Submitted:
20 September 2023
Posted:
22 September 2023
You are already at the latest version
Abstract
Nanoparticle-assisted polymerase chain reaction (nanoPCR) is a novel method for rapid detection of pathogens. A sensitive and specific multiple nanoPCR assay was developed for simultaneous detection of three subgroups avian leucosis virus (ALV). In this study, three pairs of primers that were designed based on gp85 gene sequences of subgroup A, B and J, optimizations were carried out for better performance of the nanoPCR assay and remarkably improved the detection limit of the assay. The detection limit of the assay was 101copies/μl, which was 100-fold more sensitive than conventional PCR assays. The nanoPCR assay amplified the specific 336 bp, 625 bp and 167 bp fragments of ALV-A, -B and –J respectively and no cross-reactivity were found with the genomic DNA or cDNA of Newcastle disease virus, avian infectious laryngotracheitis virus, Marek's disease virus, infectious bronchitis vaccine, infectious bursal disease virus and avian encephalomyelitis Virus. To evaluate the detection ability of the assay, 186 clinical samples were detected by nanoPCR and the detection results were further confirmed by sequencing. The sequencing results were 100% in coincidence with the nanoPCR detection results, suggesting the possibility of clinical application of the newly developed nanoPCR assay. Taking all the results together, a highly specific and sensitive nanoPCR was successfully developed, which could be useful for rapid diagnosis of ALV in poultry farming.
Keywords:
nanoPCR
; avian leukosis virus
; ALV-J
; detection
1. Introduction
Avian leucosis virus (ALV) would cause immunosuppression in chickens, making them vulnerable to viral and bacterial infection, resulting in poorly responds to vaccination, growth retardation and tumor-related mortality of chickens[1,2,3] . ALV could spread in chicken flocks through vertical and horizontal transmission, during the past decades, it had prevalent in several breeding flocks in China and caused significant economic losses to the poultry farming[4,5]. Besides, evidences showed that ALV had spread from commercial layer chicken and broiler chicken to backyard and indigenous chickens, suggesting that the pathogenicity of ALV might be enhanced and the host range of ALV had been gradually expanded[6,7]. However, no effective vaccines were available for ALV, the main control strategy relied on the eradication project initiated by the local government [8]. Therefore, the strictly monitoring of ALV was essential to the poultry industry.
Avian leukosis virus (ALV) belonged to Alpharetrovirusgenus of the Retroviridae family, it could be classified as endogenous and exogenous virus on the basis of their characteristics and routes of transmission [9]. While exogenous ALVs could be further divided into 11 different subgroups based on their host range, viral envelop interference and cross-neutralization mode. Subgroup A, B, C, D, J and K are specific to chicken, among which A, B, J subgroups were common in the field, while subgroup C, D were rarely encountered[10]. In the past decades, the frequently outbreaks of ALV in China had drawn the attention of the public, being the most widely epidemic strains, subgroup A, B, J caused huge economic damage to the poultry farms. Therefore, the detection and discrimination of subgroup A, B, J was crucial to the control and eradication of ALV.
Conventional methods such as traditional virus isolation and serum neutralization test were time consuming and labor-intensive. Besides, an additional follow-up detection was needed for final identification when virus isolation was performed. With the development of molecular technology, PCR, quantitative real-time PCR and even recombinase polymerase amplification (RPA) had been developed for rapid detection of ALV in chickens [11,12,13,14]. Despite the obvious advantages such as high specific, high sensitive and time-saving, these methods had their own shortcomings. Take real-time PCR assay for example, expensive equipment and well-trained operator were needed. Though basal RPA assay did not rely on special devices, the low detection limit restricted its applications. As for the upgraded version real-time RPA, by taking advantage of fluorescence labeling, the sensitivity of the assay as well as the detection fee were significantly improved. However, most of the owners were not willing to pay for the bill, making it impossible for the widely application of real-time RPA in the poultry farms [15]. Therefore, a rapid, sensitive and cost-effective detection method was in demand for ALV diagnosis in the field.
With the explosive revolution of nanoscience, nanotechnology-based approaches had permeated in many different industries including food, chemical, medicine and agriculture. Great efforts had been made to utilize nanomaterial (NMs) in the development of detection technique [16]. Nanoparticle-assisted polymerase chain reaction (nanoPCR) was an advanced modification of PCR in which the thermal conductivity was significantly enhanced with the help of solid gold nanometer particles [17,18]. The whole amplification process would be speeded up right after the addition of nanoparticles in reaction system, meanwhile, the total reaction time would be shortened, which would finally enhance the efficiency of PCR assay. Besides, the special nanomaterial in the reaction system might form a competitive relationship with the DNA templates, interfering the non-specific amplification of irrelevant nucleic acids, which would help to improve the specificity of PCR assay [19,20]. In fact, the better heat transfer performance of nanomaterial in nanoPCR assay would trigger higher sensitivity during amplification. In a word, the performance of PCR assay would be significantly improved when assisted with nano-particles[21].In the past few years, this technique had been widely used in pathogen identification in veterinary research, for example, nanoparticle-assisted RT-PCR assay had been established to distinguish porcine epidemic diarrhea virus (PEDV), bovine respiratory syncytial virus and canine coronaviruses [22,23,24].
In this study, a nanoparticle-assisted PCR assay was developed for rapid detection of ALV based on the gp85 gene. Three primer pairs were designed for simultaneously detection of subgroups A, B, J of ALVs. Optimal reaction conditions had been explored for better performance of the new developed nanoPCR assay. This novel assay could be a useful tool for detection and differentiation of ALV subgroups, which would be really helpful in ALV eradication.
2. Materials and methods
2.1. Viruses and sample collection
ALV-A (GD08 strain) was kindly offered by Dr. Cao Weisheng, ALV-B (SDAU09E3 strain) and ALV-J (NX0101 strain) were kindly offered by Dr. Cui Zhizhong. Newcastle disease virus (NDV), avian infectious laryngotracheitis virus (ILTV), Marek’s disease virus (MDV), Infectious bronchitis vaccine (IBV), Infectious bursal disease virus (IBDV), avian encephalomyelitis Virus (AEV) were preserved in our lab. 120 clinical specimens including kidney, liver and spleen from 6- to 7-weeks-old sick chickens were collected from the elimination group of 2 different yellow feather broiler breeders in Huizhou, Guangdong province. 66 anticoagulation blood samples were collected from 2 poultry farms under the ALV eradication program in Jiangmen, Guangdong province. All the 186 samples were preserved at -80℃ before further uses.
2.2. Primer design
Primers were designed using the Oligo 6.0 software (Molecular Biology Insights, Inc) following the manufacturer’s instruction[25]. There are three group-specific (gs) antigen of ALV encoded by gag, pol and env gene respectively. The env gene encodes two proteins, namely gp85 and gp37, among which, gp85 determines the ALV subgroup specificity, making it an ideal target for discrimination of ALV subgroups [26]. Specific primers against ALV-A, ALV-B and ALV-J were designed focused on the conserved region of gp85 gene. To differentiate three subgroups of ALV, the amplicons should be clearly distinguished by the follow-up analysis, therefore, the PCR products of different sizes were amplified using different primer pairs (Table 1). The selected primer pairs were validated by online BLAST (Nucleotide BLAST: Search nucleotide databases using a nucleotide query (nih.gov)) to guarantee the accuracy of PCR products. All the primer pairs were synthetized and purified by Sangong (Guangzhou, China).
2.3. Standard DNA template construction
Total RNA was directly extracted from DF-1 cells 5 days after infected with ALV-A, ALV-B/D or ALV-J using the automatic nucleic acid extraction instrument (Tiangen Biotech, Beijing, China) according to the manufacturer’s instruction. Reverse transcription was carried out to acquire cDNA of ALVs. Then specific target fragments were amplified using the corresponding primers and separately cloned into the pMD-18T vector (Takara Biotechnology Co., Ltd, Dalian, China) following the manufacturer’s instruction. The recombinant plasmids clones (designated 18T-A. 18T-B and 18T-J, respectively) were confirmed by PCR assay and sequencing (Sangong) before further extracted with the Plasmid Mini Kit (Omega Bio-Tek, USA). The plasmid concentrations were determined by the Nanodrop 2000 (Thermo, USA) and the copy numbers were calculated based on the corresponding molecular weight and plasmid concentration. The standard DNA templates were stored at -20℃ before they were subjected to conventional multiple PCR assay and the novel multiple nanoPCR assay as described in the following sections.
2.4. Establishment of the conventional multiple PCR assay
The conventional multiple PCR assay was performed in a 20μL volume containing 1μL of viral cDNA, respectively (for comparison with the multiple NanoPCR and for positive controls); 1μL of each primers; 2μL of dNTPs; 1μL of KOD FX Neo (1U/μL) (TOYOBO Biotechnology Company, Shanghai, China); 5μL of 2× PCR buffer for KOD FX Neo (TOYOBO Biotechnology Company, Shanghai, China). The PCR reaction conditions were as follows: initial denaturation at 94℃for 5min; followed by 30 cycles of denaturation at 94℃for 40s, annealing at 58℃for 40s, and extension at 72℃for 60s, the final extension at 72℃ last for 10min. The amplified products were analyzed by electrophoresis on 2% agarose gels.
2.5. Establishment of the multiple nanoPCR assay
2.5.1. Optimization of the multiple nanoPCR assay
Experiments were carried out to optimize the annealing temperature, primer volume and template volume for better performance of the nanoPCR assay. Briefly, the optimal annealing temperature was determined using primer concentrations and template concentrations as indicated above. Once the optimal annealing temperature was determined, optimal primer concentrations and nucleic acid templates were further determine based on the optimal annealing temperature. Specifically, the cDNA templates were tested ranging from 0.2μL to 1.4μL, while the primers at 10μM concentration were tested ranging from 0.2μL to 1.2μL per reaction volume. The reaction volume also contained 0.5μL of Taq DNA polymerase (5 U/μL) (GRED, Shandong, China), 10μL of 2× nanoPCR Buffer (GRED, Shandong, China) with ddH2O up to 20μL. PCR reaction conditions were: 94℃for 5 min; followed by 30 cycles of 94 ℃ for 40s, annealing temperatures from 52℃ to 61℃ for 30s, and 72 ℃for 60s and a final extension at 72 ℃ for 10 min. Amplified products were analyzed by 2% agarose gel electrophoresis as usual.
2.5.2. Sensitivity analysis of the multiple nanoPCR assay
To determine the sensitivity of the multiple nanoPCR assay, the 10-fold serial dilutions of standard plasmid DNA ranging from 6.31×109 copies/μL to 6.0×100 copies/μL of 18T-A, 3.51×109 copies/μL to 4×100 copies/μL of 18T-B and 3.32×109 copies/μL to 3.0×100 copies/μL of 18T-J were tested by both multiple nanoPCR assay, in the meantime, parallel experiments were carried out with conventional multiple PCR assay. All the samples were tested in triplicate and the amplified products were analyzed by electrophoresis as above.
2.5.3. Specificity of the multiple nanoPCR assay
Specificity analysis was carried out to avoid the cross-reaction with other common pathogens of chickens. DNA or cDNA of the following viruses including Newcastle disease virus (NDV), avian infectious laryngotracheitis virus (ILTV), Marek’s disease virus (MDV), Infectious bronchitis vaccine (IBV), Infectious bursal disease virus (IBDV), avian encephalomyelitis Virus (AEV) were separately subjected to the multiple nanoPCR and the conventional PCR for specificity analysis. Meanwhile, the cDNA of ALV-A, ALV-B and ALV-J were used as positive control. All the PCR products were analyzed by 2% agarose gel.
2.6. Evaluation of the multiple nanoPCR assay with clinical specimens
A total of 186 clinical samples were collected from 4 different poultry farms in Guangdong province. Among which, 120 samples belonged to the sick chickens sharing typical clinical symptoms including gray-white nodules in the liver or kidney and abnormal enlargement of liver and spleen. The rest of the samples were from healthy chickens that running through the ALV eradication projects. The reverse transcription cDNA of all these samples were subjected to the new developed nanoPCR assay. All the samples were tested in duplicate. In addition, all the PCR amplicons of ALV were sequenced and the online software BLASTN (www.ncbi.nlm.nih.gov/blast) was used for homology analysis of the sequencing results.
3. Results
3.1. Optimization of the multiple nanoPCR assay
Reverse transcription cDNA was used as templates during optimization of nanoPCR assay. The screening annealing temperatures ranged from 52 to 61℃, as showed by the results, it seemed that the higher the annealing temperature went through, the more amplified products were acquired, and this tendency was more obvious during ALV-B amplification rather than the other two subgroups. While not much difference were found when annealing at 58℃, 59℃, 60℃ and 61℃. Therefore, 58℃ was chosen as the optimal annealing temperature for the following experiments (Figure 1).
Once the annealing temperature was determined, optimal primer concentrations were further assessed. Six different concentrations were validated, as showed in Figure 2, not much difference was found when a slight increase of each primer (namely 0.2μL) during the PCR assay. Yet, the PCR assay with the highest primer concentration had acquired more amplicons than the lowest primer concentration (Figure 2). Since no visible differences were found among 0.8μL, 1.0μL and 1.2μL, 0.8μL was chosen as the optimal primer concentration for cost-saving.
As the optimal annealing temperature (58℃) and primer concentration (0.8μL) were determined, the optimal template volume was finally confirmed, according to the results, three subgroups of ALV shared different optimal templates, as showed in Figure 3, 0.8μL of ALV-B, 1μL of ALV-A and ALV-J showed the best amplified results (Figure 3).
All the subsequent experiments were carried out based on the optimized conditions, briefly, the multiple nanoPCR assay was performed in a 20μL reaction mixture that included: 0.8μL of the ALV-B and 1.0μL of the ALV-A and ALV-J extracted RNA; 10μL of 2× Nano-buffer; 0.8μL of each primer pairs (working concentration was10μM); 1.0μL of Taq DNA polymerase (5 U/μL) and ddH2O up to 20μL. PCR reaction conditions were as follow: 94℃for 5 min; 30 cycle of 94℃for 40 s, 58℃for 40 s, and 72℃for 60 s with a final extension at 72℃for 10 min.
3.2. Sensitivity of the multiple nanoPCR assay
The sensitivity of the multiple nanoPCR assay and the conventional multiple PCR assay were compared by using serial dilutions of the 18t-A, 18T-B and 18T-J as template. For ALV-A, the detection limit was 6.31 × 103 copies/μL for the conventional multiple PCR assay but 6.31 × 101 copies/μL for multiple nanoPCR assay. The detection limit of ALV-B was 3.51 × 103 copies/μL for the conventional multiple PCR assay but 3.51× 101 copies/μL for multiple nanoPCR assay. As for ALV-J, the detection limit was 3.32 × 104 copies/μL for conventional multiple PCR assay but 3.32 × 101 copies/μL for multiple nanoPCR assay (Figure 4). These results indicated that the multiple nanoPCR assay was 100 times more sensitive than the conventional multiple PCR assay.
3.3. Specificity of the multiple nanoPCR assay
Neither the multiple nanoPCR assay nor the conventional multiple PCR assay would amplify target fragments of ALV without target RNA served as template (Figure 5). As showed in the results, no cross-reaction with NDV, ILTV, MDV, IBV, AEV was found during the amplification, suggesting the high specificity of nanoPCR assay.
3.4. Evaluation of the nanoPCR assay using clinical specimens
A total of 186 clinical specimens were tested using the nanoPCR assay. Independent sequencing was performed to confirm the results of the nanoPCR assay. As showed in Table 2, 14 specimens were tested positive with ALV infection, among which 11specimens displayed ALV-J positive, only 1 specimen displayed co-infection with ALV-A and ALV-J, and 2 specimens displayed co-infection with ALV-B and ALV-J. Independent sequencing confirmed that the test results of nanoPCR assay showed 100% coincidence with sequencing, indicating the accuracy and possibility of the nanoPCR assay for clinical ALV diagnosis.
4. Discussion
Avian leucosis (AL) caused by avian leucosis virus had brought huge economic losses in the poultry breeding industry in China [27]. Since no effective vaccine was available for ALV control, the detection and differential diagnosis are essential for ALV monitoring. In the past decades, the increasing occurrence of ALV in chicken farms starving for the rapid and accurate identification of ALV in clinical samples. Traditional methods such as virus isolation had been used for ALV identification. Though virus isolation in cell culture had been the “gold standard”, it took about 7 days to present the results. Besides, the infection of ALV in DF-1cells showed no typical cytopathic effect (CPE), an extra test was needed for final conclusion. Usually, the cell cultures were subjected to a commercial enzyme-linked immunosorbent assay (ELISA) kit for analysis of ALV proliferation in the DF-1 cells. However, the detection ability of the available commercial avian leucosis virus antigen test kits was unstable, leading to the false negative results, which would hamper the eradication of ALV.
In fact, some molecular biology techniques had been applied for ALV diagnosis, taking conventional PCR for example . As we know, conventional PCR methods are usually time consuming and labor-intensive since the whole amplify procedure can last for about two hours accompanied by half an hour of agarose gel electrophoresis. Most importantly, conventional PCR methods are restricted by low sensitivity as their detection limit stays at about 1000 copies/μL or even higher. To conquer the sensitivity limitation, real-time PCR assay was developed for ALV detection. With the help of fluorescence-modified probes, the detection limit of ALV could reach as low as 10 viral DNA copies [28]. However, the performance of real-time PCR needs much more expensive instruments as well as well-trained operators, both of which are not available in most of the poultry farms. Therefore, it’s urgent to develop a sensitive and cost-effective method to meet the need of clinical sample detection in poultry farms.
Nanomaterials has been widely used as helpers during PCR amplification, thanks to the excellent thermal conductivity of gold nano-particles, the non-target temperature was greatly shortened during the amplification process[29]. As a result, the addition of solid gold nano-metal particles in nanoPCR assay would trigger a remarkable increase of amplification performance in specificity and sensitivity when compared with conventional PCR methods [30,31]. Therefore, it is a perfect substitution of conventional PCR that could not fulfill the detection needs in clinical samples.
In the present study, a multiple nanoPCR assay was established. Being the subgrouping determinant of ALVs, gp85 gene had been used as target for ALV-J identification [32]. Three primer pairs focused on the conserved region of gp85 gene from ALV-A, ALV-B and ALV-J were designed. For better discrimination of different subgroups, the length of amplicons was displayed in different sizes so that to be distinguished by agarose gel electrophoresis. In order to get better results of the multiple nanoPCR assay, Optimal reaction conditions including annealing temperature, primer concentration as well as template concentration were explored. As presented in the agarose gel electrophoresis, difference was mainly found in the longest amplicon referring to ALV-B. Unlike ALV-B, during the optimal reaction condition screening, ALV-A and ALV-J displayed no obvious difference under different conditions. Just like the markers, the lower bands tended to become blurred, the 336bp of ALV-A amplicon and 167bp of ALV-J amplicon were not clear enough to tell the difference. However, all the amplicons came from the same multiple PCR assay, it’s difficult to perform three different agarose gel electrophoresis analysis for each subgroup. Considering that, the nanoPCR were developed for simultaneously detection of three subgroups of ALVs, the optimal reaction condition carried out for each subgroup was pointless. In this case, the optimal condition of the whole multiple nanoPCR assay was mostly determined by ALV-B and the annealing temperature was 58℃. In this study, not only the annealing temperature but also the primer and template concentration were assessed, however, the optimal of template concentration was not as meaningful as annealing temperature. Usually, the PCR assay was carried out following the manufacturer’s instruction, which would recommend the volume of DNA/RNA template. Besides, the precise concentration of DNA/RNA template would not be clearly confirmed before subjected to the PCR assay.
Despite the same primer pairs were used, the detection limit of conventional PCR assay was much higher than nanoPCR assay, in this study, the detection limit of nanoPCR was about 101copies/μL for ALV-A, ALV-B and ALV-J, while the conventional PCR methods could only detect 103 copies/μL. With such excellent detection ability, the nanoPCR assay could be a strong competitor of real-time PCR since it did not rely on expensive equipment. Though nanoPCR assay and real-time PCR assay shared similar detection limit, the operation of nanoPCR was less convenient than real-time PCR. Real-time monitoring could not be achieved and the PCR products need to be analyzed by agarose gel electrophoresis analysis. Considering that the cost of nanoPCR assay was much cheaper and the real-time PCR equipment was not available in most of poultry farms, it’s believed that nanoPCR assay would be a better option for ALV detection.
In this study, a total of 186 clinical specimens were collected and tested with the nanoPCR assay. The results showed that 14 specimens were infected with ALV-A, ALV-B or ALV-J, 11 specimens displayed single infection while 3 specimens displayed co-infection. All the detection results were further confirmed by sequencing to ensure the accuracy of nanoPCR assay. As revealed by the results, the newly developed nanoPCR assay was capable of detecting ALV correctly, making it an ideal tool for ALV-A/B/J detection in clinical samples.
5. Conclusion
In conclusion, a rapid, sensitive, and specific multiple nanoPCR assay was developed. The novel assay could simultaneously detect ALV-A, ALV-B and ALV-J, which would be applied for clinical diagnosis of ALV for epidemiology and pathology study as well as infectious disease control.
Author Contributions
Miaoli Wu: sample collection and original draft writing; Shuaiqi Hu: method development; Yujun Zhu: formal analysis and validation; Feng Cong: funding acquisition and project administration; Shengwang Liu: supervision.
Acknowledgments
This study was supported by the Supported by State Key Laboratory of Veterinary Biotechnology Foundation (SKLVBF202111).
Conflicts of interest
The authors declare that they have no competing interests.
References
- Li, H.; Wang, P.; Lin, L.; Shi, M.; Gu, Z.; Huang, T.; Mo, M. L.; Wei, T.; Zhang, H.; Wei, P. The emergence of the infection of subgroup J avian leucosis virus escalated the tumour incidence in commercial Yellow chickens in Southern China in recent years. Transbound Emerg Dis 2019, 66, 312–316. [Google Scholar] [CrossRef]
- Payne, L. N.; Nair, V. The long view: 40 years of avian leukosis research. Avian Pathology 2012, 41, 11–19. [Google Scholar] [CrossRef]
- Fenton, S. P.; Reddy, M. R.; Bagust, T. J. Single and concurrent avian leukosis virus infections with avian leukosis virus-J and avian leukosis virus-A in Australian meat-type chickens. Avian Pathology 2005, 34, 48–54. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, Q.; Zhang, J.; Li, X.; Zhao, J.; Fan, W.; Ji, P.; Wang, K.; Zhou, E. M.; Zhao, Q. Co-infection with avian hepatitis E virus and avian leukosis virus subgroup J as the cause of an outbreak of hepatitis and liver hemorrhagic syndromes in a brown layer chicken flock in China. Poult Sci 2020, 99, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tan, M.; Zhang, F.; Ji, H.; Zeng, Y.; Yang, Q.; Tan, J.; Huang, J.; Su, Q.; Huang, Y.; Kang, Z. Diversity of Avian leukosis virus subgroup J in local chickens, Jiangxi, China. Sci Rep 2021, 11, 4797. [Google Scholar] [CrossRef]
- Wang, P.; Li, M.; Li, H.; Bi, Y.; Lin, L.; Shi, M.; Huang, T.; Mo, M.; Wei, T.; Wei, P. ALV-J-contaminated commercial live vaccines induced pathogenicity in Three-Yellow chickens: one of the transmission routes of ALV-J to commercial chickens. Poult Sci 2021, 100, 101153. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Sun, S.; Zhang, Z.; Meng, S. Simultaneous endemic infections with subgroup J avian leukosis virus and reticuloendotheliosis virus in commercial and local breeds of chickens. Avian Pathol 2009, 38, 443–8. [Google Scholar] [CrossRef] [PubMed]
- Borodin, A. M.; Emanuilova, Z. V.; Smolov, S. V.; Ogneva, O. A.; Konovalova, N. V.; Terentyeva, E. V.; Serova, N. Y.; Efimov, D. N.; Fisinin, V. I.; Greenberg, A. J.; Alekseev, Y. I. Eradication of avian leukosis virus subgroups J and K in broiler cross chickens by selection against infected birds using multilocus PCR. PLoS One 2022, 17, e0269525. [Google Scholar] [CrossRef] [PubMed]
- Ochi, A.; Ochiai, K.; Nakamura, S.; Kobara, A.; Sunden, Y.; Umemura, T. Molecular Characteristics and Pathogenicity of an Avian Leukosis Virus Isolated from Avian Neurofibrosarcoma. Avian Diseases 2012, 56, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, M. J. Reverse Engineering Provides Insights on the Evolution of Subgroups A to E Avian Sarcoma and Leukosis Virus Receptor Specificity. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Silva, R. F.; Fadly, A. M.; Taylor, S. P. Development of a polymerase chain reaction to differentiate avian leukosis virus (ALV) subgroups: detection of an ALV contaminant in commercial Marek’s disease vaccines. Avian Dis 2007, 51, 663–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liao, M.; Jiao, P.; Luo, K.; Zhang, H.; Ren, T.; Zhang, G.; Xu, C.; Xin, C.; Cao, W. Development of a loop-mediated isothermal amplification assay for rapid detection of subgroup J avian leukosis virus. J Clin Microbiol 2010, 48, 2116–21. [Google Scholar] [CrossRef] [PubMed]
- Hatai, H.; Ochiai, K.; Tomioka, Y.; Toyoda, T.; Hayashi, K.; Anada, M.; Kato, M.; Toda, A.; Ohashi, K.; Ono, E.; Kimura, T.; Umemura, T. Nested polymerase chain reaction for detection of the avian leukosis virus causing so-called fowl glioma. Avian Pathol 2005, 34, 473–9. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Manoharan, P.; Kathaperumal, K.; Chidambaram, B.; Divya, K. C. Differential detection of avian oncogenic viruses in poultry layer farms and Turkeys by use of multiplex PCR. J Clin Microbiol 2012, 50, 2668–73. [Google Scholar] [CrossRef]
- Chou, Q.; Russell, M.; Birch, D. E.; Raymond, J.; Bloch, W. Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications. Nucleic Acids Res 1992, 20, 1717–23. [Google Scholar] [CrossRef]
- Malik, S.; Muhammad, K.; Waheed, Y. Nanotechnology: A Revolution in Modern Industry. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Li, H.; Rothberg, L. Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles. Proc Natl Acad Sci U S A 2004, 101, 14036–9. [Google Scholar] [CrossRef]
- Sun, R.; Zhuang, H. An ultrasensitive gold nanoparticles improved real-time immuno-PCR assay for detecting diethyl phthalate in foodstuff samples. Analytical Biochemistry 2015, 480, 49–57. [Google Scholar] [CrossRef]
- Shen, C.; Yang, W.; Ji, Q.; Maki, H.; Dong, A.; Zhang, Z. NanoPCR observation: different levels of DNA replication fidelity in nanoparticle-enhanced polymerase chain reactions. Nanotechnology 2009, 20, 455103. [Google Scholar] [CrossRef]
- El-Husseini, D. M.; Helmy, N. M.; Tammam, R. H. Application of gold nanoparticle-assisted PCR for equine herpesvirus 1 diagnosis in field samples. Archives of Virology 2017, 162, 2297–2303. [Google Scholar] [CrossRef]
- Li, H.; Rothberg, L. J. DNA sequence detection using selective fluorescence quenching of tagged oligonucleotide probes by gold nanoparticles. Anal Chem 2004, 76, 5414–7. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Wang, J.; Cui, S. J. Development of a nanoparticle-assisted PCR assay to distinguish canine coronaviruses I and II. J Vet Diagn Invest 2021, 33, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yan, Y.; Wang, R.; Wang, L.; Zhou, H.; Li, Y.; Tang, L.; Xu, Y.; Jiang, Y.; Cui, W.; Qiao, X. Simultaneous Detection of Bovine Rotavirus, Bovine Parvovirus, and Bovine Viral Diarrhea Virus Using a Gold Nanoparticle-Assisted PCR Assay With a Dual-Priming Oligonucleotide System. Front Microbiol 2019, 10, 2884. [Google Scholar] [CrossRef] [PubMed]
- Wanzhe, Y.; Jianuan, L.; Peng, L.; Jiguo, S.; Ligong, C.; Juxiang, L. Development of a nano-particle-assisted PCR assay for detection of duck tembusu virus. Letters in Applied Microbiology 2016, 62, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lin, L.; Li, H.; Yang, Y.; Huang, T.; Wei, P. Diversity and evolution analysis of glycoprotein GP85 from avian leukosis virus subgroup J isolates from chickens of different genetic backgrounds during 1989-2016: Coexistence of five extremely different clusters. Arch Virol 2018, 163, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.; Tian, X.; Shao, H.; Ye, J.; Yao, Y.; Nair, V.; Qin, A. Identification of novel B-cell epitope in gp85 of subgroup J avian leukosis virus and its application in diagnosis of disease. BMC Vet Res 2018, 14, 295. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q. W.; Zhang, Z. P.; Wang, W.; Tian, J.; Xiao, Z. G. Enhanced inhibition of Avian leukosis virus subgroup J replication by multi-target miRNAs. Virol J 2011, 8, 556. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Gao, Y.; Ni, W.; Sun, M.; Wang, Y.; Yin, C.; Qi, X.; Gao, H.; Wang, X. Development and application of real-time PCR for detection of subgroup J avian leukosis virus. J Clin Microbiol 2013, 51, 149–54. [Google Scholar] [CrossRef]
- Ma, W.; Zhan, Y.; Zhang, Y.; Mao, C.; Xie, X.; Lin, Y. The biological applications of DNA nanomaterials: current challenges and future directions. Signal Transduction and Targeted Therapy 2021, 6. [Google Scholar] [CrossRef]
- Fujita, T.; Yuno, M.; Kitaura, F.; Fujii, H. A refined two-step oligoribonucleotide interference-PCR method for precise discrimination of nucleotide differences. Sci Rep 2018, 8, 17195. [Google Scholar] [CrossRef]
- Lebedev, A. V.; Paul, N.; Yee, J.; Timoshchuk, V. A.; Shum, J.; Miyagi, K.; Kellum, J.; Hogrefe, R. I.; Zon, G. Hot start PCR with heat-activatable primers: a novel approach for improved PCR performance. Nucleic Acids Res 2008, 36, e131. [Google Scholar] [CrossRef] [PubMed]
- Yehia, N.; El-Sayed, H. S.; Omar, S. E.; Amer, F. Genetic variability of the Avian leukosis virus subgroup J gp85 gene in layer flocks in Lower Egypt. Veterinary World 2020, 13, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Optimization of annealing temperature. M: DL2000 DNA marker; the annealing temperature ranging from 52℃ to 61℃. 1: 52℃; 2: 53℃; 3: 54℃; 4: 55℃; 5: 56℃; 6: 57℃; 7: 58℃; 8: 59℃; 9: 60℃; 10: 61℃; 11: Negative control with no ALV nucleic acids.
Figure 1.
Optimization of annealing temperature. M: DL2000 DNA marker; the annealing temperature ranging from 52℃ to 61℃. 1: 52℃; 2: 53℃; 3: 54℃; 4: 55℃; 5: 56℃; 6: 57℃; 7: 58℃; 8: 59℃; 9: 60℃; 10: 61℃; 11: Negative control with no ALV nucleic acids.
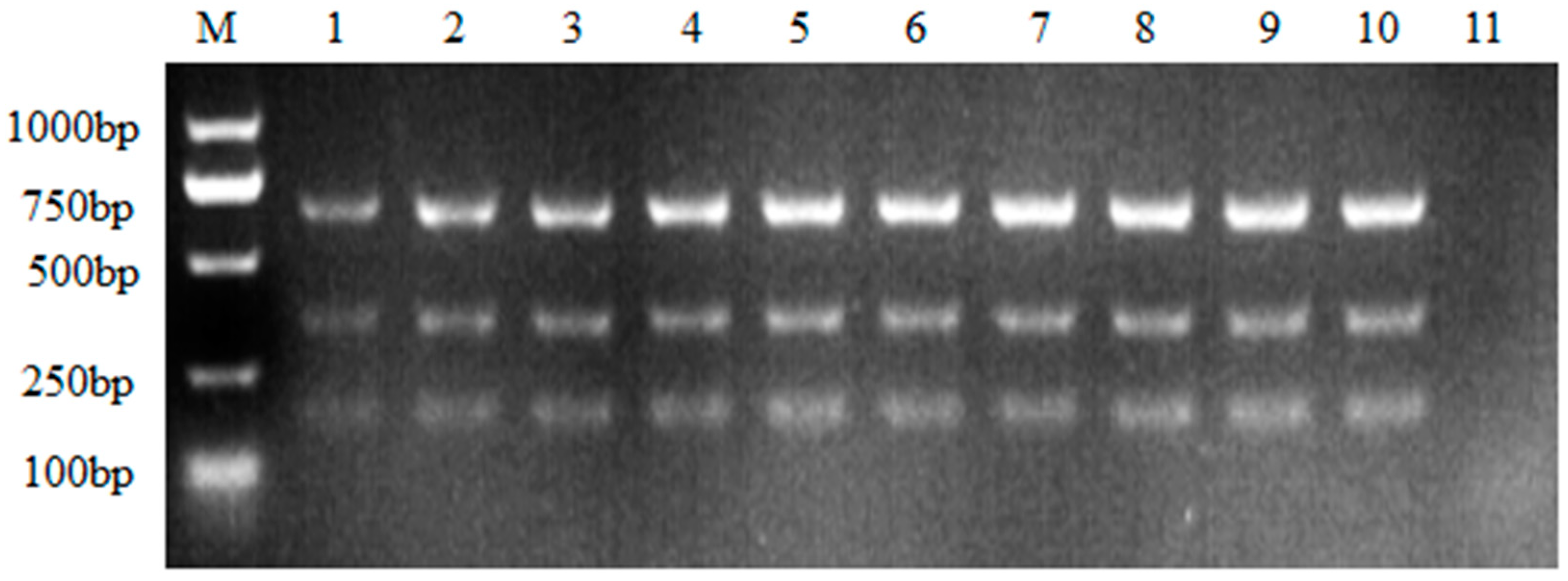
Figure 2.
Optimization of the primer concentration. M: DL2000 DNA Marker; the primer volume ranging from 0.4 μLto1.4 μL of each primer under the working concentration of 10μM. 1: 0.4μL; 2: 0.6μL; 3: 0.8μL; 4: 1.0μL; 5: 1.2μL; 6: 1.4μL; 7: Negative control.
Figure 2.
Optimization of the primer concentration. M: DL2000 DNA Marker; the primer volume ranging from 0.4 μLto1.4 μL of each primer under the working concentration of 10μM. 1: 0.4μL; 2: 0.6μL; 3: 0.8μL; 4: 1.0μL; 5: 1.2μL; 6: 1.4μL; 7: Negative control.
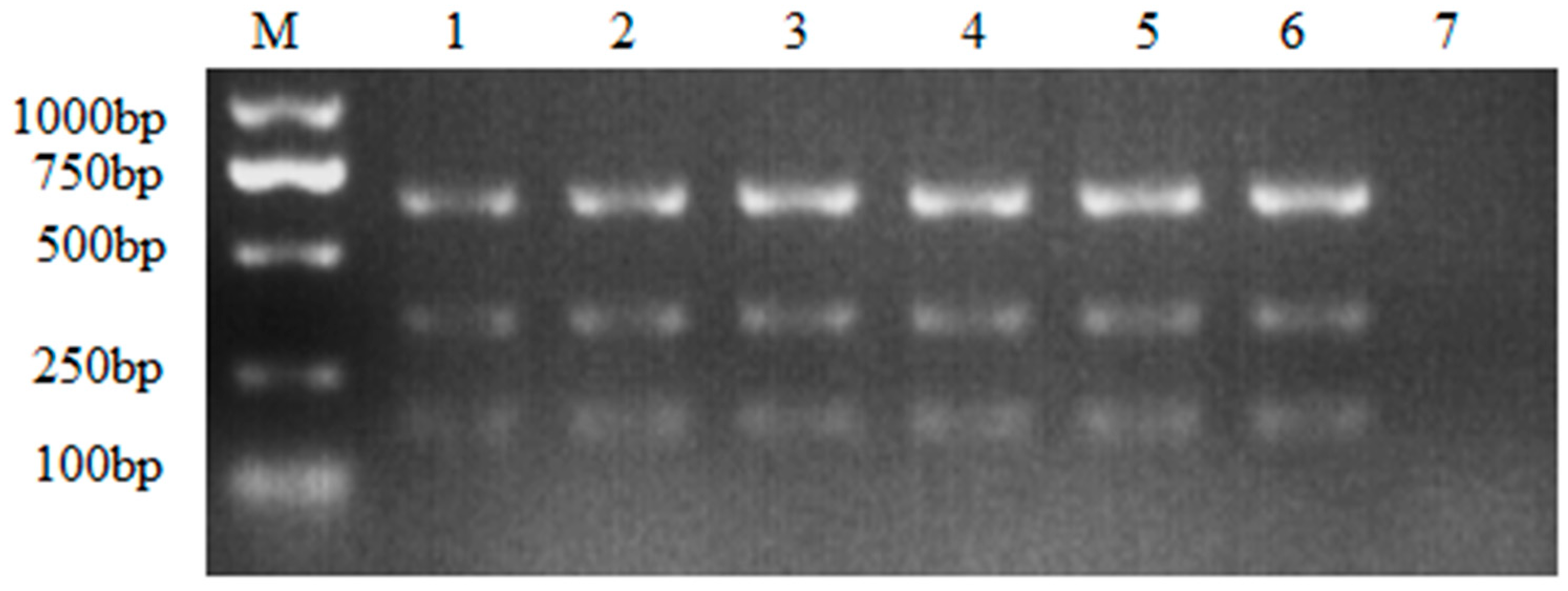
Figure 3.
Optimization of the template concentration. M: DL2000 DNA Marker; the cDNA volume ranging from 0.2 μL to 1.2 μL. 1: 0.2μL; 2: 0.4μL; 3: 0.6μL; 4: 0.8μL; 5: 1.0μL; 6: 1.2μL; 7: Negative control.
Figure 3.
Optimization of the template concentration. M: DL2000 DNA Marker; the cDNA volume ranging from 0.2 μL to 1.2 μL. 1: 0.2μL; 2: 0.4μL; 3: 0.6μL; 4: 0.8μL; 5: 1.0μL; 6: 1.2μL; 7: Negative control.
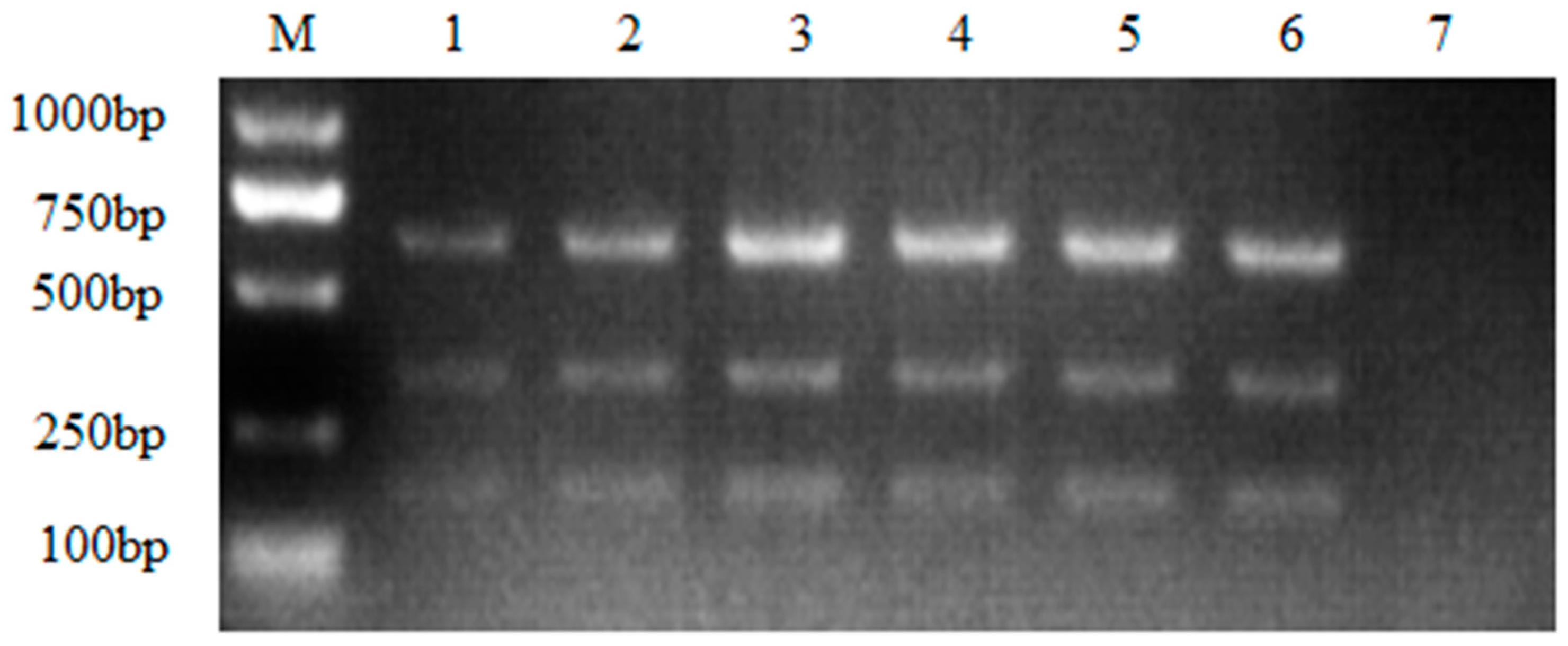
Figure 4.
Sensitivity analysis of the multiple NanoPCR assay. A: nanoPCR assay; (B) conventional multiple PCR assay. M: DL2000 marker; 1–10: 10-fold serial dilutions of standard DNA plasmids. Namely, 1: 6.31×109 copies/μL for ALV-A, 3.51×109 copies/μL for ALV-B and 3.32×109 copies/μL for ALV-J; 2: 6.31×108 copies/μL for ALV-A, 3.51×108 copies/μL for ALV-B and 3.32×108 copies/μL for ALV-J; 3: 6.31×107 copies/μL for ALV-A, 3.51×107 copies/μL for ALV-B and 3.32×107 copies/μL for ALV-J; 4: 6.31×106 copies/μL for ALV-A, 3.51×106 copies/μL for ALV-B and 3.32×106 copies/μL for ALV-J; 5: 6.31×105 copies/μL for ALV-A, 3.51×105 copies/μL for ALV-B and 3.32×105 copies/μL for ALV-J; 6: 6.31×104 copies/μL for ALV-A, 3.51×104 copies/μL for ALV-B and 3.32×104 copies/μL for ALV-J; 7: 6.31×103 copies/μL for ALV-A, 3.51×103 copies/μL for ALV-B and 3.32×103 copies/μL for ALV-J; 8: 6.31×102 copies/μL for ALV-A, 3.51×102 copies/μL for ALV-B and 3.32×102 copies/μL for ALV-J; 10: 6.31×101 copies/μL for ALV-A, 3.51×101 copies/μL for ALV-B and 3.32×101 copies/μL for ALV-J; 11: negative control.
Figure 4.
Sensitivity analysis of the multiple NanoPCR assay. A: nanoPCR assay; (B) conventional multiple PCR assay. M: DL2000 marker; 1–10: 10-fold serial dilutions of standard DNA plasmids. Namely, 1: 6.31×109 copies/μL for ALV-A, 3.51×109 copies/μL for ALV-B and 3.32×109 copies/μL for ALV-J; 2: 6.31×108 copies/μL for ALV-A, 3.51×108 copies/μL for ALV-B and 3.32×108 copies/μL for ALV-J; 3: 6.31×107 copies/μL for ALV-A, 3.51×107 copies/μL for ALV-B and 3.32×107 copies/μL for ALV-J; 4: 6.31×106 copies/μL for ALV-A, 3.51×106 copies/μL for ALV-B and 3.32×106 copies/μL for ALV-J; 5: 6.31×105 copies/μL for ALV-A, 3.51×105 copies/μL for ALV-B and 3.32×105 copies/μL for ALV-J; 6: 6.31×104 copies/μL for ALV-A, 3.51×104 copies/μL for ALV-B and 3.32×104 copies/μL for ALV-J; 7: 6.31×103 copies/μL for ALV-A, 3.51×103 copies/μL for ALV-B and 3.32×103 copies/μL for ALV-J; 8: 6.31×102 copies/μL for ALV-A, 3.51×102 copies/μL for ALV-B and 3.32×102 copies/μL for ALV-J; 10: 6.31×101 copies/μL for ALV-A, 3.51×101 copies/μL for ALV-B and 3.32×101 copies/μL for ALV-J; 11: negative control.
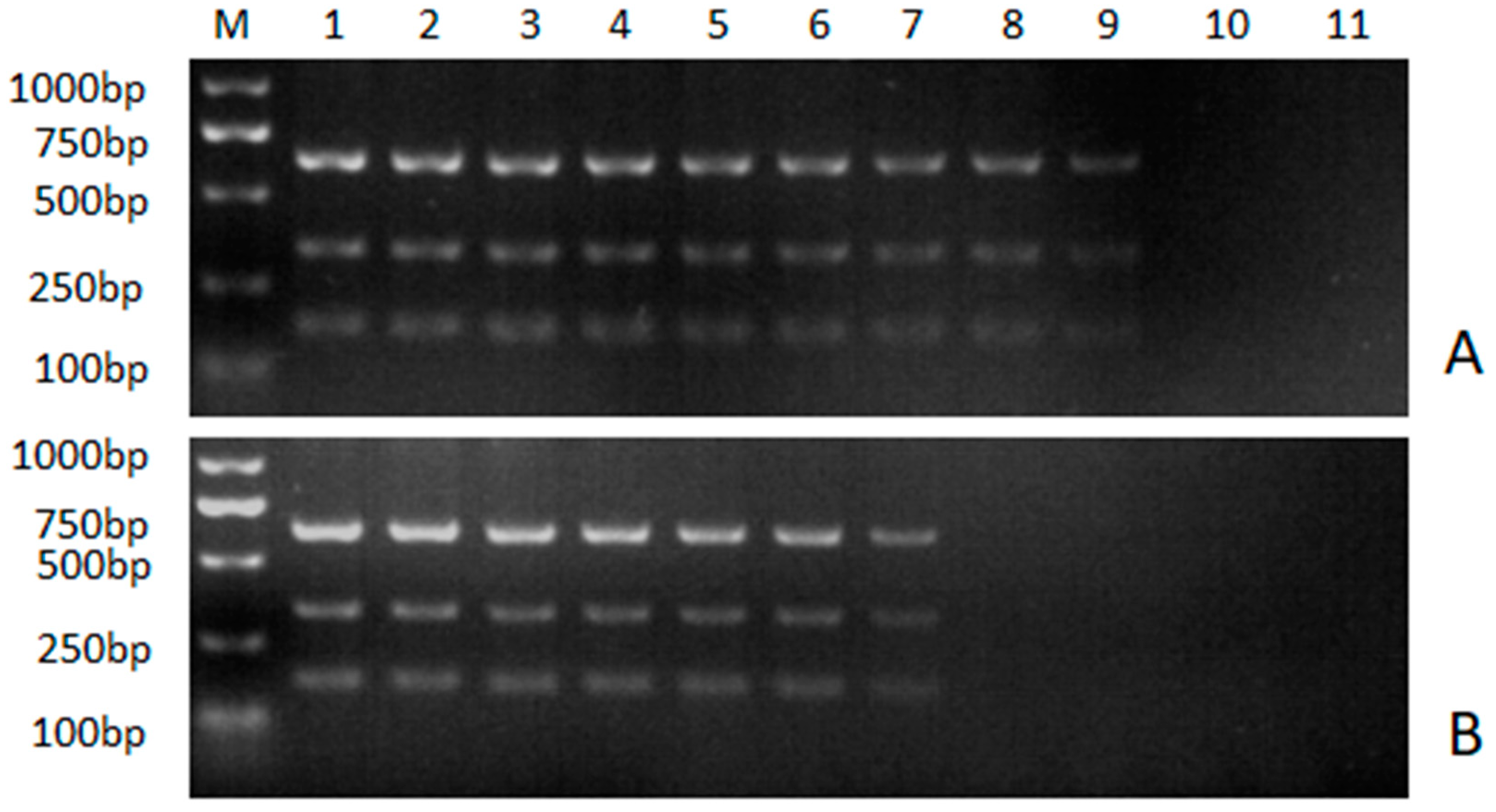
Figure 5.
Specificity analysis of nanoPCR assay. M: DL2000 DNA Marker; 1~6: nucleic acids of different viruses. Namely, 1: ALV-A, ALV-B and ALV-J; 2: NDV; 3: ILTV; 4: MDV; 5: IBV; 6: AEV; 7: Negative control.
Figure 5.
Specificity analysis of nanoPCR assay. M: DL2000 DNA Marker; 1~6: nucleic acids of different viruses. Namely, 1: ALV-A, ALV-B and ALV-J; 2: NDV; 3: ILTV; 4: MDV; 5: IBV; 6: AEV; 7: Negative control.


Table 1.
Details of primers for ALV.
| Name | Sequence | Target | Amplicon |
|---|---|---|---|
| AP1 | GATGAGGCGAGCCCTCTTTT | ALV-A | 336bp |
| AP2 | TCGGAAATAGGGGACGGGAT | ||
| BP1 | CCGGACATCACCCAAAAGGA | ALV-B | 625bp |
| BP2 | AAAGCCTATGCATCCCCCAG | ||
| JP1 | ACCTGGGCAAATAAGACGGG | ALV-J | 167bp |
| JP2 | TGGCTGGCTAAATCGGTGTT |
Table 2.
The results of nanoPCR assay for clinical specimens detection.
| Virus | Nano-PCR positive specimens |
Sequencing positive specimens |
Coincidence rate (%) |
|---|---|---|---|
| ALV-A | 0 | 0 | 100 |
| ALV-B | 0 | 0 | 100 |
| ALV-J | 11 | 11 | 100 |
| ALV-A+B | 0 | 0 | 100 |
| ALV-A+ J | 1 | 1 | 100 |
| ALV-B+J | 2 | 2 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



