
Submitted:
15 September 2023
Posted:
18 September 2023
You are already at the latest version
Abstract
Metabolic-dysfunction associated steatotic liver disease (MASLD) is a chronic liver disease that affects more than a quarter of the global population, and is increasing worldwide, due to the pandemic of obesity. Insulin resistance is closely associated with the development and progression of MASLD. Hepatic entry of increased fatty acids (FA) released from adipose tissue, increase in FA synthesis and reduced FA oxidation in the liver, and hepatic overproduction of triglyceride (TG)-rich lipoproteins may induce the development of MASLD. Since insulin resistance also induces atherosclerosis, the leading cause for death in MASLD patients is cardiovascular disease (CVD). Considering that the development of CVD determines the prognosis of MASLD patients, the ideal therapeutic interventions for MASLD should reduce body weight, improve coronary risk factors, in addition to an improvement in liver functiom. Lifestyle modification such as diet and exercise and surgical interventions such as bariatric surgery and intragastric balloons have shown to improve MASLD by reducing body weight. Sodium glucose cotransporter 2 inhibitors (SGLT2i) and glucagon-like peptide-1 receptor agonists (GLP-1RA) have been shown to improve coronary risk factors and to suppress the occurrence of CVD. Both SGLT2i and GLP-1 have been reported to improve liver enzymes, hepatic steatosis and fibrosis. We recently reported that the selective peroxisome proliferator-activated receptor-alpha (PPARα) modulator, pemafibrate, improved liver function. PPARα agonists have multiple anti-atherogenic properties. Here, we consider the pathophysiology of MASLD and the mechanisms of action of such drugs, and consider whether such drugs and the combination therapy of such drugs could be the ideal treatments for MASLD.
Keywords:
cardiovascular disease
; fatty acids
; insulin resistance
; metabolic-dysfunction associated steatotic liver disease
; pemafibrate
; triglyceride
1. Introduction
Metabolic-dysfunction associated steatotic liver disease (MASLD) is a chronic liver disease that affects more than a quarter of the global population, and is increasing worldwide [1,2,3]. The pandemic of obesity and its cardio-metabolic consequences contribute to an increased prevalence of MASLD [4]. Approximately 20–30 % of MASLD patients develop metabolic-dysfunction associated steatohepatitis (MASH), leading to liver cirrhosis and associated complications, including hepatocellular carcinoma [5]. The disease burden from liver fibrosis due to MASLD is expected to increase around two to three-fold within decade worldwide. However, it is difficult to say that an effective therapeutic strategy for MASLD has been established.
MASLD is defined as the presence of hepatic steatosis (histological, imaging or blood biomarker evidence of hepatic steatosis) plus at least one of three metabolic criteria: overweight/obesity, established type 2 diabetes or the presence of metabolic dysregulation [6]. The latter is characterized by the presence of at least 2 metabolic abnormalities including an increase in waist circumference (WC), reduced high-density lipoprotein-cholesterol (HDL-C), hypertriglyceridemia, elevated blood pressure, prediabetes, elevation of homeostasis model assessment of insulin resistance (HOMA-IR) and high-sensitivity C-reactive protein (CRP) level [6]. The diagnostic criteria of MASLD is very similar to that of the metabolic syndrome. Insulin resistance greatly contributes to the development of MASLD and MASH.
It is very useful for the establishment of effective therapeutic strategies for MASLD to understand insulin resistance-induced metabolic disorders and its effects on liver, the underlying mechanisms that drugs improve insulin resistance and/or insulin resistance-induced metabolic disorders such as type 2 diabetes and atherogenic dyslipidemia. In short, such consideration can discover the promising therapeutic interventions for MASLD.
2. The effects of insulin resistance on the development of MASLD
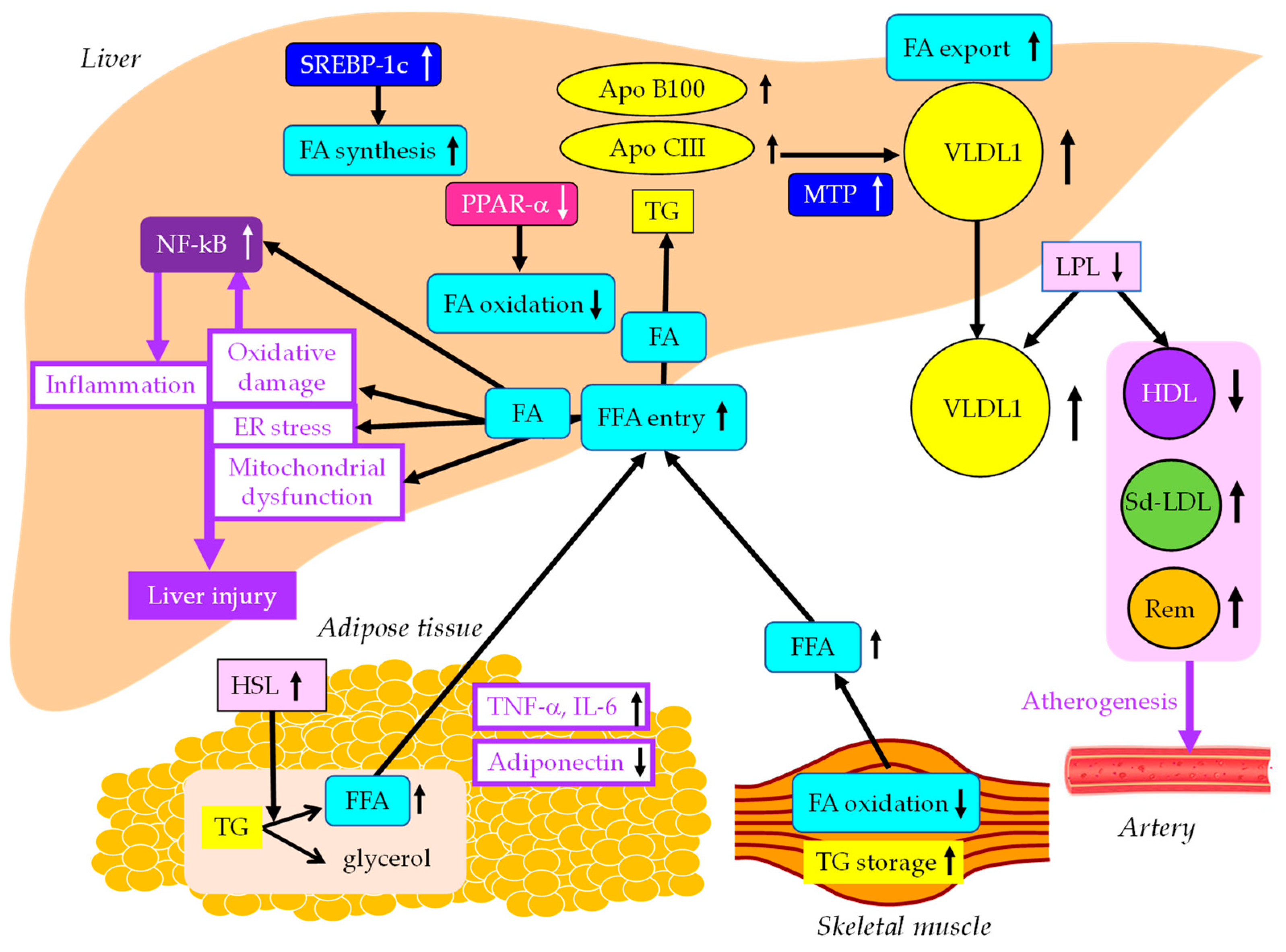
The effects of insulin resistance on the development of MASLD were shown in Figure 1. Accumulated visceral adipose tissue produces more inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6) and IL-1β, and less adiponectin, which induces systemic insulin resistance. The metabolism of free fatty acids (FFA) is altered in insulin resistance. The enzymes lipoprotein lipase (LPL) and hormone-sensitive lipase (HSL) are rate-limiting steps for FFA metabolism, because LPL hydrolyzes extracellular TG in lipoproteins and HSL hydrolyzes intracellular TG in adipocytes.
Insulin resistance enhances the expression and activity of HSL in adipose tissue. HSL catalyzes the hydrolysis of TG into FFA [7]. Insulin resistance is crossly associated with an excess TG storage within skeletal muscle [8]. Insulin resistance reduced FA oxidation, leading to diminished use of FA and storage of TG within skeletal muscle. Serum FFA increase due to increased release from the adipose tissue and decreased FA use in the skeletal muscle. Increased FFA enters the liver, leading to overproduction of TG-rich lipoprotein such as very-low-density lipoprotein (VLDL). Insulin resistance is associated with reduced apo B100 degradation [9], and is also associated with elevated hepatic apo CIII production [10], which increase VLDL because both apo B100 and apo CIII constitute VLDL. Insulin resistance increases expression of microsomal TG transfer protein (MTP), a key enzyme involved in VLDL assembly [9]. In an insulin-resistant-state, an increased FFA entry to liver, reduced degradation of apo B100 and enhanced expression of apo CIII and MTP may elevate hepatic production of VLDL. Insulin resistance also causes an increased expression of sterol regulatory element binding protein 1c (SREBP-1c), which increases FA synthesis [11]. Hepatic FA metabolism is regulated by a combination of FA uptake, FA export by VLDL secretion, de novo FA synthesis by SREBP-1c, and FA utilization by β-oxidation.
Two major physically distinct species of VLDL exist: larger TG-rich VLDL1 and smaller VLDL2 [12]. At normal TG concentrations, VLDL1 and VLDL2 circulate in approximately equal proportions. Hepatic TG accumulation and insulin resistance increase VLDL1 secretion [13,14]. MASH patients had more pronounced postprandial intestinal and hepatic VLDL1 accumulation, LDL lipid peroxidation and reduced total antioxidant status (TAS) [15]. Postprandial intestinal VLDL1 independently predicted oxidized LDL and TAS responses in MASH. Postprandial intestinal VLDL1 accumulation is associated with a pro-oxidant imbalance in MASH, and both correlate with the severity of liver disease. The Otsuka Long-Evans Tokushima Fatty (OLETF) rats showed overproduction of VLDL compared with the control rats [16]. In livers of OLETF rats, mRNA levels of TNF-α, IL-1β and IL-6 were increased, and mRNA, protein levels, and tyrosine phosphorylation of insulin receptor substrate 2 were decreased. Overproduction of VLDL in liver is significantly associated with hepatic oxidative stress, inflammation and insulin resistance. However, it remains unclear whether VLDL itself has the property of enhancing such exacerbating factors of liver fibrosis, or whether metabolic abnormalities which induce VLDL overproduction promote liver fibrosis.
Downstream of insulin signaling, the mechanistic target of rapamycin complex 1 (mTORC1), is a key regulator of lipid metabolism. Hepatic mTORC1 activity is elevated in mouse models with insulin-resistance and MASLD, but such activity is decreased in mouse models of MASH [17]. Genetic activation of mTORC1 in hepatocytes enhances lipid export from the liver by secreting VLDL while suppressing lipid synthesis to protect against MASH. In short, this study means that an increase in VLDL secretion is beneficial to prevent the development and progression of MASH. MTP is predominantly expressed in hepatocytes and enterocytes and is required for the assembly and secretion of VLDL. A rare causal variant in MTTP, encoding MTP, associated with progressive MASLD, unrelated to metabolic syndrome, was identified [18]. Hepatocyte-like cells derived from a homozygote donor had significantly lower MTP activity and lower lipoprotein apo B secretion than wild-type cells. Cytoplasmic TG accumulation in hepatocyte-like cells triggered endoplasmic reticulum (ER) stress, secretion of pro-inflammatory mediators, and production of reactive oxygen species (ROS). This MTTP variant was associated with progressive MASLD. Increased expression of MTP can be beneficial for the protection against MASLD.
FA oxidation primarily occurs in the mitochondria; however, FA oxidation commences in the peroxisomes and then is finally processed in the mitochondria [19]. In obesity, ω-oxidation by cytochrome P450 enzymes also contributes to FA oxidation. This pathway for FA oxidation generates large amounts of ROS [20]. The entry of FA into mitochondria depends on carnitine palmitoyl-transferase 1 (CPT-1). One of the major regulators of CPT-1 is the peroxisome proliferator-activated receptor-α (PPARα) [21,22,23,24]. Activation of PPARα induces transcription of genes related to FA oxidation [21,25,26]. Visceral adiposity and insulin resistance are negatively correlated with liver PPARα gene expression [26].
Overexpression of apo CIII, independent of a high-fat diet (HFD), produces MASLD-like features, including increased liver lipid content; decreased antioxidant capacity; increased expression of TNFα, IL-1β; decreased expression of adiponectin receptor [27]. HFD induced hepatic insulin resistance, marked increases in plasma TNFα (8-fold) and IL-6 (60%) in apo CIII overexpressing mice. Cell death and apoptosis were augmented in apo CIII overexpressing mice regardless of diet. Fenofibrate treatment reversed several of the effects associated with diet and apo CIII expression but did not normalize inflammatory traits even when liver lipid content was fully corrected. An increase in apo CIII plays a major role in liver inflammation and cell death in MASLD. There were no reports on adverse effects of apo CIII deficiency on MASLD, and increased apo CIII is thought to adversely affect MASLD.
An increase in FFA leads to hepatic insulin resistance by interacting with insulin signaling [28,29]. The anti-lipolytic function of insulin is impaired in insulin resistance, which may facilitate hepatic TG synthesis. Saturated FA (SFA) are stored as lipid droplets, transferred into mitochondria for β-oxidation, and secreted into blood as VLDL [30]. SFA generate lipotoxic intermediate products, such as diacylglycerols [31]. Lipotoxic intermediate products cause ER stress and ROS formation, which is a major factor in the pathogenesis of MASH [30,32]. By binding to Toll-like receptor 4, SFA induce augmentation of mitochondrial dysfunction and activation of pro-inflammatory nuclear factor-kappa B (NF-κB) [30].
Figure 1.
The effects of insulin resistance on the development of MASLD. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Solid black lines indicate the flow of substances. Purple solid lines indicate unfavorable effects on liver and artery. FA, fatty acids; FFA, free fatty acids; HDL, high-density lipoprotein; HSL, hormone sensitive lipase; IL-6, interleukin-6; LPL, lipoprotein lipase; MTP, microsomal triglyceride transfer protein; NF-κB, nuclear factor-kappa B; PPARα, peroxisome proliferator-activated receptor-α; Rem, remnant lipoproteins; Sd-LDL, small dense low-density lipoprotein; SREBP-1c, sterol regulatory element binding protein 1c; TG, triglyceride; TNF-α, tumor necrosis factor alpha; VLDL, very LDL.
Figure 1.
The effects of insulin resistance on the development of MASLD. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Solid black lines indicate the flow of substances. Purple solid lines indicate unfavorable effects on liver and artery. FA, fatty acids; FFA, free fatty acids; HDL, high-density lipoprotein; HSL, hormone sensitive lipase; IL-6, interleukin-6; LPL, lipoprotein lipase; MTP, microsomal triglyceride transfer protein; NF-κB, nuclear factor-kappa B; PPARα, peroxisome proliferator-activated receptor-α; Rem, remnant lipoproteins; Sd-LDL, small dense low-density lipoprotein; SREBP-1c, sterol regulatory element binding protein 1c; TG, triglyceride; TNF-α, tumor necrosis factor alpha; VLDL, very LDL.
3. The association of MASLD with cardiovascular diseases (CVD)
A retrospective analysis of 619 patients diagnosed with MASLD that CV events (38.3%) followed by non-liver malignancy (18.7%), and complications of liver cirrhosis (7.8%) were the three most common causes of death in MASLD patients [33], suggesting that CV events was the most crucial determinant of mortality of MASLD patients. The meta-analysis showed that MASLD was significantly associated with an increase in the development of CVD (odds ratio [OR], 2.05; 95% confidence interval [95%CI], 1.81 to 2.31; p < 0.0001) [34]. However, MASH has a higher liver-related (OR for MASH, 5.71; 95%CI, 2.31 to 14.13; OR for MASH with advanced fibrosis, 10.06; 95%CI, 4.35 to 23.25), but not cardiovascular mortality (OR, 0.91; 95%CI, 0.42 to 1.98). Therefore, MASLD can be said to be a high-risk group for CVD as well as a high-risk group for developing MASH.
A multicenter large retrospective study showed body mass index (BMI) in subjects with MASLD was significantly higher than that in those without MASLD (p < 0.01) [35]. The prevalence of MASLD showed a linear increase with the increase of BMI (BMI < 23 kg/m2, 10.5%; BMI ≥ 23 kg/m2 and < 25 kg/m2, 37.9%; BMI ≥ 25 kg/m2 and < 28 kg/m2, 58.4%; BMI ≥ 28 kg/m2, 84.2%) [35]. In short, a 7.4–11.4% increase of the prevalence of MASLD per 1 kg/m2 of BMI was observed. The prevalence of MASLD showed a linear increase with the increase of serum TG and LDL-C, and a linear decrease with the increase of HDL-C. The prevalence of MASLD was 22.8% in subjects with normal TG levels (< 150 mg/dL) and 59.5% in subjects with hypertriglyceridemia (> 150 mg/dL). The prevalence of MASLD was 27.3% in subjects with normal HDL-C levels (> 40 mg/dL) and 61.7% in subjects with hypo-HDL-C (<40 mg/dL). The prevalence of MASLD was 26.4% in subjects with normal LDL-C (< 140 mg/dL) and 38.5% in subjects with hyper-LDL-C (> 140 mg/dL).
An increased production of VLDL observed in MASLD is caused by insulin resistance as described above, and insulin resistance reduces the degradation of VLDL in the blood (Figure 1). Insulin resistance adversely affects enzymes such as LPL and hepatic TG lipase (HTGL), leading to conditions that are highly atherogenic, such as a decrease in HDL and increases in small-dense LDL (Sd-LDL) and remnant lipoproteins [36,37]. Insulin resistance reduces LPL activity. LPL is the rate-limiting enzyme for the catabolism of TG-rich lipoproteins such as VLDL [38]. The formation of HDL is related with the catabolism of TG-rich lipoproteins by LPL [39]. Therefore, reduced LPL activity increases VLDL, and reduces HDL. The activity of HTGL, the enzyme that facilitates the catabolism of HDL, is correlated with insulin resistance [40]. Low serum HDL-C may be partially due to an increased rate of clearance by HTGL [40]. LDL size are inversely proportional to HTGL activity [41], and patients with high HTGL have more Sd-LDL, as compared with subjects with low HTGL activity [42]. Increased HTGL activity due to insulin resistance may increase atherogenic lipoprotein, Sd-LDL. Remnant lipoproteins have undergone extensive intravascular remodeling. LPL, HTGL, and cholesterol ester transfer protein (CETP) induce structural and atherogenic changes that distinguish remnant lipoproteins from non-remnant lipoproteins [43]. Via the LPL-mediated removal of TG and CETP-mediated exchange of TG for cholesterol from LDL and HDL, remnant lipoproteins contain more cholesterol than nascent VLDL [44].
HDL plays a role in reverse cholesterol transport from atherosclerotic plaque which is an anti-atherogenic effect. Therefore, reduced HDL induces an atherogenic status. Since Sd-LDL is not recognized by LDL receptor, Sd-LDL stays in blood for a longer period. Sd-LDL is likely to be adhesive to endothelium and migrate into subendothelial space and lacks anti-oxidative capacity. Sd-LDL has multiple atherogenic properties. Remnant lipoproteins are up-taken by macrophages without modification such as oxidation, which is highly atherogenic property.
Weight reduction and an improvement in atherogenic lipoproteins are important to improve the prognosis of MASDL patients.
4. The therapeutic approaches for MASLD-lifestyle modification and surgical interventions
4.1. lifestyle modification
4.1.1. Diet
Weight loss by lifestyle modification is the cornerstone therapy of MASLD. Low carbohydrate diet has showed favorable effects for body weight as well as hepatic fat content in several reports. In the meta-analysis, there was no significant difference between low carbohydrate diet group and low fat diet group on the improvement of hepatic fat content and liver enzymes in MASLD [45]. In the meta-analysis of 8 randomized clinical trials (RCTs), the Mediterranean and hypocaloric dietary interventions favoring unsaturated FA result in improvements in intrahepatic lipid content and liver enzymes in patients with MASLD [46]. Another meta-analysis showed that calorie-restricted interventions had favourable effects on alanine aminotransferase (ALT) (p < 0.001), hepatic steatosis (p < 0.001) and liver stiffness (p = 0.009) [47]. The Mediterranean diet reduced ALT (p = 0.02), Fatty Liver Index (p < 0.001) and liver stiffness (p = 0.05). There was a dose-response relationship between degree of calorie restriction and beneficial effects on liver function and weight loss.
Intermittent fasting, which includes alternate-day fasting, and other forms of periodic caloric restriction have already received attention from animal research scientists [48,49]. It has been shown that fasting may benefit weight management and improve cardiovascular and metabolic risks [50]. In the meta-analysis, there were significant differences in body weight, BMI, ALT, and aspartate aminotransferase (AST) between the control and intermittent fasting group [51]. In another meta-analysis, body weight, BMI, and waist to hip ratio were significantly improved following the intermittent fasting intervention (p < 0.05) [52]. Adults with MASLD showed an improvement in serum ALT, AST, hepatic steatosis and hepatic stiffness measured by vibration-controlled transient elastography after intermittent fasting intervention (p < 0.05).
4.1.2. Exercise
Physical activity, independently from diet change, was associated with a significant reduction in intrahepatic lipid content and with reductions in ALT and AST [53]. Individuals with increasing BMI to be increasingly more likely to benefit from the intervention. Compared to standard care, exercise improved serum ALT, AST and intrahepatic fat [54]. Exercise was associated with a significant reduction in visceral (p < 0.001), subcutaneous (p < 0.001) and intrahepatic fat (p < 0.001), as well as gamma-glutamyl transferase (GGT) (p < 0.001) in pediatric obesity [55]. Supervised-exercise significantly reduced hepatic fat content compared to the control groups in youth [56]. Exercise training for about 12 weeks induced an absolute reduction in intrahepatic TG of 3.31% (95%CI, -4.41 to -2.2) [57]. Exercise reduces intrahepatic TG independent of significant weight change (-2.16%; 95%CI, -2.87 to -1.44), but benefits are substantially greater when weight loss occurs (-4.87%; 95%CI, -6.64 to -3.11). Furthermore, meta-regression identified a positive association between percentage weight loss and absolute reduction in intrahepatic TG (β, 0.99; 95%CI, 0.62 to 1.36; p < 0.001). Furthermore, exercise training also improves hepatic insulin sensitivity.
4.1.3. Diet and exercise
In the meta-analysis including RCTs assessed the effect of lifestyle-induced weight loss in MASLD, although a ≥ 5% weight loss improved hepatic steatosis, a ≥ 7% weight loss also improved non-alcoholic fatty liver disease (NAFLD) activity score (NAS), which is the sum of steatosis, hepatocellular ballooning and lobular inflammation, however, fibrosis was unchanged [58]. Interventions combining exercise and diet showed decrease in ALT (p < 0.01) and improvement in NAS [54]. In a systematic review and meta-analysis which assessed the effect of lifestyle changes on metabolic parameters in patients with MASLD, compared to conventional treatment, combined exercise with diet seems to elicit greater reductions in ALT (mean difference [MD], -13.27; 95%CI, -21.39 to -5.16), AST (MD, -7.02; 95%CI, -11.26 to -2.78) and HOMA-IR (MD, -2.07; 95%CI, -2.61 to -1.46) than diet (ALT MD, -4.48; 95%CI, -1.01 to -0.21; HOMA-IR MD, -0.61; 95%CI, -1.01 to -0.21) and exercise (ALT and AST non-significant; HOMA-IR MD, -0.46; 95% CI, -0.8 to -0.12) alone [59].
4.2. Surgical interventions
4.2.1. Bariatric surgery
Bariatric surgery has an important role in managing obesity. It can achieve significant weight loss, normalisation of glucose tolerance [60], and reduce cardiovascular risk and long-term mortality [61,62]. Bariatric surgery is associated with a significant reduction in the weighted incidence of a number of histological features of MASLD including steatosis, fibrosis, hepatocyte ballooning and lobular inflammation [63].
4.2.2. Intragastric balloons
Intragastric balloons are safe and effective in inducing weight loss in obese patients. In the meta-analysis, ALT decreased by -10.02 U/L (95 %CI, -13.2 to -6.8), GGT decreased by -9.82 U/L (95 %CI, -12.9 to -6.8), and BMI decreased by -4.98 kg/m2 (95%CI, -5.6 to -4.4) with intragastric balloons therapy [64]. Hepatic steatosis by evaluated by magnetic resonance imaging (MRI) was improved from baseline after 6 months of balloon therapy. Histological NAS was lower after 6 months of intragastric balloons versus control with sham endoscopy and diet (p = 0.03). In another meta-analysis, an improvement in steatosis was seen in 79.2% of patients, and NAS and HOMA-IR were improved in 83.5% and 64.5% of MASLD patients, respectively [65]. A reduction in liver volume by computed tomography (CT) scan was noticed in 93.9% of patients undergoing intragastric balloons placement.
5. Pharmacological interventions for MASLD
Considering that the development of CV events determines the prognosis of MASLD patients [33], the ideal therapeutic agents for MASLD should reduce body weight, improve coronary risk factors, and, if possible, reduce CV events, in addition to improving liver function. Sodium glucose cotransporter 2 inhibitors (SGLT2i) and glucagon-like peptide-1 receptor agonists (GLP-1RA) have been shown to improve coronary risk factors including body wieght and suppress the occurrence of CV events [66,67]. Here, we consider the effects of such drugs on MASLD. We also consider the effect of selective peroxisome proliferator-activated receptor-alpha (PPARα) modulator, pemafibrate, which we recently reported to improve liver function, on MASLD [68].
5.1. SGLT2i
5.1.1. Effects of SGLT2i on liver enzymes, hepatic steatosis and fiborosis.
SGLT2 mediates approximately 90% of active renal glucose reabsorption in the proximal tubule of the kidney [69]. SGLT2i decrease plasma glucose without an increase in insulin secretion by reducing renal glucose reabsorption [70], which is favorable for body weight reduction and improvement in coronary risk factors [66,71].
We previsouly reported that SGLT2i significantly reduced serum levels of AST, ALT and GGT at 3 and 6 months after the start of SGLT2i in patients with type 2 diabetes [72,73]. Hepatic fibrosis can be evaluated by using noninvasive fibrosis-4 (FIB-4) index, which was reported as a useful index in MASLD [74]. A FIB-4 ≥ 2.67 had an 80% positive predictive value for identification of advanced hepatic fibrosis [74]. We found that FIB-4 index was significantly decreased at 12 months after the start of SGLT2i in high-risk (FIB-4 ≥ 2.67) group for adavanced hepatic fibrosis [75]. The correlations between the change of FIB-4 index during 12-month SGLT2i treatment was correlated inversely with the baseline FIB-4 index. We also retrospectively studied 568 patients with MASLD and type 2 diabetes. At 96 weeks, the mean FIB-4 index had significantly decreased (from 1.79 ± 1.10 to 1.56 ± 0.75) in the SGLT2i group, but not in the pioglotazone group [76]. Another marker for hepatic fibrosis, aspartate aminotransferase to platelet ratio index (APRI) significantly decreased in both groups. The body weight of the SGLT2i group decreased by 3.2 kg, but that of the PIO group increased by 1.7 kg.
In the meta-analysis of 20 RCTs, SGLT2i induced a significant decrease in serum ALT (-7.43 U/L, 95%CI; -12.14 to -2.71; p < 0.01), AST (-2.83 U/L; 95%CI, -4.71 to -0.95; p < 0.01), GGT (-8.21 U/L; 95%CI, -9.52 to -6.91, p < 0.01), comparing with placebo or other oral antidiabetic drugs. SGLT2i treatment was associated with a decrease in liver steatosis (-3.39%; 95%CI, -6.01 to -0.77; p < 0.0.1) [77]. Improvements in such liver enzymes and liver fat content were also observed in other meta-analyses [78,79,80,81].
Type IV collagen is one of the extracellular matrices that are produced by hepatic fibroblasts. The 7S domain in the N-terminus of type IV collagen is inserted in tissues and released into the blood by turnover in connective tissues. Therefore, the serum 7S domain level increases in parallel with the amount of fibrosis and in synthesis from stellate cells and myofibroblasts following increased liver fibrosis [82]. In Japan, type IV collagen 7S is now widely used for assessing the extent of hepatic fibrosis. Elevated serum ferritin has been the main manifestation of disturbed iron homeostasis in chronic liver diseases, and was reported to be independently associated with advanced liver fibrosis in patients with MASLD [83,84,85]. The meta-analysis showed that SGLT2i significantly reduced the level of FIB-4 (MD, 0.25; 95%CI, -0.39 to -0.11; p = 0.0007); serum type Ⅳ collagen 7s (MD, 0.32; 95%CI -0.59 to -0.04; p = 0.02); and ferritin (MD, 26.7; 95%CI, 50.64 to 2.76, p = 0.03) [86].
In recent years, the use of transient elastography (TE) with Fibroscan® equipment to obtain controlled attenuation parameter (CAP) and liver stiffness measurement (LSM) has been seen as a promising tool for noninvasive quantifying hepatic steatosis and fibrosis, respectively [87,88], and showed low failure (3.2%), high reliability (> 95%), and high reproducibility [89]. In the meta-analysis, SGLT2i significantly reduced LSM level when compared with control group (SMD [standard MD], −0.50; 95 %CI, −0.99 to −0.01; p = 0.002), CAP (SMD, −0.74; 95%CI, −1.21 to −0.27; p = 0.005), serum ferritin (SMD, −1.36; 95 % CI [−2.14, −0.57], p = 0.0008), serum type IV collagen 7S (SMD, −0.66; 95%CI, −1.2 to −0.12; p = 0.0004), and FIB-4 index (SMD, −0.37; 95%CI, −0. 74 to −0.01; p = 0.03) [90].
5.1.2. The underlying mechanisms for an improvement of MASLD by SGLT2i.
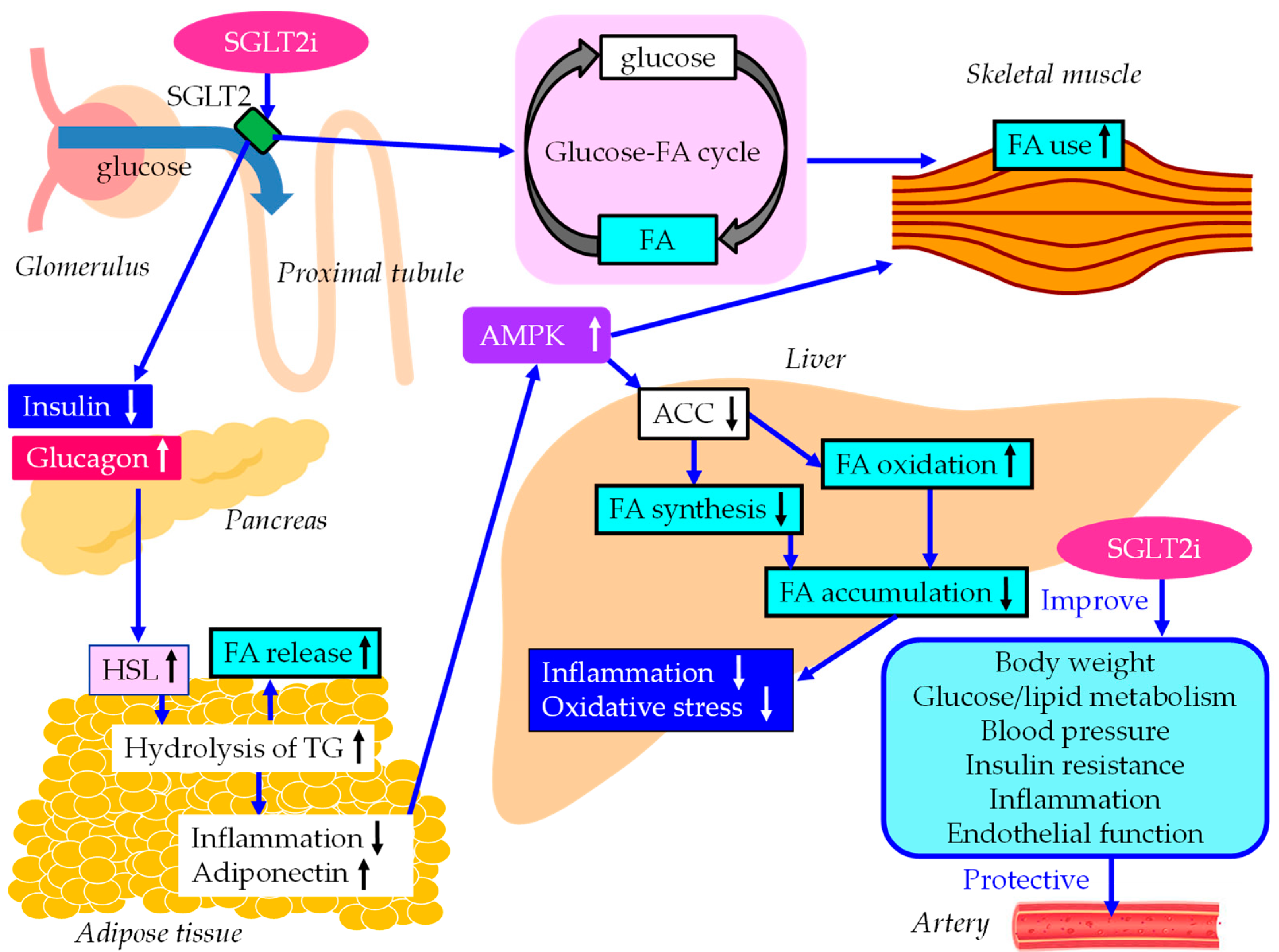
The underlying mechanisms for an improvement of MASLD and vascular protection by SGLT2i were shown in Figure 2. SGLT2i decrease plasma glucose without an increase in insulin secretion by reducing renal glucose reabsorption [70], and result in increase of the ratio of glucagon to insulin, which activates HSL in adipose tissue [91]. As a result, FA release from adipose tissue increases due to elevation in hydrolysis of stored TG, which reduces fat mass with a diminished adipocyte size, resulting in an improvement in insulin resistance. An increase in hyprolysis of TG increses serum FA, however, such incresed FA may be propmtly oxidized by skeltal muscles and liver.
Inflammatory biomarkers may play vital roles in the pathophysiology of diabetes and diabetic cardiorenal complications. The meta-analysis showed that SGLT2i reduce CRP (standard MD [SMD], 0.25; 95%CI, -0.47 to -0.03, p = 0.02) and improved adiponectin (SMD, 0.28; 95%CI, 0.15 to 0.41, p < 0.001) as compared with placebo [92]. An increase in adiponectin has beneficial effects on glucose and lipid metabolism by activation of adenosine 5'-monophosphate (AMP)-activated protein kinase (AMPK) [93]. AMPK signaling pathway plays an important role in ameliorating lipid metabolism disorders [94]. Increasing AMPK activity can inhibit FA synthesis by down-regulating the expression of acetyl-CoA carboxylase (ACC), and simultaneously, can enhance FA oxidation by increasing the expression of FA oxidation-related genes such as CPT-1. Reduced ACC activity by AMPK activation leads to the downregulation of downstream FA synthesis-related molecules and the upregulation of downstream β oxidation-associated molecules [91,95].
SGLT2i shifted energy metabolism towards FA utilization, elevated AMPK and ACC phosphorylation in skeletal muscle in diet-induced obese mice [96]. Furthermore, SGLT2i induce a negative energy balance by excreting glucose into the urine, which may induce alteration in glucose-FA cycle [97]. The fundamental concept of glucose-FA cycle is reciprocal substrate competition between glucose and FA in oxidative tissues such as skeletal muscles. SGLT2i-mediated alteration of glucose-FA cycle may increase FA metabolism in skeletal muscle. SGLT2i reduce FA accumulation in liver, which reduce inflammation and oxidative stress, resulting in an improvement of MASLD.
5.1.3. The vasculoprotective effects of SGLT2i.
The meta-analysis showed that SGLT2i reduce hemoglobin A1c (HbA1c) (MD, -0.66%; 95% CI, -0.73% to -0.58%), reduced body weight (MD, -1.80 kg; 95%CI, -3.50 to -0.11 kg) and systolic blood pressure (MD, -4.45 mmHg; 95%CI, -5.73 to -3.18 mmHg) [98]. The meta-analyses showed a significant increase in HDL-C and a significant decrease in TG [99,100]. Very recently, we reported that reduced levels of fasting apo B48, remanant lipoprotein-cholesterol, and non-HDL-C caused by SGLT2i suggest a possible beneficial effect of SGLT2i on atherogenic postprandial hyperlipidemia [101].
SGLT2i have been shown to improve endothelial dysfunction, as assessed by flow-mediated vasodilation, in individuals at high risk of CVD [102]. SGLT2i have been shown to improve oxidative stress, inflammation, mitochondrial dysfunction, glucotoxicity, such as the advanced signaling of glycation end products, and nitric oxide bioavailability. Very recently, the subanalysis of meta-analysis showed that SGLT2i significantly reduced atherosclerotic major adverse cardiovascular events (MACEs) in subjects having both chronic kidney disease and type 2 diabetes without established ASCVD [103].
Figure 2.
The underlying mechanisms for an improvement of MASLD and vascular protection by SGLT2i. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Blue solid lines indicate the effects of each metabolic event. ACC, acetyl-CoA carboxylase; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; FA, fatty acids; HSL, hormone sensitive lipase; SGLT2i, sodium glucose cotransporter 2 inhibitors; TG, triglyceride.
Figure 2.
The underlying mechanisms for an improvement of MASLD and vascular protection by SGLT2i. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Blue solid lines indicate the effects of each metabolic event. ACC, acetyl-CoA carboxylase; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; FA, fatty acids; HSL, hormone sensitive lipase; SGLT2i, sodium glucose cotransporter 2 inhibitors; TG, triglyceride.
5.2. GLP-1RA
5.2.1. Effects of GLP-1RA on liver enzymes, hepatic steatosis and fiborosis.
Recently, we reported that the 12-month dulaglutide therapy significantly improved serum GGT and NAS in patients with type 2 diabetes [104]. The meta-analyses showed that GLP-1RA improved liver enzymes [105], and liver histology scores for steatosis and fibrosis [106], and liver fat content on MRI-based techniques [107].
5.2.2. The underlying mechanisms for an improvement of MASLD by GLP-1RA.
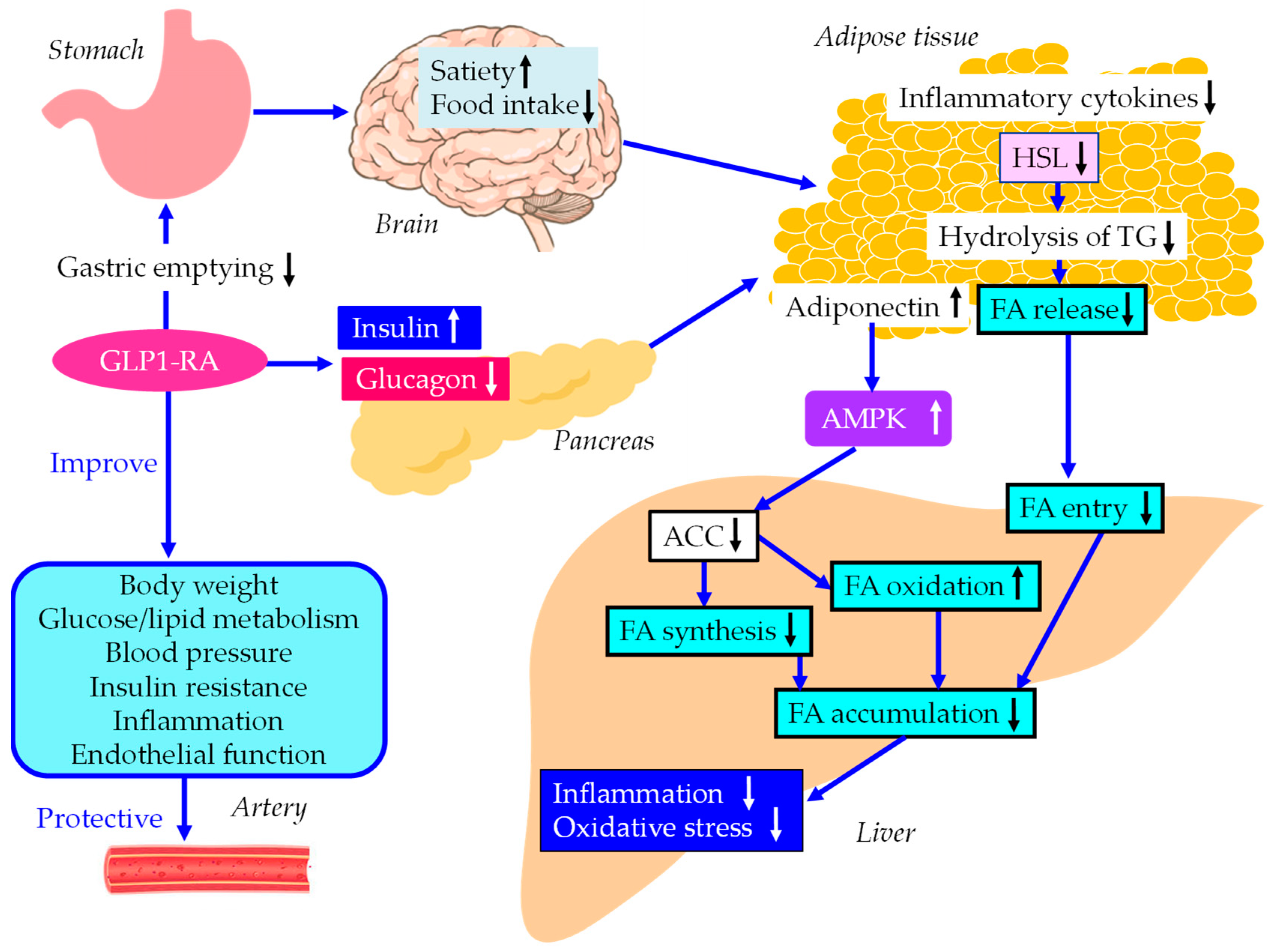
The underlying mechanisms for an improvement of MASLD and vascular protec tion by GLP-1RA were shown in Figure 3. GLP-1RA increase pancreatic insulin secretion and decrease glucagon in glucose-dependent manner, and delay gastric emptying which suppress postprandial hyperglycemia and appetite, resulting in reduction of energy intake and body weight [108,109,110]. Intestinal GLP-1 is an endogenous satiation signal, whose eating effects are primarily mediated by vagal afferents [111]. Increase in insulin secretion and decrease in glucagon secretion reduce HSL activity, resulting in decrease in hydrolysis of TG and FA release in adipose tissue, which reduces FA entry to the liver.
The meta-analysis showed that GLP-1RA showed significant reductions in CRP and TNF-α, and a significant increase in adiponectin as compared with standard diabetes therapies or placebo [112]. GLP-1RA increase adiponectin, which activates AMPK and suppresses ACC, resluting in decrease in hepatic FA synthesis and increase in hepatic FA oxidation.
5.2.3. The vasculoprotective effects of GLP-1RA.
GLP-1RA treatment achieved a greater systolic blood pressure reduction than comparator therapy (weighed MD [WMD], 2.22 mmHg; 95%CI, -2.97 to -1.47). In the pooled analysis, GLP-1RA had beneficial effects on weight loss (WMD, -2.56kg; 95%CI, -3.12 to -2.00), HbA1c reduction (WMD, -0.41%; 95%CI, -0.78 to -0.04) [113]. Compared with the patients before the treatment, the patients after the GLP-1RA treatment showed significantly reduced values of HbA1c, BMI, LDL-C and TG [114]. Furthermore, GLP-1RA improve atherogenic postprandial hyperlipidemia [101]. In addition, GLP-1RA have multiple vascular biological anti-atherogenic properties such as an improvement of endothelial function [67].
The meta-analysis showed that GLP-1 RA therapy was associated with a significantly lower risk of MACE, extended MACE, all-cause mortality, and CV mortality [115].
Figure 3.
The underlying mechanisms for an improvement of MASLD and vascular protection by GLP-1RA. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Blue solid lines indicate the effects of each metabolic event. ACC, acetyl-CoA carboxylase; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; FA, fatty acids; HSL, hormone sensitive lipase; GLP-1RA, glucagon-like peptide-1 receptor agonists; TG, triglyceride.
Figure 3.
The underlying mechanisms for an improvement of MASLD and vascular protection by GLP-1RA. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Blue solid lines indicate the effects of each metabolic event. ACC, acetyl-CoA carboxylase; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; FA, fatty acids; HSL, hormone sensitive lipase; GLP-1RA, glucagon-like peptide-1 receptor agonists; TG, triglyceride.
5.3. Pemafibrate
5.3.1. Effects of pemafibrate on liver enzymes, hepatic steatosis and fiborosis.
We previously reported that the selective PPARα modulator, pemafibrate, significantly reduced serum levels of AST, ALT and GGT and significantly increased serum albumin levels at 3, 6 and 12 months after the start of pemafibrate in patients with hypertriglyceridemia, with an improvement of atherogenic dyslipidemia [68].
Recently, we reported that pemafibrate significantly reduced hepatic steatosis index (HIS) at 12 months after the start of pemafibrate. APRI as the marker for hepatic fibrosis was significantly reduced by pemafibrate after 12 months. FIB-4 index significantly decreased in patients with baseline FIB-4 index ≥ 1.45 at 12 months after the start of pemafibrtate [116]. To our knowledge, this is the first to report that pemafibrate improved both hepatic steatosis and fibrosis indexes. Pemafibrate was reported to improve liver fibrosis assessed by MR elastography or FibroScan-aspartate aminotransferase score [117,118].
5.3.2. The underlying mechanisms for an improvement of MASLD by pemafibrate.
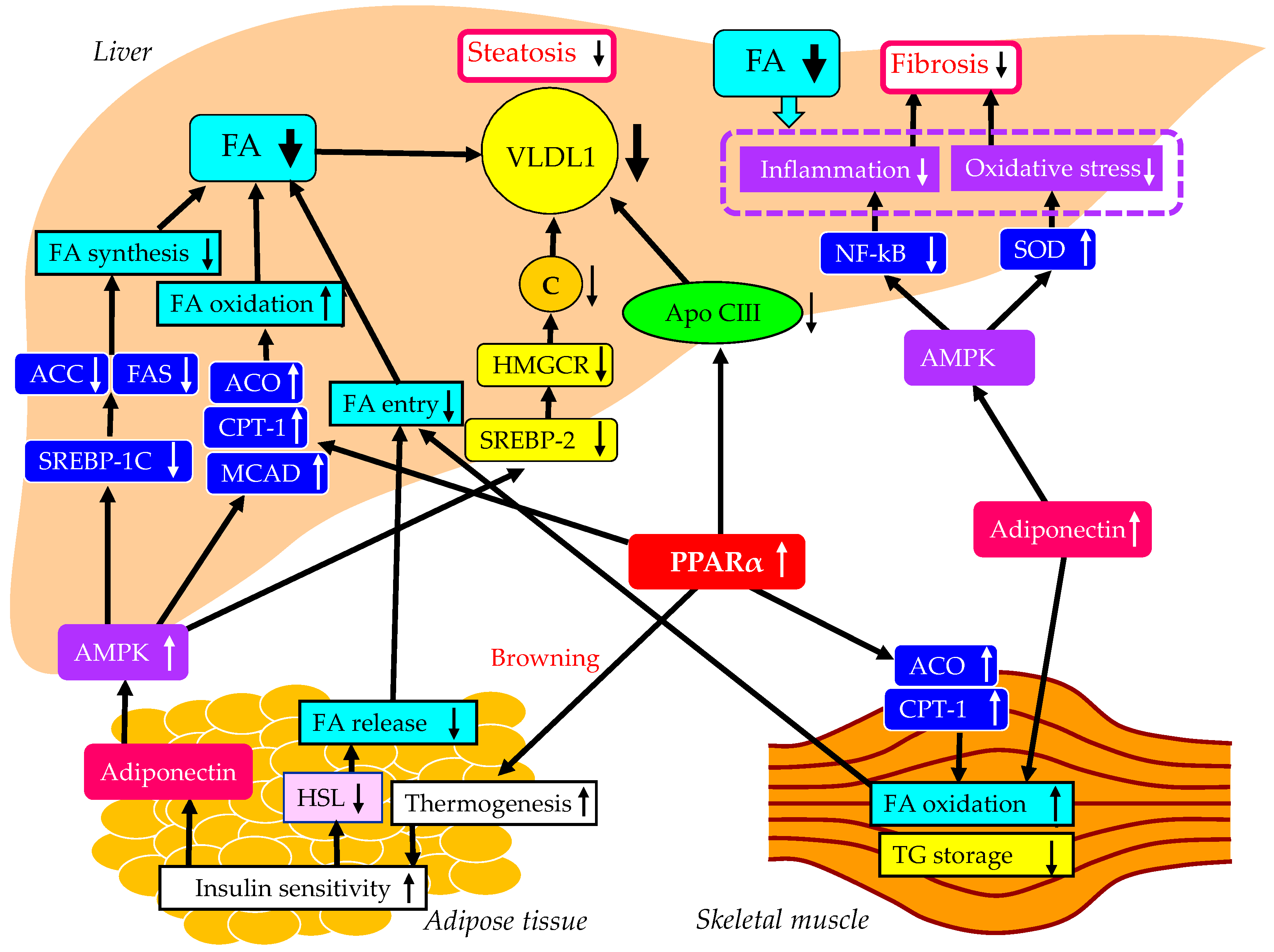
The underlying mechanisms for an improvement of MASLD by pemafibrate were shown in Figure 4. Altered properties of white adipose tissue (WAT) by obesity are associated with insulin resistance [119,120]. Brown fat increases energy expenditure by increasing thermogenesis and can utilize blood glucose and lipids, and results in improved glucose and lipid metabolism [121], which leads to reduction of FFA release from the adipose tissue. PPARα agonists can induce the browning of WAT [122], leading to an improvement of systemic insulin resistance. PPARα agonists enhance adiponectin production [123], which may be also beneficially associated with systemic insulin resistance [93]. The PPARα activation markedly stimulated the muscle and liver expression of two key enzymes involved in FA oxidation, CPT-1 and acyl-CoA oxidase (ACO) [124]. Moreover, the liver and muscle TG content were significantly reduced by the PPARα treatment [124].
Elevated FA oxidation in the skeletal muscle and the reduced FA release from the adipose tissue by PPARα agonists decrease FA entry to the liver and may result in the reduction of hepatic VLDL production. PPARα agonists reduce hepatic TG synthesis by decreasing apo CIII production [125]. Further, the treatment with PPARα agonists simulated the expression of ACO and CPT-1, leading to increase in FA oxidation and a decrease of hepatic TG storage [126].
PPARα agonists enhance adiponectin production [123], and adiponectin activates AMPK [93]. AMPK has long been regarded as a key regulator of energy metabolism, which is recognized as a critical target for MASLD treatment. AMPK activation reduces the genes related to FA synthesis such as ACC and FA synthase (FAS), by downregulating mRNA of SREBP-1c [127]. AMPK activation increases genes related to FA oxidation such as ACO, CPT-1 and medium-chain acyl-CoA dehydrogenase (MCAD) [127]. AMPK activation also inhibits the expression of SREBP-2 and its target genes such as 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) which is the key enzyme in cholesterol biosynthesis [128]. Such increased FA oxidation and reduced FA and cholesterol production in liver decrease hepatic VLDL production. Reduced hepatic VLDL accumulation may improve hepatic fibrosis by reducing inflammation and oxidative stress. Furthermore, AMPK activation improves inflammation by inhibiting NF-κB [129], and also ameliorates oxidative stress by increasing the expression of superoxide dismutase (SOD) [130], which contribute to reduction of hepatic fibrosis.
Figure 4.
The underlying mechanisms for an improvement of MASLD by pemafibrate. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Black solid lines indicate the effects of each metabolic event. ACC, acetyl-CoA carboxylase; ACO, acyl-CoA oxidase; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; C, cholesterol; CPT-1, carnitine palmitoyl-transferase 1; FA, fatty acids; FAS, FA synthase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; HSL, hormone sensitive lipase; MCAD, medium-chain acyl-CoA dehydrogenase; NF-κB, nuclear factor-kappa B; PPARα, peroxisome proliferator-activated receptor-α; SOD, superoxide dismutase; SREBP, sterol regulatory element binding protein; TG, triglyceride; VLDL, very-low-density lipoprotein.
Figure 4.
The underlying mechanisms for an improvement of MASLD by pemafibrate. Black and white arrows pointing upward and downward indicate increase and decrease in expression or activity, respectively. Black solid lines indicate the effects of each metabolic event. ACC, acetyl-CoA carboxylase; ACO, acyl-CoA oxidase; AMPK, adenosine 5'-monophosphate (AMP)-activated protein kinase; C, cholesterol; CPT-1, carnitine palmitoyl-transferase 1; FA, fatty acids; FAS, FA synthase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; HSL, hormone sensitive lipase; MCAD, medium-chain acyl-CoA dehydrogenase; NF-κB, nuclear factor-kappa B; PPARα, peroxisome proliferator-activated receptor-α; SOD, superoxide dismutase; SREBP, sterol regulatory element binding protein; TG, triglyceride; VLDL, very-low-density lipoprotein.
5.3.3. The vasculoprotective effects of pemafibrate.
PPARα agonists reduce hepatic TG synthesis by decreasing apo CIII production [125]. Further, the treatment with PPARα agonists simulated the expression of enzymes involved in FA oxidation leading to a concomitant decrease of hepatic VLDL production [126]. PPARα agonists stimulate the activity of LPL, which further reduce VLDL [125]. As a result, there is an increase in HDL levels and a decrease in Sd-LDL and remnant lipoproteins [131]. PPARα agonists elevate HDL-C levels via transcriptional induction of apo AI and apo AII formation [125]. Pemafibrate is also effective to improve atherogenic postprandial hyperlipidemia [101].
In addition, PPARα agonists promotes HDL-mediated cholesterol efflux from macrophages, via enhanced expression of ABCA1 [132]. PPARα agonists have multiple beficicial effects on vascular integrity such as anti-inflammatory effect and inhibitory effects on vasoconstriction [133]. Further, PPARα agonists inhibit smooth muscle cell proliferation, adhesion of monocytes to endothelial cells, oxidized LDL formation. PPARα agonists have a beneficial effect on procoagulant state.
In the meta-analysis to investigate the influence of fibrates on vascular risk reduc-tion in subjects with atherogenic dyslipidemia [134], compared to placebo, the greatest benefit with fibrate treatment was seen in high TG subjects, fibrate therapy reduced risk of vascular events by 25%. Very redently, the PROMINENT Trial was performed to study whether pemafibrate reduces CV risk patients with type 2 diabetes, mild-to-moderate hypertriglyceridemia, and low HDL-C and LDL-C levels [135]. The median follow-up was 3.4 years. Pemafibrate reduced serum TG by 26.2%, VLDL-C by 25.8%, remnant cholestrol by 25.6%, and apo CIII by 27.6% as compared with placebo after 4 months. However, the incidence of CV events was not lower among those who received pemafibrate than among those who received placebo. Further studies should be performed to evaluate the effect of pemafibrate on CVD in the future.
5.4. Effects of the combination therapy of SGLT2i and GLP-1RA on MASLD.
To our knowleage, there is only our study which invastigated effects of the combination therapy of SGLT2i and GLP-1RA. We found that the 12-month dulaglutide therapy significantly improved serum GGT and NAS in patients with type 2 diabetes [104]. Although a significant improvement in GGT was not observed in patients treated with GLP-1RA without SGLT2i (n = 69), a significant improvement in GGT was obtained in patients treated with GLP-1RA and SGLT2i (n = 52). Furthermore, FIB-4 index tended to decrease from 1.74 ± 1.33 to 1.62 ± 1.10 (p = 0.088) in patients treated with GLP-1RA and SGLT2i, while patients without SGLT2i showed a non-significant increase in FIB-4 index from 1.71 ± 1.02 to 1.75 ± 1.12 (p = 0.547). The combination therapy of SGLT2i and GLP-1RA may be a promissing therapeutic option for MASLD.
5.5. Effects of the combination therapy of SGLT2i and pemafibrate on MASLD.
To our knowleage, there are only two studies which invastigated effects of the combination therapy of SGLT2i and pemfibrate including our study. The other group's study was a pilot study with only seven patients. In their study, MASLD patients complicated with type 2 diabetes treated with pemafibrate for > 1 year were included, in whom prior treatment with SGLT2i > 1 year failed to normalize serum ALT levels [136]. During the one year before starting pemafibrate therapy, the therapy did not significantly change hepatic enzymes. All patients received pemafibrate 0.1 mg twice daily. During one year of pemafibrate therapy, serum levels of TG, AST, ALT, GGT, Mac-2 binding protein glycosylation isomer (M2BPGi) which is the marker for liver fibrosis, were significantly improved (p < 0.05).
We reported that pemafibrate significantly reduced serum levels of AST, ALT and GGT and significantly increased serum albumin levels at 3, 6 and 12 months after the start of pemafibrate in patients with hypertriglyceridemia (n = 246), with an improvement of atherogenic dyslipidemia [68]. We investigated the effects of combination of pemafibrate and SGLT2i in such parameters, by dividing patients into those treated with pemafibrate and SGLT2i (n = 63) and those treated with pemafibrate without SGLT2i (n = 183). There were no significant differences in changes in ALT, GGT and albumin bewteen two groups. Although a significant improvement in AST was not observed in patients treated without SGLT2i at any time, a significant improvement in AST was observed in patients treated with pemafibrate and SGLT2i at 3 and 12 months after the start of pemafibrate (Figure 5). The reversal of the AST/ALT ratio to > 1 had been consistently reported to predict the presence of more advanced liver fibrosis [137]. The marker for liver fibrosis, APRI was calculated with the formula: AST/ Upper limit of normal range of AST / platelet count x 100 [138]. Such favirable effect of the combination of pemafibrate and SGLT2i on change in AST may present that this combination therapy can be a promissing therapeutic option for MASLD.
6. Conclusion
Abnormal FA and TG metabolims induced by obesity/insulin resiatnce is closely associated with the development of MASLD. MASLD is the pathologic condition which is likely to develop CVD. Therefore, the ideal treatments for MASLD require anti-artherosclerotic effects in addition to improving liver function. Lifestyle modification such as diet and exercise and surgical interventions such as bariatric surgery and intragastric balloons have shown to improve MASLD by reducing body weight, and such interventions are also effective to reduce CVD. SGLT2i and GLP-1RA have been shown to reduce the development of CVD, and such drugs improve liver enzymes, and hepatic steatosis and fibrosis, suggesitng that such drugs can be the ideal therapeutic option for MASLD. Pemfibrate improved liver enzymes and the indexes of hepatic steatosis and fibrosis, and have multiple anti-atherogenic properties, however, the effect of pemafibrate on CVD remains to be elucidated in the clinical settings.
Author Contributions
H.Y., M.H., H.A. and H. K. conceived the review; H. Y. wrote the paper; H. K. edited the paper and provided critical guidance. All authors read and approved the final version of this paper.
Funding
This review research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest in relation to the present review paper.
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Ann Hepatol. 2023, 101133. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Le, M.H.; Yeo, Y.H.; Zou, B.; Barnet, S.; Henry, L.; Cheung, R.; Nguyen, M.H. Forecasted 2040 global prevalence of nonalcoholic fatty liver disease using hierarchical bayesian approach. Clin. Mol. Hepatol. 2022, 28, 841–850. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Fisher, E.A. The degradation of apolipoprotein B100: Multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta. 2012, 1821, 778–781. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.H. Skeletal muscle triglyceride. An aspect of regional adiposity and insulin resistance. Diabetes. Care. 2001, 24, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Taghibiglou, C.; Carpentier, A.; Van Iderstine, S.C.; Chen, B.; Rudy, D.; Aiton, A.; Lewis, G.F.; Adeli, K. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J. Biol. Chem. 2000, 275, 8416–8425. [Google Scholar]
- Chen, M.; Breslow, J.L.; Li, W.; Leff, T. Transcriptional regulation of the apoC-III gene by insulin in diabetic mice: Correlation with changes in plasma triglyceride levels. J. Lipid. Res. 1994, 35, 1918–1924. [Google Scholar] [CrossRef]
- Avramoglu, R.K.; Basciano, H.; Adeli, K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin. Chim. Acta. 2006, 368, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Duran, E.K.; Pradhan, A.D. Triglyceride-Rich Lipoprotein Remnants and Cardiovascular Disease. Clin. Chem. 2021, 67, 183–196. [Google Scholar] [CrossRef]
- Barrows, B.R.; Parks, E.J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Adiels, M.; Westerbacka, J.; Sderlund, S.; Kahri, J.; Lundbom, N.; Lundbom, J.; Hakkarainen, A.; Olofsson, S.O.; Orho-Melander, M.; et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2144–2150. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; De Michieli, F.; Biroli, G.; Fagà, E.; Pagano, G.; Cassader, M. Association of liver disease with postprandial large intestinal triglyceride-rich lipoprotein accumulation and pro/antioxidant imbalance in normolipidemic non-alcoholic steatohepatitis. Ann. Med. 2008, 40, 383–394. [Google Scholar] [CrossRef]
- Qin, B.; Anderson, R.A.; Kuzuya, T.; Kitaura, Y.; Shimomura, Y. Multiple factors and pathways involved in hepatic very low density lipoprotein-apoB100 overproduction in Otsuka Long-Evans Tokushima Fatty rats. Atherosclerosis. 2012, 222, 409–416. [Google Scholar] [CrossRef]
- Uehara, K.; Sostre-Colón, J.; Gavin, M.; Santoleri, D.; Leonard, K.A.; Jacobs, R.L.; Titchenell, P.M. Activation of Liver mTORC1 Protects Against NASH via Dual Regulation of VLDL-TAG Secretion and De Novo Lipogenesis. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1625–1647. [Google Scholar] [CrossRef]
- Grove, J.I.; Lo, P.C.K.; Shrine, N.; Barwell, J.; Wain, L.V.; Tobin, M.D.; Salter, A.M.; Borkar, A.N.; Cuevas-Ocaña, S.; Bennett, N.; et al. Identification and characterisation of a rare MTTP variant underlying hereditary non-alcoholic fatty liver disease. JHEP. Rep. 2023, 5, 100764. [Google Scholar] [CrossRef]
- Hong, S.; Gordon, D.; Stec, D.E.; Hinds, T.D. Bilirubin: A Ligand of the PPARα Nuclear Receptor. In Nuclear Receptors: The Art and Science of Modulator Design and Discovery; Badr, M.Z., Ed.; Springer International Publishing: Cham, Switzerland, 2021; pp. 463–482. [Google Scholar]
- Moreno-Fernandez, M.E.; Giles, D.A.; Stankiewicz, T.E.; Sheridan, R.; Karns, R.; Cappelletti, M.; Lampe, K.; Mukherjee, R.; Sina, C.; Sallese, A.; et al. Peroxisomal β-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI. Insight. 2018, 3, e93626. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Hosick, P.A.; Chen, S.; Tukey, R.H.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to Gilbert's syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARα. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D. Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS. One. 2016, 11, e0153427. [Google Scholar] [CrossRef]
- Hinds, T.D., Jr.; Adeosun, S.O.; Alamodi, A.A.; Stec, D.E. Does bilirubin prevent hepatic steatosis through activation of the PPARα nuclear receptor? Med. Hypotheses. 2016, 95, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; et al. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J Biol Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Paiva, A.A.; Raposo, H.F.; Wanschel, A.C.; Nardelli, T.R.; Oliveira, H.C. Apolipoprotein CIII Overexpression-Induced Hypertriglyceridemia Increases Nonalcoholic Fatty Liver Disease in Association with Inflammation and Cell Death. Oxid. Med. Cell. Longev. 2017, 2017, 1838679. [Google Scholar] [CrossRef]
- Roden, M.; Stingl, H.; Chandramouli, V.; Schumann, W.C.; Hofer, A.; Landau, B.R.; Nowotny, P.; Waldhäusl, W.; Shulman, G.I. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes. 2000, 49, 701–707. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Fuchs, M.; Sanyal, A.J. Lipotoxicity in NASH. J. Hepatol. 2012, 56, 291–293. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Sinha, R.A. Autophagy: A Cellular Guardian against Hepatic Lipotoxicity. Genes (Basel). 2023, 14, 553. [Google Scholar] [CrossRef]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2015, 149, 389–97. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Hyogo, H.; Ono, M.; Mizuta, T.; Ono, N.; Fujimoto, K.; Chayama, K.; Saibara, T.; JSG-NAFLD. Prevalence and associated metabolic factors of nonalcoholic fatty liver disease in the general population from 2009 to 2010 in Japan: a multicenter large retrospective study. J. Gastroenterol. 2012, 47, 586–595. [Google Scholar] [CrossRef]
- Yanai, H.; Hirowatari, Y.; Ito, K.; Kurosawa, H.; Tada, N.; Yoshida, H. Understanding of Diabetic Dyslipidemia by Using the Anion-Exchange High Performance Liquid Chromatography Data. J. Clin. Med. Res. 2016, 8, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Atherogenic Lipoproteins for the Statin Residual Cardiovascular Disease Risk. Int. J. Mol. Sci. 2022, 23, 13499. [Google Scholar] [CrossRef]
- Nikkila, E.A.; Huttunen, J.K.; Ehnholm, C. Postheparin plasma lipoprotein lipase and hepatic lipase in diabetes mellitus. Relationship to plasma triglyceride metabolism. Diabetes. 1977, 26, 11–21. [Google Scholar] [CrossRef]
- Nikkila, E.A.; Taskinen, M.R.; Kekki, M. Relation of plasma high-density lipoprotein cholesterol to lipoprotein-lipase activity in adipose tissue and skeletal muscle of man. Atherosclerosis. 1978, 29, 497–501. [Google Scholar] [CrossRef]
- Kasim, S.E.; Tseng, K.; Jen, K.L.; Khilnani, S. Significance of hepatic triglyceride lipase activity in the regulation of serum high density lipoproteins in type II diabetes mellitus. J. Clin. Endocrinol. Metab. 1987, 65, 183–187. [Google Scholar] [CrossRef]
- Vega, G.L.; Grundy, S.M. Effect of statins on metabolism of apo-B-containing lipoproteins in hypertriglyceridemic men. Am. J. Cardiol. 1998, 81, 36B–42B. [Google Scholar] [CrossRef]
- Zambon, A.; Austin, M.A.; Brown, B.G.; Hokanson, J.E.; Brunzell, J.D. Effect of hepatic lipase on LDL in normal men and those with coronary artery disease. Arterioscler. Thromb. 1993, 13, 147–153. [Google Scholar] [CrossRef]
- Duran, E.K.; Pradhan, A.D. Triglyceride-Rich Lipoprotein Remnants and Cardiovascular Disease. Clin. Chem. 2021, 67, 183–196. [Google Scholar] [CrossRef]
- Schwartz, E.A.; Reaven, P.D. Lipolysis of triglyceride-rich lipoproteins, vascular inflammation, and atherosclerosis. Biochim. Biophys. Acta. 2012, 1821, 858–866. [Google Scholar] [CrossRef]
- Ahn, J.; Jun, D.W.; Lee, H.Y.; Moon, J.H. Critical appraisal for low-carbohydrate diet in nonalcoholic fatty liver disease: Review and meta-analyses. Clin. Nutr. 2019, 38, 2023–2030. [Google Scholar] [CrossRef]
- Houttu, V.; Csader, S.; Nieuwdorp, M.; Holleboom, A.G.; Schwab, U. Dietary Interventions in Patients With Non-alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 716783. [Google Scholar] [CrossRef]
- Haigh, L.; Kirk, C.; El Gendy, K.; Gallacher, J.; Errington, L.; Mathers, J.C.; Anstee, Q.M. The effectiveness and acceptability of Mediterranean diet and calorie restriction in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis. Clin. Nutr. 2022, 41, 1913–1931. [Google Scholar] [CrossRef]
- Cho, Y.; Hong, N.; Kim, K.W.; Cho, S.J.; Lee, M.; Lee, Y.H.; Lee, Y.H.; Kang, E.S.; Cha, B.S.; Lee, B.W. The Effectiveness of Intermittent Fasting to Reduce Body Mass Index and Glucose Metabolism: A Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 1645. [Google Scholar] [CrossRef]
- Harris, L.; Hamilton, S.; Azevedo, L.B.; Olajide, J.; De Brún, C.; Waller, G.; Whittaker, V.; Sharp, T.; Lean, M.; Hankey, C.; et al. Intermittent fasting interventions for treatment of overweight and obesity in adults: a systematic review and meta-analysis. JBI Database System Rev Implement Rep. 2018, 16, 507–547. [Google Scholar] [CrossRef]
- Rizza, W.; Veronese, N.; Fontana, L. What are the roles of calorie restriction and diet quality in promoting healthy longevity? Ageing. Res. Rev. 2014, 13, 38–45. [Google Scholar] [CrossRef]
- Yin, C.; Li, Z.; Xiang, Y.; Peng, H.; Yang, P.; Yuan, S.; Zhang, X.; Wu, Y.; Huang, M.; Li, J. Effect of Intermittent Fasting on Non-Alcoholic Fatty Liver Disease: Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 709683. [Google Scholar] [CrossRef]
- Lange, M.; Nadkarni, D.; Martin, L.; Newberry, C.; Kumar, S.; Kushner, T. Intermittent fasting improves hepatic end points in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Hepatol. Commun. 2023, 7, e0212. [Google Scholar] [CrossRef] [PubMed]
- Orci, L.A.; Gariani, K.; Oldani, G.; Delaune, V.; Morel, P.; Toso, C. Exercise-based Interventions for Nonalcoholic Fatty Liver Disease: A Meta-analysis and Meta-regression. Clin. Gastroenterol. Hepatol. 2016, 14, 1398–1411. [Google Scholar] [CrossRef] [PubMed]
- Katsagoni, C.N.; Georgoulis, M.; Papatheodoridis, G.V.; Panagiotakos, D.B.; Kontogianni, M.D. Effects of lifestyle interventions on clinical characteristics of patients with non-alcoholic fatty liver disease: A meta-analysis. Metabolism. 2017, 68, 119–132. [Google Scholar] [CrossRef]
- González-Ruiz, K.; Ramírez-Vélez, R.; Correa-Bautista, J.E.; Peterson, M.D.; García-Hermoso, A. The Effects of Exercise on Abdominal Fat and Liver Enzymes in Pediatric Obesity: A Systematic Review and Meta-Analysis. Child. Obes. 2017, 13, 272–282. [Google Scholar] [CrossRef]
- Medrano, M.; Cadenas-Sanchez, C.; Álvarez-Bueno, C.; Cavero-Redondo, I.; Ruiz, J.R.; Ortega, F.B.; Labayen, I. Evidence-Based Exercise Recommendations to Reduce Hepatic Fat Content in Youth- a Systematic Review and Meta-Analysis. Prog. Cardiovasc. Dis. 2018, 61, 222–231. [Google Scholar] [CrossRef]
- Sargeant, J.A.; Gray, L.J.; Bodicoat, D.H.; Willis, S.A.; Stensel, D.J.; Nimmo, M.A.; Aithal, G.P.; King, J.A. The effect of exercise training on intrahepatic triglyceride and hepatic insulin sensitivity: a systematic review and meta-analysis. Obes. Rev. 2018, 19, 1446–1459. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Fernández, T.; Viñuela, M.; Vidal, C.; Barrera, F. Lifestyle changes in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. PLoS One. 2022, 17, e0263931. [Google Scholar] [CrossRef]
- Buchwald, H.; Avidor, Y.; Braunwald, E.; Jensen, M.D.; Pories, W.; Fahrbach, K.; Schoelles, K. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004, 292, 1724–1737. [Google Scholar] [CrossRef]
- Adams, T.D.; Gress, R.E.; Smith, S.C.; Halverson, R.C.; Simper, S.C.; Rosamond, W.D.; Lamonte, M.J.; Stroup, A.M.; Hunt, S.C. Long-term mortality after gastric bypass surgery. N. Engl. J. Med. 2007, 357, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Pontiroli, A.E.; Morabito, A. Long-term prevention of mortality in morbid obesity through bariatric surgery. A systematic review and meta-analysis of trials performed with gastric banding and gastric bypass. Ann. Surg. 2011, 253, 484–487. [Google Scholar] [CrossRef]
- Bower, G.; Toma, T.; Harling, L.; Jiao, L.R.; Efthimiou, E.; Darzi, A.; Athanasiou, T.; Ashrafian, H. Bariatric Surgery and Non-Alcoholic Fatty Liver Disease: a Systematic Review of Liver Biochemistry and Histology. Obes. Surg. 2015, 25, 2280–2289. [Google Scholar] [CrossRef]
- Popov, V.B.; Thompson, C.C.; Kumar, N.; Ciarleglio, M.M.; Deng, Y.; Laine, L. Effect of Intragastric Balloons on Liver Enzymes: A Systematic Review and Meta-Analysis. Dig. Dis. Sci. 2016, 61, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Chandan, S.; Mohan, B.P.; Khan, S.R.; Facciorusso, A.; Ramai, D.; Kassab, L.L.; Bhogal, N.; Asokkumar, R.; Lopez-Nava, G.; McDonough, S.; et al. Efficacy and Safety of Intragastric Balloon (IGB) in Non-alcoholic Fatty Liver Disease (NAFLD): a Comprehensive Review and Meta-analysis. Obes. Surg. 2021, 31, 1271–1279. [Google Scholar] [CrossRef]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. Multi-Organ Protective Effects of Sodium Glucose Cotransporter 2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 4416. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Glucagon-Like Peptide 1 Receptor Agonists Versus Sodium-Glucose Cotransporter 2 Inhibitors for Atherosclerotic Cardiovascular Disease in Patients With Type 2 Diabetes. Cardiol. Res. 2023, 14, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Katsuyama, H.; Hakoshima, M. Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study. Biomedicines. 2022, 10, 401. [Google Scholar] [CrossRef]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef]
- Jabbour, S.A.; Goldstein, B.J. Sodium glucose co-transporter 2 inhibitors: blocking renal tubular reabsorption of glucose to improve glycaemic control in patients with diabetes. Int. J. Clin. Pract. 2008, 62, 1279–1284. [Google Scholar] [CrossRef]
- Yanai, H.; Katsuyama, H.; Hamasaki, H.; Adachi, H.; Moriyama, S.; Yoshikawa, R.; Sako, A. Sodium-Glucose Cotransporter 2 Inhibitors: Possible Anti-Atherosclerotic Effects Beyond Glucose Lowering. J. Clin. Med. Res. 2016, 8, 10–14. [Google Scholar] [CrossRef]
- Katsuyama, H.; Hamasaki, H.; Adachi, H.; Moriyama, S.; Kawaguchi, A.; Sako, A.; Mishima, S.; Yanai, H. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters in Patients With Type 2 Diabetes: A Chart-Based Analysis. J. Clin. Med. Res. 2016, 8, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Kawaguchi, A.; Waragai, Y.; Harigae, T.; Masui, Y.; Kakuta, K.; Hamasaki, H.; Katsuyama, H.; et al. Effects of Six Kinds of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters, and Summarized Effect and Its Correlations With Baseline Data. J. Clin. Med. Res. 2017, 9, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.G.; Lydecker, A.; Murray, K.; Tetri, B.N.; Contos, M.J.; Sanyal, A.J.; Nash Clinical Research, N. Comparison of noninvasive markers of fibrosis in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2009, 7, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, H.; Hakoshima, M.; Iijima, T.; Adachi, H.; Hidekatsu Yanai, H. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Hepatic Fibrosis in Patients With Type 2 Diabetes: A Chart-Based Analysis. J. Endocrinol. Metab. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Mino, M.; Kakazu, E.; Sano, A.; Katsuyama, H.; Hakoshima, M.; Yanai, H.; Aoki, Y.; Imamura, M.; Yamazoe, T.; Mori, T.; et al. Effects of sodium glucose cotransporter 2 inhibitors and pioglitazone on FIB-4 index in metabolic-associated fatty liver disease. Hepatol. Res. 2023, 53, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Coelho, F.D.S.; Borges-Canha, M.; von Hafe, M.; Neves, J.S.; Vale, C.; Leite, A.R.; Carvalho, D.; Leite-Moreira, A. Effects of sodium-glucose co-transporter 2 inhibitors on liver parameters and steatosis: A meta-analysis of randomized clinical trials. Diabetes. Metab. Res. Rev. 2021, 37, e3413. [Google Scholar] [CrossRef]
- Mantovani, A.; Petracca, G.; Csermely, A.; Beatrice, G.; Targher, G. Sodium-Glucose Cotransporter-2 Inhibitors for Treatment of Nonalcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Metabolites. 2020, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Datta, D.; Ghosal, S. Meta-analysis of the effects of sodium glucose cotransporter 2 inhibitors in non-alcoholic fatty liver disease patients with type 2 diabetes. JGH. Open. 2020, 5, 219–227. [Google Scholar] [CrossRef]
- Wong, C.; Yaow, C.Y.L.; Ng, C.H.; Chin, Y.H.; Low, Y.F.; Lim, A.Y.L.; Muthiah, M.D.; Khoo, C.M. Sodium-Glucose Co-Transporter 2 Inhibitors for Non-Alcoholic Fatty Liver Disease in Asian Patients With Type 2 Diabetes: A Meta-Analysis. Front. Endocrinol (Lausanne). 2021, 11, 609135. [Google Scholar] [CrossRef]
- Wei, Q.; Xu, X.; Guo, L.; Li, J.; Li, L. Effect of SGLT2 Inhibitors on Type 2 Diabetes Mellitus With Non-Alcoholic Fatty Liver Disease: A Meta-Analysis of Randomized Controlled Trials. Front. Endocrinol (Lausanne). 2021, 12, 635556. [Google Scholar] [CrossRef]
- Murawaki, Y.; Ikuta, K.; Koda, M.; Kawasaki, H. Serum type III procollagen peptide, type IV collagen 7S domain, central triple-helix of type IV collagen and tissue inhibitor of metalloproteinases in patients with chronic viral liver disease: relationship to liver histology. Hepatology. 1994, 20, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Manousou, P.; Kalambokis, G.; Grillo, F.; Watkins, J.; Xirouchakis, E.; Pleguezuelo, M.; Leandro, G.; Arvaniti, V.; Germani, G.; Patch, D.; et al. Serum ferritin is a discriminant marker for both fibrosis and inflammation in histologically proven non-alcoholic fatty liver disease patients. Liver. Int. 2011, 31, 730–739. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E.; NASH Clinical Research Network. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2012, 55, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Shim, J.J.; Park, S.K.; Ryoo, J.H.; Choi, J.M.; Oh, I.H.; Jung, K.W.; Cho, H.; Ki, M.; Won, Y.J.; et al. Serum ferritin level is associated with liver steatosis and fibrosis in Korean general population. Hepatol. Int. 2019, 13, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Chen, S.; Zhao, H.; Wang, F.; Song, H.; Tian, D.; Yang, Q.; Qi, L. Meta-analysis of the effect of sodium-glucose cotransporter 2 inhibitors on hepatic fibrosis in patients with type 2 diabetes mellitus complicated with non-alcoholic fatty liver disease. Hepatol. Res. 2021, 51, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Sasso, M.; Beaugrand, M.; de Ledinghen, V.; Douvin, C.; Marcellin, P.; Poupon, R.; Sandrin, L.; Miette, V. Controlled attenuation parameter (CAP): a novel VCTE™ guided ultrasonic attenuation measurement for the evaluation of hepatic steatosis: preliminary study and validation in a cohort of patients with chronic liver disease from various causes. Ultrasound. Med. Biol. 2010, 36, 1825–1835. [Google Scholar] [CrossRef]
- Piazzolla, V.A.; Mangia, A. Noninvasive Diagnosis of NAFLD and NASH. Cells. 2020, 9, 1005. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Siddiqui, M.S.; Van Natta, M.L.; Hallinan, E.; Brandman, D.; Kowdley, K.; Neuschwander-Tetri, B.A.; Loomba, R.; Dasarathy, S.; Abdelmalek, M.; et al. Performance characteristics of vibration-controlled transient elastography for evaluation of nonalcoholic fatty liver disease. Hepatology. 2018, 67, 134–144. [Google Scholar] [CrossRef]
- Jin, Z.; Yuan, Y.; Zheng, C.; Liu, S.; Weng, H. Effects of sodium-glucose co-transporter 2 inhibitors on liver fibrosis in non-alcoholic fatty liver disease patients with type 2 diabetes mellitus: An updated meta-analysis of randomized controlled trials. J. Diabetes. Complications. 2023, 37, 108558. [Google Scholar] [CrossRef]
- Obata, A.; Kubota, N.; Kubota, T.; Iwamoto, M.; Sato, H.; Sakurai, Y.; Takamoto, I.; Katsuyama, H.; Suzuki, Y.; Fukazawa, M.; et al. Tofogliflozin Improves Insulin Resistance in Skeletal Muscle and Accelerates Lipolysis in Adipose Tissue in Male Mice. Endocrinology. 2016, 157, 1029–1042. [Google Scholar] [CrossRef]
- Wang, D.; Liu, J.; Zhong, L.; Li, S.; Zhou, L.; Zhang, Q.; Li, M.; Xiao, X. The effect of sodium-glucose cotransporter 2 inhibitors on biomarkers of inflammation: A systematic review and meta-analysis of randomized controlled trials. Front. Pharmacol. 2022, 13, 1045235. [Google Scholar] [CrossRef]
- Yanai, H.; Yoshida, H. Beneficial Effects of Adiponectin on Glucose and Lipid Metabolism and Atherosclerotic Progression: Mechanisms and Perspectives. Int. J. Mol. Sci. 2019, 20, 1190. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Pan, J.; Qu, N.; Lei, Y.; Han, J.; Zhang, J.; Han, D. The AMPK pathway in fatty liver disease. Front. Physiol. 2022, 13, 970292. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.; Lee, J.C.; Huang, H.C.; Huang, H.; Liu, H.K.; Huang, C. Delayed intervention with a novel SGLT2 inhibitor NGI001 suppresses diet-induced metabolic dysfunction and nonalcoholic fatty liver disease in mice. Br. J. Pharmacol. 2020, 177, 239–253. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Mayoux, E.; Kaneko, S.; Ota, T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine. 2017, 20, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: a systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef]
- Sanchez-Garcia, A.; Simental-Mendia, M.; Millan-Alanis, J.M.; Simental-Mendia, L.E. Effect of sodium-glucose co-transporter 2 inhibitors on lipid profile: A systematic review and meta-analysis of 48 randomized controlled trials. Pharmacol. Res. 2020, 160, 105068. [Google Scholar] [CrossRef]
- Li, D.; Wu, T.; Wang, T.; Wei, H.; Wang, A.; Tang, H.; Song, Y. Effects of sodium glucose cotransporter 2 inhibitors on risk of dyslipidemia among patients with type 2 diabetes: A systematic review and meta-analysis of randomized controlled trials. Pharmacoepidemiol. Drug. Saf. 2020, 29, 582–590. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Postprandial Hyperlipidemia: Its Pathophysiology, Diagnosis, Atherogenesis, and Treatments. Int. J. Mol. Sci. 2023, 24, 13942. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Significance of Endothelial Dysfunction Amelioration for Sodium-Glucose Cotransporter 2 Inhibitor-Induced Improvements in Heart Failure and Chronic Kidney Disease in Diabetic Patients. Metabolites. 2023, 13, 736. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; Khan, S.U.; Lone, A.N.; Ghosh, P.; Kunduru, M.; Sharma, S.; Sattur, S.; Kaluski, E. Sodium-Glucose Cotransporter-2 Inhibitors and Primary Prevention of Atherosclerotic Cardiovascular Disease: A Meta-Analysis of Randomized Trials and Systematic Review. J. Am. Heart. Assoc. 2023, 12, e030578. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, H.; Hakoshima, M.; Umeyama, S.; Iida, S.; Adachi, H.; Yanai, H. Real-World Efficacy of Glucagon-like Peptide-1 (GLP-1) Receptor Agonist, Dulaglutide, on Metabolic Parameters in Japanese Patients with Type 2 Diabetes: A Retrospective Longitudinal Study. Biomedicines. 2023, 11, 869. [Google Scholar] [CrossRef]
- Carbone, L.J.; Angus, P.W.; Yeomans, N.D. Incretin-based therapies for the treatment of non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lv, Q.; Li, S.; Wu, Y.; Li, L.; Li, J.; Zhang, F.; Sun, X.; Tong, N. Efficacy and safety of glucagon-like peptide-1 receptor agonists in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 284–295. [Google Scholar] [CrossRef]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-Like Peptide-1 Receptor Agonists for Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: An Updated Meta-Analysis of Randomized Controlled Trials. Metabolites. 2021, 11, 73. [Google Scholar] [CrossRef]
- Davies, M.J.; D'Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2018, 61, 2461–2498. [Google Scholar] [CrossRef]
- Buse, J.B.; Wexler, D.J.; Tsapas, A.; Rossing, P.; Mingrone, G.; Mathieu, C.; D'Alessio, D.A.; Davies, M.J. 2019 update to: Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2020, 63, 221–228. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes - state-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef]
- Krieger, J.P. Intestinal glucagon-like peptide-1 effects on food intake: Physiological relevance and emerging mechanisms. Peptides. 2020, 131, 170342. [Google Scholar] [CrossRef]
- Bray, J.J.H.; Foster-Davies, H.; Salem, A.; Hoole, A.L.; Obaid, D.R.; Halcox, J.P.J.; Stephens, J.W. Glucagon-like peptide-1 receptor agonists improve biomarkers of inflammation and oxidative stress: A systematic review and meta-analysis of randomised controlled trials. Diabetes Obes Metab. 2021, 23, 1806–1822. [Google Scholar] [CrossRef] [PubMed]
- Katout, M.; Zhu, H.; Rutsky, J.; Shah, P.; Brook, R.D.; Zhong, J.; Rajagopalan, S. Effect of GLP-1 mimetics on blood pressure and relationship to weight loss and glycemia lowering: results of a systematic meta-analysis and meta-regression. Am. J. Hypertens. 2014, 27, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Barchuk, M.; Lobo, M.; Nogueira, J.P. Effect of glucagon-like peptide-1 (GLP-1) analogues on epicardial adipose tissue: A meta-analysis. Diabetes. Metab. Syndr. 2022, 16, 102562. [Google Scholar] [CrossRef]
- Rahman, A.; Alqaisi, S.; Saith, S.E.; Alzakhari, R.; Levy, R. The Impact of Glucagon-Like Peptide-1 Receptor Agonist on the Cardiovascular Outcomes in Patients With Type 2 Diabetes Mellitus: A Meta-Analysis and Systematic Review. Cardiol. Res. 2023, 14, 250–260. [Google Scholar] [CrossRef]
- Katsuyama, H.; Yanai, H.; Adachi, H.; Hakoshima, M. A Significant Effect of Pemafibrate on Hepatic Steatosis and Fibrosis Indexes in Patients With Hypertriglyceridemia. Gastroenterology. Res. 2023, 16, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef]
- Morishita, A.; Oura, K.; Takuma, K.; Nakahara, M.; Tadokoro, T.; Fujita, K.; Tani, J.; Shi, T.; Himoto, T.; Tatsuta, M.; et al. Pemafibrate improves liver dysfunction and non-invasive surrogates for liver fibrosis in patients with non-alcoholic fatty liver disease with hypertriglyceridemia: a multicenter study. Hepatol Int. 2023, 17, 606–614. [Google Scholar] [CrossRef]
- Ahima, R.S. Adipose tissue as an endocrine organ. Obesity (Silver Spring) 2006, 14, 242S–249S. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Alaniz, M.H.; Takada, J.; Alonso-Vale, M.I.; Lima, F.B. Adipose tissue as an endocrine organ: From theory to practice. J. Pediatr. 2007, 83, S192–S203. [Google Scholar] [CrossRef]
- Kim, S.H.; Plutzky, J. Brown Fat and Browning for the Treatment of Obesity and Related Metabolic Disorders. Diabetes. Metab. J. 2016, 40, 12–21. [Google Scholar] [CrossRef]
- Rachid, T.L.; Silva-Veiga, F.M.; Graus-Nunes, F.; Bringhenti, I.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Differential actions of PPAR-alpha and PPAR-beta/delta on beige adipocyte formation: A study in the subcutaneous white adipose tissue of obese male mice. PLoS. One. 2018, 13, e0191365. [Google Scholar] [CrossRef]
- Maia-Fernandes, T.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F. Cardiovascular actions of adiponectin: Pathophysiologic implications. Rev. Port. Cardiol. 2008, 27, 1431–1449. [Google Scholar] [PubMed]
- Haluzík, M.M.; Haluzík, M. PPAR-alpha and insulin sensitivity. Physiol. Res. 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Haluzik, M.; Asghar, Z.; Yau, D.; Joseph, J.W.; Fernandez, A.M.; Reitman, M.L.; Yakar, S.; Stannard, B.; Heron-Milhavet, L.; et al. Peroxisome proliferator-activated receptor-alpha agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes. 2003, 52, 1770–1778. [Google Scholar] [CrossRef]
- Qiang, X.; Xu, L.; Zhang, M.; Zhang, P.; Wang, Y.; Wang, Y.; Zhao, Z.; Chen, H.; Liu, X.; Zhang, Y. Demethyleneberberine attenuates non-alcoholic fatty liver disease with activation of AMPK and inhibition of oxidative stress. Biochem. Biophys. Res. Commun. 2016, 472, 603–609. [Google Scholar] [CrossRef]
- Liu, S.; Jing, F.; Yu, C.; Gao, L.; Qin, Y.; Zhao, J. AICAR-Induced Activation of AMPK Inhibits TSH/SREBP-2/HMGCR Pathway in Liver. PLoS. One. 2015, 10, e0124951. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends. Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Yun, H.; Park, S.; Kim, M.J.; Yang, W.K.; Im, D.U.; Yang, K.R.; Hong, J.; Choe, W.; Kang, I.; Kim, S.S.; Ha, J. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS. J. 2014, 281, 4421–4438. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Wolff, D.A.; Huskin, A.L.; Helenowski, I.B.; Rademaker, A.W. Fenofibrate therapy ameliorates fasting and postprandial lipoproteinemia, oxidative stress, and the inflammatory response in subjects with hypertriglyceridemia and the metabolic syndrome. Diabetes. Care. 2007, 30, 1945–1951. [Google Scholar] [CrossRef]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Molecular Biological and Clinical Understanding of the Statin Residual Cardiovascular Disease Risk and Peroxisome Proliferator-Activated Receptor Alpha Agonists and Ezetimibe for Its Treatment. Int. J. Mol. Sci. 2022, 23, 3418. [Google Scholar] [CrossRef]
- Lee, M.; Saver, J.L.; Towfighi, A.; Chow, J.; Ovbiagele, B. Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: a meta-analysis. Atherosclerosis. 2011, 217, 492–498. [Google Scholar] [CrossRef]
- Das Pradhan, A.; Glynn, R.J.; Fruchart, J.C.; MacFadyen, J.G.; Zaharris, E.S.; Everett, B.M.; Campbell, S.E.; Oshima, R.; Amarenco, P.; Blom, D.J.; et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N. Engl. J. Med. 2022, 387, 1923–1934. [Google Scholar] [CrossRef]
- Shinozaki, S.; Tahara, T.; Miura, K.; Lefor, A.K.; Yamamoto, H. Effectiveness of One-Year Pemafibrate Therapy on Non-Alcoholic Fatty Liver Disease Refractory to Long-Term Sodium Glucose Cotransporter-2 Inhibitor Therapy: A Pilot Study. Life (Basel). 2023, 13, 1327. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Keach, J.C.; Batts, K.P.; Lindor, K.D. Independent predictors of liver fibrosis in patients with non-alcoholic steatohepatitis. Hepatology. 1999, 30, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.H.; Xin, Y.N.; Dong, Q.J.; Wang, Q.; Jiang, X.J.; Zhan, S.H.; Sun, Y.; Xuan, S.Y. Performance of the aspartate aminotransferase-to-platelet ratio index for the staging of hepatitis C-related fibrosis: an updated meta-analysis. Hepatology. 2011, 53, 726–736. [Google Scholar] [CrossRef]
Figure 5.
Changes in serum aspartate aminotransferase (AST) levels in patients treated with pemafibrate and sodium glucose cotransporter 2 inhibitors (SGLT2i) (red line) and those treated with pemafibrate without SGLT2i (blue line). This figure was made by the modificaiton of Table 3 in reference 68. Presented values indicate mean ± SE. *p < 0.05 vs. baseline.
Figure 5.
Changes in serum aspartate aminotransferase (AST) levels in patients treated with pemafibrate and sodium glucose cotransporter 2 inhibitors (SGLT2i) (red line) and those treated with pemafibrate without SGLT2i (blue line). This figure was made by the modificaiton of Table 3 in reference 68. Presented values indicate mean ± SE. *p < 0.05 vs. baseline.
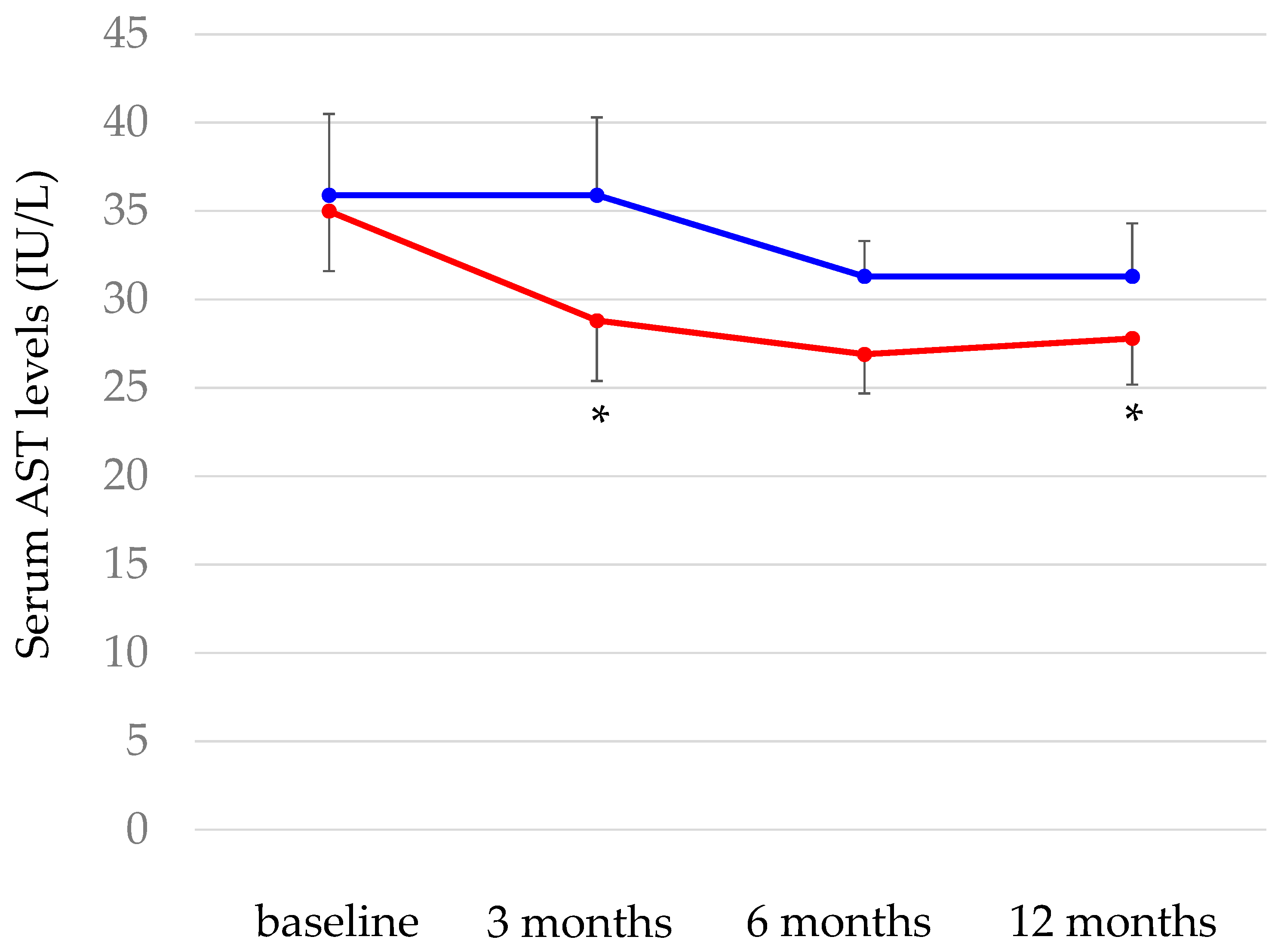
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



