
Submitted:
14 September 2023
Posted:
20 September 2023
You are already at the latest version
Abstract
Chronic myeloid leukemia (CML) is the most common form of leukemia in adults and accounts for approximately 15% of all leukemia cases. The etiology of this disease is based on the presence of the oncogenic Bcr/Abl fusion protein, which has dysregulated tyrosine kinase activity. This pathological condition is the result of a chromosomal translocation between chromosomes 9 and 22 leading to the formation of the Philadelphia chromosome (Ph). Clinical establishment of targeted therapy with Abl kinase inhibitors (AKIs) has been observed in the treatment of CML. Therapy resistance in patients with CML is due to genetic alterations in the cells of CML, specifically affecting the Bcr/Abl oncoprotein. Acquired resistance of CML patients to tyrosine kinase inhibitors (AKIs) has also been associated with the tumor microenvironment (TME). This study aimed to examine the role of hypoxic conditions in the development of chemoresistance to certain AKIs in CML. The findings of our study indicate that exposure to hypoxic conditions leads to a substantial increase in chemoresistance to crizotinib, while only resulting in a slight increase in chemoresistance to imatinib. It is worth noting that both drugs effectively block Bcr/Abl function. Furthermore, it was observed that the inclusion of the JAK1/2 inhibitor ruxolitinib resulted in an augmentation of chemoresistance to crizotinib in the context of hypoxia. Overexpression of hypoxia-inducible factor 1a (HIF1a) and silencing of JAK2 confirmed that the two proteins cooperate in mediating chemoresistance to crizotinib in CML cell lines. It is noteworthy that the compound 2-methoxy estradiol (2ME2), which is a metabolite of estradiol lacking estrogenic activity, has shown efficacy in reinstating the sensitivity of CML cells to crizotinib in the presence of hypoxic circumstances. Additionally, when used in conjunction with a JAK2 inhibitor, 2ME2 exhibited activity in restoring crizotinib sensitivity in CML cells. The findings of our study underscore the significance of selectively targeting elements of the HIF1α pathway in order to achieve total eradication of chronic myeloid leukemia (CML) cells.
Keywords:
Hypoxia
; CML
; Bcr/Abl
; chemoresistance
; Imatinib
; Crizotinib
; 2-methoxyestradiol (2ME2)
Introduction
Ph+ leukemia is characterized by the presence of the chromosomal translocation t(9;22) leading to the formation of the BCR/ABL oncogene. Increased proliferation, decreased sensitivity to various pro-apoptotic stimuli, and neoplastic transformation are the consequences of constitutive activation of Bcr/Abl [1].
Ph+ leukemia is treated with Abl kinase inhibitors (AKIs). While the treatment is initially beneficial initially [2,3,4], its clinical efficacy steadily declines with disease progression. Patients with chronic myeloid leukemia in blast crisis (BC) (CML) or acute lymphoblastic leukemia (Ph+) very rarely, if ever, benefit from AKI therapy [5].
Acquired drug resistance is the most important factor for treatment failure in cancer. Drug resistance in Ph+ leukemia may be BCR/ABL-dependent or independent. The tumor microenvironment is also thought to promote drug resistance in Ph+ leukemia. The bone marrow microenvironment (BM) is also critical for long-term hematopoiesis and stem cell regulation [6,7,8]. Conditions in the BM niche contribute to decreased drug sensitivity of cancer cells, including soluble growth factors, interleukins, stromal cells, and extracellular components [6,9,10]. The interactions between leukemia cells and the microenvironment promote leukemia cell survival and confer resistance to chemotherapy. The tumor microenvironment is characterized by regions of hypoxia and acidity, all of which may influence tumor cell sensitivity to drug treatment. In addition, the hypoxic microenvironment is associated with malignant invasion, metastasis, and epithelial-mesenchymal transition (EMT). The bone marrow microenvironment is a hypoxic niche in which normal and malignant hematopoietic stem cells, including CML. In addition, increased levels of hypoxia have been associated with disease progression and residual disease [11] and chemoresistance to therapy in several cancers [12,13].
Hypoxia-inducible factor 1α (HIF1 α), an important regulator of the cellular response to hypoxia, regulates cell growth and metabolic adaptation to hypoxia. During hypoxia, HIF is stabilized and acts as a suppressor of apoptosis by regulating the expression of many genes directly or indirectly involved in mediating chemoresistance, such as the Bak, Bax, Bcl-xL, Bcl-2, Bid, Mcl-1, NF-κB, p53, MDR, and survivin genes, which have been shown to be regulated by HIF1 [14,15,16,17,18,19,20,21]. Interestingly, the molecular nature of this phenomenon has been largely explained by HIF1 induction of anti-apoptotic target genes as well as additional molecular mechanisms that remain largely unknown [22]. Emerging data also illustrate the role of the microenvironment in mediating drug resistance to several Abl kinase inhibitors, including imatinib, nilotinib, and dasatinib, in mediating cell death [23,24,25,26], and in residual disease in Ph+ leukemia [27].
In Ph+ leukemia, apoptosis was partially suppressed under hypoxic conditions despite the activity of imatinib in inhibiting BCR/ABL kinase [28].
Hypoxia may indirectly affect chemoresistance by triggering the secretion of soluble factors by cells in the BM microenvironment such as SDF-1, VEGF, and IL -6, which are known to promote HSC maintenance [29,30]. Overcoming resistance to chemotherapy mediated by the BM microenvironment is therefore a significant therapeutic challenge in leukemia.
In this study, we show that hypoxic conditions confer significant chemoresistance to crizotinib but only minimally to imatinib, although both drugs can inhibit Bcr/Abl activity. In addition, the presence of a JAK2 inhibitor enhanced chemoresistance to crizotinib under hypoxic conditions. Overexpression of HIF1α or silencing of JAK2 in CML cells confirmed cross-talk between the two proteins in mediating chemoresistance to crizotinib in CML cell lines. Interestingly, 2ME2 was active in restoring crizotinib sensitivity of CML cells under hypoxic conditions and combination with a JAK2 inhibitor.
Our study shows that HIF1 signaling supports crizotinib chemoresistance in CML, which is independent of Bcr/Abl kinase activity. Therefore, targeting HIF1α and its signaling pathway components may be therapeutically important for complete eradication of CML.
Materials and Methods
Chemicals and reagents: Most chemicals, including cobalt chloride, were purchased from Sigma Aldrich Israel Ltd. (Rehovot, Israel); otherwise, the supplier is indicated. 2ME2 was purchased from Cayman Chemical (Ann Arbor, Michigan, USA). Imatinib, ruxolitinib, everolimus (Afinitor), and crizotinib were purchased from Selleck Chemicals LLC (Houston, TX, USA).
Cell lines: Human CML cell lines K562 and BV173 were from the American Type Culture Collection [ATCC] (VA, USA). Cells were cultured in RPMI 1640 complete medium supplemented with 10% (w/v) fetal bovine serum (Biological Industries, Israel), 1% (w/v) L-glutamine, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. All cell lines were grown at 37 °C in a humidified atmosphere containing 5% CO2.
Ectopic expression of HA-HIF1 gene: Overexpression of HIF1a in K562 was performed as described [31]. Briefly, K562 cells were infected with a retrovirus carrying HA-HIF1α P402A/P564A (Addgene plasmid #19005) [32]. Infected cells were selected for puromycin resistance and checked for the presence of HIF1α by immunoblotting with anti-HIF1α antibodies.
Silencing of JAK2 in K562 cells: Silencing of JAK2 in K562 cells was performed as previously described (31). Briefly, multiple plasmids: gRNA JAK2 (Addgene plasmid #75728), lenti-cas9blast (Addgene plasmid # 52962 ), and lenti packaging plasmids (pcMV-dR8.2 dvpr and pcMV-VSV-G (Addgene plasmid #8455 and #8454) [33] were co-transfected into HEK239T cells at 1.5 × 105 cells/mL with Fugene 6 (Roche Applied Science, Penzberg, Germany) according to the manufacturer’s instructions. The supernatant of infected cells was collected 48 hours after transfection and used to infect the selected K562 cell lines, and puromycin/blasticidin-resistant clones were selected. JAK2 protein levels were monitored by immunoblotting.
Trypan blue exclusion assay: Cells (2 × 105/well) were plated in plates with six wells. After 24 hours, cells were treated with the indicated agents. Solvent-treated samples were incubated with 1% (w/v) dimethyl sulfoxide (DMSO). Cells were harvested 72 hours later, stained with 0.4% (w/v) trypan blue solution (1:1, v/v), and counted with a hemocytometer [34].
Immunoblotting: Immunoblotting was performed as previously described [35,36]; in brief, protein analysis was performed by Western blot on an 8–12% acrylamide gel. Cell lysates were prepared for loading by adding lysis buffer (#9803 Cell Signaling Technology, MA, USA) containing protease inhibitors (P8340 and P5726, Sigma, Germany) and phosphatase inhibitor (P-1517, AG Scientific, CA, USA) to the cell pellets. After 30 minutes, samples were centrifuged and the supernatant was assayed for protein concentration using the DC™ Protein Assay (Bio Rad, USA) and determining absorbance was determined at 630 nm. Samples were lysed in lysis buffer and 50–60 μg protein per monoculture sample was loaded onto the gel. Proteins were immunoblotted onto a nitrocellulose membrane (Schleicher & Schuell BioScience GmbH, Germany), which was then blocked with 5% skim milk TBS/T and incubated with the following antibodies: Phospho-Abl (Tyr 245) (cat. 2861) and anti-cleaved PARP (cat. 5625) from Cell Signaling Technology, MA, USA. anti-α-tubulin (sc-8035, Santa Cruz, TX, USA) and anti-phospho-ERK1/2 (Thr202/Tyr204) (D13.14.4E, Cell Signaling Technology) according to the manufacturers’ instructions. Secondary antibodies, HRP-linked anti-rabbit antibody (#7074, Cell Signaling Technology) and anti-mouse antibody (NB7539 Novus, Centennial, CO, USA) were used according to the manufacturers’ instructions. Chemiluminescence was performed using SuperSignal™ West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific, MA, USA) [37].
Flow cytometry analysis: The assay was performed as described previously [31]. Briefly, K562 cells, K562/HIF1α, and K562/Si JAK2 were resuspended in PBS plus 0.5% BSA to a density of 1 × 107 cells/mL. Antibodies to CD45 KRO (#A96416, Beckman Coulter) and CD44 FITC (#9011-0441, eBioscience) were titrated or added at the manufacturer’s recommended dilutions before use. The mixture was incubated on ice for 40 minutes. After washing with PBS and 0.5% BSA, labeled cells were analyzed using a Beckman Coulter Navios cytometer.
Semi-quantitative RT-PCR: RT-PCR analysis was performed as described previously [31]. Total RNA was extracted from cells using Tri Reagent (Sigma). Single-stranded cDNA was synthesized from total RNA. Briefly, 1 μg RNA was preincubated with 1 μl oligo(dT)17 primer, and diethyl pyrocarbonate (DEPC)-treated water was added to a total volume of 15 μl at 70oC for 10 minutes, then the mixture was rapidly cooled on ice. To the annealed primer/template, 2 µl of AMV RT 5X reaction buffer, 2 µl of dNTP (25 mM), 28 units of RNasin ribonuclease inhibitor, 30 units of AMV RT and DEPC-treated water were added to a final volume of 10 µl. The reaction was incubated at 42oC for 60 minutes. The resulting cDNA was amplified using a PCR kit (Bioline, Taunton, MA, USA). The primer sets used for each gene are listed in Supplemental Table S1. A total of 35 amplification cycles were performed with an initial incubation at 94oC for 2 minutes and a final extension at 72oC for 15 minutes. Each cycle consisted of denaturation at 94oC for 30 seconds, annealing at 55–60 °C for 30 seconds, and extension at 72 °C for 2:30 minutes. To ensure that the same amount of cDNA from each sample was used in the PCR, the aliquots of reverse transcription products were used with primers for the housekeeping gene β-actin. A total of 5 µl of the PCR products were analyzed by electrophoresis on a 1.5% agarose gel.

Table 1.
Primers used in this study.
| Primers | Forward | Reverse |
|---|---|---|
| hVEGF | 5′-GTCGGGCCTCCGAAACCATG-3′ | 5′-CCTGGTGAGAGATCTGGTTC-3′ |
| hBcl2 | 5′-CTGGAGAGTGCTGAAGATTGATG-3′ | 5′-CAATCACGCGGAACACTTGATTC-3′ |
| hMcl1 | 5′-GATCAGTATATACACTTCAG-3′ | 5′-CAGGTGCAGCCTGTACTTGTC-3′ |
| hβ-actin | 5′-GCCCTGGACTTCGAGCAAGA-3′ | 5′-TGCCAGGGTACATGGTGGTG-3′ |
forward and reverse primers used for the PCR experiments.
Quantitative real-time RT-PCR: Total RNA extraction and reverse transcription were performed as described for semiquantitative RT-PCR. The cDNA of the mRNA transcripts was amplified by quantitative real-time PCR in a 15-μl reaction mixture containing 0.2 pmol/µl forward/reverse primer and 1X SYBR GREEN reaction mixture (Kappa Biosystems, Wilmington, MA, USA). The reaction was performed in a spectrofluorometric thermal cycler (Rotor-GeneTM 6000, Corbett Research, Mortlake, Australia), with an initial denaturation for 10 min at 95 °C to activate Taq DNA polymerase, followed by 55 cycles of denaturation at 95 °C for 15 s and annealing/extension at 55–60oC for 1 min. To ensure linear amplification of PCR products, different amounts of template were used in the same reaction. The β actin gene was used as an internal control and amplified in the same PCR assay. The primer sets were the same as those used in the semi-quantitative RT-PCR.
Clonigenicity assay: The Clonigenicity assay was performed as previously described [38]. Briefly, cells (1 x 104) were diluted in 1 ml of RPMI 10% FBS medium in 1 ml of 0.6% agar to obtain a final agar concentration of 0.3% agar. The cell-agar mixture was poured over a hardened agar bottom in the wells of 12-well plates and allowed to solidify. Once the top layer solidified, 1 ml of the medium containing various treatments was added on top to keep the agar moist. Cells were grown at 37 °C in a 5% CO2 humidified atmosphere until colonies were visible (2 weeks). Plates were stained with 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for 4 hours, and the dye was extracted with 1 ml solubilization buffer (20% SDS, 50% N,N-dimethylformamide, 25 mM HCL) for 24 hours. Optical density was measured at a wavelength of 570 nm with a reference wavelength of 630 nm.
Hypoxia induction by using the “OxyCycler system”: Hypoxia exposure was performed in a custom-designed computer-controlled incubation chamber connected to the BioSpherix OxyCycler (Biospherix), Redfield, NY, USA). Cells were maintained at 37 °C and 5% CO2 in the hypoxic chamber, where the O2 content was set at 3%. Cells in the control group were maintained under normoxic conditions (~21% O2 and 5% CO2) throughout the experiment. Cells were exposed to normoxia or hypoxia for 16 hours.
Immunofluorescence Imaging of cells with the Cell discoverer 7 imaging System-Zeiss: Cells were imaged using the Cell discoverer 7 imaging system from Zeiss at 20× magnification. Images were acquired using Zen blue 3.7 software. To quantify the expression of Hif-1α and phospho-c-Abl in K652 cells, we quantified the relative amounts of the specific proteins by immunofluorescence. A total of 500,000 cells were cultured in flasks, which were then transferred to an OxyCycler incubator. Cells were maintained at 37 °C and 5% CO2 in the hypoxic chamber, where the O2 level was set at 3% for a period of 16 hours. Cells in the control group were maintained under normoxic conditions (~21% O2 and 5% CO2) throughout the experiment. 4 hours before the end of the protocol, the 96-well plate was coated with poly-L-lysine (Sigma/Merck) and allowed to dry for 2 hours before the cells were seeded for an additional 2 hours and incubated in the incubator under normoxic or hypoxic conditions. At the end of the experiment, cells were washed twice with PBS and fixed with 4% paraformaldehyde in PBS at room temperature (RT) for 20 minutes. Cells were again washed twice in PBS before permeabilization in 3% FBS in PBS and blocking with 0.1% Triton X100 in 3% FBS and PBS for 60 min at RT. Cells were washed twice with 3% FBS in PBS before staining with the desired primary antibody (Hif-1α or phospho-c-Abl) in 3% FBS and 0.1% Triton X100 in PBS overnight. Cells were then stained with a secondary antibody, Alexa Fluor- AF 688 (red) (Invitrogene) overnight and DAPI (blue-nuclear) for 30 minutes, followed by 3× washes with 0.1% Triton X100 in 3% FBS and PBS.
Statistical analysis: Statistical analysis was performed by Student’s t-test, with significant values set at * P < 0.01 or ** P < 0.001.
Results
In a previous study, we showed that crizotinib, a ALK and c-Met inhibitor [39], is able to inhibit both wild-type and T315I-mutated Bcr/Abl [39]. In addition, we discovered that crizotinib, but not imatinib, was able to overcome soluble factor-mediated drug resistance in CML cells [40]. This was possible in part because crizotinib has the ability to inhibit JAK2 activity. In this study, normoxic and hypoxic conditions were used to evaluate the sensitivity of CML cell lines to Abl kinase inhibitors such as imatinib and crizotinib.
Induction of apoptosis in CML cells by imatinib and crizotinib under hypoxic conditions
Cobalt chloride (CoCl2), a known inducer of HIF1, was used to simulate hypoxic conditions in K562 and BV173 CML cell lines [41]. Immunofluorescence analysis showed that K562 cells exposed to 100 μM CoCl2 had increased levels of HIF1 (Figure 1A), comparable to exposure to low oxygen (Figure 1A). Real-time PCR was also used to monitor the expression of HIF1-responsive genes such as VEGF and Bcl2 (Figure 1B) to demonstrate the functionality of induced HIF1 in K562 cells treated with CoCl2.
Exposure to CoCl2 and growth of K562 under hypoxic conditions resulted in accumulation of HIF1a (Figure 1A), which increased the expression of VEGF and Bcl2 more than fourfold and twentyfold, respectively (Figure 1B). Figure 1C shows that CoCl2 treatment had no effect on Abl phosphorylation in K562. Furthermore, we demonstrated that imatinib and crizotinib alone or in combination with ruxolitinib inhibited phosphorylation of Abl under normoxic (Figure 1C) and hypoxic (Figure 1D) conditions.
We next examined how imatinib and crizotinib affected the proliferation of K562 cells in the presence and absence of cobalt chloride. Figure 2A shows that exposure to imatinib inhibited K562 proliferation in the presence or absence of CoCl2. In addition, we observed a significant inhibition of cell proliferation in K562 cells treated with crizotinib. However, moderate drug resistance was observed in the presence of CoCl2 in K562 cells treated with crizotinib. This suggests that the presence of CoCl2 blocks the inhibition of cell proliferation mediated by crizotinib but not imatinib (Figure 2A). Signal transducer and activator of transcription 3 (STAT3) has been associated with hypoxia-induced chemoresistance of ovarian cancer cells [42]. Therefore, we used the JAK1/2 inhibitor ruxolitinib [43], which regulates STAT3 activity, to investigate the role of JAK2 in the sensitivity of CML cells to crizotinib. Therefore, we wondered whether the addition of ruxolitinib to crizotinib in the presence of CoCl2 could restore sensitivity. Treatment with ruxolitinib alone had no effect on the proliferation of K562 cells in the presence or absence of CoCl2 (Figure 2B). Moreover, there was no significant difference in the inhibition of K562 cell proliferation between the combinations of imatinib and ruxolitinib (Figure 2B). Interestingly, the combination of ruxolitinib and crizotinib did not restore the sensitivity of K562 to crizotinib. In contrast, inclusion of ruxolitinib increased crizotinib resistance in K562 cells (Figure 2B).
The role of HIF1α and JAK2 in mediating CML chemoresistance
The above data suggest that the sensitivity of CML cells to crizotinib depends on JAK1/2 activity and hypoxic conditions (Figure 2). However, neither hypoxia nor JAK1/2 activity had a significant effect on sensitivity to imatinib (Figure 2). HIF1α is a major mediator of hypoxic conditions, modulating the transcription of a large number of genes. To directly demonstrate the role of hypoxia and JAK2 in regulating sensitivity to crizotinib, we overexpressed a stable mutant of HIF1α (Pro 402 and Pro 564 were replaced by Ala) [44] in K562. Under normoxic conditions, the HIF1 mutant Pro 402 Ala, Pro 564 Ala is not hydroxylated and therefore is not recognized by the von Hippel-Lindau tumor suppressor protein (VHL) for proteasome degradation [45]. Figure 3A shows that HA -HIF1α is overexpressed in K562/HIF1 cells under normoxic conditions. CD44 is one of the genes regulated by HIF1α [46], and monitoring of CD44 levels in K562 and K562/HIF1α cells showed upregulation of CD44 (Figure 3B).
We next examined Abl inhibitor activity in K562 cells stably expressing HIF1α. The levels of phosphorylated Bcr/Abl and Abl were essentially unaffected by overexpression of HIF1α, as shown in Figure 3C. However, the levels of phosphorylated Bcr/Abl and Abl in K562 and K562/HIF1α lysates were completely inhibited by imatinib or crizotinib alone or in combination with ruxolitinib (Figure 3C). This suggests that the ability of imatinib and crizotinib to inhibit Abl protein auto-phosphorylation is not affected in cells overexpressing HIF1α or exposed to ruxolitinib. When K562 was exposed to imatinib, PARP was significantly cleaved. Interestingly, the combination of ruxolitinib and imatinib decreased the amount of cleaved PARP in K562/HIF1α cells but not in K562 cells. The amounts of cleaved PARP in K562 cells treated with crizotinib were almost twice that in cells treated with imatinib. Moreover, the combination of ruxolitinib and crizotinib significantly (50%) reduced the amount of cleaved PARP in K562. In contrast, neither crizotinib nor crizotinib in combination with ruxolitinib induced cleavage of PARP in K562/HIF1α cells. Figure 3 shows that HIF1α is responsible for the reduction in cleaved PARP levels in K562 cells when exposed to crizotinib, although crizotinib continues to inhibit Bcr/Abl activity. Furthermore, it supports the notion that JAK1/2 inhibition contributes to the lower sensitivity of imatinib and crizotinib.
A comparison of the effect of imatinib and crizotinib was performed using a Clonigenicity assay. Figure 3D shows that imatinib inhibited colony formation in K562 and K562/HIF1α cells at concentrations of 1 and 3 uM. Crizotinib was more effective in inhibiting colony formation of K562 and significant inhibition was observed at the lowest concentration (03 μM). The ability of crizotinib to inhibit K562/HIF1α was impaired, indicating some degree of resistance. Interestingly, the addition of ruxolitinib reduced the ability of crizotinib to inhibit colony formation in both K562/HIF1-positive and negative cells.
As shown in Figure 4A, the JAK2 gene was also silenced in K562. The sensitivity of K562, K562/Si JAK2, and K562/H -HIF1α P402A/P564A to a series of Abl kinase inhibitors (AKIs) was monitored. The results shown in Figure 4B indicate that the different cells have different sensitivities to the different AKIs.
Exposure of various K562 cells to AKIs resulted in a decrease in cell viability. When K562/HIF1α cells were treated with AKIs, a marked and reproducible increase in cell viability was observed compared with K562 (Figure 4B), suggesting partial drug resistance mediated by overexpression of HIF1aα (Figure 4B). In cells treated with crizotinib, the ability of crizotinib to inhibit proliferation of K562 cells was markedly different from that of K562/Si JAK2 and K562/HIF1α cells. Crizotinib was less effective in inhibiting the proliferation of K562/Si JAK2 and K562/HIF1α cells (Figure 4B). Thus, the percentage of viable cells in K562, K562/Si JAK2, and K562/HA-HIF1α when exposed to 500nM crizotinib for 72 hours was 23.4, 57.9, and 63.4, respectively (Figure 4C). In addition, no significant differences in cell viability were observed between K562 cells exposed to other AKIs, including ponatinib and GNF-5 (Figure 4B). These results indicate that the decreased sensitivity to crizotinib is significant in cells overexpressing HIF1α or in which JAK2 has been silenced, whereas it is less evident in other AKIs.
2ME2 restores crizotinib sensitivity of CML cells under hypoxic conditions
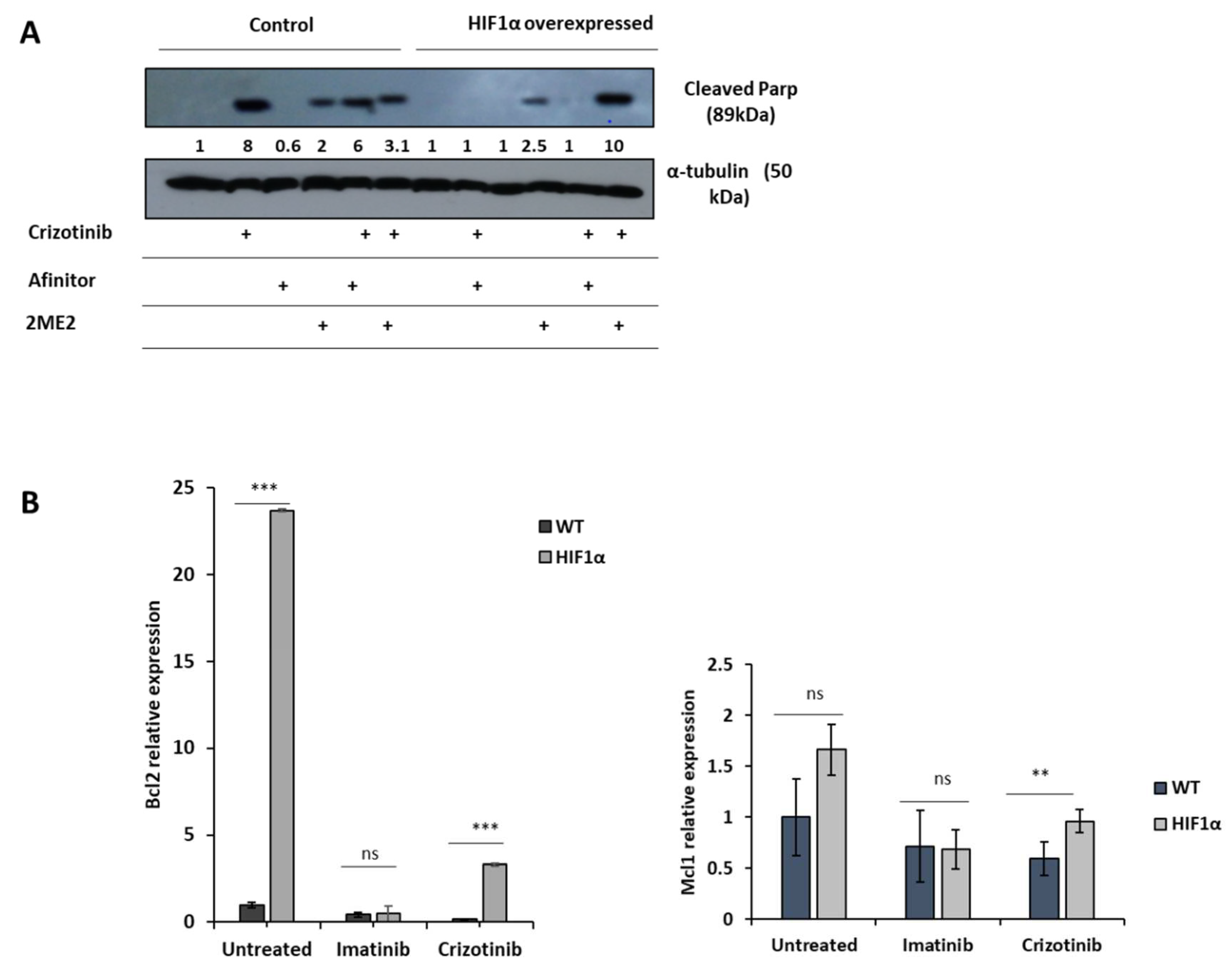
Our data suggest that HIF1α is responsible for mediating crizotinib chemoresistance in CML cells. Therefore, we investigated whether HIF1α modulators can restore sensitivity to crizotinib. Two HIF1α modulators, 2ME2 and everolimus (Afinitor), were used. 2ME2, a natural estradiol metabolite, has been shown to inhibit nuclear accumulation and activity of HIF1α in an oxygen- and proteasome-independent expression of HIF1α and restore chemosensitivity in ALL cells [48].
Figure 5A shows that crizotinib induces PARP cleavage in K562 cells but not in K562/HIF1α cells. However, 2ME2 was marginally active in inducing PARP cleavage in K562 cells, whereas Afinitor was not. The combination of Afinitor with crizotinib had little effect on the sensitivity of K562 cells to crizotinib, particularly in K562/HIF1α cells. In contrast, the combination of 2ME2 and crizotinib significantly increased the amount of cleaved PARP in K562/HIF1α cells, suggesting that 2ME2 is able to restore the sensitivity of crizotinib in K562 cells overexpressing activated HIF1α. Thus, the decreased sensitivity of CML cells to crizotinib under hypoxic conditions is primarily mediated by stabilization and increased activity of HIF1α, and inhibition of HIF1α function can restore crizotinib sensitivity in hypoxic CML cells.
To explore the possible mechanism by which activation of HIF1α confers chemoresistance to cells from CML despite complete inhibition of Bcr/Abl, we monitored changes in apoptosis-related gene expression in K562 and K562/HIF1α cells treated with crizotinib. We monitored the expression of Mcl1 and Bcl2 in K562 and k562/HIF1α cells treated with imatinib or crizotinib (Figure 5B). Treatment of K562 cells with imatinib resulted in a small reduction in the levels of both genes. Treatment with crizotinib was more effective in reducing the levels of the MCL1 and Bcl2 genes. In contrast, treatment with imatinib in K562/HIF1α produced similar reductions in the levels of the two genes as in K562 cells. However, levels of Bcl2 and MCL1 were significantly higher in crizotinib-treated K562/HIF1α cells than in imatinib-treated cells. This suggests that the levels of the two anti-apoptotic genes Bcl2 and MCL1 are relatively higher in K562/HIF1α exposed to crizotinib compared with imatinib treatment and likely contribute to the chemoresistance to crizotinib observed in CML cells under hypoxic conditions.
Discussion
Crizotinib inhibits native and mutant Bcr/Abl kinase activity and showed significant anti CML activity in vitro and in vivo, as previously reported [49]. In addition, we reported that crizotinib, unlike imatinib, was effective in overcoming tumor microenvironment (TME)-mediated chemoresistance in CML cells, which was attributed to the ability of crizotinib to inhibit JAK2 activity [40]. In this study, the anti CML activity of imatinib and crizotinib was examined under hypoxic conditions. First, we discovered that hypoxic conditions, such as reduced oxygen pressure or exposure to cobalt chloride mimicking hypoxic conditions, impaired the ability of imatinib to promote apoptosis and inhibit proliferation (Figure 2). Nevertheless, the effect of hypoxic conditions on crizotinib-treated CML cells was profound and much stronger than that of imatinib (Figure 2). The effect was not limited to inhibition of cell proliferation of CML, but also included the ability of treated CML cells to undergo apoptosis, as measured by monitoring the levels of cleaved cPARP (Figure 2C,D). The observed effect was not limited to K562 cells, but other myeloid cells such as BV -173 cells also showed impaired ability of crizotinib to induce apoptosis under hypoxic conditions compared with BV173 cells treated with imatinib. The hypoxia-induced chemoresistance of crizotinib in CML cells was further enhanced by the combination with ruxolitinib (JAK1/2 inhibitor). Our results indicate that hypoxia-mediated crizotinib resistance in CML cells is a phenomenon that is not restricted to a specific CML cell line. Hypoxia is one of the intrinsic features of solid tumors and is associated with aggressive phenotypes such as resistance to radiotherapy and chemotherapy, metastasis, and poor patient prognosis [50]. The bone marrow microenvironment (BM) is a hypoxic environment that supports hematopoietic stem cells and contributes to leukemia stem cell persistence (LSC) [28]. In CML, imatinib inhibited Bcr/Abl activity in hypoxic stem cells, but apoptosis was partially inhibited [28]. Hypoxic conditions have been reported to induce resistance to crizotinib in NSCLC cell lines with EML4- ALK rearrangement by activating the epithelial-mesenchymal transition (EMT) process [51].
Hypoxia could induce drug resistance via HIF1-dependent and HIF1-independent mechanisms [52,53]. To support our finding and provide evidence that crizotinib-induced chemoresistance in CML cells is HIF1-dependent, we overexpressed the mutant HIF1α in K562 cell lines while silencing JAK2. The mutant HIF1α (substitution of Pro 402 and Pro 564 by Ala) [44] is not subject to hydroxylation and proteasome degradation [45] and is therefore active under normoxic conditions. K562/HIF1α cells were almost completely resistant to crizotinib but not to imatinib (Figure 2 and Figure 4), whereas K562 Si JAK2 cells were partially resistant to crizotinib but not to imatinib or other AKIs (Figure 4). Remarkably, our data show that ruxolitinib, a JAK1/2 inhibitor, enhances crizotinib chemoresistance under hypoxic conditions.
To shed light on a possible mechanism responsible for resistance to crizotinib under hypoxic conditions, we measured the levels of the anti-apoptotic genes MCL-1 and Bcl2. Imatinib inhibited the expression of both anti-apoptotic genes Bcl2 and MCL1 in both cell types, although the reduction appeared to be greater in K562/HIF1α. In contrast, crizotinib significantly reduced Bcl2 and MCL1 expression in K562 cells, whereas it increased Bcl2 levels in K562/HIF1α cells. It is uncertain at this time whether the ability of crizotinib to upregulate Bcl2 contributes to its ability to promote chemoresistance in K562/HIF1α cells. Hypoxia conditions have been reported to modulate the expression of anti-apoptotic and pro-apoptotic genes [54]; hypoxic conditions inhibit the expression of pro-apoptotic proteins (Bax and Bim) and stimulate the expression of anti-apoptotic proteins (Bcl-2, Mcl-1, and XIAP) [54].
To provide further evidence for the importance of HIF1 in mediating crizotinib chemoresistance, we used the known HIF1α inhibitor 2ME2. Figure 5 shows that 2ME2 can restore sensitivity to crizotinib in K562/HIF1α cells. Our result is consistent with previous findings that 2ME2 can restore the sensitivity of medullary thyroid cancer (MTC) cells [55]. In addition, 2ME2 has been shown to reverse drug resistance in human breast tumor xenografts [56] and multiple myeloma cells [57]. 2ME2 is a natural metabolite of estradiol with estrogenic activity. The antitumor activities of 2ME2 were mediated by its pro-apoptotic activity, microtubule activity, and superoxide production [58]. In addition, 2-hydroxyestradiol, a prodrug of 2ME2, was documented to restore the sensitivity of ovarian cancer cells to platinum, which is mediated by the tumor microenvironment [59,60].
Our results showed that the combination of 2ME2 and crizotinib restored the sensitivity of crizotinib in K562 cells with active HIF1a and JAK1/2 inhibitor treatment (Figure 5). The underlying mechanism by which 2ME2 restores crizotinib sensitivity is not yet clear. The main action of 2ME2 appears to be due to disruption of cellular microtubules required for translocation of HIF1α to the nucleus. Thus, 2ME2 [47] may inhibit the accumulation and activity of HIF1α in the nucleus in a manner that is both oxygen- and proteasome-independent [47]. Consistent with previous studies, our data show that 2ME2 induces apoptosis in K562 cells [61]. In addition, 2ME2 inhibits proliferation of prostate cancer cells by modulating the activity of β-catenin [62]. Moreover, 2ME2 upregulates death receptor 5 and induces apoptosis by activating the extrinsic pathway [63]. Recent reports suggest that the antitumor activity of 2ME2 is mediated by its ability to downregulate a number of genes, including Bcl2, BCL-XL, and c-myc [61,64]. Overall, 2ME2 restores crizotinib sensitivity of K562 under hypoxic conditions by inhibiting the activity of HIF1α [47], which promotes apoptosis possibly mediated by overexpression of Bcl2, Bcl-XL, and c-myc [61].
Our data showed that ruxolitinib, a JAK1/2 inhibitor, could ameliorate the effect of hypoxia or HIF1α overexpression in mediating crizotinib resistance in CML cells. According to previous studies, JAK1/2 inhibitors such as AG490 promote the accumulation of HIF1α by inhibiting its hydroxylation [65]. Specifically, JAK1/2 is involved in the activation of PHD activity in HIF1α, which mediates the hydroxylation of HIF1α and its subsequent degradation by the proteasome [65]. Thus, as a JAK1/2 inhibitor, ruxolitinib may inhibit the degradation of HIF1a and contribute to its stabilization [66]. Other studies have shown that inactivation of JAK1 promotes proliferation of endometrial carcinoma cells by upregulating the HIF1a pathway [67]. The authors suggested that JAK1 may act as a tumor suppressor [67] and that deletion of JAK1 activates the HIF1α pathway [67]. Jeong et al. suggested that JAK1 interacts with HIF-1/2 and reduces HIF-1/2 protein expression under hypoxia; therefore, silencing of JAK1 or pharmacologic inhibition of JAK1 kinase activity by ruxolitinib results in upregulation of transcription of HIF target genes under hypoxia [67]. Crizotinib affects the stability of HIF1α in a JAK2-dependent manner, as suggested by the observation that crizotinib inhibits JAK2 activity [68]. Because imatinib does not inhibit JAK2, no stabilizing effect is observed in CML cells treated with imatinib. Moreover, the activity of crizotinib was additive to that of ruxolitinib, which may suggest that different mechanisms are involved in stimulating the stability and function of HIF1α.
In conclusion, our study demonstrated that hypoxic conditions and the presence of ruxolitinib contributed to a substantial increase in the chemoresistance of CML cells to crizotinib. Although the ability of crizotinib to inhibit Bcr/Abl was unchanged, its ability to induce apoptosis in CML cells was diminished. We found that K562 cells overexpressing HIF1a expressed higher levels of the anti-apoptotic genes Bcl2 and MCL-1, suggesting a possible mechanism of action. In addition, our study showed that the inclusion of the HIF1α inhibitor 2ME2 restored the sensitivity of crizotinib in CML cells.
Our study demonstrated that HIF1α signaling enhanced the chemoresistance of crizotinib independently of Bcr/Abl kinase activity in CML cells. Consequently, targeting HIF1a and the components of this pathway may be critical for the complete eradication of CML. In addition, targeting the cross-talk between JAK1/2 and HIF1α has been shown to be a promising strategy for cancer treatment. This intercellular communication may contribute to drug resistance in hypoxic tumor environments. Inhibitors of JAK1/2 or HIF1α or drugs targeting their downstream signaling pathways have great potential for cancer therapy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
LA and DR performed the experiments. LA, HK and DR analyzed the data. JM and JG designed the research study and wrote the paper. All authors read and approved the manuscript.
Funding
No external funding for this project.
Acknowledgments
The following plasmids were provided to us by addgene.org. pCMV-VSV-G was a gift from Bob Weinberg (Addgene plasmid # 8454; http://n2t.net/addgene:8454; RRID:Addgene_8454). pCMV-dR8.2 dvpr was a gift from Bob Weinberg (Addgene plasmid # 8455; http://n2t.net/addgene:8455; RRID:Addgene_8455). LentiCas9 blast was a gift from Feng Zhang (Addgene plasmid # 52962; http://n2t.net/addgene:52962; RRID:Addgene_52962). HA-HIF1alpha P402A/P564A-pBabe-puro was a gift from William Kaelin (Addgene plasmid # 19005; http://n2t.net/addgene:19005; RRID:Addgene_19005). JAK2 gRNA (BRDN0001149125) was a gift from John Doench & David Root (Addgene plasmid # 75728; http://n2t.net/addgene:75728 ; RRID:Addgene_75728).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Faderl S, Talpaz M, Estrov Z, O’Brien S, Kurzrock R, Kantarjian HM. N Engl J Med 1999, 341, 164–167.
- Ottmann, O.G.; Hoelzer, D. The ABL tyrosine kinase inhibitor STI571 (Glivec) in Philadelphia positive acute lymphoblastic leukemia - promises, pitfalls and possibilities. Hematol J 2002, 3, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Radich, J.P.; Dai, H.; Mao, M.; Oehler, V.; Schelter, J.; Druker, B.; Sawyers, C.; Shah, N.; Stock, W.; Willman, C.L.; et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A 2006, 103, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Silver, R.T.; Druker, B.J.; Goldman, J.M.; Gambacorti-Passerini, C.; Guilhot, F.; Schiffer, C.A.; Fischer, T.; Deininger, M.W.; Lennard, A.L.; et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood 2002, 99, 1928–1937. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.B.; Gunby, R.H.; Piazza, R.; Galietta, A.; Rostagno, R.; Scapozza, L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol 2003, 4, 75–85. [Google Scholar] [CrossRef]
- Meads, M.B.; Hazlehurst, L.A.; Dalton, W.S. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res 2008, 14, 2519–2526. [Google Scholar] [CrossRef]
- Katz, B.Z.; Polliack, A. Cancer microenvironment, extracellular matrix, and adhesion molecules: the bitter taste of sugars in chronic lymphocytic leukemia. Leuk Lymphoma 2011, 52, 1619–1620. [Google Scholar] [CrossRef] [PubMed]
- Witz, I.P. The tumor microenvironment: the making of a paradigm. Cancer Microenviron 2009, 2 Suppl 1, 9–17. [Google Scholar] [CrossRef]
- Tesfai, Y.; Ford, J.; Carter, K.W.; Firth, M.J.; O'Leary, R.A.; Gottardo, N.G.; Cole, C.; Kees, U.R. Interactions between acute lymphoblastic leukemia and bone marrow stromal cells influence response to therapy. Leuk Res 2012, 36, 299–306. [Google Scholar] [CrossRef]
- Zhang, B.; Groffen, J.; Heisterkamp, N. Increased resistance to a farnesyltransferase inhibitor by N-cadherin expression in Bcr/Abl-P190 lymphoblastic leukemia cells. Leukemia 2007, 21, 1189–1197. [Google Scholar] [CrossRef]
- Jensen, P.O.; Mortensen, B.T.; Hodgkiss, R.J.; Iversen, P.O.; Christensen, I.J.; Helledie, N.; Larsen, J.K. Increased cellular hypoxia and reduced proliferation of both normal and leukaemic cells during progression of acute myeloid leukaemia in rats. Cell Prolif 2000, 33, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Minakata, K.; Takahashi, F.; Nara, T.; Hashimoto, M.; Tajima, K.; Murakami, A.; Nurwidya, F.; Yae, S.; Koizumi, F.; Moriyama, H.; et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci 2012, 103, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A 2000, 97, 9082–9087. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Chen, X.; Huang, R.; Zeng, H.; Gong, J.; Meng, W.; Lu, Y.; Zhao, F.; Wang, L.; Zhou, Q. BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha}. J Biol Chem 2009, 284, 10004–10012. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol 2004, 24, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ahn, H.J.; Ryu, J.H.; Suk, K.; Park, J.H. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J Exp Med 2004, 199, 113–124. [Google Scholar] [CrossRef]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 2002, 62, 3387–3394. [Google Scholar]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Takahashi, F.; Nurwidya, F.; Kobayashi, I.; Minakata, K.; Hashimoto, M.; Nara, T.; Kato, M.; Tajima, K.; Shimada, N.; et al. Hypoxia increases gefitinib-resistant lung cancer stem cells through the activation of insulin-like growth factor 1 receptor. PLoS One 2014, 9, e86459. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Yarde, D.N.; Meads, M.B.; Huang, M.; Jove, R.; Hazlehurst, L.A.; Dalton, W.S. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res 2009, 69, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Tolentino, J.H.; Argilagos, R.F.; Zhang, L.; Pinilla-Ibarz, J.; Hazlehurst, L.A. Potentiation of Nilotinib-mediated cell death in the context of the bone marrow microenvironment requires a promiscuous JAK inhibitor in CML. Leuk Res 2012, 36, 756–763. [Google Scholar] [CrossRef]
- Traer, E.; Javidi-Sharifi, N.; Agarwal, A.; Dunlap, J.; English, I.; Martinez, J.; Tyner, J.W.; Wong, M.; Druker, B.J. Ponatinib overcomes FGF2-mediated resistance in CML patients without kinase domain mutations. Blood 2014, 123, 1516–1524. [Google Scholar] [CrossRef]
- Bewry, N.N.; Nair, R.R.; Emmons, M.F.; Boulware, D.; Pinilla-Ibarz, J.; Hazlehurst, L.A. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol Cancer Ther 2008, 7, 3169–3175. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S.; Tauchi, T.; Katagiri, S.; Tanaka, Y.; Ohyashiki, K. Combination of the ABL kinase inhibitor imatinib with the Janus kinase 2 inhibitor TG101348 for targeting residual BCR-ABL-positive cells. J Hematol Oncol 2014, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Manjeri, A.; Lee, K.L.; Huang, W.; Tan, S.Y.; Chuah, C.T.; Poellinger, L.; Ong, S.T. Physiologic hypoxia promotes maintenance of CML stem cells despite effective BCR-ABL1 inhibition. Blood 2014, 123, 3316–3326. [Google Scholar] [CrossRef]
- Chen, W.; Qin, Y.; Liu, S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin Transl Med 2018, 7, 27. [Google Scholar] [CrossRef]
- Jiang, M.; He, G.; Wang, J.; Guo, X.; Zhao, Z.; Gao, J. Hypoxia induces inflammatory microenvironment by priming specific macrophage polarization and modifies LSC behaviour via VEGF-HIF1α signalling. Transl Pediatr 2021, 10, 1792–1804. [Google Scholar] [CrossRef]
- Kidan, N.; Khamaisie, H.; Ruimi, N.; Roitman, S.; Eshel, E.; Dally, N.; Ruthardt, M.; Mahajna, J. Ectopic Expression of Snail and Twist in Ph+ Leukemia Cells Upregulates CD44 Expression and Alters Their Differentiation Potential. J Cancer 2017, 8, 3952–3968. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Bartz, S.; Mao, M.; Li, L.; Kaelin, W.G., Jr. The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol Cell Biol 2007, 27, 2092–2102. [Google Scholar] [CrossRef]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef]
- Najajreh, Y.; Khamaisie, H.; Ruimi, N.; Khatib, S.; Katzhendler, J.; Ruthardt, M.; Mahajna, J. Oleylamine-carbonyl-valinol inhibits auto-phosphorylation activity of native and T315I mutated Bcr-Abl, and exhibits selectivity towards oncogenic Bcr-Abl in SupB15 ALL cell lines. Mol Biol Rep 2013, 40, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Gochman, E.; Mahajna, J.; Shenzer, P.; Dahan, A.; Blatt, A.; Elyakim, R.; Reznick, A.Z. The expression of iNOS and nitrotyrosine in colitis and colon cancer in humans. Acta Histochem 2012, 114, 827–835. [Google Scholar] [CrossRef]
- Mian, A.A.; Metodieva, A.; Badura, S.; Khateb, M.; Ruimi, N.; Najajreh, Y.; Ottmann, O.G.; Mahajna, J.; Ruthardt, M. Allosteric inhibition enhances the efficacy of ABL kinase inhibitors to target unmutated BCR-ABL and BCR-ABL-T315I. BMC Cancer 2012, 12, 411. [Google Scholar] [CrossRef]
- Koren Carmi, Y.; Khamaisi, H.; Adawi, R.; Noyman, E.; Gopas, J.; Mahajna, J. Secreted Soluble Factors from Tumor-Activated Mesenchymal Stromal Cells Confer Platinum Chemoresistance to Ovarian Cancer Cells. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Dotan, N.; Wasser, S.P.; Mahajna, J. Inhibition of the androgen receptor activity by Coprinus comatus substances. Nutr Cancer 2011, 63, 1316–1327. [Google Scholar] [CrossRef]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J Cancer 2013, 2, 91–97. [Google Scholar] [CrossRef]
- Regev, O.; Kidan, N.; Nicola, M.; Khamisie, H.; Ruthardt, M.; Mahajna, J. Mesenchymal soluble factors confer imatinib drug resistance in chronic myelogenous leukemia cells. Arch Med Sci 2021, 17, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yotnda, P. Induction and testing of hypoxia in cell culture. J Vis Exp, 3791. [Google Scholar] [CrossRef]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int J Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Ostojic, A.; Vrhovac, R.; Verstovsek, S. Ruxolitinib: a new JAK1/2 inhibitor that offers promising options for treatment of myelofibrosis. Future Oncol 2011, 7, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Snell, C.E.; Turley, H.; McIntyre, A.; Li, D.; Masiero, M.; Schofield, C.J.; Gatter, K.C.; Harris, A.L.; Pezzella, F. Proline-hydroxylated hypoxia-inducible factor 1alpha (HIF-1alpha) upregulation in human tumours. PLoS One 2014, 9, e88955. [Google Scholar] [CrossRef]
- Hon, W.C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Penet, M.F.; Nimmagadda, S.; Mironchik, Y.; Raman, V.; Solaiyappan, M.; Semenza, G.L.; Pomper, M.G.; Bhujwalla, Z.M. Hypoxia regulates CD44 and its variant isoforms through HIF-1α in triple negative breast cancer. PLoS One 2012, 7, e44078. [Google Scholar] [CrossRef] [PubMed]
- Mabjeesh, N.J.; Escuin, D.; LaVallee, T.M.; Pribluda, V.S.; Swartz, G.M.; Johnson, M.S.; Willard, M.T.; Zhong, H.; Simons, J.W.; Giannakakou, P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003, 3, 363–375. [Google Scholar] [CrossRef]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1alpha signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol Ther 2012, 13, 858–870. [Google Scholar] [CrossRef]
- Mian, A.A.; Haberbosch, I.; Khamaisie, H.; Agbarya, A.; Pietsch, L.; Eshel, E.; Najib, D.; Chiriches, C.; Ottmann, O.G.; Hantschel, O.; et al. Crizotinib acts as ABL1 inhibitor combining ATP-binding with allosteric inhibition and is active against native BCR-ABL1 and its resistance and compound mutants BCR-ABL1(T315I) and BCR-ABL1(T315I-E255K). Ann Hematol 2021, 100, 2023–2029. [Google Scholar] [CrossRef]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opinion on Drug Discovery 2019, 14, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Sogabe, S.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int J Oncol 2014, 45, 1430–1436. [Google Scholar] [CrossRef]
- Adamski, J.; Price, A.; Dive, C.; Makin, G. Hypoxia-induced cytotoxic drug resistance in osteosarcoma is independent of HIF-1Alpha. PLoS One 2013, 8, e65304. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Q.; Yu, T.; Sun, S.; Wang, W.; Liu, G. Hypoxia promotes drug resistance in osteosarcoma cells via activating AMP-activated protein kinase (AMPK) signaling. J Bone Oncol 2016, 5, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.; Gouel, F.; Dubus, I.; Heuclin, C.; Roget, K.; Vannier, J.P. Hypoxia promotes chemoresistance in acute lymphoblastic leukemia cell lines by modulating death signaling pathways. BMC Cancer 2016, 16, 746. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Jiang, X.; Zhu, H.; Jiang, W.; Dong, X.; Qiao, H.; Sun, X.; Jiang, H. 2ME2 inhibits the activated hypoxia-inducible pathways by cabozantinib and enhances its efficacy against medullary thyroid carcinoma. Tumour Biol 2016, 37, 381–391. [Google Scholar] [CrossRef]
- Azab, S.S.; Salama, S.A.; Hassan, M.H.; Khalifa, A.E.; El-Demerdash, E.; Fouad, H.; Al-Hendy, A.; Abdel-Naim, A.B. 2-Methoxyestradiol reverses doxorubicin resistance in human breast tumor xenograft. Cancer Chemother Pharmacol 2008, 62, 893–902. [Google Scholar] [CrossRef]
- Chauhan, D.; Catley, L.; Hideshima, T.; Li, G.; Leblanc, R.; Gupta, D.; Sattler, M.; Richardson, P.; Schlossman, R.L.; Podar, K.; et al. 2-Methoxyestradiol overcomes drug resistance in multiple myeloma cells. Blood 2002, 100, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, N.J.; Sarkar, M.A.; Venitz, J.; Figg, W.D. 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy 2003, 23, 165–172. [Google Scholar] [CrossRef]
- Khamaisi, H.; Mahmoud, H.; Mahajna, J. 2-Hydroxyestradiol Overcomes Mesenchymal Stem Cells-Mediated Platinum Chemoresistance in Ovarian Cancer Cells in an ERK-Independent Fashion. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Koren Carmi, Y.; Khamaisi, H.; Adawi, R.; Noyman, E.; Gopas, J.; Mahajna, J. Secreted Soluble Factors from Tumor-Activated Mesenchymal Stromal Cells Confer Platinum Chemoresistance to Ovarian Cancer Cells. International Journal of Molecular Sciences 2023, 24, 7730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, H.; Shen, Y.; Zhou, X. Combined Effects of 2-Methoxyestradiol (Hypoxia-Inducible Factor 1alpha Inhibitor) and Dasatinib (A Second-Generation Tyrosine Kinase Inhibitor) on Chronic Myelocytic Leukemia Cells. J Immunol Res 2022, 2022, 6324326. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhuizen, P.J.; Ray, G.; Banerjee, S.; Dhar, G.; Kambhampati, S.; Dhar, A.; Banerjee, S.K. 2-Methoxyestradiol modulates beta-catenin in prostate cancer cells: a possible mediator of 2-methoxyestradiol-induced inhibition of cell growth. Int J Cancer 2008, 122, 567–571. [Google Scholar] [CrossRef]
- LaVallee, T.M.; Zhan, X.H.; Johnson, M.S.; Herbstritt, C.J.; Swartz, G.; Williams, M.S.; Hembrough, W.A.; Green, S.J.; Pribluda, V.S. 2-methoxyestradiol up-regulates death receptor 5 and induces apoptosis through activation of the extrinsic pathway. Cancer Res 2003, 63, 468–475. [Google Scholar] [PubMed]
- Hernandez-Luna, M.A.; Rocha-Zavaleta, L.; Vega, M.I.; Huerta-Yepez, S. Hypoxia inducible factor-1alpha induces chemoresistance phenotype in non-Hodgkin lymphoma cell line via up-regulation of Bcl-xL. Leuk Lymphoma 2013, 54, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Sanchez, R.; Berzal, S.; Sanchez-Nino, M.D.; Neria, F.; Goncalves, S.; Calabia, O.; Tejedor, A.; Calzada, M.J.; Caramelo, C.; Deudero, J.J.; et al. AG490 promotes HIF-1alpha accumulation by inhibiting its hydroxylation. Curr Med Chem 2012, 19, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, R.; Berzal, S.; Sánchez-Niño, M.D.; Neria, F.; Gonçalves, S.; Calabia, O.; Tejedor, A.; Calzada, M.J.; Caramelo, C.; Deudero, J.J.; et al. AG490 promotes HIF-1α accumulation by inhibiting its hydroxylation. Curr Med Chem 2012, 19, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.H.; Lee, H.J.; Cha, J.H.; Kim, J.H.; Kim, K.R.; Kim, J.H.; Yoon, D.K.; Kim, K.W. Hypoxia-inducible factor-1 alpha inhibits self-renewal of mouse embryonic stem cells in Vitro via negative regulation of the leukemia inhibitory factor-STAT3 pathway. J Biol Chem 2007, 282, 13672–13679. [Google Scholar] [CrossRef] [PubMed]
- Regev, O.; Kidan, N.; Khamisie, H.; Mahajna, J. Crizotinib overcomes microenvironment mediated drug resistance in Ph positive leukemia. European Journal of Cancer 2016, 61, S92–S93. [Google Scholar] [CrossRef]
Figure 1.
Expression levels HIF1α and pAbl in K562 under hypoxic conditions. K562 cells were treated with 100uM CoCl2 or grown under low oxygen conditions (2%) for 24 hours. (A) Induction of HIF1α in K562 cells by treatment with cobalt chloride and exposure to hypoxic conditions for 24 hours. (B). Real-time PCR was used to determine the relative expression of Bcl2 and VEGF in relation to β actin in K562 cells exposed to 100μM cobalt chloride (B). Immune Florence assessment of pAbl levels in K562 cells grown under normoxic (C) and hypoxic (D) conditions after 24-hour treatment with CoCl2, imatinib, ruxolitinib, imatinib/ruxolitinib, crizotinib, and crizotinib/ruxolitinib.
Figure 1.
Expression levels HIF1α and pAbl in K562 under hypoxic conditions. K562 cells were treated with 100uM CoCl2 or grown under low oxygen conditions (2%) for 24 hours. (A) Induction of HIF1α in K562 cells by treatment with cobalt chloride and exposure to hypoxic conditions for 24 hours. (B). Real-time PCR was used to determine the relative expression of Bcl2 and VEGF in relation to β actin in K562 cells exposed to 100μM cobalt chloride (B). Immune Florence assessment of pAbl levels in K562 cells grown under normoxic (C) and hypoxic (D) conditions after 24-hour treatment with CoCl2, imatinib, ruxolitinib, imatinib/ruxolitinib, crizotinib, and crizotinib/ruxolitinib.
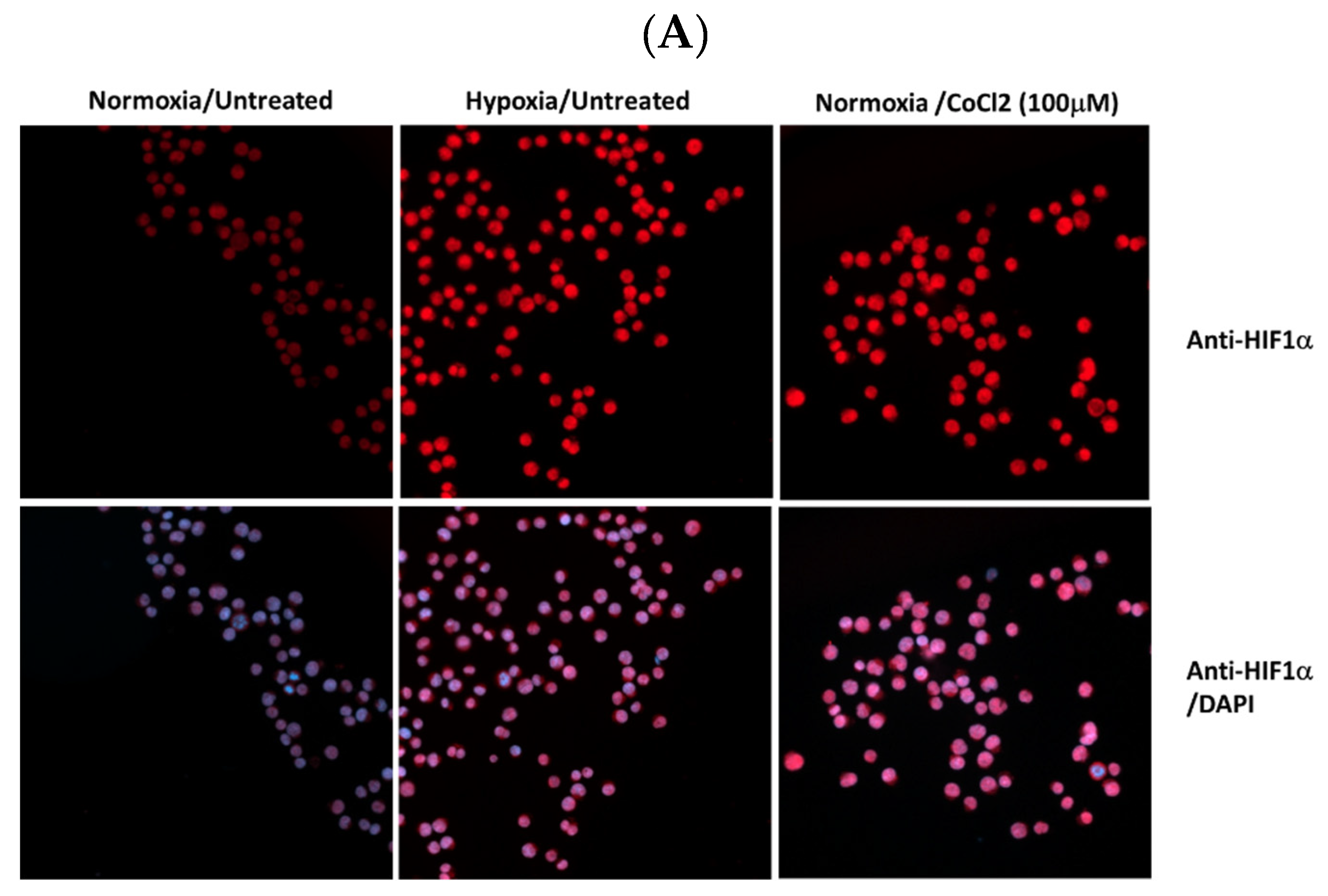
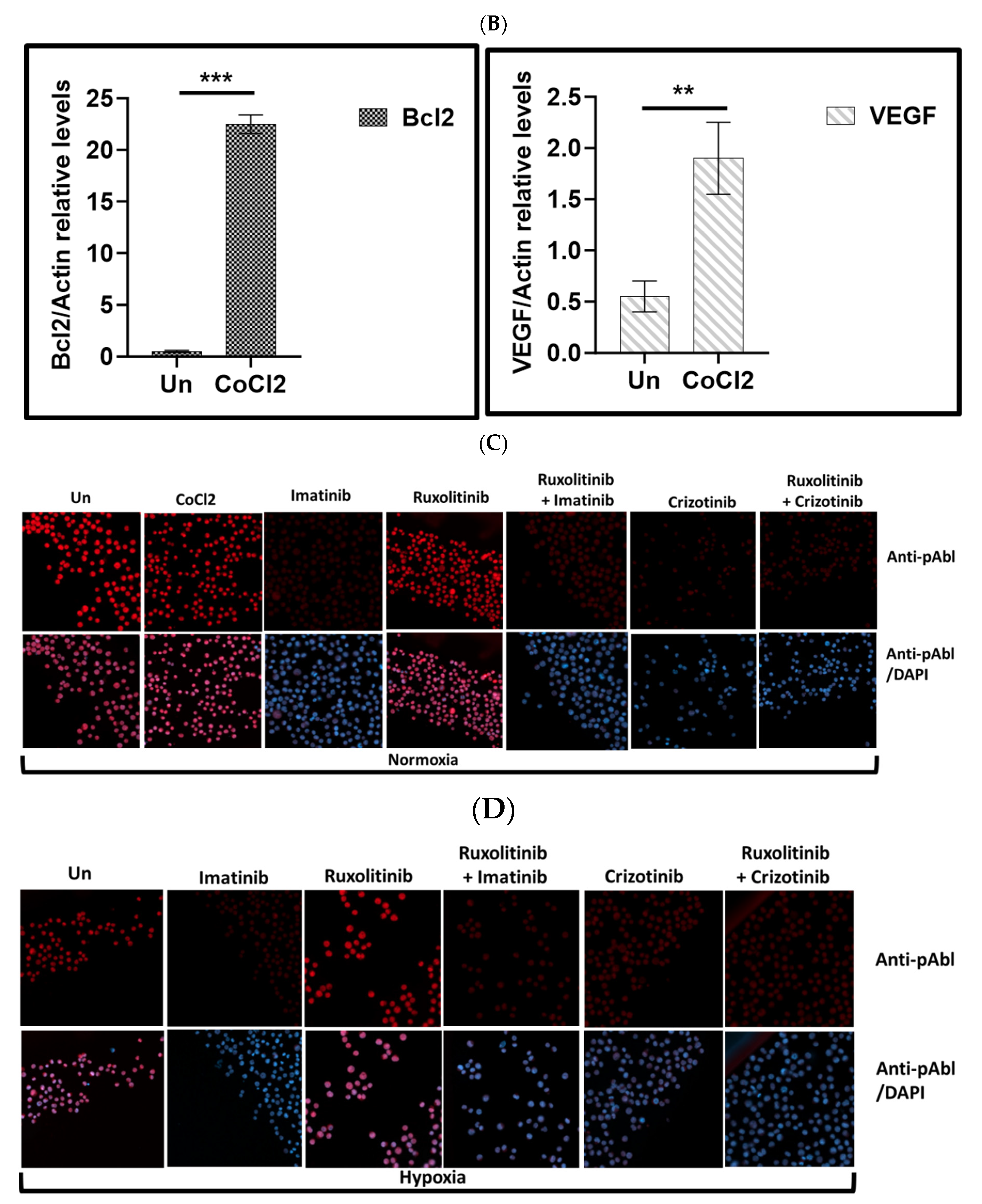
Figure 2.
Effect of CoCl2 and ruxolitinib on the sensitivity of K562 and BV -173 cells to treatment with imatinib or crizotinib. K562 or BV-173 cells exposed to 100μM cobalt chloride and treated with 1%DMSO, 1μM imatinib, crizotinib or ruxolitinib for 24 hours (A) Measurement of viability of K562 with the trypan blue exclusion assay using a two-sample t-test. (B,C) Immunoblot of K562 or BV-173 cells exposed to 1μM imatinib, crizotinib or ruxolitinib in the presence or absence of 100µM CoCl2. Filters were probed with anti-c-PARP and α-tubulin antibodies. Numbers below blot traces represent relative expression normalized to α-tubulin. Uncropped WB Figures S1.
Figure 2.
Effect of CoCl2 and ruxolitinib on the sensitivity of K562 and BV -173 cells to treatment with imatinib or crizotinib. K562 or BV-173 cells exposed to 100μM cobalt chloride and treated with 1%DMSO, 1μM imatinib, crizotinib or ruxolitinib for 24 hours (A) Measurement of viability of K562 with the trypan blue exclusion assay using a two-sample t-test. (B,C) Immunoblot of K562 or BV-173 cells exposed to 1μM imatinib, crizotinib or ruxolitinib in the presence or absence of 100µM CoCl2. Filters were probed with anti-c-PARP and α-tubulin antibodies. Numbers below blot traces represent relative expression normalized to α-tubulin. Uncropped WB Figures S1.
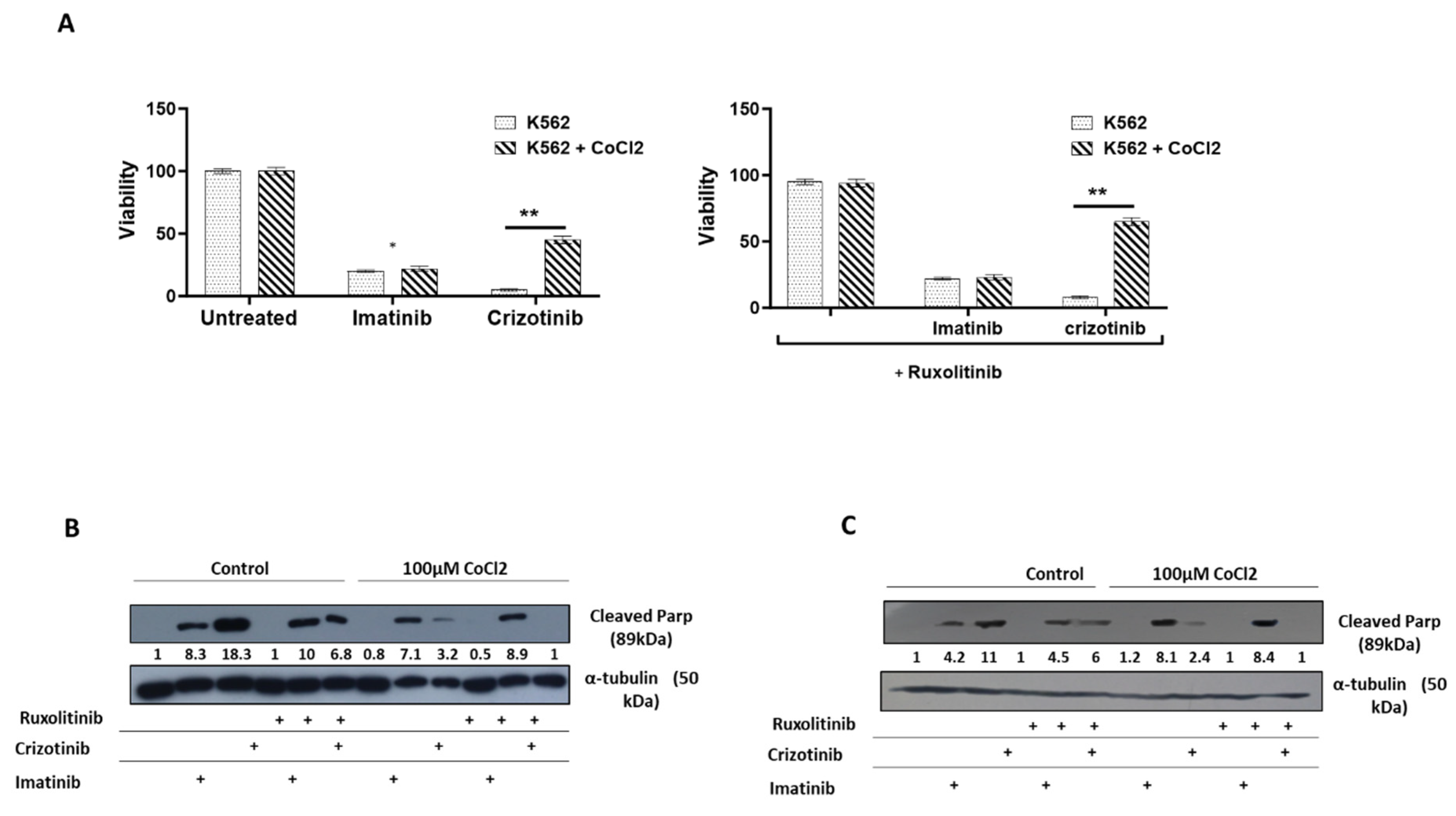
Figure 3.
Effect of overexpression of HIFα on chemoresistance of K562. (A). Immunoblot of HIF1α in K562 and K562/HIF1α cells. (B). Expression level of CD44 in K562 and K562/HIF1α by FACS analysis. (C). Levels of pAbl and cleaved PARP in K562 and K562/HIF1α exposed to imatinib (1μM), crizotinib (1μM), or ruxolitinib (1μM) for 24 hours as indicated in Materials and Methods. (D). Clonigenicity of K562 and K562/HIF1α cells under semi-solid conditions in the presence of imatinib (0.3, 1 and 3μM), crizotinib (0.3, 1 and 3μM) in the presence and absence of ruxolitinib (1μM). (E). Heatmap of colony absorption after dye extraction of stained colonies from the different samples.
Figure 3.
Effect of overexpression of HIFα on chemoresistance of K562. (A). Immunoblot of HIF1α in K562 and K562/HIF1α cells. (B). Expression level of CD44 in K562 and K562/HIF1α by FACS analysis. (C). Levels of pAbl and cleaved PARP in K562 and K562/HIF1α exposed to imatinib (1μM), crizotinib (1μM), or ruxolitinib (1μM) for 24 hours as indicated in Materials and Methods. (D). Clonigenicity of K562 and K562/HIF1α cells under semi-solid conditions in the presence of imatinib (0.3, 1 and 3μM), crizotinib (0.3, 1 and 3μM) in the presence and absence of ruxolitinib (1μM). (E). Heatmap of colony absorption after dye extraction of stained colonies from the different samples.
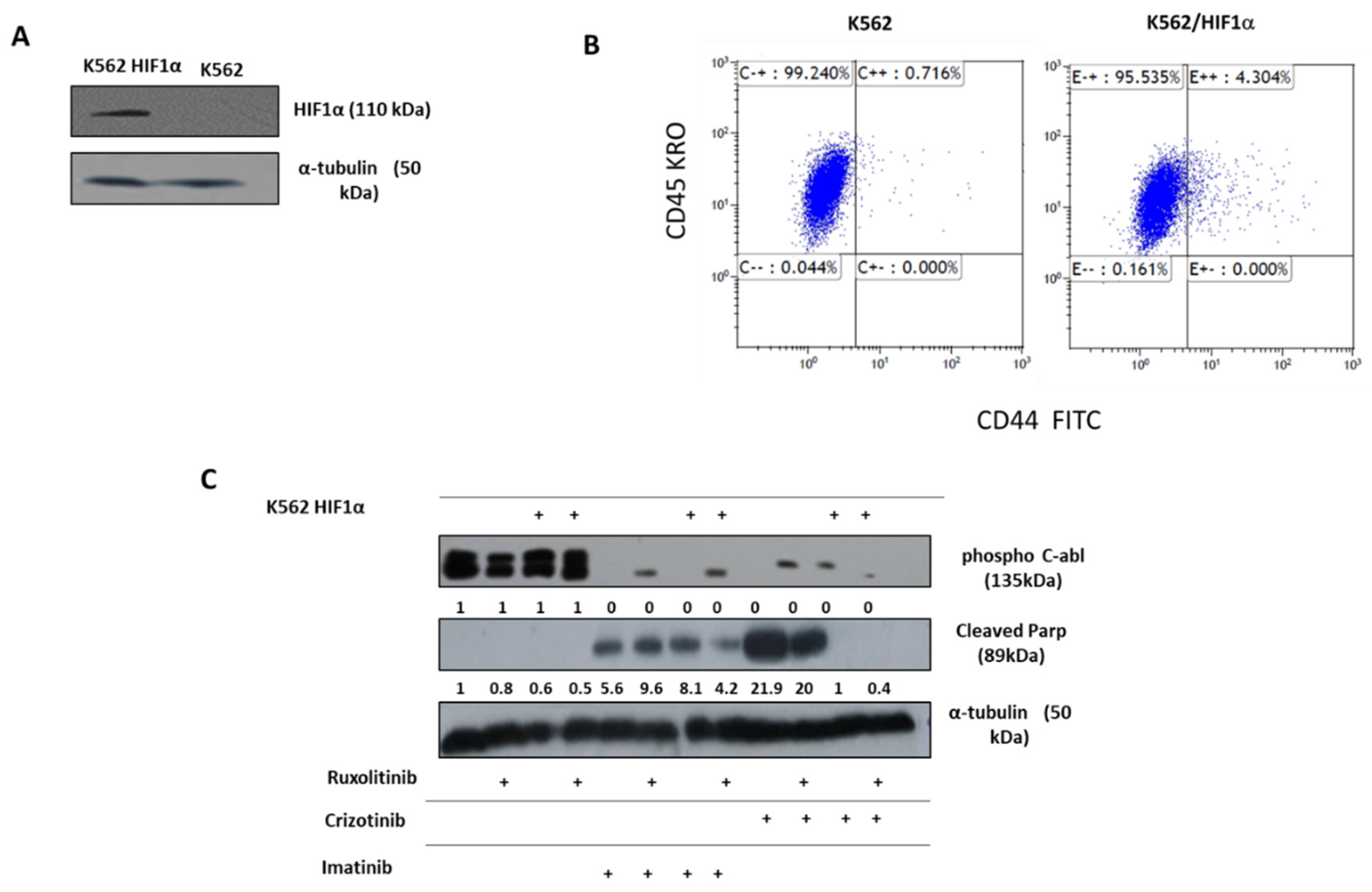
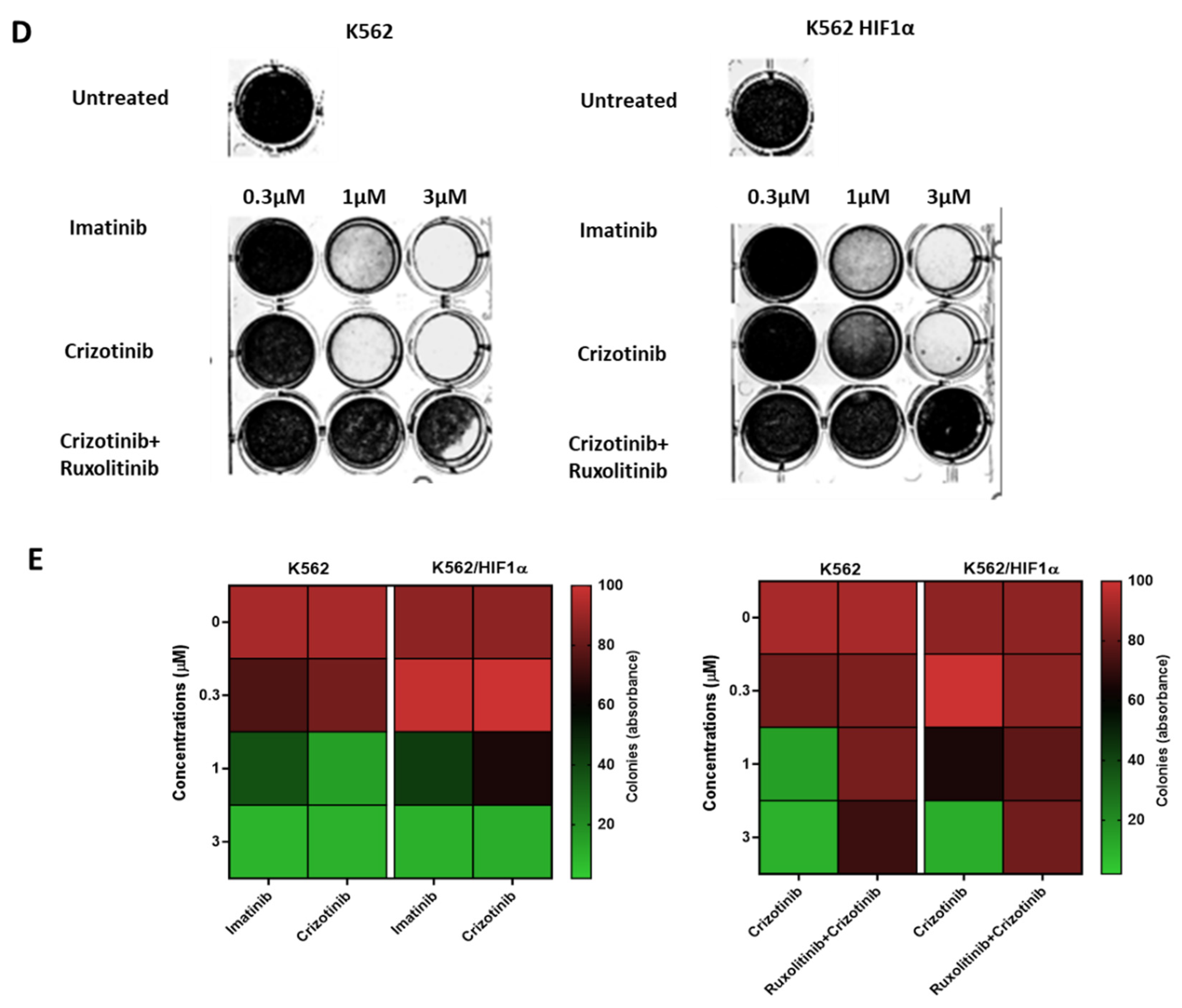
Figure 4.
Effect of JAK2 silencing and HIF1α overexpression on the sensitivity of K562 to different AKI treatments. (A) Immunoblot of JAK2 in K562 and K562 SiJAK2 cells (B) K562, K562 Si JAK2 and K562 HIF1α cells were treated with 1000nM imatinib, crizotinib, ponatinib or GNF-5. (C) Cells were treated with different concentrations of crizotinib. Cell viability was monitored as described in Materials and Methods. * p ≤0.05, ** p ≤0.05, *** p ≤ 0.001 and p ≤ 0.0001.
Figure 4.
Effect of JAK2 silencing and HIF1α overexpression on the sensitivity of K562 to different AKI treatments. (A) Immunoblot of JAK2 in K562 and K562 SiJAK2 cells (B) K562, K562 Si JAK2 and K562 HIF1α cells were treated with 1000nM imatinib, crizotinib, ponatinib or GNF-5. (C) Cells were treated with different concentrations of crizotinib. Cell viability was monitored as described in Materials and Methods. * p ≤0.05, ** p ≤0.05, *** p ≤ 0.001 and p ≤ 0.0001.
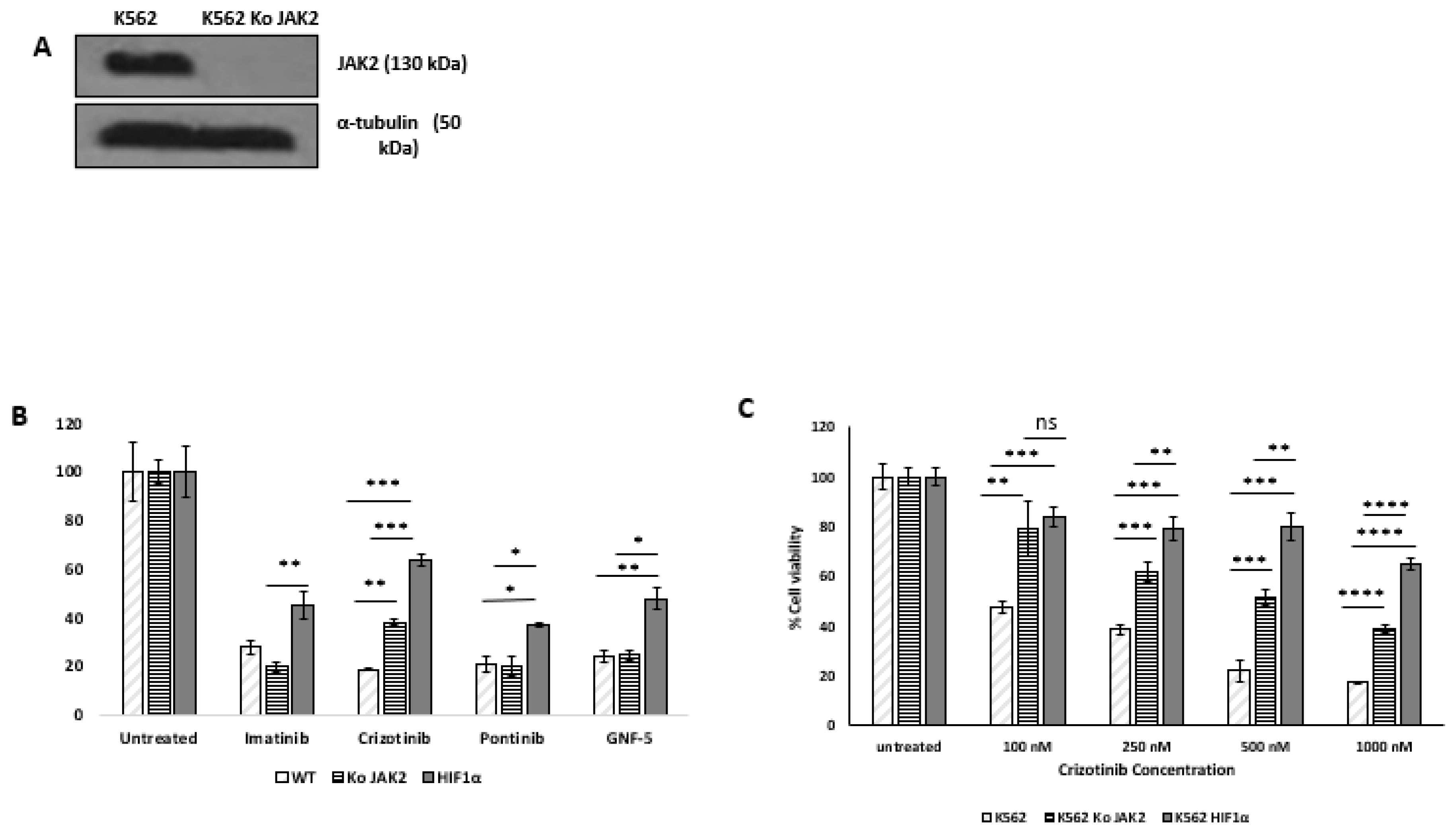
Figure 5.
Effect of 2ME2 on HIF1α-mediated resistance to crizotinib. K562 or K562 HIF1α cells were treated with 1%DMSO, imatinib, crizotinib, Afinitor, or 2ME2 for 24 hours. (A) Immunoblot of K562 or HIF1α cells exposed to 1μM crizotinib, Afinitor or 2ME2. Filters were probed with anti-c-PARP and α-tubulin antibodies. Numbers below blot traces represent relative values normalized to α-tubulin. Uncropped WB Figures S1. (B) Relative expression of Mcl1 and Bcl2 relative to β-actin was determined by real-time PCR in K562 and K562-HIF1α cells treated with 1% DMSO, imatinib, or crizotinib for 24 hours. Concentrations of PCR amplicons were analyzed with a two-sample t test. Shown is the mean relative expression (log2) ± s.e.m. * p ≤0.05, ** p ≤ 0.01 and *** p ≤ 0.001.
Figure 5.
Effect of 2ME2 on HIF1α-mediated resistance to crizotinib. K562 or K562 HIF1α cells were treated with 1%DMSO, imatinib, crizotinib, Afinitor, or 2ME2 for 24 hours. (A) Immunoblot of K562 or HIF1α cells exposed to 1μM crizotinib, Afinitor or 2ME2. Filters were probed with anti-c-PARP and α-tubulin antibodies. Numbers below blot traces represent relative values normalized to α-tubulin. Uncropped WB Figures S1. (B) Relative expression of Mcl1 and Bcl2 relative to β-actin was determined by real-time PCR in K562 and K562-HIF1α cells treated with 1% DMSO, imatinib, or crizotinib for 24 hours. Concentrations of PCR amplicons were analyzed with a two-sample t test. Shown is the mean relative expression (log2) ± s.e.m. * p ≤0.05, ** p ≤ 0.01 and *** p ≤ 0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



