
Submitted:
15 September 2023
Posted:
18 September 2023
You are already at the latest version
Abstract
Over the past two decades, a growing body of evidence observations have shown group two innate lymphoid cells (ILC2) to be critical drivers of Type 2 (T2) inflammatory responses associated with allergic inflammatory conditions such as asthma. ILC2 release copious amounts of pro-inflammatory T2 cytokines – interleukin (IL)-4, IL-5, IL-9, and IL-13. This review provides a comprehensive overview of the newly discovered regulatory subtype of ILC2 described in murine and human mucosal tissue and blood. These KLRG1+ILC2 have the capacity to produce the anti-inflammatory cytokine, IL-10. Papers compiled in this review were based on query of PubMed and Google Scholar for articles published from 2000-2023 using keywords “IL-10” and “ILC2”. Studies with topical relevance to IL-10 production by ILC2 were included. ILC2 respond to microenvironmental cues including retinoic acid (RA), IL-2, IL-4, IL-10, IL-33, as well as neuropeptide mediators such as neuromedin-U (NMU), prompting a shift towards IL-10 and away from T2 cytokine production. In contrast, TGF-β attenuates IL-10 production by ILC2. Immune regulation provided by IL-10+ILC2s holds potential significance for management of T2 inflammatory conditions. The observation of context-specific cues which alter phenotype of ILC warrants examining characteristics of ILC subsets to determine the extent of plasticity or whether the current classification of ILCs requires refinement.
Keywords:
Allergic Inflammatory Disease
; Asthma
; Type 2 Inflammation
; Interleukin-10
1. Introduction
Innate lymphoid cells (ILC) are a family of lineage-negative cells that are comprised of natural killer (NK) cells, lymphoid tissue inducers (LTi) and three subgroups: ILC1, ILC2, ILC3 (Figure 1) [1,2,3,4]. This nomenclature was proposed in 2013 [5]. Since their discovery, ILC have been recognized as primarily tissue resident cells [6], particularly at the mucosal interface tissue of the airways, gastrointestinal tract, and skin [1,2], but ILC are also found in blood [6]. The estimates regarding the frequency of these cells put them at around 0.09% (0 -1.5%), of total CD45+ cells in healthy individuals [7,8]. The earliest progenitor cells that give rise to the ILC family is the common lymphoid progenitor (CLP), which expresses inhibitor of DNA binding 2 (Id2) [9]. Id2 is a fate-determining transcription factor that plays an indispensable role in the development of ILC as an inhibitor of E-protein transcription factors which are important in adaptive immune cells [3,10]. CLP develop into early or common innate lymphoid progenitors (EILP/CILPs) which express transcription factor nuclear factor interleukin 3 (NFIL3) [11], thymocyte selection-associated HMG box (Tox), and maintain Id2 [12,13,14]. Natural killer progenitors (NKP) continue to express Id2, NFIL3 [11], and Tox, in addition to a transcription factor from the ETS family, ETS1, and give rise to mature NK cells [13,15,16]. Next, common helper innate lymphoid cell progenitor (CHILP) fails to generate NK cells but have been shown to give rise to all other ILCs, including LTi cells (Figure 1) [10]. All CHILPs still express Id2 [13], but promyelocytic leukemia zinc finger (PLZF) containing CHILP also express GATA3 and give rise to groups ILC1-3, while PLZF lacking CHILP develop into LTi cells (Figure 1). CHILPs also lack expression of Tox, NFIL3, and ETS1 [10].
Functionally, ILC1-3 are classified as the lineage-negative cells innate immune counterparts of CD4+ T helper cell that lack recombinase activation gene (RAG) and so do not respond in an antigen selective manner. Like CD4+ T cells, ILC1s produce interferon-gamma (IFNγ) in response to IL-12, IL-15, and IL-18, which parallels T helper 1 (Th1) cells [17,18,19]. ILC2 produce several type 2 cytokines such as IL-5, IL-9 and IL-13 in response to TSLP, IL-25, and IL-33 and are analogous to type 2 T helper (Th2) cells [8,17,20]. Likewise, ILC3 are like T helper 17 (Th17) cells, and mainly produce IL-17 and IL-22 in response to IL-23 and IL-1β (Figure 1) [17,21]. The expression of fate-determining transcription factors in ILC1-3 are also paralleled to their T cell counterparts. T-bet is mainly seen in ILC1 [14], while ILC2 and ILC3 express GATA3 [22,23,24] and RORγt [14,25], respectively (Figure 1). In terms of surface receptors, in both humans and mice, ILC are broadly defined as lin−CD94−CD45+CD127+ cells, meaning these cells lack the classic lineage markers that T and B cells have which generally include CD3+CD14+CD16+CD19+ CD20+CD56+FcER1+ hence they are known as innate cells as opposed to adaptive cells [26]. Human ILC1 are further defined as CD103+CRTH2−NKp44−/+C-kit− cells [27], while human ILC2s are broadly described as ST2+CD25+CysLTR1+CRTH2+KLRG1+/- cells [28] and ILC3s are generally CRTH2–CD117+ NKp44+/-C-kit+ [26,29].
NK cells and LTis are included in this family as they share features with ILC1 and ILC3, respectively, in development, phenotype, and function. While ILC1 and NK cells are primarily activated in response to viruses, intracellular microbes, and cancers, ILC2 are implicated in defence against helminth [30] and viral infections [31], as well as having a role in allergic diseases [32,33]. Thus far, ILC2 have been implicated in several allergic diseases such as asthma [20], and chronic rhinosinusitis [34], atopic dermatitis [35,36], nasal polyposis [34,37], and food allergy [38,39,40], as having a central role in driving Type 2 (T2) inflammation. ILC3 are proposed to drive recruitment of neutrophils through the Th17 immunity pathway, in response to epithelial tissue alarmins released during extracellular microbes or fungi infections. The IL-17 produced by ILC3 may have a role in chronic obstructive pulmonary disease (COPD) [41] and driving neutrophilic bronchitis in asthma, characterized by >64% neutrophils in sputum differential cell counts [42]. The focus of this review will be on immunobiology of ILC2.
2. Asthma and Type 2 Inflammation
The immunological response triggered by allergens generally triggers a Type 2 (T2) inflammatory cascade (Figure 2). In allergic asthma, atopic dermatitis, and allergic rhinitis, otherwise innocuous allergens including grass or tree pollens, house-dust mite, cockroach, or animal dander will stimulate the classic adaptive immune pathway to produce IgE direct specifically against the allergen [43]. This process is orchestrated by maturation of CD4+ Th2 cells in lymph nodes and subsequent production and release of T2 cytokines by these cells, such as IL-4, IL-9, IL-5 and IL-13, within tissues [43]. In contrast, ILC respond to microenvironmental cues from epithelial and other immune cells as opposed to direct allergen presentation in an innate immunity path (Figure 2). Discovered almost simultaneously in 2010 by Moro et al.[44], Neill et al.[45], and Price et al.[46], ILC2 feature prominently as effector cells of airway inflammation in asthma. Epithelial cells at mucosal tissues can be damaged by protease activity of allergens or environmental elemental such as pollution and viral infection triggering alarmin production including thymic stromal lymphopoietin (TSLP), IL-33 and IL-25 [43,47]. These alarmins prompt ILC2 to secrete T2 cytokines. Additionally, lipid mediators such as PGD2 and Cysteinyl leukotrienes (CysLTs) stimulate ILC2 [42,48]. Overall, T2 cytokines prompt the features of allergic diseases which in the context of asthma is a substantial influx of eosinophils to mucosal sites [49,50]. Eosinophils also release T2 cytokines as well as cytotoxic granules that damage the integrity of the epithelial barrier, leading to greater release of alarmins, thereby driving a chronic inflammatory response [51]. Airway eosinophilia is characterized by >3% eosinophils in sputum [52]. Processes triggered by T2 cytokine expression ultimately lead to mucus hypersecretion in the airways, variable airflow obstruction, and airway/bronchial hyperresponsiveness [51,53], which contribute to symptoms of asthma such as coughing, wheeze, and shortness of breath [53,54,55]. Consequently, identification of regulatory mechanisms that dampen this vicious feedforward loop is crucial for management of T2 inflammatory disease.
Although ILC2 are numerically fewer than CD4+ Th2 cells in asthmatic lungs (10,000-fold fewer), the cell produces up-to 500-fold more T2 cytokines than the latter on a cell per cell basis and likely contribute to persistence of eosinophilia in the airways [8,20,56]. Additionally, ILC2 may be the main drivers of eosinophilia and asthma symptoms in conditions where CD4+ T cell activity has been dampened by steroid therapy recapitulated as knocking out recombinase activating gene (RAG-/-) in mice models [47,57,58]. Additionally, ILC2 are significantly increased in the blood of patients with severe (7844±2350 cells/mL) compared to mild (310±74 cell/mL) asthma and similarly in sputum of severe (8715±2404 cells/mL) vs. mild (182±44 cells/mL) of asthma [8,59] patients, where ILC2 levels corelate with disease severity [55,60,61,62]. It is now postulated that the co-operative interaction of CD4+ Th2 and ILC2 function to drive inflammation in allergic respiratory diseases [63].
Importantly, it has recently been realized that considering only T2 inflammation in asthma is an oversimplification, and asthma endotypes are now broadly categorized as the classic T2 high and a T2 low endotype that presents through Th1 and Th17 immune pathways [43]. T2 low asthma is often accompanied by neutrophilia (>64% neutrophils in sputum differential cell counts) mainly driven by IL-17, as opposed to the eosinophilia [43]. There is also a mixed granulocytic endotype of asthma, which is the simultaneous presence of eosinophilia and neutrophilia, and the paucigranuloytic endotype, characterized by <3% eosinophils and <64% neutrophils in sputum differential cell counts [64,65]. This complex landscape underscores the multitude of innate and adaptive immune cells and interactions within the tissue microenvironment that collectively contribute to pathogenesis of asthma. Therefore, understanding and harnessing the body’s intrinsic regulatory pathways responsible for controlling inflammation is paramount to the management of allergic diseases such as asthma.
3. Plasticity of ILC
ILCs were initially considered to be terminally differentiated cells, distinguished by the receptors, transcription factors, and panel of cytokines listed above. However, there has been a paradigm shift based on recent evidence that ILCs are plastic. These cells can transdifferentiate and inter-convert between the defined subgroups and can produce a spectrum of overlapping cytokine profiles depending on signals from the local tissue milieu [66]. For example, Cai et al. [67] found that through induction with IL-25, IL-33 and lipid mediators including cysteinyl leukotrienes (CysLT) and prostaglandin D4 (PGD4), mice lung ILC2 can be induced to produce IL-17 thereby shifting to display an ILC3-like profile. Other studies have implicated cues such as IL-1β, IL-23, and TGF-β in driving plasticity of ILC2 to ILC3 [68,69]. More recently, these ILC-3 like ILC2 which are C-kit+ and IL-17 producing, have been shown to contribute to Th17 mediated pathologies and are implicated in causing neutrophilic inflammation in airway diseases [67,68,69,70,71].
Plasticity has also been reported among ILC1 and ILC3 in the human intestines, especially in relation to Crohn’s disease, where there is a marked decline in ILC3 in favor of ILC1, while healthy individuals have more ILC3s and fewer ILC1s [72]. Tissue microenvironmental cues are driving this trans-differentiation from ILC3 towards ILC1, specifically through IL-12 and IL-23 [73,74]. Furthermore, in vitro studies have shown ILC2 and ILC3 shift towards an ILC1 phenotype under influence with IL-1β and IL-12 [75,76]. In vivo, conversion of ILC2 to ILC1s occurred in the lungs of mice infected with influenza A virus, which led to rapid downregulation of GATA3, characteristic to ILC2 [76]. Meanwhile, IL-4 seems to be critical in maintaining typical ILC2 phenotype, since it was able to reverse conversion of ILC3-like ILC2 back towards T2 cytokine production [68,70]. ILC2 to ILC1 trans-differentiation also appears to be reversed in the presence of IL-4, which favors ILC2 phenotype by activating the STAT6 pathway, leading to the expression of GATA3 and CRTH2 [69,77].
Of specific interest to this paper are recent studies reporting a novel subtype of regulatory CRTH2+/-KLRG1+ILC2 that express the anti-inflammatory cytokine, IL-10, which downregulates T2 cytokine production and decreases eosinophil recruitment [78,79]. Furthermore, the presence of IL-10+ILC2 had clinical benefit in inflammatory and allergic conditions such as grass pollen allergy [80,81,82]. These findings not only demonstrate plasticity within the major ILC subgroups, but posit that within each subset, ILC produce variable cytokine profiles according to local microenvironmental prompts, which in turn contribute to immune regulation or dysregulation.
4. Interleukin-10
Interleukin (IL)-10 is an important anti-inflammatory or immune-regulatory cytokine that maintains homeostasis and prevents tissue damage due to immune hyperreactivity [83]. IL-10 binds to a heterodimeric receptor and activates the JAK1/STAT3 pathway promoting upregulation of itself, along with anti-inflammatory molecules such as suppressor of cytokine signaling 3 (SOCS-3) [84,85]. The overall effect of IL-10 is downregulation of cytokine production and pro-inflammatory genes in their targets T cells, B cells, NK cells, and granulocytes, which in turn may downregulate airway inflammatory responses as seen in asthma [79,84],. Dysfunction or dysregulation of IL-10 contributes to numerous human autoimmune diseases and allergic conditions [84,86,87]. Consequences of impaired IL-10 function in experimental disease models include chronic inflammatory bowel disease, rheumatoid arthritis, psoriasis, lupus, multiple sclerosis, transplant rejection, cancer, and infection [86]. Moreover, mutations of IL10 that decrease IL-10 production are correlated with the incidence of allergy [79]. Regulatory T (Treg) [88] and B (Breg) cells are known to be primary producers of immunoregulatory cytokines such as IL-10 and TGF-β [1,79,81]. In addition, a newly described subset of ILC2 that contribute to homeostasis at mucosal sites demonstrate autologous production and secretion of IL-10 [79,81,89]. Given its wide array of targets and its spectrum of anti-inflammatory properties, therapeutic manipulation to increase endogenous IL-10 production by various immune cells is the focus of several human disease conditions [90,91].
5. The Role of IL-10 Expressing ILC2
Seehus et al. [79] were the first to identify this a subclass of ILC2 capable of IL-10 production. These cells have regulatory functions, but do not express Foxp3 [92], making them molecularly and phenotypically distinct from Tregs [79]. Additionally, it has been noted that ILC2 produce IL-10 more readily than ILC1s and ILC3s, supporting the existence of IL-10+ILC2 as a distinct sub-class [2]. IL-10+ILC2 are characterized by their high expression of the transcription factor Id3 and tend to express surface receptor Killer Cell Lectin-Like Receptor G1 (KLRG1) [2,79]. Here we will investigate emerging evidence that resident and circulating naive ILC2 recruited to mucosal sites can differentiate into mature ILC2 that steadily produce IL-10 in response to molecules such as IL-2, IL-4, IL-10, IL-33, neuromedin-u (NMU) and retinoic acid (RA) (Table 1).
Firstly, IL-10+ILC2 have been detected in the lungs of healthy mice after chronic treatment with the allergen papain or IL-33 [79,93]. In humans, IL-10+ILC2 were rarer in individuals with grass pollen and house dust mite allergies compared to healthy individuals, indicating that these cells could facilitate tolerance to allergens [81]. A recent study assessed IL-10+ILC2 in regulating grass-pollen induced inflammatory responses in sensitized mice [81]. It was found that IL-10+ILC2 attenuated the T2 response and maintained the integrity of the epithelial barrier, effectively inhibiting the main drivers of eosinophilic bronchitis [81]. Furthermore, using murine models of asthma, Howard et al.[80] observed the regulatory role of IL-10+ILC2 in dampening airway hyperresponsiveness and eosinophilia in the lungs. As such, perhaps the airway hyperresponsiveness and bronchial hyperinflammation in asthma may be attributable to inadequate downregulation of immune responses due to diminished numbers of IL-10+ILC2. Thus, it would also be expected that IL-10+ILC2 levels are increased in atopic subjects following successful allergen immunotherapy (AIT). [81]. In line with this, in a recent prospective, double-blind, placebo-controlled study, Golebski et al. found sublingual and subcutaneous AIT for grass pollen allergy increased anti-inflammatory IL-10+ILC2 to levels equivalent to those found in non-atopic individuals [81,94]. The frequency of IL-10+ILC2 was found to increase with improvement of clinical symptoms in atopic subjects. In a cohort control study that examined AIT efficacy in individuals with allergic rhinitis in response to house dust mite (HDM), Boonpiyathad et al. [94] found that the percentages of IL-10+CTLA-4+ILCs in individuals who responded to treatment was increased by 3.2% (95% CI) after 2 years. Concurrently, IL-10+ILC2s were greater in individuals that responded to treatment than non-responders and those in the placebo control group [94].
Levels of IL-10+ILC2 cells in mice declined within 14 days following stoppage of stimulated with allergen or IL-33 challenge [79]. However, a small number of these cells were maintained and quickly re-mobilized upon one restimulation injection with IL-33, as late as 30 days post initial allergen challenge. This shows that ILC2 can be permanently polarized into IL-10+ILC2 and maintained to rapidly respond to subsequent allergen exposure, analogous to T memory cells. Together, these findings suggest that IL-10+ILC2 have an essential influence on tolerance to allergens and may be a good biomarker to assess AIT effectiveness.
6. Positive Modulators of IL-10 Expression by ILC2
Retinoic acid (RA) is a potent inducer of IL-10+ILC2. In 2021, Golebski et al. [81] categorized ILC2 isolated from human peripheral blood into four groups including KLRG1+CRTH2+, KLRG1–CRTH2+, KLRG1+CRTH2– and KLRG1–CRTH2– and analyzed these in cell cultures. The data showed that KLRG1+CRTH2– ILC2 produced the greatest quantity of IL-10 following in vitro incubation with IL-2, IL-7, and IL-33, in the presence of RA [81]. IL-7 was included as a growth factor for ILC2 to maintain viability of cell cultures and is not directly correlated with IL-10 production. The production of IL-5 and IL-13 was detected in all conditions, while only ILC2 cultured in the presence of RA produced IL-10, but not ILC2 stimulated with IL-2, IL-7 and IL-33 alone [81]. Therefore, both in combination with IL-2 and IL-33, as well as alone, RA is a powerful stimulus of IL-10 production by ILC2 in a dose-dependent manner [79,81]. Other studies have also found RA to be necessary in the conversion of inflammatory ILC2 to IL-10+ILC2 [94,95]. One distinction between ILC2 and the Th2 counterparts is the presence of retinoid-related orphan receptor alpha (RORα) in ILC2 [96]. RORα is critical for ILC2 development but has a limited role in Th2 cells [42]. RA itself is a vitamin A metabolite and CD103+ dendritic cells (DCs) are a major source of RA within the lung [97]. Mice that are deficient in CD103+DCs, mimicking an RA-deficient microenvironment, showed reduction in the Il10 gene expression by ILC2 previously activated in vivo with IL-33 injections [79].
As discussed, epithelial cell derived IL-33 is a potent inducer of proliferation and differentiation in ILC2 and induces T2 cytokine production by these cells. However, one study reported that exposure to IL-33 (four daily injections) and chronic exposure to allergen papain (five daily injections, one week rest, then five more daily injections) caused blood ILC2s of mice to secret IL-10 [79]. Meanwhile, long-term clonal expansion experiments showed that once KLRG1+ILC2 are polarized toward IL-10 production, they continue to produce IL-10 upon activation with IL-33 and TSLP alone, even when RA was subsequently absent [81]. In addition, after IL-10GFP mice were given 3 days of intranasal IL-33 to activate ILC2, culturing the isolated ILC2 for just 2 days with IL-2, IL-4, and IL-7 led to about half the total ILC2 population to become IL-10+ [80]. These IL-10+ILC2 maintained IL-5 production but had attenuated IL-13 production as compared to ILC2 only activated with IL-33 [80]. Therefore, epithelial derived alarmins IL-33 and TSLP can be potential enhancers of previously polarized IL-10+ILC2, but do not seem sufficient inducers of IL-10 production [79].
Contrastingly, IL-2 is another inducer of IL-10+ILC2 generation. The administration of IL-2 to healthy mice was effective at inhibiting eosinophilia in the lungs, which was associated with increased IL-10+ILC2 numbers, even in the absence of adaptive immune cells [79]. Some production of IL-5 and IL-13 from these IL-10+ILC2 was maintained [79]. In addition, the incubation of IL-10+ILC2 with IL-2 in culture has been shown to increase the number of IL-10+ILC2 as well as expanding secretion IL-10 on a per cell basis [21,79]. Interestingly, IL-10 production was optimally induced in culture by the addition of IL-2 and RA, as compared to IL-2 or RA, alone [79]. Moreover, in the brain meninges of both WT and Rag1 deficient mice who lack adaptive immunity, intracisternal administration of IL-33, which activates AMP activated protein kinase (AMPK), led to suppressed ILC1 and ILC3, while IL-10+ILC2 numbers increased [98]. Interestingly, mice that were IL-2 deficient did not show this effect, speaking to the importance of IL-2 signaling in promoting regulation and attenuation of anti-inflammatory cells [98]. There is some evidence that combined stimulation of ILC2 with IL-2 and RA indirectly exerts an effect by increasing the efficiency of IL-33 to induce IL-10 production [2,95], but this requires further investigation. The potential source of IL-2 in vivo is activated lung mast cells, that are also known to produce RA [95]. Seehus et al. [79] noted that mice deficient in mast cells had reduced IL-10 secretion capacity by ILC2, pointing to a role for mast-cell-derived IL-2 and RA in IL-10+ILC2 activation.
The significance of IL-2 for IL-10+ILC2 was confirmed by Bando et al. [21] who also showed that stimulation with IL-4, IL-10, and neuropeptide NMU strongly induced IL-10 production by intestinal ILC2 of mice. In addition to this, cultures using specific blocking antibodies revealed that IL-10 production was effectively stimulated by both IL-2 and IL-4 independently [21]. Furthermore, ILC2 express the IL-10 receptor, and when stimulated with IL-10 show a marked reduction in T2 cytokine production, namely IL-5 and IL-13, [7,21]. IL-10 secretion itself further induced IL-10 production by ILC2 through an autocrine effect [21,79,80]. In support of this, ILC2 cultures with IL-10 inducers (IL-2 and IL-4) treated with IL-10–blocking antibodies showed that IL-10-eGFP expression was blunted by inhibiting IL-10 [21].
A different study by Howard et al. [80] provided further support that IL-4 alone can induce IL-10+ILC2. Supernatants from culturing IL-33 activated ILC2 with IL-4 producing Th2 cells demonstrated a dramatic increase in the level of IL-10, which was significantly reduced when an anti-IL-4 antibody was added [80]. Because Th2 cells are dominant sources of IL-4 in lung inflammation, Th2 cell derived IL-4 may be important for stimulating IL-10 production by ILC2. However, Th2-derived IL-4 is also necessary for the accumulation of inflammatory ILC2 within the lungs and IL-4 produced by ILC2 is important to Th2 cell development and function (Figure 2) [99,100]. Thus, like IL-2 it is not exactly clear in which contexts IL-4 would stimulate more T2 cytokine or IL-10 production. Notably, IL-4 stimulation can induce IL-10 production in other cell types, namely Th1 cells. In vitro, antibodies blocking IL-4 inhibited IL-10 production by Th1 while addition of exogenous IL-4 to Th1 enhanced IL-10 [101]. Furthermore, IL-4 derived from Th2 cells, specifically, have been shown to induce IL-10 production by Th1 cells in culture [101]. Zhu et al. [102] has shown that when DCs are stimulated with IL-4 and RA, shift from a pro-inflammatory to an anti-inflammatory profile of cytokines. This, in conjunction with the recognized significance of RA to IL-10 production by ILC2, suggests that the combination of IL-4 and/or IL-2 with RA in the local milieu leads to induction of an immune regulatory profile of ILC2.
7. Inhibitors of IL-10 Expression By ILC2
The multitude of stimulating factors indicate that there is a complex interaction between cells within the local tissue that support IL-10+ILC2 differentiation and proliferation. Conversely, tumour necrosis factor like peptide 1A (TL1A), a member of the tumour necrosis factor super family (TNFSF15), has been found to suppress IL-10 production by ILC2 stimulated with IL-2 or IL-4 in culture [21]. Human ILC2 mainly express CRTH2, CD127, ST2, and CD25 [17], but also express death receptor 3 (DR3), a member of the TNF-receptor superfamily (TNFRSF25), which is a receptor specific to TL1A [103]. The TL1A/DR3 axis activates conventional ILC2, contributing to the optimization of T2 responses [103]. TL1A has been specifically shown to induces production of IL-5 and IL-13 by ILC2 and promote allergic inflammation [2,104,105]. Alongside TL1A, experiments adding TGF-β to ILC2 isolated from peripheral blood of subjects with systemic sclerosis that were cultured on dermal fibroblast showed decreased IL-10 production and dramatically decreased KLRG1 expression on ILC2, associated with IL-10 generation [106]. Addition of TFG-β also attenuated IL-10 production by lung ILC2 that was evoked by IL-2 and RA in culture [79].
8. Receptors, Sex Disparities & Neuroimmune Control OF IL-10+ILC2
In examining surface markers, Golebski et al. [81] found that ILC2 in blood expressing KLRG1+ but not KLRG1- produced IL-10, regardless of CRTH2 expression. KLRG1, or killer cell lectin-like receptor G1, is a co-inhibitory receptor that binds its ligand E-cadherin with low affinity [107]. E-cadherin is expressed on epithelial cells (ECs), dendritic cells (DCs), and other antigen-presenting cells [108]. The KLRG1/E-cadherin interaction has previously been proposed to inhibit IL-2 stimulated proliferation of ILC2, thereby reducing or controlling numbers of mature ILC2 within tissues [81]. In line with these findings, Bando et al. [21] noted that only KLRG1+ILC2 were capable of IL-10 production while no other lineage negative cells produced IL-10. In systemic sclerosis, skin biopsies show increased overall ILC2 numbers as compared to healthy donors and KLRG1-ILC2 specifically were elevated as compared to KLRG1+ILC2 [106]. The percentage of KLRG1+ILC2 was negatively correlated to extent of cutaneous fibrosis in these patients, implicating this surface receptor as indicative of the anti-inflammatory properties of ILC2 [106]. This also point towards ILC2 plasticity, as it signifies that CRTH2+ILC2 with a pro-inflammatory cytokine profile may be modulated by the local tissue microenvironmental cues to develop an anti-inflammatory property associated with increased KLRG1 expression and IL-10 secretion.
Notably, recent studies [109,110,111] have also pointed toward KLRG1 and IL-10 production as one of the main drivers of phenotypic differences in ILC2 that contribute to sex disparities in asthma where prevalence is greater in females compared to males post-puberty. Growing evidence indicates that ILC2 from males express more KLRG1 than ILC2 from females [109,110,111,112], while females generally have more ILC2 than males. Additionally, 5alpha-DHT or testosterone can attenuate pro-inflammatory T2 cytokine secretion by ILC2 [113]. Specifically, male lung ILC2 stimulated with IL-2 and IL-33 showed decreased IL-5 and IL-13 production by ILC2 compared to female lung ILC2, proposed to be mediated by testosterone [113]. Kadel et al. [112] identified a subset of KLRG1-ILC2 in the lungs of female mice as the major contributor to sex bias in ILC2 numbers and disease. Also, they found that elevated androgens led to decreased cytokine production by lung ILC2 in both sexes of mice, while removal of estrogen did not have a major effect on ILC2 [112]. Thus, testosterone potentially has an immune-protective effect by promoting KLRG1+IL-10+ILC2. To date, the effects of KLRG1 on ILC2 in driving sex differences has only been studied in mouse models. Human studies are needed to evaluate the relevance of KLRG1+ and KLRG1-ILC2 subsets and IL-10 production in asthma pathophysiology and sex disparity.
Besides KLRG1, studies have implicated cytotoxic T lymphocyte associated protein 4 (CTLA4) and CD25 expression with the production of IL-10 by ILC2 post-AIT [94,95]. The Ctla4 gene was also upregulated in IL-10+ILC2 [79]. Conversely, the surface intercellular adhesion molecule, ICAM-1, has been shown to play a particularly important role in the development and function of pulmonary ILC2 [105], and has been implicated in ILC2-dependent airway hyperreactivity and inflammation in mice [114]. Impairment in the ERK signaling pathway in ICAM-1-/-ILC2 led to an upregulation of IL-10 production and a decline in GATA3 levels in these cells [2,114], which in turn reduced output of T2 cytokines (IL-5, IL-9, and IL-13) upon allergen challenge [2]. Furthermore, presence of ICAM-1 directly inhibited IL-10 production in murine lung ILC2, and anti-ICAM-1 treatment downregulated IL-5 and IL-13 secretion, whilst triggering greater IL-10 expression [2,113,114]. This effect was also observed in ILC2 from T- and B- cell deficient mice [114]. Therefore, it is possible that IL-10-producing ILC2 do not express ICAM-1 on their surface like conventional ILC2, although this remains to be investigated in humans.
The neuropeptide, neuromedin U (NMU), which is the ligand of NMUR1, has demonstrated the capacity to rapidly activate ILC2 and presents an important neuroimmune axis in inflammatory conditions [89,115]. As well, in vivo co-administration of NMU with IL-25 strongly amplified allergic inflammation [89]. However, Bando et al. [21] found NMU to induce IL-10 production in murine intestinal ILC2 in culture experiments. On the other hand, Semaphoring (Sema6D) is an axonal guidance cue that is produced by mesenchymal cells, and lung ILC2s from Sema6d−/− mice showed increased KLRG1 and PD-1 expression. All the while, Sema6D deficiency did not affect Id2 or GATA3 expression in ILC2 [116]. Also, ILC2s from the lungs of both Sema6d−/− and WT mice cultured with IL-2 and IL-33 exhibited decreased IL-5 and IL-13 but augmented IL-10 synthesis [116]. Concurrently, Sema6d−/− mice showed significantly diminished eosinophilia and ILC2 numbers in bronchoalveolar lavage fluid (BALF) after challenge with IL-33, as well as elevated IL-10 and less IL-5 and IL-13 in BALF compared to WT mice [116]. Extended inquiry to clarify the function of NMU and Sema6d in relation to IL-10+ILC2 is warranted.
9. Mechanisms Associated with IL-10 Expressing ILC2
Although IL-10+ILC2 are found in several tissues, the precise cellular mechanisms which enhance IL-10 synthesis by ILC2 remain unclear. For example, it is known that activated ILC2 utilize fatty acid oxidation for fuel and that their metabolic activity is controlled by presence of fatty acids in the cells’ microenvironment [80]. Remarkably, it was observed that IL-4 stimulated IL-10+ILC2 demonstrated a switch towards the glycolytic pathway and shifted away from the traditional fatty acid oxidation in ILC2 that promotes pro-inflammatory functions [80]. Furthermore, in a study with subjects who have traumatic brain injury (TBI), ILCs were found to be elevated in cerebrospinal fluid (CSF) along with an energy sensing enzyme, AMPK [98]. AMPK is a serine/threonine kinase, which functions as a master metabolic switch, implicated in gluconeogenesis pathways. Compellingly, in the CSF of TBI patients activation of AMPK attenuated the proinflammatory ILC1 and ILC3 and raised the frequency of regulatory IL-10+ILC2 [98]. Metformin, which is a common treatment drug for type 2 diabetes due to its effects on glucose metabolism, can activate AMPK [98]. TBI model mice treated with metformin saw increased IL-10+ILC2 and decreased ILC1 and ILC3 levels in the brain meninges, which was accompanied by improved neurological outcomes [98]. These findings suggest that glucose may be necessary for the generation and function of IL-10+ILC2 in addition to RA. It is yet to be determined whether this metabolic switch towards the glycolytic pathway and away from fatty acid metabolism contributes to the transition towards regulatory ILC2.
Furthermore, Miyamoto et al. [93] reported that the absence of the transcription factor Cbfβ or its receptor proteins Runx1 or Runx3 in ILC2, both in vitro and in vivo, increased the presence of TIGIT+IL-10+ILC2 and reduced airway inflammation. In severe airway anaphylaxis, a group of TIGIT+IL-10+ILC2 was identified as seemingly “exhausted” cells, since they expressed the inhibitory receptor, TIGIT, characteristic of exhausted CD8+ T cells [2]. However, a different study concluded that IL-10 production is not because of exhausted ILC2, since Sema6d−/− mice, which had increased expression of IL-10, had comparable expression of exhaustion markers like TIGIT with WT ILC2 [116]. As of now, it is unclear whether regulatory ILC2 are more phenotypically like Tregs or exhausted CD8+ T cells.
It has also been reported that transcription factors cMaf and Blimp-1 can regulate IL-10 generation in several immune cells such as CD4+ and CD8+ T cells [80,117,118]. These transcription factors have previously been implicated in suppressing inflammation [117,119]. In several experiments, IL-4 stimulation resulted in the activation of cMaf and Blimp-1 within the nucleus that regulated IL-10 production in Th1, Th2, Th17, Treg and regulatory B (Breg) cells [117,118,119,120]. Recently, it has been discovered that these transcription factors are also engaged in IL-10+ILC2, as knockdown models of cMaf and Blimp-1 led to a significant decline in IL-10+ILC2 numbers [80].

Table 1.
Summary of the inducers and inhibitors of IL-10 production by ILC2 that have been identified in the included studies.
Table 1.
Summary of the inducers and inhibitors of IL-10 production by ILC2 that have been identified in the included studies.
| Study | Models and Methods | Inducers of IL-10 |
Inhibitors of IL-10 |
|---|---|---|---|
| Bando J. et al. [21] | ILC2 from small intestine lamina propria of WT mice subjected to intestinal inflammation. | IL-2, IL-4, IL-10, IL-27, and NMU | TL1A |
| Seehus C. et al. [79] | WT murine blood and lung ILC2, pre-activated +/- intraperitoneal IL-33. | IL-2, IL-33, RA | TGF-β |
| Howard E. et al. [80] | Blood ILC2 from WT and RAG deficient (Rag2–/– γc–/–) in a mouse model of asthma. | IL-2, IL-4, cMaf, Blimp-1 | - |
| Golebski K. et al. [81] | Murine lung-tissue-extracted ILC2 using a model for grass pollen allergic rhinitis. Blood ILC2 from healthy controls and grass pollen allergic rhinitics. | IL-2, IL-33, RA, KLRG1 | - |
| Wang S. et al. [82] | Intestinal ILC2 from healthy and Rag1−/−Il10−/− mice and human intestinal tissue. | TGF-β | - |
| Boonpiyathad T. et al. [94] | PBMC from subjects with allergic rhinitis undergoing AIT. | RA, CD25, CTLA-4 | - |
| Baban B. et al. [98] | Examined CSF of traumatic brain injury patients and model mice and analyzed effect of metformin. | KLRG1, IL-2, IL-33, AMPK |
- |
| Laurent P. et al. [106] | Skin biopsies and peripheral blood of systemic sclerosis patients. | KLRG1 | TGF-β |
| Hurrell BP. et al. [114] | Asthma model mice, and ICAM-1 deficient mice challenged intranasally with IL-33. | - | ICAM-1 |
| Naito M. et al. [116] | Isolated lung ILC2 from WT, allergic lung inflammation model mice and Sema6D deficient mice. | - | Sema6D |
| Morita H. et al. [121] | Isolated ILC2 from blood and nasal tissue of healthy and chronic rhinosinusitis subjects and HDM sensitized mice. | IL-2, IL-33, RA, CTLA-4 | TGF-β |
10. Conclusion
Current literature suggests that a number of cytokines including IL-2, IL-4, IL-10, IL-33, RA, NMU alone or in combination are the most promising inducers of IL-10 production by ILC2 (Figure 3). Specifically, KLRG1 and CTLA-4 surface receptors are also associated with induction of IL-10 expression by ILC2. Additionally, ILC2-derived IL-10 can perpetuate the anti-inflammatory functions of ILC2 in an autocrine fashion. Conversely, TL1A, TGF-β, Sema6D, and presence of surface receptor ICAM-1 have inhibitory effects on IL-10-generating ILC2. There seems to be shift in the metabolic environment towards glycolytic pathways in vivo further enhancing the regulatory phenotype of ILC2. Given the capacity of ILC2 for abundant secretion of cytokines relative to their cell numbers, promoting endogenous, persistent IL-10 may be a basis for targeted treatment of inflammatory conditions. In tandem, the ratio of conventional ILC2 to IL-10+ILC2 may serve as a biomarker for evaluation of AIT efficacy. Understanding the tissue microenvironmental cues and intracellular mechanisms involved in generating regulatory ILC2 is essential for uncovering allergen tolerance and immunomodulation. Finally, recognizing the plasticity of ILC and the extent of ILC responsiveness to the local tissue milieu raises the question of whether ILC classifications require refinement.
Author Contributions
Conceptualization NEF, MX. and RS; resources, NEF and MX; writing—original draft preparation, NEF and MX; writing—review and editing, RS; visualization, NEF. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
All authors contributed to the content of this manuscript, reviewed, and approved the final version for submission to Cells. Authors confirm that neither the manuscript nor any parts of its content are currently under consideration or published in another journal. Authors report no conflicts of interest.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Komlósi, Z.I.; van de Veen, W.; Kovács, N.; Szűcs, G.; Sokolowska, M.; O’Mahony, L.; Akdis, M.; Akdis, C.A. Cellular and Molecular Mechanisms of Allergic Asthma. Mol Aspects Med 2022, 85, 100995. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, Y.; Zhang, Y.; Ni, B. IL-10-Producing ILCs: Molecular Mechanisms and Disease Relevance. Frontiers in Immunology 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Boos, M.D.; Yokota, Y.; Eberl, G.; Kee, B.L. Mature Natural Killer Cell and Lymphoid Tissue–Inducing Cell Development Requires Id2-Mediated Suppression of E Protein Activity. Journal of Experimental Medicine 2007, 204, 1119–1130. [Google Scholar] [CrossRef]
- Bartemes, K.R.; Kita, H. Roles of Innate Lymphoid Cells (ILCs) in Allergic Diseases: The 10-Year Anniversary for ILC2s. Journal of Allergy and Clinical Immunology 2021, 147, 1531–1547. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells — a Proposal for Uniform Nomenclature. Nat Rev Immunol 2013, 13, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue Residency of Innate Lymphoid Cells in Lymphoid and Nonlymphoid Organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef]
- Bonne-Année, S.; Bush, M.C.; Nutman, T.B. Differential Modulation of Human Innate Lymphoid Cell (ILC) Subsets by IL-10 and TGF-β. Sci Rep 2019, 9, 14305. [Google Scholar] [CrossRef]
- Smith, S.G.; Chen, R.; Kjarsgaard, M.; Huang, C.; Oliveria, J.-P.; O’Byrne, P.M.; Gauvreau, G.M.; Boulet, L.-P.; Lemiere, C.; Martin, J.; et al. Increased Numbers of Activated Group 2 Innate Lymphoid Cells in the Airways of Patients with Severe Asthma and Persistent Airway Eosinophilia. Journal of Allergy and Clinical Immunology 2016, 137, 75–86.e8. [Google Scholar] [CrossRef]
- Yang, Q.; Saenz, S.A.; Zlotoff, D.A.; Artis, D.; Bhandoola, A. Cutting Edge: Natural Helper Cells Derive from Lymphoid Progenitors. The Journal of Immunology 2011, 187, 5505–5509. [Google Scholar] [CrossRef]
- Zook, E.C.; Kee, B.L. Development of Innate Lymphoid Cells. Nat Immunol 2016, 17, 775–782. [Google Scholar] [CrossRef]
- Seillet, C.; Rankin, L.; Groom, J.; Mielke, L.; Tellier, J.; Chopin, M.; Huntington, N.; Belz, G.; Carotta, S. Nfil3 Is Required for the Development of All Innate Lymphoid Cell Subsets. The Journal of experimental medicine 2014, 211. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, F.; Harly, C.; Xing, S.; Ye, L.; Xia, X.; Wang, H.; Wang, X.; Yu, S.; Zhou, X.; et al. TCF-1 Upregulation Identifies Early Innate Lymphoid Progenitors in the Bone Marrow. Nat Immunol 2015, 16, 1044–1050. [Google Scholar] [CrossRef]
- Seehus, C.R.; Aliahmad, P.; de la Torre, B.; Iliev, I.D.; Spurka, L.; Funari, V.A.; Kaye, J. The Development of Innate Lymphoid Cells Requires TOX-Dependent Generation of a Common Innate Lymphoid Cell Progenitor. Nat Immunol 2015, 16, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, K.; Chandler, K.J.; Spaulding, C.; Zandi, S.; Sigvardsson, M.; Graves, B.J.; Kee, B.L. Gene Deregulation and Chronic Activation in Natural Killer Cells Deficient in the Transcription Factor ETS1. Immunity 2012, 36, 921–932. [Google Scholar] [CrossRef]
- Male, V.; Nisoli, I.; Kostrzewski, T.; Allan, D.S.J.; Carlyle, J.R.; Lord, G.M.; Wack, A.; Brady, H.J.M. The Transcription Factor E4bp4/Nfil3 Controls Commitment to the NK Lineage and Directly Regulates Eomes and Id2 Expression. Journal of Experimental Medicine 2014, 211, 635–642. [Google Scholar] [CrossRef]
- Panda, S.K.; Colonna, M. Innate Lymphoid Cells in Mucosal Immunity. Frontiers in Immunology 2019, 10. [Google Scholar] [CrossRef]
- Cope, A.; Le Friec, G.; Cardone, J.; Kemper, C. The Th1 Life Cycle: Molecular Control of IFN-γ to IL-10 Switching. Trends in Immunology 2011, 32, 278–286. [Google Scholar] [CrossRef]
- Hsieh, C.-S.; Macatonia, S.E.; Tripp, C.S.; Wolf, S.F.; O’Garra, A.; Murphy, K.M. Development of TH1 CD4+ T Cells Through IL-12 Produced by Listeria-Induced Macrophages. Science 1993, 260, 547–549. [Google Scholar] [CrossRef]
- Chen, R.; Smith, S.G.; Salter, B.; El-Gammal, A.; Oliveria, J.P.; Obminski, C.; Watson, R.; O’Byrne, P.M.; Gauvreau, G.M.; Sehmi, R. Allergen-Induced Increases in Sputum Levels of Group 2 Innate Lymphoid Cells in Subjects with Asthma. Am J Respir Crit Care Med 2017, 196, 700–712. [Google Scholar] [CrossRef]
- Bando, J.K.; Gilfillan, S.; Di Luccia, B.; Fachi, J.L.; Sécca, C.; Cella, M.; Colonna, M. ILC2s Are the Predominant Source of Intestinal ILC-Derived IL-10. Journal of Experimental Medicine 2019, 217, e20191520. [Google Scholar] [CrossRef]
- Klein Wolterink, R.G.J.; Serafini, N.; Van Nimwegen, M.; Vosshenrich, C.A.J.; De Bruijn, M.J.W.; Fonseca Pereira, D.; Veiga Fernandes, H.; Hendriks, R.W.; Di Santo, J.P. Essential, Dose-Dependent Role for the Transcription Factor Gata3 in the Development of IL-5 + and IL-13 + Type 2 Innate Lymphoid Cells. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10240–10245. [Google Scholar] [CrossRef] [PubMed]
- Hoyler, T.; Klose, C.S.N.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The Transcription Factor GATA3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.; Bernink, J.; Golebski, K.; Karrich, J.J.; Peters, C.P.; Blom, B.; te Velde, A.A.; Fokkens, W.J.; van Drunen, C.M.; Spits, H. The Transcription Factor GATA3 Is Essential for the Function of Human Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; d’Hargues, Y.; Göppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-Bet Gradient Controls the Fate and Function of CCR6−RORγt+ Innate Lymphoid Cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef]
- Salter, B.M.; Aw, M.; Sehmi, R. The Role of Type 2 Innate Lymphoid Cells in Eosinophilic Asthma. Journal of Leukocyte Biology 2019, 106, 889–901. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human Type 1 Innate Lymphoid Cells Accumulate in Inflamed Mucosal Tissues. Nat Immunol 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-Responsive Type 2 Innate Lymphoid Cells Are Defined by Expression of CRTH2 and CD161. Nat Immunol 2011, 12, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Bennstein, S.B.; Uhrberg, M. Biology and Therapeutic Potential of Human Innate Lymphoid Cells. The FEBS Journal 2022, 289, 3967–3981. [Google Scholar] [CrossRef]
- Fallon, P.G.; Ballantyne, S.J.; Mangan, N.E.; Barlow, J.L.; Dasvarma, A.; Hewett, D.R.; McIlgorm, A.; Jolin, H.E.; McKenzie, A.N.J. Identification of an Interleukin (IL)-25–Dependent Cell Population That Provides IL-4, IL-5, and IL-13 at the Onset of Helminth Expulsion. J Exp Med 2006, 203, 1105–1116. [Google Scholar] [CrossRef]
- Fonseca, W.; Lukacs, N.W.; Elesela, S.; Malinczak, C.-A. Role of ILC2 in Viral-Induced Lung Pathogenesis. Frontiers in Immunology 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.; Steer, C.; Mathä, L.; Gold, M.; Martinez-Gonzalez, I.; Mcnagny, K.; McKenzie, A.N.J.; Takei, F. Group 2 Innate Lymphoid Cells Are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity 2014, 40. [Google Scholar] [CrossRef] [PubMed]
- Bartemes, K.R.; Iijima, K.; Kobayashi, T.; Kephart, G.M.; McKenzie, A.N.; Kita, H. IL-33-Responsive Lineage−CD25+CD44hi Lymphoid Cells Mediate Innate Type-2 Immunity and Allergic Inflammation in the Lungs. J Immunol 2012, 188, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Bailey, M.; Zaunders, J.; Mrad, N.; Sacks, R.; Sewell, W.; Harvey, R.J. Group 2 Innate Lymphoid Cells (ILC2s) Are Increased in Chronic Rhinosinusitis with Nasal Polyps or Eosinophilia. Clin Exp Allergy 2015, 45, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-Specific Expression of IL-33 Activates Group 2 Innate Lymphoid Cells and Elicits Atopic Dermatitis-like Inflammation in Mice. Proc Natl Acad Sci U S A 2013, 110, 13921–13926. [Google Scholar] [CrossRef]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP Elicits IL-33–Independent Innate Lymphoid Cell Responses to Promote Skin Inflammation. Sci Transl Med 2013, 5, 170ra16. [Google Scholar] [CrossRef]
- Shaw, J.L.; Fakhri, S.; Citardi, M.J.; Porter, P.C.; Corry, D.B.; Kheradmand, F.; Liu, Y.-J.; Luong, A. IL-33–Responsive Innate Lymphoid Cells Are an Important Source of IL-13 in Chronic Rhinosinusitis with Nasal Polyps. Am J Respir Crit Care Med 2013, 188, 432–439. [Google Scholar] [CrossRef]
- Lee, J.-B.; Chen, C.-Y.; Liu, B.; Mugge, L.; Angkasekwinai, P.; Facchinetti, V.; Dong, C.; Liu, Y.-J.; Rothenberg, M.E.; Hogan, S.P.; et al. IL-25 and CD4+ TH2 Cells Enhance Type 2 Innate Lymphoid Cell–Derived IL-13 Production, Which Promotes IgE-Mediated Experimental Food Allergy. Journal of Allergy and Clinical Immunology 2016, 137, 1216–1225.e5. [Google Scholar] [CrossRef]
- Krempski, J.W.; Kobayashi, T.; Iijima, K.; McKenzie, A.N.; Kita, H. Group 2 Innate Lymphoid Cells Promote Development of T Follicular Helper (Tfh) Cells and Initiate Allergic Sensitization to Peanuts. J Immunol 2020, 204, 3086–3096. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Burton, O.T.; Oettgen, H.C.; Chatila, T. IL-4 Production by Group 2 Innate Lymphoid Cells Promotes Food Allergy by Blocking Regulatory T-Cell Function. Journal of Allergy and Clinical Immunology 2016, 138, 801–811.e9. [Google Scholar] [CrossRef]
- Ju, X.; Miromoto, T.; Beaudin, S.; Salter, B.; Wattie, J.; Howie, K.J.; O’Byrne, P.M.; Gauvreau, G.M.; Sehmi, R. Enumeration of IL-17A/F Producing Innate Lymphoid Cells in Subjects with COPD. Journal of Allergy and Clinical Immunology 2019, 143, AB218. [Google Scholar] [CrossRef]
- Doherty, T.A.; Broide, D.H. Airway Innate Lymphoid Cells in the Induction and Regulation of Allergy. Allergol Int 2019, 68, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. The Basic Immunology of Asthma. Cell 2021, 184, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate Production of TH2 Cytokines by Adipose Tissue-Associated c-Kit+Sca-1+ Lymphoid Cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef]
- Price, A.E.; Liang, H.-E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically Dispersed Innate IL-13–Expressing Cells in Type 2 Immunity. Proc Natl Acad Sci U S A 2010, 107, 11489–11494. [Google Scholar] [CrossRef]
- Christianson, C.A.; Goplen, N.P.; Zafar, I.; Irvin, C.; Good, J.T.; Rollins, D.R.; Gorentla, B.; Liu, W.; Gorska, M.M.; Chu, H.; et al. The Persistence of Asthma Requires Multiple Feedback Circuits Involving ILC2 and IL33. J Allergy Clin Immunol 2015, 136, 59–68.e14. [Google Scholar] [CrossRef]
- Fajt, M.L.; Gelhaus, S.L.; Freeman, B.; Uvalle, C.E.; Trudeau, J.B.; Holguin, F.; Wenzel, S.E. Prostaglandin D2 Pathway Upregulation: Relation to Asthma Severity, Control, and TH2 Inflammation. Journal of Allergy and Clinical Immunology 2013, 131, 1504–1512.e12. [Google Scholar] [CrossRef]
- Sehmi, R.; Smith, S.G.; Kjarsgaard, M.; Radford, K.; Boulet, L.-P.; Lemiere, C.; Prazma, C.M.; Ortega, H.; Martin, J.G.; Nair, P. Role of Local Eosinophilopoietic Processes in the Development of Airway Eosinophilia in Prednisone-Dependent Severe Asthma. Clin Exp Allergy 2016, 46, 793–802. [Google Scholar] [CrossRef]
- Busse, W.W.; Sedgwick, J.B. Eosinophils in Asthma. Ann Allergy 1992, 68, 286–290. [Google Scholar]
- Nair, P.; Martin, J.G.; Cockcroft, D.C.; Dolovich, M.; Lemiere, C.; Boulet, L.-P.; O’Byrne, P.M. Airway Hyperresponsiveness in Asthma: Measurement and Clinical Relevance. The Journal of Allergy and Clinical Immunology: In Practice 2017, 5, 649–659.e2. [Google Scholar] [CrossRef]
- Nair, P. What Is an “Eosinophilic Phenotype” of Asthma? Journal of Allergy and Clinical Immunology 2013, 132, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Hargreave, F.E.; Nair, P. The Definition and Diagnosis of Asthma. Clinical & Experimental Allergy 2009, 39, 1652–1658. [Google Scholar] [CrossRef]
- O’Byrne, P.M.; Inman, M.D.; Parameswaran, K. The Trials and Tribulations of IL-5, Eosinophils, and Allergic Asthma. Journal of Allergy and Clinical Immunology 2001, 108, 503–508. [Google Scholar] [CrossRef]
- Jia, Y.; Fang, X.; Zhu, X.; Bai, C.; Zhu, L.; Jin, M.; Wang, X.; Hu, M.; Tang, R.; Chen, Z. IL-13+ Type 2 Innate Lymphoid Cells Correlate with Asthma Control Status and Treatment Response. Am J Respir Cell Mol Biol 2016, 55, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Hochdörfer, T.; Israelsson, E.; Hasselberg, A.; Cavallin, A.; Thörn, K.; Muthas, D.; Shojaee, S.; Lüer, K.; Müller, M.; et al. Activation of Group 2 Innate Lymphoid Cells after Allergen Challenge in Asthmatic Patients. Journal of Allergy and Clinical Immunology 2019, 144, 61–69.e7. [Google Scholar] [CrossRef]
- Klein Wolterink, R.G.J.; Kleinjan, A.; van Nimwegen, M.; Bergen, I.; de Bruijn, M.; Levani, Y.; Hendriks, R.W. Pulmonary Innate Lymphoid Cells Are Major Producers of IL-5 and IL-13 in Murine Models of Allergic Asthma. Eur J Immunol 2012, 42, 1106–1116. [Google Scholar] [CrossRef]
- Verma, M.; Liu, S.; Michalec, L.; Sripada, A.; Gorska, M.M.; Alam, R. Experimental Asthma Persists in IL-33 Receptor Knockout Mice Because of the Emergence of Thymic Stromal Lymphopoietin–Driven IL-9+ and IL-13+ Type 2 Innate Lymphoid Cell Subpopulations. Journal of Allergy and Clinical Immunology 2018, 142, 793–803.e8. [Google Scholar] [CrossRef]
- Bartemes, K.; Kephart, G.; Fox, S.J.; Kita, H. Enhanced Innate Type 2 Immune Response in Peripheral Blood from Patients with Asthma. J Allergy Clin Immunol 2014, 134, 671–678.e4. [Google Scholar] [CrossRef]
- Yu, Q.-N.; Tan, W.-P.; Fan, X.-L.; Guo, Y.-B.; Qin, Z.-L.; Li, C.-L.; Chen, D.; Lin, Z.B.; Wen, W.; Fu, Q.-L. Increased Group 2 Innate Lymphoid Cells Are Correlated with Eosinophilic Granulocytes in Patients with Allergic Airway Inflammation. Int Arch Allergy Immunol 2018, 176, 124–132. [Google Scholar] [CrossRef]
- Liu, T.; Wu, J.; Zhao, J.; Wang, J.; Zhang, Y.; Liu, L.; Cao, L.; Liu, Y.; Dong, L. Type 2 Innate Lymphoid Cells: A Novel Biomarker of Eosinophilic Airway Inflammation in Patients with Mild to Moderate Asthma. Respir Med 2015, 109, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Nagakumar, P.; Denney, L.; Fleming, L.; Bush, A.; Lloyd, C.M.; Saglani, S. Type 2 Innate Lymphoid Cells in Induced Sputum from Children with Severe Asthma. Journal of Allergy and Clinical Immunology 2016, 137, 624–626.e6. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, C.J.; Barlow, J.L.; McKenzie, A.N.J. Insights into the Initiation of Type 2 Immune Responses. Immunology 2011, 134, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Surette, M.G.; Virchow, J.C. Neutrophilic Asthma: Misconception or Misnomer? The Lancet Respiratory Medicine 2021, 9, 441–443. [Google Scholar] [CrossRef]
- Svenningsen, S.; Nair, P. Asthma Endotypes and an Overview of Targeted Therapy for Asthma. Frontiers in Medicine 2017, 4. [Google Scholar] [CrossRef]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of Innate Lymphoid Cell Subsets. Nat Rev Immunol 2020, 20, 552–565. [Google Scholar] [CrossRef]
- Cai, T.; Qiu, J.; Ji, Y.; Li, W.; Ding, Z.; Suo, C.; Chang, J.; Wang, J.; He, R.; Qian, Y.; et al. IL-17–Producing ST2+ Group 2 Innate Lymphoid Cells Play a Pathogenic Role in Lung Inflammation. Journal of Allergy and Clinical Immunology 2019, 143, 229–244.e9. [Google Scholar] [CrossRef]
- Bernink, J.H.; Ohne, Y.; Teunissen, M.B.M.; Wang, J.; Wu, J.; Krabbendam, L.; Guntermann, C.; Volckmann, R.; Koster, J.; van Tol, S.; et al. C-Kit-Positive ILC2s Exhibit an ILC3-like Signature That May Contribute to IL-17-Mediated Pathologies. Nat Immunol 2019, 20, 992–1003. [Google Scholar] [CrossRef]
- Golebski, K.; Ros, X.R.; Nagasawa, M.; van Tol, S.; Heesters, B.A.; Aglmous, H.; Kradolfer, C.M.A.; Shikhagaie, M.M.; Seys, S.; Hellings, P.W.; et al. IL-1β, IL-23, and TGF-β Drive Plasticity of Human ILC2s towards IL-17-Producing ILCs in Nasal Inflammation. Nat Commun 2019, 10, 2162. [Google Scholar] [CrossRef]
- Chen, X.-J.; Liu, C.; Zhang, S.; Zhang, L.-F.; Meng, W.; Zhang, X.; Sun, M.; Zhang, Y.; Wang, R.-Z.; Yao, C.-F. ILC3-like ILC2 Subset Increases in Minimal Persistent Inflammation after Acute Type II Inflammation of Allergic Rhinitis and Inhibited by Biminkang: Plasticity of ILC2 in Minimal Persistent Inflammation. Journal of Leukocyte Biology 2022, 112, 1445–1455. [Google Scholar] [CrossRef]
- Wen, Y.; Zeng, Q.; Luo, X.; Ma, R.; Tang, Y.; Liu, W. Leptin Promoted IL-17 Production from ILC2s in Allergic Rhinitis. Mediators Inflamm 2020, 2020, 9248479. [Google Scholar] [CrossRef]
- Simoni, Y.; Fehlings, M.; Kløverpris, H.N.; McGovern, N.; Koo, S.-L.; Loh, C.Y.; Lim, S.; Kurioka, A.; Fergusson, J.R.; Tang, C.-L.; et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 2017, 46, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127+ Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Doty, A.L.; Tang, Y.; Berrie, D.; Iqbal, A.; Tan, S.A.; Clare-Salzler, M.J.; Wallet, S.M.; Glover, S.C. Enrichment of IL-17A+IFN-Γ+ and IL-22+IFN-Γ+ T Cell Subsets Is Associated with Reduction of NKp44+ILC3s in the Terminal Ileum of Crohn’s Disease Patients. Clinical and Experimental Immunology 2017, 190, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Mazzurana, L.; Forkel, M.; Rao, A.; Van Acker, A.; Kokkinou, E.; Ichiya, T.; Almer, S.; Höög, C.; Friberg, D.; Mjösberg, J. Suppression of Aiolos and Ikaros Expression by Lenalidomide Reduces Human ILC3-ILC1/NK Cell Transdifferentiation. Eur J Immunol 2019, 49, 1344–1355. [Google Scholar] [CrossRef]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory Triggers Associated with Exacerbations of COPD Orchestrate Plasticity of Group 2 Innate Lymphoid Cells in the Lungs. Nat Immunol 2016, 17, 626–635. [Google Scholar] [CrossRef]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P.; et al. IL-1β, IL-4 and IL-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat Immunol 2016, 17, 636–645. [Google Scholar] [CrossRef]
- Ogasawara, N.; Poposki, J.A.; Klingler, A.I.; Tan, B.K.; Weibman, A.R.; Hulse, K.E.; Stevens, W.W.; Peters, A.T.; Grammer, L.C.; Schleimer, R.P.; et al. IL-10, TGF-β and Glucocorticoid Prevent the Production of Type 2 Cytokines in Human Group 2 Innate Lymphoid Cells. J Allergy Clin Immunol 2018, 141, 1147–1151.e8. [Google Scholar] [CrossRef]
- Seehus, C.R.; Kadavallore, A.; Torre, B. de la; Yeckes, A.R.; Wang, Y.; Tang, J.; Kaye, J. Alternative Activation Generates IL-10 Producing Type 2 Innate Lymphoid Cells. Nat Commun 2017, 8, 1900. [Google Scholar] [CrossRef]
- Howard, E.; Lewis, G.; Galle-Treger, L.; Hurrell, B.P.; Helou, D.G.; Shafiei-Jahani, P.; Painter, J.D.; Muench, G.A.; Soroosh, P.; Akbari, O. IL-10 Production by ILC2s Requires Blimp-1 and cMaf, Modulates Cellular Metabolism, and Ameliorates Airway Hyperreactivity. J Allergy Clin Immunol 2021, 147, 1281–1295.e5. [Google Scholar] [CrossRef]
- Golebski, K.; Layhadi, J.A.; Sahiner, U.; Steveling-Klein, E.H.; Lenormand, M.M.; Li, R.C.Y.; Bal, S.M.; Heesters, B.A.; Vilà-Nadal, G.; Hunewald, O.; et al. Induction of IL-10-Producing Type 2 Innate Lymphoid Cells by Allergen Immunotherapy Is Associated with Clinical Response. Immunity 2021, 54, 291–307.e7. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Chen, Y.; Qu, Y.; Xiong, Z.; Ye, B.; Du, Y.; Tian, Y.; Yin, Z.; Xu, Z.; et al. Regulatory Innate Lymphoid Cells Control Innate Intestinal Inflammation. Cell 2017, 171, 201–216.e18. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; Vieira, P.; Fiorentino, D.F.; Trounstine, M.L.; Khan, T.A.; Mosmann, T.R. Homology of Cytokine Synthesis Inhibitory Factor (IL-10) to the Epstein-Barr Virus Gene BCRFI. Science 1990, 248, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the Interleukin-10 Receptor. Annual Review of Immunology 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The Primary Mechanism of the IL-10-Regulated Antiinflammatory Response Is to Selectively Inhibit Transcription. Proc Natl Acad Sci U S A 2005, 102, 8686–8691. [Google Scholar] [CrossRef]
- Iyer, S.S.; Cheng, G. Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit Rev Immunol 2012, 32, 23–63. [Google Scholar] [CrossRef]
- Glocker, E.-O.; Kotlarz, D.; Klein, C.; Shah, N.; Grimbacher, B. IL-10 and IL-10 Receptor Defects in Humans. Annals of the New York Academy of Sciences 2011, 1246, 102–107. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Bond, M.W.; Mosmann, T.R. Two Types of Mouse T Helper Cell. IV. Th2 Clones Secrete a Factor That Inhibits Cytokine Production by Th1 Clones. J Exp Med 1989, 170, 2081–2095. [Google Scholar] [CrossRef]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.-E.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The Neuropeptide NMU Amplifies ILC2-Driven Allergic Lung Inflammation. Nature 2017, 549, 351–356. [Google Scholar] [CrossRef]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb Perspect Biol 2019, 11, a028548. [Google Scholar] [CrossRef]
- Saraiva, M.; Vieira, P.; O’Garra, A. Biology and Therapeutic Potential of Interleukin-10. J Exp Med 2019, 217, e20190418. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, C.; Kojo, S.; Yamashita, M.; Moro, K.; Lacaud, G.; Shiroguchi, K.; Taniuchi, I.; Ebihara, T. Runx/Cbfβ Complexes Protect Group 2 Innate Lymphoid Cells from Exhausted-like Hyporesponsiveness during Allergic Airway Inflammation. Nat Commun 2019, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Boonpiyathad, T.; Tantilipikorn, P.; Ruxrungtham, K.; Pradubpongsa, P.; Mitthamsiri, W.; Piedvache, A.; Thantiworasit, P.; Sirivichayakul, S.; Jacquet, A.; Suratannon, N.; et al. IL-10-Producing Innate Lymphoid Cells Increased in Patients with House Dust Mite Allergic Rhinitis Following Immunotherapy. J Allergy Clin Immunol 2021, 147, 1507–1510.e8. [Google Scholar] [CrossRef]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A.; et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef]
- Ferreira, A.C.F.; Szeto, A.C.H.; Heycock, M.W.D.; Clark, P.A.; Walker, J.A.; Crisp, A.; Barlow, J.L.; Kitching, S.; Lim, A.; Gogoi, M.; et al. RORα Is a Critical Checkpoint for T Cell and ILC2 Commitment in the Embryonic Thymus. Nat Immunol 2021, 22, 166–178. [Google Scholar] [CrossRef]
- Khare, A.; Krishnamoorthy, N.; Oriss, T.B.; Fei, M.; Ray, P.; Ray, A. Inhaled Antigen Upregulates Retinaldehyde Dehydrogenase In Lung CD103+ But Not Plasmacytoid Dendritic Cells To Induce Foxp3 De Novo in CD4+ T Cells and Promote Airway Tolerance. J Immunol 2013, 191, 25–29. [Google Scholar] [CrossRef]
- Baban, B.; Braun, M.; Khodadadi, H.; Ward, A.; Alverson, K.; Malik, A.; Nguyen, K.; Nazarian, S.; Hess, D.C.; Forseen, S.; et al. AMPK Induces Regulatory Innate Lymphoid Cells after Traumatic Brain Injury. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Coomes, S.M.; Kannan, Y.; Pelly, V.S.; Entwistle, L.J.; Guidi, R.; Perez-Lloret, J.; Nikolov, N.; Müller, W.; Wilson, M.S. CD4+ Th2 Cells Are Directly Regulated by IL-10 during Allergic Airway Inflammation. Mucosal Immunol 2017, 10, 150–161. [Google Scholar] [CrossRef]
- Pelly, V.S.; Kannan, Y.; Coomes, S.M.; Entwistle, L.J.; Rückerl, D.; Seddon, B.; MacDonald, A.S.; McKenzie, A.; Wilson, M.S. IL-4-Producing ILC2s Are Required for the Differentiation of TH2 Cells Following Heligmosomoides Polygyrus Infection. Mucosal Immunology 2016, 9, 1407–1417. [Google Scholar] [CrossRef]
- Mitchell, R.E.; Hassan, M.; Burton, B.R.; Britton, G.; Hill, E.V.; Verhagen, J.; Wraith, D.C. IL-4 Enhances IL-10 Production in Th1 Cells: Implications for Th1 and Th2 Regulation. Sci Rep 2017, 7, 11315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Buttrick, T.; Bassil, R.; Zhu, C.; Olah, M.; Wu, C.; Xiao, S.; Orent, W.; Elyaman, W.; Khoury, S.J. IL-4 and Retinoic Acid Synergistically Induce Regulatory Dendritic Cells Expressing Aldh1a2. J Immunol 2013, 191, 3139–3151. [Google Scholar] [CrossRef] [PubMed]
- Meylan, F.; Hawley, E.T.; Barron, L.; Barlow, J.L.; Penumetcha, P.; Pelletier, M.; Sciumè, G.; Richard, A.C.; Hayes, E.T.; Gomez-Rodriguez, J.; et al. The TNF-Family Cytokine TL1A Promotes Allergic Immunopathology through Group 2 Innate Lymphoid Cells. Mucosal Immunol 2014, 7, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Aw, M.; Salter, B.M.A.; Ju, X.; Mukherjee, M.; Gauvreau, G.M.; O’Byrne, P.M.; Nair, P.; Sehmi, R. The Role of the TL1A/DR3 Axis in the Activation of Group 2 Innate Lymphoid Cells in Subjects with Eosinophilic Asthma. Am J Respir Crit Care Med 2020, 202, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Lei, A.-H.; Xiao, Q.; Liu, G.-Y.; Shi, K.; Yang, Q.; Li, X.; Liu, Y.-F.; Wang, H.-K.; Cai, W.-P.; Guan, Y.-J.; et al. ICAM-1 Controls Development and Function of ILC2. J Exp Med 2018, 215, 2157–2174. [Google Scholar] [CrossRef]
- Laurent, P.; Allard, B.; Manicki, P.; Jolivel, V.; Levionnois, E.; Jeljeli, M.; Henrot, P.; Izotte, J.; Leleu, D.; Groppi, A.; et al. TGFβ Promotes Low IL10-Producing ILC2 with Profibrotic Ability Involved in Skin Fibrosis in Systemic Sclerosis. Annals of the Rheumatic Diseases 2021, 80, 1594–1603. [Google Scholar] [CrossRef]
- Nakamura, S.; Kuroki, K.; Ohki, I.; Sasaki, K.; Kajikawa, M.; Maruyama, T.; Ito, M.; Kameda, Y.; Ikura, M.; Yamamoto, K.; et al. Molecular Basis for E-Cadherin Recognition by Killer Cell Lectin-like Receptor G1 (KLRG1). J Biol Chem 2009, 284, 27327–27335. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1--More than a Marker for T Cell Senescence. Age (Dordr) 2009, 31, 285–291. [Google Scholar] [CrossRef]
- Laffont, S.; Blanquart, E.; Savignac, M.; Cénac, C.; Laverny, G.; Metzger, D.; Girard, J.-P.; Belz, G.T.; Pelletier, L.; Seillet, C.; et al. Androgen Signaling Negatively Controls Group 2 Innate Lymphoid Cells. Journal of Experimental Medicine 2017, 214, 1581–1592. [Google Scholar] [CrossRef]
- Loering, S.; Cameron, G.J.; Bhatt, N.P.; Belz, G.T.; Foster, P.S.; Hansbro, P.M.; Starkey, M.R. Differences in Pulmonary Group 2 Innate Lymphoid Cells Are Dependent on Mouse Age, Sex and Strain. Immunol Cell Biol 2021, 99, 542–551. [Google Scholar] [CrossRef]
- Zhao, H.; Moarbes, V.; Gaudreault, V.; Shan, J.; Aldossary, H.; Cyr, L.; Fixman, E.D. Sex Differences in IL-33-Induced STAT6-Dependent Type 2 Airway Inflammation. Frontiers in Immunology 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kadel, S.; Ainsua-Enrich, E.; Hatipoglu, I.; Turner, S.; Singh, S.; Khan, S.; Kovats, S. A Major Population of Functional KLRG1- ILC2s in Female Lungs Contributes to a Sex Bias in ILC2 Numbers. Immunohorizons 2018, 2, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Cephus, J.; Stier, M.; Fuseini, H.; Yung, J.; Toki, S.; Bloodworth, M.; Zhou, W.; Goleniewska, K.; Zhang, J.; Garon, S.; et al. Testosterone Attenuates Group 2 Innate Lymphoid Cell-Mediated Airway Inflammation. Cell Rep 2017, 21, 2487–2499. [Google Scholar] [CrossRef]
- Hurrell, B.P.; Howard, E.; Galle-Treger, L.; Helou, D.G.; Shafiei-Jahani, P.; Painter, J.D.; Akbari, O. Distinct Roles of LFA-1 and ICAM-1 on ILC2s Control Lung Infiltration, Effector Functions, and Development of Airway Hyperreactivity. Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.; Chesné, J.; Ribeiro, H.; García-Cassani, B.; Carvalho, T.; Bouchery, T.; Shah, K.; Barbosa-Morais, N.L.; Harris, N.; Veiga-Fernandes, H. Neuronal Regulation of Type 2 Innate Lymphoid Cells via Neuromedin U. Nature 2017, 549, 277–281. [Google Scholar] [CrossRef]
- Naito, M.; Nakanishi, Y.; Motomura, Y.; Takamatsu, H.; Koyama, S.; Nishide, M.; Naito, Y.; Izumi, M.; Mizuno, Y.; Yamaguchi, Y.; et al. Semaphorin 6D–Expressing Mesenchymal Cells Regulate IL-10 Production by ILC2s in the Lung. Life Sci. Alliance 2022, 5, e202201486. [Google Scholar] [CrossRef]
- Neumann, C.; Heinrich, F.; Neumann, K.; Junghans, V.; Mashreghi, M.-F.; Ahlers, J.; Janke, M.; Rudolph, C.; Mockel-Tenbrinck, N.; Kühl, A.A.; et al. Role of Blimp-1 in Programing Th Effector Cells into IL-10 Producers. J Exp Med 2014, 211, 1807–1819. [Google Scholar] [CrossRef]
- Sun, J.; Dodd, H.; Moser, E.K.; Sharma, R.; Braciale, T.J. CD4+ T Cell Help and Innate-Derived IL-27 Induce Blimp-1-Dependent IL-10 Production by Antiviral CTLs. Nat Immunol 2011, 12, 327–334. [Google Scholar] [CrossRef]
- Xu, M.; Pokrovskii, M.; Ding, Y.; Yi, R.; Au, C.; Harrison, O.J.; Galan, C.; Belkaid, Y.; Bonneau, R.; Littman, D.R. C-MAF-Dependent Regulatory T Cells Mediate Immunological Tolerance to a Gut Pathobiont. Nature 2018, 554, 373–377. [Google Scholar] [CrossRef]
- Rutz, S.; Ouyang, W. Regulation of Interleukin-10 Expression. In Regulation of Cytokine Gene Expression in Immunity and Diseases; Ma, X., Ed.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, Netherlands, 2016; pp. 89–116. ISBN 978-94-024-0921-5. [Google Scholar]
- Morita, H.; Kubo, T.; Rückert, B.; Ravindran, A.; Soyka, M.B.; Rinaldi, A.O.; Sugita, K.; Wawrzyniak, M.; Wawrzyniak, P.; Motomura, K.; et al. Induction of Human Regulatory Innate Lymphoid Cells from Group 2 Innate Lymphoid Cells by Retinoic Acid. J Allergy Clin Immunol 2019, 143, 2190–2201.e9. [Google Scholar] [CrossRef]
Figure 1.
Representation of the major innate lymphoid cell types (Natural Killer cells, Innate Lymphoid Types 1-3, Lymphoid Tissue Inducers) along with their progenitor cells, key cytokine inducers, and main cytokine products as well as identifying transcription factors. Innate lymphoid cells (ILC) are defined as lin−CD94−CD127+. ILC1 are generally identified as lin−CD127+CD117−CD56+/-CRTH2−NKp44−, ILC2s as lin−CD127+CD117+/−CRTH2+ and ILC3s as lin−CD94−CD56+/−CD127+CD117+CRTH2-.
Figure 1.
Representation of the major innate lymphoid cell types (Natural Killer cells, Innate Lymphoid Types 1-3, Lymphoid Tissue Inducers) along with their progenitor cells, key cytokine inducers, and main cytokine products as well as identifying transcription factors. Innate lymphoid cells (ILC) are defined as lin−CD94−CD127+. ILC1 are generally identified as lin−CD127+CD117−CD56+/-CRTH2−NKp44−, ILC2s as lin−CD127+CD117+/−CRTH2+ and ILC3s as lin−CD94−CD56+/−CD127+CD117+CRTH2-.
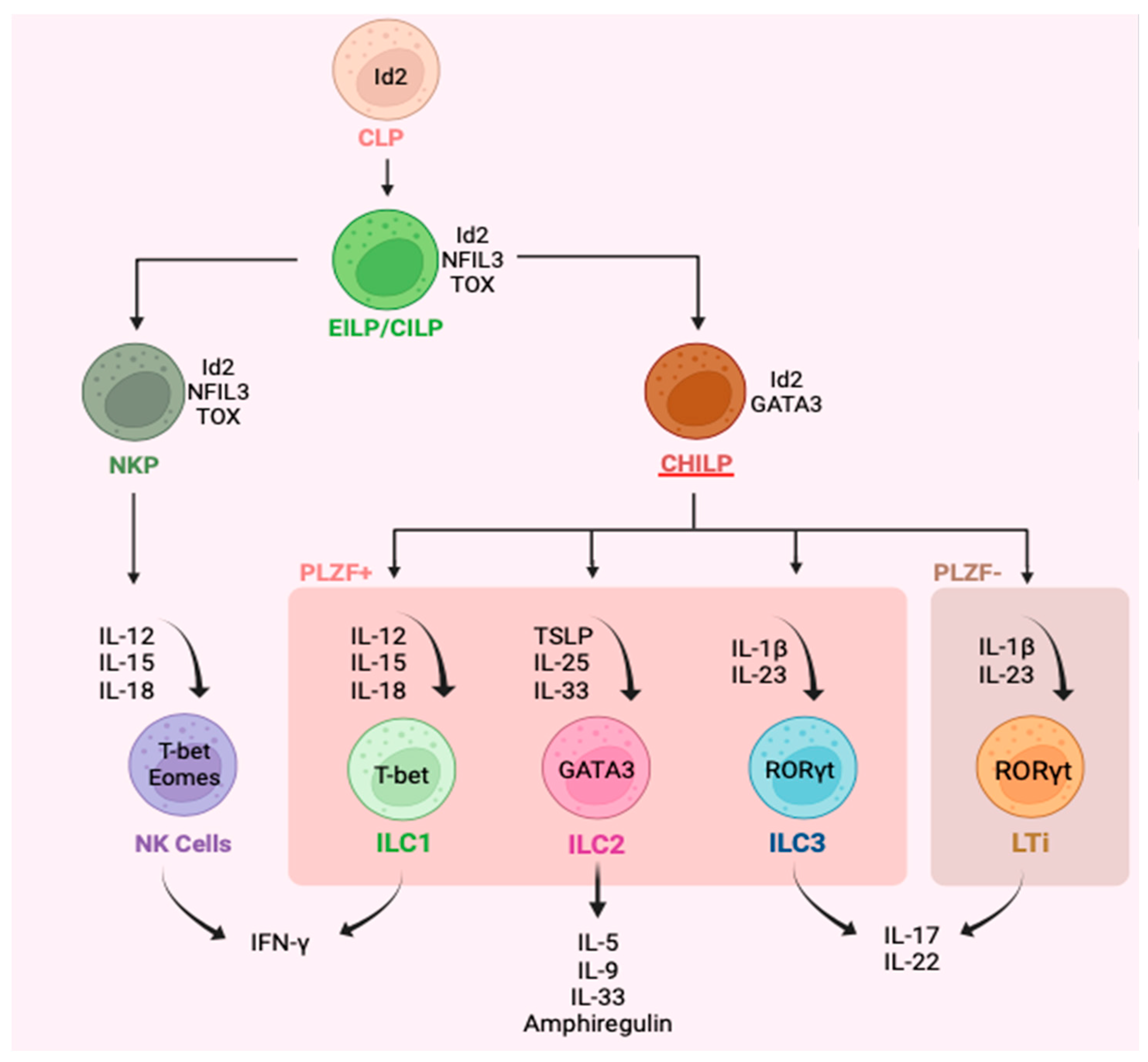
Figure 2.
The primary role of ILC2 in the type 2 inflammatory response within the lungs in eosinophilic allergic asthma. Disruption of bronchial epithelial barrier results in cell activation and release of alarmin cytokines that activate type 2 innate lymphoid cells triggering the release type 2 cytokines and cascade of type 2 inflammatory responses characteristic of eosinophilic asthma.
Figure 2.
The primary role of ILC2 in the type 2 inflammatory response within the lungs in eosinophilic allergic asthma. Disruption of bronchial epithelial barrier results in cell activation and release of alarmin cytokines that activate type 2 innate lymphoid cells triggering the release type 2 cytokines and cascade of type 2 inflammatory responses characteristic of eosinophilic asthma.

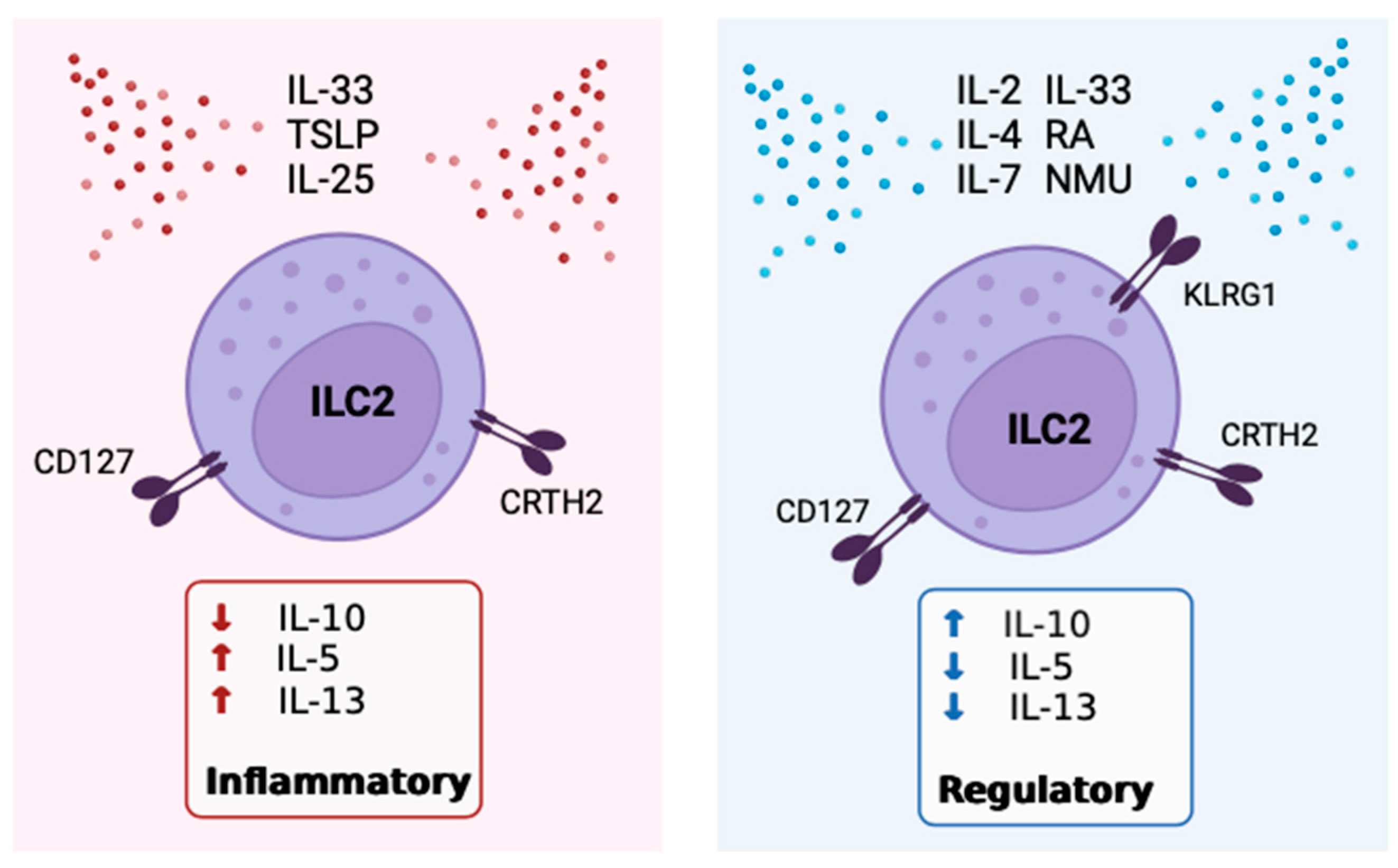
Figure 3.
Inflammatory vs. regulatory ILC2 main inducers, characteristic receptors, and cytokine products. The figure details the currently known stimuli that promote either the regulatory or inflammatory phenotype of ILC2.
Figure 3.
Inflammatory vs. regulatory ILC2 main inducers, characteristic receptors, and cytokine products. The figure details the currently known stimuli that promote either the regulatory or inflammatory phenotype of ILC2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



