
Submitted:
17 September 2023
Posted:
18 September 2023
You are already at the latest version
Abstract
Systemic autoimmune diseases (SAIDs) such as systemic lupus erythematosus (SLE), systemic sclerosis (SSc) and rheumatoid arthritis (RA) are fully related to the unregulated innate and adaptive immune systems involved in their pathogenesis. They have similar pathogenic characteristics including the interferon signature, loss of tolerance to self-nuclear antigens, and enhanced tissue damage like necrosis and fibrosis. Glucocorticoids and immunosuppressants, which have limited specificity and are prone to tolerance, are used as the first-line therapy. A plethora of novel immunotherapies have been developed including monoclonal and bispecific antibodies, and other biological agents to target cellular and soluble factors involved in disease pathogenesis such as B cells, co-stimulatory molecules, cytokines or their receptors, and signalling molecules. Many of these have shown encouraging results in clinical trials. CAR-T cell therapy is considered the most promising technique for curing autoimmune diseases, with recent successes in the treatment of SLE and SSc.
Keywords:
Immunotherapy
; autoimmune diseases
; monoclonal antibodies
; bispecific antibodies
; CAR-T cells
Introduction
Approximately 5–10% of the world's population suffers from autoimmune diseases [1]. In these patients a highly complex network of cytokines and their receptors on immune cells destroys healthy tissues and becomes overactive. Many various physiological systems, including the joints, skin, kidneys, blood cells, heart, and lungs, might be affected by the symptoms of these disorders. Conventional treatment, mainly with glucocorticoids and immunosuppressants, has been the mainstay of therapy for moderate to severe disease for many decades, but has poor specificity and is prone to tolerance [2]. There are patients who have persistently active disease despite treatment, and those who suffer from side effects of drugs [3]. It wasn't until 2011 that the US Food and Drug Administration (FDA) authorized the first biologic drug (belimumab) for the treatment of people with active SLE [4].
Particularly for individuals who do not react to traditional therapies, targeted therapy has recently become a more promising option [5]. Novel biologic agents with different mechanisms of action including monoclonal (mAb) and bispecific antibodies (BsAb) can target B cells, co-stimulatory molecules, cytokines or their receptors, demonstrating the clinical efficacy and safety [6,7]. Some agents such as rituximab, tocilizumab, anifrolumab and abatacept have shown activity in several autoimmune diseases including SLE, SSc, and RA, and are currently in clinical trials. Furthermore, the main aspects of SAIDs pathogenesis including the activation of autoreactive B cell clones and suppression of cytotoxic or regulatory T cells imply the possibility of therapeutic application of chimeric antigen receptor T (CAR-T) cells to treatment autoreactive immune cells.
The main progress of CAR-T cells has been realized in oncology, although improvements in SAIDs have also been made. According to recent evidence, treating SLE using CD19 CAR-T cells is viable and highly effective process [8]. Similar data were obtained in patient with severe systemic sclerosis demonstrating the high potential of this method in clinical practice [9]. Progress in manufacturing of CAR-T and other cell immunotherapies allowed obtaining more affordable products for both cancer and autoimmune diseases [10]. Recent developments in CAR-T therapy are anticipated to usher in a new age of autoimmune disease treatment. This article discusses current advances in immunotherapy and future research in this field for the successful treatment of autoimmune diseases, using SLE, SSc and RA as examples.
Systemic lupus erythematosus (SLE)
Lupus erythematosus is a collection of autoimmune diseases, and SLE is the most severe form that affects predominantly young women. With a variety of clinical symptoms and blood autoantibodies that target DNA, RNA, and associated proteins, this illness exhibits great heterogeneity [11]. Other form of lupus is cutaneous lupus erythematosus (CLE) divided into several subsets including chronic cutaneous LE (CCLE) and discoid LE (DLE), the most common type of CCLE with inflamed skin.
The pathogenesis involving genetic factors, epigenetics, and environmental factors resulting in the immune abnormalities leads to the loss of tolerance and sustained autoantibody production. The abnormal activation of B cells and T cells with abundant autoantibodies in tissues and organs results in damage and inflammation [12]. Since first-line therapy including glucocorticoids and immunosuppressants are known to be addictive and because significant side effects, novel targeted therapy with biological agents may be a good alternative [13].
The pathophysiology of SLE is largely governed by B lymphocytes. Autoantibodies produced by autoreactive B cells accumulate in tissues and organs, where they cause inflammation and damage. Effector cells such as plasma cells are differentiated B-lymphocytes that produce immunoglobulins essential for humoral immune response. Other than production of autoantibodies, B cells also secrete cytokines and have antigen presenting capability in the processing and presentation of self-antigens to autoreactive T cells leading to their activation and differentiation [14]. In this connection, the high-priority strategy of targeted therapy is blocking of B-cell-related cytokines and signal molecules by monoclonal antibodies and other biological agents. The main targeted pathways include BAFF/APRIL and inhibition [15].
The B cell activating factor (BAFF or BLyS) is a survival factor that regulates B cell maturation, differentiation, and antibody production by binding to three different receptors, BAFF-R, TACI and BCMA [16]. The level of BAFF was found to correlate with disease activity [17]. And its homolog, APRIL (a proliferating inducing ligand) forms heterotrimers with BAFF and enhance BAFF-like activity [18,19].
Belimumab is the first biological agent to be authorized by the FDA for SLE. This is a fully humanized mAb that binds to soluble BAFF and prevents it from attaching to the three receptors [20]. This antibody was shown to be efficacious in active seropositive patients with low complement and positive anti-dsDNA antibodies by two large phase III trials [21,22]. Other agents targeted BAFF/APRIL family are shown in Table 1.
CD20, a transmembrane calcium channel is involved in the activation, proliferation and differentiation of B cells [23] as well as CD22, a receptor on the surface of the B cell membrane promotes the proliferation and differentiation of B cells by regulating the signal transduction of the B cell receptor (BCR) [24]. Blocking of these two B-cell membrane receptors to inhibit B cell proliferation and reduce the inflammatory response is also one of the targets for biological agents [25,26].
Mature B cells, Tfh cells, and Treg cells all express CXCR5, which is also important in B cell migration, the development of germinal cells (GCs), and the direction of disease-causing double negative T-cells towards lymphoid organs and the kidneys [27]. Also follicular dendritic cells and macrophages express CXCL13, that is a ligand of CXCR5 [28]. Both molecules are crucial for the development and movement of B lymphocytes. Some studies show that circulating CXCL13 levels rise in SLE patients, suggesting a new target for the disease's therapy [29]. MAb5261 is a humanized IgG mAb against CXCL13 in preclinical stage currently [30].
The interaction of costimulatory signals with T cells, particularly CD40/40L, CD28, CD80/CD86, and inducible T cell co-stimulator ligand (ICOSL) is necessary for the immune activation of B cells. CD40 on antigen-presenting cell (APC) binds to its ligand CD40L expressed on T cell resulting in upregulation of CD80/CD86 and other co-stimulatory molecules on APC [31]. T cell activation results from CD28, which is constitutively expressed on T cells, binding to CD80/CD86 on APC. This is followed by an increase in CTLA-4 expression on the activated T cell, which has a greater affinity for binding to CD80/CD86 than CD28 providing negative signals to T cell activation [32]. By obstructing this route, one may indirectly stop the growth and activation of B cells as well as the creation of antibodies thus achieving a therapeutic effect [33].
The pathogenesis of autoimmune diseases involves releasing of many pro- and anti-inflammatory cytokines such as IL-10, IL-12, IL-18 and those involved in the IL-23/IL-17 pathway and IL-33/ST2 axis. The elevated levels of these cytokines in serum of SLE patients were found to correlate with the disease activity [34,35,36]. Targeting of cytokines is one more approach in addition to above-mentioned of SLE immunotherapies. Novel biological agents inhibited such cytokines like IL-6, IL-17, IFNɑ, TNFɑ, IL21/IL23 were developed last years and are objects for clinical trials (Table 1). Anifrolumab, mAb, was approved for medical use in the United States in July 2021, and in the European Union in February 2022. It binds to the type I interferon receptor, blocking the activity of type I interferons such as interferon-α and interferon-β [37].
Bispecific antibodies (BsAbs) are a subclass of monoclonal antibodies (mAbs) that can bind two antigens at once. Non-IgG-like BsAbs and IgG-like BsAbs are the two groups of BsAbs that may be separated based on the presence or absence of Fc segments. Non-IgG-like BsAbs exert their effects via structural domains attached to antigens. They have low molecular weights, and have strong tissue penetration. However, the antibodies are unable to mediate complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) owing to their modest molecular weights and absence of receptor-binding Fc structures [38,39]. The creation of BsAbs that resemble IgG has been facilitated by the advancement of genetic engineering techniques [40]. A subtype of BsAbs called bispecific T cell engagers (BiTEs) has been developed to simultaneously bind tumor-expressed antigens (e.g. BCMA, CD19) and CD3 on T cells [41]. Blinatumomab was the first BiTE approved by the US FDA to treat acute lymphoblastic leukemia [42].
Bispesific antibodies (BsAbs) are also currently being studied in SLE and other autoimmune diseases (Table 1). The advantage of BsAbs is blocking multiple activation pathways, providing a broader scope of inhibition. This can lead to a more comprehensive and effective treatment for SAID patients, where multiple pathways with cytokines contribute to disease activity. This approach holds immense potential for treating various autoimmune diseases providing tailored treatment options. Finding more specific key targets is critical in the development of BsAbs. Also further research efforts should be aimed at reducing the side effects of the use of BsAbs, especially immunogenicity. Furthermore, as research and development technology continues to advance, the production cost of BsAbs is expected to decrease significantly. This reduction in cost will make it more accessible for clinicians to customize treatment plans based on individual patient needs.
Despite significant advances in the treatment of SLE with antibodies, the CD20-targeting mAb rituximab has been shown to deplete B cells in tissues incompletely and a significant proportion of B cells survive depletion, suggesting limitations of this approach in certain severe forms of the disease [43,44]. Additionally, rituximab is not effective in depleting plasmablasts and long-lived plasma cells, which are implicated in the generation of autoantibodies in SLE, because they do not express CD20 [45]. The profound reduction of plasmablasts and CD19+ B lymphocytes in tissues theoretically might improve immunosuppressive therapy. In this respect employment of CD19-CAR T cells therapy would be a proper decision. Because they may reach deep tissues, anti-CD19 CAR T cells provide an additional benefit over anti-CD20 mAb treatment. CAR-T cells are bioengineered redirected T lymphocytes that have a specialized membrane receptor to recognize a specific antigen of a target cell without MHC restriction [46]. T cells from peripheral blood of a patient are separated and then CAR genes are inserted into T cell genome to form CAR-T cells. The created CAR-T cells are then expanded and infused back into the patient [47].
Targeting B cells and their malignant offspring using their highly specific and prevalent surface antigen CD19 is currently the most advanced method of CAR T cell therapy, notably in the treatment of cancer [46,48] (Figure 1). Two preclinical studies in lupus-prone mice support the efficacy of CD19 CAR T cells in SLE. A CD19-targeting CAR T cell treatment was found to decrease B cells, stop the formation of autoantibodies, and restore organ symptoms [49,50]. Also, the rapid disappearance of dsDNA autoantibodies and clinical remission were achieved in a single patient with severe and refractory SLE presented with active lupus nephritis, suggesting the principle feasibility of CD19-CAR T cell therapy for treatment SLE and other autoimmune diseases [51].
Based on these researches five patients with SLE were enrolled in a special CAR T cell program. This study revealed the deep depletion of B cells, improvement of clinical symptoms and normalization of laboratory parameters including seroconversion of anti-double-stranded DNA antibodies. After three months, all five patients had achieved stable remission of SLE according to DORIS criteria. These data indicate that CD19 CAR T cell transfer is feasible, tolerable and highly effective in SLE [8].
Systemic Sclerosis (SSc)
SSc is an autoimmune disease characterized by excessive connective tissue deposition and fibrosis, vasculopathy and a dysregulated immune system [52].
B cells were found to play a significant role in the pathogenesis of severe SSc, contributing to the development of fibrosis. The existing data include the increased levels of B-cell stimulating substances, altered B-cell homeostasis with expansion of naïve and reduction in memory B cells, and antifibrotic effects of B-cell depletion in murine fibrosis models [53]. The CD20-targeting antibody rituximab showed promising effectiveness in the first randomized controlled trials, however its therapeutic importance in SSc is not verified later. While rituximab slowed forced vital capacity decline in one research, another experiment showed considerable skin improvement [54,55].
Besides B cells the role of T cells in SSc is also discussed. The activation of T cells is verified by the early presence of IgG autoAbs, the presence of T cell infiltrates in skin prior to fibrosis, and the production of T cell cytokines IL-4 and Il-13 [56]. The pathophysiology of the illness also involves other cells, such as macrophages and profibrotic growth factors like TGFb. Through the generation of profibrotic, inflammatory, and vasoconstrictive mediators, platelets contribute to the pathophysiology of SSc [57]. Immunotherapy of SSc includes general immunosuppression and therapies targeting specific molecules involved in T cell and B cell survival and function. MAbs and BsAbs currently in clinical trials for the treatment of SSc are listed in Table 1.
As in instances of SLE, CD20+ B-cell depletion was speculated to be insufficient in SSc, moreover plasmablasts, which may also be responsible for autoantibody production, are targeted via CD19 [58]. Nowadays, the major use of CAR-T cells is in the treatment of various cancers. However, the recent study in SLE is not only a potential game-changer for the treatment of SLE with CD-19-targeted CAR-T cells, but it is also very pertinent to SSc [8].
The first treatment of a severe refractory SSc patient with CD19-CAR T cells was reported recently. T-cells have been genetically modified outside of the body to produce an artificial T cell receptor targeting CD19 expressed on B-cells, and then injected into the patient (Figure 1). CAR T-cells then find CD19-expressing cells and kill them [58]. A 60-year-old man presented with diffuse cutaneous SSc with first non-Raynaud’s disease manifestation (skin, lung and heart fibrosis) demonstrated significant improvement in carpal arthritis 3 months after CAR T-cell therapy. Skin, heart and joint fibrosis also showed a tendency of improvement, and the patients subjectively reported less frequent and less severe attacks of Raynaud’s phenomenon. These findings indicate that B cell-mediated autoimmunity plays a significant role in severe SSc and support the early idea that CD19-targeting CAR T-cell therapy may be useful in treating SAIDs.
Rheumatoid arthritis (RA)
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease that primarily affects the lining of the synovial joints and is associated with progressive disability, premature death, and socioeconomic burdens. The current treatment of RA consists of nonsteroidal anti-inflammatory drugs, glucocorticoids, conventional disease-modifying antirheumatic drugs, biologic and targeted synthetic drugs. The use of biological agents in the treatment of RA over the past two decades has fundamentally altered the clinical course of this chronic deforming disease. A number of druggable targets has been identified and developed to treat RA, such as B cells and fibroblast-like synoviocytes (FLS), cytokines (e.g. tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-6 and granulocyte-macrophage colony-stimulating factor (GM-CSF)), signaling pathways (e.g. Janus kinase family (JAK)) and epigenetic regulators (e.g. DNA methylation, histone modifications, and noncoding RNAs) [59,60,61].
Along with other autoimmune diseases the main B cell depletion strategy is anti-CD20 antibody therapy [62]. Rituximab has demonstrated efficacy in patients with RA [63]. However, its effectiveness in other SAIDs was not proven. This applies to both SLE [43,44] and SSc [54,55].
Other critical targets in the pathogenesis of RA are IL-6 and TNF-α (Table 1). IL-6 stimulates neutrophil recruitment to the joints during the early stages of RA, which facilitates the following infiltration of monocytes into the synovial fluid [64]. Excessive and persistent joint inflammation damages tissue via bone erosion, activation of osteoclasts, and cartilage damage. Additionally, IL-6 directly promotes osteoclast activity by causing synovial cells to produce RANKL [65].
Anti-IL-6 mAb tocilizumab has shown beneficial efficacy in refractory RA patients [66]. Moreover, monotherapy with tocilizumab was revealed to have a greater effect in decreasing RA activity compared with monotherapy with adalimumab, a TNF-α antagonist [67]. Thus, tocilizumab was approved for RA treatment in Japan, the European Union (EU), and the United States in 2008, 2009, and 2010, respectively. An alternative anti-IL-6R antibody, sarilumab, approved for the treatment of RA in 2017 in Japan, the EU, and the United States is a human monoclonal antibody that has higher affinity than tocilizumab [68]. Clinical trials of other monoclonal antibodies against IL-6, including sirukumab, clazakizumab, and olokizumab, for RA are ongoing.
Infliximab, the first tumor necrosis factor-alpha (TNF-α) inhibitor, has been approved by FDA for the treatment of RA in 1998 [69]. The development of several new targeted therapies with different modes of action that are effective in controlling inflammatory arthritis with delayed structural damage and a favorable side-effect profile followed [70] (Table 1).
Recently, the CD-19 CAR-T cells were successfully utilized in treating of patients with SLE and SSc [8,58]. According to the previous data, two different approaches can be used in therapy of RA by chimeric modification of the T cell receptor additionally. One of them is chimeric autoantibody receptor T cells (CAAR-T cells) which have a high affinity for an autoantibody on the B cell surface and generate a cytotoxic effect. And another one is chimeric antigen receptor in regulatory T lymphocytes (CAR-Tregs) that bind to a specific antigen in a target cell to execute activation and regulatory function of Tregs [71] (Figure 1).
Targeting autoreactive B cells, CAAR-T comprises of a particular antigen, a transmembrane domain, and an intracellular signaling domain. It is a variant of modified CAR (Figure 1). Unlike the scFv domain, the CAAR determines the selective cytotoxicity of CAAR-T cells solely against immune cells that carry receptors to certain autoantigens without overall immunosuppression. When the specific antigen of the CAAR-T cells binds to the specific auto-antibodies expressed on B cells, the B cells will be destroyed, thus reducing the generation of autoantibodies [72].
Since Tregs are usually suppressed in SAIDs, including RA, another effectual approach for the restoration of immune tolerance implies “switching” T cell phenotype from cytotoxic to regulatory. Compared with CAR-T and CAAR-T, CAR-Tregs are more commonly investigated in autoimmune diseases. CAR redirects Tregs to the site where autoimmune activity happens, increasing their suppressive efficiency without systemic immunosuppression [73]. CAR-Tregs represent CAR-T cells converted to Tregs by transduction of FOXP3 (along with CAR), which is a member of the FOX protein family that controls pathways responsible for the development and function of regulatory T cells [74]. FOXP1 knockout in CAR-T-cells may improve their expansion and persistence, also the reduction of FOXP1 levels may help alleviate autoimmune diseases driven by abnormal B-cell responses [75]. CAR-Tregs recognize and regulate autoimmune T lymphocytes through induction of anergy, immunological ignorance, and clonal deletion [76]. CAR-Tregs generate immunosuppressive cytokines such as IL-10, IL-35, and TGF-β and induce apoptosis of Teff cells via Fas-ligand, granzyme B/A, and perforin, thus suppressing Teff cells [77].
Therefore, CAAR-T, and CAR-Treg cells may be promising approaches in patients affected by RA additionally to the CD-19 CAR-T cells therapy, which has already been proven effective in treatment of SLE and SSc [8,58].
Since a CAR or CAAR only targets a single cell type, its applicability to RA, which often exhibits a variety of autoreactive responses, is limited. The idea was to develop a customized therapeutic strategy using a universal CAR-T cell system that allows targeting of various types of autoreactive B cell subsets by T cells expressing a single scFv combined with known autoantigen peptides according to patient’s specific auto-antigen profiles [78]. The authors used an approach previously developed for cancer therapy [79]. Anticitrullinated protein antibodies (ACPAs) are one of the most specific serological markers for RA that is linked to the onset of the illness [80]. Four autoantibody-positive citrullinated peptides, including citrullinated vimentin, citrullinated type II collagen, citrullinated fibrinogen and tenascin C, were selected as mediators for targeting autoreactive B cells by redirection of CAR-Ts. For this purpose, mediators were conjugated with fluorescein isothiocyanate (FITC) and anti-FITC CAR-Ts were prepared as described in an earlier study [81]. The results showed that the engineered T cells effectively targeted and eliminated autoreactive B cells expressing autoantibodies to citrullinated vimentin, citrullinated type II collagen, citrullinated fibrinogen, and tenascin C. The specificity of the T cells was confirmed by their recognition of the FITC-labeled auto-antigenic peptide epitopes. This approach holds promise for the development of targeted therapies for autoimmune diseases characterized by the presence of autoreactive B cells. By selectively eliminating these pathogenic B cells, it may be possible to mitigate the immune response and reduce disease severity. Further studies are warranted to optimize this strategy and evaluate its efficacy in preclinical models and eventually in clinical trials. Overall, not only RA but other autoimmune diseases may benefit from this innovative approach.
Conclusion and Perspective
Despite substantial advances in the treatment of SAIDs, some patients do not respond to the current conventional therapies and are at high risk for organ failure and even death. For this reason, new options have emerged, including immunotherapy. SAIDs such as SLE, SSc, and RA share common molecular targets for antibody treatment including B cells, T-cell or T-cell co-stimulator blockers and pro-inflammatory cytokines or cytokine receptors (Table 1). Some biological agents such as rituximab, tocilizumab, anifrolumab and abatacept have shown activity in several autoimmune diseases including SLE, SSc, and RA, discussed above, and are currently in clinical trials (Figure 2). But even therapeutics using biological agent was found to be not entirely efficacious. The development of CAR T cells represents an innovative way to counteract several of the limitations of biologic therapies, including adverse effects from repeated injections, incomplete autoantibodies depletion, or immunogenicity. Recently, the CD-19 CAR-T cells were successfully utilized in treating patients with SLE and SSc clearing the way for treating RA and other SAIDs. Advances with this therapy have shown very encouraging results. This approach holds great potential for improving the lives of patients with autoimmune diseases. CAAR-T cells could be also used to deplete pathogenic B cells specifically, and CAR-Treg cells may be a promising therapy to promote localized tolerance in organs affected by RA and other autoimmune diseases. Furthermore, the use of different targets for these modified T cells allows for fine-tuning of their function, providing a personalized approach to treatment. This innovative approach holds great promise for improving outcomes for patients affected by SAID. Several CD-19 CAR-T cell preclinical approaches have been studied or are ongoing in patients with autoimmune diseases, including SLE and SSc. Further research and clinical trials are needed to fully explore the potential of CAAR-T and CAR-Treg cell therapy in this context, but the initial results are highly encouraging. By understanding the unique characteristics of each therapeutic agent, healthcare professionals can optimize therapy outcomes and alleviate symptoms more effectively. The future of immunotherapy in precision medicine is indeed promising, offering hope for improved management of SAIDs and enhanced patient outcomes.
Funding
This work was funded by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (project number FZSM-2023-0011).
Acknowledgments
The study has been performed according to the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030).
List of abbreviations
| ADCC | Antibody-dependent cell-mediated cytotoxicity |
| ACPA | Anticitrullinated protein antibody |
| APRIL | A proliferating inducing ligand |
| BAFF | B cell activating factor |
| BsAb | Bispecific antibody |
| CAR-T cell | Chimeric antigen receptor T cell |
| CAAR-T cell | Chimeric autoantibody receptor T cell |
| CCLE | Chronic cutaneous lupus erythematosus |
| CDC | Complement-dependent cytotoxicity |
| CLE | Cutaneous lupus erythematosus |
| CTLA-4 | Cytotoxic T lymphocyte-associated protein 4 |
| DLE | Discoid lupus erythematosus |
| FDA US | Food and Drug Administration |
| FITC | Fluorescein isothiocyanate |
| FLS | Fibroblast-like synoviocytes |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| JAK | Janus kinase family |
| mAb | Monoclonal antibody |
| SAIDs | Systemic autoimmune diseases |
| SLE | Systemic lupus erythematosus |
| TNF | Tumor necrosis factor |
| RA | КRheumatoid arthritis |
References
- Wang, L.; Wang, F.; Gershwin, M.E. Human Autoimmune Diseases: A Comprehensive Update. J. Intern. Med. 2015, 278, 369–395. [CrossRef]
- Dima, A.; Jurcut, C.; Arnaud, L. Hydroxychloroquine in Systemic and Autoimmune Diseases: Where Are We Now? Jt. Bone Spine 2021, 88, 105143. [CrossRef]
- Emamikia, S.; Gentline, C.; Chatzidionysiou, K.; Arnaud, L.; Vollenhoven, R. van Relationship between Glucocorticoid Dose and Adverse Events in Systemic Lupus Erythematosus: Data from a Randomized Clinical Trial. Scand. J. Rheumatol. 2018, 47, 131–140. [CrossRef]
- Dubey, A.K.; Handu, S.S.; Dubey, S.; Sharma, P.; Sharma, K.K.; Ahmed, Q.M. Belimumab: First Targeted Biological Treatment for Systemic Lupus Erythematosus. J. Pharmacol. Pharmacother. 2011, 2, 317–319. [CrossRef]
- Felten, R.; Dervovic, E.; Chasset, F.; Gottenberg, J.-E.; Sibilia, J.; Scher, F.; Arnaud, L. The 2018 Pipeline of Targeted Therapies under Clinical Development for Systemic Lupus Erythematosus: A Systematic Review of Trials. Autoimmun. Rev. 2018, 17, 781–790. [CrossRef]
- Hafeez, U.; Gan, H.K.; Scott, A.M. Monoclonal Antibodies as Immunomodulatory Therapy against Cancer and Autoimmune Diseases. Curr. Opin. Pharmacol. 2018, 41, 114–121. [CrossRef]
- Zhao, Q. Bispecific Antibodies for Autoimmune and Inflammatory Diseases: Clinical Progress to Date. BioDrugs 2020, 34, 111–119. [CrossRef]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Wilhelm, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T Cell Therapy for Refractory Systemic Lupus Erythematosus. Nat. Med. 2022, 28, 2124–2132. [CrossRef]
- Bergmann, C.; Müller, F.; Jörg, D.; Györfi, D.M.H.; Völkl, S.; Aigner, M.; Harrer, T.; Bayerl, N.; Atzinger, A.; Taubmann, J.; et al. AB0816 TREATMENT OF A PATIENT WITH SEVERE DIFFUSE SYSTEMIC SCLEROSIS (SSC) USING CD19-TARGETING CAR-T-CELLS. Ann. Rheum. Dis. 2023, 82, 1621.1-1621. [CrossRef]
- Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Kudriaeva, A.; Miftakhova, R.; Rybalov, A.; Bulatov, E. Recent Advances in the Development of Bioreactors for Manufacturing of Adoptive Cell Immunotherapies. Bioengineering 2022, 9, 808. [CrossRef]
- Yaniv, G.; Twig, G.; Shor, D.B.-A.; Furer, A.; Sherer, Y.; Mozes, O.; Komisar, O.; Slonimsky, E.; Klang, E.; Lotan, E.; et al. A Volcanic Explosion of Autoantibodies in Systemic Lupus Erythematosus: A Diversity of 180 Different Antibodies Found in SLE Patients. Autoimmun. Rev. 2015, 14, 75–79. [CrossRef]
- Mohan, C.; Putterman, C. Genetics and Pathogenesis of Systemic Lupus Erythematosus and Lupus Nephritis. Nat. Rev. Nephrol. 2015, 11, 329–341. [CrossRef]
- Mok, M.Y.; Shoenfeld, Y. Recent Advances and Current State of Immunotherapy in Systemic Lupus Erythematosus. Expert Opin. Biol. Ther. 2016, 16, 927–939. [CrossRef]
- Tanaka, Y.; Kubo, S.; Iwata, S.; Yoshikawa, M.; Nakayamada, S. B Cell Phenotypes, Signaling and Their Roles in Secretion of Antibodies in Systemic Lupus Erythematosus. Clin. Immunol. 2018, 186, 21–25. [CrossRef]
- Yang, B.; Zhao, M.; Wu, H.; Lu, Q. A Comprehensive Review of Biological Agents for Lupus: Beyond Single Target. Front. Immunol. 2020, 11, 539797. [CrossRef]
- Bossen, C.; Schneider, P. BAFF, APRIL and Their Receptors: Structure, Function and Signaling. Semin. Immunol. 2006, 18, 263–275. [CrossRef]
- Zollars, E.; Bienkowska, J.; Czerkowicz, J.; Allaire, N.; Ranger, A.M.; Magder, L.; Petri, M. BAFF (B Cell Activating Factor) Transcript Level in Peripheral Blood of Patients with SLE Is Associated with Same-Day Disease Activity as Well as Global Activity over the next Year. Lupus Sci. Med. 2015, 2, e000063. [CrossRef]
- Mackay, F.; Ambrose, C. The TNF Family Members BAFF and APRIL: The Growing Complexity. Cytokine Growth Factor Rev. 2003, 14, 311–324. [CrossRef]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The BAFF/APRIL System in SLE Pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [CrossRef]
- Baker, K.P.; Edwards, B.M.; Main, S.H.; Choi, G.H.; Wager, R.E.; Halpern, W.G.; Lappin, P.B.; Riccobene, T.; Abramian, D.; Sekut, L.; et al. Generation and Characterization of LymphoStat-B, a Human Monoclonal Antibody That Antagonizes the Bioactivities of B Lymphocyte Stimulator. Arthritis Rheum. 2003, 48, 3253–3265. [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and Safety of Belimumab in Patients with Active Systemic Lupus Erythematosus: A Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2011, 377, 721–731. [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A Phase III, Randomized, Placebo-controlled Study of Belimumab, a Monoclonal Antibody That Inhibits B Lymphocyte Stimulator, in Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [CrossRef]
- Dörner, T.; Lipsky, P.E. Beyond Pan-B-Cell-Directed Therapy — New Avenues and Insights into the Pathogenesis of SLE. Nat. Rev. Rheumatol. 2016, 12, 645–657. [CrossRef]
- Macauley, M.S.; Pfrengle, F.; Rademacher, C.; Nycholat, C.M.; Gale, A.J.; Drygalski, A. von; Paulson, J.C. Antigenic Liposomes Displaying CD22 Ligands Induce Antigen-Specific B Cell Apoptosis. J. Clin. Investig. 2013, 123, 3074–3083. [CrossRef]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-Generation Anti-CD20 Monoclonal Antibodies in Autoimmune Disease Treatment. Autoimmun. Highlights 2017, 8, 12. [CrossRef]
- Daridon, C.; Blassfeld, D.; Reiter, K.; Mei, H.E.; Giesecke, C.; Goldenberg, D.M.; Hansen, A.; Hostmann, A.; Frölich, D.; Dörner, T. Epratuzumab Targeting of CD22 Affects Adhesion Molecule Expression and Migration of B-Cells in Systemic Lupus Erythematosus. Arthritis Res. Ther. 2010, 12, R204. [CrossRef]
- Valentine, K.M.; Hoyer, K.K. CXCR5+ CD8 T Cells: Protective or Pathogenic? Front. Immunol. 2019, 10, 1322. [CrossRef]
- Da, Z.; Li, L.; Zhu, J.; Gu, Z.; You, B.; Shan, Y.; Shi, S. CXCL13 Promotes Proliferation of Mesangial Cells by Combination with CXCR5 in SLE. J. Immunol. Res. 2016, 2016, 2063985. [CrossRef]
- Bao, Y.-Q.; Wang, J.-P.; Dai, Z.-W.; Mao, Y.-M.; Wu, J.; Guo, H.-S.; Xia, Y.-R.; Ye, D.-Q. Increased Circulating CXCL13 Levels in Systemic Lupus Erythematosus and Rheumatoid Arthritis: A Meta-Analysis. Clin. Rheumatol. 2020, 39, 281–290. [CrossRef]
- Klimatcheva, E.; Pandina, T.; Reilly, C.; Torno, S.; Bussler, H.; Scrivens, M.; Jonason, A.; Mallow, C.; Doherty, M.; Paris, M.; et al. CXCL13 Antibody for the Treatment of Autoimmune Disorders. BMC Immunol. 2015, 16, 6. [CrossRef]
- Schoenberger, S.P.; Toes, R.E.M.; Voort, E.I.H. van der; Offringa, R.; Melief, C.J.M. T-Cell Help for Cytotoxic T Lymphocytes Is Mediated by CD40–CD40L Interactions. Nature 1998, 393, 480–483. [CrossRef]
- McCoy, K.D.; Gros, G.L. The Role of CTLA-4 in the Regulation of T Cell Immune Responses. Immunol. Cell Biol. 1999, 77, 1–10. [CrossRef]
- Sharabi, A.; Tsokos, G.C. T Cell Metabolism: New Insights in Systemic Lupus Erythematosus Pathogenesis and Therapy. Nat. Rev. Rheumatol. 2020, 16, 100–112. [CrossRef]
- Koenig, K.F.; Groeschl, I.; Pesickova, S.S.; Tesar, V.; Eisenberger, U.; Trendelenburg, M. Serum Cytokine Profile in Patients with Active Lupus Nephritis. Cytokine 2012, 60, 410–416. [CrossRef]
- MOK, M.Y.; WU, H.J.; LO, Y.; LAU, C.S. The Relation of Interleukin 17 (IL-17) and IL-23 to Th1/Th2 Cytokines and Disease Activity in Systemic Lupus Erythematosus. J. Rheumatol. 2010, 37, 2046–2052. [CrossRef]
- Mok, M.Y.; Huang, F.P.; Ip, W.K.; Lo, Y.; Wong, F.Y.; Chan, E.Y.T.; Lam, K.F.; Xu, D. Serum Levels of IL-33 and Soluble ST2 and Their Association with Disease Activity in Systemic Lupus Erythematosus. Rheumatology 2010, 49, 520–527. [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti–Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The Making of Bispecific Antibodies. mAbs 2017, 9, 182–212. [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative Molecular Formats and Therapeutic Applications for Bispecific Antibodies. Mol. Immunol. 2015, 67, 95–106. [CrossRef]
- Baeuerle, P.A.; Kufer, P.; Bargou, R. BiTE: Teaching Antibodies to Engage T-Cells for Cancer Therapy. Curr Opin Mol Ther 2009, 11, 22–30.
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory Therapy of Cancer with T Cell-Engaging BiTE Antibody Blinatumomab. Exp Cell Res 2011, 317, 1255–1260. [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.; et al. Efficacy and Safety of Rituximab in Moderately-to-severely Active Systemic Lupus Erythematosus: The Randomized, Double-blind, Phase Ii/Iii Systemic Lupus Erythematosus Evaluation of Rituximab Trial. Arthritis Rheum. 2010, 62, 222–233. [CrossRef]
- Kamburova, E.G.; Koenen, H.J.P.M.; Borgman, K.J.E.; Berge, I.J. ten; Joosten, I.; Hilbrands, L.B. A Single Dose of Rituximab Does Not Deplete B Cells in Secondary Lymphoid Organs but Alters Phenotype and Function. Am. J. Transplant. 2013, 13, 1503–1511. [CrossRef]
- Tedder, T.F.; Engel, P. CD20: A Regulator of Cell-Cycle Progression of B Lymphocytes. Immunol. Today 1994, 15, 450–454. [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive Cell Transfer as Personalized Immunotherapy for Human Cancer. Science 2015, 348, 62–68. [CrossRef]
- Valiullina, A.Kh.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomed 2023, 11, 626. [CrossRef]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B Cell Depletion by CD19-Targeted CAR T Cells Is a Highly Effective Treatment for Murine Lupus. Sci. Transl. Med. 2019, 11. [CrossRef]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic Efficacy of Anti-CD19 CAR-T Cells in a Mouse Model of Systemic Lupus Erythematosus. Cell. Mol. Immunol. 2021, 18, 1896–1903. [CrossRef]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [CrossRef]
- Pattanaik, D.; Brown, M.; Postlethwaite, B.C.; Postlethwaite, A.E. Pathogenesis of Systemic Sclerosis. Front. Immunol. 2015, 6, 272. [CrossRef]
- Sato, S.; Fujimoto, M.; Hasegawa, M.; Takehara, K. Altered Blood B Lymphocyte Homeostasis in Systemic Sclerosis: Expanded Naive B Cells and Diminished but Activated Memory B Cells. Arthritis Rheum. 2004, 50, 1918–1927. [CrossRef]
- Maher, T.M.; Tudor, V.A.; Saunders, P.; Gibbons, M.A.; Fletcher, S.V.; Denton, C.P.; Hoyles, R.K.; Parfrey, H.; Renzoni, E.A.; Kokosi, M.; et al. Rituximab versus Intravenous Cyclophosphamide in Patients with Connective Tissue Disease-Associated Interstitial Lung Disease in the UK (RECITAL): A Double-Blind, Double-Dummy, Randomised, Controlled, Phase 2b Trial. Lancet Respir. Med. 2023, 11, 45–54. [CrossRef]
- Ebata, S.; Yoshizaki, A.; Oba, K.; Kashiwabara, K.; Ueda, K.; Uemura, Y.; Watadani, T.; Fukasawa, T.; Miura, S.; Yoshizaki-Ogawa, A.; et al. Safety and Efficacy of Rituximab in Systemic Sclerosis (DESIRES): A Double-Blind, Investigator-Initiated, Randomised, Placebo-Controlled Trial. Lancet Rheumatol. 2021, 3, e489–e497. [CrossRef]
- Sakkas, L.I.; Chikanza, I.C.; Platsoucas, C.D. Mechanisms of Disease: The Role of Immune Cells in the Pathogenesis of Systemic Sclerosis. Nat. Clin. Pr. Rheumatol. 2006, 2, 679–685. [CrossRef]
- Ntelis, K.; Solomou, E.E.; Sakkas, L.; Liossis, S.-N.; Daoussis, D. The Role of Platelets in Autoimmunity, Vasculopathy, and Fibrosis: Implications for Systemic Sclerosis. Semin. Arthritis Rheum. 2017, 47, 409–417. [CrossRef]
- Bergmann, C.; Müller, F.; Distler, J.H.W.; Györfi, A.-H.; Völkl, S.; Aigner, M.; Kretschmann, S.; Reimann, H.; Harrer, T.; Bayerl, N.; et al. Treatment of a Patient with Severe Systemic Sclerosis (SSc) Using CD19-Targeted CAR T Cells. Ann. Rheum. Dis. 2023, 82, 1117–1120. [CrossRef]
- Huang, J.; Fu, X.; Chen, X.; Li, Z.; Huang, Y.; Liang, C. Promising Therapeutic Targets for Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 686155. [CrossRef]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [CrossRef]
- Nygaard, G.; Firestein, G.S. Restoring Synovial Homeostasis in Rheumatoid Arthritis by Targeting Fibroblast-like Synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [CrossRef]
- Barnas, J.L.; Looney, R.J.; Anolik, J.H. B Cell Targeted Therapies in Autoimmune Disease. Curr. Opin. Immunol. 2019, 61, 92–99. [CrossRef]
- Edwards, J.C.W.; Szczepański, L.; Szechiński, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-Cell–Targeted Therapy with Rituximab in Patients with Rheumatoid Arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [CrossRef]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A Regulator of the Transition from Neutrophil to Monocyte Recruitment during Inflammation. Trends Immunol. 2003, 24, 25–29. [CrossRef]
- Hashizume, M.; Hayakawa, N.; Mihara, M. IL-6 Trans-Signalling Directly Induces RANKL on Fibroblast-like Synovial Cells and Is Involved in RANKL Induction by TNF-α and IL-17. Rheumatology 2008, 47, 1635–1640. [CrossRef]
- Maini, R.N.; Taylor, P.C.; Szechinski, J.; Pavelka, K.; Bröll, J.; Balint, G.; Emery, P.; Raemen, F.; Petersen, J.; Smolen, J.; et al. Double-blind Randomized Controlled Clinical Trial of the Interleukin-6 Receptor Antagonist, Tocilizumab, in European Patients with Rheumatoid Arthritis Who Had an Incomplete Response to Methotrexate. Arthritis Rheum. 2006, 54, 2817–2829. [CrossRef]
- Gabay, C.; Emery, P.; Vollenhoven, R. van; Dikranian, A.; Alten, R.; Pavelka, K.; Klearman, M.; Musselman, D.; Agarwal, S.; Green, J.; et al. Tocilizumab Monotherapy versus Adalimumab Monotherapy for Treatment of Rheumatoid Arthritis (ADACTA): A Randomised, Double-Blind, Controlled Phase 4 Trial. Lancet 2013, 381, 1541–1550. [CrossRef]
- Genovese, M.C.; Fleischmann, R.; Kivitz, A.J.; Rell-Bakalarska, M.; Martincova, R.; Fiore, S.; Rohane, P.; Hoogstraten, H. van; Garg, A.; Fan, C.; et al. Sarilumab Plus Methotrexate in Patients With Active Rheumatoid Arthritis and Inadequate Response to Methotrexate: Results of a Phase III Study. Arthritis Rheumatol. 2015, 67, 1424–1437. [CrossRef]
- Schaible, T.F. Long Term Safety of Infliximab. Can. J. Gastroenterol. 2020, 14, 29C-32C. [CrossRef]
- Nam, J.L.; Ramiro, S.; Gaujoux-Viala, C.; Takase, K.; Leon-Garcia, M.; Emery, P.; Gossec, L.; Landewe, R.; Smolen, J.S.; Buch, M.H. Efficacy of Biological Disease-Modifying Antirheumatic Drugs: A Systematic Literature Review Informing the 2013 Update of the EULAR Recommendations for the Management of Rheumatoid Arthritis. Ann. Rheum. Dis. 2014, 73, 516. [CrossRef]
- Santamaria-Alza, Y.; Vasquez, G. Are Chimeric Antigen Receptor T Cells (CAR-T Cells) the Future in Immunotherapy for Autoimmune Diseases? Inflamm. Res. 2021, 70, 651–663. [CrossRef]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Zenzo, G.D.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering Chimeric Antigen Receptor T Cells for Targeted Therapy of Autoimmune Disease. Science 2016, 353, 179–184. [CrossRef]
- Li, Y.-J.; Chen, Z. Cell-Based Therapies for Rheumatoid Arthritis: Opportunities and Challenges. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X221100294. [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T Cells Engineered with a Novel Insulin-Specific Chimeric Antigen Receptor as a Candidate Immunotherapy for Type 1 Diabetes. J. Autoimmun. 2019, 103, 102289. [CrossRef]
- Kaminskiy, Y.; Kuznetsova, V.; Kudriaeva, A.; Zmievskaya, E.; Bulatov, E. Neglected, yet Significant Role of FOXP1 in T-Cell Quiescence, Differentiation and Exhaustion. Front. Immunol. 2022, 13, 971045. [CrossRef]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9. [CrossRef]
- Boroughs, A.C.; Larson, R.C.; Choi, B.D.; Bouffard, A.A.; Riley, L.S.; Schiferle, E.; Kulkarni, A.S.; Cetrulo, C.L.; Ting, D.; Blazar, B.R.; et al. Chimeric Antigen Receptor Costimulation Domains Modulate Human Regulatory T Cell Function. JCI Insight 2019, 4. [CrossRef]
- Zhang, B.; Wang, Y.; Yuan, Y.; Sun, J.; Liu, L.; Huang, D.; Hu, J.; Wang, M.; Li, S.; Song, W.; et al. In Vitro Elimination of Autoreactive B Cells from Rheumatoid Arthritis Patients by Universal Chimeric Antigen Receptor T Cells. Ann. Rheum. Dis. 2021, 80, 176–184. [CrossRef]
- Minutolo, N.G.; Hollander, E.E.; Powell, D.J. The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front. Oncol. 2019, 9, 176. [CrossRef]
- Sakkas, L.I.; Bogdanos, D.P.; Katsiari, C.; Platsoucas, C.D. Anti-Citrullinated Peptides as Autoantigens in Rheumatoid Arthritis—Relevance to Treatment. Autoimmun. Rev. 2014, 13, 1114–1120. [CrossRef]
- Kim, M.S.; Ma, J.S.Y.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of Genetically Engineered CAR-T Cells Using Bifunctional Small Molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [CrossRef]
Figure 1.
Three different approaches in CAR-T cell therapy for treatment of SAIDs. Panel A shows CAR-T which recognizes CD19 on the target B-cell and induces a cytotoxic effect. Panel B shows CAAR-T cell that recognizes autoantibody on the surface of the target B-cell and induces a cytotoxic effect. Panel C shows CAR-Treg that recognizes an antigen on the target cell and induces a regulatory response.
Figure 1.
Three different approaches in CAR-T cell therapy for treatment of SAIDs. Panel A shows CAR-T which recognizes CD19 on the target B-cell and induces a cytotoxic effect. Panel B shows CAAR-T cell that recognizes autoantibody on the surface of the target B-cell and induces a cytotoxic effect. Panel C shows CAR-Treg that recognizes an antigen on the target cell and induces a regulatory response.
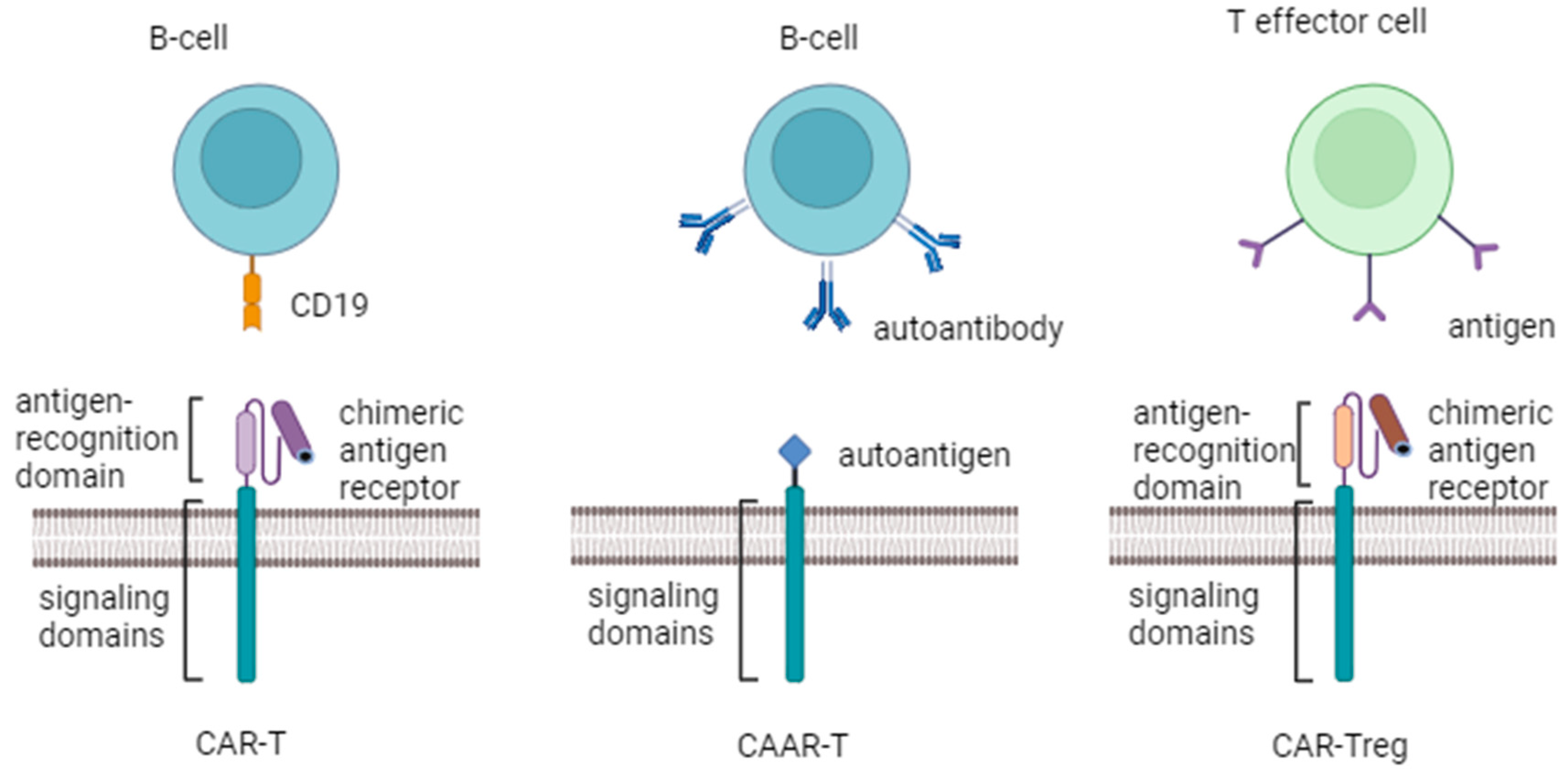
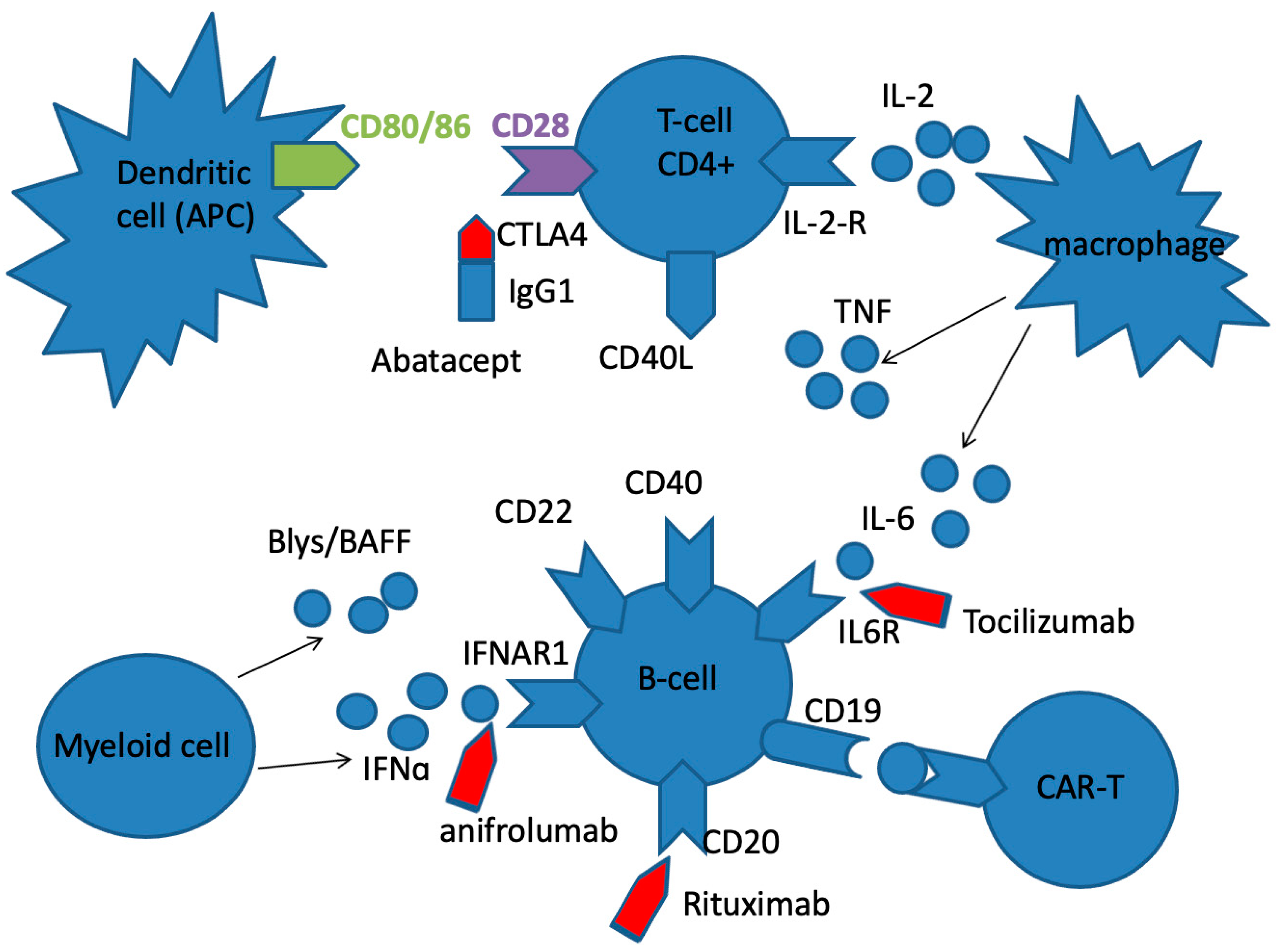
Figure 2.
The basic targets for the treatment of SLE, SSc and RA with biologic agents. Rituximab is chimeric anti-CD20 mAb, that eliminates autoreactive B cells, and a subset of T cells. Abatacept is fusion protein, a soluble CTLA-4 analog that inhibits T cell activation by binding to CD80 and CD86 receptors on APC, selectively blocking the specific interaction of CD80/CD86 receptors to CD28 and, therefore, inhibiting T cell proliferation and B cell immunological response. Anifrolumab is an immunoglobulin gamma 1 kappa (IgG1κ) mAb that selectively binds to subunit 1 of INFAR1, inhibiting receptor activity and reducing downstream signalling and gene transcription of inflammatory mediators. Tocilizumab is anti-IL-6R mAb, that binds soluble and membrane bound IL-6 receptors, preventing IL-6 mediated inflammation.
Figure 2.
The basic targets for the treatment of SLE, SSc and RA with biologic agents. Rituximab is chimeric anti-CD20 mAb, that eliminates autoreactive B cells, and a subset of T cells. Abatacept is fusion protein, a soluble CTLA-4 analog that inhibits T cell activation by binding to CD80 and CD86 receptors on APC, selectively blocking the specific interaction of CD80/CD86 receptors to CD28 and, therefore, inhibiting T cell proliferation and B cell immunological response. Anifrolumab is an immunoglobulin gamma 1 kappa (IgG1κ) mAb that selectively binds to subunit 1 of INFAR1, inhibiting receptor activity and reducing downstream signalling and gene transcription of inflammatory mediators. Tocilizumab is anti-IL-6R mAb, that binds soluble and membrane bound IL-6 receptors, preventing IL-6 mediated inflammation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



