
Submitted:
12 November 2023
Posted:
13 November 2023
You are already at the latest version
Abstract
Clinical trials often involve new substances characterized or developed for a specific pathology or class of pathologies. At other times, a clinical trial is observational; in other words, a known sub-stance has a range of effects on a given cohort of subjects. The types of trials are different and complex, and often, young clinical novice and the inexperienced researchers may struggle to evaluate how to proceed correctly. In this paper, we aim to create a mini-guide for beginners in clinical trials and provide elements for designing a clinical trial. Our intention is to highlight where to find the materials and how to adapt, even in the absence of sponsors, so that even the most inexperienced and under-resourced researcher can gain an understqanding of the complexi-ty of the process andaddress it appropriately. We do not claim to be exhaustive or cover all ele-ments, but rather, we aim to create a checklist. With this approach, starting discussioms about the experimental phases of the drug and concluding with the clinical trial, we aim to assist those facing this challenging "world."
Keywords:
clinical trials
; study design
; law procedures
; statistical approach
; health
; Patients
; Non-Commercial Clinical Trials
; enrollment
; regulatory agencies
; packaging
1. Introduction
Developing a new drug involves multiple research stages to ensure the compound's safety, effectiveness, and practicality. This process begins with pre-clinical research and concludes with the four fundamental phases of clinical research.
Preclinical development, also referred to as preclinical studies or non-clinical studies, constitutes a pivotal research phase in drug development that precedes human clinical trials. Throughout this stage, researchers collect essential data to assess the viability of the drug and conduct numerous tests to ensure its safety [1].
Initially, this phase is carried out in vitro, particularly on cell cultures or in silico organs, and subsequently on laboratory animals, starting with simpler species and progressively advancing to small mammals within the rodent family [1]. Preclinical studies involving animals play a crucial role in evaluating the effectiveness of therapeutic drugs and strategies before advancing to clinical trials. However, it's important to note that discussing these studies in detail is beyond the scope of this work.
While in vitro preclinical studies are not subject to specific legislation, studies involving animals in vivo demand careful consideration of the animals as sentient beings. They are subject to various laws and regulations designed to safeguard the well-being of the animal species.
Namely, the four phases of clinical research start with phase 1, which requires proving the drug's safety on a small number of healthy volunteers. During this phase, researchers investigate the side effects of the drug and suggest a modality of administration for the compound [2,3].
Next, phase 2 involves using a placebo compound to compare the drug's beneficial effects with a non-active compound and fine-tuned doses. A group of patients is involved in this phase so that researchers can evaluate safety and efficacy in unhealthy conditions [2,4].
Phase 3 requires more patients than Phase 2 and is necessary to confirm/retract the drug’s efficacy compared to the placebo [2,5]. Usually, at the end of phase 3, the drug is approved/rejected by the competent authority (see Table 1).
After the drug has been approved for use in the general population, a post-marketing surveillance named phase 4 begins, allowing the evaluation of the long-term risks and benefits in a real scenario [2,6].
The process for drug repositioning, meaning using an existing drug for a different therapeutic purpose, is different. This approach reduces the cost of experimentation and makes the compound readily available because researchers do not test the safety and functionality of the compound, as all the information on the drug can be obtained from life science databases. The candidate drug can be tested on a new group of patients alone or in addition to existing therapies. Comparison with placebo may be facultative [7].
Figure 1.
Drug development and clinical trial stages. The time required for the whole process is estimated at 15-20 years. From 1-2 to 6 years are required in the preclinical stage; then from 3 to 10 years are necessary to develop the central clinical stages (1-3), Phase 4 becomes active after the drug has been approved and is active for 10-20 years [8].
Figure 1.
Drug development and clinical trial stages. The time required for the whole process is estimated at 15-20 years. From 1-2 to 6 years are required in the preclinical stage; then from 3 to 10 years are necessary to develop the central clinical stages (1-3), Phase 4 becomes active after the drug has been approved and is active for 10-20 years [8].
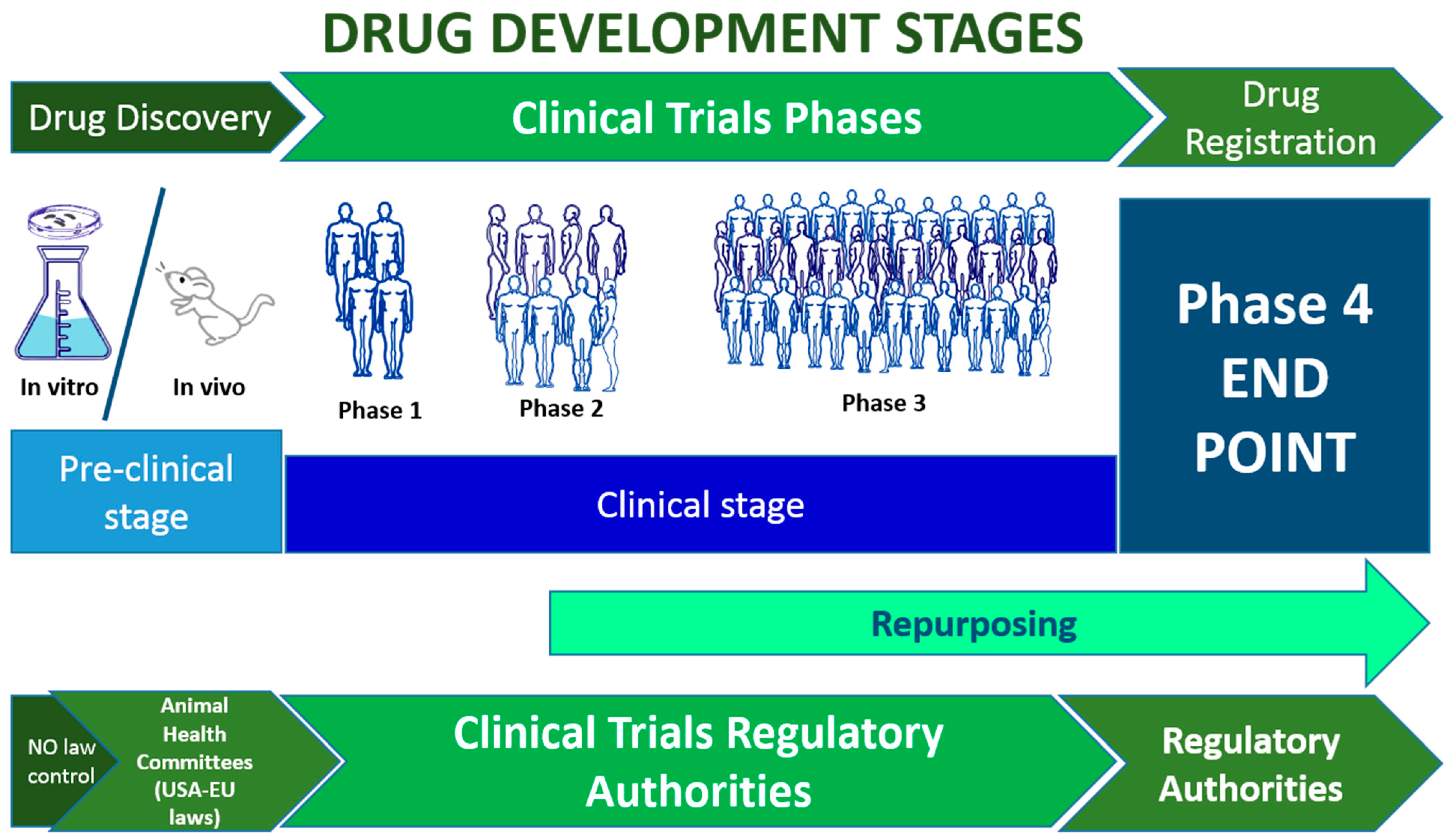
Overall, each clinical study must be planned carefully since every stage plays a vital role in the quality, execution, and interpretation of biomedical research [9]. The choice of the study design is a crucial part of the study goal,relying on many variables, including previous research, availability of study enrollees, funding, and time limitations. In general, two main types of clinical research can be identified: i) observational studies and ii) interventional studies. Other types of clinical studies are summarized in Figure 2.
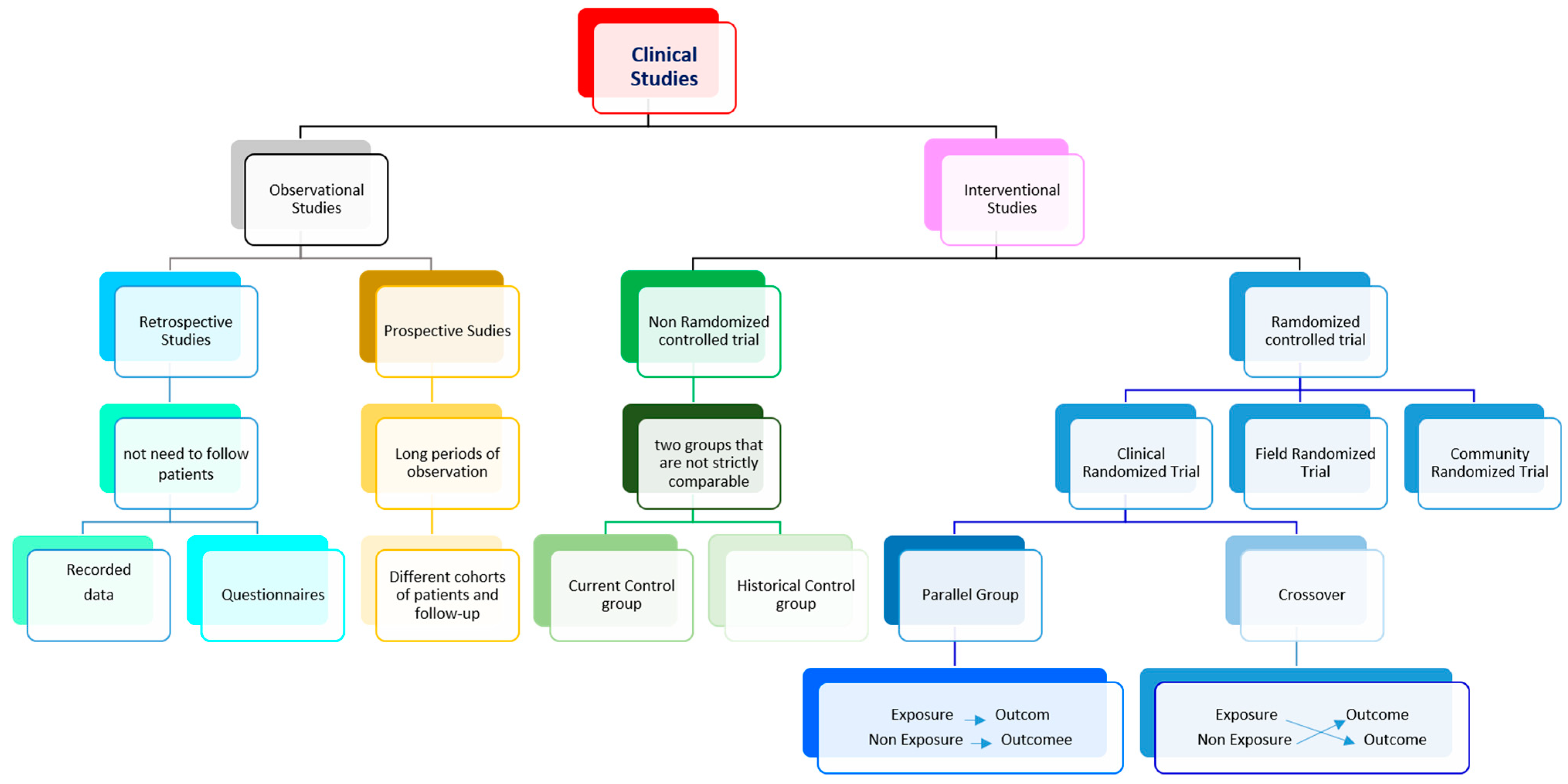
In epidemiological studies, researchers merely collect data from participants without acting directly on them, identifying relationships between factors and results. The data may concern the participants' health, habits, or environments. In experimental studies, participants receive one or more interventions, in cases of pharmacological substances, this happens during the experimental clinical phase [10]. These interventions can involve drugs, medical devices, procedures, or participant behavior changes (e.g., diet or exercise). Such interventions may be related to control (no medication), to placebo (no-active medication), or, in other cases, to comparisons of several available interventions between them. According to the temporality of data collection, observational studies can be designed as i) retrospective studies, where data are collected from the past through logs or by asking participants to recall exposures or outcomes [11]; or ii) prospective studies, where participants are followed over time, collecting data during the trial. Nevertheless, interventional studies have many variants in how they are designed [12]. Concerning how the data were collected, studies can be classified as i) randomized control trials or as controlled selection (i.e. non-randomized controlled trials). In the latter, the studied drug is administered to one group and the outcomes are compared with an alternative treatment/placebo administered to another group. Finally, according to the role of the researcher, the study may be i) open, where the subject and researcher know the phase of the study; ii) blind, where only the researcher knows; or iii) double-blind, when neither subject nor researcher knows [12]. All these experimental studies are used to evaluate study questions relating either to therapeutic agents, such as treatments, surgical approaches, or diagnostic tests; or to prevention, which may include changes in protective equipment, engineering controls, management, policy, or any element that needs to be evaluated as a potential cause of illness or injury.
This short manuscript describes an interventional clinical trial and the procedures needed for authorization.
2. Detailed procedures
Since not all clinical studies are pharmacological, we will focus our attention on the realization of a clinical study that is non-drug dependent (e.g., a study with well-known drugs or with a device).
The first step in a clinical trial is to clearly define all the research steps and draw up a document in which objectives, duration, and the number of patients involved are clarified. Subsequently, a trial protocol needs to be developed, in which the researcher must outline the aims, design, methods, statistical concerns, and organizational details of the study, including the rationale for carrying out the trial and its ethical issues [13]. Trial protocols are required to fulfill the standards of good clinical practice and are necessary for approval from local ethics committees[13]. A key role in promoting rigorous and transparent reporting practices, thereby elevating the overall quality of health research, can be played by the Equator Network, a global initiative committed to promoting quality and transparency in health research through its meticulously developed guidelines. Its most important contributions are the CONSORT (Consolidated Standards of Reporting Trials) and STROBE (STrengthening the Reporting of OBservational studies in Epidemiology) guidelines. These guidelines provide a comprehensive framework designed to advance the reporting standards of clinical trials and observational studies, respectively. By providing researchers with structured protocols for transparently presenting methodologies, results, and conclusions, Equator's guidelines contribute significantly to the credibility and reliability of scientific evidence in health research.
There are some rules in designing a clinical study that needs to be taken into account and that regard i) subject selection, ii) subject evaluation, iii) variables taken into account in the study, and 4) the statistics used for their analysis [13,14,15].
First of all, i) it is important to be clear about the subjects to be studied, selecting patients with certain characteristics similar to each other, and excluding subjects who show clinical symptoms that may deviate from the correct answers [13,14,16]. Then, ii) subject evaluation is performed through a working method, a tool that allows researchers to test the hypothesis. It can be an objective method using a tool or a drug [14,15]. iii) Measures taken into account during the study can be objective, such as the speed of stride or the response latency of an electrophysiological stimulus, or subjective, such as the rating on an evaluation scale or answers to a subjective questionnaire [13,14]. Finally, (iv) the statistical analyses need to be accurately chosen to be suitable for the characteristics of the variables that need to be characterized, considering the researchers' role in the study (i.e. if the study is open, blind, or double-blind). Therefore, the actual statistical analysis makes use of personal knowledge enriched by specialists who deal with the evaluation of clinical variables [13,14,15]. (https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-8-general-considerations-clinical-trials-step-5_en.pdf). The number of patients should be estimated according to the restrictive parameters by power analysis. Inclusion and exclusion criteria should be considered for enlistment, and any criteria for removing or replacing subjects. Additionally, a number of participants at least 5-10% larger should be estimated depending on possible adverse effects during the procedure. Note that all the parameters of the actual court size can be obtained with many available software (e.g.: https://clincalc.com/stats/samplesize.aspx). The estimated number of subjects recruited, recruitment criteria, and recruitment methods should be listed in any groups in which the subjects will be divided and how to administer the drug [17].
Then, it is important to indicate how the entire process will be monitored, who will be available to the subject, how they will be contacted, and what time latency the responses provide. In addition, the researchers should indicate the procedure for emergencies and difficulties that may be deferred, prioritizing the well-being of the subjects involved in the study[17]. A post-trial observation period should also be estimated for each subject. The subject must be covered by health insurance for the entire study duration and its follow-up (Registries for Evaluating Patient Outcomes: A User’s Guide [Internet]. 3rd edition.).
The document should also include the periodicity of the visits and the type of evaluations, instrumental and not, to be performed on the patient.
Finally, it is the responsibility of the researcher to register the authorized clinical trial in the clinical trial registration system operated by the government or another organization. For example, ClinicalTrials.gov is a database of records and results of privately and publicly funded clinical trials conducted worldwide and managed by the US National Library of Medicine. Each clinical trial registration system provides detailed instructions and explanations that are also useful for submitting the document to the ethics committee (https///classic.clinicaltrialials.gov/ct2/manage-recs/present).
The last step for the drafting of the document is to point out i) the maximum benefit from the study, ii) which procedures will be performed to minimize harm to the parties involved, iii) the expected adverse effects and rescue procedures, iv) future resources if the study should be successful, and v) minimum resources allowed in case of failure.
The end of the study is represented by the deep development of all design sections: subjects, methods, and statistical procedures. This document must be submitted to the approval of the ethical committee. (https://health.ec.europa.eu/system/files/2020-02/12_ec_guideline_20060216_en_0.pdf)
How do you choose the ethical committee to present the trial? Ethics Committees/Institutional Review Boards (IRBs) shall be chosen in relation to the trial's location. Suppose the hospital or clinical site where the trial and enlistment will be held does not have an ethics committee. In that case, reference will be made to the one reported by itself or to the one competent for local headquarters or competent administrative health area.
The documentation prepared for the ethical committee and the written request for its acceptance will be the starting point. Usually, the presentation to the ethical committee includes health insurance for the study. Otherwise, the researchers would need to look for an insurance company experienced in clinical trials, which should provide compensation in the event of physical damage occurring during the trial. CROs usually offer this service, but if researchers want to do it autonomously, it is sufficient to contact a latge company that provides one with a local organization dedicated to this procedure.
Contacting a pharmaceutical company, a licensed pharmacy, or a company authorized to package the drug is also necessary. Researchers should provide drugs with the package leaflet of the medical product. Several multinationals and non-CROs can carry out the process of packaging and making related documentation: (i.e.: https://www.catalent.com/; https//stmpharmapro.it/it/https:///stmpharmapro.it/it).
Documents should include the i) manufacturing site (bulk production, primary packaging, secondary packaging, quality control, batch certification), (ii) manufacturing authorization (MIA), and other (iii) specifications (control processes of drug procedures).
It is also possible to have a sponsor for the clinical trial, as suggested by the FDA guideline (https://www.fda.gov/media/75398/download).
Once all the authorizations and documents are ready, if a sponsor is missing and the intention is that of repositioning a substance that has already passed the four experimental stages, researchers need to think only about how to administer the drug.
This step requires carefully evaluating the possible statistical permutations of events between drug and placebo, considering the different groups of patients, stratified by age, sex, economic conditions, pathological conditions, etc.
To realize a good strategy of substance administration and provide a compound that is simple to recognize and administrate, it is crucial to avoid possible errors. For this reason, researchers must implement a system of labeling and randomizing the drug vs. placebo in the various subgroups of enrolled subjects. Labels, barcodes progressively numbered, should identify the patient, the year and month of administration of the substance, and the type of administered substance. The process must be done without the researcher knowing the details but under their direction to indicate the possible permutations.
To better understand all the procedures for repositioning a drug, which is beyond the scope of this short article, we point out the work of Zamami and collaborators 2021, and of Jourdan and collaborators 2020 [7,18].

Table 1.
major authorities for clinical trials.
| Country | Regulatory Body | Actually under control of |
|---|---|---|
| Austria | Austrian Agency for Health and Food Safety | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Belgium | Federal Agency for Medicines and Health Products (FAMHP) | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Bulgaria | Bulgarian Drug Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Croatia | Agency for medicinal products and medical devices of Croatia | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Cyprus | Ministry of Health - Pharmaceutical Services | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Czechia | State Institute for Drug Control | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Denmark | Danish Medicines Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Estonia | State Agency of Medicines | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Finland | Finnish Medicines Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| France | National Agency for the Safety of Medicine and Health Products | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Germany | Federal Institute for Drugs and Medical Devices | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Greece | National Organization for Medicines | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Hungary | National Institute of Pharmacy and Nutrition | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Iceland | Icelandic Medicines Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Ireland | Health Products Regulatory Authority (HPRA) | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Italy | Italian Medicines Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Latvia | State Agency of Medicines | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Liechtenstein | Office of Health / Department of Pharmaceuticals | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Lithuania | State Medicines Control Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Luxembourg | Ministry of Health | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Malta | Malta Medicines Authority (MMA) | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Netherlands | Medicines Evaluation Board | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Netherlands | Healthcare and Youth Care Inspectorate, Ministry of Health, Welfare and Sport | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Norway | Norwegian Medicines Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Poland | Office for Registration of Medicinal Products, Medical Devices and Biocidal Products | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Portugal | National Authority of Medicines and Health Products | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Romania | National Authority of Medicines and Medical Devices of Romania | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Slovakia | State Institute for Drug Control | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Slovenia | Agency for Medicinal Products and Medical Devices of the Republic of Slovenia | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Spain | Spanish Agency for Medicines and Health Products | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Sweden | Swedish Medical Products Agency | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
| Albania; | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Bosnia | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Kosovo | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Republic of Macedonia; | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Montenegro; | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Serbia; | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Turkey. | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Herzegovina; | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Yugoslav | The European Medicines Agency (EMA) 2022 https://www.ema.europa.eu/en |
|
| Bangladesh | Directorate General of Drug Administration (DGDA) | https://dgdagov.info/ |
| China | State Food and Drug Administration (SFDA), National Medical Products Administration (NMPA) | http://www.sfda.com/ http://english.nmpa.gov.cn/ |
| Hong Kong | Drug Office - Department of Health | https://www.dh.gov.hk/english/main/main_ps/main_ps.html |
| India | Central Drug Standards Control Organization (CDSCO) | https://cdsco.gov.in/opencms/opencms/en/Home/ |
| Japan | Ministry of Health, Labor and Welfare (MHLW), Japanese Pharmaceuticals and Medical Devices Agency (PMDA) | https://www.mhlw.go.jp/english/ https://cdx-japan.mblbio.com/ |
| Malaysia | Ministry of Health (MOH) | https://www.moh.gov.my/ |
| Philippines | Department of Health (DOH) | https://lawphil.net/administ/doh/doh.html |
| Singapore | Health Sciences Authority (HSA) | https://www.hsa.gov.sg/ |
| South Korea | Ministry of Food and Drug Safety (MFDS) | https://www.mfds.go.kr/eng/index.do |
| Thailand | Food and Drug Administration of Thailand (FDATA) | https://en.fda.moph.go.th/ |
| Taiwan | Taiwan Food and Drug Administration (TFDA) | https://www.fda.gov.tw/eng/ |
| Vietnam | Drug Administration of Vietnam | https://dav.gov.vn/dich-vu-cong-ce5.html |
| Brazil | National Health Surveillance Agency (ANVISA) | The HHS Office for Human Research Protections (2021) https://www.hhs.gov/ohrp/index.html |
| Colombia | Ministry of Health | The HHS Office for Human Research Protections (2021) https://www.hhs.gov/ohrp/index.html |
| Perù | Ministry of Health | The HHS Office for Human Research Protections (2021) https://www.hhs.gov/ohrp/index.html |
| Cile | Ministry of Health | The HHS Office for Human Research Protections (2021) https://www.hhs.gov/ohrp/index.html |
| Canada | CanadaFDA, Health Canada (HC) reviews | https://www.canada.ca/en/health-canada/services/drugs-health-products |
| USA | The United States Food and Drug Administration (FDA or US FDA) | https://www.fda.gov/ |
| New Zealand | Medsafe is the Medicines and Medical Devices Regulatory Authorit | https://www.medsafe.govt.nz/ |
| Australia | The Therapeutic Goods Administration (TGA) | https://www.tga.gov.au/therapeutic-goods-administration-tga |
3. Conclusion
The purpose of this document is to provide newly graduated students or inexperienced researchers with the initial elements to understand how to conduct a clinical study, taking into consideration the type of study they intend to undertake and the regulations in force.
On the web, there is an overwhelming amount of information that can confuse the reader, and often, researchers at the beginning of their career do not have an extensive portfolio to guide their experimentation. For this reason, we have decided to create a list for easy orientation in designing a clinical study to better grasp its focal points. This manuscript does not claim to be exhaustive and should only be considered as a collection medium for valuable insights.
Author Contributions
Conceptualization, G.M; A.P; and G.M; Resources G.M; Methnalisys, M.M; A.P. S.D; and G.M; Writing – Original Draft Preparation, M.M; A.P; P.I; and G.M; Writing – Review & Editing, S.D; M.M; PB; and G.M; Visualization, P.I; M.M; A.P; and G.M.; Supervision, A.P; G.M; Project Administration, M.M; S.D; A.P; and G.M.
Funding
This work was partially supported by the Italian Ministry of Health “Ricerca Finalizzata” grants: RF-2021-12374979 to A.P. The funding source was not involved in the design or writing of the report and in the decision to submit the article for publication.
Data Availability Statement
All the data shown in this paper are available in the PubMed Library. The authors created all representative draws appositely and are available on request.
Acknowledgments
All authors thank Massimo Tolu, and Massimiliano Di Virgilio for their excellent technical assistance.
Conflicts of Interest
The authors declare no conflict of interest. No sponsors participate in the choice of the items; the design of the paper; the collection of literature, the interpretation of analyzed papers; the writing of the manuscript; or in the decision to publish in the NueroSci journal.
References
- Huang W, Percie du Sert N, Vollert J, Rice ASC. General Principles of Preclinical Study Design. Handb Exp Pharmacol. 2020;257:55-69. [CrossRef]
- Mohs, R. C.; Greig, N. H. Drug Discovery and Development: Role of Basic Biological Research. Alzheimers Dement. Transl. Res. Clin. Interv. 2017, 3 (4), 651–657. [CrossRef]
- Seyhan, A. A. Lost in Translation: The Valley of Death across Preclinical and Clinical Divide – Identification of Problems and Overcoming Obstacles. Transl. Med. Commun. 2019, 4 (1), 18. [CrossRef]
- Torres-Saavedra, P. A.; Winter, K. A. An Overview of Phase 2 Clinical Trial Designs. Int. J. Radiat. Oncol. 2022, 112 (1), 22–29. [CrossRef]
- Salloum, N. C.; Fava, M.; Ball, S.; Papakostas, G. I. Success and Efficiency of Phase 2/3 Adjunctive Trials for MDD Funded by Industry: A Systematic Review. Mol. Psychiatry 2020, 25 (9), 1967–1974. [CrossRef]
- Cesana, B. M.; Biganzoli, E. M. Phase IV Studies: Some Insights, Clarifications, and Issues. Curr. Clin. Pharmacol. 2018, 13 (1), 14–20. [CrossRef]
- Zamami, Y.; Hamano, H.; Niimura, T.; Aizawa, F.; Yagi, K.; Goda, M.; Izawa-Ishizawa, Y.; Ishizawa, K. Drug-Repositioning Approaches Based on Medical and Life Science Databases. Front. Pharmacol. 2021, 12, 752174. [CrossRef]
- David, S.; Kim, P. Y. Drug Trials. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023.
- Thiese, M. S. Observational and Interventional Study Design Types; an Overview. Biochem. Medica 2014, 24 (2), 199–210. [CrossRef]
- Bero, L.; Chartres, N.; Diong, J.; Fabbri, A.; Ghersi, D.; Lam, J.; Lau, A.; McDonald, S.; Mintzes, B.; Sutton, P.; Turton, J. L.; Woodruff, T. J. The Risk of Bias in Observational Studies of Exposures (ROBINS-E) Tool: Concerns Arising from Application to Observational Studies of Exposures. Syst. Rev. 2018, 7 (1), 242. [CrossRef]
- Hess, D. R. Retrospective Studies and Chart Reviews. Respir. Care 2004, 49 (10), 1171–1174.
- Song, J. W.; Chung, K. C. Observational Studies: Cohort and Case-Control Studies: Plast. Reconstr. Surg. 2010, 126 (6), 2234–2242. [CrossRef]
- Al-Jundi, A.; Sakka, S. Protocol Writing in Clinical Research. J. Clin. Diagn. Res. JCDR 2016, 10 (11), ZE10–ZE13. [CrossRef]
- Evans, S. R. Fundamentals of Clinical Trial Design. J. Exp. Stroke Transl. Med. 2010, 3 (1), 19–27. [CrossRef]
- An, M.-W.; Duong, Q.; Le-Rademacher, J.; Mandrekar, S. J. Principles of Good Clinical Trial Design. J. Thorac. Oncol. 2020, 15 (8), 1277–1280. [CrossRef]
- An, M.-W.; Duong, Q.; Le-Rademacher, J.; Mandrekar, S. J. Principles of Good Clinical Trial Design. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15 (8), 1277–1280. [CrossRef]
- Chaudhari, N.; Ravi, R.; Gogtay, N.; Thatte, U. Recruitment and Retention of the Participants in Clinical Trials: Challenges and Solutions. Perspect. Clin. Res. 2020, 11 (2), 64. [CrossRef]
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug Repositioning: A Brief Overview. J. Pharm. Pharmacol. 2020, 72 (9), 1145–1151. [CrossRef]
Figure 2.
Types of Clinical studies. The flowchart summarizes the major categories of clinical trials. The right choice is essential before designing a study protocol to increase scientific quality and clinical value. The Regulatory Authorities can apply different protocols for each type of study to oversee and evaluate the clinical trial.
Figure 2.
Types of Clinical studies. The flowchart summarizes the major categories of clinical trials. The right choice is essential before designing a study protocol to increase scientific quality and clinical value. The Regulatory Authorities can apply different protocols for each type of study to oversee and evaluate the clinical trial.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



