
Submitted:
18 September 2023
Posted:
22 September 2023
You are already at the latest version
Abstract
Various 4-aminotetrahydropyridinylidene salts were treated with aldehydes in alkaline medium. Their conversion to 5-substituted -hydroxyketones in a one-step reaction succeeded only with an aliphatic aldehyde. Instead, aromatic aldehydes gave 5-substituted -aminoketones or a single -diketone. The new compounds were characterized by spectroscopic methods and a single crystal structure analysis. Some of them showed anticancer and antibacterial properties.
Keywords:
antibacterial
; anticancer activity
; dihydropyridin-4(1H)-ones
; tetrahydropyridinylidene salts
1. Introduction
Recently, we described the synthesis and antiprotozoal potency of 4-aminotetrahydropyridinylidene salts [1] and of 1-benzyl derivatives of these salts with enhanced antiprotozoal activities [2,3]. The insertion of an additional benzyl group in ring position 3 via alkylation under basic conditions yielded 1,3-disubstituted compounds with increased antiplasmodial actvity [4]. This paper reports the synthesis of 5-substituted derivatives. Surprisingly, the reaction afforded mainly β-aminoketones, less frequently δ-diketones or the expected β-hydroxyketones, which are usually obtained from α,β-unsaturated ketones under Morita-Baylis-Hillmann conditions [5,6,7]. Similar Mannich-like products were obtained from formaldehyde and secondary amines with quinolin-4(1H)-ones [8,9] or pyridin-4(1H)-ones [10,11]. However, their synthesis from 4-amino-tetrahydropyridinylidene salts or from their hydrolysis products, the 2,3-dihydropyridin-4(1H)-ones have not yet been reported. Unsubstituted 2,3-dihydropyridin-4(1H)-one has been prepared from ethyl 4-oxo-1,4-dihydropyridin-1-carboxylate [12]. Its 2,2-dimethyl analogue has been synthesized from 2-methylhexa-3,5-diyn-2-amine [13] as well as from 1-ethenyl-4,4-dimethylazetidin-2-one via UV-irradiation [14] (Scheme 1). Further cyclic enaminones have been used as convenient synthons for numerous reactions including photocycloadditions [15,16,17,18,19] and alkaloid synthesis [20]. The structural and electronic properties of a series of 2,3-dihydropyridin-4-ones have been a subject of a theoretical study [21]. So far, only the insertion of an allyl or an alkenyl substituent in ring position 5 of a dihydropyridine-4-one was reported [22,23].
2,3-Dihydropyridin-4(1H)-one is a partial structure of cenocladamide, an alkaloid from Piper cenocladum [24]. This alkaloid and derivatives thereof have previously been investigated for their anticancer potency [25]. Furthermore, the dihydropyridin-4(1H)-one moiety is part of derivatives of piperlongumine, which have been investigated for their anticancer activity [26] as well as of the potent antibacterial MRX-I [27] (Scheme 1). Therefore, we investigated the new compounds for their anticancer activities as well as their antibacterial potency in vitro.
2. Results and Discussion
2.1. Chemistry
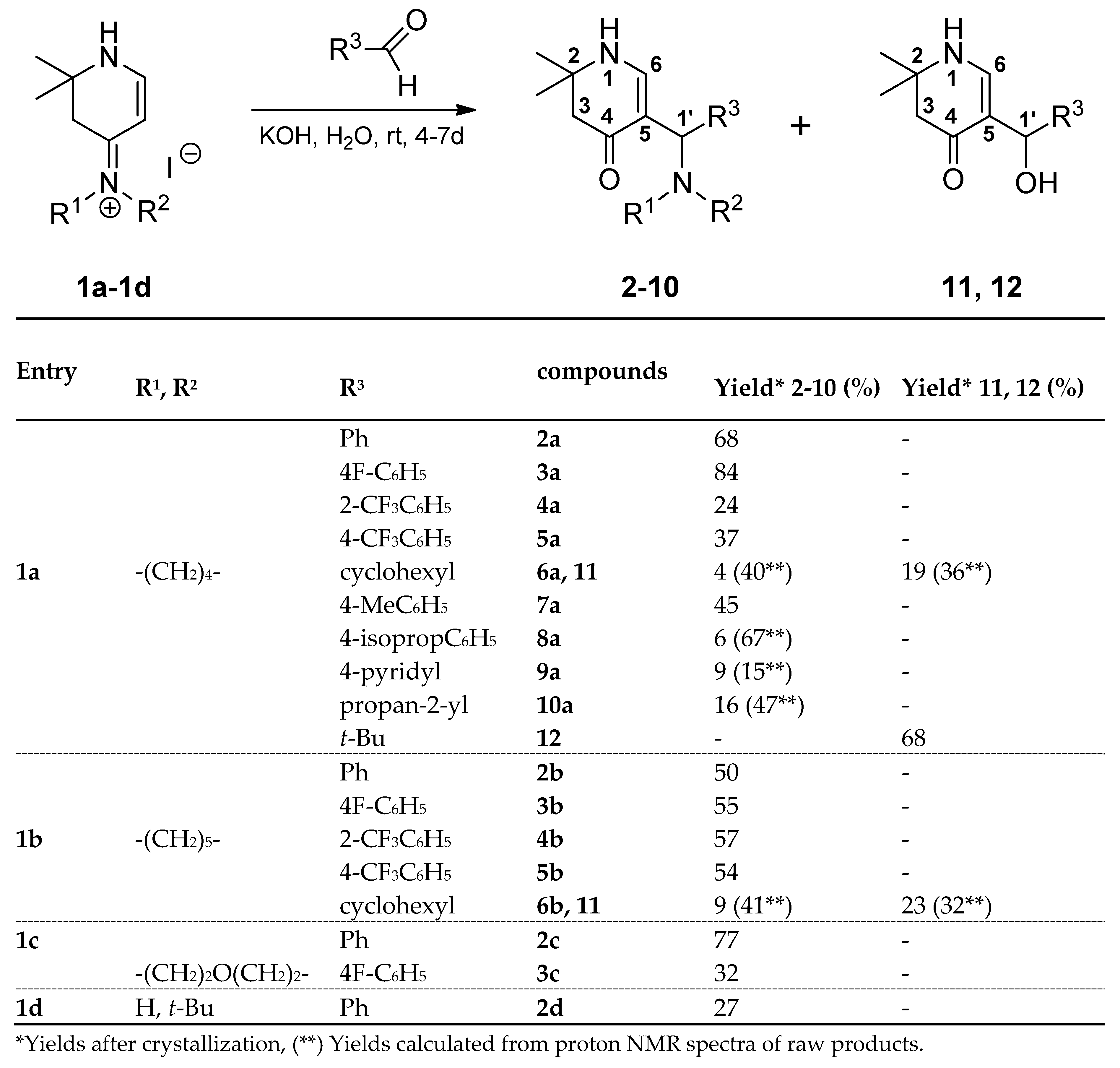
The different 4-dialkylaminotetrahydropyridinylidene salts 1a-1d used as starting products were prepared as described earlier [1]. They were exposed for several days to aromatic aldehydes or aliphatic aldehydes in alkaline medium at room temperature. Hydrolysis of 1a-1d in ring position 4 was taken into account, but the expected β-hydroxyketones 11 and 12 were only formed as main product from the aliphatic cyclohexane carbaldehyde and pivalaldehyde, whereas the β-aminoketones 6a and 6b were isolated as by-products. Reaction of salts 1a-1d with aromatic aldehydes afforded mainly β-aminoketones (compounds 2-5 and 7-10) (Scheme 2).
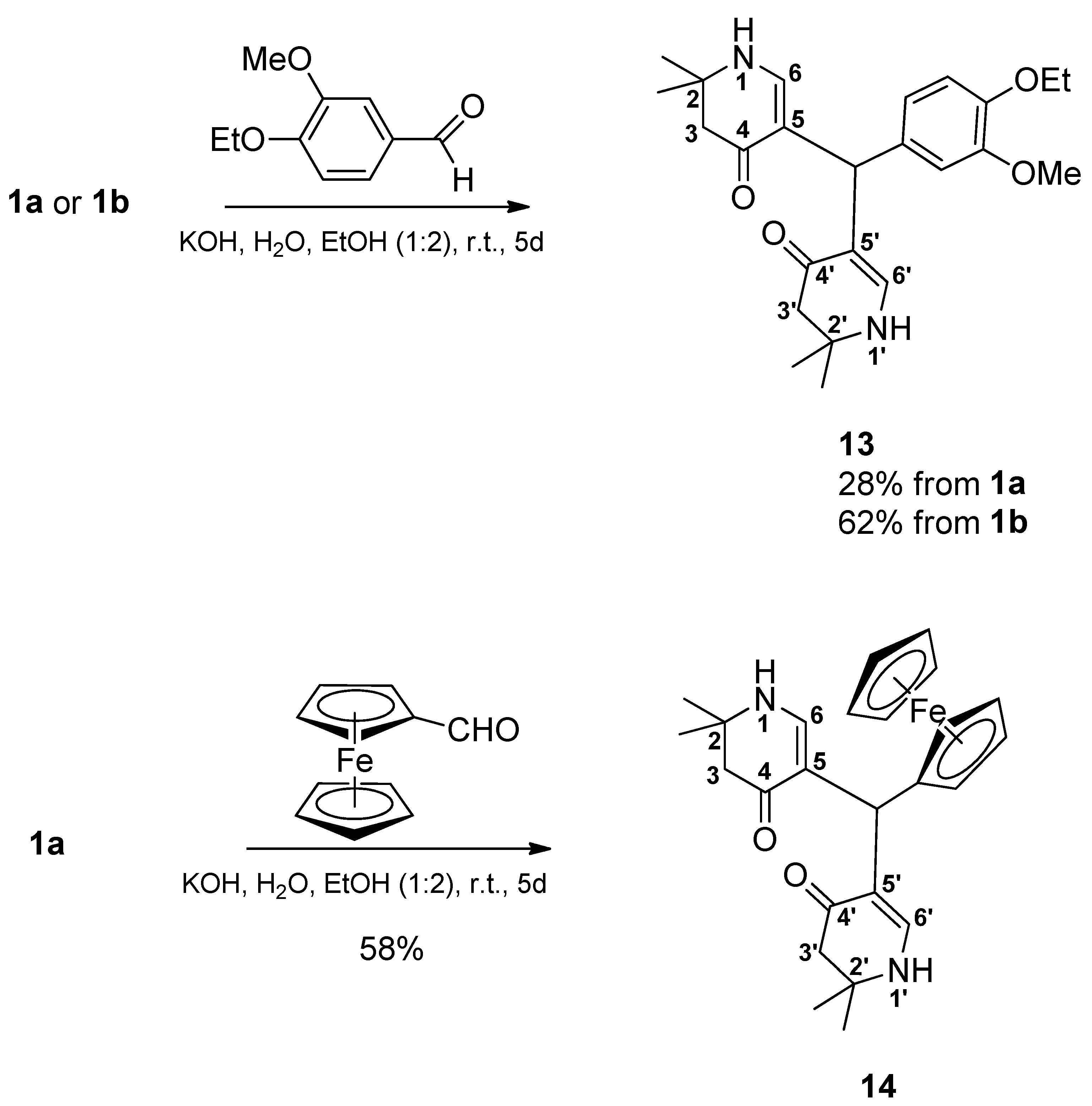
Due to the low solubility in water of solid ferrocenyl carbaldehyde and 4-ethoxy-3-methoxybenzaldehyde, ethanol was added to the reaction mixture. Other than the so far reported reactions, these electron sufficient aldehydes gave dimers 13 and 14 (Scheme 3).
In order to confirm the initial steps of the proposed mechanism, we treated the 4-dialkylaminotetrahydropyridinylidene salt 1a with potassium hydroxide 4 days to yield dihydropyridone 15, quantitatively. Then a mixture of pyrrolidine and benzaldehyde in alkaline solution was added and considerable amounts of 2a were formed overnight. The reaction of 15 without secondary amine gave the hydroxy analogue 11 in high yield (Scheme 4).
The reaction mechanism can be considered as following: From the 4-dialkylaminotetrahydropyridinylidene salts 1 their corresponding bases 16 are set free in a first step. These are hydrolyzed to ketone 15 which is deprotonated to the resonance-stabilized anion 17. It is readily attacked by aldehydes giving aldols 18,19 which can tautomerize to compounds 11,12. Moreover, a proton can be abstracted from α-position. Cleavage of a hydroxy from the formed resonance-stabilized anions 20,21 leads to α,β-unsaturated ketones 22,23. Addition of secondary amines in β-position yields β-aminoketones 2-10. This is the main reaction for the less electrophilic aromatic aldehydes as well as isobutyric aldehyde. Alternatively, δ-diketones 13, 14 are obtained via a Michael reaction (Scheme 5).
2.2. Structure Elucidation
The structures of the obtained compounds were elucidated using NMR spectroscopy: In 13C-spectra, a signal shift from 162 to 189 ppm was observed for C-4 due to ketone formation. Furthermore, the resonance of the proton in position 5 disappeared in 1H spectra. The remaining olefinic proton showed a coupling to the proton in ring position 1. Connectivity was proven by cross peaks of the newly formed methine proton to C-4 and C-5 as well as to NCH2 and aromatic carbons in HMBC spectra. Through space couplings were detected in NOE experiments (Nuclear Overhauser experiments) (Figure 1). Both can be seen in the supplementary information.
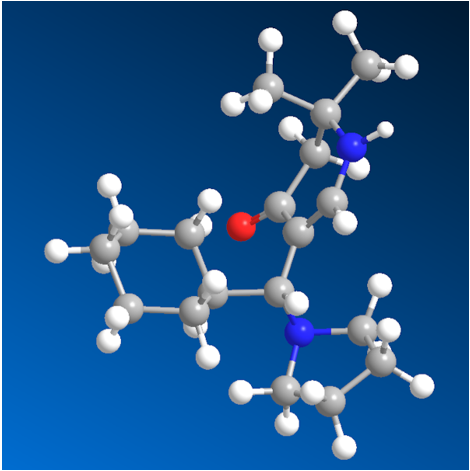
Finally, evidence of the structure of compound 6a was achieved by a single X-ray crystal analysis which confirmed compound as 5-[cyclohexyl(pyrrolidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one. This is the first determination of a structure containing a (pyrrolidin-1-yl) substituent in a 2,3-dihydropyridin-4(1H)-one (Figure 2). All atoms lie on general positions. The compound is a racemate.
2.3. Biological Activities
Most of the new compounds were tested for their anti-proliferative activity against human leukemia cells (CCRF-CEM) as well as against non-tumorigenic human lung fibroblasts (MRC-5) using an XTT-assay. Cells were exposed to compounds at concentrations of 5 and 50 µM for a time period of 72h. The results are presented in Figure 3 A and B.
At concentrations of 5 µM only the 2-(trifluoromethyl)phenyl aminoketone 4a showed moderate activity against a leukemia cell line. All other test compounds were inactive at this concentration. At 50µM the tert-butylaminoketone 2d as well as the dimer 11 were still non-effective and the 4-(trifluoromethyl)phenyl aminoketones 5a and 5b had moderate activity. All other compounds showed a selective anti-leukemic effect. However, the aminoketones 6b and 8a, dimer 12 and the β-hydroxyketone 13 were comparably toxic against lung fibroblasts. The most promising selectivity was observed for the 2-(trifluoromethyl)phenyl aminoketone 4a, which showed good anti-leukemic effect paired with low cytotoxicity in fibroblasts at 5 µM.
The results of the antibacterial assay against Gram negative (E. coli) and Gram-positive bacteria (B. sub.) are listed in Table 1.
The most active compounds against Escherichia coli were aminoketones 3a, 4b, 5a, 5b with fluorophenyl or (trifluoromethyl)phenyl substitution as well as their cyclohexyl analogue 6a. The most active compound against Bacillus subtilis was the β-hydroxyketone 11.
3. Materials and Methods
3.1. Instrumentation and Chemicals
Solvents were used without further purification. Aldehydes were purified by chromatographic separation on a small column filled with aluminum oxide 60 active basic (activity I) (Merck) to remove acidic impurities. For thin-layer chromatography (TLC), TLC plates silica gel 60 F254 (Merck) were used. Melting points were obtained on an Electrothermal IA 9200 melting point apparatus. IR spectra: Bruker Alpha Platinum ATR FTIR spectrometer (KBr discs); frequencies are reported in cm-1. The structures of all newly synthesized compounds were determined by one- and two-dimensional NMR spectroscopy. NMR spectra were acquired with Varian UnityInova 400 (298 K) or Bruker Avance Neo 400 instruments in 5 mm tubes. Some spectra were acquired in CDCl3 containing 0.03% TMS. Chemical shifts were recorded in parts per million (ppm), for 1H spectra TMS (0.00) was used as internal standard, and for 13C spectra the central peak of the CDCl3 peak was used as internal reference (77.0). Most of the spectra were acquired in DMSO-d6. In this case, the central peaks of the solvent signal at 2.49 ppm in 1H spectra, and at 39.7 ppm in 13C spectra served as internal reference. Shifts in 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra are reported in ppm; 1H- and 13C-resonances were assigned using 1H,1H- and 1H,13C-correlation spectra and are numbered as given in Scheme 1. Abbreviations: aromatic H, ArH; aromatic C, ArC, quaternary aromatic C, ArCq. Signal multiplicities are abbreviated as follows: s, singlet; d, doublet; dd, double doublet; dsept, double septet; quin, quintet; t, triplet; m, multiplet; br, broad. Coupling constants (J) are reported in Hertz (Hz). Assignments marked with an asterisk are interchangeable. HRMS: Micromass Tofspec 3E spectrometer (MALDI) and GCT Premier, Waters (EI, 70eV) or Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer, Thermo Fisher Scientific (HESI, capillary voltage 3.5 kV). Electron ionisation (EI+, 70 eV, source at 250 °C) mass spectra were acquired on a JMS-T2000GC (AccuTOFTM GC-Alpha) from JEOL Ltd. (Tokyo, Japan) equipped with a direct insertion probe (DIP). 1H NMR and 13C NMR spectra of new compounds are available in Supplementary Materials.
3.2. Syntheses
Compounds 1a-1d have been prepared earlier and their NMR data are in accordance with reported literature [1].
rac-2,2-Dimethyl-5-[(R)-phenyl(pyrrolidin-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (2a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (16 mL) and a solution of KOH (1.466 g (26.13 mmol)) in water (16 mL) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5-10 min). Then benzaldehyde (0.693 g (6.53 mmol)) was added. The reaction mixture was stirred for 4 days at r.t.. The separated crystalline solid was filtered with suction, washed with water and acetone and dried in vacuo yielding 2a (1.261g (68%)) as orange-yellow solid. For analytical purposes it was recrystallized from ethyl acetate/cyclohexane giving a white solid.
From 15: Compound 15 (0.671 g (5.36 mmol)) was dissolved in a solution of KOH (1.213 g (21.62 mmol)) in distilled water (26 mL). The mixture was sonicated and stirred at r.t. until all was dissolved. To the resulting solution, pyrrolidine (0.409 g (5.75 mmol)) and benzaldehyde (0.582 g (5.48 mmol)) were added. The reaction mixture was stirred for 16h at r.t.. The separated crystalline solid was filtered with suction, washed clean with water and dried in vacuo giving 2a (1.129 g (74%)) of as a pale pink solid. For analytical purposes it was recrystallized from ethyl acetate giving colorless needles.
Rf (CH2Cl2:MeOH = 10:1): 0.09; mp: 147-148°C; IR = 3232, 2967, 2783, 1623, 1568, 1534, 1395, 1231, 700; 1H NMR (CDCl3, 400 MHz) δ = 1.19 (s, 3H, CH3), 1.29 (s, 3H, CH3), 1.75 (br, s, 4H, (CH2)2), 2.37 (s, 2H, 3-H), 2.50 (br, s, 4H, 2NCH2), 4.52 (s, 1H, 1′-H), 4.60 (d, J = 5.5 Hz, 1H, 1-H), 7.14 – 7.38 (m, 6H, 6-H, ArH); 13C NMR (CDCl3, 100 MHz) δ = 23.49 ((CH2)2), 26.57, 27.57 (2CH3), 49.18 (C-3), 53.17 (2NCH2), 53.80 (C-2), 63.98 (C-1′), 111.43 (C-5), 126.09, 127.14, 128.03 (5ArC), 144.87 (ArCq), 149.23 (C-6), 189.50 (C-4); HRMS (EI): calcd. (C18H24N2O+) [M]+: 284.1889; found: 284.1874.
rac-2,2-Dimethyl-5-[(R)-phenyl(piperidin-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (2b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (16 mL) and a solution of KOH (1.470 g (26.20 mmol)) in water (16 mL) was added. The mixture was stirred at r.t. until a solution was formed (ca. 10-20 minutes). Then benzaldehyde (0.695 g (6.55 mmol) was added. The reaction mixture was stirred for 5 days at r.t.. The separated crystalline solid was filtered with suction, washed with water and acetone and dried in vacuo giving 2b (0.972 g (50%)) as orange-yellow solid. For analytical purposes it was recrystallized from ethyl acetate giving a white solid. Rf (CH2Cl2:MeOH = 1:1): 0.86; mp: 158°C; IR = 2925, 1623, 1571, 1527, 1255, 1232, 700; 1H NMR (DMSO-d6, 400 MHz) δ = 1.09 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.31 – 1.51 (m, 6H, (CH2)3), 2.13 – 2.22 (m, 4H, 3-H, NCH2), 2.28 – 2.38 (m, 2H, NCH2), 4.37 (s, 1H, 1′-H), 6.95 (d, J = 6.8 Hz, 1H, 6-H), 7.09 – 7.14 (m, 1H, ArH), 7.20 – 7.26 (m, 4H, ArH), 7.35 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 24.62 (CH2), 26.11 (2CH2), 26.19, 26.78 (2CH3), 48.90 (C-3), 52.34 (2NCH2), 53.03 (C-2), 64.17 (C-1′), 106.75 (C-5), 125.98, 127.30, 128.17 (5ArC), 144.62 (ArCq), 149.38 (C-6), 188.49 (C-4); HRMS (HESI): calcd. (C19H27N2O+) [M+H]+: 299.2123; found: 299.2114.
rac-2,2-Dimethyl-5-[(R)-phenyl(morpholin-4-yl)methyl]-2,3-dihydropyridin-4(1H)-one (2c): Compound 1c (2.000 g (6.22 mmol)) was suspended in water (30 mL) and KOH (1.397 g (24.89 mmol)) was added. To the resulting yellow solution benzaldehyde (0.660 g (6.22 mmol)) was added. The reaction mixture was stirred for 4 days at r.t.. The separated crystalline solid was filtered with suction, washed with water and acetone and dried over phosphorus pentoxide in vacuo giving 2c (1.436 g (77%)) as off-white solid. For analytical purposes it was recrystallized from ethyl acetate giving white needles. Rf (CH2Cl2:MeOH = 1:1): 0.89; mp: 165°C; IR = 3257, 2961, 1623, 1571, 1522, 1395, 1291, 1231, 1181, 1116, 702; 1H NMR (DMSO-d6, 400 MHz) δ = 1.09 (s, 3H, CH3), 1.17 (s, 3H, CH3), 2.15 – 2.25 (m, 4H, 3-H, NCH2), 2.29 – 2.39 (s, 2H, NCH2), 3.49 – 3.60 (br, s, 4H, 2OCH2), 4.36 (s, 1H, 1′-H), 7.00 (d, J = 6.8 Hz, 1H, 6-H), 7.13 – 7.16 (m, 1H, ArH), 7.25 – 2.26 (m, 4H, ArH), 7.42 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.20, 26.77 (2CH3), 48.82 (C-3), 52.11 (2NCH2), 53.04 (C-2), 64.25 (C-1′), 66.61 (2OCH2), 106.04 (C-5), 126.26, 127.42, 128.33 (5ArC), 143.71 (ArCq), 149.51 (C-6), 188.47 (C-4); HRMS (HESI): calcd. (C14H16ON+) [M+H-C4H9NO]+: 214.1232; found: 214.1223.
rac-5-[(R)-(tert-Butylamino)(phenyl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (2d): Compound 1d (2.013 g (6.53 mmol)) was suspended in water (30 mL) and KOH 1.400 g (25 mmol)) was added. Then benzaldehyde (0.693 g (6.53 mmol)) was added and the reaction mixture was stirred for 6 days at r.t.. From the resulting orange resin, the aqueous solution was decanted and the resin washed with water repeatedly. Then it was dried overnight in vacuo over phosphorus pentoxide. The dry resin was dissolved in the minimum amount of hot ethyl acetate and left for crystallization at r. t. The solid was sucked off and dried giving 2d (0.512 g (27%)) as off-white needles. Rf (MeOH): 0.07; mp: 136°C; IR = 2964, 1633, 1550, 1477, 1454, 1412, 1367, 1357, 1195, 1174, 947, 875, 828, 727, 713, 697, 678; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.10 (s, 3H, CH3), 1.20 (s, 9H 3CH3), 2.31 (d, J = 15.0 Hz, 1H, 3-H), 2.36 (d, J = 15.0 Hz, 1H, 3-H), 5.21 (s, 1H, 1′-H), 5.98 (br, s, 1H, NH*), 6.25 (br, s, 1H, 6-H*), 6.94 (br, s, 1H, 1-H*), 7.12 – 7.34 (m, 5H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 26.64, 26.81 (2CH3), 31.66 (3CH3), 41.37 (C-3), 50.97 (C-2), 53.45 (C(CH3)3), 73.61 (C-1′), 109.18 (C-5), 126.17, 126.43, 127.59 (5ArC), 138.93 (C-6), 146.02 (ArCq), 161.70 (C-4); HRMS (HESI): calcd. (C18H27N2O+) [M+H]+: 287.2123; found: 287.2114.
rac-5-[(R)-(4-Fluorophenyl)(pyrrolidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (3a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (16 mL) and a solution of KOH (1.466 g (26.13 mmol)) in water (16 mL) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5-10 min). Then 4-fluorobenzaldehyde (0.810 g (6.53 mmol)) was added and the reaction mixture was stirred for 6 days at r.t.. The separated crystalline solid was filtered with suction, washed with water and dried in vacuo over phosphorus pentoxide giving 3a (1.658 g (84 %)) as beige solid. It was recrystallized from ethyl acetate giving white needles. Rf (CH2Cl2:MeOH = 1:1): 0.29; mp: 169°C; IR = 3231, 2970, 2783, 1623, 1603, 1568, 1531, 1507, 1395, 1220, 1182; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.62 – 1.65 (m, 4H, (CH2)2), 2.14 (s, 2H, 3-H), 2.25 – 2.39 (m, 2H, NCH2), 2.33 – 2.39 (m, 2H, NCH2), 4.29 (s, 1H, 1′-H), 7.00 – 7.05 (m, 2H, ArH), 7.10 (d, J = 6.8 Hz, 1H, 6-H), 7.25 – 7.29 (m, 2H, ArH), 7.36 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 23.34 ((CH2)2), 26.02, 26.85 (2CH3), 48.92 (C-3), 52.79 (2NCH2), 53.03 (C-2), 63.35 (C-1′), 108.89 (C-5), 114.74 (d, 2J(C,F) = 21.0 Hz, ArC), 128.66 (d, 3J(C,F) = 7.9 Hz, ArC), 141.72 (d, 4J(C,F) = 3.1 Hz, ArCq), 149.08 (C-6), 160.69 (d, 1J(C,F) = 241.3 Hz, ArCq), 187.86 (C-4); HRMS (HESI): calcd. (C18H24FN2O+) [M+H]+: 303.1873; found: 303.1863.
rac-5-[(R)-(4-Fluorophenyl)(piperidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (3b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.470 g (26.20 mmol)) was added. Then 4-fluorobenzaldehyde (0.810 g (6.53 mmol)) was added and the reaction mixture was stirred for 4 days at r.t.. The separated crystalline solid was filtered with suction, washed with water and dried in vacuo over phosphorus pentoxide yielding 3b as a yellowish solid. It was dissolved in hot ethyl acetate and the insoluble part was removed by filtration. The product crystallized overnight as needles, was sucked off and washed with ice cold ethyl acetate giving 3b (1.137 g (55%)) as yellowish needles. Rf (CH2Cl2:MeOH = 1:1): 0.30; mp: 183°C; IR = 3250, 3031, 2965, 2938, 1624, 1571, 1525, 1506, 1393, 1380, 1290, 1255, 1231, 1220, 1180; 1H NMR (DMSO-d6, 400 MHz) δ = 1.10 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.34 – 1.48 (m, 6H, (CH2)3), 2.14 – 2.23 (m, 4H, 3-H, NCH2), 2.26 – 2.36 (m, 2H, NCH2), 4.37 (s, 1H, 1′-H), 6.95 (d, J = 6.8 Hz, 1H, 6-H), 7.05 (t, J = 8.9 Hz, 2H, ArH), 7.25 (dd, J = 8.5, 5.8 Hz, 2H, ArH), 7.38 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 24.56 (CH2), 26.09 (2CH2), 26.15, 26.72 (2CH3), 48.88 (C-3), 52.22 (2NCH2), 53.02 (C-2), 63.60 (C-1′), 106.41 (C-5), 114.78 (d, 2J(C,F) = 21.0 Hz, ArC), 128.96 (d, 3J(C,F) = 7.8 Hz, ArC), 140.55 (d, 4J(C,F) = 3.0 Hz, ArCq), 149.31 (C-6), 160.66 (d, 1J(C,F) = 241.6 Hz, ArCq), 188.51 (C-4); HRMS (HESI): calcd. (C19H26FN2O+) [M+H]+: 317.2029; found: 317.2020.
rac-5-[(R)-4-Fluorophenyl)(morpholin-4-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (3c): Compound 1c (0.700 g (2.18 mmol)) was suspended in water (11 mL) and KOH (0.489 g (8.71 mmol)) was added. 4-Fluorobenzaldehyde (0.271 g (2.18 mmol)) was added and the reaction mixture was stirred for 6 days at r.t.. The separated resin was washed with water and dried in vacuo. It was triturated with hot ethyl acetate and the insoluble parts were filtered off and a part of the solvent removed by evaporation. The formed amorphous precipitate was filtered off and the filtrate concentrated in vacuo. The product crystallized and was sucked off, washed with cold ethyl acetate and dried yielding 3c (0.225 g (32%)). Rf (CH2Cl2:MeOH = 1:1): 0.90; mp: 162°C; IR = 3244, 2969, 2957, 2842, 2809, 1651, 1621, 1571, 1508, 1394, 1292, 1263, 1249, 1224, 1182, 1118; 1H NMR (DMSO-d6, 400 MHz) δ = 1.09 (s, 3H, CH3), 1.17 (s, 3H, CH3), 2.14 – 2.23 (m, 4H, 3-H, NCH2), 2.30 – 2.39 (m, 2H, NCH2), 3.51 – 3.56 (m, 4H, 2OCH2), 4.35 (s, 1H, 1′-H), 7.00 (d, J = 6.8 Hz, 1H, 6-H), 7.07 (t, J = 8.8 Hz, 2H, ArH), 7.27 (dd, J = 8.5, 5.8 Hz, 2H, ArH), 7.45 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.18, 26.73 (2CH3), 48.82 (C-3), 52.02 (2NCH2), 53.05 (C-2), 63.72 (C-1′), 66.60 (2OCH2), 105.78 (C-5), 114.99 (d, 2J(C,F) = 21.0 Hz, ArC), 129.15 (d, 3J(C,F) = 7.7 Hz, ArC), 139.72 (d, 4J(C,F) = 3.0 Hz, ArCq), 149.46 (C-6), 160.81 (d, 1J(C,F) = 241.8 Hz, ArCq), 188.51 (C-4); HRMS (HESI): calcd. (C14H15FNO+) [M+H-C4H9NO]+: 232.1138; found: 232.1129.
rac-2,2-Dimethyl-5-{(R)-(pyrrolidin-1-yl)[(2-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (4a): Compound 1a (1.000 g (3.27 mmol)) was suspended in water (16 mL) and KOH (0.733 g (13.1 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5-10 minutes). Then 2-(trifluoromethyl)benzaldehyde (0.569 g (3.27 mmol)) was added and the reaction mixture was stirred for 4 days at r.t.. The separated resinous solid was washed with water and dried in vacuo over phosphorus pentoxide. It was dissolved in hot ethyl acetate and cyclohexane was added till first turbidity appeared. An oil separated which solidified. This amorphous precipitate was filtered off and discarded. The filtrate was evaporated and dissolved in hot ethyl acetate. It was allowed to stand for some days till crystallization took place giving 4a (0.282 g (24%)) as white needles. Rf (CH2Cl2:MeOH = 1:1): 0.39; mp: 139°C; IR = 3281, 2968, 1623, 1606, 1577, 1530, 1392, 1311, 1262, 1158, 1124, 1034, 773; 1H NMR (DMSO-d6, 400 MHz) δ = 1.08 (s, 3H, CH3), 1.14 (s, 3H, CH3), 1.58 – 1.67 (m, 4H, (CH2)2), 2.12 – 2.21 (m, 2H, 3-H), 2.28 – 2.46 (m, 4H, 2NCH2), 4.81 (s, 1H, 1′-H), 6.70 (d, J = 6.9 Hz, 1H, 6-H), 7.25 (d, J = 6.9 Hz, 1H, 1-H), 7.38 (t, J = 7.6 Hz, 1H, ArH), 7.60 (d, J = 7.9 Hz, 1H, ArH), 7.64 (t, J = 7.6 Hz, 1H, ArH), 8.04 (d, J = 7.8 Hz, 1H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 23.31 ((CH2)2), 26.33, 26.74 (2CH3), 48.89 (C-3), 52.05 (2NCH2), 52.95 (C-2), 57.93 (C-1′), 107.40 (C-5), 124.46 (q, 1J(C,F) = 274 Hz, CF3), 125.93 (q, 3J(C,F) = 6.1 Hz, ArC), 125.99 (q, 2J(C,F) = 29.5 Hz, ArCq), 126.68 (ArC), 129.67 (ArC), 132.26 (ArC), 143.16 (ArCq), 149.61 (C-6), 187.94 (C-4); HRMS (HESI): calcd. (C19H24F3N2O+) [M+H]+: 353.1841; found: 353.1830.
rac-2,2-Dimethyl-5-{(R)-(piperidin-1-yl)[(2-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (4b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.400 g (25 mmol) was added. Then 2-(trifluoromethyl)benzaldehyde (1.137 g (6.53 mmol)) was added and the reaction mixture was stirred for 5 days at r.t.. It was poured into a separatory funnel and was extracted 5 times with CH2Cl2. The combined organic phases were dried over sodium sulfate, filtered and the solvent was evaporated. The orange resinous residue was dissolved in the minimum amount of hot ethyl acetate and left for crystallization over the weekend. The precipitate was sucked off, washed with cold ethyl acetate and dried at 100°C in vacuo giving 4b (1.370 g (57%)) as white powder. Rf (CH2Cl2:MeOH = 1:1): 0.83; mp: 168°C; IR = 2922, 1626, 1575, 1531, 1451, 1386, 1311, 1295, 1266, 1247, 1162, 1153, 1112, 1087, 1058, 1034; 1H NMR (DMSO-d6, 400 MHz) δ = 1.06 (s, 3H, CH3), 1.14 (s, 3H, CH3), 1.27 – 1.50 (m, 6H, (CH2)3), 2.09 – 2.28 (m, 4H, 3-H, NCH2), 2.30 – 2.40 (m, 2H, NCH2), 4.75 (s, 1H, 1′-H), 6.62 (d, J = 6.8 Hz, 1H, 6-H), 7.27 (d, J = 6.9 Hz, 1H, 1-H), 7.36 (t, J = 7.6 Hz, 1H, ArH), 7.60 (d, J = 7.9 Hz, 1H, ArH), 7.63 (t, J = 7.6 Hz, 1H, ArH), 8.05 (d, J = 7.9 Hz, 1H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 24.60 (CH2), 26.17 (2CH2), 26.58 (2CH3), 48.81 (C-3), 52.14 (2NCH2), 52.94 (C-2), 59.18 (C-1′), 105.58 (C-5), 124.49 (q, 1J(C,F) = 274 Hz, CF3), 125.98 (q, 3J(C,F) = 6.0 Hz, ArC), 126.65 (ArC), 126.73 (q, 2J(C,F) = 29.4 Hz, ArCq), 129.13 (ArC), 132.26 (ArC), 143.57 (ArCq), 149.81 (C-6), 188.42 (C-4); HRMS (HESI): calcd. (C20H26F3N2O+) [M+H]+: 367.1997; found: 367.1986.
rac-2,2-Dimethyl-5-{(R)-(pyrrolidin-1-yl)[(4-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (5a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.400 g (25 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5-10 minutes). Then 4-(trifluoromethyl)benzaldehyde (1.137 g (6.53 mmol)) was added. The reaction mixture was stirred for 7 days at r.t.. It was poured into a separatory funnel and extracted 5 times with CH2Cl2. The combined organic phases were dried over sodium sulfate, filtered and the solvent was evaporated. The orange resinous residue was dissolved in the minimum amount of hot ethyl acetate and left for crystallization overnight. The solid was sucked off and washed with cold ethyl acetate and dried at 100°C in vacuo giving 5a (0.856 g (37%)) as yellowish needles. Rf (CH2Cl2:MeOH = 1:1): 0.45; mp: 167°C; IR = 2967, 1568, 1532, 1259, 1391, 1324, 1226, 1151, 1113, 1104, 1064, 1015, 906, 665; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.62 – 1.69 (m, 4H, (CH2)2), 2.11 – 2.19 (m, 2H, 3-H ), 2.26 – 2.33 (m, 2H, NCH2), 2.35 – 2.41 (m, 2H, NCH2), 4.40 (s, 1H, 1′-H), 7.11 (d, J = 6.8 Hz, 1H, 6-H), 7.45 (d, J = 6.7 Hz, 1H, 1-H), 7.47 (d, J = 8.1 Hz, 2H, ArH), 7.58 (d, J = 8.1 Hz, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 23.36 ((CH2)2), 25.98, 26.84 (2CH3), 48.82 (C-3), 52.73 (2NCH2), 53.07 (C-2), 63.78 (C-1′), 108.22 (C-5), 124.61 (q, 1J(C,F) = 272 Hz, CF3), 125.10 (q, 3J(C,F) = 3.8 Hz, ArC), 126.78 (q, 2J(C,F) = 31.5 Hz, ArCq), 127.62 (ArC), 149.36 (C-6), 150.40 (ArCq), 187.82 (C-4); HRMS (HESI): calcd. (C19H24F3N2O+) [M+H]+: 353.1841; found: 353.1830.
rac-2,2-Dimethyl-5-{(R)-(piperidin-1-yl)[(4-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (5b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.400 g (25 mmol)) was added. Then 2-(trifluoromethyl)benzaldehyde (1.137 g (6.53 mmol)) was added and the reaction mixture was stirred for 7 days at r.t.. It was poured into a separatory funnel and extracted 5 times with CH2Cl2. The combined organic phases were dried over sodium sulfate, filtered and the solvent was evaporated. The orange resin was dissolved in the minimum amount of hot ethyl acetate and left for crystallization overnight. The solid was sucked off, washed with cold ethyl acetate and dried at 100°C in vacuo giving 5b (1.304 g (54%)) as white needles. Rf (CH2Cl2:MeOH = 1:1): 0.63; mp: 180°C; IR = 2933, 1619, 1562, 1516, 1380, 1323, 1292, 1226, 1181, 1156, 1104, 1062, 1035, 1014, 986; 1H NMR (DMSO-d6, 400 MHz) δ = 1.10 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.31 – 1.52 (m, 6H, (CH2)3), 2.14 – 2.24 (m, 4H, 3-H, NCH2), 2.30 – 2.39 (m, 2H, NCH2), 4.46 (s, 1H, 1′-H), 6.96 (d, J = 6.8 Hz, 1H, 6-H), 7.44 – 7.48 (m, 3H, 1-H, ArH), 7.59 (d, J = 8.1 Hz, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 24.53 (CH2), 26.06 (2CH2), 26.16, 26.69 (2CH3), 48.81 (C-3), 52.25 (2NCH2), 53.09 (C-2), 64.23 (C-1′), 105.73 (C-5), 124.64 (q, 1J(C,F) = 272 Hz, CF3), 125.11 (q, 3J(C,F) = 3.8 Hz, ArC), 126.70 (q, 2J(C,F) = 31.6 Hz, ArCq), 127.91 (ArC), 149.62 (ArCq), 149.67 (C-6), 188.47 (C-4); HRMS (HESI): calcd. (C20H26F3N2O+) [M]+: 367.1997; found: 367.1986.
rac-5-[(R)-Cyclohexyl(pyrrolidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (6a) and rac-[(R)-Cyclohexyl(hydroxy)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (13): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.501 g (26.75 mmol)) was added. The mixture was stirred and sonicated at r.t. until most anything was dissolved. Then, cyclohexane carbaldehyde (0.749 g (6.68 mmol)) was added and the reaction mixture was stirred for 7 days at r.t.. The separated white precipitate was filtered with suction, washed with water and dried in vacuo giving a white solid. This was treated with dichloromethane and filtered. The filtrate was evaporated and the residue crystallized overnight in hot ethyl acetate in form of sparkling prisms of 6a (68 mg (4%)). The solid of the dichloromethane filtration was dissolved in hot ethyl acetate. Overnight, compound 11 (0.355 g (19%)) crystallized as silky needles. Compound 6a: Rf (CH2Cl2:MeOH = 1:1): 0.19; mp: 161°C; IR (KBr) = 3191, 3023, 2962, 2925, 2849, 2776, 1619, 1585, 1561, 1543, 1409, 1293, 1270, 1241, 1210, 1179; 1H NMR (DMSO-d6, 400 MHz) δ = 0.62 – 0.78 (m, 2H, CH2), 0.95 – 1.02 (m, 1H, CH2), 1.05 – 1.20 (m, 7H, CH2, 2CH3), 1.52 – 1.71 (m, 11H, CH, CH2), 2.18 (br, s, 2H, 3-H), 2.22 - 2.26 (m, 2H, NCH2), 2.28 – 2.33 (m, 2H, NCH2), 3.23 (d, J = 6.5 Hz, 1H, 1′-H), 6.95 (d, J = 6.6 Hz, 1H, 6-H), 7.20 (d, J = 6.6 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 22.99 ((CH2)2), 26.03 (CH2), 26.10 (CH3), 26.26 (CH2), 26.55 (CH3), 26.83, 27.79, 31.26 (3CH2), 40.45 (CH), 49.13 (C-3), 50.45 (2NCH2), 52.78 (C-2), 61.81 (C-1′), 103.50 (C-5), 149.44 (C-6), 189.39 (C-4); HRMS (HESI): calcd. (C18H31N2O+) [M+H]+: 291.2436; found: 291.2426.
Crystal Structure Determination of6a: All the measurements were performed using monochromatized Mo Kα radiation at 100K: C18H30N2O, Mr 290.44, orthorhombic, space group P b c a, a = 10.5346(4)Å, b = 11.7374(4)Å, c = 28.1853(11)Å, V = 3485.1(2)Å3, Z = 8, dcalc = 1.107g cm-3, μ = 0.068mm-1. A total of 125061 reflections were collected (Θmax = 30.0°), from which 5084 were unique (Rint = 0.0642), with 4213 having I > 2σ(I). The structure was solved by direct methods (SHELXS-97) [29] and refined by full-matrix least-squares techniques against F2 (SHELXL-2014/6) [30]. The non-hydrogen atoms were refined with an-isotropic displacement parameters without any constraints. The position of the H atom bonded to N1 was taken from a difference Fourier map, the N–H distance was fixed to 0.88Å, and this H atom was refined with an individual isotropic displacement parameter without any constraints to the bond angles. The H atom bound to C6 was put at the external bisector of the N–C–C angle at a C–H distance of 0.95Å and an individual isotropic displacement parameter was refined for it. The H atoms of the tertiary C–H groups were refined with individual isotropic displacement parameters and all X–C–H angles equal at a C–H distance of 1.00Å. The H atoms of the CH2 groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometry with approximately tetrahedral angles and C–H distances of 0.99Å. The H atoms of the methyl groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometries with tetrahedral angles, enabling rotations around the C–C bonds, and C–H distances of 0.98Å. For 211 parameters, final R indices of R1 = 0.0431 and wR2 = 0.1167 (GOF = 1.035) were obtained. The largest peak in a difference Fourier map was 0.417eÅ-3. The final atomic parameters, as well as bond lengths and angles have been deposited at the Cambridge Crystallographic Data Centre (CCDC 2193723).
- Compound 11 from 15:
Compound 15 (0.412 g (3.29 mmol)) was dissolved in a solution of KOH (0.752 g (13.4 mmol)) in distilled water (15 mL) with sonication. To this solution, cyclohexyl carbaldehyde (0.375 g (3.24 mmol)) was added and the mixture stirred at r.t. After a few minutes a white precipitate was formed. To complete the reaction, the mixture was stirred for further 4 days at r.t. The white precipitate was sucked off, washed with water and dried over phosphorous pentaoxide in a desiccator under reduced pressure. Yield: 0.712 g (91%) of pure alcohol 11.
Data of compound 11: Rf (CH2Cl2:MeOH = 1:1): 0.79; mp: 180°C; IR = 3283, 3221, 3042, 2926, 2849, 1557, 1519, 1409, 1309, 1264, 1244, 1183, 1121; 1H NMR (DMSO-d6, 400 MHz) δ = 0.79 – 0.95 (m, 2H, CH2), 0.99 – 1.12 (m, 3H, CH2), 1.15 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.26 – 1.35 (m, 1H, CH), 1.42 (br, d, J = 12.9 Hz, 1H, CH2), 1.54 – 1.67 (m, 3H, CH2), 1.77 (br, d, J = 13.0 Hz, 1H, CH2), 2.11 – 2.20 (m, 2H, 3-H), 4.07 (dd, J = 6.7, 5.0 Hz, 1H, 1′-H), 4.21 (d, J = 5.0 Hz, 1H, OH), 7.04 (d, J = 6.6 Hz, 1H, 6-H), 7.28 (d, J = 6.7 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.02 (CH2), 26.04 (CH3), 26.16 (CH2), 26.56 (CH2), 26.92 (CH3), 28.63, 29.33 (2CH2), 44.23 (CH), 49.17 (C-3), 52.90 (C-2), 70.05 (C-1′), 109.10 (C-5), 148.23 (C-6), 188.81 (C-4); HRMS (HESI): calcd. (C14H22NO2-) [M-H]-: 236.1651; found: 236.1658.
rac-5-[(R)-Cyclohexyl(piperidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (6b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30mL) and KOH (1.449 g (25.82 mmol)) was added. The mixture was stirred and sonicated at r.t. until most anything was dissolved. Then, cyclohexane carbaldehyde (0.749 g (6.68 mmol)) was added and the reaction mixture was stirred for 7 days at r.t.. The separated off-white solid was filtered with suction, washed with water and dried in vacuo. It was treated with dichloromethane and the suspension was filtered. The filtrate was evaporated and the residue dissolved in hot ethyl acetate. After a first crystallization, a solid mixture was sucked off. From the filtrate, 6b crystallized as yellowish prisms (0.170 g (9%)). The solid from the treatment with dichloromethane was dissolved in the minimum amount of hot ethyl acetate and left for crystallization overnight. The solid was sucked off and dried yielding 11 (0.355 mg (23%)) as white needles. Compound 6b: Rf (CH2Cl2:MeOH = 1:1): 0.84; mp: 154°C; IR = 3203, 3026, 2927, 2848, 1616, 1562, 1397, 1383, 1293, 1276, 1259, 1242; 1H NMR (DMSO-d6, 400 MHz) δ = 0.57 – 0.66 (m, 1H, CH2), 0.74 – 0.84 (m, 1H, CH2), 1.00 – 1.16 (m, 3H, CH2), 1.18, 1.19 (2s, 6H, 2CH3), 1.22 – 1.75 (m, 11H, CH, CH2), 1.92 – 2.00 (m, 1H, CH2), 2.00 – 2.09 (m, 2H, NCH2), 2.19 (s, 2H, 3-H), 2.22 - 2.29 (m, 2H, NCH2), 3.14 (br, s, 1H, 1′-H), 6.90 (d, J = 6.6 Hz, 1H, 6-H), 7.18 (d, J = 6.6 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 24.95 (CH2), 25.84 (CH3), 25.96, 26.03 (2CH2), 26.35 (2CH2), 26.65 (CH3), 26.78, 30.50, 31.33 (3CH2), 36.58 (CH), 49.14 (C-3), 50.24 (2NCH2), 52.77 (C-2), 64.45 (C-1′), 101.84 (C-5), 148.69 (C-6), 190.15 (C-4); HRMS (HESI): calcd. (C14H22NO+) [M+H-C5H11N]+: 220.1701; found: 220.1693.
rac-2,2-Dimethyl-5-[(R)-(4-methylphenyl)(pyrrolidin-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (7a): Compound 1a (1.000 g (3.27 mmol)) was suspended in water (16 mL) and KOH (0.733 g (13.1 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5-10 min). Then 4-methylbenzaldehyde (0.393 g (3.27 mmol)) was added and the reaction mixture was stirred for 6 days at r.t.. The separated beige crystalline solid was filtered with suction, washed with water and dried in vacuo over phosphorus pentoxide. It was treated with hot ethyl acetate. The insoluble parts were removed by filtration and the filtrate was concentrated in vacuo. Crystallization took place overnight yielding 7a (0.440 g (45%)) as colorless plates. Rf (CH2Cl2:MeOH = 1:1): 0.25; mp: 167°C; IR = 3240, 2968, 1622, 1571, 1536, 1394, 1240; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.57 – 1.67 (m, 4H, (CH2)2), 2.09 – 2.17 (m, 2H, 3-H), 2.22 (s, 3H, ArCH3), 2.24 – 2.40 (m, 4H, 2NCH2), 4.27 (s, 1H, 1′-H), 7.02 (d, J = 7.8 Hz, 2H, ArH), 7.08 (d, J = 6.8 Hz, 1H, 6-H), 7.13 (d, J = 7.8 Hz, 2H, ArH), 7.30 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 20.79 (ArCH3), 23.35 ((CH2)2), 26.04, 26.92 (2CH3), 48.98 (C-3), 52.81 (2NCH2), 53.01 (C-2), 63.58 (C-1′), 109.27 (C-5), 126.91, 128.69 (ArC), 134.90, 142.63 (ArCq), 149.13 (C-6), 187.83 (C-4); HRMS (HESI): calcd. (C19H27N2O+) [M+H]+: 299.2123; found: 299.2116.
rac-5-[(R)-(4-Isopropylphenyl)(pyrrolidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (8a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (32 mL) and KOH (1.466 g (26.13 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5-10 min). Then, 4-isopropylbenzaldehyde (0.968 g (6.53 mmol)) was added and the reaction mixture was stirred for 5 days at r.t.. The separated resinous beige product was dried in vacuo over phosphorus pentoxide and then treated with hot ethyl acetate. The insoluble part was filtered off. Cyclohexane was added to the solution and the mixture was allowed to stand overnight. The precipitate was filtered off and discarded, the filtrate was evaporated to dryness and the residue was recrystallized from ethyl acetate giving an almost pure product. Further crystallization from ethyl acetate afforded pure 8a (0.118 g (6%)) as white needles. Rf (CH2Cl2:MeOH = 1:1): 0.91; mp: 152°C; IR = 3240, 3042, 2963, 1622, 1568, 1461, 1395, 1290, 1239, 1181; 1H NMR (DMSO-d6, 400 MHz) δ = 1.09 (s, 3H, CH3), 1.15 – 1.16 (m, 9H, CH(CH3)2, CH3), 1.61 – 1.65 (m, 4H, (CH2)2), 2.14 (s, 2H, 3-H), 2.25 – 2.37 (m, 4H, 2NCH2), 2.80 (quin, J = 6.9 Hz, 1H, CH(CH3)2), 4.28 (s, 1H, 1′-H), 7.07 – 7.09 (m, 3H, 6-H, 2ArH), 7.16 (d, J = 8.0 Hz, 2H, 2ArH), 7.31 (d, J = 6.7 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 23.33 ((CH2)2), 24.09, 24.12 (CH(CH3)2), 25.94, 27.10 (2CH3), 33.16 (CH(CH3)2), 48.95 (C-3), 52.86 (2NCH2), 53.02 (C-2), 63.50 (C-1′), 109.23 (C-5), 125.99, 126.90 (4ArC), 142.98, 145.90 (ArCq), 149.23 (C-6), 187.84 (C-4); HRMS (HESI+): calcd. (C21H31N2O+) [M+H]+: 327.2436; found: 327.2426.
rac-2,2-Dimethyl-5-[(R)-pyrrolid-4-yl)(pyrrolidine-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (9a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (32 mL) and KOH (1.466 g (26.13 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5-10 min). Then pyridine-4-carbaldehyde (0.700 g (6.53 mmol)) was added and the reaction mixture was stirred for 4 days at r.t.. The solution was extracted 5 times with dichloromethane and the combined organic layers were dried over sodium sulfate, filtered and the solvents evaporated in vacuo giving a yellow oil. This was dissolved in hot ethyl acetate and cooled to r.t.. Then cyclohexane was added dropwise until the mixture was opacified permanently. After stirring overnight at r.t. the formed precipitate was sucked off and dissolved in hot ethyl acetate, filtered from insoluble parts and left for crystallization yielding 9a (0.170 g (9%)) as yellowish needles. Rf (CH2Cl2:MeOH = 1:1): 0.37; mp: 169°C; IR = 3270, 3026, 2800, 1624, 1594, 1574, 1505, 1386, 1292, 1226, 1180, 1115; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.64 – 1.67 (m, 4H, (CH2)2), 2.16 (s, 2H, 3-H ), 2.28 – 2.50 (m, 4H, 2NCH2), 4.33 (s, 1H, 1′-H), 7.10 (d, J = 6.8 Hz, 1H, 6-H), 7.24 (dd, J = 4.5, 1.6 Hz, 2H, ArH), 7.49 (d, J = 6.8 Hz, 1H, 1-H), 8.40 (dd, J = 4.5, 1.6 Hz, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 23.34 ((CH2)2), 26.01, 26.76 (2CH3), 48.76 (C-3), 52.56 (2NCH2), 53.08 (C-2), 63.14 (C-1′), 107.57 (C-5), 122.24 (ArC), 149.49, 149.57 (C-6, ArC), 154.17 (ArCq), 187.77 (C-4); HRMS (HESI): calcd. (C17H24N3O+) [M+H]+: 286.1919; found: 286.1909.
rac-2,2-Dimethyl-5-[(R)-2-methyl-1-(pyrrolidin-1-yl)propyl]-2,3-dihydropyridin-4(1H)-one (10a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.473 g (26.25 mmol)) was added. To the mixture isobutyric aldehyde (475 mg (6.59 mmol)) was added and it was stirred and sonicated at r.t. until nearly all was dissolved. The reaction mixture was stirred for 7 days at r.t.. The raw product contained small amounts of Morita-Baylis-Hillman product, which was not isolated. The separated red oil was exhaustively extracted with dichloromethane, the combined organic layers dried over sodium sulfate, filtered and the solvent evaporated in vacuo giving a red oil. This was dissolved in the minimum amount of hot dichloromethane. The product crystallized in form of yellowish needles overnight, was sucked off and dried. Yield: 0.255 g (16%). Rf (CH2Cl2:MeOH = 1:1): 0.14; mp: 148°C; IR = 2955, 2771, 1617, 1584, 1562, 1537, 1456, 1408, 1382, 1364, 1268, 1239, 1211, 1181, 1125, 1110, 667; 1H NMR (DMSO-d6, 400 MHz) δ = 0.67 (d, J = 6.6 Hz, 3H, CH(CH3)2), 0.73 (d, J = 6.6 Hz, 3H, CH(CH3)2), 1.17 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.52 – 1.64 (m, 4H, 2CH2), 1.94 (dsept, J = 6.6 Hz, 1H, CH(CH3)2), 2.14 – 2.23 (m, 2H, 3-H), 2.22 – 2.36 (m, 4H, 2NCH2), 3.18 (d, J = 6.3 Hz, 1H, 1‘-H), 6.98 (d, J = 6.7 Hz, 1H, 6-H), 7.24 (d, J = 6.7 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 17.21 (CH(CH3)2), 20.83 (CH(CH3)2), 23.05 (2CH2), 25.90, 26.72 (2CH3), 30.12 (CH(CH3)2), 49.12 (C-3), 50.84 (2NCH2), 52.75 (C-2), 62.71 (C-1‘), 103.16 (C-5), 149.56 (C-6), 189.34 (C-4); HRMS (HESI): calcd. (C15H27N2O+) [M+H]+: 251.2118; found: 251.2114.
rac-2,2-Dimethyl-5-[(R)-(1-Hydroxy-2,2-dimethylpropyl)-2,3-dihydropyridin-4(1H)-one (12) Compound 1a (2.413 g (7.88 mmol)) was suspended in water (32 mL) and KOH (1.496 g (26.66 mmol) was added. To the mixture pivalaldehyde (0.678 g (7.88 mmol)) was added. The reaction mixture was stirred 4 days at r.t.. The formed white fluffy solid was sucked off and dried overnight in a desiccator over phosphorous pentaoxide. Yield: 1.129 g (68%) Rf (CH2Cl2:MeOH = 1:1): 0.80; mp: 168°C; IR = 3249, 2950, 1526, 1402, 1389, 1366, 1258, 1239, 1211, 1177, 1000; 1H NMR (DMSO-d6, 400 MHz) δ = 0.75 (s, 9H, 3CH3), 1.16 (s, 3H, CH3), 1.20 (s, 3H, CH3), 2.12 (d, J = 16.0 Hz, 1H, 3-H), 2.19 (d, J = 16.0 Hz, 1H, 3-H), 4.15 (d, J = 4.6 Hz, 1H, 1′-H), 4.43 (d, J = 4.8 Hz, 1H, OH), 7.07 (d, J = 6.6 Hz, 1H, 6-H), 7.39 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 25.45 (CH3), 26.13 (3CH3), 27.23 (CH3), 36.55 (C(CH3)3), 49.11 (C-3), 52.62 (C-2), 72.25 (C-1′), 107.94 (C-5), 149.17 (C-6), 188.70 (C-4); HRMS (DIP EI): calcd. (C12H21NO2) [M+]: 211.1567; found: 211.1573.
5,5’-[(4-Ethoxy-3-methoxyphenyl)methylene]bis(2,2-dimethyl-2,3-dihydropyridin-4(1H)-one) (13): From 1a: Compound 1a (2.000 g (6.53 mmol)) was suspended in water (15 mL) and ethanol (30 mL). Then a solution of KOH (1.488 mg (26.52 mmol) in a mixture of water (15 mL) and ethanol (30 mL) was added. Finally, 4-ethoxy-3-methoxybenzaldehyde (1.177 g (6.53 mmol)) was added and the reaction mixture was stirred for 5 days. Most of ethanol was evaporated in vacuo. The remaining solution was diluted with water, transferred into a separatory funnel and then extracted 4 times with dichloromethane. The combined organic layers were dried over sodium sulfate, filtered, and the solvent was evaporated yielding a yellow oil. This was dissolved in a minimum amount of hot ethyl acetate. Then, cyclohexane was added dropwise until the mixture was opacified permanently. The formed beige precipitate was sucked off, washed with a cold mixture of cyclohexane and ethyl acetate, and dried at 100°C at reduced pressure giving 11 (0.375 g (28%)). For analytical purposes, it was recrystallized from ethyl acetate giving the product as off-white precipitate.
From 1b: Compound 1b (2.091 g (6.53 mmol)) was suspended in water (15 mL) and ethanol (30 mL). Then a solution of KOH (1.579 g (28.14 mmol) in a mixture of water (15 mL) and ethanol (30 mL) was added. Finally, 4-ethoxy-3-methoxybenzaldehyde (1.177 g (6.53 mmol) was added and the reaction mixture was stirred for 5 days. Most of ethanol was evaporated in vacuo. The remaining solution was diluted with water, transferred into a separatory funnel and then extracted 4 times with dichloromethane. The combined organic layers were dried over sodium sulfate, filtered, and the solvent was evaporated giving a yellow oil. This was dissolved in a minimum amount of hot ethyl acetate. Cyclohexane was added dropwise until the mixture was opacified permanently. The formed beige precipitate was sucked off, washed with a cold mixture of cyclohexane and ethyl acetate and dried at 100°C at reduced pressure giving 13 (0.830 g (62%)). For analytical purposes it was recrystallized from ethyl acetate giving the product as off-white precipitate. Rf (CH2Cl2:MeOH = 1:1): 0.80; mp: 125°C; IR = 2968, 1578, 1512, 1378, 1294, 1241, 1182, 1135; 1H NMR (DMSO-d6, 400 MHz) δ = 1.17 (s, 6H, 2CH3), 1.18 (s, 6H, 2CH3), 1.28 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.14 – 2.22 (m, 4H, 3-H, 3′-H), 3.65 (s, 3H, OCH3), 3.92 (q, J = 7.0 Hz, 2H, OCH2CH3), 4.84 (s, 1H, CH), 6.50 (dd, J = 8.5, 2.0 Hz, 1H, ArH), 6.55 – 6.62 (m, 3H, 6-H, 6′-H, ArH), 6.78 (d, J = 8.2 Hz, 1H, ArH), 6.99 (d, J = 6.7 Hz, 2H, 1-H, 1′-H); 13C NMR (DMSO-d6, 100 MHz) δ = 15.08 (OCH2CH3), 26.36, 26.80 (4CH3), 37.99 (CH), 49.35 (C-3, C-3′), 53.12 (C-2, C-2′), 55.42 (OCH3), 63.90 (OCH2), 109.22 (C-5, C-5′), 112.38, 112.89, 119.87 (ArC), 138.27, 145.83, 148.65 (ArCq), 148.81 (C-6, C-6′), 188.74 (C-4, C-4′); HRMS (HESI): calcd. (C24H33N2O4+) [M+H]+: 413.2440; found: 413.2433.
5,5’-(Ferrocenylmethylene)bis(2,2-dimethyl-2,3-dihydropyridin-4(1H)-one) (14): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (15 mL) and ethanol (30 mL). Then a solution of KOH (1.435 g (25.57 mmol)) in a mixture of water (15 mL) and ethanol (30 mL) was added. Finally, ferrocene carbaldehyde (1.398 g (6.53 mmol)) was added. The reaction mixture was stirred for 5 days. The formed precipitate was sucked off and dried in vacuo over phosphorus pentoxide. It was treated with hot ethyl acetate and filtered. The filtrate was discarded. The precipitate was suspended in a mixture of dichloromethane and ethanol (4:1) and filtered off. The filtrate was evaporated and the residue was dissolved in a hot mixture of ethyl acetate and ethanol (1:1). The mixture was filtered and the filtrate was left for crystallization overnight. The formed solid was sucked off and dried at 150°C in vacuo giving 14 (0.850 g (58%)) as a yellow solid. Rf (CH2Cl2:MeOH = 1:1): 0.83; mp: 262°C (decomp.); IR = 3441, 3288, 2965, 1620, 1569, 1528, 1382, 1297, 1243, 1177; 1H NMR (DMSO-d6, 400 MHz) δ = 1.14 (s, 6H, 2CH3), 1.17 (s, 6H, 2CH3), 2.09 – 2.18 (m, 4H, 3-H, 3′-H), 3.85 (br, s, 2H, ArH), 4.01 (br, s, 2H, ArH), 4.07 (s, 5H, ArH), 4.63 (s, 1H, CH), 6.80 (d, J = 6.7 Hz, 2H, 6-H, 6′-H), 6.92 (d, J = 6.7 Hz, 2H, 1-H, 1′-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.32, 26.48 (4CH3), 32.76 (CH), 49.49 (C-3, C-3′), 53.01 (C-2, C-2′), 66.50 (2ArC), 68.01 (2ArC), 68.41 (5ArC), 94.48 (ArCq), 110.75 (C-5, C-5′), 148.78 (C-6, C-6′), 188.07 (C-4, C-4′); HRMS (HESI): calcd. (C25H31FeN2O2+) [M+H]+: 447.1735; found: 447.1721.
2,2-Dimethyl-2,3-dihydropyridin-4(1H)-one (15): Compound 1b (2.50 g (7.81 mmol)) was suspended in 37.5 mL water and KOH (2.127g (37.91 mmol)) was added. The mixture was stirred at r.t. for 4 days. The solution was transferred into a separatory funnel and extracted 10 times with dichloromethane. The combined organic phases were dried over sodium sulfate, filtered and the solvent evaporated. The remaining oil crystallized spontaneously giving 15 as an off white solid. Yield: 812 mg (6.49 mmol; 83%). The melting point (93°) corresponds well with that reported (93-94°) by Gusev [9] for this compound. 1H NMR (DMSO-d6, 400 MHz) δ = 1.17 (s, 6H, 2CH3), 2.16 (br, s, 2H, 3-H), 4.62 (dd, J = 7.1, 1.2 Hz, 1H, 5-H), 7.13 (dd, J = 7.0, 6.9 Hz, 1H, 6-H), 7.41 (br, s, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.36 (2CH3), 49.25 (C-3), 53.01 (C-2), 95.00 (C-5), 150.22 (C-6), 190.60 (C-4).
3.3. Bioassays
3.3.1. Cytotoxicity against Human (Cancer) Cell Lines
The human leukemia cell line CCRF-CEM was cultured in RPMI 1640 medium containing 2 mM L-glutamine (Gibco®, Thermo Fisher Scientific Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin and 100 µg/mL streptomycin (all obtained from Gibco ®). Human MRC-5 lung fibroblasts were cultivated in Minimum Essential Medium (MEM; Gibco ®) containing 4 mM L-glutamine, 10% FBS, 100 units/mL penicillin and 100 µg/mL streptomycin. MRC-5 cells were sub-cultured at 90% confluence by trypsinization using a 0.25% trypsin-EDTA solution (Gibco®).
Cytotoxicity was measured via cell metabolic activity, which was determined using XTT (2,3-Bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide). Cells in the logarithmic growth phase were seeded into 96-well plates (flat bottom; 100µl/well) at a density of 10,000 cells/well. CCRF-CEM cells were immediately used for experiments, whereas MRC-5 cells were incubated overnight before being used for experiments. Both cell lines were treated with compounds (5 and 50 µM) for 72 h before cell metabolic activity was determined using a commercial kit (Cell proliferation kit II, Roche) according to manufacturer’s instructions. CCRF-CEM and MRC-5 cells were incubated with the XTT-solution for 4 h and 2 h, respectively. Then absorbance was measured at 490 nm using a Hidex Sense microplate reader (Hidex, Turku, Finland). Experiments were performed as two independent experiments carried out in triplicates. Cell viability was calculated relative to mock-treated cells (0.1% DMSO) using blank-corrected values. Vinblastine (100 nM) served as positive control.
3.3.2. Antibacterial Activity
All compounds were dissolved in DMSO at a concentration of 0.01mg/µL, disc plate method [31] was used to detect bioactivity against Gram positive (B. subtilis) and Gram negative (E. coli) bacteria. Results were noted as the ability of the compound to inhibit the growth of the corresponding test organism by noting the zone of inhibition (ZOI) around a disc (6mm) as follows: mild clearance= detectable activity (±) (ZOI = 7-9 mm), clearance=active (+) (ZOI = 10 mm) and clear clearance = higher activity (++) (ZOI > 10mm). Compound 6a has cyclohexane moiety that could have interfere with its ability to penetrate bacterial outer membrane (lipopolysaccharide layer), disrupting this layer that is a structurally significant in Gram-negative bacteria [32], while the addition of fluorine has improved Gram-negative activity in 3a, 4b, 5a, 5b. Fluorine has the ability to change the electron distribution of a molecule, leading to modifications in the molecule’s pKa, dipole moment, chemical reactivity, and stability. The introduction of fluorine can reduce the basicity of compounds, resulting in improved bioavailability through better permeation through cellular membranes.
4. Conclusions
Exposition of 4-amino-tetrahydropyridinylidene salts to a series of aldehydes in alkaline medium afforded 5-substituted dihydropyridin-4(1H)-ones. The expected β-hydroxyketone was only isolated after reaction with cyclohexane carbaldehyde. Aromatic aldehydes yielded in most cases β-aminoketones and less frequently δ-diketones. Most of the aminoketones exhibited anti-proliferative activity against human leukemia cells. A few of these compounds showed high inhibitory activity against Escherichia coli, whereas the β-hydroxyketone was the most active against Bacillus subtilis. These compounds could be a base for further investigation to turn into leading compounds that could be further refined and optimized for possible applications.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figures S1–S20: 1H-, 13C-NMR and MS-spectra. Tables S1–S7: Crystal data.
Author Contributions
Conceptualization, W.S. and M.H.; investigation, W.S., M.H., F.B., T.P., M.A., E.-M.P.-W., R.S., and R.W.; methodology W.S. and M.H.; data curation, W.S., M.H., F.B., T.P., M.A., E.-M.P.-W., and R.S.; writing—original draft preparation, W.S. and R.W.; writing—review and editing, W.S., E.-M.P.-W., T.P., R.B. and R.W.; supervision, W.S. and R.B.; project administration, W.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Acknowledgments
The authors acknowledge open access funding by the University of Graz. Furthermore, NAWI Graz is acknowledged for supporting the Graz Central Lab Environmental, Plant & Microbial Metabolomics. The support by the “Jeol Application Lab” at Graz University of Technology is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of selected compounds are available from the authors.
References
- Seebacher, W.; Faist J.; Belaj, F.; Saf, R.; Kaiser, M.; Brun, R.; Weis, R. Synthesis of new tetrahydropyridinylidene ammonium salts and their antiprotozoal potency. Monatsh. Chem. 2015, 146, 1299-1308. [CrossRef]
- Mohsin, N.-ul-A.; Seebacher, W.; Faist, J.; Hochegger, P.; Kaiser, M.; Mäser, P.; Belaj, F.; Saf, R.; Kretschmer, N.; Alajlani, M.; Turek, I.; Brantner, A.; Bauer, R.; Bucar, F.; Weis, R. Synthesis of new 1-benzyl tetrahydropyridinylidene ammonium salts and their antimicrobial and anticellular activities. Eur. J. Med. Chem. 2018, 143, 97-106. [CrossRef]
- Mohsin, N.-ul-A.; Seebacher, W.; Hochegger, P.; Faist, J.; Saf, R.; Kaiser, M.; Mäser, P.; Weis, R. Synthesis of new 1-benzyl tetrahydro-4-ylidene piperidinium salts and their antiplasmodial and antitrypanosomal activities. Med. Chem. Res. 2019, 28, 742-753. [CrossRef]
- Petrisch, M.; Seebacher, W.; Mohsin, N.-ul-A.; Dolensky, J.; Hochegger, P.; Kaiser, M.; Mäser, P.; Belaj, F.; Saf, R.; Kretschmer, N.; Alajlani, M.; Brantner, A.; Bauer, R.; Schühly, W.; Weis, R. Preparation of new 1,3-dibenzyl tetrahydropyridinylidene ammonium salts and their antimicrobial and anticellular activities. Eur. J. Med. Chem. 2021, 210, 112969. [CrossRef]
- Sui, Z.; Flores, C.; Winters, M.P. Cyclopentylpyrazoles as n-type calcium channel blockers. Patent WO2014/28801, 2014, A1.
- Luo, S.; Wang, P.G.; Cheng, J.-P. Remarkable Rate Acceleration of Imidazole-Promoted Baylis−Hillman Reaction Involving Cyclic Enones in Basic Water Solution. J. Org. Chem. 2004, 69, 2, 555–558. [CrossRef]
- Innocenti, R.; Menchi, G.; Trabocchi, A. Dual Iminium- and Lewis Base Catalyzed Morita-Baylis-Hillman Reaction on Cyclopent-2-enone. Synlett 2018, 29, 820 – 824. [CrossRef]
- Kamal, A.A; Petrera, L.; Eberhard, J.; Hartmann, R.W. Structure-functionality relationship and pharmacological profiles of Pseudomonas aeruginosa alkylquinolone quorum sensing modulators. Org. Biomol. Chem. 2017, 15, 4620-4630. [CrossRef]
- Funk, P.; Motyka, K.; Soural, M.; Malon, M.; Koshino, H.; Kusz, J.; Hlavac, J. Study of 2-aminoquinolin-4(1H)-one under Mannich and retro-Mannich reaction. PLoS ONE 2017, 12, e0175364. [CrossRef]
- Ma, Y.; Luo, W.; Quinn, P.J.; Liu, Z.; Hider, R.C. Design, synthesis, physicochemical properties, and evaluation of novel iron chelators with fluorescent sensors. J. Med. Chem. 2004, 47, 6349-6362. [CrossRef]
- Lörinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthetic-and DFT modelling studies on regioselective modified Mannich reactions of hydroxy-KYNA derivatives. RSC Adv. 2021, 11, 543-554. [CrossRef]
- Haider, A.; Cornuz, G.; Wyler, H. Synthesis of 4-Oxo-1,2,3,4-tetrahydropyridine (2,3-Dihydro-4(1H)pyridinone). Helv. Chim. Acta. 1975, 58, 1287-1292. [CrossRef]
- Gusev, B.P.; Él’perina, E.A.; Kucherov, V.F. Chemistry of polyene and polyyne compounds. Chem. Heterocycl. Compd. 1971, 7, 1424-1426. [CrossRef]
- Buhr, G. 4-Aza-cycloalk-2-en-one und Verfahren zu ihrer Herstellung. German Patent DT 2013761, 21 March 1970.
- Jones, T.N.; McClure, C.K. Synthesis of 4-Ethoxy-2,3-dihydropyridinium Tetrafluoroborate Salts and 4-Ethoxy-2,3-dihydropyridines. Synlett. 2009, 8, 1289-1290. [CrossRef]
- Niphakis, M.J.; Turunen, B.J.; Georg, G.I. Synthesis of 6- and 7- Membered Cyclic Enaminones: Scope and Mechanism. J. Org. Chem. 2010, 75, 6793 – 6805. [CrossRef]
- Bi, L.; Georg, G.I. Direkt Hiyama Cross-Coupling of Enaminones with Triethoxy(aryl)silanes and Dimethylphenylsilanol. Org. Lett. 2011, 13, 5413-5415. [CrossRef]
- Brimioulle R.; Bach, T.; Enantioselective Lewis Acid Catalysis of Intramolecular Enone [2+2] Photocycloaddition Reactions. Science. 2013, 342, 840-843. [CrossRef]
- Brimioulle, R.; Bauer, A.; Bach, T. Enantioselective Lewis Acid Catalysis in Intramolecular [2+2] Photocycloaddition Reactions: A Mechanistic Comparison between Representative Coumarin and Enone Substrates. J. Am. Chem. Soc. 2015, 137, 5170-5176. [CrossRef]
- Chattopadhyay, A.K.; Hanessian, S. Cyclic enaminones. Part II: applications as versatile intermediates in alkaloid synthesis. Chem. Commun. 2015, 51, 16450-16467. [CrossRef]
- Saeed, B.A.; Elias, R.S. A DFT study for the structures and electronic spectra of 2,3-dihydropyridine-4-ones. Eur. J. Chem. 2011, 2, 469-474. [CrossRef]
- Donohoe, T.J.; Johnson, D.J.; Mace, L.H.; Thomas, R.E.; Chiu, J.Y. K.; Rodrigues, J.S.; Compton, R.G.; Banks, C.E.; Tomcik, P.; Bamford, M.J.; Ichihara, O. The ammonia-free partial reduction of substituted pyridinium salts. Org. Biomol. Chem. 2006, 4, 1071-1084. [CrossRef]
- Yu, Y.-Y.; Niphakis, M.J.; Georg, G.I. Palladium(II)-Catalyzed Dehydrogenative Alkenylation of Cyclic Enaminones via the Fujiwara–Moritani Reaction. Org. Lett. 2011, 13, 5932-5935. [CrossRef]
- Dodson, C.D.; Dyer, L.A.; Searcy, J.; Wright, Z.; Letourneau, D.K. Cenocladamide, a dihydropyridone alkaloid from Piper cenocladum. Phytochemistry, 2000, 53, 51-54. [CrossRef]
- Santos, C.C.F.; Paradela, L.S.; Novaes, L.F.T.; Dias, S.M.G.; Pastre, J.C. Design and synthesis of cenocladamide analogues and their evaluation against breast cancer cell lines. Med. Chem. Comm. 2017, 8, 755-766. [CrossRef]
- Qian, J.; Xu, Z.; Meng, C.; Liu, J.; Hsu, P.-L.; Li, Y.; Zhu, W.; Yang, Y.; Morris-Natschke, S.L.; Lee, K.-H.; Zhang, Y.; Ling, Y. Design and synthesis of benzylidenecyclohexenones as TrxR inhibitors displaying high anticancer activity including ROS, apoptosis, and autophagy. Eur. J. Med. Chem. 2020, 204, 112610. [CrossRef]
- Li, C.R.; Zhai, Q.-Q.; Wang, X.-K.; Hu, X.-X.; Li, G.-Q.; Zhang, W.-X.; Pang, J.; Lu, X.; Yuan, H.; Gordeev, M.F.; Chen, L.-T.; Yang, X.-Y.; You, X.-F. In vivo Antibacterial Activity of MRX-I, a new Oxazolidinone. Antimicrob. Agents Chemother. 2014, 58, 2418-2421. [CrossRef]
- Johnson, C.K. ORTEP Report ORNL-3794; Publisher: Oak Ridge National Laboratory, Tennessee, USA, 1965.
- Sheldrick, G.M.; A short history of SHELX. Acta Cryst 2008, 64, 112-122. [CrossRef]
- Sheldrick, G.M.; Crystal structure refinement with SHELXL. Acta Cryst 2015, 71, 3-8. [CrossRef]
- Alajlani, M.; Shiekh, A.; Hasnain, S.; Brantner, A.; Purification of Bioactive Lipopeptides produced by Bacillus subtilis strain BIA. Chromatographia. 2016, 79, 1527-1532. [CrossRef]
- Klobucar, K.; Côté, J. P.; French, S.; Borrillo, L.; Guo, A. B. Y.; Serrano-Wu, M. H.; Lee, K. K.; Hubbard, B.; Johnson, J. W.; Gaulin, J. L.; Magolan, J.; Hung, D. T.; Brown, E. D.; Chemical Screen for Vancomycin Antagonism Uncovers Probes of the Gram-Negative Outer Membrane. ACS Chem Biol. 2021, 21,16(5), 929-942. [CrossRef]
Scheme 1.
Syntheses of 2,3-dihydropyridin-4(1H)-ones and compounds containing this motif.
Scheme 2.
Synthesis of compounds 2-12.
Scheme 3.
Synthesis of δ-diketones 13, 14 with electron sufficient aldehydes.
Scheme 4.
Control experiments with dihydropyridone 15.
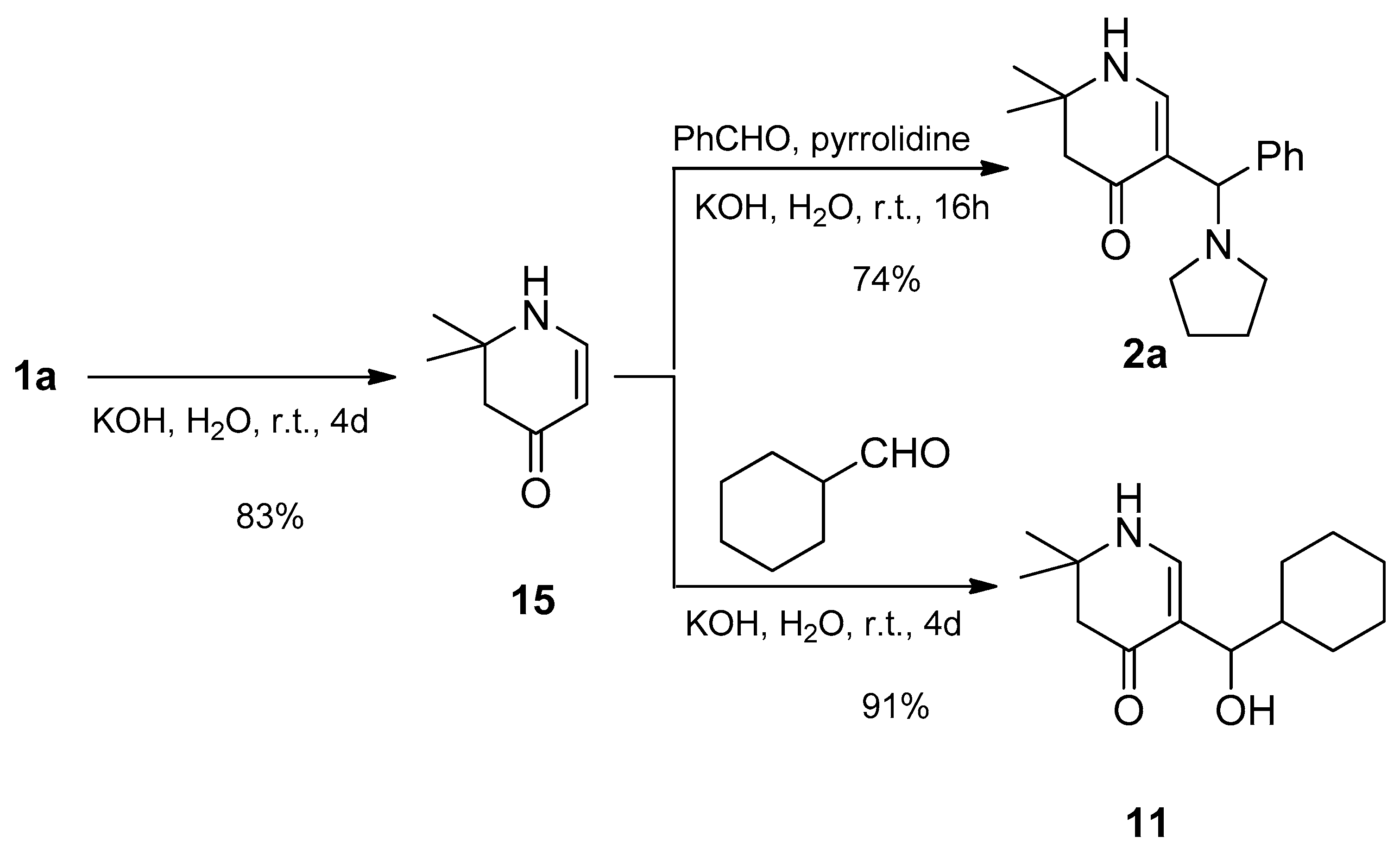
Scheme 5.
Possible mechanisms of the formation of compounds 2-14.
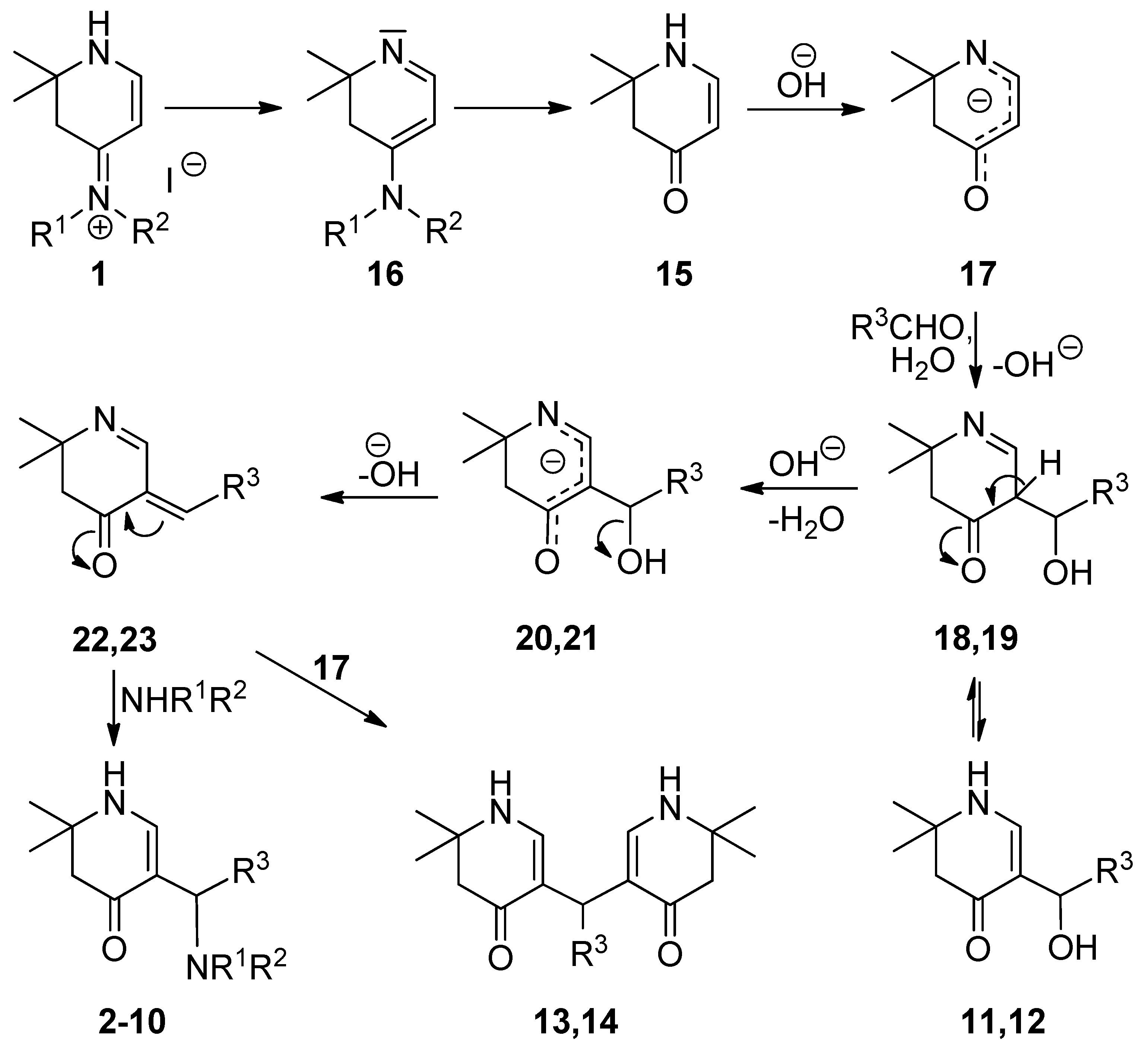
Figure 1.
Through space couplings indicated as arrows in compound 2a.
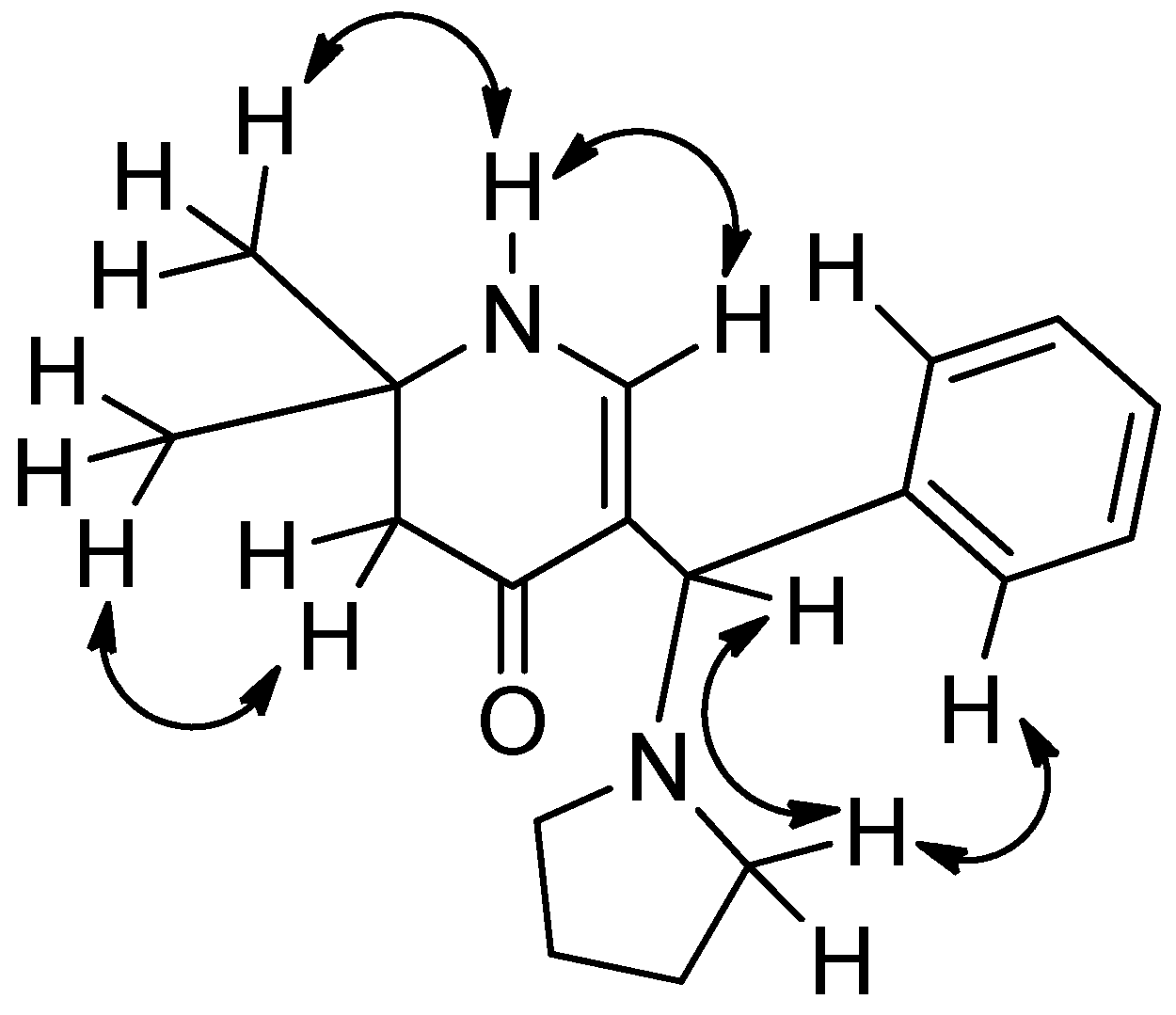
Figure 2.
Stereoscopic ORTEP [28] plot of 6a showing the atomic numbering scheme. The probability ellipsoids are drawn at the 50% probability level. The H atoms are drawn with arbitrary radii.
Figure 2.
Stereoscopic ORTEP [28] plot of 6a showing the atomic numbering scheme. The probability ellipsoids are drawn at the 50% probability level. The H atoms are drawn with arbitrary radii.
Figure 3.
Anti-proliferative activity of compounds 2b – 14 against human leukemia cell line CCRF-CEM (A) and against non-tumorigenic MRC-5 lung fibroblasts (B) expressed as mean ± SEM. Cells were treated with 50 and 5 µM of the compounds for 72h. Cell viability was measured via XTT assay. Viability rates were expressed as percentage of vehicle-treated control cells (0.1% DMSO). Vinblastine (VB) at a concentration of 100nM served as positive control.
Figure 3.
Anti-proliferative activity of compounds 2b – 14 against human leukemia cell line CCRF-CEM (A) and against non-tumorigenic MRC-5 lung fibroblasts (B) expressed as mean ± SEM. Cells were treated with 50 and 5 µM of the compounds for 72h. Cell viability was measured via XTT assay. Viability rates were expressed as percentage of vehicle-treated control cells (0.1% DMSO). Vinblastine (VB) at a concentration of 100nM served as positive control.
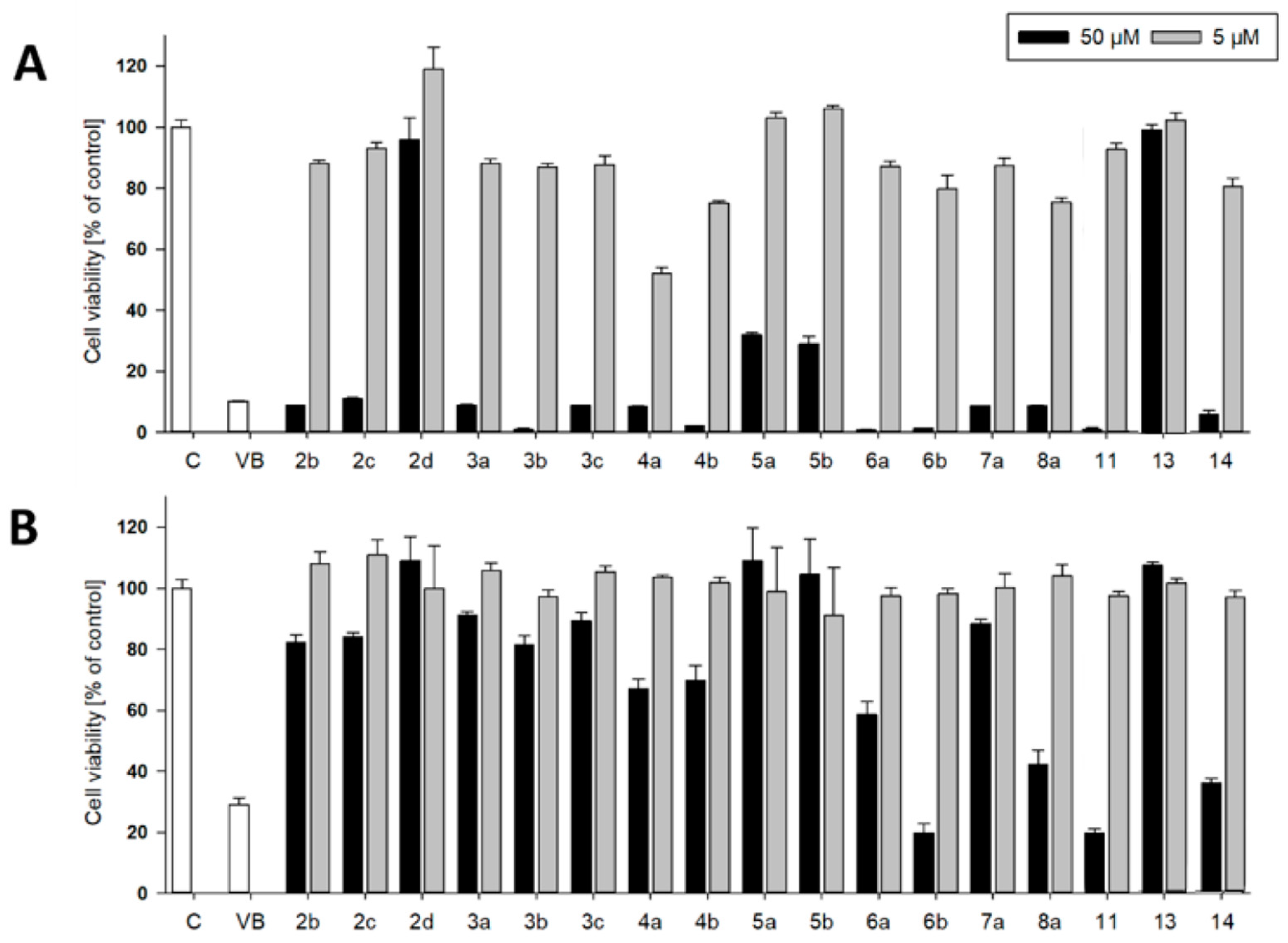

Table 1.
Antibacterial activity of compound 2b - 14.
| Substance | Escherichia coli | Bacillus subtilis |
|---|---|---|
| 2b | +* | - |
| 2c | + | + |
| 3a | ++ | - |
| 3b | + | + |
| 3c | + | - |
| 4a | ± | + |
| 4b | ++ | ± |
| 5a | ++ | + |
| 5b | ++ | + |
| 6a | ++ | ± |
| 6b | + | ± |
| 7a | + | + |
| 8a | + | - |
| 9a | + | - |
| 11 | + | ++ |
| 13 | + | + |
| 14 | ± | ± |
* Antimicrobial activity monitored as mild /detectable activity (±); active (+); higher activity (++); no activity (-).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



