
Submitted:
12 September 2023
Posted:
18 September 2023
You are already at the latest version
Abstract
The black-necked crane is the only species that lives in the plateau. At present, there is little re-search on viral diseases of the black-necked crane. In this study, a virus metagenomics approach was employed to investigate the viral composition of black-necked cranes in Saga County, Shi-gatse City, Tibet, China. The identified virus families carried by black-necked cranes mainly in-clude Genomoviridae, Parvoviridae, and Picornaviridae. Among them, one picornavirus genome is characterized as a novel species in the genus Grusopivirus of the family Picornaviridae, four new parvoviruses genome were obtained and classified into four different novel species within the genus Chaphamaparvovirus of the subfamily Hamaparvovirinae, and four novel genomoviruses ge-nome were also acquired and identified as members of three different species including Ge-mykroznavirus haeme1, Gemycircularvirus ptero6, and Gemycircularvirus ptero10. All of these viruses were firstly detected in fecal samples of black-necked cranes. This study provides valuable in-formation for understanding the viral community composition in the digestive tract of black-necked cranes in Tibet and for monitoring, preventing, and treating black-necked cranes viral diseases.
Keywords:
viral metagenomics
; black-necked crane
; genomic structure
; phylogenetic analysis
1. Introduction
The black-necked crane (Grus nigricollis) is a vulnerable species and the only one that inhabits the plateau. It is classified as a vulnerable species (VU) by the International Union for Conservation of Nature (IUCN), with a global population ranging from 6,600 to 6,800 individuals [1]. The Tibet Plateau is one of the main habitats for black-necked cranes and has one of the largest populations of this species in the world. However, human activities, including agriculture, mining, and tourism, are increasingly threatening the black-necked crane population [2,3,4]. Additionally, viruses that infect birds and other crane species potentially endanger their lives. In recent years, emerging and re-emerging viruses such as avian influenza virus, Marek's disease virus, West Nile virus, and Crane hepatitis herpesviruses. have been found to infect cranes and pose health risks [5,6,7,8]. Some of these viruses may also have the potential to cross species barriers and infect humans. However, there have been limited virological studies conducted on black-necked cranes.
Viral metagenomics is a powerful tool for exploring both new and known viruses, and it has been widely utilized to understand viral compositions in diverse samples [9,10]. However, the viral composition in fecal samples from black-necked crane remains poorly understood. Therefore, the objective of this study is to investigate the viral composition of fecal samples collected from black-necked cranes in their natural habitat in Sa'gya County, Tibet Province, China, using a viral metagenomics approach. The findings of this study will provide valuable information for the prevention and treatment of viral diseases in this vulnerable species.
2. Materials and Methods
2.1. Sample Collection and Preparation
In December 2020, 10 fecal samples were collected from healthy black-necked cranes in Sakya County, Shigatse City, Tibet Province, China. About two gram of each fecal sample was re-suspended in 2 ml phosphate-buffered saline (PBS) and vigorously vortexed for 10 min, then centrifuged at 12, 000 rpm for 10 min. Finally every fecal supernatant was collected in a new 1.5 ml centrifuge tube and stored at -80℃ for further use.
2.2. Viral Nucleic Acid Extraction
A total of 500 μl fecal suspension was filtered through a 0.45-µm filter (Merck Millipore, MA, USA) to remove particles of bacterial and eukaryotic cell sizes. The filtrates were then treated with a mixture of nuclease enzymes to digest unprotected nucleic acids at 37℃ for 90 minutes. Viral RNA and DNA were extracted by using the QIAamp MinElute Virus Spin Kit (Qiagen, HQ, Germany) according to the improved manufacturer's protocol.
2.3. Library construction and bioinformatics analysis
The cDNA of viral RNA was synthesized by reverse transcription using six-base random primers. The complementary chain of cDNA was generated using Klenow Fragment DNA polymerase (M0210L, New England Biolabs, MA, USA). Next, libraries were constructed using the Nextera XT DNA Sample Preparation Kit (Illumina, CA, USA) and were sequenced using the NovaSeq Illumina platform with 250 bases paired ends with dual barcoding for each pool.
For bioinformatics analysis, the paired-end reads of 250 bp generated by NovaSeq were debarcoded using the vendor software from Illumina. We used an in-house analysis pipeline running on a 32-nodes Linux cluster to process the data. Low sequencing quality tails were trimmed using a Phred quality score threshold of 10. Adaptors were trimmed using the default parameters of VecScreen, which is an NCBI BLASTn program with specialized parameters designed for adaptor removal. Bacterial reads were subtracted by mapping to bacterial nucleotide sequences from the BLAST NT database using Bowtie2 v2.2.4. The cleaned reads were then de novo assembled by SOAPdenovo2 version r240 using a Kmer size of 63 with default settings [11]. The assembled contigs, along with singlets, were aligned to an in-house viral proteome database using BLASTx (v.2.2.7) with an E-value cutoff of <10-5. The candidate viral hits were compared to an in-house non-virus non-redundant (NVNR) protein database to remove false positive viral hits. The NVNR database was compiled using non-viral protein sequences extracted from the NCBI nr fasta file, based on annotation taxonomy excluding the Virus Kingdom.
2.4. Phylogenetic analysis
The analysis of evolutionary relationships was carried out using amino acid sequences predicted from the genomic data, with reference to the closest viral relatives determined by the best BLASTx hit, and representative members of related viral species or genera. Sequence alignment was conducted with Clustal W in MEGA version X with default settings [12]. Phylogenetic trees were constructed using MrBayes v3.2.7, with the parameters “lset nst = 6 rates = invgamma”. This setting applied the GTR substitution model with gamma-distributed rate variation across sites and a proportion of invariable sites ("GTR + I+Г"). Additionally, “prset aamodelpr = mixed” was employed to enable the program to use the ten built-in amino acid models. The maximum number of generations was set to be ten million, and sampling occurred at every 50 generations, with the first 25% of Markov chain Monte Carlo (mcmc) samples being discarded as burn-in. Convergence was confirmed when the standard deviation of split frequencies was below 0.01. Bootstrap values were assigned to each node.
2.5. Sequence alignment and ORF prediction
The pairwise comparison of viral amino acid sequences was conducted using the SDTv1.2 software with default settings. Putative open reading frames (ORFs) in the genome were predicted using Geneious 11.1.2 software and the NCBI ORF finder. For genomoviruses, putative exons and introns were predicted by Netgenes2 at https://services.healthtech.dtu.dk/services/NetGene2-2.42/.
2.6. Nucleotide Sequence Accession Number
The viral genome sequences were deposited in the GenBank with the accession numbers: OR532946 to OR532954. The raw sequence reads from the metagenomic library were deposited in the Shirt Read Archive of the GenBank database under accession number: SRR25662272
3. Results
3.1. Viral metagenomic overview
The library generated a total of 4,837,834 raw sequence reads on the Illumina NovaSeq platform. After conducting bioinformatics analysis, it was found that 139,584 of these sequence reads had the best matches with viral proteins. This accounted for 2.9% of the total number of raw data reads. Further analysis was conducted to determine the percentage of viral reads belonging to different virus families. Among them, sequence reads from the Picornaviridae family accounted for the largest proportion, representing 93.7% of the total analyzed virus reads. This was followed by the Parvoviridae family at 3.1% and the Genomoviridae family at 1.6%. The remaining virus families, including Herpesviridae, Circoviridae, Baculoviridae, Flaviviridae, Ascoviridae, and Reoviridae, accounted for a small proportion (Figure 1).
3.2. A novel picornavirus belonging to the genus Grusopivirus of the family Picornaviridae
In this study, 79,484 sequence reads belonging to the family Picornaviridae were found in the library, where one nearly complete genome of picornavirus was obtained using the assemble sequences program in Geneious 11.1.2 and tentatively named grusopivirus D. The genome of grusopivirus D is 8,205 nt in length, which includes a 789-nt 5' UTR, a 7,155-nt polyprotein ORF and a 261-nt 3' UTR. The GC content of grusopivirus D is 41.8%. Similar to the members of avihepatoviruses and parechoviruses, the polyprotein of grusopivirus D could be divided into VP0, VP3, VP1, 2A-2C, and 3A-3D by comparison with the polyprotein of Avihepatovirus A strain (NC_008250) and Parechovirus A strain (AB084913) (Figure. 2a). The P1 polypeptide is 761-aa in length and cleaved at VP0/VP3 (Q261/G262), VP3/VP1 (Q516/T517). It shared the highest amino acid identity of 40.81% with that of Grusopivirus A1 strain (NC_075281). The P2 polypeptide of 870-aa contains three non-structural proteins including 2A (cleavage site: Q1180/G1181), 2B (cleavage site: Q1296/G1297), and 2C. BLASTp showed that the P2 region had 96.21% amino acid sequence identity to that of Grusopivirus A1 strain (NC_075281). A conserved NPGP motif function as mediating cotranslational termination-reinitiation of RNA translation was present in the 2A protein, while the conserved NTPase motif “GAPGVGKS” was also found in its 2C protein (Figure 2a). The P3 polypeptide is 753-aa in length and cleaves into four non-structural proteins including 3A, 3B, 3Cpro (protease), and 3Dpol (RNA dependent RNA polymerase) at sites 3A/3B (Q1719/S1720), 3B/3C (Q1749/G1750), and 3C/3D (Q1930/G1931). The P3 of grusopivirus D had the highest amino acid sequence identity of 98.41% with that of Grusopivirus A1 strain (NC_075281). Multiple conserved proteinase and polymerase motifs including GXCGX10-15GXH, KDE, PSG, YGDD, and FLKR were found in 3C and 3D proteins separately (Figure. 2a).
Two phylogenetic trees were respectively constructed based on the P1 region and 3CD of grusopivirus D and other 36 representative strains belonging to the subfamily Paavivirinae of the family Picornaviridae (Figure 2b). The results showed that grusopivirus D clustered with those strains NC_075281, KY312542, NC_075445, and NC_075282 formed a separately clade. Among them, grusopivirus D had the closest genetic distance with the strain NC_075281, which was isolated from the fecal sample of red-crowned crane in 2014. The P1 region of grusopivirus D shares 40.81% amino acid sequence identity to the strain NC_075281, and the its polyprotein is 178-aa longer than that of the strain NC_075281. According to the International Committee on Taxonomy of Viruses (https://ictv.global/report/chapter/picornaviridae/picornaviridae), the members of a picornavirus species should share a significant degree of amino acid identity of P1, 2C, 3C, and 3D proteins. Our result indicate that grusopivirus D should defined as a novel species in the genus Grusopivirus.
3.3. Four new parvoviruses belonging to the genus Chaphamaparvovirus of the family Parvoviridae
Here, 2,618 sequence reads assigning to the Parvoviridae family was detected in the library, where four nearly complete genome of parvoviruses were acquired using the assemble sequences program in Geneious 11.1.2 and temporarily named Chaphamaparvovirus c2, c5, c7 and c11. The nearly genomes of Chaphamaparvovirus c2, c5, c7 and c11 are 4,246-nt, 4,392-nt, 4,158-nt, and 4,198-nt in length, both of which have three ORFs encoded two non-structural proteins (NS1 and NS2) and one structural protein (VP) (Figure 3a). The length of the NS1 protein is 654-aa for Chaphamaparvovirus c2, 657-aa for c5, 648-aa for c7, and 678-aa for c11, while the NS2 of these four parvoviruses is 139-aa, 195-aa, 218-aa, and 219-aa in length respectively. The conserved NTPase/helicase motifs (Walker A, B, B' and C) were found in all of these four NS1 protein, however, there are differences in some amino acid sites in this conserved domain comparing with that of the reference chaphamaparvoviruses (Figure 3b). The VP protein of these four viruses is different in length. Among them, the longest VP is in Chaphamaparvovirus c2 (526 aa), followed by Chaphamaparvovirus c11 (499 aa), Chaphamaparvovirus c7 (496 aa), and Chaphamaparvovirus c5 (490 aa). The conserved phospholipase A2 (PLA2) motif that is often present in members of the subfamily Parvovirinae was absent in the VP protein of these parvoviruses [13].
Phylogenetic tree was constructed using the NS1 proteins of the mentioned parvoviruses, as well as their closest viral relatives based on best BLASTp hits and other representative strains belonging to the genus Chaphamaparvovirus of the subfamily Hamaparvovirinae. The result showed that Chaphamaparvovirus c2, Chaphamaparvovirus c7 and Chaphamaparvovirus c11 clustered with the strain NC_075278, which was detected in the fecal sample of red-crowned crane, forming a separately clade. Additionally, Chaphamaparvovirus c5 clustered with the strain MT138318, which was found in the anal swab of wild bird, forming another separately clade (Figure 3c). To determine the species or genus of these parvoviruses, pairwise comparison of NS1 was conducted. The result of showed that Chaphamaparvovirus c2 shared >87% amino acid sequence identity with strain NC_075278, which is a unclassified chaphamaparvovirus, while Chaphamaparvovirus c5, Chaphamaparvovirus c7, and Chaphamaparvovirus c11 shared <60% but >35% amino acid sequence identity with other members of the genus Chaphamaparvovirus or with each other (Figure S1). Based on the novel demarcation criteria proposed by Judit J. Pénzes and co-workers, viruses can be considered members of the same species if the NS1 proteins share more than 85% amino acid sequence identity [14]. Therefore, the four parvoviruses identified here belong to four different novel species within the genus Chaphamaparvovirus of the subfamily Hamaparvovirinae.
3.4. Four novel genomoviruses belonging to the family Genomoviridae
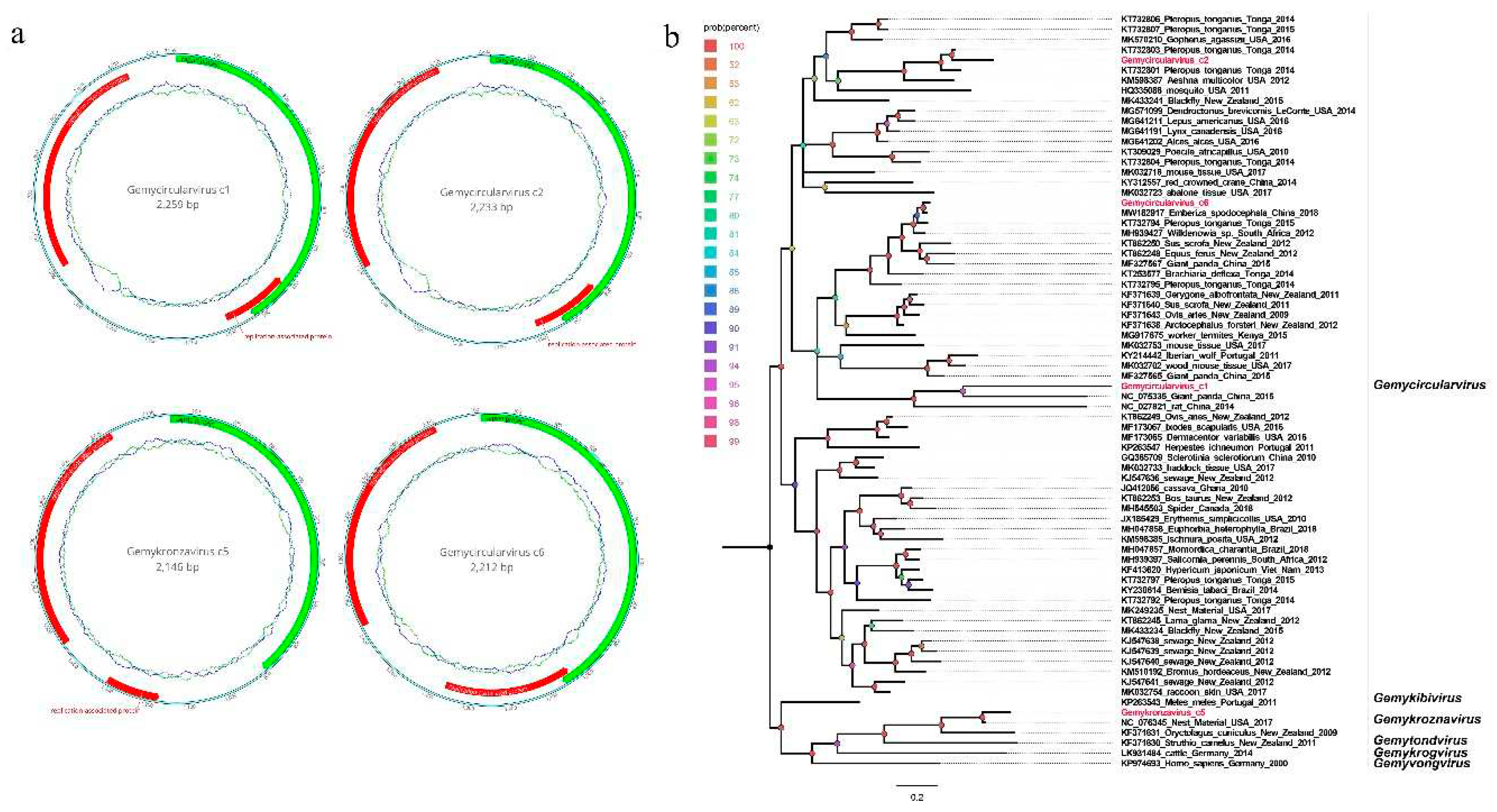
In the present study, 1,341 sequence reads belonging to the family Genomoviridae were found in the library, where four complete genome of genomoviruses was obtained using the assemble sequences program in Geneious 11.1.2 and named Gemycircularvirus c1, Gemycircularvirus c2, Gemykronzavirus c5, and Gemycircularvirus c6. The genomes of them are 2,259-nt, 2,233-nt, 2,146-nt, and 2,212-nt in length, both of which have two bidirectional ORFs encoded a capsid protein (CP) and a replicase-associated protein (Rep). An intron lies within the Rep gene, which is similar to those in some genomovirus (Figure 4a). The length of the CP protein is 309-aa for both Gemycircularvirus c1 and c2, 314-aa for Gemycircularvirus c6, and 285-aa for Gemykronzavirus c5 in length. The Rep protein of these four genomoviruses is 269-aa for Gemycircularvirus c1, 261-aa for Gemycircularvirus c2, 239-aa for Gemykronzavirus c5, and 304-aa for Gemycircularvirus c6. The conserved rolling circle replication (PCR) motifs I, II, and III were present in the Rep proteins of Gemycircularvirus c2 (FTYSQ, HFHVFTD, and YAIKD), Gemykronzavirus c5 (LTYSQ, HYHVVAQ, and YCLKD), and Gemycircularvirus c6 (LTYSQ, HLHVFCD, and YATD), while in the Rep protein of Gemycircularvirus c1, only PCR motif I (FTYSQ) was found, not motif II and III (data not shown).
Phylogenetic tree was constructed based on Rep proteins of these, as well as their closest viral relatives based on best BLASTp hits and other representative strains belonging to several different genera of the family Genomoviridae. The results showed that these four genomoviruses were located on four separate branches, among them, Gemycircularvirus c2 clustered with Pteropus associated gemycircularvirus (GenBank no. KT732801), which was detected from fecal sample of Pteropus tonganus in Tonga, Gemycircularvirus c6 clustered with Emberiza spodocephala gemycircularvirus (GenBank no. MW182917), which was isolated from anal swab of Emberiza spodocephala in China, Gemycircularvirus c1 clustered with Giant panda associated gemycircularvirus (GenBank no. NC_075335), which was detected from fecal sample of Giant panda in China, while Gemykronzavirus c5 clustered with Finch associated genomovirus (GenBank no. NC_076345), which was found from Nest Material of Finch in USA (Figure 4b). To determine the species or genus of these genomoviruses, pairwise comparison of genome-wide was conducted. The result showed that Gemykronzavirus c5 shared >78% genome-wide identity with the representative strain NC_076345, which belongs to the species of Gemykroznavirus haeme1, Gemycircularvirus c1 and c2 shared >78% genome-wide identity with the representative strain KT732803, belonging to the species of Gemycircularvirus ptero6, while Gemycircularvirus c6 shared >78% genome-wide identity with the representative strain KT732794, belonging to the species of Gemycircularvirus ptero10 (Figure S2). According to the novel demarcation criteria proposed by Arvind Varsani and co-workers, a genome-wide pairwise identity of 78% was chosen as a species demarcation threshold for genomoviruses, whereas Rep sequence phylogeny was used to define genera [15]. Based on these criteria, the four genomoviruses found here are classified into three species belonging to two different genera: Gemycircularvirus and Gemykroznavirus. Specifically, the three species are Gemykroznavirus haeme1, Gemycircularvirus ptero6, and Gemycircularvirus ptero10.
4. Discussion
The black-necked crane, as a vulnerable species, is highly susceptible to viral diseases. Previous studies have identified various viruses that can infect cranes and cause diseases. For instance, Lee and co-workers isolated a low pathogenic H7N7 avian influenza virus from a red-crowned crane in a zoo in South Korea [5]. Ozawa and co-workers reported the widespread prevalence of crane-associated adenovirus 1 in cranes overwintering on the Izumi plain, Japan [16]. Taniguchi and co-workers demonstrated that crane herpesvirus can induce hemagglutination [17]. In our previous study, viral metagenomics revealed the presence of multiple viruses in the fecal samples of red-crowned cranes [18]. However, limited research has been conducted on viral diseases in black-necked cranes. Hence, in this study, we employed high-throughput sequencing to investigate the virome of black-necked crane fecal samples. Our results showed that multiple viruses were firstly detected in black-necked crane and classified into novel virus species.
Members of the Picornaviridae family are small, single-stranded RNA viruses with genome lengths ranging from approximately 7.2 to 9.4 kb. The Picornaviridae family currently consists of 158 species grouped into 68 genera [19]. Different genera of Picornaviridae infect various animals and humans, leading to a variety of diseases [20,21,22,23,24]. For instance, Enterovirus is the most common genus of Picornaviridae virus and can cause diseases such as hand, foot, and mouth disease, poliomyelitis, and myocarditis [25,26,27]. Hepatovirus includes human hepatitis A virus, which can cause acute hepatitis [28]. Recently, viruses in the genus Senecavirus have garnered attention from both veterinary and public health communities because they have been found to cause swine hand, foot, and mouth disease [29]. Grusopivirus is a novel genus of the Picornaviridae family, which was first discovered in the fecal sample of a red-crowned crane in 2014 by our lab [18]. In this study, a novel grusopivirus was detected for the first time from fecal samples of black-necked cranes. This virus has a similar genomic structure and potential cleavage sites to the members of avihepatoviruses, which can cause poultry diseases. It suggests that the novel grusopivirus has the potential to be pathogenic to black-necked cranes. By amino acid sequence alignment, we found that P1 of grusopivirus D only had amino acid sequence identity of 40.81% with that of Grusopivirus A1 strain (NC_075281), while P2 and P3 of grusopivirus D shared over 96% amino acid sequence identity with that of Grusopivirus A1. Considering that the capsid protein, encoded by P1 of picoranvirus, plays an important role as a ligand in virus infection within the body, we speculate that these differences in the P1 polypeptide are related to differences in host receptors. Currently, all discovered grusopiviruses have been isolated from members of the Gruiforms family. This prompts us to think that grusopiviruses can only infect cranes. However, the epidemiological and pathological characteristics of Grusopiviruses are not well understood. Therefore, further experimental and epidemiological studies are needed to understand its pathogenesis and transmission mechanisms.
Parvovirus is non-enveloped, icosahedral, single-stranded DNA virus with a genome approximately 4 kb and 6 kb in length [30]. Historically, the Parvoviridae family has two subfamilies including Densoviridae and Parvovirinae, which infect vertebrates and invertebrates, respectively. Recently, a new subfamily called Hamaparvovirinae has been identified, which includes the genera Brevihamaparvovirus, Chaphamaparvovirus, Hepanhamaparvovirus, Ichthamaparvovirus, and Penstylhamaparvovirus [31]. Members of the genus Chaphamaparvovirus can infect various animals, including dogs, wolves, chickens, pheasants, Larus delawarensis (a species of gull), bats, Sarcophilus harrisii (Tasmanian devil), Pavo cristatus (peacock), Cebusimitator (white-headed capuchin), parrots, and rodents [32,33,34,35,36,37]. Some of these infections can cause diseases in their respective hosts. For example, a study by Michael et al. reported that chaphamaparvovirus is the cause of hepatitis outbreaks in pheasants (Phasianus colchicus), which are characterized by high mortality [34]. Subir Sarker also found that galliform chaphamaparvovirus is associated with spotty liver disease in chickens [38]. Additionally, dogs, especially puppies infected with carnivore chaphamaparvovirus, exhibit clinical signs such as diarrhea, fever, and cough [39]. In this study, four novel chaphamaparvoviruses were characterized for the first time. They were classified into four different novel species within the genus Chaphamaparvovirus of the subfamily Hamaparvovirinae. Surprisingly, phylogenetic analysis and pairwise alignment indicated that these four chaphamaparvoviruses have a higher genetic relationship with representative strains that were detected from fecal samples of red-crowned cranes. We speculate that chaphamaparvovirus is widely distributed among members of the Gruiforms family. Further epidemiological investigation is needed to determine whether these viruses can cause black-necked crane disease and whether interspecies transmission is possible.
Members of the family Genomoviridae are small, icosahedral, non-enveloped single-stranded circular DNA viruses. Their genomes are approximately 1.8-2.4 kb in length and encode a rolling-circle replication initiation protein (Rep) and a capsid protein (CP) in an ambisense orientation [40]. The family Genomoviridae is currently classified into nine genera: Gemycircularvirus, Gemyduguivirus, Gemygorvirus, Gemykibivirus, Gemykolovirus, Gemykrogvirus, Gemykroznavirus, Gemytondvirus, and Gemyvongvirus [15]. The first discovered genomovirus was Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1 (SsHADV-1), which infects phytopathogenic fungus Sclerotinia sclerotiorum [41]. Recently, multiple genomoviruses have been detected in diverse samples taken from various organisms, including Actinopterygii, Arachnida, Aves, Embryophyte, Gastropoda, Insecta, Leotiomycetes, Mammalia, Reptilia, and the environment [15,42,43,44,45,46]. In the present study, we detected four novel genomoviruses for the first time in fecal samples from black-necked cranes. From a genome evolution perspective, these four genomoviruses are genetically related to strains isolated from nest material of finches in the USA, or from feces of Pteropus tonganus in Tonga. This indicates that the same species of genomovirus can infect different bird species, even if they live in different locations. However, since our samples were collected from healthy individuals, we cannot be certain if these novel genomoviruses will cause disease in black-necked cranes. It is also possible that these novel genomoviruses originated from foodborne insects. Therefore, further epidemiological investigations, including large-scale collection of blood samples, will be beneficial to clarify whether these genomoviruses are true pathogens of black-necked cranes. For genomovirus, the three conserved PCR motifs (I, II, and III) in Rep protein is crucial important for its replication. To our surprise, only PCR motif I was found, not motif II and III in the Rep protein of Gemycircularvirus c1. However, all other genomoviruses have three conserved PCR motifs. It is unknown whether the absence of two PCR motifs will affect the proliferation of Gemycircularvirus c1. Further experiments on virus proliferation in infected cells will help answer this question. Additionally, phylogenetic analysis based on the Rep proteins revealed that Gemycircularvirus c1 and Gemycircularvirus c2 are located in different branches. Therefore, we believe that phylogenetic trees constructed based on the Rep proteins can be effectively utilized for clustering virus genera, but may not be suitable for determining virus species.
Although multiple new viruses were identified in black-necked crane fecal samples using viral metagenomic method in this study, we are unable to determine whether these new viruses can cause disease in black-necked cranes. This limitation stems from the small number of samples collected and the exclusive focus on fecal samples. Moreover, we are uncertain about the exact sources of these viruses and their potential for cross-species transmission. Consequently, conducting a comprehensive epidemiological investigation in the region is crucial, necessitating the collection of various sample types from numerous animals on a larger scale
5. Conclusions
In summary, this study presents an overview of the viral community found in the feces of the black-necked crane and significantly enhances our understanding of the viral composition in their stool samples. The prevalence of viruses related to the black-necked crane, as described in this study, offers valuable information for the prevention and treatment of viral diseases in birds in the local region.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Pairwise comparison of NS1 amino acid sequences of four chaphamaparvoviruses identified in this study with the representative strains of different species of the genus Chaphamaparvovirus. The pairwise identity (%) was marked with different colors. Figure S2: pairwise comparison of genome-wide nucleotide sequences of four genomoviruses identified in this study with the representative strains of different genera of the family Genomoviridae. The pairwise identity (%) was marked with different colors.
Author Contributions
S.Y., Y.W. and W.Z. conceived and designed the study. Q.Z., Y.S., L.J., R.Z., Y.X. and Y.W. carried out the study. X.W., L.J. and Q.S. analyzed the data. S.Y. and Q.Z. contributed to writing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Key Research and Development Programs of China (No. 2022YFC2603801), National Natural Science Foundation of China (No. 32102682), and Science Foundation of Higher Education of Jiangsu Province (No. 21KJB230006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The viral genome sequences were deposited in the GenBank with the accession numbers: OR532946 to OR532954. The raw sequence reads from the metagenomic library were deposited in the Shirt Read Archive of the GenBank database under accession number: SRR25662272.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- International), B.I. (BirdLife IUCN Red List of Threatened Species: Grus Nigricollis. IUCN Red List Threat. Species 2020.
- Yang, Y.; Xie, C.; Shen, C.; Tian, B.; Wang, S.; Bian, X.; Guo, Y.; Zhu, Y.; Fang, H. Changes in the Landscape Patterns of Black-Necked Crane Habitat and Its Correlation with Their Individual Population Numbers during the Past 40 Years in China. Ecol. Evol. 2023, 13, e10125. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yu, H.; Geng, Y.; Liu, W.; Zheng, S.; Yang, N.; Meng, Y.; Dou, L.; Price, M.; Ran, J.; et al. A High-Quality Draft Genome Assembly of the Black-Necked Crane (Grus Nigricollis) Based on Nanopore Sequencing. Genome Biol. Evol. 2019, 11, 3332–3340. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, Y.; DuBay, S.G.; Zhao, C.; Wang, B.; Ran, J. Dung-Associated Arthropods Influence Foraging Ecology and Habitat Selection in Black-Necked Cranes (Grus Nigricollis) on the Qinghai-Tibet Plateau. Ecol. Evol. 2019, 9, 2096–2105. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.-J.; Lee, Y.-N.; Cheon, S.-H.; Park, Y.-R.; Baek, Y.-G.; Kye, S.-J.; Lee, M.-H.; Lee, Y.-J. Isolation and Characterization of Low Pathogenic H7N7 Avian Influenza Virus from a Red-Crowned Crane in a Zoo in South Korea. BMC Vet. Res. 2020, 16, 432. [Google Scholar] [CrossRef]
- Olsen, G.H.; Miller, K.J.; Docherty, D.E.; Bochsler, V.S.; Sileo, L. Pathogenicity of West Nile Virus and Response to Vaccination in Sandhill Cranes (Grus Canadensis) Using a Killed Vaccine. J. Zoo Wildl. Med. 2009, 40, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Ming, X.; Xu, J.; Cheng, W.; Zhang, X.; Chen, H.; Ding, C.; Jung, Y.-S.; Qian, Y. First Molecular Detection and Characterization of Marek’s Disease Virus in Red-Crowned Cranes (Grus Japonensis): A Case Report. BMC Vet. Res. 2018, 14, 122. [Google Scholar] [CrossRef] [PubMed]
- Foerster, S.; Chastel, C.; Kaleta, E.F. Crane Hepatitis Herpesviruses. Zentralbl. Veterinarmed. B 1989, 36, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Yang, S.; Wang, H.; Wang, H.; Zhang, J.; Gong, G.; Xiao, Y.; Yang, J.; Wang, X.; Lu, J.; et al. Virome in the Cloaca of Wild and Breeding Birds Revealed a Diversity of Significant Viruses. Microbiome 2022, 10, 60. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, D.; Ji, Z.; Zhang, Y.; Wang, Y.; Chen, X.; He, Y.; Lu, X.; Li, R.; Guo, Y.; et al. Viral Metagenomics Reveals Diverse Viruses in Tissue Samples of Diseased Pigs. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. Gigascience 2012. [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.-T.; Giménez-Lirola, L.G.; Jiang, Y.-H.; Halbur, P.G.; Opriessnig, T. Characterization of a Novel Porcine Parvovirus Tentatively Designated PPV5. PLoS ONE 2013, 8, e65312. [Google Scholar] [CrossRef]
- Pénzes, J.J.; Söderlund-Venermo, M.; Canuti, M.; Eis-Hübinger, A.M.; Hughes, J.; Cotmore, S.F.; Harrach, B. Reorganizing the Family Parvoviridae: A Revised Taxonomy Independent of the Canonical Approach Based on Host Association. Arch. Virol. 2020, 165, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Krupovic, M. Family Genomoviridae: 2021 Taxonomy Update. Arch. Virol. 2021, 166, 2911–2926. [Google Scholar] [CrossRef]
- Esaki, M.; Ito, G.; Tokorozaki, K.; Matsui, T.; Masatani, T.; Amano, K.; Ozawa, M. Prevalence and Organ Tropism of Crane-Associated Adenovirus 1 in Cranes Overwintering on the Izumi Plain, Japan. Transbound. Emerg. Dis. 2022, 69, e2800–e2807. [Google Scholar] [CrossRef] [PubMed]
- Uwatoko, K.; Inaba, Y.; Sasaki, T.; Sano, Y.; Pan, I.J.; Suzuki, H.; Yamaguchi, S.; Taniguchi, T. Hemagglutination with Crane Herpesvirus. J. Vet. Med. Sci. 1998, 60, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, S.; Liu, D.; Zhou, C.; Li, W.; Lin, Y.; Wang, X.; Shen, Q.; Wang, H.; Li, C.; et al. The Fecal Virome of Red-Crowned Cranes. Arch. Virol. 2019, 164, 3–16. [Google Scholar] [CrossRef]
- Zell, R.; Knowles, N.J.; Simmonds, P. A Proposed Division of the Family Picornaviridae into Subfamilies Based on Phylogenetic Relationships and Functional Genomic Organization. Arch. Virol. 2021, 166, 2927–2935. [Google Scholar] [CrossRef]
- Bian, L.; Wang, Y.; Yao, X.; Mao, Q.; Xu, M.; Liang, Z. Coxsackievirus A6: A New Emerging Pathogen Causing Hand, Foot and Mouth Disease Outbreaks Worldwide. Expert Rev. Anti. Infect. Ther. 2015, 13, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Huaman, J.L.; Pacioni, C.; Sarker, S.; Doyle, M.; Forsyth, D.M.; Pople, A.; Carvalho, T.G.; Helbig, K.J. Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Yinda, C.K.; Zell, R.; Deboutte, W.; Zeller, M.; Conceição-Neto, N.; Heylen, E.; Maes, P.; Knowles, N.J.; Ghogomu, S.M.; Van Ranst, M.; et al. Highly Diverse Population of Picornaviridae and Other Members of the Picornavirales, in Cameroonian Fruit Bats. BMC Genomics 2017, 18, 249. [Google Scholar] [CrossRef] [PubMed]
- Philipps, A.; Dauber, M.; Groth, M.; Schirrmeier, H.; Platzer, M.; Krumbholz, A.; Wutzler, P.; Zell, R. Isolation and Molecular Characterization of a Second Serotype of the Encephalomyocarditis Virus. Vet. Microbiol. 2012, 161, 49–57. [Google Scholar] [CrossRef]
- Pankovics, P.; Boros, Á.; Kiss, T.; Reuter, G. Identification and Complete Genome Analysis of Kobuvirus in Faecal Samples of European Roller (Coracias Garrulus): For the First Time in a Bird. Arch. Virol. 2015, 160, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Fort, M.K.; Downing, C.; Doan, H.Q.; Benoist, F.; Oberste, M.S.; Khan, F.; Tyring, S.K. Coxsackievirus A6 Associated Hand, Foot and Mouth Disease in Adults: Clinical Presentation and Review of the Literature. J. Clin. Virol. 2014, 60, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Uprety, P.; Graf, E.H. Enterovirus Infection and Acute Flaccid Myelitis. Curr. Opin. Virol. 2020, 40, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lu, H.; Ma, L.; Zhu, S.; Yan, D.; Han, J.; Zhang, Y. Molecular Characteristics of Enterovirus B83 Strain Isolated from a Patient with Acute Viral Myocarditis and Global Transmission Dynamics. Viruses 2023, 15. [Google Scholar] [CrossRef]
- Bauer, D.; Farthofer, A.; Chromy, D.; Simbrunner, B.; Steininger, L.; Schmidbauer, C.; Binter, T.; Trauner, M.; Mandorfer, M.; Schmidt, R.; et al. Recent Outbreaks of Severe Hepatitis A Virus Infections in Vienna. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 335–344. [Google Scholar] [CrossRef]
- Montiel, N.; Buckley, A.; Guo, B.; Kulshreshtha, V.; VanGeelen, A.; Hoang, H.; Rademacher, C.; Yoon, K.-J.; Lager, K. Vesicular Disease in 9-Week-Old Pigs Experimentally Infected with Senecavirus A. Emerg. Infect. Dis. 2016, 22, 1246–1248. [Google Scholar] [CrossRef] [PubMed]
- Mietzsch, M.; Pénzes, J.J.; Agbandje-McKenna, M. Twenty-Five Years of Structural Parvovirology. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Pénzes, J.J.; de Souza, W.M.; Agbandje-McKenna, M.; Gifford, R.J. An Ancient Lineage of Highly Divergent Parvoviruses Infects Both Vertebrate and Invertebrate Hosts. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Palombieri, A.; Di Profio, F.; Lanave, G.; Capozza, P.; Marsilio, F.; Martella, V.; Di Martino, B. Molecular Detection and Characterization of Carnivore Chaphamaparvovirus 1 in Dogs. Vet. Microbiol. 2020, 251, 108878. [Google Scholar] [CrossRef]
- Cui, H.; Pan, S.; Xu, X.; Ji, J.; Ma, K.; Yao, L.; Kan, Y.; Bi, Y.; Xie, Q. Molecular Characteristics of Novel Chaphamaparvovirus Identified in Chickens. Poult. Sci. 2023, 102, 102449. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Bilic, I.; Viloux, N.; Palmieri, N.; Albaric, O.; Chatenet, X.; Tvarogová, J.; Dinhopl, N.; Heidl, S.; Liebhart, D.; et al. A Novel Chaphamaparvovirus Is the Etiological Agent of Hepatitis Outbreaks in Pheasants (Phasianus Colchicus) Characterized by High Mortality. Transbound. Emerg. Dis. 2022, 69, e2093–e2104. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.d.S.F.; Abreu, W.U.; Rodrigues, L.R.R.; Marinho, L.F.; Morais, V.D.S.; Villanova, F.; Pandey, R.P.; Araújo, E.L.L.; Deng, X.; Delwart, E.; et al. Novel Chaphamaparvovirus in Insectivorous Molossus Molossus Bats, from the Brazilian Amazon Region. Viruses 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S. Molecular and Phylogenetic Characterisation of a Highly Divergent Novel Parvovirus (Psittaciform Chaphamaparvovirus 2) in Australian Neophema Parrots. Pathog. (Basel, Switzerland) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Roediger, B.; Lee, Q.; Tikoo, S.; Cobbin, J.C.A.; Henderson, J.M.; Jormakka, M.; O’Rourke, M.B.; Padula, M.P.; Pinello, N.; Henry, M.; et al. An Atypical Parvovirus Drives Chronic Tubulointerstitial Nephropathy and Kidney Fibrosis. Cell 2018, 175, 530–543. [Google Scholar] [CrossRef]
- Sarker, S. Characterization of a Novel Complete-Genome Sequence of a Galliform Chaphamaparvovirus from a Free-Range Laying Chicken Clinically Diagnosed with Spotty Liver Disease. Microbiol. Resour. Announc. 2022, 11, e0101722. [Google Scholar] [CrossRef]
- Fahsbender, E.; Altan, E.; Seguin, M.A.; Young, P.; Estrada, M.; Leutenegger, C.; Delwart, E. Chapparvovirus DNA Found in 4% of Dogs with Diarrhea. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Krupovic, M.; Ghabrial, S.A.; Jiang, D.; Varsani, A. Genomoviridae: A New Family of Widespread Single-Stranded DNA Viruses. Arch. Virol. 2016, 161, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Tang, Y.; Zhang, S.; She, X.; Lan, G.; Varsani, A.; He, Z. Identification and Molecular Characterization of a Single-Stranded Circular DNA Virus with Similarities to Sclerotinia Sclerotiorum Hypovirulence-Associated DNA Virus 1. Arch. Virol. 2014, 159, 1527–1531. [Google Scholar] [CrossRef]
- Li, W.; Gu, Y.; Shen, Q.; Yang, S.; Wang, X.; Wan, Y.; Zhang, W. A Novel Gemycircularvirus from Experimental Rats. Virus Genes 2015, 51, 302–305. [Google Scholar] [CrossRef]
- da Silva Assis, M.R.; Vieira, C.B.; Fioretti, J.M.; Rocha, M.S.; de Almeida, P.I.N.; Miagostovich, M.P.; Fumian, T.M. Detection and Molecular Characterization of Gemycircularvirus from Environmental Samples in Brazil. Food Environ. Virol. 2016, 8, 305–309. [Google Scholar] [CrossRef]
- Schmidlin, K.; Sepp, T.; Khalifeh, A.; Smith, K.; Fontenele, R.S.; McGraw, K.J.; Varsani, A. Diverse Genomoviruses Representing Eight New and One Known Species Identified in Feces and Nests of House Finches (Haemorhous Mexicanus). Arch. Virol. 2019, 164, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Hanna, Z.R.; Runckel, C.; Fuchs, J.; DeRisi, J.L.; Mindell, D.P.; Van Hemert, C.; Handel, C.M.; Dumbacher, J.P. Isolation of a Complete Circular Virus Genome Sequence from an Alaskan Black-Capped Chickadee (Poecile Atricapillus) Gastrointestinal Tract Sample. Genome Announc. 2015, 3. [Google Scholar] [CrossRef]
- Chiumenti, M.; Greco, C.; Antelmi, I.; Sion, V.; Altamura, G.; Nigro, F.; Saldarelli, P. Molecular Characterisation of a Novel Gemycircularvirus Associated with Olive Trees in Italy. Virus Res. 2019, 263, 169–172. [Google Scholar] [CrossRef]
Figure 1.
The composition of fecal virome detected in black-necked crane. The percentage of virus sequences in different virus family were shown in pie chart.
Figure 1.
The composition of fecal virome detected in black-necked crane. The percentage of virus sequences in different virus family were shown in pie chart.
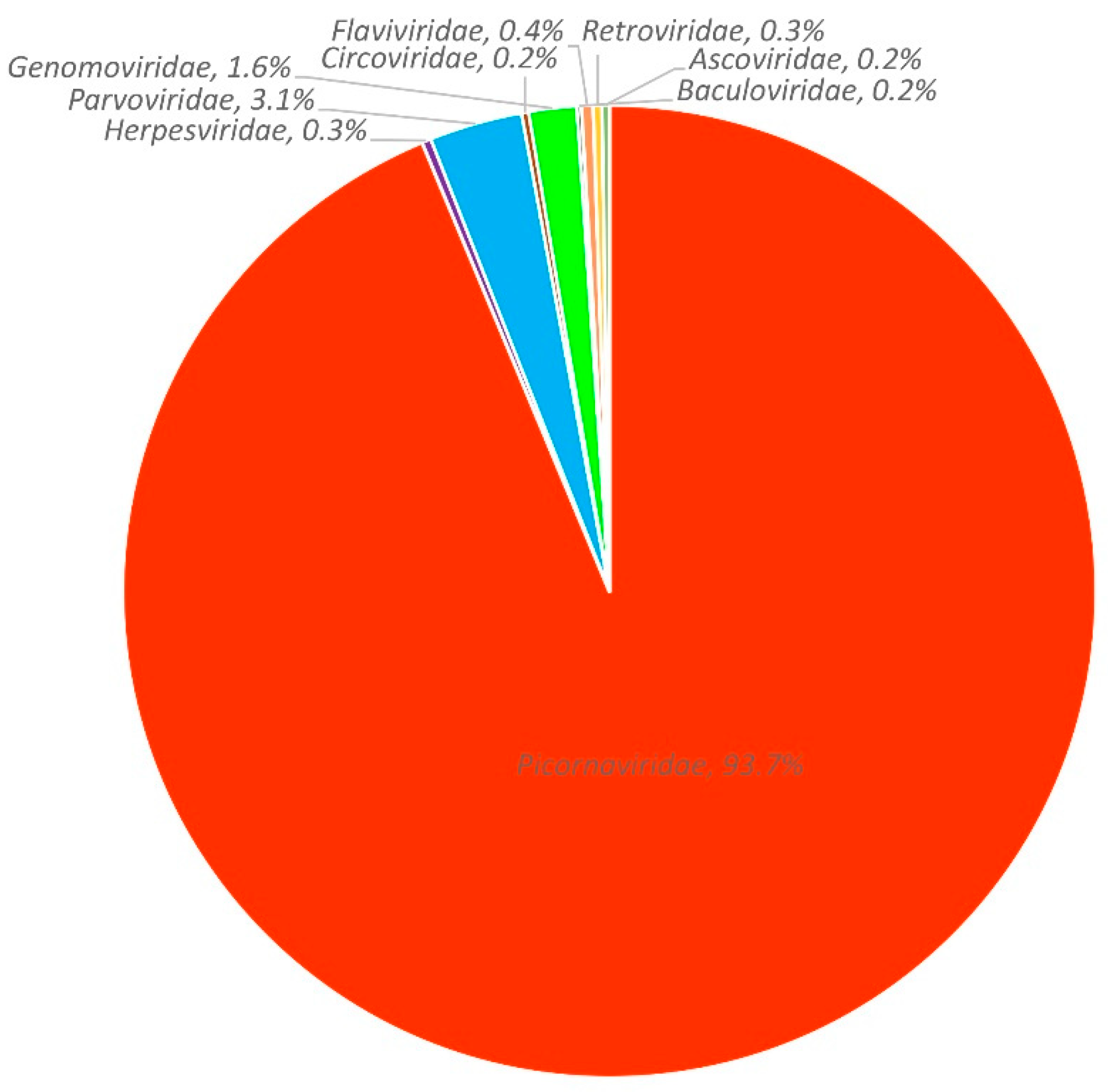
Figure 2.
The genomic organization, conserved motifs, and phylogenetic analysis of the grusopivirus identified in black-necked crane. (a) The genomic organization of one grusopivirus strain. The ORF and viral encoding proteins of grusopivirus were marked with different colors. The conserved motifs were also shown. (b), (c) The phylogenetic analysis based on the P1 region and 3CD of grusopivirus, which identified in this study, and other reference strains belonging to the subfamily Paavivirinae of the family Picornaviridae. Grusopivirus D identified in this study was marked with red solid circle.
Figure 2.
The genomic organization, conserved motifs, and phylogenetic analysis of the grusopivirus identified in black-necked crane. (a) The genomic organization of one grusopivirus strain. The ORF and viral encoding proteins of grusopivirus were marked with different colors. The conserved motifs were also shown. (b), (c) The phylogenetic analysis based on the P1 region and 3CD of grusopivirus, which identified in this study, and other reference strains belonging to the subfamily Paavivirinae of the family Picornaviridae. Grusopivirus D identified in this study was marked with red solid circle.
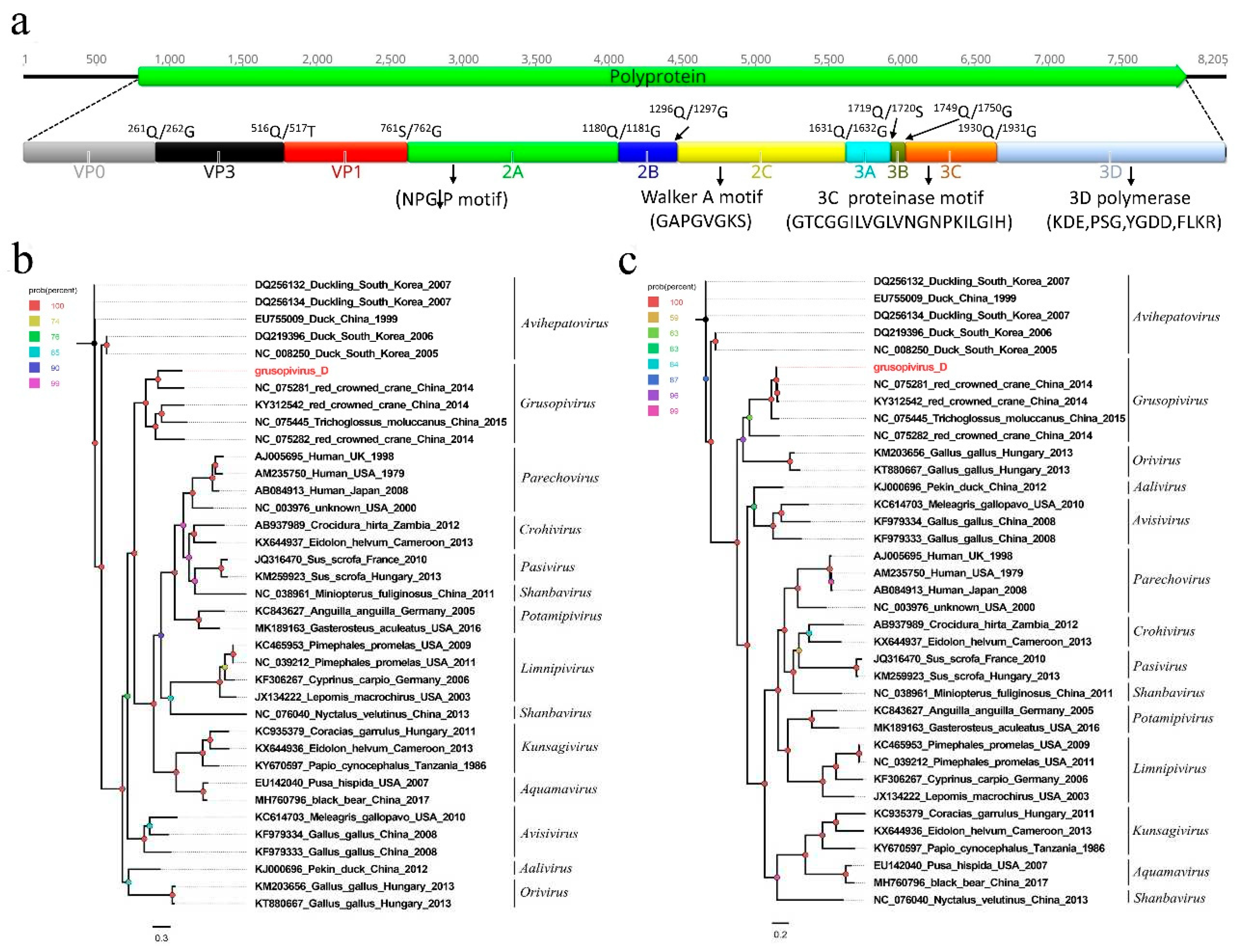
Figure 3.
The genomic organization, conserved helicase domain motifs, and phylogenetic analysis of four chaphamaparvoviruses identified in black-necked crane. (a) The genomic organization of four chaphamaparvoviruses identified in black-necked crane. Viral encoding proteins of four chaphamaparvoviruses were marked with different colors. (b) The conserved NTPase/helicase motif (Walker A, B, B' and C) of the four parvoviruses and other reference strains were shown. The conserved motifs were marked with orange frame. (c) The phylogenetic analysis based on NS1 proteins of the mentioned parvoviruses, as well as their closest viral relatives based on best BLASTp hits and other representative strains belonging to the genus Chaphamaparvovirus of the subfamily Hamaparvovirinae. Four chaphamaparvoviruses identified in this study were marked with red solid circle.
Figure 3.
The genomic organization, conserved helicase domain motifs, and phylogenetic analysis of four chaphamaparvoviruses identified in black-necked crane. (a) The genomic organization of four chaphamaparvoviruses identified in black-necked crane. Viral encoding proteins of four chaphamaparvoviruses were marked with different colors. (b) The conserved NTPase/helicase motif (Walker A, B, B' and C) of the four parvoviruses and other reference strains were shown. The conserved motifs were marked with orange frame. (c) The phylogenetic analysis based on NS1 proteins of the mentioned parvoviruses, as well as their closest viral relatives based on best BLASTp hits and other representative strains belonging to the genus Chaphamaparvovirus of the subfamily Hamaparvovirinae. Four chaphamaparvoviruses identified in this study were marked with red solid circle.
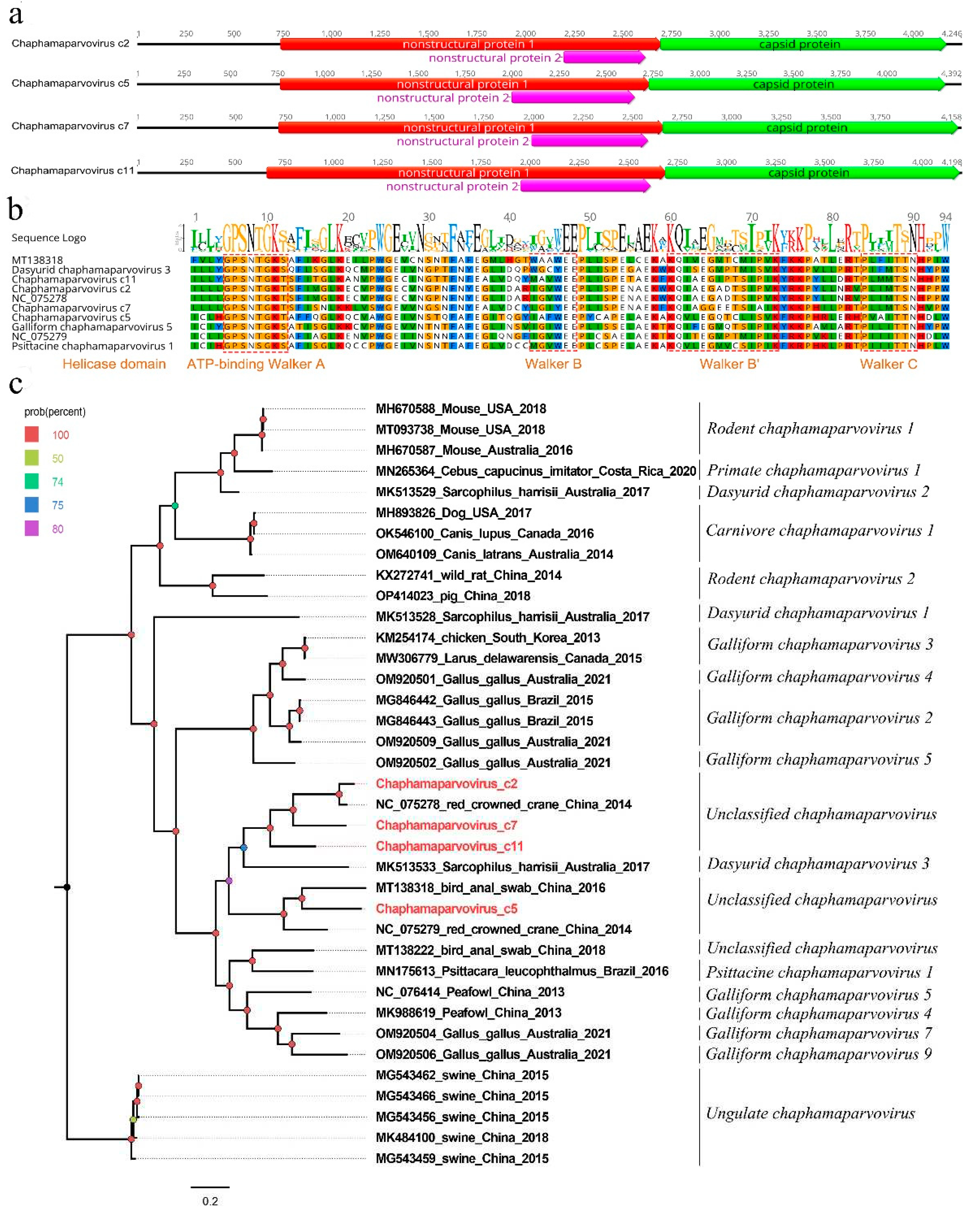
Figure 4.
The genomic organization and phylogenetic analysis of genomoviruses identified in black-necked crane. (a) The genomic organization of four genomoviruses identified in black-necked crane. Viral encoding proteins of four genomoviruses were marked with different colors. (b) The phylogenetic analysis based on the Rep proteins of four genomoviruses which identified in this study, and reference strains of other genomoviruses. Four genomoviruses identified in this study were marked with red solid circle.
Figure 4.
The genomic organization and phylogenetic analysis of genomoviruses identified in black-necked crane. (a) The genomic organization of four genomoviruses identified in black-necked crane. Viral encoding proteins of four genomoviruses were marked with different colors. (b) The phylogenetic analysis based on the Rep proteins of four genomoviruses which identified in this study, and reference strains of other genomoviruses. Four genomoviruses identified in this study were marked with red solid circle.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



