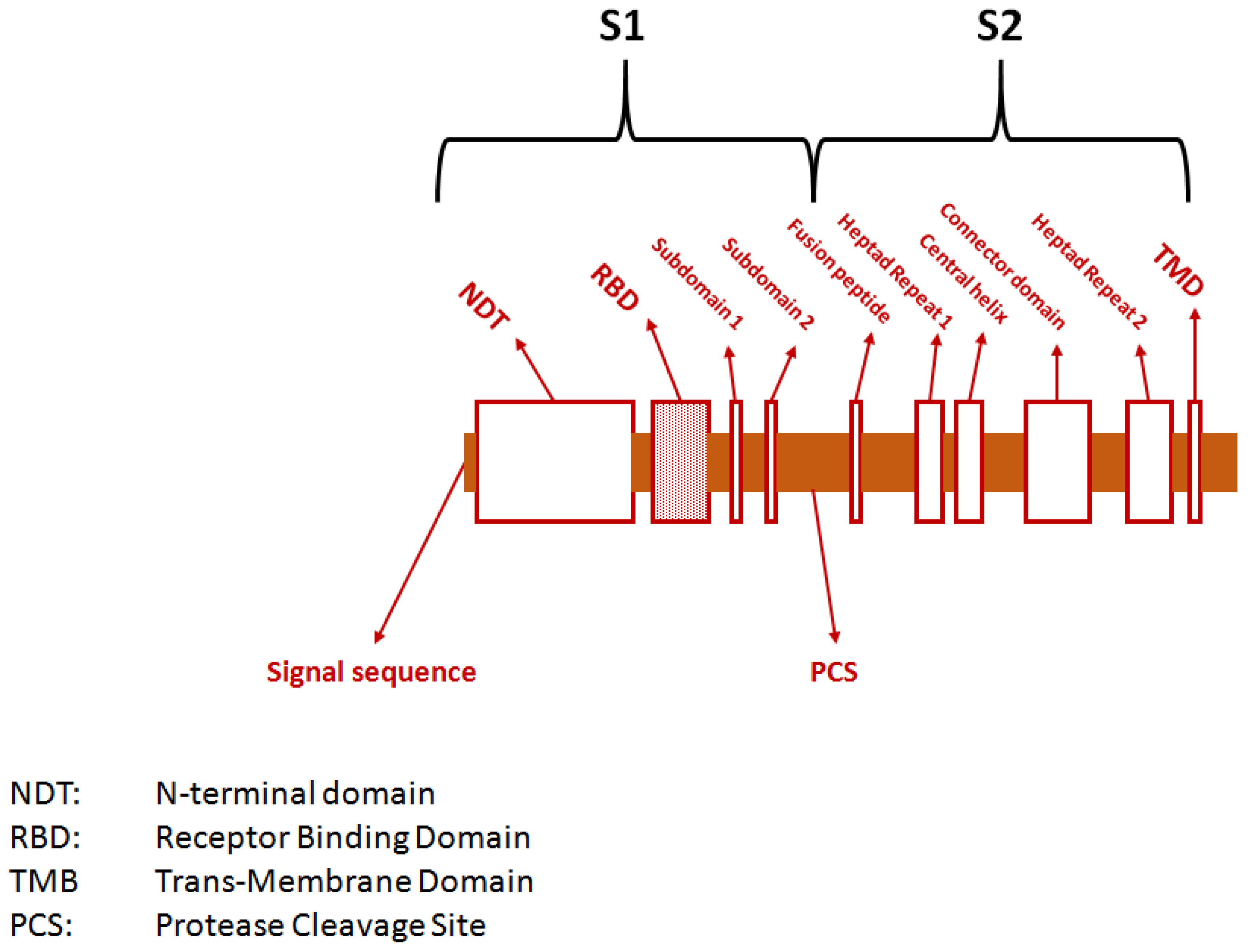
Figure 3.
Potential mechanisms for SARS-CoV-2 entry into host cells: (1) direct attachment to receptors expressed on the surface of host cells, (2) cathepsin-dependent endocytosis, (3) fusion of SARS-CoV-2-bearing extracellular particles with the host cell membrane, and (4) cell-to-cell transmission.
Figure 3.
Potential mechanisms for SARS-CoV-2 entry into host cells: (1) direct attachment to receptors expressed on the surface of host cells, (2) cathepsin-dependent endocytosis, (3) fusion of SARS-CoV-2-bearing extracellular particles with the host cell membrane, and (4) cell-to-cell transmission.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



