
Submitted:
18 September 2023
Posted:
20 September 2023
You are already at the latest version
Abstract
The dengue virus (DENV) is an arbovirus belonging to the Flaviviridae family; the species comprises four antigenically distinct serotypes (DENV–1, DENV–2, DENV–3, and DENV–4), which are further subdivided into genotypes. The virus is transmitted to humans through mosquito bites, primarily from the Aedes spp. genus. Dengue is endemic in various parts of the world, including tropical and subtropical regions of Asia, Latin America, Africa, and Oceania. In Brazil, the state of Tocantins, located in north-central Brazil, has experienced a significant number of arboviral disease cases, particularly dengue. This study aimed to monitor DENV circulation within the state by conducting full genome sequencing of viral genomes recovered from 61 patients between June 2021 and July 2022. During this period, both DENV-1 and DENV-2 serotypes were identified. Our findings confirm the circulation of DENV serotypes 1 and 2 in Tocantins, affecting males and females equally, with younger age groups (4 to 43 years old) being the most susceptible. Phylogenetic analysis revealed that the circulating viruses belong to DENV–1 genotype V American and DENV–2 genotype III Southeast Asian/American. The Bayesian analysis of DENV-1 Genotype V sequenced here is closely related to genomes previously sequenced in the state of São Paulo. Regarding DENV-2 genotype III genomes, these are clustered in a distinct, well-supported subclade, along with previously reported isolates from the states of Goiás and São Paulo. In both cases, our results suggest that multiple introductions of these genotypes occurred in the Tocantins state. This observation highlights the significant impact of major population centers in Brazil on virus dispersion, including other Latin-American countries and the USA. In the SNP analysis, DENV-1 displayed 122 distinct missense mutations, while DENV-2 had 44, with significant mutations predominantly occurring in the envelope and NS5 proteins. The analyses performed here reveal the concomitant circulation of distinct DENV-1 and -2 genotypes in some Brazilian states, underscoring the dynamic evolution of the DENV and the ongoing significance of surveillance efforts in supporting public health policies.
Keywords:
DENV
; Genomic surveillance
; Epidemiology
; molecular clock
; SNPs
; arboviruses
1. Introduction
The Dengue virus (DENV) is an arbovirus transmitted to humans via the bite of infected Aedes aegypti mosquitoes [1]. Dengue is an endemic disease in numerous regions worldwide, including tropical and subtropical areas of Asia, Latin America, Africa, and Oceania [2,3]. Taxonomically, the virus belongs to the Flaviviridae family, within the genus Flavivirus [4]. DENV encompasses four distinct serotypes (DENV-1, DENV-2, DENV-3, and DENV-4) [5]. Within each serotype, the number of genotypes varies; presently, DENV-1 has five genotypes (I-V), DENV-2 has six genotypes (I-VI), DENV-3 has four genotypes (I-IV), and DENV-4 has two genotypes (I-II) [6,7].
The four serotypes of DENV are globally distributed [2,3]. Infections by one serotype grant immunity against that specific serotype but not against the others [8]. Subsequent infections by different serotypes elevate the risk of developing severe forms of the disease [9]. Monitoring the circulating DENV genotypes can offer valuable insights into the virus’s evolutionary history and geographic distribution [10]. Understanding the potential co-circulation of various virus serotypes can assist in preventing the emergence of more severe diseases [11].
DENV-1 genotype V predominates in Brazil and probably was introduced in the Americas from Southeast Asia or India [12]. Genotype V has been linked to numerous dengue fever outbreaks in Southeast Asia, including Malaysia, Singapore, and Thailand [12]. Like other DENV-1 genotypes, genotype V can cause a spectrum of symptoms, ranging from mild flu-like illness to severe dengue fever, which can be life-threatening [13]. DENV-2 genotype III, previously named Southern Asian-American, predominates in Brazil [14] and has been associated with various dengue fever outbreaks in South America, especially in Brazil, Venezuela, and Colombia [15]. Similar to other DENV-2 genotypes, genotype III can manifest as a wide range of symptoms, from mild to severe illness, and in certain cases, it can progress to dengue hemorrhagic fever (DHF) or dengue shock syndrome (DSS), which can be life-threatening [16].
Historically, the state of Tocantins in northern-central Brazil has grappled with a high incidence of dengue cases, dating back to the first documented transmission of DENV in the municipality of Araguaína in 1991 [17]. Concurrently, with the advent of the COVID-19 pandemic, SARS-CoV-2 was initially detected in Brazil in late February 2020, reaching the state of Tocantins by March of the same year [18]. Interestingly, alongside the surge of the pandemic virus, there was a noticeable decline in the reporting of suspected dengue cases within the state [19]. However, in 2022, a significant resurgence in dengue cases emerged in Tocantins, with the notification of over 22,000 cases, reinstating the state’s unfortunate status as the leader in dengue cases in the northern region of the country [20].
Just as in other Brazilian states, dengue surveillance in Tocantins states falls under the purview of local health authorities, particularly the state and municipal health departments [21]. These agencies are responsible for gathering, consolidating, and analyzing data on reported dengue cases sourced from healthcare providers and laboratories [22]. They also oversee the implementation of measures aimed at controlling the spread of the virus [22]. However, it’s important to note that the sequencing of viral genomes typically does not feature within the mandatory protocols established by health authorities [23]. To identify the circulating viral serotypes and genotypes, as well as their dispersion patterns, a more targeted approach involving the sequencing of recovered dengue genomes becomes necessary [11], mainly when dengue vaccine has been introduced in a country, as happened at moment in Brazil [24].
In endemic regions, genomic surveillance significantly contributes to identifying the most probable sources of infection, comprehending geographical distribution, and monitoring the movement of infected vectors [25]. Furthermore, this information plays a pivotal role in establishing early epidemiological markers that guide critical decision-making by competent authorities [23]. Additionally, it aids in determining whether the circulating genotypes are linked to an increased risk of severe dengue (DHF/DSS) within the population [26,27]. Recognizing the importance of this type of analysis, the present study was conducted with the objective of performing the complete genome sequencing of DENV strains retrieved from Tocantins State between June 2021 and July 2022. Additionally, we conducted Bayesian phylogenetic and molecular clock analyses to explore the viral dispersion in Brazil, aiming to provide insight into its spread among Brazilian states and American countries and pinpoint the introduction timing of DENV-1 and DENV-2 into Tocantins.
2. Materials and Methods
2.1. Ethical aspects
This project is part of a collaborative initiative between the Central Public Health Laboratory of the Tocantins State Health Department (LACEN-TO/SES-TO) and the Federal University of Tocantins. We used DENV-positive samples generously provided by LACEN/TO, collected between June 2021 and July 2022. All protocols and procedures strictly followed Resolution 466/2012, and the project is registered on Plataforma Brasil under CAAE number: 21010719.7.0000.
2.2. Sample collected and RNA isolation
Sample collections occurred when individuals with suspected dengue sought assistance at a primary healthcare facility. We collected serum samples from individuals exhibiting febrile illness and clinical symptoms indicative of dengue. These samples were subsequently sent to LACEN-TO. Serum aliquots were then extracted to obtain RNA using the Maxwell® RSC miRNA Plasma and Serum Kit (Promega, Madison, WI, USA).
2.3. Synthesis of cDNA and multiplex PCR
The extracted RNA was subjected to reverse transcription using Luna Script RT SuperMix (5×) (New England Biolabs, Ipswich, MA, USA). The resulting cDNAs served as templates for whole-genome amplification through multiplex PCR, utilizing the DENV sequencing primer scheme [28], which was divided into two separate pools [29]. Amplicons originating from the two primer pools were consolidated and purified using AMPure XP beads (Beckman Coulter, Brea, CA, USA). The concentrations of the purified PCR products were quantified utilizing a Qubit dsDNA HS Assay Kit with a Qubit 3.0 fluorometer (ThermoFisher Scientific Corporation, Waltham, MA, USA).
2.4. Minion whole genome sequencing
The MinION library preparation was conducted employing an SQK-LSK-109 Ligation Sequencing Kit, along with EXP-NBD104 and EXP-NBD114 Native Barcoding Kits (Oxford Nanopore, Oxford, UK). Subsequently, the resultant library was loaded onto Oxford MinION R9.4 flow cells (FLO-MIN106) and subjected to sequencing using a MinION Mk1B device. Raw data collection was facilitated through the ONT MinKNOW software. For high-accuracy base calling of raw FAST5 files and barcode demultiplexing, Guppy (v6.0.1) was employed. Consensus sequences were then generated through de novo assembly utilizing the online tool Genome Detective [30].
2.5. Phylogenetic Analyses
The 57 genome sequences of DENV-1 and the four DENV-2 genomes reported in this study were initially assigned to genotypes using the Dengue Virus Typing Tool [30]. To compare the phylogenetic relationships of the newly generated genome sequences, two datasets were constructed. The dataset for the comparison of DENV-1 genomes comprised 3,956 reference sequences of all known five genotypes (I-V) (Table S1), while the dataset for the comparison of DENV-2 genomes comprised 3,211 genomes reference sequences of all known six genotypes (I-VI) (Table S2). Both datasets were sourced from the National Center for Biotechnology Information [31]. In the selection process, only genomic sequences exceeding 9,000 bases in length and accompanied by recorded dates and sampling locations were considered. Sequence alignments were conducted using MAFFT v7.490 with default settings and further reviewed manually using AliView v1.28. Maximum likelihood (ML) phylogenies were constructed using IQ-TREE v2.2.0 [32]. The ML analyses utilized the transition model (TIM2) for nucleotide substitution, incorporating empirical base frequencies (+F) along with the FreeRate model (+R8), which were selected by the ModelFinder software [32]. The analysis comprised 1000 ultrafast bootstrap replicates (−B 1000) and an SH-aLRT branch test (−alrt 1000). Tree visualization was executed using FigTree v1.4.4 [33].
2.5.1. Bayesian Evolutionary Analysis
To explore spatial and temporal diffusion patterns, we selected sequences from three distinct DENV-1 clades (Table S3) and one DENV-2 clade (Table S4). We conducted multiple sequence alignments using MAFFT with default settings, followed by manual inspection in AliView. ML phylogenies were constructed using IQ-TREE2 [34], utilizing the TIM2+F+R8 substitution model, which was determined as the best-fit model by ModelFinder. The temporal signal assessment was performed with TempEst v1.5.3 [35] through root-to-tip genetic distance regression analysis relative to sampling dates (Figures S1 to S4). Once we confirmed sufficient temporal signal in terms of genetic divergence from the root to tip concerning sampling dates, we subjected the aligned sequences to analysis using the BEAST v1.10.4 package [36]. For molecular clock model selection, we employed stringent path-sampling (PS) and stepping stone (SS) procedures, running 100 path steps of 1 million interactions each [37].
We employed two molecular clock models: (i) the strict molecular clock model and (ii) the more flexible uncorrelated log-normal relaxed molecular clock model. These were coupled with two non-parametric population growth models: (i) the Bayesian skygrid coalescent model and (ii) the standard Bayesian skyline plot (BSP; 10 groups) [38-40]. Both SS and PS estimators consistently favored the Bayesian skyline plot with an uncorrelated log-normal relaxed molecular clock as the most appropriate model for our dataset (Table S5). We ran three independent MCMC runs, each comprising 50 million states, with sampling every 5000 steps to ensure stationarity and achieve adequate effective sample sizes (ESS) for all statistical parameters. The results from the three independent runs were consolidated using Log Combiner V1.10.4, and MCMC chain convergence was assessed using Tracer v1.7.2 [41]. The maximum clade trees from the MCMC samples were summarized by TreeAnnotator V1.10.4, with 10% burn-in removal, and the resulting MCMC phylogenetic tree was visualized using the ggtree R package [42,43].
We also employed a discrete phylogeographical model [44] to reconstruct the spatial diffusion of the virus across the sampled locations in our dataset. Phylogeographic analyses were performed using an asymmetric model of location transitions. We estimated location diffusion rates through the Bayesian stochastic search variable selection (BSSVS) model, discretized based on location (Brazilian states and countries of the Americas). To ensure reliable results, the MCMC was run for an adequate duration to achieve stationarity and an ESS exceeding 200.
2.6. Single Nucleotide Polymorphisms (SNPs) Analysis
To identify missense mutations in the newly sequenced DENV genomes from Tocantins, we initially aligned them using the Minimap2 aligner [45] against the oldest genome within each genotype and serotype. Specifically, DENV-1 genomes were aligned against JQ922544 (DENV-1 Genotype V from India, collected in 1963), while DENV-2 genomes were aligned against OK469346 (DENV-2 Genotype Southern Asian-American from Jamaica, collected in 1981). The resulting SAM files from the alignments were sorted, converted to BAM format, and indexed using Samtools V1.9 [46]. Subsequently, the BAM file underwent variant detection and genomic VCF file generation through the use of bcftools’ mpileup and bcftools call functions, both integral components of the Samtools framework [46]. Finally, we applied the bcftools filter to refine the called variations and produce the final VCF file.
3. Results
In this study, we were able to recover either partial or near-complete nucleotide sequences of 57 DENV-1 genomes (average coverage: 98%; range: 70.1 - 99.1%) (Table S6) and 4 DENV-2 genomes (average coverage: 90.7%; range: 89.8 - 91.3%) (Table S7). These genomes were recovered from DENV-infected patients in the state of Tocantins, sampled between June 2021 and July 2022. Among the 57 sequenced DENV-1 genomes, 22 (38.6%) were from patients residing in Porto Nacional municipality, while the rest were from patients in Palmas (n = 13; 22.8%) and other locations (Table S6 and Figure 1). For DENV-2, patients from Araguaína, Bandeirantes do Tocantins, and Colinas do Tocantins (Table S7 and Figure 1). No significant difference could be detected between in the gender of affected patients, since males (50.8%) and females (49.2%), with younger age groups (4 to 43 years old) being the most susceptible. The mean Ct value for RT-qPCR in the 57 samples with DENV-1 genomes was 17.8 (range: 12.1 to 23.1) (Table S6). For DENV-2 (4 genomes), the mean Ct value for RT-qPCR was 21.6 (range: 19.2 to 23.5) (Table S7).
3.1. Phylogenetic Analysis
All 57 DENV-1 genome sequences belonged to genotype V (Figure 2). Additionally, the DENV-1 genomes from Tocantins were observed to group into three distinct clades suggesting that at least three independent introduction events occurred in the Tocantins state during 2021-2022. Similarly, the four DENV-2 genomes recovered here group inside the Genotype III - Southern Asian-American clade (Figure 3).
3.1.1. Bayesian evolutionary analysis
The mean evolutionary rate estimated for Clade I (Clade Americas according to Fritsch et al., 2023 [47]) of DENV-1 Genotype V was 7.85 × 10−4 substitutions per site per year (95% highest posterior density interval: 5.96 × 10−4 to 8.63 × 10−4). The time of the most recent common ancestor (TMRCA) was estimated to be December 20, 2004 (95% HDP: October 12, 2001, to October 15, 2007). According to the Bayesian phylogeny, the genomes within Clade I from the Tocantins state formed a single subclade with strong support (posterior probability = 0.99). This subclade was part of a larger cluster that included sequences from São Paulo, Amazonas, Peru, Cuba, and even a sequence from the USA, isolated from a patient who had traveled to Cuba (OQ445881). The estimated TMRCA for this cluster was October 28, 2010 (95% HDP: September 15, 2007, to November 1, 2018), while for the Tocantins subclade, it was determined to be April 12, 2019 (95% HDP: January 18, 2018, to June 22, 2013) (Figure 4).
Clade II (Clade III according to Fritsch et al., 2023 [47]) of DENV-1 Genotype V exhibited a mean evolutionary rate of 9.18 × 10−4 substitutions per site per year (95% HDP: 7.55 × 10−4 to 1.09 × 10−3). The TMRCA for this clade was estimated to be January 22, 2003 (95% HDP: August 12, 2001, to May 20, 2004). The Bayesian phylogeny revealed a close relationship between isolates from Clade II in the Tocantins state and isolates from the São Paulo state (ON426305 and ON632048) (Figure 5). The estimated TMRCA for this clade was May 16, 2017 (95% HDP: September 18, 2014, to November 1, 2018), while the Tocantins subclade’s TMRCA was estimated to be April 12, 2019 (95% HDP: January 18, 2018, to January 13, 2020) (Figure 5). The Bayesian stochastic search variable selection (BSSVS) procedure detected supported rates of diffusion from São Paulo state to Tocantins with a Bayes Factor of 17.6 and a posterior probability of 0.65.
For Clade III (Clade IV according to Fritsch et al., 2023 [47]) of DENV-1 Genotype V, the estimated mean evolutionary rate was 1.06 × 10−3 substitutions per site per year (95% HDP: 8.69 × 10−4 to 1.16 × 10−3). The TMRCA for this clade was estimated to be November 20, 1998 (95% HDP: September 29, 1998, to December 31, 1998). The Bayesian phylogeny indicated that isolates from the Tocantins state formed a single, well-supported clade with a posterior probability of 0.99 (Figure 6). The TMRCA for the Tocantins clade was estimated to be March 1, 2019 (95% HDP: April 3, 2018, to February 29, 2020). Notably, Tocantins strains were closely related to sequences from São Paulo, including one sequence from Florida, USA, associated with travel cases (OQ445880) (Figure 6). The BSSVS procedure identified well-supported rates of diffusion from Goiás state to São Paulo (Bayes Factor: 15.1; Posterior Probability: 0.62) and Tocantins (Bayes Factor: 18.6; Posterior Probability: 0.67).
For DENV-2 clade (lineage BR-4 according to Fritsch et al., 2023 [47] and Santos et al., 2023 [14]), the estimated mean evolutionary rate was 1.25 × 10−3 substitutions per site per year (95% HDP: 8.83 × 10−4 to 1.51 × 10−3). The TMRCA was estimated to be March 20, 2016 (95% HDP: February 8, 2015, to February 22, 2017). The Bayesian phylogeny revealed that isolates from the Tocantins state formed a distinct subclade with strong support, clustering with isolates from the Goiás and São Paulo states (MT929748 and ON634741, respectively) (Figure 7). The estimated TMRCA for this clade was July 26, 2017 (95% HDP: August 5, 2016, to July 22, 2018), with the Tocantins subclade’s TMRCA estimated to be February 16, 2020 (95% HDP: September 14, 2019, to August 19, 2020). The BSSVS procedure indicated well-supported rates of DENV-2 diffusion from Goiás to Tocantins state (Bayes Factor: 75.6; Posterior Probability: 0.92).
3.2. SNPs Analysis
The genomic analysis of DENV-1 revealed a total of 122 distinct missense mutations, with 45 of these mutations being shared among the 57 sequenced genomes obtained in this study (Table S8). The majority of the shared missense mutations were located in the envelope and NS5 proteins. In clade I, which includes two genomes, exclusive point missense mutations were observed in NS1, NS2A, NS4A, 2K, NS4B, and NS5 (Table 1). Clade II, consisting of eight genomes, exhibited exclusive point missense mutations shared among them in the anchored capsid protein, NS1, NS2A, NS3, NS4A, and NS5 (Table 1). In clade III, encompassing forty-seven genomes, exclusive point missense mutations shared among them were observed in protein pr, envelope protein E, NS1, NS2A, NS4B, and NS5 (Table 1).
The genomic analysis of DENV-2 revealed a total of 44 distinct missense mutations shared among the four sequences obtained in this study (Table S9). In addition, some mutations were detected exclusively within specific proteins in particular genomes, such as those highlighted in proteins NS4A and NS5 (Table 2). Notably, a substantial portion of the identified missense mutations in DENV-2 were concentrated within the NS5 protein, 15 of those shared among all four genomes reported here. It is worth emphasizing that NS5, with approximately 2,700 bases, represents the largest protein within DENV genomes. Additionally, significant mutations were observed within the envelope protein, with eight shared missense mutations identified (Table 2).
4. Discussion
Genome sequencing has become pivotal in epidemiological analyses; yet its availability in developing countries, especially remote regions with limited facilities (though often with frequent outbreaks), remains a challenge [48]. In such a context, samples from suspected DENV cases may endure extended storage in suboptimal conditions and undergo lengthy transportation to reach adequately equipped laboratories, often resulting in sample degradation [49]. To tackle these hurdles, in this study, a portable and relatively less expensive tool, the MinION sequencer, was used throughout. In this study, 57 DENV-1 genomes and 4 DENV-2 genomes were efficiently recovered and fully or near-fully sequenced between June 2021 and July 2022, immediately upon sample collection from patients.
The recent identification of two DENV serotypes in Tocantins aligns with the Ministry of Health’s findings. Out of 25,368 tests conducted in Brazil to detect DENV serotypes up to week 44 of 2022, 84.2% (21,350) were positive for DENV-1, and 15.8% (4,018) were positive for DENV-2 [20]. However, in that study, data were primarily gathered to determine prevalent subtypes by focusing on specific regions along the genome. Furthermore, in the State of Tocantins in 2022 [50] a significant rise in dengue cases was detected within the state (18,903 cases in 124 municipalities), underscoring the need for more detailed investigations into the virus’s circulation. Thus, DENV whole-genome sequencing offers numerous advantages, including identifying strain variants, tracking viral evolution, conducting epidemiological investigations that involve studying the dispersion of viruses, and enhancing public health preparedness to address the emergence of new lineages.
Since the first dengue epidemic in Brazil in 1981 and the initial reports on the detection of DENV in Tocantins in 1991, the virus has been able to perpetuate itself across the state, with reported cases of dengue in humans predominantly concentrated in urban areas [51]. The Brazilian Ministry of Health reported that approximately 70% of documented dengue infections occur in urban areas of municipalities with more than 50,000 inhabitants experiencing economic development [52]. This phenomenon is attributed to commercial exchanges that facilitate the transmission of Aedes aegypti and DENV infections [53]. The high population density, availability of mosquito food sources, adaptability of mosquitoes to urban environments, and neglect in controlling mosquito breeding sites are potential factors contributing to this trend [54]. The findings of this study further corroborate that municipalities with populations exceeding 50,000 are the most affected by DENV-1 and DENV-2. Moreover, similar results were observed by Santos et al. (2009) [55] in their investigation of the epidemiological profile of dengue in a neighboring city, Anápolis, in the state of Goiás.
Our study revealed that DENV infection can affect people of all age groups, but individuals aged 4 to 43 years had a higher incidence rate. These findings are consistent with those of Valadares et al. (2012) [56], who analyzed the epidemiological and environmental characteristics of dengue in the two largest cities in the State of Tocantins over an 11-year period (2000 to 2010). Valadares et al. (2012) [56] found that young adults between 20 and 39 years of age were the most affected. In our study, children and young adults were the age groups where the infection was most prevalent. Monteiro et al. (2009) [57] found similar results when they examined the epidemiological and vector aspects of dengue in the city of Teresina, Piauí, Brazil, from 2002 to 2006. They found that the age group most affected was 15 to 49 years old. Therefore, our study suggests a tendency for dengue infections to occur in younger age groups (4 to 13 and 14 to 23 years old), which could be due to the fact that young people generally lead more active lifestyles and spend more time outdoors, increasing their exposure to a positive mosquito with vector competence for the DENV.
4.1. Phylogenetic Analysis of DENV-1 and DENV-2
DENV-1 was initially detected in the 1980s in the Northern Region of Brazil, specifically in the state of Roraima. Subsequently, this serotype has spread extensively throughout Brazil [22], including the state of Tocantins, as well as numerous other states across the nation [58]. The sequenced genomes of DENV-1 in this study are classified as Genotype V, encompassing strains from the Americas, West Africa, Asia, and the Indian Ocean archipelagos (Comoros, La Reunion, and Seychelles), according to Chen and Vasilakis’ (2011) [59] classification. Similar investigations have also confirmed the circulation of genotype V in the Americas [60-62]. Genotype V has remained the predominant DENV-1 genotype in circulation across the American continent, with genetic makeup reflecting geographical associations [47]. Carvalho et al. (2010) [63] observed that the genetic makeup of DENV-1 strains exhibits geographical associations with the countries from which they were isolated or recovered, given the relatively infrequent movement between countries compared to dissemination within specific countries. In the study conducted by Bruycker-Nogueira et al. (2018) [12], continuous molecular surveillance spanning 30 years of DENV-1 circulation in Brazil affirmed the ongoing prevalence of genotype V in the country.
In our study, the analysis of 57 DENV-1 genome sequences from the Tocantins state has provided valuable insights into the evolution and transmission dynamics of these dengue virus serotypes. A significant discovery emerged as these sequences clustered into three distinct clades, suggesting the occurrence of at least three independent introduction events in Tocantins during the period of 2021-2022. The phylogenetic analysis not only shed light on the relationships between virus strains within Tocantins but also established connections with sequences from other states in Brazil. This demonstrates the complex transmission dynamics of dengue viruses and their potential for long-distance spread.
Additionally, we successfully recovered and fully sequenced four genomes of DENV-2, classifying them as Southeast Asian/American genotype III. These genomes fall into two distinct clades within the phylogenetic tree. One clade comprises strains from Southeast Asia, while the other encompasses strains from Central America, South America, and the Caribbean over the past three decades [59]. The Southeast Asian/American genotype III has been associated with epidemics of DHF/DSS in Latin American countries upon its introduction [64]. Furthermore, this genotype has had a significant epidemiological impact, typically leading to the co-circulation of distinct genotypes of the same serotype in the regions where it is introduced [65]. DENV-2 of the Southeast Asian/American genotype III has been circulating in Brazil since at least the 1990s [66] and has been responsible for several DHF/DSS epidemics in the Americas [67]. This genotype predominates in Brazil at the moment [14,47,68].
4.1.1. Bayesian evolutionary analysis
The analysis of the phylogenetic tree confirms the distinct presence of DENV-1 Genotype V and shows a dynamic viral evolution within the different clades [69,70]. The DENV-1 genomes recovered here exhibit a clear clustering within genotype V, affirming its distinct presence. Additionally, DENV-1 genomes from Tocantins were shown to exhibit a diverse distribution across three separate clades, suggesting a dynamic viral evolution within the region, with several distinct DENV-1 introductions occurring into the state. Similarly, the alignment of DENV-2 genomes with representatives of Genotype III - Southern Asian-American clade reinforces previous findings [71,72]. Notably, the evolutionary rates within Clade I of DENV-1 Genotype V were estimated, revealing significant genetic dynamics. In the analysis of genomic sequences from Tocantins within Clade I, supported by Bayesian analysis, we observe a broader connection with sequences from various South American regions, including likely travel-related cases. Moving on to Clade II of DENV-1 Genotype V, the genetic proximity between Tocantins and São Paulo isolates, along with detected diffusion rates, highlights possible dissemination patterns. Furthermore, the strong clade formation by Tocantins isolates in Clade III underscores their genetic coherence and shared history with São Paulo, and even an international sequence. A similar pattern emerges for DENV-2, as Bayesian phylogeny shows a distinct Tocantins subclade with strong support, linked to Goiás and São Paulo. Diffusion rates between these states, revealed by the BSSVS procedure, emphasize the complex dynamics of DENV-2 transmission [73]. In summary, these findings shed light on the intricate evolutionary pathways and geographical spread of DENV-1 and DENV-2 to Brazilian states and other Latin American countries, including the USA.
Regarding the timing of these events, the Bayesian phylogeny shows that the genomes within Clade I of DENV-1 Genotype V from Tocantins state formed a single subclade within a larger cluster, with an estimated TMRCA of 2010. Clade II of DENV-1 Genotype V exhibits a close relationship between isolates from Tocantins and São Paulo, with a TMRCA estimated in 2017, while the Tocantins subclade’s TMRCA was estimated to be in 2019. For Clade III of DENV-1 Genotype V, isolates from the Tocantins state formed a single clade, with a TMRCA in 2019. Notably, Tocantins strains were closely related to sequences from São Paulo, including one sequence from Florida, USA, associated with travel cases. In the case of DENV-2, isolates from Tocantins formed a distinct subclade with an estimated TMRCA of 2017, and the dating of the TMRCA DENV-2 Tocantins subclade was estimated to be 2020. In summary, the temporal analysis of DENV-1 and -2 corroborated with recent studies [14,47,71]. Additionally, these findings highlight the intricate evolutionary pathways and geographical spread of DENV-1 and DENV-2 to Brazilian states and other Latin American countries, including the USA. The diversity and dispersion of these viruses to neighboring states and countries emphasize the ongoing genetic diversity and complexity, underscoring the critical need for continuous genomic surveillance to inform dengue control and prevention strategies.
4.2. SNPs analysis
In the genomic analysis of DENV-1, we identified a total of 122 distinct missense mutations, of which 45 were shared among the 57 genomes sequenced in this study. The majority of such mutations were found in the envelope protein (seven mutations: Glu10Asp, Leu31Val, His32Thr, Gln36Lys, Thr157Glu, Ala369Thr, Ile454Thr). The envelope protein is important in mediating viral entry and assembly of progeny virus during cellular infection [74]. The gene coding for the NS5 protein also exhibited seven mutations in 45 genomes (Thr135Met, Ala370Thr, Met408Ile, Ser585Asn, Lys732Glu, Lys814Arg, Asp834Glu). This is very important because NS5 is the most highly conserved among the viral NS proteins and is responsible for the capping, methylation, and replication of the RNA genome [75]. Additionally, we observed eleven exclusive point missense mutations in the two genomes grouped in clade I and fifteen exclusive point missense mutations in the eight genomes in clade II. For the forty-seven genomes classified in clade III, thirteen exclusive point missense mutations were observed. In contrast, in the genomic analyses of DENV-2, 44 distinct missense mutations were found, with three additional exclusive mutations in individual genomes. Notably, a significant number of these mutations were concentrated in the region coding for NS5, with 15 mutations shared among the four genomes. Furthermore, we observed eight shared missense mutations in the gene coding for the envelope protein of DENV-2. These findings highlight the genomic diversity and mutation patterns in both DENV-1 and DENV-2, with potential implications for viral evolution and pathogenicity [14,47].
In light of the imminent large-scale utilization of a Dengue virus vaccine, it is imperative to emphasize the importance of providing comprehensive genomic data regarding viral diversity within specific regions and elucidating its spatial distribution, while identifying potential risk zones. Finding high risk areas and identifying vulnerable demographic groups is pivotal for devising effective preventive strategies and conducting awareness campaigns. Brazil is currently adopting the Dengue vaccine TAK-003, as highlighted by Rivera et al. (2022) [76]. It is conceivable that specific SNPs within this vaccine may persist and perpetuate through subsequent generations of the virus. Given the immunological pressure exerted by the Dengue vaccine on the vaccinated population, the emergence of new viral mutations is expected [24]. Consequently, the study of these mutations assumes paramount importance in the context of monitoring vaccine efficacy and comprehending the intricate landscape of viral-host co-evolution.
Some limitations need to be considered. Firstly, the accessibility of genome sequencing tools remains a significant challenge, particularly in developing countries and remote areas with limited facilities. Second, the specimens collected from suspected DENV cases are often subjected to unfavorable storage conditions and long transportation distances to better-equipped laboratories, leading to sample degradation that hampers successful sequencing efforts [49]. While the study highlights the use of portable sequencers like the MinION to address these challenges, it’s important to note that portable sequencers might have their own technical limitations that could impact data quality and accuracy. Additionally, the study focuses on a specific time frame for sample collection (June 2021 to July 2022), which might not fully capture the broader temporal dynamics of DENV spread and evolution. Furthermore, the study’s sample size is relatively small, with 57 DENV-1 genomes and 4 DENV-2 genomes, limiting the generalizability of the findings. Despite these limitations, the study provides valuable insights into the genomic characteristics and spread of DENV-1 and DENV-2 between Brazilian states and other American countries, contributing to the understanding of disease epidemiology and aiding in the formulation of control strategies.
5. Conclusions
The current study involved the full or nearly full genome sequencing of DENV samples from patients from the State of Tocantins, north-central Brazil, providing valuable information on genotype classification and viral dispersion. Compared to usual DENV typing protocols that do not involve full genome sequencing, our approach was shown to be more reliable for genotype identification. Our findings confirm the circulation of DENV serotypes 1 and 2 in Tocantins, affecting males and females equally, with younger age groups (4 to 43 years old) being the most susceptible. The phylogenetic analysis of the full/nearly full genomes revealed that the DENV-1 genomes belong to genotype V American, while the DENV-2 genomes belong to the genotype III Southeast Asian/American clade. In the phylogenetic analysis of DENV-1 Genotype V, isolates from Tocantins closely clustered with sequences from São Paulo. Additionally, in the case of the DENV-2 clade, Tocantins isolates formed a distinct, well-supported subclade, grouping together with isolates from Goiás and São Paulo states. This observation highlights the significant impact of major population centers in Brazil on virus dispersion, including other Latin-American countries and the USA. In the SNP analysis, DENV-1 displayed 122 distinct missense mutations, while DENV-2 had 44, with significant mutations predominantly occurring in the envelope and NS5 proteins. Thus, our analyses reveal the presence of distinct DENV-1 and -2 genotypes and dispersion among Brazilian states, underscoring the dynamic evolution of the DENV and the ongoing significance of surveillance efforts in supporting public health policies.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1. Root-to-tip regression of genetic distances and sampling dates for 76 complete and near-complete Clade I of DENV-1 Genotype V sequences sampled in Brazil, South America, and North America. The correlation coefficient (r) and r squared are depicted above the graph. Figure S2. Root-to-tip regression of genetic distances and sampling dates for 63 complete and near-complete Clade II of DENV-1 Genotype V sequences sampled in Brazil and South America. The correlation coefficient (r) and r squared are depicted above the graph. Figure S3. Root-to-tip regression of genetic distances and sampling dates for 170 complete and near-complete Clade III of DENV-1 Genotype V sequences sampled in Brazil, South America, and North America. The correlation coefficient (r) and r squared are depicted above the graph. Figure S4. Root-to-tip regression of genetic distances and sampling dates for 63 complete and near-complete DENV-2 sequences sampled in Brazil. The correlation coefficient (r) and r squared are depicted above the graph. Table S1. DENV-1 Genomes of All Known Five Genotypes (I-V) Obtained from GenBank. Table S2. DENV-2 Genomes of All Known Six Genotypes (I-VI) Obtained from GenBank. Table S3. DENV-1 sequences clades I to III Obtained from GenBank. Table S4. DENV-2 sequences clades I Obtained from GenBank. Table S5. Model comparison of the strict molecular clock. uncorrelated relaxed clock and demographic growth models through path sampling (PS) and stepping stone (SS) methods. Blue numbers indicate the best-fitting model. Table S6. Summary of data of patients from which DENV-1 genomes were recovered, Cts, and coverage of the RT-qPCR of samples sequenced in this study. Table S7. Summary of data of patients from which DENV-2 genomes were recovered, Cts, and coverage of the RT-qPCR of samples sequenced in this study. Table S8. Missense mutations found in the newly sequenced genomes of DENV-1 from Tocantins were reported in this study. Table S9. Missense mutations found in the newly sequenced genomes of DENV-9 from Tocantins were reported in this study.
Author Contributions
Conceived and designed the experiments: U.J.B.d.S and F.S.C. Performed the experiments: U.J.B.d.S. and F.D.P.C. Analyzed the data: U.J.B.d.S and Y.S.M.M. Contributed to the writing of the manuscript: U.J.B.d.S., F.S.C., F.D.P.C and Y.S.M.M. Reviewed the manuscript: U.J.B.d.S., Y.S.M.M., R.N.S., F.D.P.C., J.D.G., E.G.G., A.C.F., P.M.R., F.R.S. and F.S.C. All authors have read and agreed to the published version of the manuscript. A.C.F., F.S.C, F.R.S., and P.M.R. are CNPq research fellows.
Funding
This work was partially supported by grants from Rede Corona-ômica BR MCTI/FINEP (http://www.corona-omica.br-mcti.lncc.br, accessed on 12 January 2023) affiliated by RedeVírus/MCTI (FINEP = 01.20.0029.000462/20, CNPq = 404096/2020-4). Fundação de Amparo à Pesquisa of Tocantins states (FAPT) paid for the DENV-1 and -2 primers. National Council for Scientific and Technological Development (CNPq Process number: 443215/2019-7) and the Federal University of Tocantins help to pay publishing charges for Open Access articles.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper. The DENV-1 genome sequences have been deposited in GenBank under the accession numbers OR518216 to OR518272. The DENV-2 genome sequences have also been deposited in GenBank with the following accession numbers: OR486032 to OR486035.
Acknowledgments
We thank the entire team at Lacen-TO for their tireless work during the SARS-CoV-2 pandemic and during another epidemic outbreak of arboviruses. U.J.B.d.S. was granted a post-doctoral scholarship (DTI-A) from CNPq. A.C.F., P.M.R., F.R.S., and F.S.C. are CNPq research fellows.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siler, J.F.; Hall, M.W.; Hitchens, A. Dengue, its history, epidemiology, mechanism of transmission, etiology, clinical manifestations, immunity and prevention. Philipp J. Sci. 1926, 29, 1–304. [Google Scholar]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Gubler, D.J. Dengue and dengue hemorrhagic fever in the Americas. P R Health Sci J. 1987, 6, 107–111. [Google Scholar]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; Stapleton, J.T. ; ICTV Virus Taxonomy Profile: Flaviviridae. J Gen Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Lewis, J.G.; Gubler, D.J.; Trent, D.W. Molecular evolution and epidemiology of dengue-3 viruses. J. Gen. Virol. 1994, 75, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Gubler, D.J.; Trent, D.W. Molecular evolution and phylogeny of dengue-4 viruses. J. Gen. Virol. 1997, 78, 2279–2286. [Google Scholar] [CrossRef]
- Reich, N.G.; Shrestha, S.; King, A.A.; Rohani, P.; Lessler, J.; Kalayanarooj, S.; Yoon, I.K.; Gibbons, R.V.; Burke, D.S.; Cummings, D.A. Interactions between serotypes of dengue highlight epidemiological impact of cross-immunity. J. R. Soc. Interface. 2013, 10, 20130414. [Google Scholar] [CrossRef]
- Sabin, A.B. Research on dengue during World War II. Am. J. Trop. Med. Hyg. 1952, 1, 30–50. [Google Scholar] [CrossRef]
- Kyle, J.L.; Harris, E. Global spread and persistence of dengue. Annu. Rev. Microbiol. 2008, 62, 71–92. [Google Scholar] [CrossRef]
- Weaver, S.C.; Vasilakis, N. Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Infect. Genet. Evol. 2009, 9, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Bruycker-Nogueira, F.; Souza, T.M.A.; Chouin-Carneiro, T.; Costa Faria, N.R.; Santos, J.B.; Torres, M.C.; Ramalho, I.L.C.; Aguiar, S.F.; Nogueira, R.M.R.; Filippis, A.M.B.; Santos, F.B. DENV-1 Genotype V in Brazil: Spatiotemporal dispersion pattern reveals continuous co-circulation of distinct lineages until 2016. Sci Rep. 2018, 8, 17160. [Google Scholar] [CrossRef] [PubMed]
- San Martín, J.L.; Brathwaite, O.; Zambrano, B.; Solórzano, J.O.; Bouckenooghe, A.; Dayan, G.H.; Guzmán, M.G. The epidemiology of dengue in the Americas over the last three decades: a worrisome reality. Am. J. Trop. Med. Hyg. 2010, 82, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.M.; Pavon, J.A.R.; Dias, L.S.; Viniski, A.E.; Souza, C.L.C.; de Oliveira, E.C.; de Azevedo, V.C.; da Silva, S.P.; Cruz, A.C.R.; Medeiros, D.B.A.; Nunes, M.R.T.; Slhessarenko, R.D. Dengue virus serotype 2 genotype III evolution during the 2019 outbreak in Mato Grosso, Midwestern Brazil. Infect. Genet. Evol. 2023, 113, 105487. [Google Scholar] [CrossRef]
- Yenamandra, S.P.; Koo, C.; Chiang, S.; Lim, H.S.J.; Yeo, Z.Y.; Ng, L.C.; Hapuarachchi, H.C. Evolution, heterogeneity and global spread of the cosmopolitan genotype of Dengue virus type 2. Sci. Rep. 2021, 11, 13496. [Google Scholar] [CrossRef]
- Torres, M.C.; Lima de Mendonça, M.C.; Damasceno Dos Santos Rodrigues, C.; Fonseca, V.; Ribeiro, M.S.; Brandão, A.P.; Venâncio da Cunha, R.; Dias, A.I.; Santos Vilas Boas, L.; Felix, A.C.; Alves Pereira, M.; de Oliveira Pinto, L.M.; Sakuntabhai, A.; On Behalf Of ZikAction Consortium. Dengue Virus Serotype 2 Intrahost Diversity in Patients with Different Clinical Outcomes. Viruses 2021, 13, 349. [Google Scholar] [CrossRef]
- Vasconcelos, P.F.C.; Travassos da Rosa, E.S.; Travassos da Rosa, J.F.S.; Freitas, R.B. .; Dégallier, N.; Rodrigues, S.G.; Travassos da Rosa, A.P.A. Epidemia de febre clássica de dengue causada pelo sorotipo 2 em Araguaiana, Tocantins, Brasil. Rev. Inst. Med. Trop. Sao Paulo 1993, 35, 141–148. [Google Scholar] [CrossRef]
- Souza, U.J.B.; Dos Santos, R.N.; Campos, F.S.; Lourenço, K.L.; Fonseca, F.G.; Spilki, F.R.; Corona-Ômica Br/McTi Network. High Rate of Mutational Events in SARS-CoV-2 Genomes across Brazilian Geographical Regions, February 2020 to June 2021. Viruses 2021, 13, 1806. [Google Scholar] [CrossRef]
- TOCANTINS. Secretaria de Saúde do Tocantins. Monitoramento das Arboviroses Urbanas (dengue, chikungunya e Zika) e Silvestre (febre amarela) no ano de 2020. Palmas – TO. Available online: https://central.to.gov.br/download/250434 (accessed on 6 October 2022).
- Brasil. Ministério da Saúde. Monitoramento dos casos de arboviroses até a semana epidemiológica 52 de 2022. Available online: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/boletins/epidemiologicos/edicoes/2023/boletim-epidemiologico-volume-54-no-01/ (accessed on 6 February 2023).
- Brasil. Ministério da Saúde. Diretrizes Nacionais para a Prevenção e Controle de Epidemias de Dengue. Brasilia – DF. Secretaria de Vigilância em Saúde. 2009. Available online: https://bvsms.saude.gov.br/bvs/publicacoes/diretrizes_nacionais_prevencao_controle_dengue.pdf (accessed on 5 December 2022).
- Brasil. Ministério da Saúde. Departamento de Vigilância Epidemiológica. Guia de Vigilância Epidemiológica. Brasilia – DF. Secretaria de Vigilância em Saúde. 2005. Available online: https://bvsms.saude.gov.br/bvs/publicacoes/Guia_Vig_Epid_novo2.pdf (accessed on 6 February 2023).
- Brazil. Ministry of Health of Brazil. Secretariat of Health Surveillance. Department of Noncommunicable Diseases Surveillance and Health Promotion. Health Brazil 2015/2016: an analysis of health situation and the epidemic caused by Zika virus and other diseases transmitted by Aedes aegypti, 2017. Available online: https://bvsms.saude.gov.br/bvs/publicacoes/health_brazil_2015_2016.pdf (accessed on 9 September 2023).
- Abrão, E.P.; Espósito, D. L.; Lauretti, F.; da Fonseca, B. A. Dengue vaccines: what we know, what has been done, but what does the future hold? Rev. Saude Publica 2015, 49, 60. [Google Scholar] [CrossRef]
- Gonçalves, C.M.; Melo, F.F.; Bezerra, J.M.; Chaves, B.A.; Silva, B.M.; Silva, L.D.; Pessanha, J.E.M.; Arias, J.R.; Secundino, N.F.C.; Norris, D.E.; Pimenta, P.F.P. Distinct variation in vector competence among nine field populations of Aedes aegypti from a Brazilian dengue-endemic risk city. Parasites Vectors 2014, 7, 320. [Google Scholar] [CrossRef]
- Holmes, E.C.; Twiddy, S.S. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 2003, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Áñez, G.; Evolución molecular del virus dengue: un área de investigación prioritaria. Investigación Clínica, 2007 48(3):273-276. Available online: http://ve.scielo.org/scielo.php?script=sci_arttext&pid=S0535-51332007000300001&lng=es (accessed on 6 February 2023).
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, S.C.B.; Blacklaws, B.A.; Yohan, B.; Yudhaputri, F.A.; Hayati, R.F.; Schwem, B.; Salvaña, E.M.; Destura, R.V.; Lester, J.S.; Myint, K.S.; Sasmono, R.T.; Frost, S.D.W. Assessment of a multiplex PCR and Nanopore-based method for dengue virus sequencing in Indonesia. Virol. J. 2020, 17, 24. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.M.; Deforche, K.; de Oliveira, T. Genome Detective: an automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Figtree v1.4.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 9 September 2023).
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using tempest (formerly path-o-gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M. A.; Alekseyenko, A. V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.T.; Zhu, H.; Guan, Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef] [PubMed]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinformatics 2020, 69, e96. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Fritsch, H.; Moreno, K.; Lima, I.A.B.; Santos, C.S.; Costa, B.G.G.; de Almeida, B.L.; Dos Santos, R.A.; Francisco, M.V.L.O.; Sampaio, M.P.S.; de Lima, M.M.; Pereira, F.M.; Fonseca, V.; Tosta, S.; Xavier, J.; de Oliveira, C.; Adelino, T.; de Mello, A.L.E.S.; Gräf, T.; Alcantara, L.C.J.; Giovanetti, M.; de Siqueira, I.C. Phylogenetic Reconstructions Reveal the Circulation of a Novel Dengue Virus-1V Clade and the Persistence of a Dengue Virus-2 III Genotype in Northeast Brazil. Viruses 2023, 15, 1073. [Google Scholar] [CrossRef]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, A.; Manoharan, M. Chapter 16 - Dengue Virus. Emerging and Reemerging Viral Pathogens. Ed. Academic Press: Cambridge, MA, USA, 2020; pp. 281-359. [CrossRef]
- TOCANTINS. Secretaria de Saúde do Tocantins. Monitoramento dos casos de dengue até a semana epidemiológica 43. Palmas – TO. Boletins Epidemiológicos. 2022. Available online: https://central.to.gov.br/download/307814 (accessed on 3 January 2023).
- Salles, T.S.; da Encarnação Sá-Guimarães, T.; de Alvarenga, E.S.L.; Guimarães-Ribeiro, V.; de Meneses, M.D.F.; de Castro-Salles, P.F.; Dos Santos, C.R.; do Amaral Melo, A.C.; Soares, M.R.; Ferreira, D.F.; Moreira, M.F. History, epidemiology and diagnostics of dengue in the American and Brazilian contexts: a review. Parasit. Vectors 2018, 11, 264. [Google Scholar] [CrossRef] [PubMed]
- Siqueira Jr., J. B.; Martelli, C.M.; Coelho, G.E.; Simplicio, A.C.; Hatch, D.L. Dengue and dengue hemorrhagic fever, Brazil, 1981-2002. Emerg. Infect. Dis. 2005, 11, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Brady, O.J.; Scott, T.W.; Zou, C.; Pigott, D.M.; Duda, K.A.; Bhatt, S.; Katzelnick, L.; Howes, R.E.; Battle, K.E.; Simmons, C.P.; Hay, S.I. Global spread of dengue virus types: mapping the 70 year history. Trends Microbiol. 2014, 22, 138–146. [Google Scholar] [CrossRef]
- Araújo, H.R.; Carvalho, D.O.; Ioshino, R.S.; Costa-da-Silva, A.L.; Capurro, M.L. Aedes aegypti Control Strategies in Brazil: Incorporation of New Technologies to Overcome the Persistence of Dengue Epidemics. Insects. 2015, 11, 576–94. [Google Scholar] [CrossRef]
- Santos, C.H.; Sousa, F.Y.; Lima, L.R.; Stival, M.M. Perfil Epidemiológico do Dengue em Anápolis-GO, 2001 – 2007. Rev. Pat. Trop. 2009, 38, 249–259. [Google Scholar] [CrossRef]
- Valadares, A.F.; Rodrigues, C. Filho, J.; Peluzio, J.M. Impacto da dengue em duas principais cidades do Estado do Tocantins: infestação e fator ambiental (2000 a 2010). Epidemiol. Serv. Saúde 2013, 22, 59–66. [Google Scholar] [CrossRef]
- Monteiro, E.S.C.; Coelho, M.E.; Cunha, I.S.; Cavalcante, M.A.S.; Carvalho, F.A.A. Aspectos epidemiológicos e vetoriais da dengue na cidade de Teresina, Piauí - Brasil, 2002 a 2006. Epidemiol. Serv. Saúde 2009, 18, 365–374. [Google Scholar] [CrossRef]
- Nogueira, R.M.; de Araújo, J.M.; Schatzmayr, H.G. Dengue viruses in Brazil, 1986-2006. Rev. Panam. Salud Publica 2007, 22, 358–363. [Google Scholar] [CrossRef]
- Chen, R.; Vasilakis, N. Dengue--quo tu et quo vadis? Viruses 2011, 3, 1562–1608. [Google Scholar] [CrossRef]
- Áñez, G.; Heisey, D.A.; Espina, L.M.; Stramer, S.L.; Rios, M. Phylogenetic analysis of dengue virus types 1 and 4 circulating in Puerto Rico and Key West, Florida, during 2010 epidemics. Am. J. Trop. Med. Hyg. 2012, 87, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.R.; Cruz, A.C.R.; Vallinoto, M.; Melo, D.V.; Ramos, R.T.J.; Medeiros, D.B.A.; Silva, E.V.P.; Vasconcelos, P.F.C. Molecular characterisation of dengue virus type 1 reveals lineage replacement during circulation in Brazilian territory. Mem. Inst. Oswaldo Cruz 2012, 107, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Drumond, B.P.; Mondini, A.; Schmidt, D.J.; Bosch, I.; Nogueira, M.L. Population dynamics of DENV-1 genotype V in Brazil is characterized by co-circulation and strain/lineage replacement. Arch. Virol. 2012, 157, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.E.; Martin, D.P.; Oliveira, L.M.; Ribeiro, B.M.; Nagata, T. Comparative analysis of American Dengue virus type 1 full-genome sequences. Virus Genes 2010, 40, 60–66. [Google Scholar] [CrossRef]
- Rico-Hesse, R.; Harrison, L.M.; Salas, R.A.; Tovar, D.; Nisalak, A.; Ramos, C.; Boshell, J.; de Mesa, M.T.; Nogueira, R.M.; da Rosa, A.T. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 1997, 230, 244–251. [Google Scholar] [CrossRef]
- Rico-Hesse, R. Dengue virus evolution and virulence models. Clin. Infect. Dis. 2007, 44, 1462–1466. [Google Scholar] [CrossRef]
- Torres, M.C.; de Bruycker Nogueira, F.; Fernandes, C.A.; Louzada Silva Meira, G.; Ferreira de Aguiar, S.; Chieppe, A.O.; Bispo de Filippis, A.M. Re-introduction of dengue virus serotype 2 in the state of Rio de Janeiro after almost a decade of epidemiological silence. PLoS One 2019, 14, e0225879. [Google Scholar] [CrossRef]
- Nogueira, R.M.; Miagostovich, M.P.; Lampe, E.; Schatzmayr, H.G. Isolation of dengue virus type 2 in Rio de Janeiro. Mem. Inst. Oswaldo Cruz 1990, 85, 253. [Google Scholar] [CrossRef]
- Jesus, J.G.; Dutra, K.R.; Sales, F.C. da S.; Claro, I.M.; Terzian, A.C.; Candido, D. da S.; et al. Genomic detection of a virus lineage replacement event of dengue virus serotype 2 in Brazil, 2019. Mem. Inst. Oswaldo Cruz 2020, 115, e190423. [Google Scholar] [CrossRef]
- Carrillo-Valenzo, E.; Danis-Lozano, R.; Velasco-Hernández, J.X.; Sánchez-Burgos, G.; Alpuche, C.; López, I.; Rosales, C.; Baronti, C.; de Lamballerie, X.; Holmes, E.C.; Ramos-Castañeda, J. Evolution of dengue virus in Mexico is characterized by frequent lineage replacement. Arch Virol. 2010, 155, 1401–1412. [Google Scholar] [CrossRef]
- Zhang, C.; Mammen, M.P. Jr.; Chinnawirotpisan, P.; Klungthong, C.; Rodpradit, P.; Monkongdee, P.; Nimmannitya, S.; Kalayanarooj, S.; Holmes, E.C. Clade replacements in dengue virus serotypes 1 and 3 are associated with changing serotype prevalence. J. Virol. 2005, 79, 15123–15130. [Google Scholar] [CrossRef]
- Giovanetti, M.; Pereira, L.A.; Santiago, G.A.; Fonseca, V.; Mendoza, M.P.G.; de Oliveira, C.; de Moraes, L.; Xavier, J.; Tosta, S.; Fritsch, H.; et al. Emergence of Dengue Virus Serotype 2 Cosmopolitan Genotype, Brazil. Emerg. Infect. Dis. 2022, 28, 1725–1727. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Mayer, S.V.; Johnson, W.L.; Chen, R.; Volkova, E.; Vilcarromero, S.; Widen, S.G.; Wood, T.G.; Suarez-Ognio, L.; Long, K.C.; et al. Lineage II of Southeast Asian/American DENV-2 is associated with a severe dengue outbreak in the Peruvian Amazon. Am. J. Trop. Med. Hyg. 2014, 91, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M. dos P.; Guimarães, V.N.; Souza, M.; de Paula Cardoso, D.; de Almeida, T.N.; de Oliveira, T. S.; Fiaccadori, F.S. Phylodynamics of DENV-1 reveals the spatiotemporal co-circulation of two distinct lineages in 2013 and multiple introductions of dengue virus in Goiás, Brazil. Infect. Genet. Evol. 2016, 43, 130–134. [Google Scholar] [CrossRef]
- Wispelaere, M.; Yang, P.L. Mutagenesis of the DI/DIII linker in dengue virus envelope protein impairs viral particle assembly. J. Virol. 2012, 86, 7072–83. [Google Scholar] [CrossRef]
- Tay, M.Y.; Smith, K.; Ng, I.H.; Chan, K.W.; Zhao, Y.; Ooi, E.E.; Lescar, J.; Luo, D.; Jans, D.A.; Forwood, J.K.; Vasudevan, S.G. The C-terminal 18 Amino Acid Region of Dengue Virus NS5 Regulates its Subcellular Localization and Contains a Conserved Arginine Residue Essential for Infectious Virus Production. PLoS Pathog 2012, 12, e1005886. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.; Biswal, S.; Sáez-Llorens, X.; Reynales, H.; López-Medina, E.; Borja-Tabora, C.; Bravo, L.; Sirivichayakul, C.; Kosalaraksa, P.; Martinez Vargas, L.; et al. Three-year Efficacy and Safety of Takeda’s Dengue Vaccine Candidate (TAK-003). Clin. Infect. Dis. 2022, 75, 107–117. [Google Scholar] [CrossRef]
Figure 1.
Political map (MOU1) of Brazilian regions with highlights on the Tocantins state, showing locations of the origin of the patients in which the DENV-1 and DENV-2 genomes reported in this study were collected.
Figure 1.
Political map (MOU1) of Brazilian regions with highlights on the Tocantins state, showing locations of the origin of the patients in which the DENV-1 and DENV-2 genomes reported in this study were collected.
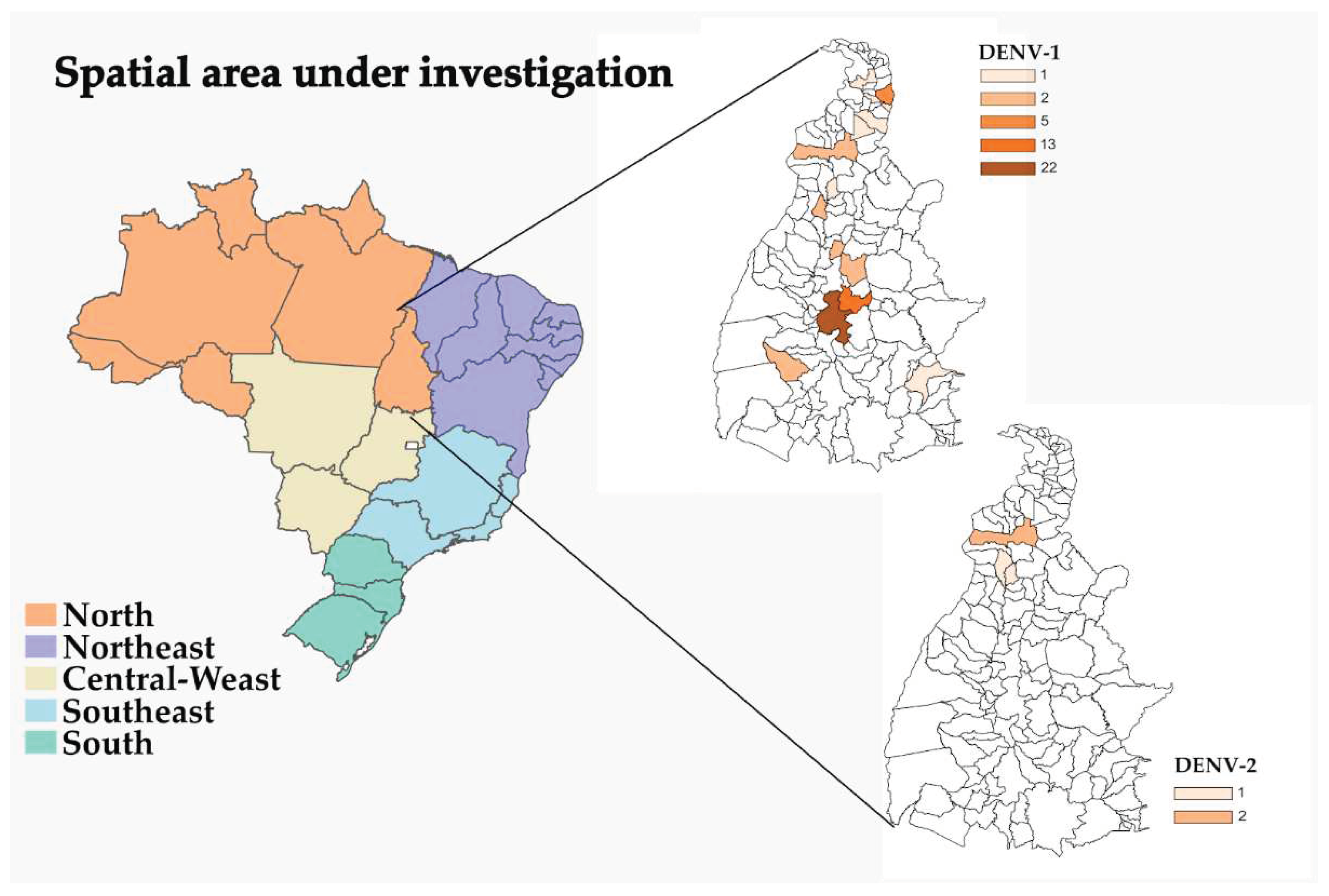
Figure 2.
Phylogenetic tree with genomes of all DENV-1 genotypes available at GenBank (access 2023/04/14). Colors indicate the main DENV genotypes. The 57 genomes of DENV-1 reported in the present study are marked by red dots.
Figure 2.
Phylogenetic tree with genomes of all DENV-1 genotypes available at GenBank (access 2023/04/14). Colors indicate the main DENV genotypes. The 57 genomes of DENV-1 reported in the present study are marked by red dots.
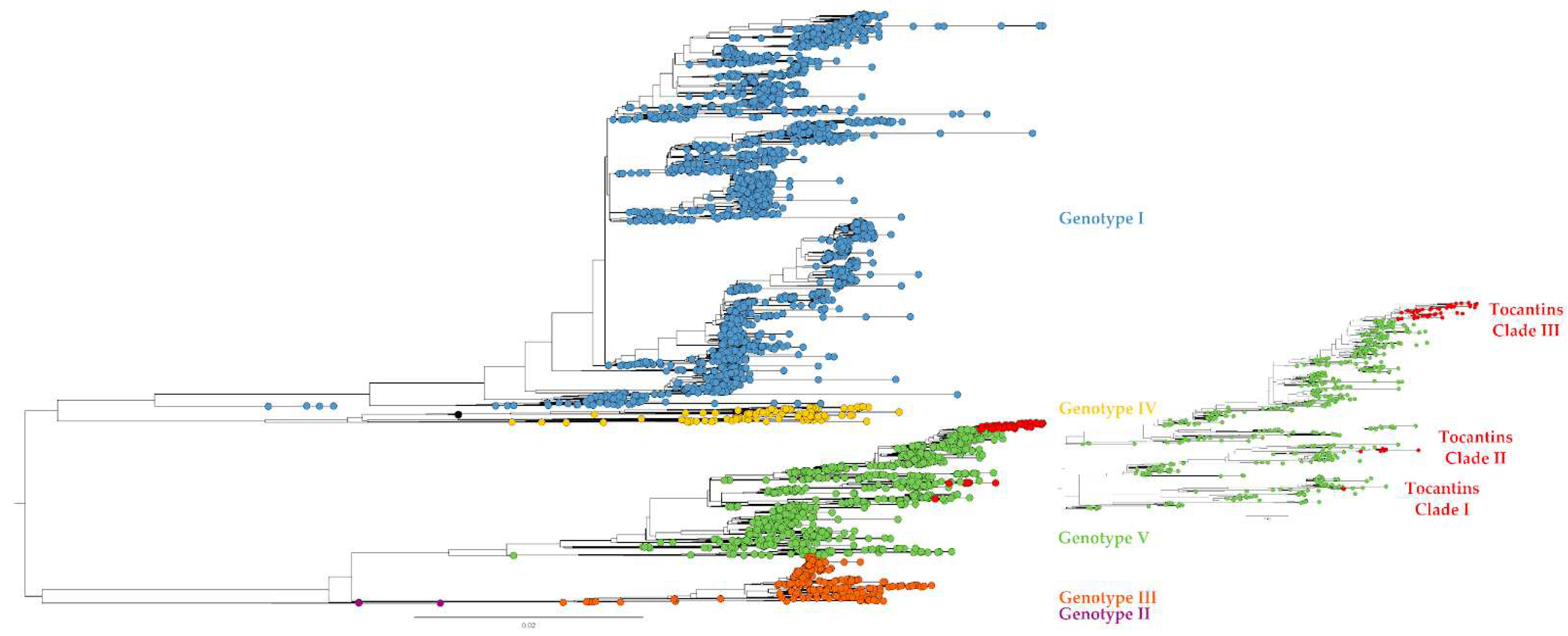
Figure 3.
Phylogenetic tree with genomes of all DENV-2 genotypes available at GenBank. Colors indicate the different genotypes. The four genomes of DENV-2 reported in the present study are marked by red dots.
Figure 3.
Phylogenetic tree with genomes of all DENV-2 genotypes available at GenBank. Colors indicate the different genotypes. The four genomes of DENV-2 reported in the present study are marked by red dots.
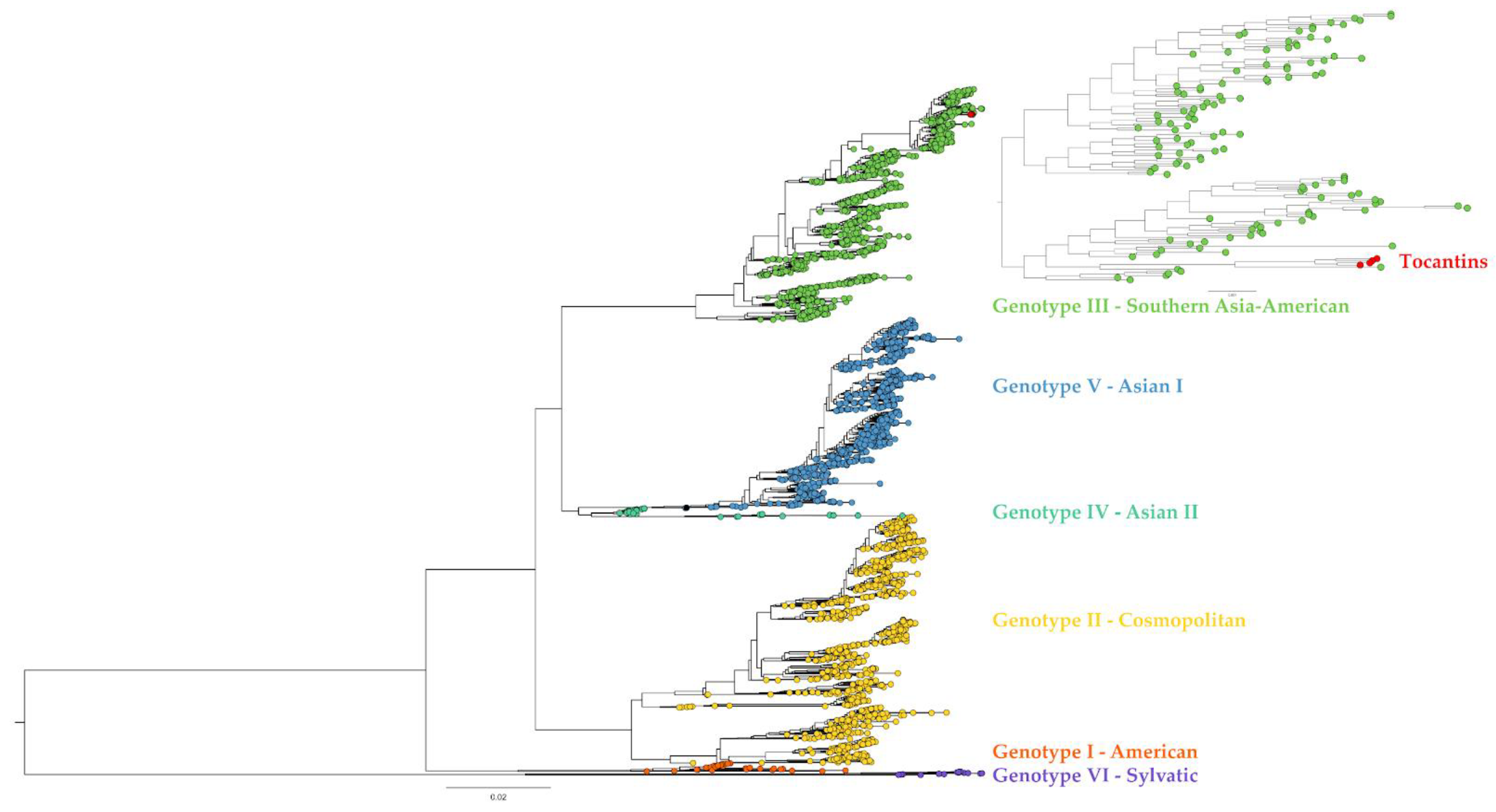
Figure 4.
Time-scaled phylogenetic tree of 76 complete and near-complete Clade I of DENV-1 Genotype V sequences sampled in Brazil, South America, and North America. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
Figure 4.
Time-scaled phylogenetic tree of 76 complete and near-complete Clade I of DENV-1 Genotype V sequences sampled in Brazil, South America, and North America. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
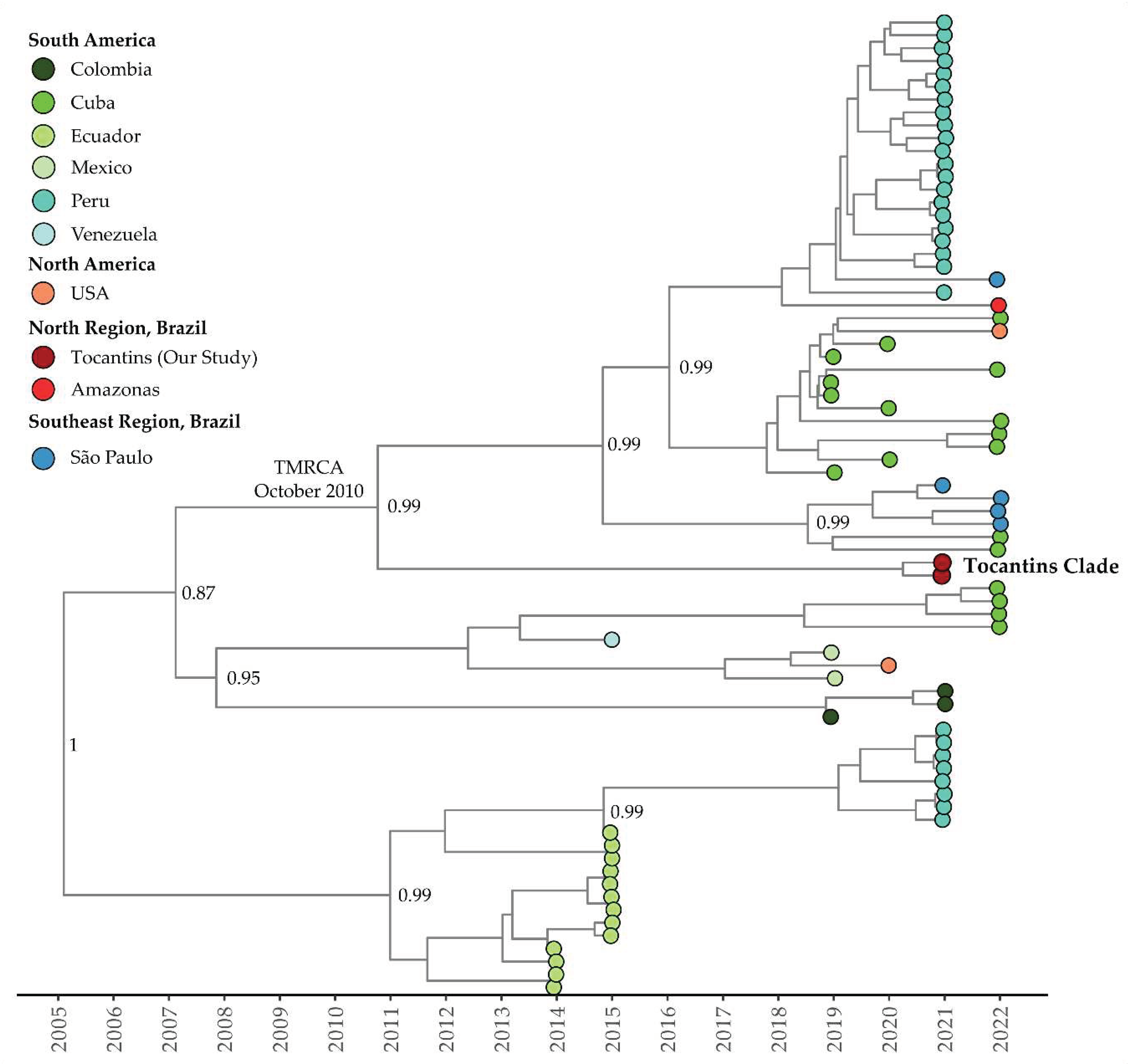
Figure 5.
Time-scaled phylogenetic tree of 63 complete and near-complete Clade II of DENV-1 Genotype V sequences sampled in Brazil and South America. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
Figure 5.
Time-scaled phylogenetic tree of 63 complete and near-complete Clade II of DENV-1 Genotype V sequences sampled in Brazil and South America. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
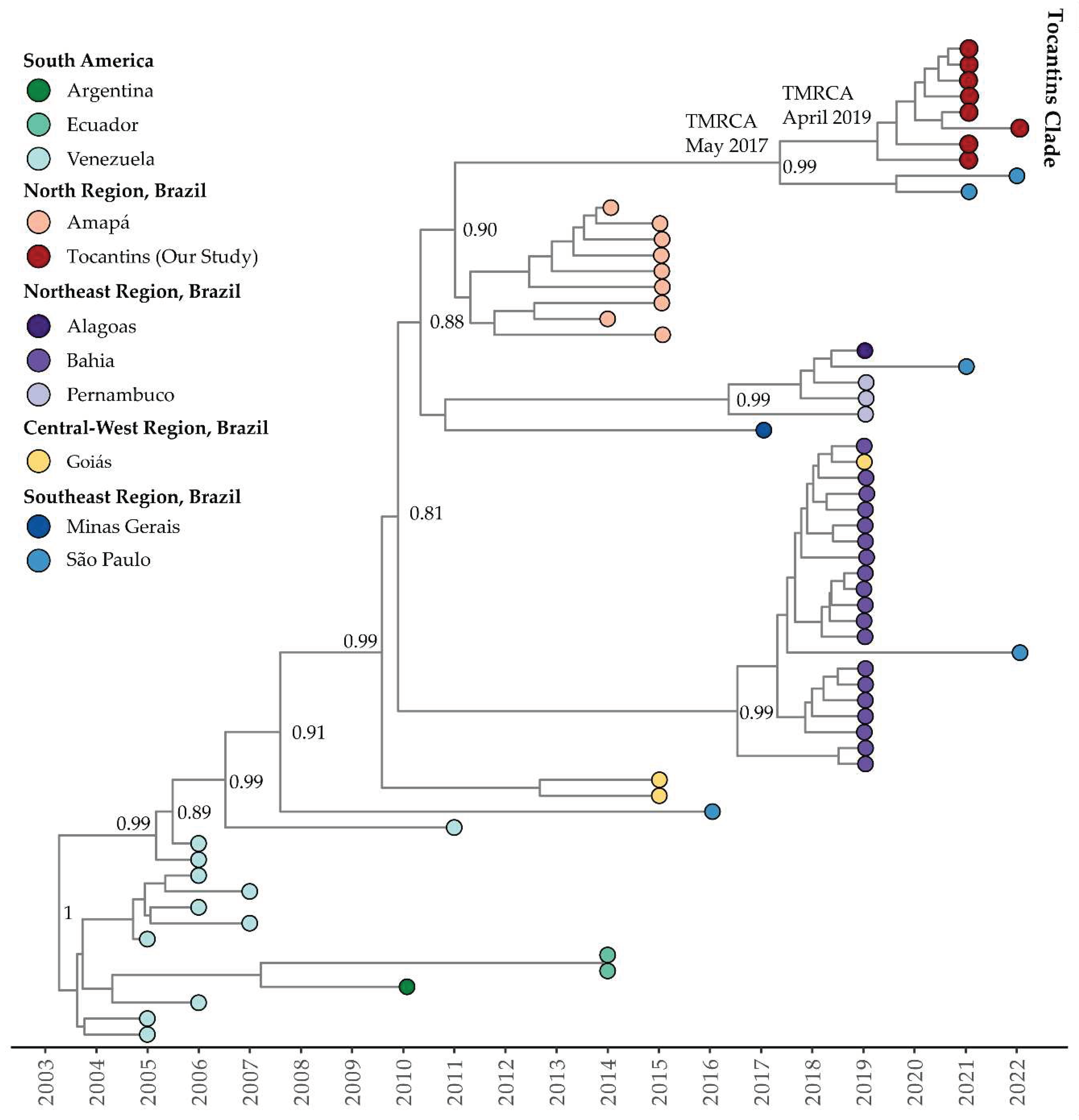
Figure 6.
Time-scaled phylogenetic tree of 170 complete and near-complete Clade III of DENV-1 Genotype V sequences sampled in Brazil, South America, and North America. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
Figure 6.
Time-scaled phylogenetic tree of 170 complete and near-complete Clade III of DENV-1 Genotype V sequences sampled in Brazil, South America, and North America. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
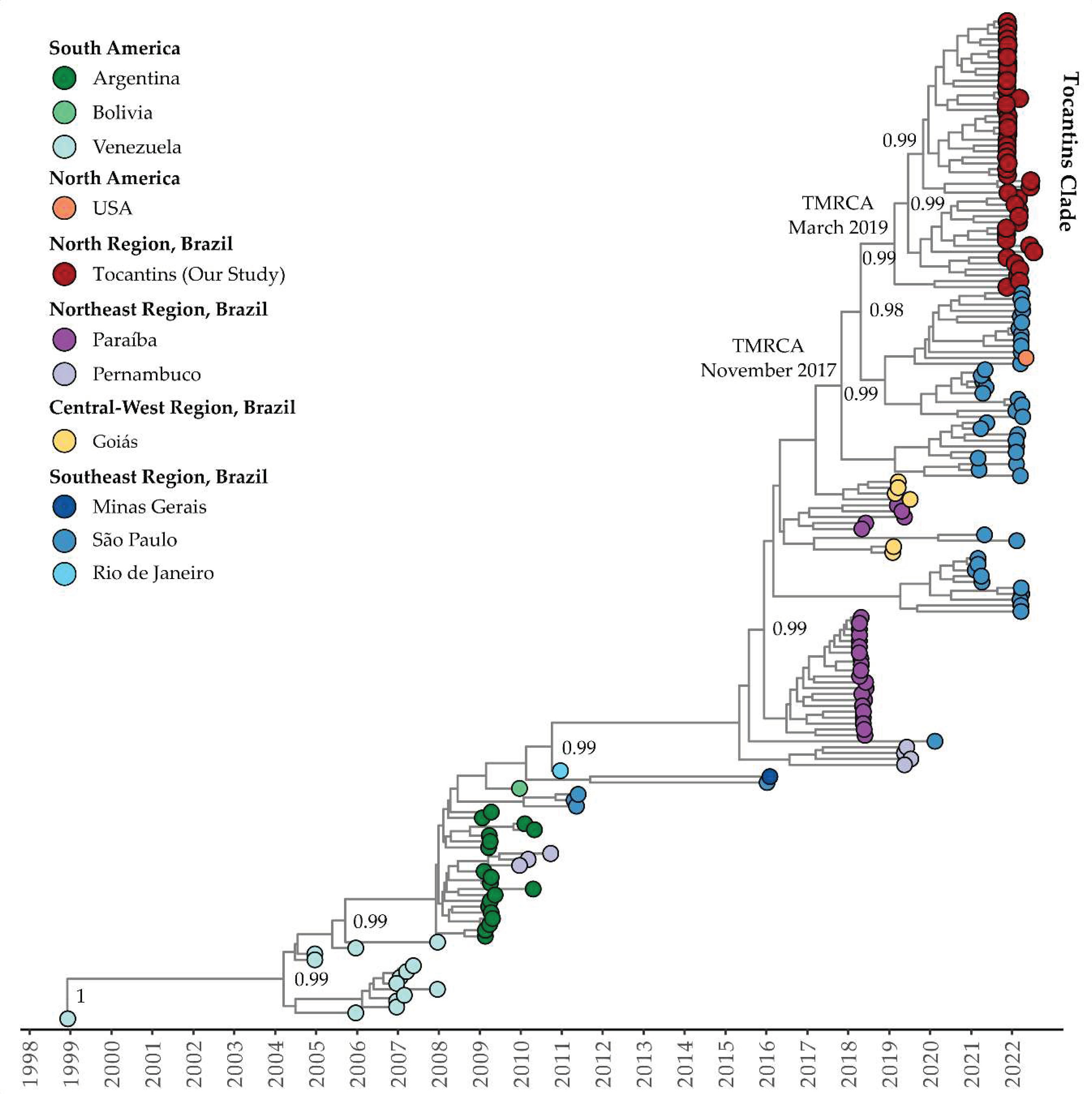
Figure 7.
Time-scaled phylogenetic tree of 63 complete and near-complete DENV-2 sequences sampled in Brazil. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
Figure 7.
Time-scaled phylogenetic tree of 63 complete and near-complete DENV-2 sequences sampled in Brazil. Colors represent different sampling locations according to the legend on the left of the tree. Genomes reported in the presented study are colored in red.
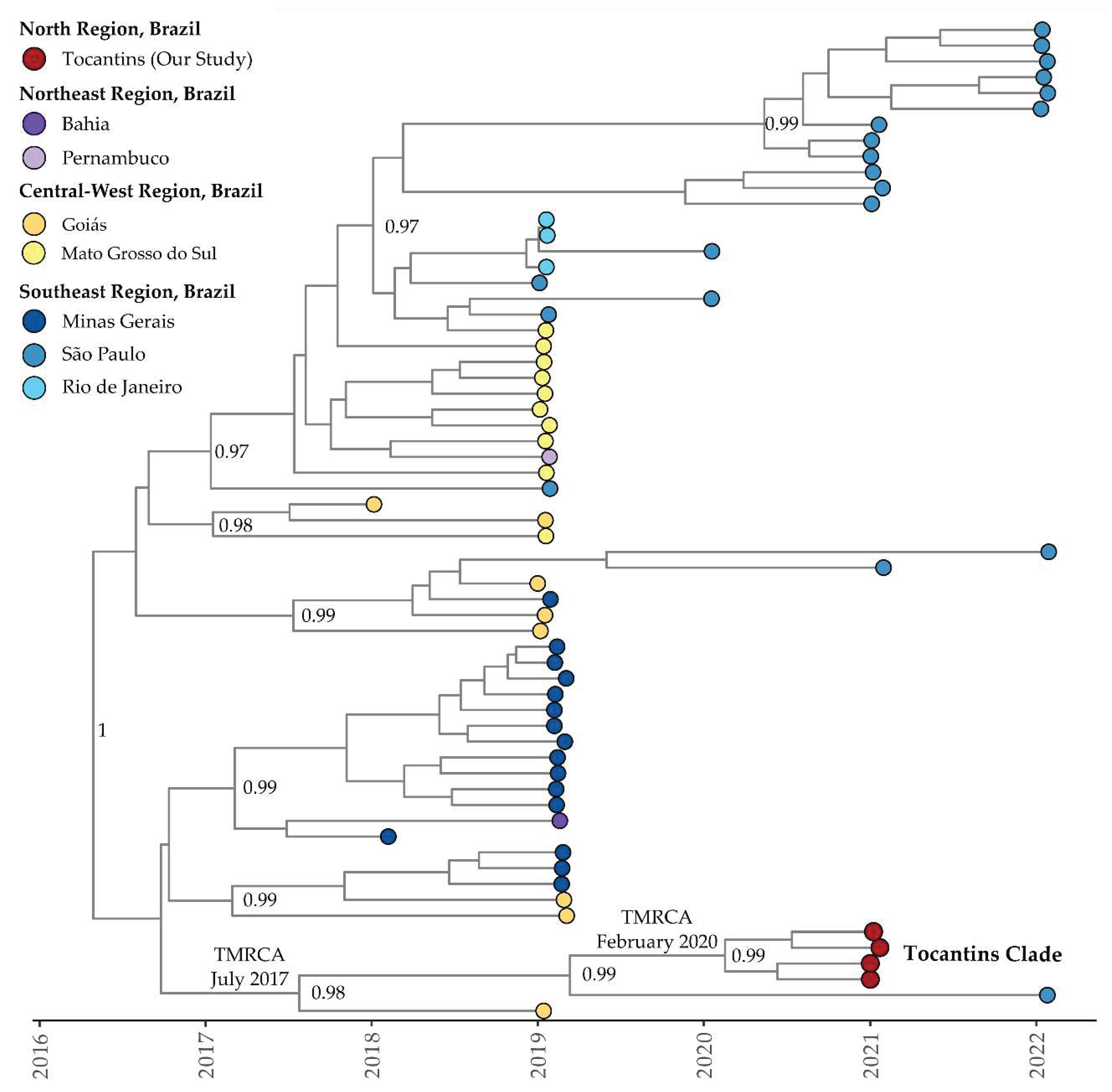

Table 1.
Clade-specific DENV-1 mutations found in genomes of clades I, II, and III. Common row for each gene/protein mutation and specify their occurrence in each clade.
Table 1.
Clade-specific DENV-1 mutations found in genomes of clades I, II, and III. Common row for each gene/protein mutation and specify their occurrence in each clade.
| Mutations | |||
|---|---|---|---|
| Gene/Protein | Clade I | Clade II | Clade III |
| Anchored Capsid Protein | Arg100Lys | ||
| Protein pr | Asp89Glu | ||
| Envelope Protein (E) | Glu10Asp, Leu31Val, His32Thr, Gln36Lys, Thr157Glu, Ala369Thr, Ile454Thr | Glu10Asp, Leu31Val, His32Thr, Gln36Lys, Thr157Glu, Ala369Thr, Ile454Thr | Glu10Asp, Leu31Val, His32Thr, Gln36Lys, Thr157Glu, Ala369Thr, Ile454Thr, Ile480Val* |
| NS1 | Ala60Val, Ile128Thr | Lys227Arg, Tyr247Phe | |
| NS2A | Met18Ile | Ser28Asn | |
| NS3 | His252Thr | ||
| NS4A | Ser1Cys, Lys24Glu, Cys92Arg, Arg97His | Cys48Arg, Pro77Leu, Ser82Phe, Pro107Ser | Val18Ala, Arg34His |
| 2K | Met17Leu | ||
| NS4B | Ala20Thr | ||
| NS5 | Thr135Met, Ala370Thr, Met408Ile, Ser585Asn, Asn637Asp*, Thr675Ala*, Lys732Glu, Lys814Arg, Asp834Glu | Thr135Met, Ala370Thr, Met408Ile, Ser585Asn, Lys732Glu, Lys814Arg, Asp834Glu | Thr135Met, Gln195Arg*, Lys311Arg*, Ala370Thr, Met408Ile, Ser585Asn, Val642Ala*, His649Tyr*, Lys732Glu, Val784Ile*, Thr789Ala*, Lys814Arg, Asp834Glu |
*Mutations detected specifically within that a particular clade.
Table 2.
DENV-2 mutations in each sample. Common row for each gene/protein mutation and specify their occurrence in each sample.
Table 2.
DENV-2 mutations in each sample. Common row for each gene/protein mutation and specify their occurrence in each sample.
| DENV-2 | Id of Sample | |||
|---|---|---|---|---|
| Gene/Protein | TO-UFT-2200 | TO-UFT-2201 | TO-UFT-689 | TO-UFT-6960 |
| Envelope protein | Met6Ile, Leu91Ile, Val129Ile, Leu131Gln, Ile170Thr, Met340Thr, Ser363Ala, Ile380Val | |||
| NS4A | Ile18Thr* | |||
| NS5 | Thr254Pro, Ile271Val, Ile276Val, Lys338Glu, Ser429Gly, Kys514Arg, Asp521Glu, Ser523Gly, Val553Ile, Glu558Lys, Arg586Lys, Val637Ala, Gln644His, Leu670Ile, Glu895Lys | Thr254Pro, Ile271Val, Ile276Val, Lys338Glu, Ser429Gly, Kys514Arg, Asp521Glu, Ser523Gly, Val553Ile, Glu558Lys, Arg586Lys, Val637Ala, Gln644His, Leu670Ile, Ile874Val*, Glu895Lys | Thr254Pro, Ile271Val, Ile276Val, Lys338Glu, Ser429Gly, Kys514Arg, Asp521Glu, Ser523Gly, Val553Ile, Glu558Lys, Arg586Lys, Val637Ala, Gln644His, Leu670Ile, Glu895Lys | Thr254Pro, Ile271Val, Ile276Val, Lys338Glu, Val335Ile*, Ser429Gly, Kys514Arg, Asp521Glu, Ser523Gly, Val553Ile, Glu558Lys, Arg586Lys, Val637Ala, Gln644His, Leu670Ile, Glu895Lys |
*Mutations detected specifically in each sample.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



