
Submitted:
18 September 2023
Posted:
19 September 2023
You are already at the latest version
Abstract
Indoleamine 2,3-dioxygenase 2 (IDO2) is a paralogue of IDO1, a tryptophan-degrading enzyme producing immunomodulatory molecules. However, the two proteins are unlikely to carry out the same functions. IDO2 shows little or no tryptophan catabolic activity and exerts contrasting immunomodulatory roles in a context-dependent manner, in both cancer and autoimmune diseases. The recently described potential non-enzymatic activity of IDO2 has suggested its possible involvement in alternative pathways resulting in either pro- or anti-inflammatory effects in different models. In a previous study on non-small cell lung cancer (NSCLC) tissues, we found that IDO2 expression, revealed at the plasma membrane level of tumor cells, was significantly associated with poor prognosis. In this study, the A549 human cell line, basally expressing IDO2, was used as an in vitro model of human lung adenocarcinoma to gain more insights on a possible alternative function of IDO2, different from the catalytic one. In these cells, immunocytochemistry and isopycnic sucrose gradient analyses confirmed the IDO2 protein localization in the cell membrane compartment, and immunoprecipitation of tyrosine-phosphorylated proteins revealed that IDO2 can be targeted by kinase activities. The different localization than the cytosolic one and the phosphorylation state are the first indications for the signaling function of IDO2, suggesting that the IDO2 non-enzymatic role in cancer cells is worthy of deeper understanding.
Keywords:
IDO2
; lung adenocarcinoma
; A549
; membrane localization
; tyrosine-phosphorylation
1. Introduction
The immunoregulatory metabolite kynurenine (Kyn) can be produced through tryptophan (Trp) degradation operated by the heme-containing enzymes tryptophan 2,3-dioxygenase 2 (TDO2) and indoleamine 2,3-dioxygenase 1 (IDO1). TDO2, selectively expressed in the liver, is the main enzyme responsible for the metabolism of dietary tryptophan and controls the homeostasis of this essential amino acid. IDO1, acting on a large variety of indoleamine-containing substrates, has a high affinity for Trp and an enzymatic biological function based on the immunosuppressive effect caused by Trp depletion and Kyn accumulation, inhibiting effector T cell responses and promoting Treg and dendritic cell (DC) tolerance [1,2,3]. Another enzyme potentially able to convert Trp into Kyn is the IDO1 paralogue indoleamine 2,3-dioxygenase 2 (IDO2), discovered in 2007 by Ball and coworkers [4].
IDO2, mapped to chromosome 8 in humans and mice, is encoded by a gene located adjacent to and downstream of IDO1. The two genes were generated from a more ancient proto-IDO gene duplication that occurred before the divergence of marsupial and eutherian (placental) mammals [4,5,6]. IDO1 is widely expressed in different cell types like endothelial cells of the placenta and lung, or lymphoid tissues, mostly in DCs. On the contrary, IDO2 mRNA expression is confined in the liver, cerebral cortex, and kidney. Multiple pro-inflammatory stimuli have been proposed as IDO2 regulators, though with relatively less strength compared to IDO1 [7,8,9,10]. IDO1 and IDO2 share 43% identity in their amino acid sequence, including conserved residues involved in the catalytic activity. Despite displaying high sequence homology, the two enzymes have different affinities for Trp. IDO1 has a high rate of Trp catalysis (Km around 7–22 μM), while IDO2 has been reported to have a very low catalytic efficiency with a high Km value (6.8–9.4 mM), 100-fold higher than the physiological l-tryptophan concentrations [11,12]. Based on this evidence, several possible hypotheses have been made for the IDO2 activity and function, including the erroneous Km evaluation due to the interference of reducing reagents commonly used to dose indoleamine 2,3-dioxygenase activity [13], the existence of a natural substrate different from Trp and a functional role distinct from Trp catabolism [14,15]. In support of the latter hypothesis, it has been found that Ido2 deletion does not decrease the Kyn systemic levels in knockout mice, whereas affects IDO1-dependent T cell suppression and Treg induction [16], suggesting an Ido1–Ido2 genetic interaction and a possible functional role of Ido2 in the modulation of immune responses, which, however, remains ambiguous. Moreover, untargeted analysis of metabolites produced by cells overexpressing human or murine IDO2 revealed that no amino acid, nor any other compound commonly found in the cell line culture medium, is specifically metabolized by IDO2 (data obtained in our laboratory and not shown), thus confirming that the similarity of IDO1 and IDO2 in the amino acid sequence does not necessarily result in the same metabolic activity. For IDO2 a pro-inflammatory function has been described in B cells in the initiation, progression, and severity of autoimmune arthritis by both the in vivo model of KRN.g7 mice, genetically deficient for the Ido2 gene, and the specific silencing of Ido2 [17,18]. Nevertheless, in other studies, Ido2 appears to have an anti-inflammatory role in a psoriasis-like inflammation model, since its deletion exacerbates the disease symptoms and increases the number of IL-17–positive lymphocytes infiltrating the dermis [19].
The expression of IDO2 is upregulated in a variety of cancers, including non-small cell lung cancer (NSCLC) [20], pancreatic [21,22], colon, gastric and renal tumors [23,24], and medullary thyroid carcinoma [25]. Liu et al. assessed the biological value of IDO2 in mouse B16/BL6 melanoma cells, which showed a significantly reduced proliferation with a cell cycle arrest in the G1 phase, high apoptosis rate, and reduced cell migration when constitutively highly expressed Ido2 gene was silenced. In the same study, in vivo onset of tumor growth was delayed in mice injected with Ido2-silenced cells [26]. IDO2 overexpression is recurrent in pancreatic ductal adenocarcinoma (PDAC) tumors and is involved in tumorigenesis mechanisms, as demonstrated by the improved disease-free survival recorded in adjuvant-radiotherapy treated PDAC patients featuring single nucleotide polymorphisms (SNPs) thought to completely inactivate the already negligible Trp-catalytic activity of IDO2 [21,27]. To evaluate cancer risk, the involvement of IDO2 SNPs was investigated also in a large cohort of NSCLC patients and healthy matched controls, and a highly significant incidence of R248W genotype in NSCLC patients was found, compared to the control group [28]. Strong evidence of a significant correlation between IDO2 expression and poor NSCLC prognosis was found in a recent study assessing IDO2 presence and localization in tumor cells by an extensive immunohistochemical analysis of NSCLC specimens [20]. Among analyzed histotypes, the adenocarcinoma showed the highest IDO2 expression, associated with high intratumoral/mixed tumor-infiltrating lymphocyte localization. In the same study, 83% of tumors showed a membrane reinforcement staining of IDO2 that, in 51% of the cases, localized at the basolateral side of the cell membrane, between tumor and stromal tissue [20].
In the present study, starting from the findings mentioned above and to gain more insights on its possible alternative function (i.e., different from the catalytic one), we analyzed IDO2 in the human lung adenocarcinoma cell line A549, which basally expresses IDO2 and is widely used as lung carcinoma/infectious model and for drug discovery. In particular, in vitro experiments aimed to investigate the localization of IDO2 protein and its possible interplay with molecular partners to better explain the results observed in NSCLC specimens.
2. Results
2.1. A549 Cells Basally Express IDO2
In previous studies, we found that IDO2 protein was detectable by immunostaining in NSCLC specimens, most of which presented a staining reinforcement at the membrane level [20]. To get more insights into the cellular localization of IDO2 in lung tumor cells, we have used the lung adenocarcinoma human cell line A549 as an in vitro cell model and the A549/hIDO2 transfectant, as a positive control overexpressing the human IDO2 gene. First, IDO2 expression was verified both at the transcript and protein level. After 40 cycles of end-point RT-PCR, the IDO2 product amplified from A549 cDNA was of the same size, but less abundant, compared to that obtained from A549/hIDO2 positive control. The human actin beta (ACTB), amplified by 30 cycles, was used as a housekeeping gene for sample normalization (Figure 1A). By immunoblot analysis, the IDO2 protein was found basally expressed in this primary lung tumor-derived cell line and gave a signal at the same molecular weight detected for the IDO2 protein overexpressed in the A549/hIDO2 transfectant. Immunoblot with anti–β-tubulin provided the loading control (Figure 1B). To investigate whether IDO2 endogenously expressed by A549 cells was capable to degrade Trp, Kyn production was measured in the culture supernatant of either A549 cells or A549/hIDO2 transfectant. In line with the very low Trp catalytic activity already reported for IDO2 [11,12], Kyn concentrations in the supernatants of both the A549 cell line and A549/hIDO2 transfectant were comparable to that recorded in the medium incubated without cells (Figure 1C). Incidentally, A549 cells did not express IDO1 nor TDO2 enzymes (data not shown). Thus, because in these cells IDO2 protein is well detectable at its basal expression level and not active in catabolizing Trp into Kyn, the human lung carcinoma cells A549 were chosen as a suitable model to explore IDO2 cellular localization and possible alternative function, different from the enzymatic one.
2.2. IDO2 Is Localized at the Membrane Level in A549 Cells
Cell localization of IDO2 in A549 cells was investigated by immunostaining of paraffin-embedded cells. The IDO2-specific staining, absent in the control samples incubated without the primary antibody (Figure 2A, upper and lower left panels), was instead visible at the plasma membrane level in the A549 cells (Figure 2A, upper right panel), in agreement with what was reported in NSCLC patients’ tissues (20), and appeared much stronger in the A549/hIDO2 transfectant overexpressing IDO2 (Figure 2A, lower right panel).
To get more insights into the specific cell compartment where IDO2 was localized, we used a sucrose isopycnic gradient analysis to fractionate subcellular organelles from A549 cells. The immunoblot by the anti-IDO2 specific antibody on the obtained different-density cell fractions revealed that IDO2 was completely absent in cytosol low-density (#1-3) and highest-density (#14-17) fractions. In contrast, IDO2 was detectable in medium-density fractions (#9-13), with a very strong signal in fraction #10 (Figure 2B). The re-immunoblotting with an antibody recognizing E-cadherin, a specific epithelial cell adhesion molecule mainly located on the plasma membrane, indicated that IDO2+ and E-cadherin+ fractions were overlapping both showing the stronger signal at fraction #10, thus suggesting a likely cellular colocalization of the two molecules. Therefore, as a whole, these data indicated that, in A549 cells, IDO2 is not confined to the cytosol, but rather localized at the membrane level, a peculiar position suggesting its potential role as a signaling molecule.
2.3. IDO2 Is Tyrosine-Phosphorylated in A549 Cells
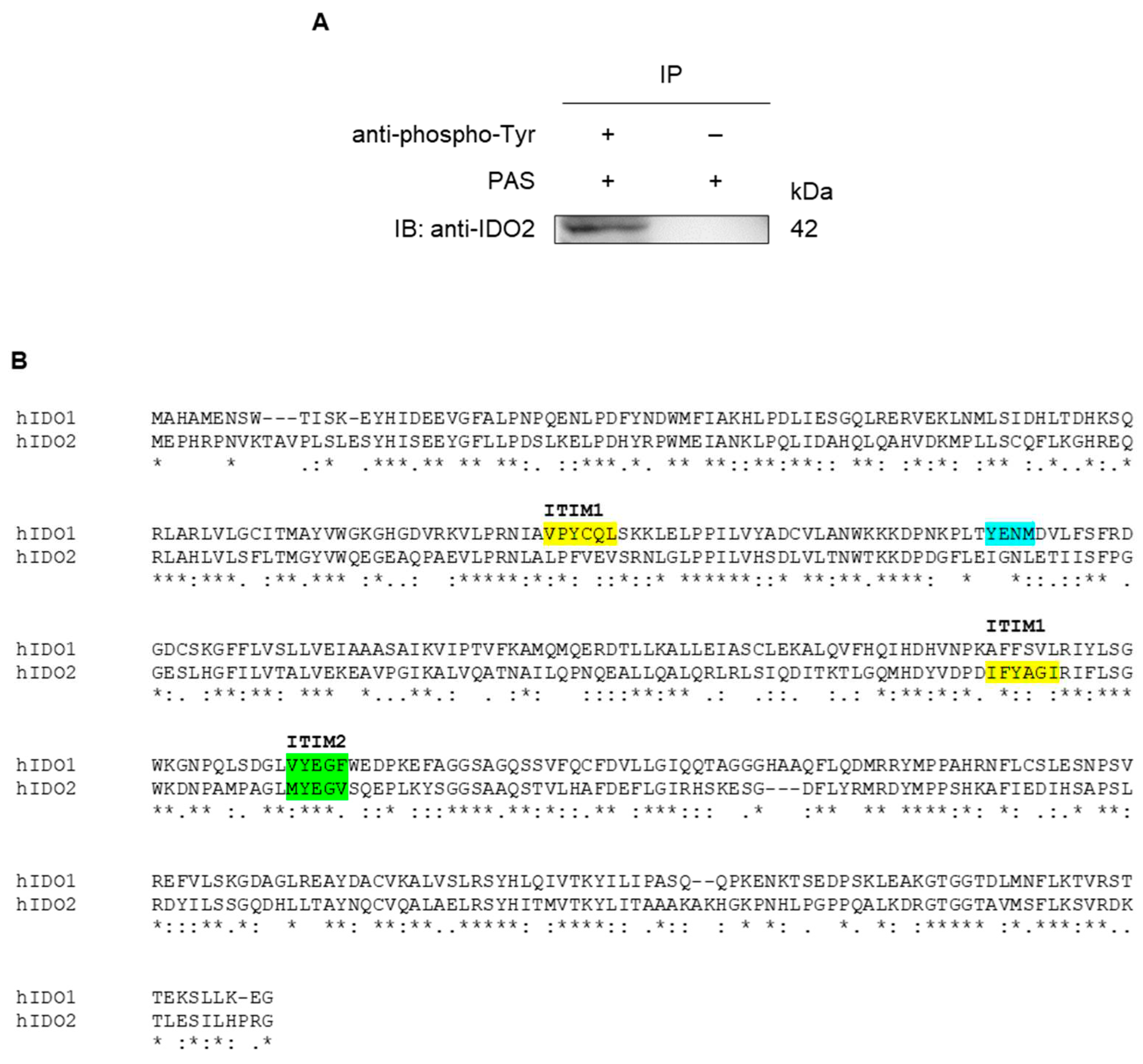
Since the IDO2 polypeptide contains tyrosine phosphorylation consensus sequences that might be possible targets of kinase activities triggering the putative signaling function of membrane-localized IDO2, the tyrosine-phosphorylation of this molecule has been investigated by immunoprecipitation experiments. After immunoprecipitation from A549 whole cell lysate, either in the presence or in the absence of an anti-phosphotyrosine antibody, tyrosine-phosphorylated proteins were immunoblotted with the anti-hIDO2 specific antibody I23O2 (Figure 3A). The analysis revealed that IDO2 could actually exist in a tyrosine-phosphorylated state in A549 cells. Indeed, similarly to IDO1, human IDO2 possesses two ITIM domains: while the so-called ITIM2 shows a high homology sequence and a similar position in both the IDO proteins (VYEGF in human IDO1, MYEGV in human IDO2), the putative IDO2’s ITIM1 motif (IFYAGI) differs from that of IDO1 (VPYCQL); moreover, the YENM motif—useful for the binding of IDO1 to the phosphoinositide 3-kinase (PI3K) [29]—is absent in IDO2 [28] (Figure 3B). The findings of IDO2 membrane localization and tyrosine-phosphorylation support the hypothesis of a prevalent—if not unique—signaling, rather than metabolic, activity of IDO2 in A549 cells.
3. Discussion
Since its discovery in 2007 [4], many efforts have been made to understand the function of IDO2. Although it was evident from the first studies that IDO2 was endowed with a negligible Trp-metabolizing activity compared to IDO1 and TDO2, it has been counted for many years as one of the three enzymes involved in Trp catabolism. This assumption, the high amino acid homology between IDO1 and IDO2, either human and murine, and the absence of a crystal structure of IDO2 prompted the investigators to hypothesize for IDO2 an enzymatic immunoregulatory role, functionally related to its paralog IDO1. Nowadays, though there is much evidence connecting IDO1 and IDO2, similar functions for the two proteins have been not always determined. IDO1 and IDO2 appear to play opposite roles in B-cell mediated immunity, with IDO1 inhibiting and IDO2 driving inflammatory responses of B cells [30]. In particular, in the KRN.g7 mouse model of autoimmune arthritis, IDO1 and IDO2 appear to have contrasting roles in regulating B cell-mediated immune responses. By Ido1 and Ido2 single and double knockout mice, it was clarified that IDO1 is responsible for T cell suppression, while IDO2, directly acting in B cells, mediates the proinflammatory response of autoimmune processes in an IDO1-independent manner [18]. Moreover, while IDO1 expression is described to have a protective value in the multiple sclerosis animal models, IDO2 deletion does not seem to affect the course of the pathology in experimental autoimmune encephalomyelitis [31]. In contrast, in a psoriasis-like dermatitis model, Ido2, but not Ido1, knockout mice were associated with exacerbated symptoms with higher CD4+ and CD8+ T cell infiltration and IL-17 positive cells [19]. In addition, the Ido2 deletion was associated with higher mortality in the LPS-induced endotoxin shock model and correlated to increased production of inflammatory cytokines (including IL-6) in serum [32].
Besides its role in murine models of autoimmune and inflammatory diseases, IDO2 appears to have a prominent role in the onset, development, and prognosis of many types of human cancers [23,28], in striking analogy with IDO1 [15,33,34]. The upregulation of IDO1 in several cancer cell types is well demonstrated, acting as a mediator of tumor immune escape and therefore, often associated with a worse outcome [7,35]. As a result, a variety of compounds targeting IDO1 catalytic activity have been developed and considered as possible drug candidates in cancer immunotherapy [36,37], but with no evident beneficial effect, so far [38]. The failure of IDO1 catalytic inhibitors can be explained by the observation that the signaling activity of IDO1 is retained either in dendritic cells or in tumor cells [39] despite the selective loss of the IDO1’s Trp-metabolizing function. Nowadays it is widely accepted that IDO1 protein is not only endowed with its well-defined catalytic activity, but it can also act as a non-enzymatic signaling platform via its phosphorylation domains, i.e., the ITIM1 and ITIM2 motifs, as well as the YENM sequence [29]. Interestingly, while the Trp-degrading IDO1 enzyme is mostly located in the cytosol, the signaling IDO1 protein is localized to the early endosomes which are active subcellular signaling locations [37]. Although the precise subcellular localization of human IDO2 is still to be defined and the function of its ITIM motifs is still unknown, it could be speculated that IDO2 proximity to the plasma membrane is functional to a signaling activity and ITIMs act as connection points for the recruitment of adaptor proteins containing SH2 domains, as it occurs in IDO1. Indeed, it has been previously demonstrated that SH2-containing tyrosine phosphatases (SHPs) and SOCS3 can bind IDO1’s phosphorylated ITIM1 and ITIM2, respectively [40,41] and that Src kinase is involved in the activation of IDO1 signaling in DCs [40,42,43,44]. If analog mechanisms might operate also in the IDO2 molecule is something to be still completely investigated.
Our previous findings, reported by immunohistochemical analysis of NSCLC specimens, unveiled a membrane reinforcement of the IDO2 staining in the majority of analyzed tumors, half of which presented higher IDO2 expression (i.e., stain reinforcement) on the basolateral side of the tumor cell plasma membrane, at the interface between tumor and stromal tissue and without an apical immunolabel [20]. These findings prompted us to deepen our knowledge about the cell compartment where IDO2 localizes and unveil a possible signaling pathway initiated by the association of IDO2 with putative molecular partners. The human lung adenocarcinoma cell line A549, a well-accepted in vitro lung carcinoma model, allowed us to both reproduce some observations reported by our previous immunohistochemical study on surgical specimens from NSCLC patients and find additional evidence for a possible IDO2 signaling role. IDO2 protein, basally well detectable by end-point RT-PCR and immunoblot experiments, displayed negligible tryptophan activity in A549 cells so that Kyn concentration was similar to that recorded in the medium incubated without cells. The plasma membrane localization of IDO2, already observed in surgical specimens from NSCLC patients, was confirmed by immunostaining of both paraffin-embedded A549 cells and A549/hIDO2 transfectants. For the first time, documented by a sucrose isopycnic gradient analysis, importantly proving the pick overlapping of both IDO2 and plasma membrane protein E-cadherin at fraction #10, thus suggesting that IDO2 could be a peripheral or integral membrane protein. The never previously described evidence of IDO2 tyrosine-phosphorylation in unstimulated A549 cells here corroborated our hypothesis that the IDO2 membrane localization is functional for itself signaling activity.
Based on the here documented IDO2 membrane localization and phosphorylation state in A549 cells, we believe that also IDO2 could possess a still not elucidated signaling activity, explaining its presence and role in several human tumors [23,28]. Intriguingly, even at a low level, we found IDO2 transcript and protein expression in many human cell lines, such as HepG2, HEK293, H1975, H1650, SKOV-3, and Caco2 (data not shown). Considering the observation reported in our previous study documenting the staining of IDO2 in cancer, but not in normal, lung cells [20], this finding suggests a potential relationship between the expression of IDO2 and a possible control loss in the common mechanisms of cell cycle regulation and proliferation. Moreover, the study of IDO2 polymorphism supports the hypothesis of an IDO2 biological function alternative to the catalytic one. In the human IDO2 sequence, a high prevalence of two SNPs has been described. The first is rs10109853, corresponding to a substitution of arginine to tryptophan in position 248 of the human amino acid sequence (R248W), which leads to a further >90% reduction of its catalytic activity. The second is rs4503083, a nonsense mutation that generates a premature stop codon (Y359X) [6]. The absence of enzymatic activity as a consequence of the presence of one or both of the above-mentioned SNPs has been elegantly demonstrated [16,27], thus further suggesting that IDO2 has a still unexplored non-enzymatic function.
4. Materials and Methods
4.1. Cell Culture and End-Point RT-PCR
The human lung adenocarcinoma cell line A549, obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), was cultured according to standard procedures in RPMI-1640 medium, supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS), 2 mM of l-glutamine and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin). Lipofectamine 3000 reagent (Thermo Scientific, Waltham, MA, USA) and a pEF-BOS–based vector containing Ido2 wild-type gene, generated as previously described [40], were used for the transfection of A549 cells according to the manufacturer’s instructions. After puromycin selection (0.5 μg/ml), A549/hIDO2 transfectant was characterized for IDO2 protein expression. Cells were incubated at 37 °C in a 5% CO2 and humidified atmosphere. In the present study, in Figure 1 and Figure 2 the A549/hIDO2 transfectant was used as an IDO2-overexpressing control of the untransfected A549 cell line.
For the End-point RT-PCR, RNA was extracted with TRIzol Reagent (Thermo Scientific, Waltham, MA, USA) and retro-transcribed with QuantiTect® Reverse Transcription kit (QIAGEN, Hilden, Germany). cDNA was amplified using Platinum Taq DNA polymerase (Thermo Scientific, Waltham, MA, USA) by 40 cycles for IDO2, or 30 cycles for the human actin beta (ACTB) housekeeping gene, used for sample normalization. Primers: IDO2 FORW 5’- TGG GAT AAA GGC TCT TGT TC-3’; IDO2 REV 5’-TGG TGA TGT CCT GAA TAG AC-3’; ACTB FORW 5'-CTC GTC GTC GAC AAC GGC T-3'; ACTB REV 5'- TCA GGG TGA GGA TGC CTC TC-3‘.
4.2. Kyn Determination
The enzymatic activity catabolizing Trp into Kyn was measured in vitro in culture supernatants of A549 cells and A549/hIDO2 transfectant (2×105 cells/ml per well in a 12-well plate) after 48 hrs of incubation. Kyn concentration was detected using a Perkin Elmer (Waltham, MA, USA), series 200, HPLC instrument, combined with a Kinetex® C18 column (250×4.6 mm, 5 μm, 100 A; Phenomenex, Torrance, CA, USA), maintained at the temperature of 25 °C and pressure of 1800 PSI. A mobile phase containing 10 mM NaH2PO4 pH 3.0 (99%) and methanol (1%) (Sigma-Aldrich, Merk, Darmstadt, Germany), with a flow rate of 1 ml/min, was used and an UV detector identified Kyn and Trp at 360 nm and 220 nm, respectively. The software TotalChrom v. 6.3.1 was used for evaluating the concentrations of Kyn in samples by means of a calibration curve. The detection limit of the analysis for Kyn was 0.05 μM.
4.3. Western Blotting and Immunoprecipitation
In the immunoblot experiment, the A549 whole-cell lysate was prepared from 2×105 cells and analyzed by SDS-PAGE. As a positive control, the whole cell lysate from A549/hIDO2 transfectant, overexpressing the human IDO2 gene, was also included. IDO2 protein expression was assessed by Western blotting with the rabbit polyclonal anti-hIDO2 antibody I23O2 (Aviva Systems Biology, San Diego, CA, USA) in combination with appropriate horseradish peroxidase-conjugated antibody (Merk Millipore, Burlington, MA, USA), followed by enhanced chemiluminescence (ECL, Bio-Rad, Hercules, CA, USA). As a loading control, β-tubulin was revealed with a specific mouse monoclonal antibody (clone AA2, Merk Sigma-Aldrich, Darmstadt, Germany).
For the immunoprecipitation (IP) of phosphorylated IDO2, 12×106 A549 cells were lysed for 30 min on ice in 400 µl of lysis buffer (20 mM Tris-HCl pH 7.4, 50 mM NaCl, 1% Triton-100) containing cOmplete™ Protease Inhibitor Cocktail (Roche, Merk, Darmstadt, Germany) and phosphatase inhibitors (Thermo Scientific, Waltham, MA, USA). After centrifugation, the supernatant was incubated overnight at 4 °C under rotary agitation with phospho-tyrosine (P-Tyr-1000) MultiMab™ Rabbit mAb mix (Cell Signaling, Danvers, MA, USA) (2 µl of Ab in 400 µl of lysate/sample), or in the absence of it in the negative control sample. After the addition of Protein-A Sepharose beads (Sigma-Aldrich, Merk, Darmstadt, Germany) (30 µl/sample), samples were incubated for 1 h at 4 °C under rotary agitation. Negative control was incubated only with Protein-A Sepharose beads, without previous incubation with P-Tyr-1000 mAb mix. To eliminate possible non-specific interactions, IP samples were centrifuged at 800 rpm for 5 min at 4 °C and washed 3 times with washing buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% Triton-100). IDO2 immunoblot detection was performed with the rabbit polyclonal anti-hIDO2 antibody I23O2 (Aviva Systems Biology, San Diego, CA, USA) as above described in Western blotting analysis.
4.4. Immunocytochemical Analysis
A549 and A549/hIDO2 cells (5 ×106 each) were collected and fixed in 5 ml of 4% formaldehyde (10% formalin, neutral buffered) (Sigma, Merk, Darmstadt, Germany). Subsequently, a cell block was obtained through the Hologic protocol relating to the Cellient™ Automated Cell Block System (Hologic, Marlborough, MA, USA): in detail, the formalin-fixed cell cultures were at first centrifuged to 1727 rpm for 10 min. After supernatant removal, the cell precipitate was transferred into a vial of Preservcyt™ solution (buffered methanol-based solution) of the ThinPrep™ system (Hologic, Marlborough, MA, USA) and processed through the Cellient™ instrumentation for 45 min, concentrating the cells and distributing them in a thin layer into a paraffin cell block. Subsequently, sections of 4 µm were prepared and placed on positively charged slides, for the immunocytochemical staining through the BOND III fully automated immunohistochemistry stainer (Leica Biosystems, Nußloch, Germany). In detail, heat-induced antigen retrieval was obtained by incubating slides for 20 min with the ready-to-use citrate-based pH 6.0 Bond™ Epitope Retrieval Solution 1 (Leica Biosystems, Newcastle Upon Tyne, United Kingdom). Subsequent incubations were performed with the polyclonal anti-hIDO2 primary antibody I23O2 for 15 min (dilution 1: 500, Aviva Systems Biology, San Diego, CA, USA) and the ready-to-use Bond™ Polymer Refine Detection System (Leica Biosystems, Newcastle Upon Tyne, United Kingdom). This reagent includes a peroxide block, the secondary antibody (rabbit anti-mouse IgG), the anti-rabbit Poly–HRP-IgG to localize the post-primary rabbit antibodies, the substrate chromogen 3,3’-diaminobenzidine tetrahydrochloride hydrate (DAB) to visualize the complex by a brown precipitate, and the hematoxylin for nuclear counterstaining. An appropriate negative control was included during the test, using the same immunostaining protocol, but without the primary antibody incubation.
4.5. Immunocytochemical Analysis
For subcellular fractionation, the isopycnic sucrose gradient was performed as previously described [29]. Briefly, 5×106 A549 cells were resuspended in 1 ml of Sucrose Buffer (12% sucrose, 10 mM KCl, 2 mM MgCl2, and 100 mM Tris-HCl pH 7.2) and homogenized. Then, the cell homogenate was loaded at the top of a continuous sucrose gradient (16-55% w/w) and after ultracentrifugation at 141.000×g for 4 hrs at 4 °C, seventeen fractions were recovered. From each fraction, proteins were precipitated by TCA and resuspended in 50 µl of 1X Loading Dye. Samples were analyzed by SDS-PAGE and immunoblot for the presence of IDO2 (I23O2 rabbit polyclonal antibody, Aviva Systems Biology, San Diego, CA, USA) and E-cadherin (rabbit mAb clone SP64, Abcam, Cambridge, UK) in combination with appropriate horseradish peroxidase-conjugated antibody (Merk Millipore, Burlington, MA, USA), and followed by enhanced chemiluminescence (ECL, Bio-Rad, Hercules, CA, USA).
4.6. Statistical Analysis
For statistical analysis, GraphPad Prism 8.0.1 software for Windows (GraphPad) was used. Data are expressed as the mean ± S.D. and statistical significance was determined by the ANOVA one-way analysis. A p-value ≤ 0.05 was considered statistically significant.
5. Conclusions
In conclusion, we have demonstrated that the human lung adenocarcinoma cell line A549 is a suitable in vitro model to study IDO2 cellular localization and still not completely elucidated function, different from the catalytic one. The evidence that IDO2 protein, basally expressed in A549 cells, localizes at the plasma membrane level and is tyrosine-phosphorylated suggests a possible role for this protein as a signaling molecule in tumor cells. Further studies on kinases targeting IDO2 and molecular adaptors mediating the putative IDO2 signaling activity are worthy of being carried forward, aiming to define IDO2 as a suitable drug target in cancer therapy.
Author Contributions
CS: End-point RT-PCR, immunoblot and immunoprecipitation analysis, conceptualization, and original draft preparation; FDM: isopycnic gradient experiments; MM: immunocytochemical analysis; SA: HPLC analysis; SR and EP: sample processing support; GM, MG, and MTP: provided support for result analysis and revised final version of the manuscript; CO: optimized HPLC analysis, revised final version of the manuscript, obtained funding; MLB: analyzed results, drafted and edited the manuscript; CV: conceived and supervised the study, drafted the manuscript, obtained funding. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondazione AIRC per la Ricerca sul Cancro (IG #23084 to CV), Italian Ministry of Education, University, and Research (PRIN2017-20173EAZ2Z to CV) and Italian Ministry of Education, University, and Research (PRIN2020-2020L45ZW4 to CO).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author without undue reservation.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Fiore, A.; Murray, P.J. Tryptophan and indole metabolism in immune regulation. Current opinion in immunology 2021, 70, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, U.; Mondanelli, G.; Belladonna, M.L.; Orabona, C.; Pallotta, M.T.; Iacono, A.; Puccetti, P.; Volpi, C. Amino-acid sensing and degrading pathways in immune regulation. Cytokine & growth factor reviews 2017, 35, 37–45. [Google Scholar]
- Gargaro, M.; Scalisi, G.; Manni, G.; Briseño, C.G.; Bagadia, P.; Durai, V.; Theisen, D.J.; Kim, S.; Castelli, M.; Xu, C.A.; et al. Indoleamine 2,3-dioxygenase 1 activation in mature cDC1 promotes tolerogenic education of inflammatory cDC2 via metabolic communication. Immunity 2022, 55, 1032–1050.e14. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.J.; Sanchez-Perez, A.; Weiser, S.; Austin, C.J.; Astelbauer, F.; Miu, J.; McQuillan, J.A.; Stocker, R.; Jermiin, L.S.; Hunt, N.H. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 2007, 396, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.J.; Takubo, M.; Takahashi, A.; Hasegawa, T.; Noma, H.; Suzuki, T. Evolution of vertebrate indoleamine 2,3-dioxygenases. Journal of molecular evolution 2007, 65, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Metz, R.; Duhadaway, J.B.; Kamasani, U.; Laury-Kleintop, L.; Muller, A.J.; Prendergast, G.C. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer research 2007, 67, 7082–7087. [Google Scholar] [CrossRef]
- Théate, I.; van Baren, N.; Pilotte, L.; Moulin, P.; Larrieu, P.; Renauld, J.C.; Hervé, C.; Gutierrez-Roelens, I.; Marbaix, E.; Sempoux, C.; et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer immunology research 2015, 3, 161–172. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Metz, R.; Muller, A.J.; Merlo, L.M.; Mandik-Nayak, L. IDO2 in Immunomodulation and Autoimmune Disease. Frontiers in immunology 2014, 5, 585. [Google Scholar] [CrossRef]
- Jusof, F.F.; Bakmiwewa, S.M.; Weiser, S.; Too, L.K.; Metz, R.; Prendergast, G.C.; Fraser, S.T.; Hunt, N.H.; Ball, H.J. Investigation of the tissue distribution and physiological roles of indoleamine 2, 3-dioxygenase-2. International Journal of Tryptophan Research 2017, 10, 1178646917735098. [Google Scholar] [CrossRef]
- Fukunaga, M.; Yamamoto, Y.; Kawasoe, M.; Arioka, Y.; Murakami, Y.; Hoshi, M.; Saito, K. Studies on Tissue and Cellular Distribution of Indoleamine 2,3-Dioxygenase 2: The Absence of IDO1 Upregulates IDO2 Expression in the Epididymis. Journal of Histochemistry & Cytochemistry 2012, 60, 854–860. [Google Scholar]
- Pantouris, G.; Serys, M.; Yuasa, H.J.; Ball, H.J.; Mowat, C.G. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids 2014, 46, 2155–2163. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Yang, D.; Hu, N.; Guo, Z.; Kuang, C.; Yang, Q. Establishment of a human indoleamine 2, 3-dioxygenase 2 (hIDO2) bioassay system and discovery of tryptanthrin derivatives as potent hIDO2 inhibitors. European journal of medicinal chemistry 2016, 123, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.J.; Stocker, R. Methylene blue and ascorbate interfere with the accurate determination of the kinetic properties of IDO2. The FEBS journal 2021, 288, 4892–4904. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.J.; Ball, H.J.; Ho, Y.F.; Austin, C.J.D.; Whittington, C.M.; Belov, K.; Maghzal, G.J.; Jermiin, L.S.; Hunt, N.H. Characterization and evolution of vertebrate indoleamine 2, 3-dioxygenases: IDOs from monotremes and marsupials. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 2009, 153, 137–144. [Google Scholar] [CrossRef] [PubMed]
- van Baren, N.; Van den Eynde, B.J. Tryptophan-degrading enzymes in tumoral immune resistance. Frontiers in immunology 2015, 6, 34. [Google Scholar] [CrossRef]
- Metz, R.; Smith, C.; DuHadaway, J.B.; Chandler, P.; Baban, B.; Merlo, L.M.; Pigott, E.; Keough, M.P.; Rust, S.; Mellor, A.L.; et al. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. International immunology 2014, 26, 357–367. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Pigott, E.; DuHadaway, J.B.; Grabler, S.; Metz, R.; Prendergast, G.C.; Mandik-Nayak, L. IDO2 is a critical mediator of autoantibody production and inflammatory pathogenesis in a mouse model of autoimmune arthritis. Journal of immunology (Baltimore, Md. : 1950) 2014, 192, 2082–2090. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; DuHadaway, J.B.; Montgomery, J.D.; Peng, W.D.; Murray, P.J.; Prendergast, G.C.; Caton, A.J.; Muller, A.J.; Mandik-Nayak, L. Differential Roles of IDO1 and IDO2 in T and B Cell Inflammatory Immune Responses. Frontiers in immunology 2020, 11, 1861. [Google Scholar] [CrossRef]
- Fujii, K.; Yamamoto, Y.; Mizutani, Y.; Saito, K.; Seishima, M. Indoleamine 2,3-Dioxygenase 2 Deficiency Exacerbates Imiquimod-Induced Psoriasis-Like Skin Inflammation. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Mandarano, M.; Bellezza, G.; Belladonna, M.L.; Vannucci, J.; Gili, A.; Ferri, I.; Lupi, C.; Ludovini, V.; Falabella, G.; Metro, G.; et al. Indoleamine 2,3-Dioxygenase 2 Immunohistochemical Expression in Resected Human Non-small Cell Lung Cancer: A Potential New Prognostic Tool. Frontiers in immunology 2020, 11, 839. [Google Scholar] [CrossRef]
- Nevler, A.; Muller, A.J.; Sutanto-Ward, E.; DuHadaway, J.B.; Nagatomo, K.; Londin, E.; O'Hayer, K.; Cozzitorto, J.A.; Lavu, H.; Yeo, T.P.; et al. Host IDO2 Gene Status Influences Tumor Progression and Radiotherapy Response in KRAS-Driven Sporadic Pancreatic Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 2019, 25, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Costantino, C.L.; Metz, R.; Muller, A.J.; Prendergast, G.C.; Yeo, C.J.; Brody, J.R. Genotyping and expression analysis of IDO2 in human pancreatic cancer: a novel, active target. Journal of the American College of Surgeons 2009, 208, 781–787; discussion 787-9. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xu, W.; Liu, F.; Zhu, H.; Zhang, L.; Ding, Z.; Liang, H.; Song, J. The emerging roles of IDO2 in cancer and its potential as a therapeutic target. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2021, 137, 111295. [Google Scholar]
- Löb, S.; Königsrainer, A.; Zieker, D.; Brücher, B.L.; Rammensee, H.G.; Opelz, G.; Terness, P. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer immunology, immunotherapy CII 2009, 58, 153–157. [Google Scholar] [CrossRef]
- Gu, P.; Ling, B.; Ma, W.; Zhang, J.; Zhang, W.; Zeng, Y.; Liu, Y.; Chi, J.; Ruan, X.; Zheng, X.; et al. Indoleamine 2,3-dioxygenase 2 immunohistochemical expression in medullary thyroid carcinoma: implications in prognosis and immunomodulatory effects. BMC cancer 2022, 22, 1116. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.; Zheng, X.; Zhang, X.; Wang, H.; Li, Q.; Yuan, K.; Zhou, N.; Yu, Y.; Song, N.; et al. Gene silencing of indoleamine 2,3-dioxygenase 2 in melanoma cells induces apoptosis through the suppression of NAD+ and inhibits in vivo tumor growth. Oncotarget 2016, 7, 32329–32340. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Peng, W.; DuHadaway, J.B.; Montgomery, J.D.; Prendergast, G.C.; Muller, A.J.; Mandik-Nayak, L. The Immunomodulatory Enzyme IDO2 Mediates Autoimmune Arthritis through a Nonenzymatic Mechanism. Journal of immunology (Baltimore, Md. : 1950) 2022, 208, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Mandarano, M.; Belladonna, M.L.; Suvieri, C.; Pelliccia, C.; Bellezza, G.; Sidoni, A.; Carvalho, A.; Grohmann, U.; Volpi, C. Current Challenges for IDO2 as Target in Cancer Immunotherapy. Frontiers in immunology 2021, 12, 679953. [Google Scholar] [CrossRef]
- Iacono, A.; Pompa, A.; De Marchis, F.; Panfili, E.; Greco, F.A.; Coletti, A.; Orabona, C.; Volpi, C.; Belladonna, M.L.; Mondanelli, G.; et al. Class IA PI3Ks regulate subcellular and functional dynamics of IDO1. EMBO reports 2020, 21, e49756. [Google Scholar] [CrossRef]
- Merlo, L.M.F.; Peng, W.; Mandik-Nayak, L. Impact of IDO1 and IDO2 on the B Cell Immune Response. Frontiers in immunology 2022, 13, 886225. [Google Scholar] [CrossRef]
- Wetzel, L.A.; Hurtado, M.; MacDowell Kaswan, Z.A.; McCusker, R.H.; Steelman, A.J. Deletion of indoleamine 2,3 dioxygenase (Ido)1 but not Ido2 exacerbates disease symptoms of MOG35-55-induced experimental autoimmune encephalomyelitis. Brain, Behavior, & Immunity - Health 2020, 7, 100116. [Google Scholar]
- Yamamoto, Y.; Yamasuge, W.; Imai, S.; Kunisawa, K.; Hoshi, M.; Fujigaki, H.; Mouri, A.; Nabeshima, T.; Saito, K. Lipopolysaccharide shock reveals the immune function of indoleamine 2,3-dioxygenase 2 through the regulation of IL-6/stat3 signalling. Scientific Reports 2018, 8, 15917. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nature medicine 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Kato, S.; Nesline, M.K.; Conroy, J.M.; DePietro, P.; Pabla, S.; Kurzrock, R. Indoleamine 2,3-dioxygenase (IDO) inhibitors and cancer immunotherapy. Cancer treatment reviews 2022, 110, 102461. [Google Scholar] [CrossRef]
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Frontiers in immunology 2020, 11, 531491. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. International review of cell and molecular biology 2018, 336, 175–203. [Google Scholar]
- Pallotta, M.T.; Rossini, S.; Suvieri, C.; Coletti, A.; Orabona, C.; Macchiarulo, A.; Volpi, C.; Grohmann, U. Indoleamine 2,3-dioxygenase 1 (IDO1): an up-to-date overview of an eclectic immunoregulatory enzyme. The FEBS journal 2022, 289, 6099–6118. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Yang, L.Q.; Shi, Z.H.; Li, X.M.; Qiu, H.Y. An updated patent review of IDO1 inhibitors for cancer (2018-2022). Expert opinion on therapeutic patents 2022, 32, 1145–1159. [Google Scholar] [CrossRef]
- Orecchini, E.; Belladonna, M.L.; Pallotta, M.T.; Volpi, C.; Zizi, L.; Panfili, E.; Gargaro, M.; Fallarino, F.; Rossini, S.; Suvieri, C.; et al. The signaling function of IDO1 incites the malignant progression of mouse B16 melanoma. Oncoimmunology 2023, 12, 2170095. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nature immunology 2011, 12, 870–878. [Google Scholar] [CrossRef]
- Albini, E.; Rosini, V.; Gargaro, M.; Mondanelli, G.; Belladonna, M.L.; Pallotta, M.T.; Volpi, C.; Fallarino, F.; Macchiarulo, A.; Antognelli, C.; et al. Distinct roles of immunoreceptor tyrosine-based motifs in immunosuppressive indoleamine 2,3-dioxygenase 1. Journal of cellular and molecular medicine 2017, 21, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Volpi, C.; Mondanelli, G.; Pallotta, M.T.; Vacca, C.; Iacono, A.; Gargaro, M.; Albini, E.; Bianchi, R.; Belladonna, M.L.; Celanire, S.; et al. Allosteric modulation of metabotropic glutamate receptor 4 activates IDO1-dependent, immunoregulatory signaling in dendritic cells. Neuropharmacology 2016, 102, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
IDO2 transcript and protein are basally detectable in A549 cells and associated with negligible Kyn production. IDO2 expression and catalytic activity were investigated in the A549 cell line and A549/hIDO2 transfectant, used as a positive control overexpressing the human IDO2 gene. (A) IDO2 expression was analyzed by end-point RT-PCR (40 cycles). The human actin beta (ACTB) was the housekeeping gene for sample normalization (30 cycles). NC, negative control (i.e., without cDNA). One experiment representative of three. (B) In A549 and A549/hIDO2 whole-cell lysates, IDO2 protein expression was assessed by Western blotting with the rabbit polyclonal anti-hIDO2 antibody I23O2. Anti–β-tubulin immunoblot served as a loading control. One experiment representative of three. (C) The catalytic activity degrading Trp was measured by HPLC as Kyn released in the culture supernatant by either A549 cells or A549/hIDO2 transfectant. Data (mean ± SD) are the results of three independent measurements. The culture medium incubated without cells is also shown. One-way ANOVA was used for the analysis. ns, not significant.
Figure 1.
IDO2 transcript and protein are basally detectable in A549 cells and associated with negligible Kyn production. IDO2 expression and catalytic activity were investigated in the A549 cell line and A549/hIDO2 transfectant, used as a positive control overexpressing the human IDO2 gene. (A) IDO2 expression was analyzed by end-point RT-PCR (40 cycles). The human actin beta (ACTB) was the housekeeping gene for sample normalization (30 cycles). NC, negative control (i.e., without cDNA). One experiment representative of three. (B) In A549 and A549/hIDO2 whole-cell lysates, IDO2 protein expression was assessed by Western blotting with the rabbit polyclonal anti-hIDO2 antibody I23O2. Anti–β-tubulin immunoblot served as a loading control. One experiment representative of three. (C) The catalytic activity degrading Trp was measured by HPLC as Kyn released in the culture supernatant by either A549 cells or A549/hIDO2 transfectant. Data (mean ± SD) are the results of three independent measurements. The culture medium incubated without cells is also shown. One-way ANOVA was used for the analysis. ns, not significant.
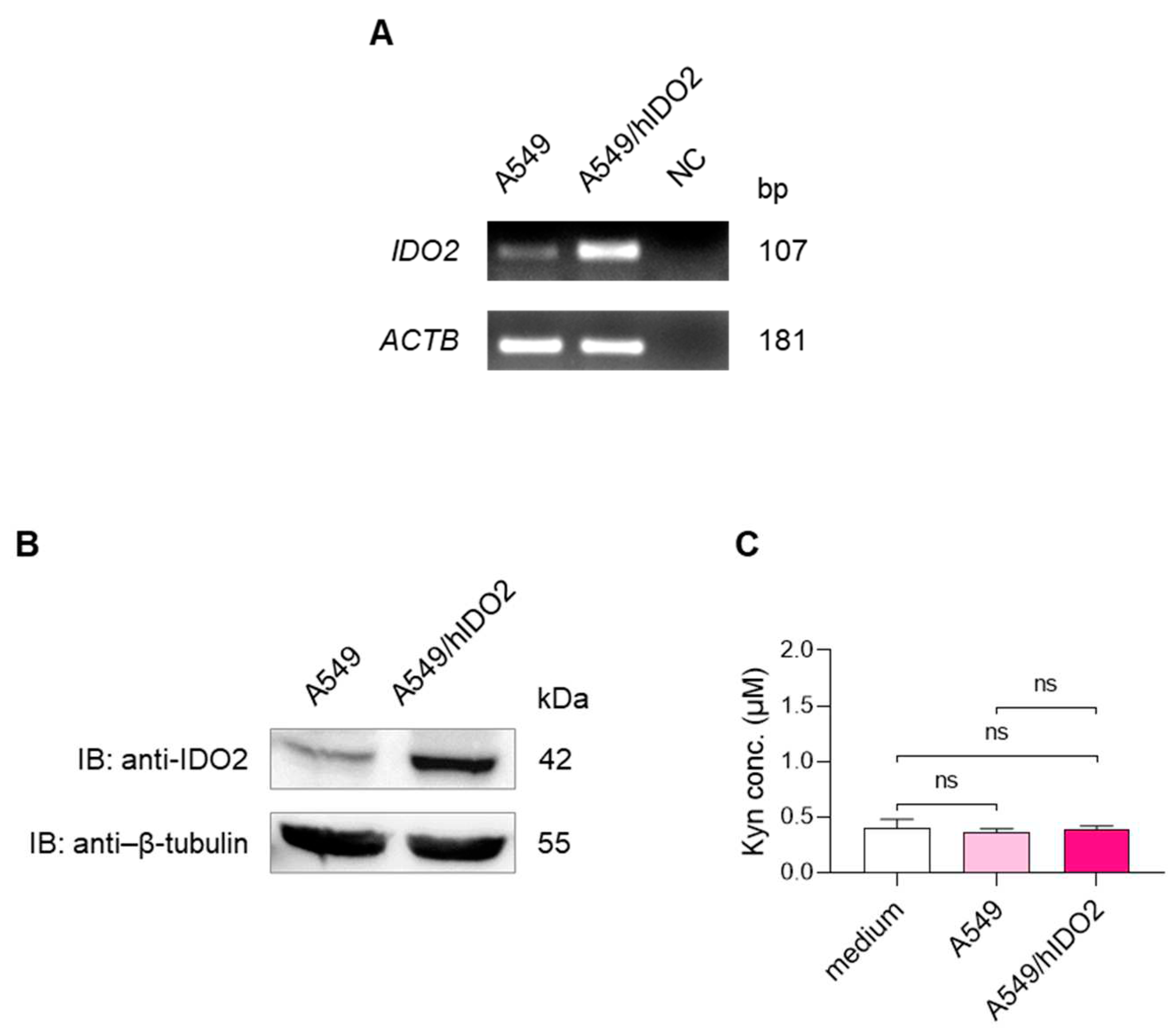
Figure 2.
Membrane localization of IDO2 protein in A549 cells. (A) Immunocytochemical analysis of IDO2 in paraffin-embedded A549 cells and A549/hIDO2 transfectant. Representative images of membranous staining in sections incubated in the presence of I23O2 anti-IDO2 antibody (upper and lower right panels), or in the absence of it (ctrl, negative control) (upper and lower left panels). A549/hIDO2 transfectant was included as a positive control overexpressing the human IDO2 gene. Original magnification 400X; scale bar: 20 μm. (B) Analysis of IDO2 cellular localization by isopycnic sucrose gradient of A549 fractions prepared from whole cell lysate. Fractions were immunoblotted with anti-IDO2 I23O2 and anti–E-cadherin as a marker of the plasma membrane compartment.
Figure 2.
Membrane localization of IDO2 protein in A549 cells. (A) Immunocytochemical analysis of IDO2 in paraffin-embedded A549 cells and A549/hIDO2 transfectant. Representative images of membranous staining in sections incubated in the presence of I23O2 anti-IDO2 antibody (upper and lower right panels), or in the absence of it (ctrl, negative control) (upper and lower left panels). A549/hIDO2 transfectant was included as a positive control overexpressing the human IDO2 gene. Original magnification 400X; scale bar: 20 μm. (B) Analysis of IDO2 cellular localization by isopycnic sucrose gradient of A549 fractions prepared from whole cell lysate. Fractions were immunoblotted with anti-IDO2 I23O2 and anti–E-cadherin as a marker of the plasma membrane compartment.
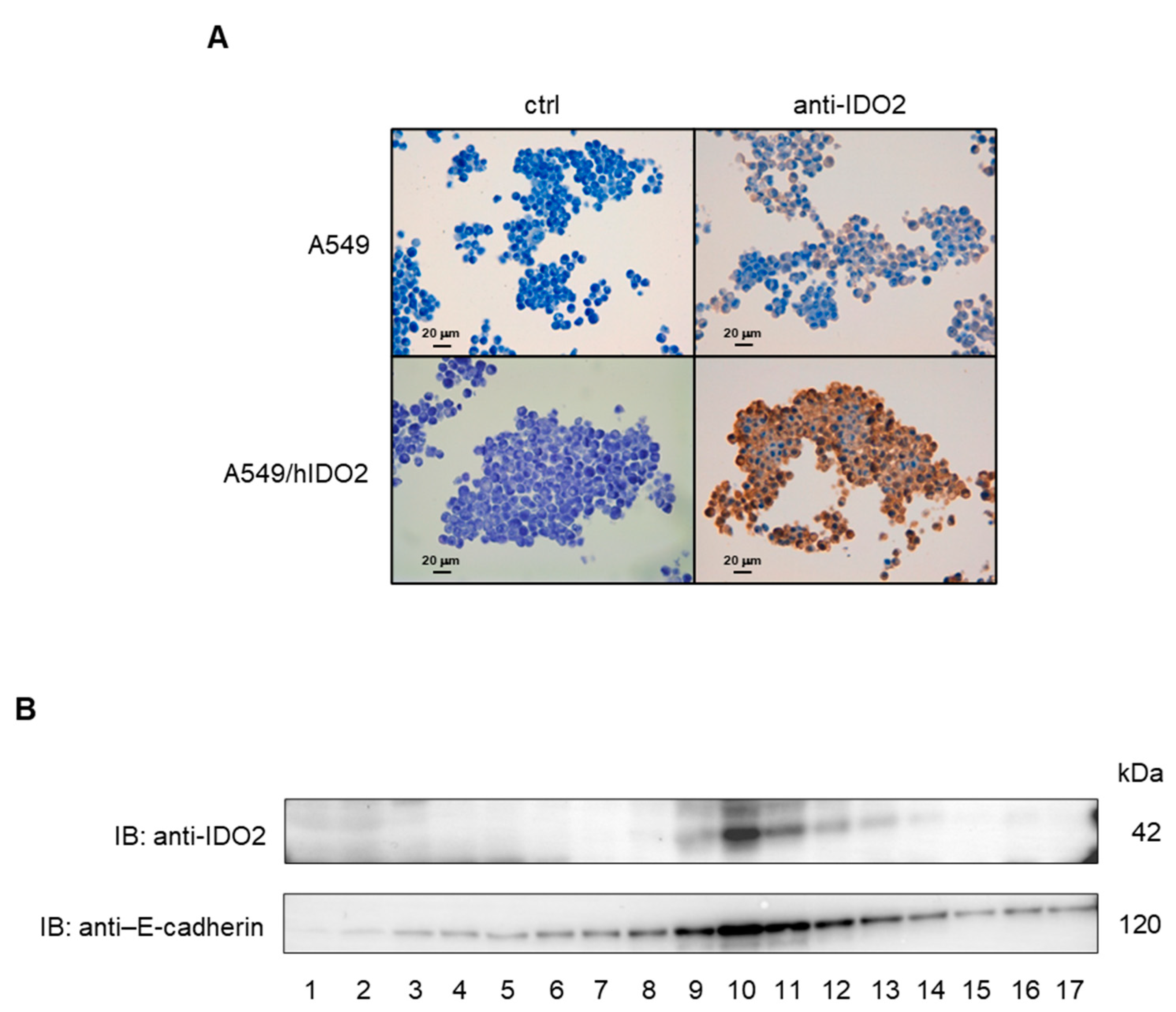
Figure 3.
Basal phosphorylation of IDO2 tyrosine residues in A549 cells. (A) Whole lysate from 12×106 untreated A549 cells was immunoprecipitated by anti–phospho-tyrosine mAb mix (2 µl in 400 µl of cell lysate, overnight at 4 °C) and 30 µl of Protein-A Sepharose beads (PAS) (1 h at 4 °C under rotary agitation). Negative control was prepared by incubation of the whole cell lysate only with PAS, avoiding pre-incubation with the anti–phospho-tyrosine mAb mix. Washed immunoprecipitated samples were run on SDS-PAGE and analyzed by Western blotting. Tyrosine-phosphorylated IDO2 detection was achieved by the specific rabbit polyclonal anti-hIDO2 antibody I23O2. One experiment representative of three. (B) Human IDO1 and IDO2 amino acid sequences, containing putative phosphorylation sites. ITIM1 (yellow) and ITIM2 (green) motifs, are indicated in both IDO1 (accession number: NP_002155.1) and IDO2 (accession number: NP_919270.3) sequences aligned by T-coffee (https://tcoffee.crg.eu/apps/tcoffee/index.html). Phosphoinositide 3-kinase (PI3K) binding domain (YENM, cyan) is indicated in IDO1 protein.
Figure 3.
Basal phosphorylation of IDO2 tyrosine residues in A549 cells. (A) Whole lysate from 12×106 untreated A549 cells was immunoprecipitated by anti–phospho-tyrosine mAb mix (2 µl in 400 µl of cell lysate, overnight at 4 °C) and 30 µl of Protein-A Sepharose beads (PAS) (1 h at 4 °C under rotary agitation). Negative control was prepared by incubation of the whole cell lysate only with PAS, avoiding pre-incubation with the anti–phospho-tyrosine mAb mix. Washed immunoprecipitated samples were run on SDS-PAGE and analyzed by Western blotting. Tyrosine-phosphorylated IDO2 detection was achieved by the specific rabbit polyclonal anti-hIDO2 antibody I23O2. One experiment representative of three. (B) Human IDO1 and IDO2 amino acid sequences, containing putative phosphorylation sites. ITIM1 (yellow) and ITIM2 (green) motifs, are indicated in both IDO1 (accession number: NP_002155.1) and IDO2 (accession number: NP_919270.3) sequences aligned by T-coffee (https://tcoffee.crg.eu/apps/tcoffee/index.html). Phosphoinositide 3-kinase (PI3K) binding domain (YENM, cyan) is indicated in IDO1 protein.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



