
Submitted:
17 September 2023
Posted:
19 September 2023
You are already at the latest version
Abstract
Epitranscriptomics refers to post-transcriptional regulation of gene expression via RNA modifications and editing that affect RNA functions. Many kinds of modifications of mRNA have been described, among which N6-Methyladenosine (m6A), N1-methyladenosine (m1A), 7-methylguanosine (m7G), pseudouridine (Ψ), 5-methylcytidine (m5C). They alter mRNA structure and consequently stability, localization and translation efficiency. Perturbation of the epitranscriptome is associated with human diseases, thus opening the opportunity for potential manipulations as therapeutic approach. In this review, we overview the functional roles of epitranscriptomic marks in the skeletal muscle system, in particular in embryonic myogenesis, muscle cell differentiation and muscle homeostasis processes. Further, we explored high throughput epitranscriptome sequencing data to identify RNA chemical modifications in muscle-specific genes and we discuss the possible functional role and the potential therapeutic applications.
Keywords:
epitranscriptomics
; RNA modifications
; m6A
; skeletal muscle
; gene expression
1. Epitranscriptomics at a glance
Epitranscriptomics is a recently born field that studies the multitude of biochemical post-transcriptional RNA modifications and editing, which give rise to functionally relevant changes in the transcriptome [1]. The first discovery of RNA modification dates back to 1957, with the identification of pseudouridine (ψ), a nucleotide variant with respect to the four standard bases of RNA [2]. Since then, thin layer chromatography and high-performance liquid chromatography coupled to mass spectrometry (HPLC-MS) have allowed to identify more than 170 chemical modifications for both coding and non-coding RNAs in various species, humans included (listed at https://iimcb.genesilico.pl/modomics/) [3]. The variety of chemical modifications observed in RNAs is much wider than epigenetic modifications detected in genomic DNA, possibly because they confer a high grade of versatility to the RNA molecules that can perform a large number of regulatory and catalytic functions through the assumption of specific folds. RNA structures are generated by the three-dimensional organization of small structural motifs determined by base pairing between complementary sequences. To expand the structural flexibility of RNA, folding is based on different types of base pairings, which are established through hydrogen bonding interactions between different edges of the base molecules: the Watson-Crick edge, the Hoogsteen edge and the Sugar edge. Chemical modifications of RNA are installed on all three edges, further increasing the functional diversification of RNA species. Furthermore, non-canonical base pairing generates functional variation in the exposed surfaces for interacting protein ligands [4]. Thus, chemical modifications have an impact on RNAs’ base pairing potential and ultimately on their folding and protein-RNA interactions.
In most cases, the functional importance of RNA modifications has been demonstrated and correlated with the function of the modified RNA species. For example, base modifications were first identified in abundant RNA species like tRNAs and rRNAs at the end of the 1960s, and their possible role in RNA processing and size reduction has been inferred. Thereafter, it became evident that chemical modifications occur in all types of RNAs, including mRNAs, small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), long non-coding RNAs, microRNAs, and circular RNAs.
At first, it was assumed that RNA modifications were constitutive and always present in the same positions, fine-tuning the chemo-physical properties, the structure, and the catalytic function of RNAs. However, in the last decade, it has been shown that many RNA decorations are reversible, dynamic, and sensitive to external stimuli; in some cases, it has also been suggested a possible crosstalk between diverse RNA modifications [5,6]. Epitranscriptomics studies revealed that post-transcriptional RNA modifications regulate all facets of RNA metabolism, comprising RNA processing, export, translation, and stability, thus representing an additional layer of gene regulation. Recent evidence also implies a role for RNA modifications in DNA double-strand break repair [7].
RNA modifications are the result of the activities of highly conserved enzymes involved in the establishment of the modifications (the writers or effectors) and enzymes that reverse the modifications (the erasers). RNA-binding proteins (the readers, also known as binders) recognize and bind to the modified RNAs, thus modulating their fate and function. The RNA Modification Enzyme (RNAME) database (https://chenweilab.cn/rname/) has been developed to provide a comprehensive resource for the enzymatic machineries responsible for RNA modifications [8]. These three classes of proteins are distributed throughout the nuclei, cytoplasm and mitochondria and control several biological processes; hence, their disruption is linked to a variety of diseases [9]. The isolation of antibodies directed against specific RNA modifications, combined with next-generation sequencing, have strongly fed the epitrascriptomics field, especially to probe RNA modifications in the sequence context also in low-abundant RNA species like mRNA. It is also possible to identify RNA modification at a single-nucleotide resolution by using labeling strategies that take advantage of the unique chemistry of modified bases. Due to space limitations, we do not discuss the techniques utilized to map nucleosides modifications, which have been extensively reviewed elsewhere [10,11,12]. A further great impetus to this field of study came from the discovery that some chemical modifications are implicated in critical biological processes and the RNA modification machinery is often dysregulated in human diseases, including cancers, suggesting that the modulation of the enzymatic activities involved has a potential therapeutic application in oncological diseases [13].
About ten modifications have been described in mammalian mRNA, among which the most represented and characterized is N⁶-Methyladenosine (m6A), which regulates most aspects of mRNA metabolism and is implicated in several biological processes. The latest advances in epitranscriptomics have revealed the importance of RNA modifications other than m6A in regulating many features of RNA processing.
This review focuses on epitranscriptomics in skeletal muscle, where, to date, only m6A modification has been described. Of note, an analysis performed on data collected in the ENCORE (The Encyclopaedia of RNA Epitranscriptome) database [14] underscored the presence of six additional RNA modifications in the transcripts encoding proteins that are implicated in skeletal muscle homeostasis [15,16]. In the first part of this review, we will provide an overview of the current knowledge about m6A and the six supplementary RNA modifications reported in the ENCORE dataset. In the second part, we aim to overview the literature regarding the functional meaning of m6A in skeletal myogenesis. Lastly, we will speculatively discuss the possible functional role of RNA chemical modifications in muscle-specific gene expression and the consequent therapeutic applications.
1.1. N⁶-Methyladenosine (m6A)
(Figure 1A, Table 1). m6A is the most prevalent modification in the 5’ CAP of mRNA where it has a key role in its stabilization, its deposition depends on the activity of the 5’-CAP specific adenosine N6 methyl transferase (CAPAM) [17,18]. Adenosine methylation has also been observed in internal sites of mRNAs in the 1970s. Since then, it has been found to be the most prevalent chemical modification in the transcripts of many species, including mammals (0.2-0.6% of total adenosines) [19,20,21]. In addition to mRNA, m6A is observed in all RNA types, including tRNA, rRNA and non-coding RNA (ncRNA). At the molecular level, m6A results in the regulation of stability, conformation or interaction of RNA with binding proteins. m6A does not impede canonical Watson–Crick A:U base pairing but can disrupt the formation of trans-sugar-Hoogsteen G:A base pairs, which are abundant within the core of kink turns (k-turns), the RNA structural motifs that function as protein binding sites [22,23]. m6A is a selective modification, which occurs inside the consensus sequence DRACH (D = A, G, or U; R = G or A; H = A, C or U) [24,25]. High-throughput sequencing revealed that m6A is statistically more represented near the distal end of mRNAs, around the stop codon, and in the 3′ untranslated region (UTR) [26,27]. Moreover, m6A has been reported within the coding region and in the 5’UTR of mRNA where it can enhance CAP-independent translation initiation [28]. Several transcription-based and chromatin-based hypotheses have been made about the mechanisms underlying the selection of specific transcripts by the methyltransferase complex, ranging from the intervention of transcription factors that mediate the interaction between the target RNA and the catalytic complex, to the number of CpG islands in the upstream promoter or the type of histone modifications [29].
Adenosine methylation is transcript-specific and highly heterogeneous from cell to cell. Its abundance is mediated by external stimuli and depends on the balance between the activities of two classes of enzymes: methylases and demethylases. A great stimulus in the study of the functional role of m6A modification came from the identification of the nuclear multisubunit methyltransferase complex, which includes the methyltransferase-like 3 (METTL3)– methyltransferase-like 14 (METTL14) heterodimer, with METTL3 being the catalytic subunit and METTL14 being its allosteric activator, the Wilms Tumor 1-Associating Protein (WTAP), the Vir Like M6A Methyltransferase Associated (VIRMA) protein, also indicated as KIAA1429 and the Zinc Finger Protein 217 (ZFP217) [30]. The METTL3–METTL14 methyltransferase complex accounts for the deposition of m6A in newly synthesized RNA Polymerase II-dependent transcripts. More recently, an additional m6A mRNA methyltransferase, METTL16, has been isolated, which regulates the cellular levels of S-Adenosyl-Methionine (SAM), a substrate of methylases that functions as methyldonor, and targets the U6 small nuclear RNA (U6 snRNA), which regulates mRNA splicing [31]. The reversibility of m6A is ensured by the activity of the demethylases α-ketoglutarate-dependent dioxygenase AlkB homolog 5 (ALKBH5) and fat mass and obesity associated protein (FTO) [32,33]. Methylated transcripts are subsequently regulated by m6A readers that modulate various aspects of their processing. Accordingly, the main mechanism underlying the regulatory effect of m6A on target RNA function is based on its ability to recruit reader proteins. The major class of m6A readers is represented by the family of YT521-B homology (YHT) proteins that contain a ~150 amino acid-YTH domain that allows RNA recognition in an m6A-dependent manner through a “tryptophan cage”, in which two or three tryptophan residues wrap the methyl group. YTH proteins interact with m6A-containing RNAs both in the nucleus (YTHDC1 and YTHDC2) and in the cytoplasm (YTHDF1, YTHDF2, and YTHDF3). YTH proteins may also contain a low-complexity region, which triggers a mechanism of phase-separation of YTH proteins, especially when bound to m6A mRNA, thus targeting methylated transcripts to non-membranous P bodies, stress granules, and other RNA–protein complexes where they are processed. The effects of the YTH binders are disparate and dependent on the specific reader that binds the modification. Additional m6A binders have been isolated: Proline rich coiled-coil 2 A (Prrc2a) binds and stabilizes Olig2 mRNA, which controls oligodendrocyte specification, this m6A-dependent interaction involves a consensus GGACU motif in the coding sequence of Olig2 [34]. Other readers can bind to unfolded RNA induced by the presence of m6A. While the methyl group at the N6 position of adenosine does not alter Watson–Crick A-U base pairing, the steric hindrance of the methyl group on the Watson-Crick edge stabilizes unpaired bases, thus facilitating the exposure of binding sites for like insulin-like growth factor 2 binding proteins (IGF2BPs) and fragile X mental retardation protein (FMRP) [35,36,37]. IGF2BP1, 2, and 3 are single-stranded RNA-binding proteins that contain six RNA-binding domains: two RNA recognition motif (RRM) domains and four K homology (KH) domains. Upon binding to m6A-modified transcripts, IGF2BPs promote their stability [35]. Accordingly, the first function identified for m6A was its ability to regulate mRNA stability, both negatively through interaction with YTHDF2 and positively by the mediation of IGFBPs [23]. Another m6A reader is represented by the heterogeneous nuclear ribonucleoprotein C (HNRNPC), an abundant nuclear RNA-binding protein responsible for pre-mRNA processing. This interaction explains the ability of m6A to modulate alternative splicing of pre-mRNAs [38]. Similarly, heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) is a m6A reader involved in primary microRNA processing and alternative splicing [39]. Regulation of alternative splicing by m6A is also linked to its interaction with the YTHDC1 reader that can recruit the splicing regulators and arginine-rich splicing factors 3 (SRSF3) and 10 (SRSF10) [38,40]. In addition, m6A can promote mRNA nuclear export and regulate translation in different ways depending on the position of the chemical modification [23,37,41,42].
The analysis of the distribution of m6A shows that this modification is enriched in the transcripts of genes that regulate development and cell fate specification, while it is rare in the transcripts of housekeeping genes [26,43]. Accordingly, m6A is implicated in a wide range of key biological functions and aberrant m6A distribution, together with mutations in genes encoding proteins involved in RNA modification, are often associated with severe diseases, including cancer, metabolic, neurological and cardiac disorders [44,45,46,47,48,49].
1.2. N1-methyladenosine (m1A)
(Figure 1B, Table 1). m1A is an m6A isomer that results from a methylation occurring on N1 of adenosine. This chemical modification confers a positive electrostatic charge under physiological conditions and impairs the canonical Watson-Crick base pairing through electrostatic and steric effects. This is an ancient modification that is conserved across species and is present in all kinds of RNAs, including cellular and mitochondrial mRNAs [50,51,52]. The presence of m1A has been well characterized in tRNA and rRNA , where it has a role in maintaining the tertiary structure [53,54] in mRNAs m1A is 10-fold less represented than m6A (0.01–0.05% of total adenosines). Several transcriptome-wide mapping experiments have been undertaken to define the m1A methylome [50,51,55,56,57]. The first studies were based on next-generation sequencing of antibody-mediated immunoprecipitated RNAs. These works identified many m1A sites in mRNA, enriched around both canonical and alternative translational initiation sites and in GC rich highly structured regions around the start codons, and these were linked to enhanced translation [50,51]. Other studies instead described m1A in a low number of cytosolic mRNAs, mostly within the codon region and 3’UTR, as having a repressive role on translation when detected in coding sequences (CDS) [55,57]. The divergent results have been interpreted because of the low specificity of the antibody directed against m1A. The altered physical-chemical properties of m1A can regulate protein translation in different ways that depend on the location of m1A. When m1A is allocated in the 5’UTR, translation is enhanced possibly through the destabilization of the secondary structure in the 5’ UTR, while m1A in the CDS interferes with translation [58]. Like m6A, m1A is a dynamic modification that is regulated by external stimuli: for example, it is promoted by oxidative damage and starvation [50,51]. m1A might have a protective role on RNA that accumulates in stress granules upon heat shock [59]. Deposition of m1A in tRNA is ensured by the transfer tRNA transferases TRMT6-TRMT61A complex, which also catalyzes methylation of adenosine in mRNAs that contain a tRNA T-loop structure [56]. A methyltransferase complex including TRMT61B and TRMT10C is responsible for the deposition of m1A marks in mitochondrial RNA [60]. Ablation of m1A is catalyzed by the AlkB family of demethylases, and specific demethylation of m1A in mRNA is catalyzed by ALKBH3 [51]. The cellular readers of m1A are still under study, but in vitro data indicate that m1A can be recognized and bound by YTH proteins (YTHDC1, YTHDF1, YTHDF2, YTHDF3) [61], this suggesting a crosstalk between m6A and m1A RNA modifications.
1.3. 5-methylcytidine (m5C)
(Figure 1C, Table 1). In m5C modifications, a methyl group is attached to the fifth carbon of the cytosine ring, a chemical imprint that was first detected on DNAs and later on RNAs in the 1970s [62]. The development of bisulfite-based techniques to identify m5C mark in RNAs has allowed the detection of its deposition in all types of RNA molecules. In mRNAs, it is 3–10-fold rarer than m6A (0.03–0.1% of cytines), and it is enriched in the 5′ or 3′ UTRs or next to translational start sites [63,64,65]. m5C deposition does not alter the base pairing edges, but it significantly changes the physicochemical properties of the original nucleobase, modulating its ability to interact with proteins [66]. m5C deposition in RNAs is catalyzed by the tRNA aspartic acid methyltransferase 1 (TRDMT1), also called S-adenosyl-methionine-dependent methyltransferase 2 (DNMT2), and the nucleolar protein 1 (NOL1)/NSUN protein family, which includes seven proteins in humans (NSUN1 to NSUN7), with NSUN2 and NSUN6 being the responsible members for m5C deposition in mRNAs [52,67,68,69]. To date, the erasers of mRNA m5C marks are still under debate. While a demethylation pathway for m5C modifications of DNA has been described that involves the ten–eleven translocation (TET) demethylases, the involvement of TET demethylases as m5C erasers is still unclear [70,71].
Interestingly, it has also been shown a positive reciprocal interaction between m5C and m6A [72]. m5C can increase mRNA stability and promote mRNA export from the nucleus to the cytoplasm; furthermore, m5C modulates protein translation, both positively and negatively [73,74,75]. These activities are ensured by the interaction with the readers Aly/REF export factor (ALYREF) and Y-box binding protein 1 (YBX1). Also, RAD52 is a reader of m5C marks, which is involved in the DNA damage pathway by recognizing hybrid strands containing m5C-marked RNAs and DNAs [76]. Aberrant m5C deposition on RNA has been linked to several diseases [77] and 5-hydroxymethylcytosine (hm5C) has been suggested to play an important role in the regulation of embryonic stem cells differentiation [78,79,80].
1.4. 7-methylguanosine (m7G)
(Figure 1D, Table 1). m7G was first identified as a component of the CAP structure of RNA polymerase II (Pol II) transcripts. The m7G CAP modification was first identified on eukaryotic mRNAs, and subsequent studies have shown that this is an evolutionarily conserved modification in all organisms. It is installed co-transcriptionally on nearly all RNA polymerase II target genes and it represents a critical feature required for stability, splicing, and efficient translation of mRNAs [81,82,83,84]. m7G deposition in the CAP of mRNA is catalyzed by the mRNA CAP methyltransferase RNMT in complex with the RNMT-activating mini-protein (RAM) [85,86]. m7G is also found internally in the variable loop of tRNAs and in eukaryotic 18S rRNAs, where it modulates RNA processing and function. Aberrant m7G has been linked to human diseases, such as microcephalic primordial dwarfism [87]. Installation of m7G in tRNAs and rRNAs is catalyzed by a complex of methyltransferase-like 1 (METTL1) with WD repeat domain 4 (WDR4) and a complex of Williams-Beuren syndrome chromosome region 22 (WBSCR22), also known as BUD23, with tRNA methyltransferase activator subunit 11–2 (TRMT112), respectively [54,88,89,90]. Besides its ubiquitous presence in the CAP, m7G is also found internally in in mRNAs as it has been first evidenced by differential enzymatic digestion combined with liquid chromatography-tandem mass spectrometry analysis [91]. Subsequent works allowed the mapping of thousands of m7G marks in mammalian mRNAs enriched both in the 3′ UTR and in the 5′ UTR, by m7G-methylated RNA antibody-based immunoprecipitation sequencing (MeRIP-seq) and chemical-based methods [92,93]. These studies revealed that internal mRNA m7G promotes mRNA translation and is dynamically regulated upon heat shock and oxidative stress [94]. m7G has also been detected in human mature miRNAs and miRNA precursors, where it is important to produce mature miRNAs and maintain high levels of mature let-7e miRNAs [95]. Writers, erasers, and readers of internal mRNA m7G are still unidentified, even if a subset of m7G marks is deposed by the METTL1-WDR4 complex [92]. Mechanistically, this chemical modification takes place in the Hoogsteen edge of guanosine and, similarly to m1A, introduces a positive charge that could potentially modulate protein–RNA interactions and reorganize local secondary structures in RNAs, and consequently their biological functions. For example, it has been suggested that m7G could inhibit the formation of G-quadruplex structures, which are four-stranded structures based on the Hoogsteen base pairing of guanosines that play key roles in the control of gene expression [96]. Prevention of G-quadruplex is important to guarantee let-7e pri-miRNA processing [95].
1.5. Pseudouridine (Ψ)
(Figure 1E, Table 1). Pseudourydilation is an isomerization reaction whereby uridine undergoes a 180° rotation, resulting in the formation of a glycosidic bond between the C5 of uridine and-C1’ of the ribose sugar, which substitutes the N1-C1’ glycosidic bond of uridine. This modification is conserved in various species; it is the most represented post-transcriptional modification in all types of RNAs, so much so that it earned the designation of “fifth ribonucleotide” [2]. Installation of Ψ increases the hydrogen bonding potential of the ribonucleotide that exhibits an extra hydrogen donor (N1 imino proton) and does not change Watson-Crick base pairing property. Consequently, the presence of Ψ influences the physical-chemical properties of the modified RNA. In tRNAs, Ψ is important for maintaining its structure, while in rRNAs, it influences the interaction with tRNAs and mRNAs, thus regulating translation fidelity [97,98]. Spliceosomal snRNAs are pseudouridylated, suggesting a role in the assembly of the spliceosomal machinery [99]. This modification has been observed also in mRNAs (0.2-0.6% of the total uridines in mRNA), both in the CDS and in the 3’UTR. The number of Ψs is dynamically regulated by external stimuli [27,100]. In mRNAs, Ψ promotes read-through at Ψ containing stop codons through non-canonical base pairing [101]. Another function is related to the regulation of RNA splicing: insertion of Ψ in the pre-mRNAs may happen in intronic sequences that are critical for the interaction with the spliceosomal machinery; furthermore, Ψ influences alternative splicing when it decorates retained introns and cassette exons, or RNA-binding protein (RBP) binding sites critical for splicing [102,103]. The Ψ writers belong to a large family of pseudouridine synthases (PUS), which are generally RNA-independent enzymes that modify uridine by recognizing a consensus sequence and/or secondary structural elements of the target RNA rev in [52]. On the opposite, Diskerin pseudouridine synthase 1 (DKC1) is an RNA-dependent enzyme that is recruited to the target RNA by a small nucleolar RNA guide. Among the PUS family of enzymes, PUS1, PUS7 and RNA pseudouridine synthase 4 (RPUSD4) can depose Ψ cotranscriptionally [103]. There are currently no known erasers or readers for this RNA modification. Aberrant pseudouridylation has been associated with human cancers and other diseases like mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA), an autosomal recessive disease in humans characterized by disorders of the oxidative phosphorylation and iron metabolism in skeletal muscle and bone marrow [104].
1.6. 2’-O-methylation (Nm or 2’-O-Me)
(Figure 1F, Table 1). This is a conserved RNA modification that is characterized by the methylation of the 2′ hydroxyl (–OH) group of the ribose. O-Me is not limited to a specific base, therefore it is indicated as Nm, where N stands for any nucleoside (Am, Um, Cm, or Gm). Nm sites have been detected in all types of RNAs: they increase the hydrophobicity of target RNAs, protect them from nuclease attacks, regulate RNA folding, and impact the ability of modified RNA to interact with proteins or other RNAs (reviewed in [105]. rRNA contains several Nm sites located in key functional sites of the ribosome [106,107,108,109]. 2’O methylation is also located at fixed positions in tRNAs, and it is linked to tRNA stability and translation efficiency [110,111]. 2’-O methylation in mRNA is observed in the CAP region, on the first and second transcribed nucleotides [112]. In addition, there are also internal Nm sites that have been identified with high throughput mapping: they are widespread, without preference for any region of the mRNA, representing roughly 0.01% of their unmodified counterparts [113,114]. Nm sites in mRNA regulate mRNA stability, translation, and its ability to interact with other RNAs; in addition, 2’-O methylation of mRNAs has been shown to regulate gene expression in vivo [115]. There are two classes of Nm writers: independent methyltransferases and small nucleolar ribonucleoprotein (snoRNP) complexes which include the C/D-box small nucleolar RNAs (C/D snoRNAs), these complexes are indicated C/D snoRNP. Small RNAs mark the target nucleotide sequence to be methylated by the enzyme fibrillarin (FBL) through sequence complementarity. Unbiased motif search of Nm sites has revealed the presence of a consensus sequence next to the Nm which contain an AGAUC sequence followed by a 5-nt-long AG-rich stretch, suggesting the existence of a specific methyltransferase that might function independently from snoRNPs [113]. The independent enzymes CMTR1 and CMTR2 catalyze Nm modifications that occur in the CAP structure [105,116,117].
1.7. Nucleoside editing
(Figure 1G, Table 1). The first description of RNA editing in mammals came after the discovery of cytosine-to-uridine (C-to-U) conversion in the mRNA encoding apolipoprotein-B100 that causes a premature stop codon, leading to the production of the shorter apolipoprotein-B48 isoform in vertebrate intestine [118]. C-to-U mRNA editing is catalyzed by cytidine deaminases that belong to the activation-induced cytidine deaminase/apolipoprotein B editing complex (AID/APOBEC) family [119]. In the same year, adenosine-to-inosine (A-to-I) RNA editing was described in the oocytes of Xenopus Leavis and shown to be crucial in the unwinding of double-stranded RNAs (dsRNAs) [120]. Subsequent studies have demonstrated that A-to-I is the most common type of RNA editing. This RNA modification is a highly conserved process that underlies multiple cellular processes [121,122,123,124]. A-to-I editing is a hydrolytic deamination catalyzed by adenosine deaminases acting on dsRNAs (ADARs). In mammals, there are three ADAR genes: ADAR1 and 2 are catalytically active, while ADAR3 is inactive and may function as a dominant negative [125]. In Drosophila, mice, and humans, A-to-I editing events are strongly enriched in the brain [126,127,128,129]. Millions of A-to-I editing events have been identified in humans, and they are enriched in genes involved in neurological disorders and cancer [130]. An atlas of A-to-I editing is available (https://omictools.com/the-rna-editing-atlas -tool). In human mRNAs, A-to-I editing is detected principally in introns and UTRs; the preferred target sequences are intronic retrotransposon elements like Alu repeats, but it is also observed in coding regions [131,132,133,134]. It has been reported that m6A negatively regulates A-to-I RNA editing [135](Xiang, et al. 2018). Inosine has distinct base-pairing abilities in comparison to adenosine; it can base pair with any natural bases, but it preferentially pairs with cytosine rather than with uridine, leading to variations in the secondary structure of target RNAs and modifications of the encoded information. Inosines are read as guanosines by the ribosome, therefore A-to-I editing can change the amino acid sequence of proteins [125]. MicroRNA A-to-I editing can have an impact on RNA splicing, when it takes place in sites like the RNA splicing sites [136], or it can change the target specificity of miRNAs, thus impacting RNA degradation [125,137].
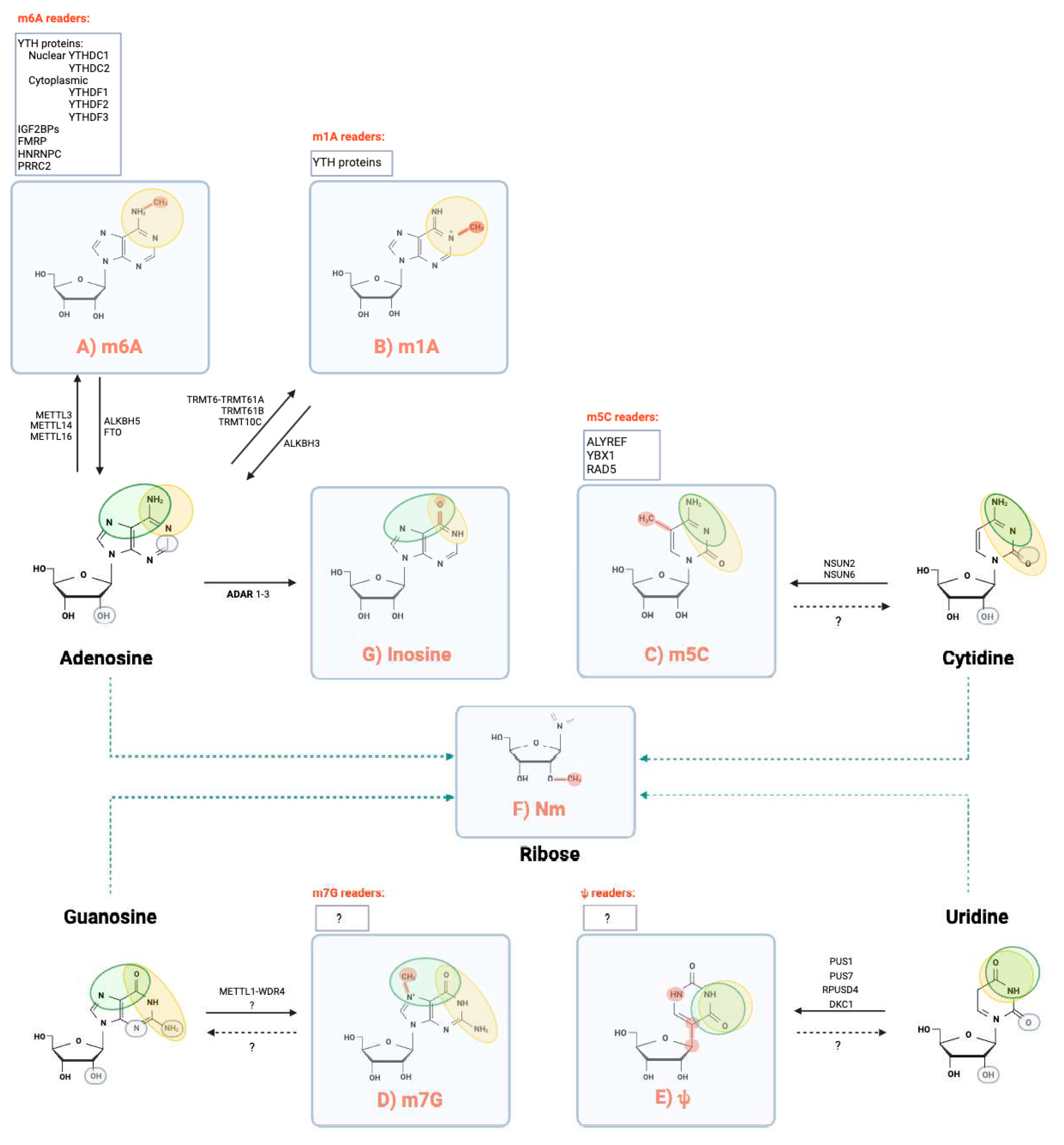
Figure 1.
Chemical structure of mRNA modifications reported in eukaryotic. The four standard ribonucleotides are reported with ovals indicating the Watson Creek (green), Hoogsteen (orange) and sugar (grey) edges of the ribonucleotides. In the squares are indicated the chemical modifications for Adenosine (N⁶-Methyladenosine m6A), N1-Methyladenosine (m1A) Inosine), 5-methylcytidine (m5C), 7-methylguanosine (m7G), 2’O-methylation (Nm or 2’-O-Me), Pseudouridine (Ψ). A summary of the known writers, erasers and readers ) is also reported in the grey squares.
Figure 1.
Chemical structure of mRNA modifications reported in eukaryotic. The four standard ribonucleotides are reported with ovals indicating the Watson Creek (green), Hoogsteen (orange) and sugar (grey) edges of the ribonucleotides. In the squares are indicated the chemical modifications for Adenosine (N⁶-Methyladenosine m6A), N1-Methyladenosine (m1A) Inosine), 5-methylcytidine (m5C), 7-methylguanosine (m7G), 2’O-methylation (Nm or 2’-O-Me), Pseudouridine (Ψ). A summary of the known writers, erasers and readers ) is also reported in the grey squares.

Table 1.
Stoichiometry, consensus sequence and mRNA preferred regions of modified ribonucleosides. N stands for any base, D = A, G, or U; H = A, C or U; R = G or A; Y = C or U. Underlined nucleotides in the consensus sequences are chemically modified.
Table 1.
Stoichiometry, consensus sequence and mRNA preferred regions of modified ribonucleosides. N stands for any base, D = A, G, or U; H = A, C or U; R = G or A; Y = C or U. Underlined nucleotides in the consensus sequences are chemically modified.
| Modified ribonucleoside | Stoichiometry | Target sequence | mRNA preferred regions | |
|---|---|---|---|---|
| m6A | 0.2-0.6% m6A/A | DRACH | 5’ UTR, near the stop codon, 3’ UTR | |
| m1A | 0,01-0,05% m1A/A | GC-rich and GA-rich sequence, GUUCNANNC | All segments of the transcripts, enriched in: first splice site, GC rich highly structured regions in the 5’ UTRs, translation start site | |
| m5C | 0,03-0,1% m5C/C | CTCCA | 5′/3′ UTRs, next to Argonaute-binding regions | |
| m7G | 0.002-0.05% m7G/G | GA- or GG-enriched sequence motifs | 5′UTR near the start codon and near the stop codon, | |
| Ψ | 0.2-0.6% Ψ/U | AU (PUS1-specific)GUUCNANYCY(PUS4)UGUAGPUS7) | Coding sequence and 3’UTR. | |
| Nm | 0,01% Nm/N | NmAGAUC followed by a 5-nt-long AG-rich stretch | CDS, near splice sites, 5’/3’ UTR, introns, alternatively spliced regions | |
| A to I | Not available | dsRNA, preference for U in the -1 position | Introns (Alu sequences) and UTRs; coding sequence | |
2. Epitranscriptomics in skeletal muscle
2.1. N⁶-Methyladenosine modification in embryonic myogenesis
The importance of m6A in modulating embryogenesis comes from several studies in which the deletion of the m6A methyltransferase Mettl3 is embryonic lethal in mice [138], due to a compromised transition from self-renewal to differentiation state in embryonic stem cells and to the targeting of several transcripts encoding pluripotency transcription factors [138,139,140]. Besides METTL3, another N6-methyltransferase, METTL16, is important in embryonic development [141]. Indeed, Mettl16 knockout in mice causes developmental arrest around the time of implantation by influencing the mRNA levels of the SAM synthetase MAT2a [141]. Similarly, embryonic neural stem cells lacking METTL14 display markedly decreased proliferation and premature differentiation, suggesting that m6A modification affects embryonic neural stem cell self-renewal [142]. In addition to methyltransferases, ZFP217, a factor that modulates m6A deposition, is crucial for embryonic stem cell fate, since its knockdown results in compromised cell growth and lineage differentiation by regulating the transcripts of pluripotency-associated factors [143]. These findings confirm that m6A modification is important for embryonic stem cell self-renewal maintenance and mouse development.
Numerous studies point to the m6A modification as an important epigenetic mechanism regulating muscle development during embryogenesis. Highly dynamic changes in RNA m6A modification have been profiled across different stages of skeletal muscle development [144,145], finding m6A modification on transcripts of important genes for skeletal muscle development. For instance, MeRIP-seq analysis identified several differentially methylated genes enriched in pathways related to porcine skeletal muscle development [146]. Similarly, the analysis of m6A distribution in Dingan goose [147], goat [148], and duck [149] embryonic skeletal muscle revealed differentially regulated m6A peaks in important developmental pathways, including the MAPK and Wnt signaling, or in muscle-related genes during key embryonic stages. Moreover, RNA IP assay revealed that the m6A methylation reader IGF2BP1 targets many embryonic myogenic genes in porcine skeletal muscle, including the myogenic marker Myogenin and the terminal differentiation gene Myosin Heavy Chain 2 [146]. Coherently, the deletion of the m6A demethylase FTO in mice during the pregnancy period results in fewer and smaller myofibers, if compared to controls, confirming its crucial role in the skeletal muscle development of the offspring [150].
Taken together, these studies clearly demonstrated that m6A modification affects skeletal muscle development in numerous species. The biggest limitation of these findings is that they originate from methods based on RNAs extracted from a bulk of different cells, thus losing spatial localization information. Analytical techniques for m6A RNA with single-cell resolution and spatial information will be more informative. Recently, a m6A-specific in situ hybridization mediated proximity ligation assay (m6AISH-PLA) has been developed, which allows to visualize cellular m6A RNA at single-molecule resolution and could be used to investigate cell-to-cell variation and spatial pattern [151]. By applying this technology to histological sections, it would be possible to follow the spatial-temporal dynamics of m6A modification during myogenesis.
2.2. N⁶-Methyladenosine modification in myoblast differentiation
In vitro studies clarified that RNA m6A methylation finely regulates the transition from myoblast proliferation to differentiation (Figure 2) [144,152,153,154]. Moreover, co-localization and interaction between the transcriptional (m5C) and posttranscriptional (m6A) modifications have been described during the differentiation process of the murine myoblast cell line C2C12 [155], revealing a cooperative regulation of m5C and m6A modifications in spatiotemporal gene expression during myogenesis.
The first publication on an epitranscriptomic modifier enzyme in myoblast differentiation goes back to 2017, when Wang and coworkers started clarifying the role of the m6A demethylase FTO in C2C12 and primary myoblast differentiation [150]. FTO expression raises during myoblast differentiation, paralleling m6A demethylation. FTO silencing in primary myoblasts suppresses myogenic differentiation by affecting mitochondrial biogenesis through the mTOR-PGC-1α axis, while FTO overexpression does not affect myotube formation [150]. A limitation of this study is the analysis of primary myoblasts in which FTO was silenced by siRNA technology, instead of knocking-out FTO in vitro in primary myoblasts isolated from the inducible skeletal muscle-specific FTO mice. Moreover, it is still unclear why FTO overexpression does not affect myoblast differentiation. Similar results were reported by other studies: indeed, FTO deficiency has been associated with a reduction in fat and lean mass in mice [156], while FTO overexpression leads to obesity without affecting skeletal muscle mass [157]. FTO knockdown in primary goat myoblasts increases CCND1 and GADD45B mRNA m6A modification, thereby decreasing their stability and leading to impaired myoblast proliferation and myogenic differentiation [148].
Contrary to FTO, the expression levels of the methyltransferases METTL3, METTL14, and WTAP decrease during myoblast differentiation in vitro and in vivo [158]. Another pioneering study in 2017 clarified the pivotal role of the m6A methyltransferase METTL3 in maintaining the mRNA levels of a master myogenic regulator of skeletal muscle, i.e. MyoD, and therefore the myogenic potential throughout the cell cycle in proliferating myoblasts [159]. Indeed, METTL3 knock-down inhibits myotube formation, similarly to MyoD deletion [159]. Numerous recent studies focused on the role of the m6A writer METTL3 in muscle differentiation. The silencing of METTL3 in proliferating myoblasts clearly demonstrates its pivotal role in regulating myoblast cell transitions during the myogenic process, since it induces premature C2C12 differentiation in vitro and reduces the capacity of serial transplantation in vivo [153]. Consistently, deletion of METTL3 specifically in muscle stem cells inhibits their proliferation and skeletal muscle regeneration upon injury, by regulating the expression of several genes involved in the Notch signaling pathway [160] required for muscle stem cell self-renewal, differentiation, and muscle regeneration [161]. Conversely, METTL3 overexpression in a muscle stem cell-specific METTL3 conditional knock-in mouse increases muscle stem cell proliferation and muscle regeneration in vivo following injury [160].
A faster muscle stem cell differentiation due to an altered transition from proliferation to differentiation state upon METTL3 or METTL14 knockdown was also confirmed by Kudou K. and colleagues in [158], while overexpression of either METTL3 or METTL14 inhibits myotube formation. The m6A writers METTL3 and METTL14, together with the m6A reader YTHDF1, finely regulate the mRNA of mitogen-activated protein kinase (MAPK) interacting protein kinases 2 (MNK2), a critical regulator of ERK/MAPK signaling, thus controlling myoblast differentiation [158]. Similarly, silencing of the m6A reader IGF2BP1 promotes C2C12 proliferation, altering their transition into a differentiating state, thus inhibiting their differentiation [146].
Overall, consistent conclusions were drawn regarding the functions of RNA methyltransferases in regulating the transition from the proliferative to the differentiating state of muscle stem cells, mainly via loss-of-function studies. On the contrary, apparent contradictory data were reported in gain-of-function studies: while muscle regeneration was increased in METTL3 muscle stem cell-specific knock-in mice [160], myotube differentiation was inhibited in C2C12 overexpressing METTL3 [158]. However, it is difficult to arrive to final considerations on METTL3 overexpression by analyzing different experimental models and having the underlying molecular mechanisms still uncharacterized.
More recently, it has been shown that METTL3, together with the m6A reader YTHDF1, post-transcriptionally regulates the mRNA of the Mef2C gene, encoding a transcription factor that promotes muscle differentiation in concert with MyoD, thus affecting bovine myoblast differentiation [162]. Moreover, a positive feedback loop has been reported, since MEF2C directly binds to and activates the METTL3 promoter region, further regulating bovine skeletal myoblast differentiation [162]. Indeed, the expression levels of METTL3 and MEF2C positively correlate in loss- and gain-of-function experiments: METTL3 knockdown decreases MEF2C expression, while METTL3 overexpression increases MEF2C protein levels. Coherently, knockdown of the demethylase FTO in myoblasts induces a significant upregulation of MEF2C protein expression, whereas FTO overexpression results in a decrease of MEF2C, further proving the direct involvement of m6A modification in the regulation of MEF2C expression during myoblast differentiation.
Moreover, METTL3 represses the expression of skeletal muscle-specific miRNAs during myoblast differentiation, by indirectly regulating the expression of either transcription factors or epigenetic regulators, which in turn affect miRNA expression [163]. In addition to coding mRNA, gain- and loss-of-function experiments clarified that METTL3 positively regulates the abundance of long-non-coding RNAs, thereby affecting the expression of their adjacent mRNAs, during myoblast differentiation [164].
In conclusion, an increasing number of studies in recent years clarified the importance of m6A modification in the orchestrated regulation of muscle stem cell differentiation and myogenesis. A deeper investigation is needed to explore the key upstream and downstream factors that regulate m6A modifications, and their cooperation with other epigenetic regulators, to elucidate the specific functional mechanism of m6A modification in regulating myogenic differentiation.
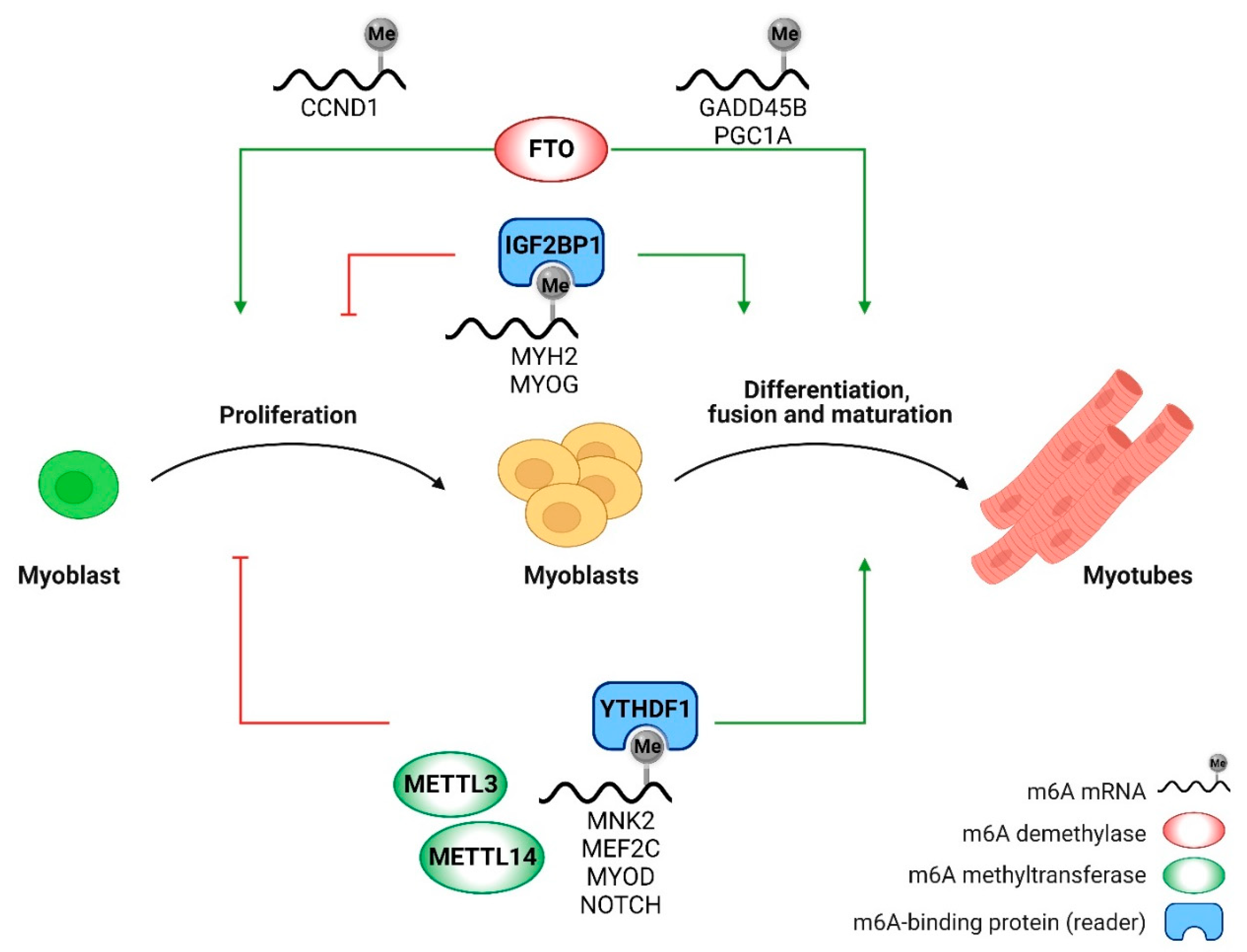
Figure 2.
m6A RNA modification controls myoblasts proliferation and differentiation through post-transcriptional regulation of genes involved in myogenesis.
Figure 2.
m6A RNA modification controls myoblasts proliferation and differentiation through post-transcriptional regulation of genes involved in myogenesis.
2.3. N⁶-Methyladenosine modification in skeletal muscle homeostasis
Besides muscle differentiation, METTL3 has been shown to control muscle mass growth and homeostasis in post-natal life by targeting the Activin receptor, thereby modulating the TGF-beta signaling [165]. Short-term deletion of Mettl3 in muscle fibers clarified that METTL3 is indispensable for the hypertrophic response of skeletal muscle to mechanical overload, while long-term deletion of Mettl3 leads to a progressive decline in skeletal muscle mass [165]. Conversely, METTL3 overexpression in newborn mice with adeno-associated viruses induces a hypertrophic response, and METTL3 overexpression in adult muscles, by electroporation, induces an enhanced hypertrophic response to overload [165].
A proof of the importance of m6A modification in maintaining skeletal muscle homeostasis comes also from a very recent paper, which claims that among liver, heart, and skeletal muscle, the latter is the most susceptible to m6A decrease with aging, which positively correlates with a decreased expression of METTL3 [166]. METTL3 deficiency leads to smaller myotubes and affected senescence of myotubes. As for the mechanism, METTL3 targets and stabilizes the mRNA of Nephronectin, a matrix protein involved in cell-matrix adhesion and important for myotube fusion. Coherently, knockdown of Nephronectin in myoblasts phenocopies the METTL3 deficiency defects [166]. Like in aging, the m6A mRNA global levels in skeletal muscles significantly decrease upon denervation, parallel to an increase in the expression of the m6A demethylase ALKBH5 [167]. Importantly, ALKBH5 promotes neurogenic muscle atrophy by demethylating and thus stabilizing Hdac4 mRNA. The HDAC4 protein in turn interacts with and deacetylates FoxO3, resulting in the activation of FoxO3 signaling [167].
The research has moved very fast in the last three years, clarifying the role of m6A modification in muscle differentiation and homeostasis. No information is currently available regarding the other less abundant mRNA modifications in skeletal muscle, such as the m5C and hm5C modifications in tRNAs, or the pseudouridylation of coding and non-coding RNAs, which play important roles in the regulation of non-muscle stem cell fate [168,169,170]. Future studies should address their functions to better understand skeletal muscle physiology and to provide new insights for possible therapeutic approaches aimed at maintaining muscle mass.
3. Epitranscriptomics as novel pathogenetic mechanism and potential therapeutic approach for muscular disorders
Muscular disorders include a wide-range of inherited or acquired diseases affecting the muscular system. Genes encoding proteins implicated in numerous processes, such as contractility, membrane integrity, gene regulation, and metabolism, are altered in muscular diseases. Over 650 genes have been associated with monogenic neuromuscular disorders [171]. Genetic information has allowed the understanding of the molecular pathogenesis of muscular disorders, such as muscular dystrophies (MDs), which have been mapped to at least 29 different genetic loci. Despite this knowledge, MDs are still undertreated, and steroid medications represent the standard treatment to slow down disease progression.
The identification of the molecular basis of RNA modifications may provide new pathogenetic mechanisms at the basis of muscular diseases and hence drive the discovery of new therapeutic targets. Since epitranscriptomics is a dynamic and reversible regulatory mechanism, the manipulation of RNA modifications represents a promising approach for the treatment of MDs and other muscular diseases. The field of epitranscriptome-targeting drugs is still in its beginnings, but efforts are directed towards drug discovery research on RNA-epitranscriptomics and low molecular weight compounds that have been shown to potentially revert defects, at least in cancer [172].
The rapid progress of new technologies, such as high-throughput sequencing, has allowed the collection of multiple data. To predict potential RNA modifications in MD-related genes, we exploited the ENCORE (The Encyclopaedia of RNA Epitranscriptome) database [14], which integrates epitranscriptome sequencing data for the investigation of post-transcriptional modifications of RNAs. Interestingly, the vast majority of RNAs encoded by genes associated with MD [171] can be methylated at different sites, according to our research, although in cellular contexts other than muscle (Table 2).
Table 2.
List of RNA modifications identified from high throughput epitranscriptome sequencing data collected in the ENCORE (The Encyclopaedia of RNA Epitranscriptome) database [14] (https://rna.sysu.edu.cn/encore/index.php). Genes related to MD were obtained from the 2023 GeneTable of Neuromuscular Disorders [171] (http://www.musclegenetable.fr, accessed on May 30th,2023). The numbers represent the number of RNA modification sites of a specific modification type on the gene.
Table 2.
List of RNA modifications identified from high throughput epitranscriptome sequencing data collected in the ENCORE (The Encyclopaedia of RNA Epitranscriptome) database [14] (https://rna.sysu.edu.cn/encore/index.php). Genes related to MD were obtained from the 2023 GeneTable of Neuromuscular Disorders [171] (http://www.musclegenetable.fr, accessed on May 30th,2023). The numbers represent the number of RNA modification sites of a specific modification type on the gene.
| Gene | Gene ID | m6A | m1A | m5C | m7G | PseudoU | 2'-O-Me | RNA-editing, A-I sites | Total |
|---|---|---|---|---|---|---|---|---|---|
| ACTA1 | ENSG00000143632.14 | 1 | 0 | 0 | 0 | 0 | 0 | 2 | 3 |
| ANO5 | ENSG00000171714.11 | 23 | 0 | 0 | 0 | 0 | 0 | 3 | 26 |
| B3GALNT2 | ENSG00000162885.13 | 13 | 0 | 1 | 0 | 0 | 1 | 8 | 23 |
| B4GAT1 | ENSG00000174684.7 | 27 | 1 | 1 | 0 | 0 | 0 | 0 | 29 |
| BVES | ENSG00000112276.14 | 11 | 0 | 0 | 0 | 0 | 0 | 1 | 12 |
| CACNA1S | ENSG00000081248.11 | 6 | 0 | 2 | 0 | 0 | 0 | 0 | 8 |
| CAPN3 | ENSG00000092529.24 | 17 | 0 | 0 | 0 | 0 | 0 | 2 | 19 |
| CAV3 | ENSG00000182533.6 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 7 |
| CAVIN1 | ENSG00000177469.13 | 45 | 0 | 1 | 0 | 0 | 0 | 0 | 46 |
| CHKB | ENSG00000100288.19 | 25 | 0 | 0 | 0 | 0 | 0 | 1 | 26 |
| COL12A1 | ENSG00000111799.21 | 203 | 0 | 0 | 0 | 0 | 0 | 0 | 203 |
| COL6A1 | ENSG00000142156.14 | 93 | 0 | 0 | 0 | 0 | 0 | 0 | 93 |
| COL6A2 | ENSG00000142173.15 | 88 | 0 | 0 | 0 | 0 | 0 | 3 | 91 |
| COL6A3 | ENSG00000163359.15 | 144 | 0 | 1 | 0 | 0 | 0 | 5 | 150 |
| DAG1 | ENSG00000173402.11 | 138 | 0 | 2 | 0 | 0 | 1 | 6 | 147 |
| DES | ENSG00000175084.11 | 35 | 0 | 1 | 0 | 0 | 0 | 0 | 36 |
| DMD | ENSG00000198947.15 | 116 | 0 | 2 | 0 | 0 | 0 | 19 | 137 |
| DNAJB6 | ENSG00000105993.15 | 86 | 0 | 14 | 0 | 0 | 0 | 8 | 108 |
| DNM2 | ENSG00000079805.16 | 118 | 0 | 13 | 0 | 1 | 0 | 29 | 161 |
| DPM1 | ENSG00000000419.12 | 25 | 0 | 0 | 0 | 0 | 1 | 16 | 42 |
| DPM2 | ENSG00000136908.17 | 54 | 0 | 0 | 0 | 0 | 0 | 0 | 54 |
| DPM3 | ENSG00000179085.7 | 6 | 0 | 1 | 0 | 0 | 0 | 0 | 7 |
| DYSF | ENSG00000135636.14 | 33 | 0 | 4 | 0 | 0 | 0 | 1 | 38 |
| EMD | ENSG00000102119.10 | 22 | 0 | 10 | 0 | 0 | 0 | 0 | 32 |
| FHL1 | ENSG00000022267.17 | 76 | 0 | 0 | 0 | 4 | 0 | 2 | 82 |
| FKRP | ENSG00000181027.10 | 64 | 0 | 0 | 0 | 0 | 0 | 12 | 76 |
| FKTN | ENSG00000106692.14 | 49 | 0 | 0 | 0 | 1 | 0 | 4 | 54 |
| GAA | ENSG00000171298.13 | 69 | 0 | 6 | 0 | 2 | 0 | 0 | 77 |
| GGPS1 | ENSG00000152904.11 | 83 | 0 | 5 | 0 | 0 | 0 | 1 | 89 |
| GMPPB | ENSG00000173540.12 | 76 | 0 | 0 | 0 | 0 | 0 | 1 | 77 |
| GOLGA2 | ENSG00000167110.17 | 81 | 0 | 3 | 0 | 0 | 0 | 10 | 94 |
| GOSR2 | ENSG00000108433.16 | 139 | 0 | 0 | 0 | 0 | 0 | 3 | 142 |
| HNRNPDL | ENSG00000152795.17 | 94 | 0 | 3 | 0 | 0 | 1 | 0 | 98 |
| INPP5K | ENSG00000132376.20 | 62 | 0 | 1 | 0 | 0 | 0 | 1 | 64 |
| ITGA7 | ENSG00000135424.16 | 39 | 0 | 0 | 0 | 0 | 0 | 0 | 39 |
| JAG2 | ENSG00000184916.9 | 48 | 0 | 4 | 1 | 0 | 0 | 0 | 53 |
| LAMA2 | ENSG00000196569.12 | 21 | 0 | 0 | 0 | 0 | 1 | 3 | 25 |
| LARGE1 | ENSG00000133424.20 | 62 | 0 | 0 | 0 | 0 | 1 | 63 | 126 |
| LIMS2 | ENSG00000072163.19 | 27 | 0 | 0 | 0 | 0 | 0 | 2 | 29 |
| LMNA | ENSG00000160789.20 | 72 | 0 | 5 | 0 | 0 | 1 | 22 | 100 |
| LRIF1 | ENSG00000121931.16 | 72 | 0 | 0 | 0 | 0 | 0 | 0 | 72 |
| MPDU1 | ENSG00000129255.16 | 36 | 0 | 2 | 0 | 0 | 0 | 0 | 38 |
| MSTO1 | ENSG00000125459.15 | 33 | 0 | 1 | 0 | 1 | 0 | 0 | 35 |
| MYOT | ENSG00000120729.9 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 7 |
| PLEC | ENSG00000178209.15 | 167 | 0 | 26 | 1 | 2 | 0 | 21 | 217 |
| POGLUT1 | ENSG00000163389.12 | 37 | 0 | 1 | 0 | 1 | 0 | 1 | 40 |
| POMGNT1 | ENSG00000085998.14 | 59 | 0 | 1 | 0 | 0 | 0 | 0 | 60 |
| POMGNT2 | ENSG00000144647.6 | 49 | 0 | 1 | 0 | 0 | 0 | 1 | 51 |
| POMK | ENSG00000185900.9 | 24 | 0 | 0 | 0 | 0 | 0 | 0 | 24 |
| POMT1 | ENSG00000130714.16 | 53 | 0 | 4 | 0 | 0 | 0 | 16 | 73 |
| POMT2 | ENSG00000009830.11 | 66 | 0 | 2 | 0 | 0 | 0 | 5 | 73 |
| POPDC3 | ENSG00000132429.10 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 25 |
| PYROXD1 | ENSG00000121350.16 | 48 | 0 | 1 | 0 | 2 | 0 | 0 | 51 |
| RXYLT1 | ENSG00000118600.11 | 23 | 0 | 2 | 0 | 0 | 0 | 1 | 26 |
| RYR1 | ENSG00000196218.12 | 32 | 0 | 3 | 0 | 0 | 1 | 1 | 37 |
| SELENON | ENSG00000162430.17 | 59 | 0 | 4 | 0 | 0 | 0 | 15 | 78 |
| SGCA | ENSG00000108823.16 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| SGCB | ENSG00000163069.12 | 61 | 0 | 17 | 0 | 0 | 0 | 2 | 80 |
| SGCG | ENSG00000102683.7 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 6 |
| SMCHD1 | ENSG00000101596.15 | 153 | 0 | 2 | 0 | 0 | 0 | 16 | 171 |
| SYNE1 | ENSG00000131018.23 | 339 | 0 | 4 | 0 | 0 | 2 | 50 | 399 |
| SYNE2 | ENSG00000054654.16 | 482 | 0 | 2 | 0 | 0 | 0 | 22 | 506 |
| TCAP | ENSG00000173991.5 | 11 | 0 | 4 | 0 | 0 | 0 | 0 | 15 |
| TMEM43 | ENSG00000170876.7 | 89 | 0 | 2 | 0 | 0 | 0 | 0 | 91 |
| TNPO3 | ENSG00000064419.13 | 94 | 0 | 1 | 0 | 0 | 0 | 8 | 103 |
| TOR1AIP1 | ENSG00000143337.18 | 90 | 0 | 5 | 0 | 0 | 0 | 3 | 98 |
| TRAPPC11 | ENSG00000168538.16 | 67 | 0 | 0 | 0 | 0 | 0 | 3 | 70 |
| TRIM32 | ENSG00000119401.10 | 38 | 0 | 0 | 0 | 1 | 0 | 0 | 39 |
| TRIP4 | ENSG00000103671.9 | 23 | 0 | 0 | 0 | 0 | 0 | 11 | 34 |
| TTN | ENSG00000155657.26 | 249 | 0 | 5 | 0 | 0 | 1 | 0 | 255 |
| VCP | ENSG00000165280.16 | 84 | 0 | 10 | 0 | 0 | 6 | 0 | 100 |
In addition to the genes listed above that are directly related to MDs because of existing genetic defects (Table 2), we investigated RNA modifications of key genes regulating myogenesis and skeletal muscle regeneration in response to injury and genetic dystrophies, such as transcription factors and epigenetic regulators (Table 3). Their central role in muscle development and maintenance makes them excellent candidates for druggable epitranscriptome therapies in muscular diseases.
Table 3.
Lists of genes encoding for myogenic regulatory factors and related RNA modifications from the ENCORE (The Encyclopaedia of RNA Epitranscriptome) database [14] (https://rna.sysu.edu.cn/encore/index.php). The numbers represent the number of RNA modification sites of a specific modification type on the gene.
Table 3.
Lists of genes encoding for myogenic regulatory factors and related RNA modifications from the ENCORE (The Encyclopaedia of RNA Epitranscriptome) database [14] (https://rna.sysu.edu.cn/encore/index.php). The numbers represent the number of RNA modification sites of a specific modification type on the gene.
| Gene | Gene ID | m6A | m1A | m5C | m7G | PseudoU | 2'-O-Me | RNA-editing, A-I sites | Total |
|---|---|---|---|---|---|---|---|---|---|
| EZH2 | ENSG00000106462.10 | 52 | 0 | 0 | 0 | 0 | 1 | 4 | 57 |
| HDAC4 | ENSG00000068024.16 | 80 | 0 | 17 | 0 | 0 | 0 | 12 | 109 |
| MEF2A | ENSG00000068305.17 | 128 | 0 | 1 | 0 | 2 | 0 | 49 | 180 |
| MEF2C | ENSG00000081189.15 | 41 | 0 | 0 | 0 | 0 | 0 | 3 | 44 |
| MEF2D | ENSG00000116604.18 | 38 | 0 | 2 | 0 | 0 | 0 | 8 | 48 |
| MYF5 | ENSG00000111049.4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| MYH1 | ENSG00000109061.10 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| MYH2 | ENSG00000125414.19 | 2 | 0 | 1 | 0 | 0 | 0 | 0 | 3 |
| MYOD1 | ENSG00000129152.4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| NFYA | ENSG00000001167.14 | 79 | 0 | 6 | 1 | 0 | 0 | 1 | 87 |
| PAX3 | ENSG00000135903.19 | 59 | 0 | 0 | 0 | 0 | 0 | 0 | 59 |
| PAX7 | ENSG00000009709.12 | 37 | 0 | 0 | 0 | 0 | 0 | 0 | 37 |
| SIRT1 | ENSG00000096717.12 | 76 | 0 | 0 | 0 | 0 | 0 | 27 | 103 |
| SMAD4 | ENSG00000141646.13 | 104 | 0 | 4 | 0 | 0 | 0 | 5 | 113 |
Also in this case, the majority of myogenic genes show RNA modifications, in particular m6A. This is not surprising, taking into consideration that m6A regulates gene expression through different mechanisms, as described above[168]. m6A enrichment in muscle genes is also consistent with the results from the muscle stem cell-specific Mettl3 conditional knockout mouse model, which affects stem cells fate and muscle regeneration after injury [173].
2′-O-methylation and adenosine to inosine RNA-editing (A-to-I) represent two of the most common RNA modifications provided by RNA-guided mechanisms and, consequently, potentially exploitable for therapeutic application. For example, site-specific box C/D-directed methylation of the branchpoint adenosine could inhibit the splicing of an intron, or site-specific methylation of a central nucleotide within a sense codon may trigger premature termination of translation [174], thus inhibiting the expression of non-functional or aberrant proteins associated with muscular diseases. One 2′-O-methylation site has been identified in Ezh2 (Table 2), the subunit of the Polycomb PRC2 and PRC3 complexes with histone lysine methyltransferase (HKMT) activity, that triggers transcriptional repression and controls the expression of muscle genes and the differentiation of satellite-cell-derived myoblasts following muscle injury [175,176]. EZH2 overexpression represses muscle gene expression and differentiation, therefore additional 2′-O-methylation could represent a strategy to interfere with its increased expression.
The inosine RNA modification that results from the hydrolytic deamination of adenosines (A-to-I) catalyzed by the adenosine deaminase ADAR represents another interesting tool to recode transcripts and alter splicing events, thus correcting disorders at the mRNA level and restoring protein function [177]. This could be relevant for diseases resulting from G-to-A genomic single point mutations. For example, one of the most common nucleotide changes at the first intronic nucleotide of the DMD gene is a G-to-A, which disrupts the splice site consensus sequence, thus producing an abnormal transcript, as reported for Duchenne and Becker MDs [178].
Hdac4, Sirt1, Mef2 and Smad4 transcripts show the higher number of RNA modification sites, with m6A, m5C and A-I being more represented. Both Hdac4 and Sirt1 genes encode for histone deacetylase enzymes, which exert a key role in the control of gene transcription and homeostasis in skeletal muscle. SIRT1 enzymatic activity is deeply correlated with the differentiation of muscle fibers, energy homeostasis and muscle cell fate signaling. In addition to SIRT1-mediated effects on the transcription of key genes, among which MyoD [179], SIRT1 is intimately linked to nutrient availability in muscle cells and controls energy metabolism [180]. Moreover, as an inhibitor of NF-κB signaling, SIRT1 activation ameliorates muscle pathology in MD [181,182].
HDAC4 is crucial in maintaining muscle integrity upon different stimuli [183]. HDAC4 expression is stabilized by the ALKBH5 demethylates upon skeletal muscle denervation [167]. ALKBH5-mediated m6A modification of HDAC4 triggers neurogenic muscle atrophy, since HDAC4 interacts with and activates FoxO3 [167]. Although the ALKBH5-HDAC4-FoxO3 axis is of great interest for the maintenance of skeletal muscle mass, ALKBH5 cannot be considered a potential therapeutic target for the treatment of neurogenic muscle atrophy, since HDAC4 inhibition is protective upon short-term denervation [184], but it is deleterious after long-term denervation [185] or in a chronic condition of denervation, such as in Amyotrophic Lateral Sclerosis (ALS) [186,187]. On the contrary, HDAC4 functions in DMD need to be preserved, since HDAC4 depletion is detrimental to dystrophic muscles[188]; thus, the stabilization of HDAC4 mRNA may be a useful therapeutic approach. Similarly, another study claimed the importance of m6A modification in HDAC4 mRNA stabilization in sepsis-induced myocardial injury. A METTL3/IGF2BP1/m6A/HDAC4 axis has been described in cardiomyocytes, where the m6A reader IGF2BP1 enhances HDAC4 mRNA stability and thus regulates the inflammatory damage of cardiomyocytes induced by lipopolysaccharide[189]. The identification of RNA modifications in genes encoding for chromatin modifiers, such as EZH2, SIRT1 and HDAC4, provides a proof of a crosstalk between epitranscriptional and epigenetic mechanisms in muscle physiology and pathology.
Among the transcription factors that regulate muscle gene expression, a key role is played by the MEF2 family of proteins which work in concert with MYOD and other myogenic factors (rev in [190]). In vertebrates there are four MEF2 paralogs: MEF2A-D, each encoded by a distinct gene. MEF2 proteins share a N-terminal DNA-binding and dimerization domain, while the transcripts are highly diversified and undergo extensive alternative splicing within their C-terminal transactivation domain, which produce multiple isoforms. MEF2 splice variants differently participate in early commitment to muscle differentiation and maintenance of the differentiated state in vertebrates [191,192,193,194,195]. In addition to alternative splicing, the activity of MEF2 factors is finely modulated by various means that directly involve their transcripts. It has indeed been shown that translation of Mef2ca in zebrafish is negatively regulated by interaction of the transcript with eukaryotic Initiation Factor 4E Binding Proteins (eIF4EBPs) in response to inactivity [196]. Also, the murine Mef2a transcript is subjected to a translational control mechanism that is mediated by the Mef2a 3’UTR which is relieved during muscle cell differentiation [197]. The molecular details of these regulatory processes are still waiting to be clarified, we can hypothesize that dynamic chemical modifications of Mef2 transcripts might play a role, possibly modulating the interaction with trans-acting RNA binding factors in response to external stimuli. The hypothesis that the Mef2 transcripts undergo a modulation of their chemical modification profile during muscle differentiation is further strengthened by literature data which demonstrate METTL3 stabilizes Mef2c RNA and increases its translation in bovine and quail muscle cells [198,199]. Accordingly, multiple m6A and A-to-I sites have been retrieved within the Mef2 transcripts (Table 3). Interestingly, only the Mef2a transcript shows two pseudouridine (ψ) modifications. It is well known that ψ is the most abundant modified nucleoside in non-coding RNAs, stabilizing the tRNA and rRNA structure and enhancing their function [200]. In addition, ψ regulates the splicing process by modifying specific snRNAs, while its role in mRNA remains essentially unknown [201]. Mutations in genes encoding for pseudouridine synthases have been identified in patients with neurodevelopmental disorders [202,203], thus reinforcing the possible role of ψ as a regulator also in the expression of neuromuscular genes, such as Mef2a. Recently, ψ modifications have been identified in nascent pre-mRNA at sites associated with alternative splicing [204], therefore it would be useful to investigate whether a correlation between ψ and Mef2a splicing exists. Moreover, since ψ content in 3′-UTR mRNA is regulated in response to environmental signals [200], flexible adaptation to continuous neighborhood environmental factors in pathological muscle may be induced through ψ mRNA modifications. Dysregulated expression and splicing of Mef2a, Mef2c and Mef2d genes occur in several neuromuscular disorders, including Becker syndrome and myotonic dystrophy (DM), which is characterized by the expression of MEF2 embryonic isoforms [205,206,207]. Therefore, clarifying the impact of RNA modifications on MEF2 function will potentially open new therapeutic options for these pathologies.
Another gene that controls muscle cell fate is Nf-ya, which encodes for the NF-YA DNA binding subunit of the transcription factor NF-Y. Expression levels and alternative splicing of NF-YA are crucial in muscle cell proliferation and differentiation, and muscle stem cell-specific knock out studies highlighted that NF-YA expression is fundamental to preserve the pool of muscle stem cells and ensures muscle regeneration upon injury [208,209,210]. Although NF-YA mutations have not been observed in muscular diseases, we cannot exclude that alterations in its expression or splicing may participate in muscle pathogenetic mechanisms, as demonstrated in cancer disease [211,212]. Indeed, two alternative splice isoforms are generated from the Nf-ya gene (NF-YAs and NF-YAl) and are not functionally equivalent in various types of cells, myoblasts included [213]. In mouse embryonic myoblasts, the expression of both NF-YA variants is high and drops in post-natal muscles, with only NF-YAl being expressed at low levels [214]. Gain of function studies highlighted that NF-YAs enhances cell proliferation, in opposition to NF-YAl that improves cell differentiation [213], therefore it would be key investigating whether epitranscriptomics mechanisms participate to Nf-ya splicing or can be exploited to restore altered events. Finally, an interesting crosstalk between m6A and NF-Y came to light from studies on myeloid leukaemia [215]. The interrogation of sequences under METTL3 peaks for enriched motifs identified the NF-Y binding site as the top hit, suggesting a cooperative binding between METTL3 and NF-Y on promoter-bound METTL3 to maintain m6A-dependent translation control.
SMAD4 has been included in our analysis because of its role in muscle stem cells activity: it is a downstream cofactor for canonical TGFβ superfamily signaling, and Smad4 specific deletion in adult mouse stem cells triggers terminal myogenic commitment associated with impaired proliferative potential. Consistently, adult skeletal muscle regeneration is evidently compromised following Smad4 abrogation [216]. Through the interaction with AR, SMAD4 chromatin binding orchestrates a muscle hypertrophy transcriptional program that is altered in the spinal and bulbar muscular atrophy (SBMA) mouse model [217]. Understanding the possible diverse set of modifications or rewiring the modifications in Smad4 transcript may be crucial in the fine-tune of AR-SMAD4 functional complex, which has been proposed as a promising target for SBMA and other conditions associated with muscle loss.
Overall, deep analysis of RNA modifications of muscle-related gene transcripts in pathological contexts may provide new mechanisms at the basis of altered expression and splicing events. The most challenging question before the potential application of therapies targeting epitranscription in muscular diseases is to monitor the dynamic changes of RNA modifications, such as m6A, under physiological and pathological conditions.
Author Contributions
Conceptualization, C.I., V.M., S.M.; methodology, S.B.; formal analysis, S.B.; writing—original draft preparation, C.I., A. R., G.C., V.M., S.B., S.M.; writing—review and editing, C.I., V.M., S.B., S.M.; visualization, E.M.; funding acquisition, C.I., S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by University of Modena and Reggio Emilia: Finanziamento FAR_DIP 2022 to S.M. and Finanziamento FAR_DIP 2021 to C.I.
References
- He, C. Grand Challenge Commentary: RNA epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Ribonucleic Acids from Yeast Which Contain a Fifth Nucleotide - PubMed Available online:. Available online: https://pubmed.ncbi.nlm.nih.gov/13463012/ (accessed on 12 September 2023).
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2021, 50, D231–D235. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Bhattacharyya, D. RNA structure and dynamics: A base pairing perspective. Prog. Biophys. Mol. Biol. 2013, 113, 264–283. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- He, C. Grand Challenge Commentary: RNA epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, S.; Balestra, F.R.; Huertas, P. The Emerging Role of RNA Modifications in DNA Double-Strand Break Repair. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Nie, F.; Tang, Q.; Liu, Y.; Qin, H.; Liu, S.; Wu, M.; Feng, P.; Chen, W. RNAME: A comprehensive database of RNA modification enzymes. Comput. Struct. Biotechnol. J. 2022, 20, 6244–6249. [Google Scholar] [CrossRef]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, L.; Li, X. Detection technologies for RNA modifications. Exp. Mol. Med. 2022, 54, 1601–1616. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef]
- Wiener, D.; Schwartz, S. The epitranscriptome beyond m6A. Nat. Rev. Genet. 2020, 22, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.-J.; Sun, W.-J.; Lin, P.-H.; Zhou, K.-R.; Liu, S.; Zheng, L.-L.; Qu, L.-H.; Yang, J.-H. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2017, 46, D327–D334. [Google Scholar] [CrossRef]
- Imbriano, C.; Molinari, S. Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology. Genes 2018, 9, 107. [Google Scholar] [CrossRef]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2023 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2022, 33, 76–117. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.-J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Akichika, S.; Hirano, S.; Shichino, Y.; Suzuki, T.; Nishimasu, H.; Ishitani, R.; Sugita, A.; Hirose, Y.; Iwasaki, S.; Nureki, O.; et al. Cap-specific terminal N 6 -methylation of RNA by an RNA polymerase II–associated methyltransferase. Science 2019, 363, 141–141. [Google Scholar] [CrossRef]
- Wei, C.-M.; Gershowitz, A.; Moss, B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell 1975, 4, 379–386. [Google Scholar] [CrossRef]
- Sommer, S.; Lavi, U.; Darnell, J.E. The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J. Mol. Biol. 1978, 124, 487–499. [Google Scholar] [CrossRef]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Ashraf, S.; Huang, L.; Lilley, D.M.J. Effect of methylation of adenine N6 on kink turn structure depends on location. RNA Biol. 2019, 16, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Boulias, K.; Greer, E.L. Biological roles of adenine methylation in RNA. Nat. Rev. Genet. 2022, 24, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.-M.; Moss, B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 1977, 16, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Schibler, U.; Kelley, D.E.; Perry, R.P. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J. Mol. Biol. 1977, 115, 695–714. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR m6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Slobodin, B.; Han, R.; Calderone, V.; Vrielink, J.A.O.; Loayza-Puch, F.; Elkon, R.; Agami, R. Transcription Impacts the Efficiency of mRNA Translation via Co-transcriptional N6-adenosine Methylation. Cell 2017, 169, 326–337. [Google Scholar] [CrossRef]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Human MRNA (N6-Adenosine)-Methyltransferase Purification and CDNA Cloning of the AdoMet-Binding Subunit of The. 1997, 3, 1233–1247.
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m 6 A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, A.; Sun, B.; Sun, J.-G.; Zhang, J.; Zhang, T.; Chen, Y.; Xiao, Y.; Gao, Y.; Zhang, Q.; et al. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2018, 29, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Edupuganti, R.R.; Geiger, S.; Lu, Z.; Wang, S.-Y.; Baltissen, M.P.A.; Jansen, P.W.T.C.; Rossa, M.; Müller, M.; Stunnenberg, H.G.; He, C.; et al. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017, 24, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Shi, H.; Zhu, A.C.; Lu, Z.; Miller, N.; Edens, B.M.; Ma, Y.C.; He, C. The RNA-binding protein FMRP facilitates the nuclear export of N6-methyladenosine–containing mRNAs. J. Biol. Chem. 2019, 294, 19889–19895. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m6A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef]
- Haussmann, I.U.; Bodi, Z.; Sanchez-Moran, E.; Mongan, N.P.; Archer, N.; Fray, R.G.; Soller, M. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 2016, 540, 301–304. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Luo, G.-Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 2017, 6, e31311. [Google Scholar] [CrossRef]
- Edens, B.M.; Vissers, C.; Su, J.; Arumugam, S.; Xu, Z.; Shi, H.; Miller, N.; Ringeling, F.R.; Ming, G.-L.; He, C.; et al. FMRP Modulates Neural Differentiation through m6A-Dependent mRNA Nuclear Export. Cell Rep. 2019, 28, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Garbo, S.; Zwergel, C.; Battistelli, C. m6A RNA methylation and beyond – The epigenetic machinery and potential treatment options. Drug Discov. Today 2021, 26, 2559–2574. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef]
- Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 2021, 22, 375–392. [Google Scholar] [CrossRef]
- Leptidis, S.; Papakonstantinou, E.; Diakou, K.I.; Pierouli, K.; Mitsis, T.; Dragoumani, K.; Bacopoulou, F.; Sanoudou, D.; Chrousos, G.P.; Vlachakis, D. Epitranscriptomics of cardiovascular diseases (Review). Int. J. Mol. Med. 2021, 49, 1–21. [Google Scholar] [CrossRef]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Yin, L.; Zhu, X.; Novák, P.; Zhou, L.; Gao, L.; Yang, M.; Zhao, G.; Yin, K. The epitranscriptome of long noncoding RNAs in metabolic diseases. Clin. Chim. Acta 2021, 515, 80–89. [Google Scholar] [CrossRef]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef]
- Sun, H.; Li, K.; Liu, C.; Yi, C. Regulation and functions of non-m6A mRNA modifications. Nat. Rev. Mol. Cell Biol. 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Saikia, M.; Fu, Y.; Pavon-Eternod, M.; He, C.; Pan, T. Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs. RNA 2010, 16, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Warda, A.S.; Sharma, S.; Entian, K.-D.; Lafontaine, D.L.J.; Bohnsack, M.T. Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol. 2016, 14, 1138–1152. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.-W.; et al. Base-Resolution Mapping Reveals Distinct m1A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol. Cell 2017, 68, 993–1005. [Google Scholar] [CrossRef]
- Zhou, H.; Rauch, S.; Dai, Q.; Cui, X.; Zhang, Z.; Nachtergaele, S.; Sepich, C.; He, C.; Dickinson, B.C. Evolution of a reverse transcriptase to map N1-methyladenosine in human messenger RNA. Nat. Methods 2019, 16, 1281–1288. [Google Scholar] [CrossRef]
- Zhou, H.; Kimsey, I.J.; Nikolova, E.N.; Sathyamoorthy, B.; Grazioli, G.; McSally, J.; Bai, T.; Wunderlich, C.H.; Kreutz, C.; Andricioaei, I.; et al. m1A and m1G disrupt A-RNA structure through the intrinsic instability of Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2016, 23, 803–810. [Google Scholar] [CrossRef]
- Alriquet, M.; Calloni, G.; Martínez-Limón, A.; Ponti, R.D.; Hanspach, G.; Hengesbach, M.; Tartaglia, G.G.; Vabulas, R.M. The protective role of m1A during stress-induced granulation. J. Mol. Cell Biol. 2020, 12, 870–880. [Google Scholar] [CrossRef]
- Chujo, T.; Suzuki, T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA 2012, 18, 2269–2276. [Google Scholar] [CrossRef]
- Dai, X.; Wang, T.; Gonzalez, G.; Wang, Y. Identification of YTH Domain-Containing Proteins as the Readers for N1-Methyladenosine in RNA. Anal. Chem. 2018, 90, 6380–6384. [Google Scholar] [CrossRef]
- Dubin, D.T.; Taylor, R.H. The methylation state of poly A-containing-messenger RNA from cultured hamster cells. Nucleic Acids Res. 1975, 2, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, W.; Liu, J.; Gu, N.; Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 2019, 26, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.D.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Legrand, C.; Tuorto, F.; Hartmann, M.; Liebers, R.; Jacob, D.; Helm, M.; Lyko, F. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res. 2017, 27, 1589–1596. [Google Scholar] [CrossRef]
- Hajnic, M.; Alonso-Gil, S.; Polyansky, A.A.; de Ruiter, A.; Zagrovic, B. Interaction preferences between protein side chains and key epigenetic modifications 5-methylcytosine, 5-hydroxymethycytosine and N6-methyladenine. Sci. Rep. 2022, 12, 1–12. [Google Scholar] [CrossRef]
- Reid, R.; Greene, P.J.; Santi, D.V. Exposition of a family of RNA m5C methyltransferases from searching genomic and proteomic sequences. Nucleic Acids Res. 1999, 27, 3138–3145. [Google Scholar] [CrossRef]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.-L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNA Asp by the DNA Methyltransferase Homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef]
- Selmi, T.; Hussain, S.; Dietmann, S.; Heiß, M.; Borland, K.; Flad, S.; Carter, J.-M.; Dennison, R.; Huang, Y.-L.; Kellner, S.; et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res. 2020, 49, 1006–1022. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, W.; Zhao, Y.; Yang, Y. Dynamic transcriptomic m5C and its regulatory role in RNA processing. Wiley Interdiscip. Rev. RNA 2021, 12, e1639. [Google Scholar] [CrossRef]
- Shen, Q.; Zhang, Q.; Shi, Y.; Shi, Q.; Jiang, Y.; Gu, Y.; Li, Z.; Li, X.; Zhao, K.; Wang, C.; et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 2018, 554, 123–127. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Tang, H.; Jiang, B.; Dou, Y.; Gorospe, M.; Wang, W. NSUN2-Mediated m5C Methylation and METTL3/METTL14-Mediated m6A Methylation Cooperatively Enhance p21 Translation. J. Cell. Biochem. 2017, 118, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, A.; Sun, B.-F.; Yang, Y.; Han, Y.-N.; Yuan, X.; Chen, R.-X.; Wei, W.-S.; Liu, Y.; Gao, C.-C.; et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nature 2019, 21, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Sun, B.-F.; Chen, Y.-S.; Xu, J.-W.; Lai, W.-Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export — NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [PubMed]
- Schumann, U.; Zhang, H.-N.; Sibbritt, T.; Pan, A.; Horvath, A.; Gross, S.; Clark, S.J.; Yang, L.; Preiss, T. Multiple links between 5-methylcytosine content of mRNA and translation. BMC Biol. 2020, 18, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, H.; Zhu, X.; Yadav, T.; Ouyang, J.; Truesdell, S.S.; Tan, J.; Wang, Y.; Duan, M.; Wei, L.; et al. m5C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Xue, C.; Zhao, Y.; Li, L. Advances in RNA cytosine-5 methylation: detection, regulatory mechanisms, biological functions and links to cancer. Biomark. Res. 2020, 8, 1–13. [Google Scholar] [CrossRef]
- Huber, S.M.; van Delft, P.; Mendil, L.; Bachman, M.; Smollett, K.; Werner, F.; Miska, E.A.; Balasubramanian, S. Formation and Abundance of 5-Hydroxymethylcytosine in RNA. ChemBioChem 2015, 16, 752–755. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Minerva Anestesiol. 2011, 25, 2436–2452. [Google Scholar] [CrossRef]
- Yang, J.; Bashkenova, N.; Zang, R.; Huang, X.; Wang, J. The roles of TET family proteins in development and stem cells. Development 2020, 147. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Both, G.W.; Furuichi, Y.; Shatkin, A.J. 5′-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 1975, 255, 33–37. [Google Scholar] [CrossRef]
- Murthy, K.; Park, P.; Manley, J. A nuclear micrococcal-sensitive, ATP-dependent exoribonuclease degrades uncapped but not capped RNA substratesx. Nucleic Acids Res. 1991, 19, 2685–2692. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, Y. Discovery of m7G-cap in eukaryotic mRNAs. Proc. Jpn. Acad. Ser. B 2015, 91, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H. Regulation of mRNA cap methylation. Biochem. J. 2009, 425, 295–302. [Google Scholar] [CrossRef]
- Pillutla, R.C.; Yue, Z.; Maldonado, E.; Shatkin, A.J. Recombinant Human mRNA Cap Methyltransferase Binds Capping Enzyme/RNA Polymerase IIo Complexes. J. Biol. Chem. 1998, 273, 21443–21446. [Google Scholar] [CrossRef] [PubMed]
- Gonatopoulos-Pournatzis, T.; Dunn, S.; Bounds, R.; Cowling, V.H. RAM/Fam103a1 Is Required for mRNA Cap Methylation. Mol. Cell 2011, 44, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Abdel-Salam, G.M.H.; Guy, M.P.; Alomar, R.; Abdel-Hamid, M.S.; Afifi, H.H.; Ismail, S.I.; Emam, B.A.; Phizicky, E.M.; Alkuraya, F.S. Mutation in WDR4 impairs tRNA m7G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 2015, 16, 1–11. [Google Scholar] [CrossRef]
- Alexandrov, A.; Martzen, M.R.; Phizicky, E.M. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA 2002, 8, 1253–1266. [Google Scholar] [CrossRef]
- Haag, S.; Kretschmer, J.; Bohnsack, M.T. WBSCR22/Merm1 is required for late nuclear pre-ribosomal RNA processing and mediates N7-methylation of G1639 in human 18S rRNA. RNA 2014, 21, 180–187. [Google Scholar] [CrossRef]
- Leulliot, N.; Chaillet, M.; Durand, D.; Ulryck, N.; Blondeau, K.; van Tilbeurgh, H. Structure of the Yeast tRNA m7G Methylation Complex. Structure 2008, 16, 52–61. [Google Scholar] [CrossRef]
- Chu, J.-M.; Ye, T.-T.; Ma, C.-J.; Lan, M.-D.; Liu, T.; Yuan, B.-F.; Feng, Y.-Q. Existence of Internal N7-Methylguanosine Modification in mRNA Determined by Differential Enzyme Treatment Coupled with Mass Spectrometry Analysis. ACS Chem. Biol. 2018, 13, 3243–3250. [Google Scholar] [CrossRef]
- Zhang, L.-S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.-L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X.; et al. Transcriptome-wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian mRNA. Mol. Cell 2019, 74, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Malbec, L.; Zhang, T.; Chen, Y.-S.; Zhang, Y.; Sun, B.-F.; Shi, B.-Y.; Zhao, Y.-L.; Yang, Y.; Yang, Y.-G. Dynamic methylome of internal mRNA N7-methylguanosine and its regulatory role in translation. Cell Res. 2019, 29, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Qing, Y.; Dong, L.; Han, L.; Wu, D.; Li, Y.; Li, W.; Xue, J.; Zhou, K.; Sun, M.; et al. QKI shuttles internal m7G-modified transcripts into stress granules and modulates mRNA metabolism. Cell 2023, 186, 3208–3226. [Google Scholar] [CrossRef]
- Pandolfini, L.; Barbieri, I.; Bannister, A.J.; Hendrick, A.; Andrews, B.; Webster, N.; Murat, P.; Mach, P.; Brandi, R.; Robson, S.C.; et al. METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol. Cell 2019, 74, 1278–1290. [Google Scholar] [CrossRef] [PubMed]
- Dumas, L.; Herviou, P.; Dassi, E.; Cammas, A.; Millevoi, S. G-Quadruplexes in RNA Biology: Recent Advances and Future Directions. Trends Biochem. Sci. 2020, 46, 270–283. [Google Scholar] [CrossRef]
- Deb, I.; Popenda. ; Sarzyńska, J.; Małgowska, M.; Lahiri, A.; Gdaniec, Z.; Kierzek, R. Computational and NMR studies of RNA duplexes with an internal pseudouridine-adenosine base pair. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Jack, K.; Bellodi, C.; Landry, D.M.; Niederer, R.O.; Meskauskas, A.; Musalgaonkar, S.; Kopmar, N.; Krasnykh, O.; Dean, A.M.; Thompson, S.R.; et al. rRNA Pseudouridylation Defects Affect Ribosomal Ligand Binding and Translational Fidelity from Yeast to Human Cells. Mol. Cell 2011, 44, 660–666. [Google Scholar] [CrossRef]
- Morais, P.; Adachi, H.; Yu, Y.-T. Spliceosomal snRNA Epitranscriptomics. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef]
- Karijolich, J.; Yu, Y.-T. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 2011, 474, 395–398. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, X.; Kierzek, R.; Yu, Y.-T. A Flexible RNA Backbone within the Polypyrimidine Tract Is Required for U2AF65 Binding and Pre-mRNA Splicing InVivo. Mol. Cell. Biol. 2010, 30, 4108–4119. [Google Scholar] [CrossRef]
- Martinez, N.M.; Su, A.; Burns, M.C.; Nussbacher, J.K.; Schaening, C.; Sathe, S.; Yeo, G.W.; Gilbert, W.V. Pseudouridine synthases modify human pre-mRNA co-transcriptionally and affect pre-mRNA processing. Mol. Cell 2022, 82, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Casas, K.; Mengesha, E.; Inbal, A.; Fischel-Ghodsian, N. Missense Mutation in Pseudouridine Synthase 1 (PUS1) Causes Mitochondrial Myopathy and Sideroblastic Anemia (MLASA). Am. J. Hum. Genet. 2004, 74, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, D.G.; Teysset, L.; Carré, C. RNA 2’-O-Methylation (Nm) Modification in Human Diseases. Genes 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Monaco, P.L.; Marcel, V.; Diaz, J.-J.; Catez, F. 2′-O-Methylation of Ribosomal RNA: Towards an Epitranscriptomic Control of Translation? Biomolecules 2018, 8, 106. [Google Scholar] [CrossRef]
- Decatur, W.A.; Fournier, M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002, 27, 344–351. [Google Scholar] [CrossRef]
- Nachmani, D.; Bothmer, A.H.; Grisendi, S.; Mele, A.; Bothmer, D.; Lee, J.D.; Monteleone, E.; Cheng, K.; Zhang, Y.; Bester, A.C.; et al. Germline NPM1 mutations lead to altered rRNA 2′-O-methylation and cause dyskeratosis congenita. Nat. Genet. 2019, 51, 1518–1529. [Google Scholar] [CrossRef]
- Sharma, S.; Marchand, V.; Motorin, Y.; Lafontaine, D.L.J. Identification of sites of 2′-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Wilkinson, M.L.; Crary, S.M.; Jackman, J.E.; Grayhack, E.J.; Phizicky, E.M. The 2′-O-methyltransferase responsible for modification of yeast tRNA at position 4. RNA 2007, 13, 404–413. [Google Scholar] [CrossRef]
- Endres, L.; Rose, R.E.; Doyle, F.; Rahn, T.; Lee, B.; Seaman, J.; McIntyre, W.D.; Fabris, D. 2'-O-ribose methylation of transfer RNA promotes recovery from oxidative stress in Saccharomyces cerevisiae. PLOS ONE 2020, 15, e0229103. [Google Scholar] [CrossRef]
- Picard-Jean, F.; Brand, C.; Tremblay-Létourneau, M.; Allaire, A.; Beaudoin, M.C.; Boudreault, S.; Duval, C.; Rainville-Sirois, J.; Robert, F.; Pelletier, J.; et al. 2'-O-methylation of the mRNA cap protects RNAs from decapping and degradation by DXO. PLOS ONE 2018, 13, e0193804. [Google Scholar] [CrossRef]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Indrisiunaite, G.; DeMirci, H.; Ieong, K.-W.; Wang, J.; Petrov, A.; Prabhakar, A.; Rechavi, G.; Dominissini, D.; He, C.; et al. 2′-O-methylation in mRNA disrupts tRNA decoding during translation elongation. Nat. Struct. Mol. Biol. 2018, 25, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.A.; Ho, H.-T.; Ranganathan, S.V.; Vangaveti, S.; Ilkayeva, O.; Assi, H.A.; Choi, A.K.; Agris, P.F.; Holley, C.L. Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Bélanger, F.; Stepinski, J.; Darzynkiewicz, E.; Pelletier, J. Characterization of hMTr1, a Human Cap1 2′-O-Ribose Methyltransferase*. J. Biol. Chem. 2010, 285, 33037–33044. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Purta, E.; Kaminska, K.H.; Cymerman, I.A.; Campbell, D.A.; Mittra, B.; Zamudio, J.R.; Sturm, N.R.; Jaworski, J.; Bujnicki, J.M. 2′-O-ribose methylation of cap2 in human: function and evolution in a horizontally mobile family. Nucleic Acids Res. 2011, 39, 4756–4768. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef]
- Bass, B. A developmentally regulated activity that unwinds RNA duplexes. Cell 1987, 48, 607–613. [Google Scholar] [CrossRef]
- Keegan, L.P.; McGurk, L.; Palavicini, J.P.; Brindle, J.; Paro, S.; Li, X.; Rosenthal, J.J.C.; O'Connell, M.A. Functional conservation in human and Drosophila of Metazoan ADAR2 involved in RNA editing: loss of ADAR1 in insects. Nucleic Acids Res. 2011, 39, 7249–7262. [Google Scholar] [CrossRef]
- Slavov, D.; Crnogorac-Jurčević, T.; Clark, M.; Gardiner, K. Comparative analysis of the DRADA A-to-I RNA editing gene from mammals, pufferfish and zebrafish. Gene 2000, 250, 53–60. [Google Scholar] [CrossRef]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA Editing of Embedded Alu Elements in the Human Transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef]
- Tan, M.H.; GTEx Consortium; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Mazloomian, A.; Meyer, I.M. Genome-wide identification and characterization of tissue-specific RNA editing events inD. melanogasterand their potential role in regulating alternative splicing. RNA Biol. 2015, 12, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.; Park, C.-K.; Leung, A.K.; Gao, Y.; Hyde, T.M.; Kleinman, J.E.; Rajpurohit, A.; Tao, R.; Shin, J.H.; Weinberger, D.R. Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 2016, 19, 1093–1099. [Google Scholar] [CrossRef]
- Huntley, M.A.; Lou, M.; Goldstein, L.D.; Lawrence, M.; Dijkgraaf, G.J.P.; Kaminker, J.S.; Gentleman, R. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genom. 2016, 17, 61. [Google Scholar] [CrossRef]
- Wahlstedt, H.; Daniel, C.; Ensterö, M.; Öhman, M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009, 19, 978–986. [Google Scholar] [CrossRef]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. J. Biol. Chem. 2017, 292, 4326–4335. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiao, X. Genome sequence–independent identification of RNA editing sites. Nat. Methods 2015, 12, 347–350. [Google Scholar] [CrossRef]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Lagergren, J.; Öhman, M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie 2015, 117, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Eggington, J.M.; Greene, T.; Bass, B.L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011, 2, 319. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.-F.; Yang, Q.; Liu, C.-X.; Wu, M.; Chen, L.-L.; Yang, L. N6-Methyladenosines Modulate A-to-I RNA Editing. Mol. Cell 2018, 69, 126–135. [Google Scholar] [CrossRef]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef]
- Brümmer, A.; Yang, Y.; Chan, T.W.; Xiao, X. Structure-mediated modulation of mRNA abundance by A-to-I editing. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.F.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. M6A MRNA Methylation Facilitates Resolution of Naïve Pluripotency toward Differentiation. Science (1979) 2015, 347, 1002–1006. [Google Scholar]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m6A RNA Modification Controls Cell Fate Transition in Mammalian Embryonic Stem Cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nature 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Mendel, M.; Chen, K.-M.; Homolka, D.; Gos, P.; Pandey, R.R.; McCarthy, A.A.; Pillai, R.S. Methylation of Structured RNA by the m6A Writer METTL16 Is Essential for Mouse Embryonic Development. Mol. Cell 2018, 71, 986–1000. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Yue, M.; Wang, J.; Kumar, S.; Wechsler-Reya, R.J.; Zhang, Z.; Ogawa, Y.; Kellis, M.; Duester, G.; et al. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 2018, 21, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, F.; Zhang, F.; Sancho, A.; Fidalgo, M.; Di Cecilia, S.; Vashisht, A.; Lee, D.-F.; Chen, C.-H.; Rengasamy, M.; Andino, B.; et al. Coordination of m 6 A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell Stem Cell 2015, 17, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pei, Y.; Zhou, R.; Tang, Z.; Yang, Y. Regulation of RNA N6-methyladenosine modification and its emerging roles in skeletal muscle development. Int. J. Biol. Sci. 2021, 17, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Liu, J.; Zhang, J.; Mu, T.; Feng, X.; Ma, R.; Gu, Y. Regulatory role of RNA N6-methyladenosine modifications during skeletal muscle development. Front. Cell Dev. Biol. 2022, 10, 929183. [Google Scholar] [CrossRef]
- Zhang, X.; Yao, Y.; Han, J.; Yang, Y.; Chen, Y.; Tang, Z.; Gao, F. Longitudinal epitranscriptome profiling reveals the crucial role of N6-methyladenosine methylation in porcine prenatal skeletal muscle development. J. Genet. Genom. 2020, 47, 466–476. [Google Scholar] [CrossRef]
- Xu, T.; Xu, Z.; Lu, L.; Zeng, T.; Gu, L.; Huang, Y.; Zhang, S.; Yang, P.; Wen, Y.; Lin, D.; et al. Transcriptome-wide study revealed m6A regulation of embryonic muscle development in Dingan goose (Anser cygnoides orientalis). BMC Genom. 2021, 22, 1–16. [Google Scholar] [CrossRef]
- Deng, K.; Fan, Y.; Liang, Y.; Cai, Y.; Zhang, G.; Deng, M.; Wang, Z.; Lu, J.; Shi, J.; Wang, F.; et al. FTO-mediated demethylation of GADD45B promotes myogenesis through the activation of p38 MAPK pathway. Mol. Ther. - Nucleic Acids 2021, 26, 34–48. [Google Scholar] [CrossRef]
- Chen, B.; Liu, S.; Zhang, W.; Xiong, T.; Zhou, M.; Hu, X.; Mao, H.; Liu, S. Profiling Analysis of N6-Methyladenosine mRNA Methylation Reveals Differential m6A Patterns during the Embryonic Skeletal Muscle Development of Ducks. Animals 2022, 12, 2593. [Google Scholar] [CrossRef]
- Wang, X.; Huang, N.; Yang, M.; Wei, D.; Tai, H.; Han, X.; Gong, H.; Zhou, J.; Qin, J.; Wei, X.; et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1α pathway-mediated mitochondria biogenesis. Cell Death Dis. 2017, 8, e2702–e2702. [Google Scholar] [CrossRef]
- Ren, X.; Deng, R.; Zhang, K.; Sun, Y.; Li, Y.; Li, J. Single-Cell Imaging of m 6 A Modified RNA Using m 6 A-Specific In Situ Hybridization Mediated Proximity Ligation Assay (m 6 AISH-PLA). Angew. Chem. Int. Ed. 2021, 60, 22646–22651. [Google Scholar] [CrossRef]
- Chen, J.N.; Chen, Y.; Wei, Y.Y.; Raza, M.A.; Zou, Q.; Xi, X.Y.; Zhu, L.; Tang, G.Q.; Jiang, Y.Z.; Li, X.W. Regulation of m6A RNA Methylation and Its Effect on Myogenic Differentiation in Murine Myoblasts. Mol. Biol. 2019, 53, 384–392. [Google Scholar] [CrossRef]
- Gheller, B.J.; Blum, J.E.; Fong, E.H.H.; Malysheva, O.V.; Cosgrove, B.D.; Thalacker-Mercer, A.E. A defined N6-methyladenosine (m6A) profile conferred by METTL3 regulates muscle stem cell/myoblast state transitions. Cell Death Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef]
- Yang, X.; Mei, C.; Ma, X.; Du, J.; Wang, J.; Zan, L. M6 A Methylases Regulate Myoblast Proliferation, Apoptosis and Differentiation. Animals 2022, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, S.; Zhang, X.; Ren, S.; Tang, Z.; Gao, F. Coordinated transcriptional and post-transcriptional epigenetic regulation during skeletal muscle development and growth in pigs. J. Anim. Sci. Biotechnol. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Gao, X.; Shin, Y.-H.; Li, M.; Wang, F.; Tong, Q.; Zhang, P. The Fat Mass and Obesity Associated Gene FTO Functions in the Brain to Regulate Postnatal Growth in Mice. PLOS ONE 2010, 5, e14005. [Google Scholar] [CrossRef]
- Church, C.; Moir, L.; McMurray, F.; Girard, C.; Banks, G.T.; Teboul, L.; Wells, S.; Brüning, J.C.; Nolan, P.M.; Ashcroft, F.M.; et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat. Genet. 2010, 42, 1086–1092. [Google Scholar] [CrossRef]
- Xie, S.J.; Lei, H.; Yang, B.; Diao, L.T.; Liao, J.Y.; He, J.H.; Tao, S.; Hu, Y.X.; Hou, Y.R.; Sun, Y.J.; et al. Dynamic M6A MRNA Methylation Reveals the Role of METTL3/14-M6A-MNK2-ERK Signaling Axis in Skeletal Muscle Differentiation and Regeneration. Front Cell Dev Biol 2021, 9. [Google Scholar] [CrossRef]
- Kudou, K.; Komatsu, T.; Nogami, J.; Maehara, K.; Harada, A.; Saeki, H.; Oki, E.; Maehara, Y.; Ohkawa, Y. The requirement of Mettl3-promoted MyoD mRNA maintenance in proliferative myoblasts for skeletal muscle differentiation. Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Liang, Y.; Han, H.; Xiong, Q.; Yang, C.; Wang, L.; Ma, J.; Lin, S.; Jiang, Y.-Z. METTL3-Mediated m6A Methylation Regulates Muscle Stem Cells and Muscle Regeneration by Notch Signaling Pathway. Stem Cells Int. 2021, 2021, 1–13. [Google Scholar] [CrossRef]
- Brack, A.S.; Conboy, I.M.; Conboy, M.J.; Shen, J.; Rando, T.A. A Temporal Switch from Notch to Wnt Signaling in Muscle Stem Cells Is Necessary for Normal Adult Myogenesis. Cell Stem Cell 2008, 2, 50–59. [Google Scholar] [CrossRef]
- Yang, X.; Ning, Y.; Raza, S.H.A.; Mei, C.; Zan, L. MEF2C Expression Is Regulated by the Post-transcriptional Activation of the METTL3-m6A-YTHDF1 Axis in Myoblast Differentiation. Front. Veter- Sci. 2022, 9, 900924. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.-T.; Xie, S.-J.; Lei, H.; Qiu, X.-S.; Huang, M.-C.; Tao, S.; Hou, Y.-R.; Hu, Y.-X.; Sun, Y.-J.; Zhang, Q.; et al. METTL3 regulates skeletal muscle specific miRNAs at both transcriptional and post-transcriptional levels. Biochem. Biophys. Res. Commun. 2021, 552, 52–58. [Google Scholar] [CrossRef]
- Xie, S.-J.; Tao, S.; Diao, L.-T.; Li, P.-L.; Chen, W.-C.; Zhou, Z.-G.; Hu, Y.-X.; Hou, Y.-R.; Lei, H.; Xu, W.-Y.; et al. Characterization of Long Non-coding RNAs Modified by m6A RNA Methylation in Skeletal Myogenesis. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, J.M.; Hinger, S.A.; Golubeva, V.A.; Barajas, J.M.; Dorn, L.E.; Iyer, C.C.; Sun, H.-L.; Arnold, W.D.; He, C.; Accornero, F. The m6A methyltransferase METTL3 regulates muscle maintenance and growth in mice. Nat. Commun. 2022, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, M.; Liu, D.; Shi, Y.; Ren, J.; Wang, S.; Jing, Y.; Zhang, S.; Zhao, Q.; Li, H.; et al. m6A epitranscriptomic regulation of tissue homeostasis during primate aging. Nat. Aging 2023, 3, 705–721. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, T.; Wang, Q.; Fu, R.; Zhang, Z.; Chen, N.; Li, Z.; Gao, G.; Peng, S.; Yang, D. m6A demethylase ALKBH5 drives denervation-induced muscle atrophy by targeting HDAC4 to activate FoxO3 signalling. J. Cachex- Sarcopenia Muscle 2022, 13, 1210–1223. [Google Scholar] [CrossRef]
- Blanco, S.; Kurowski, A.; Nichols, J.; Watt, F.M.; Benitah, S.A.; Frye, M. The RNA–Methyltransferase Misu (NSun2) Poises Epidermal Stem Cells to Differentiate. PLOS Genet. 2011, 7, e1002403. [Google Scholar] [CrossRef]
- Trixl, L.; Amort, T.; Wille, A.; Zinni, M.; Ebner, S.; Hechenberger, C.; Eichin, F.; Gabriel, H.; Schoberleitner, I.; Huang, A.; et al. RNA cytosine methyltransferase Nsun3 regulates embryonic stem cell differentiation by promoting mitochondrial activity. Cell. Mol. Life Sci. 2017, 75, 1483–1497. [Google Scholar] [CrossRef]
- Blanco, S.; Bandiera, R.; Popis, M.; Hussain, S.; Lombard, P.; Aleksic, J.; Sajini, A.; Tanna, H.; Cortés-Garrido, R.; Gkatza, N.; et al. Stem cell function and stress response are controlled by protein synthesis. Nature 2016, 534, 335–340. [Google Scholar] [CrossRef]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2023 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2022, 33, 76–117. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Towards a druggable epitranscriptome: Compounds that target RNA modifications in cancer. Br. J. Pharmacol. 2021, 179, 2868–2889. [Google Scholar] [CrossRef]
- Liang, Y.; Han, H.; Xiong, Q.; Yang, C.; Wang, L.; Ma, J.; Lin, S.; Jiang, Y.-Z. METTL3-Mediated m6A Methylation Regulates Muscle Stem Cells and Muscle Regeneration by Notch Signaling Pathway. Stem Cells Int. 2021, 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Hengesbach, M.; Yu, Y.-T.; Morais, P. From Antisense RNA to RNA Modification: Therapeutic Potential of RNA-Based Technologies. Biomedicines 2021, 9, 550. [Google Scholar] [CrossRef] [PubMed]
- Rugowska, A.; Starosta, A.; Konieczny, P. Epigenetic modifications in muscle regeneration and progression of Duchenne muscular dystrophy. Clin. Epigenetics 2021, 13, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Caretti, G.; DI Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004, 18, 2627–2638. [Google Scholar] [CrossRef]
- Reardon, S. Step aside CRISPR, RNA editing is taking off. Nature 2020, 578, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef]
- Fulco, M.; Schiltz, R.L.; Iezzi, S.; King, M.T.; Zhao, P.; Kashiwaya, Y.; Hoffman, E.; Veech, R.L.; Sartorelli, V. Sir2 Regulates Skeletal Muscle Differentiation as a Potential Sensor of the Redox State. Mol. Cell 2003, 12, 51–62. [Google Scholar] [CrossRef]
- Molinari, S.; Imbriano, C.; Moresi, V.; Renzini, A.; Belluti, S.; Lozanoska-Ochser, B.; Gigli, G.; Cedola, A. Histone deacetylase functions and therapeutic implications for adult skeletal muscle metabolism. Front. Mol. Biosci. 2023, 10. [Google Scholar] [CrossRef]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol Ameliorates Muscular Pathology in the Dystrophic mdx Mouse, a Model for Duchenne Muscular Dystrophy. Experiment 2011, 338, 784–794. [Google Scholar] [CrossRef]
- Tonkin, J.; Villarroya, F.; Puri, P.L.; Vinciguerra, M. SIRT1 signaling as potential modulator of skeletal muscle diseases. Curr. Opin. Pharmacol. 2012, 12, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Marroncelli, N.; Bianchi, M.; Bertin, M.; Consalvi, S.; Saccone, V.; De Bardi, M.; Puri, P.L.; Palacios, D.; Adamo, S.; Moresi, V. HDAC4 regulates satellite cell proliferation and differentiation by targeting P21 and Sharp1 genes. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Williams, A.H.; Meadows, E.; Flynn, J.M.; Potthoff, M.J.; McAnally, J.; Shelton, J.M.; Backs, J.; Klein, W.H.; Richardson, J.A.; et al. Myogenin and Class II HDACs Control Neurogenic Muscle Atrophy by Inducing E3 Ubiquitin Ligases. Cell 2010, 143, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Pigna, E.; Renzini, A.; Greco, E.; Simonazzi, E.; Fulle, S.; Mancinelli, R.; Moresi, V.; Adamo, S. HDAC4 preserves skeletal muscle structure following long-term denervation by mediating distinct cellular responses. Skelet. Muscle 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Pigna, E.; Simonazzi, E.; Sanna, K.; Bernadzki, K.M.; Proszynski, T.; Heil, C.; Palacios, D.; Adamo, S.; Moresi, V. Histone deacetylase 4 protects from denervation and skeletal muscle atrophy in a murine model of amyotrophic lateral sclerosis. EBioMedicine 2019, 40, 717–732. [Google Scholar] [CrossRef]
- Renzini, A.; Pigna, E.; Rocchi, M.; Cedola, A.; Gigli, G.; Moresi, V.; Coletti, D. Sex and HDAC4 Differently Affect the Pathophysiology of Amyotrophic Lateral Sclerosis in SOD1-G93A Mice. Int. J. Mol. Sci. 2022, 24, 98. [Google Scholar] [CrossRef]
- Renzini, A.; Marroncelli, N.; Cavioli, G.; Di Francescantonio, S.; Forcina, L.; Lambridis, A.; Di Giorgio, E.; Valente, S.; Mai, A.; Brancolini, C.; et al. Cytoplasmic HDAC4 regulates the membrane repair mechanism in Duchenne muscular dystrophy. J. Cachex- Sarcopenia Muscle 2022, 13, 1339–1359. [Google Scholar] [CrossRef]
- Shen, H.; Xie, K.; Li, M.; Yang, Q.; Wang, X. N6-methyladenosine (m6A) methyltransferase METTL3 regulates sepsis-induced myocardial injury through IGF2BP1/HDAC4 dependent manner. Cell Death Discov. 2022, 8, 1–8. [Google Scholar] [CrossRef]
- Taylor, M.V.; Hughes, S.M. Mef2 and the skeletal muscle differentiation program. Semin. Cell Dev. Biol. 2017, 72, 33–44. [Google Scholar] [CrossRef]
- Nakka, K.; Ghigna, C.; Gabellini, D.; Dilworth, F.J. Diversification of the muscle proteome through alternative splicing. Skelet. Muscle 2018, 8, 1–18. [Google Scholar] [CrossRef]
- Zhu, B.; Ramachandran, B.; Gulick, T. Alternative Pre-mRNA Splicing Governs Expression of a Conserved Acidic Transactivation Domain in Myocyte Enhancer Factor 2 Factors of Striated Muscle and Brain. J. Biol. Chem. 2005, 280, 28749–28760. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Faralli, H.; Yao, Z.; Rakopoulos, P.; Palii, C.; Cao, Y.; Singh, K.; Liu, Q.-C.; Chu, A.; Aziz, A.; et al. Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. 2013, 27, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Baruffaldi, F.; Montarras, D.; Basile, V.; De Feo, L.; Badodi, S.; Ganassi, M.; Battini, R.; Nicoletti, C.; Imbriano, C.; Musarò, A.; et al. Dynamic Phosphorylation of the Myocyte Enhancer Factor 2Cα1 Splice Variant Promotes Skeletal Muscle Regeneration and Hypertrophy. STEM CELLS 2016, 35, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Ganassi, M.; Badodi, S.; Polacchini, A.; Baruffaldi, F.; Battini, R.; Hughes, S.; Hinits, Y.; Molinari, S. Distinct functions of alternatively spliced isoforms encoded by zebrafish mef2ca and mef2cb. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2014, 1839, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Williams, V.C.; Hinits, Y.; Hughes, S.M. eIF4EBP3L Acts as a Gatekeeper of TORC1 In Activity-Dependent Muscle Growth by Specifically Regulating Mef2ca Translational Initiation. PLOS Biol. 2013, 11, e1001679. [Google Scholar] [CrossRef]
- Black, B.L.; Lu, J.; Olson, E.N. The MEF2A 3′ Untranslated Region Functions as a cis-Acting Translational Repressor. Mol. Cell. Biol. 1997, 17, 2756–2763. [Google Scholar] [CrossRef]
- Yang, X.; Ning, Y.; Raza, S.H.A.; Mei, C.; Zan, L. MEF2C Expression Is Regulated by the Post-transcriptional Activation of the METTL3-m6A-YTHDF1 Axis in Myoblast Differentiation. Front. Veter- Sci. 2022, 9, 900924. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, W.; Luo, W.; Liu, S.; Jiang, H.; Liu, S.; Xu, J.; Chen, B. Cloning of the RNA m6A Methyltransferase 3 and Its Impact on the Proliferation and Differentiation of Quail Myoblasts. Veter- Sci. 2023, 10, 300. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef]
- Adachi, H.; De Zoysa, M.D.; Yu, Y.-T. Post-transcriptional pseudouridylation in mRNA as well as in some major types of noncoding RNAs. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2018, 1862, 230–239. [Google Scholar] [CrossRef]
- Lin, T.; Smigiel, R.; Kuzniewska, B.; Chmielewska, J.J.; Kosińska, J.; Biela, M.; Biela, A.; Kościelniak, A.; Dobosz, D.; Laczmanska, I.; et al. Destabilization of mutated human PUS3 protein causes intellectual disability. Hum. Mutat. 2022, 43, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Tasak, M.; Maddirevula, S.; Abdel-Salam, G.M.H.; Sayed, I.S.M.; Alazami, A.M.; Al-Sheddi, T.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum. Genet. 2019, 138, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.M.; Su, A.; Burns, M.C.; Nussbacher, J.K.; Schaening, C.; Sathe, S.; Yeo, G.W.; Gilbert, W.V. Pseudouridine synthases modify human pre-mRNA co-transcriptionally and affect pre-mRNA processing. Mol. Cell 2022, 82, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Bachinski, L.L.; Sirito, M.; Böhme, M.; Baggerly, K.A.; Udd, B.; Krahe, R. Altered MEF2 isoforms in myotonic dystrophy and other neuromuscular disorders. Muscle Nerve 2010, 42, 856–863. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Patel, R.; Iakova, P.; Cai, Z.-J.; Quan, L.; Timchenko, L.T. Overexpression of CUG Triplet Repeat-binding Protein, CUGBP1, in Mice Inhibits Myogenesis. J. Biol. Chem. 2004, 279, 13129–13139. [Google Scholar] [CrossRef]
- Lee, K.-S.; Cao, Y.; Witwicka, H.E.; Tom, S.; Tapscott, S.J.; Wang, E.H. RNA-binding Protein Muscleblind-like 3 (MBNL3) Disrupts Myocyte Enhancer Factor 2 (Mef2) β-Exon Splicing. J. Biol. Chem. 2010, 285, 33779–33787. [Google Scholar] [CrossRef]
- Basile, V.; Baruffaldi, F.; Dolfini, D.; Belluti, S.; Benatti, P.; Ricci, L.; Artusi, V.; Tagliafico, E.; Mantovani, R.; Molinari, S.; et al. NF-YA splice variants have different roles on muscle differentiation. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2016, 1859, 627–638. [Google Scholar] [CrossRef]
- Rigillo, G.; Basile, V.; Belluti, S.; Ronzio, M.; Sauta, E.; Ciarrocchi, A.; Latella, L.; Saclier, M.; Molinari, S.; Vallarola, A.; et al. The transcription factor NF-Y participates to stem cell fate decision and regeneration in adult skeletal muscle. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Libetti, D.; Bernardini, A.; Sertic, S.; Messina, G.; Dolfini, D.; Mantovani, R. The Switch from NF-YAl to NF-YAs Isoform Impairs Myotubes Formation. Cells 2020, 9, 789. [Google Scholar] [CrossRef]
- Rigillo, G.; Belluti, S.; Campani, V.; Ragazzini, G.; Ronzio, M.; Miserocchi, G.; Bighi, B.; Cuoghi, L.; Mularoni, V.; Zappavigna, V.; et al. The NF–Y splicing signature controls hybrid EMT and ECM-related pathways to promote aggressiveness of colon cancer. Cancer Lett. 2023, 567, 216262. [Google Scholar] [CrossRef]
- Belluti, S.; Semeghini, V.; Rigillo, G.; Ronzio, M.; Benati, D.; Torricelli, F.; Bonetti, L.R.; Carnevale, G.; Grisendi, G.; Ciarrocchi, A.; et al. Alternative splicing of NF-YA promotes prostate cancer aggressiveness and represents a new molecular marker for clinical stratification of patients. J. Exp. Clin. Cancer Res. 2021, 40, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Basile, V.; Baruffaldi, F.; Dolfini, D.; Belluti, S.; Benatti, P.; Ricci, L.; Artusi, V.; Tagliafico, E.; Mantovani, R.; Molinari, S.; et al. NF-YA splice variants have different roles on muscle differentiation. Biochim. et Biophys. Acta (BBA) - Gene Regul. Mech. 2016, 1859, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Rigillo, G.; Basile, V.; Belluti, S.; Ronzio, M.; Sauta, E.; Ciarrocchi, A.; Latella, L.; Saclier, M.; Molinari, S.; Vallarola, A.; et al. The transcription factor NF-Y participates to stem cell fate decision and regeneration in adult skeletal muscle. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef]
- Paris, N.D.; Soroka, A.; Klose, A.; Liu, W.; Chakkalakal, J.V. Smad4 restricts differentiation to promote expansion of satellite cell derived progenitors during skeletal muscle regeneration. Abstract 2016, 5. [Google Scholar] [CrossRef]
- Forouhan, M.; Lim, W.F.; Zanetti-Domingues, L.C.; Tynan, C.J.; Roberts, T.C.; Malik, B.; Manzano, R.; Speciale, A.A.; Ellerington, R.; Garcia-Guerra, A.; et al. AR cooperates with SMAD4 to maintain skeletal muscle homeostasis. Acta Neuropathol. 2022, 143, 713–731. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



