
Submitted:
22 September 2023
Posted:
25 September 2023
You are already at the latest version
Abstract
The molecular basis of Down syndrome (DS) predisposition to leukemia is not fully understood but involves various factors such as chromosomal abnormalities, oncogenic mutations, epigenetic alterations, and changes in selection dynamics.
Myeloid leukemia associated with DS (ML-DS) is preceded by a preleukemic phase called transient abnormal myelopoiesis driven by GATA1 gene mutations and progresses to ML-DS through additional mutations in cohesin genes, CTCF, RAS, or JAK/STAT pathway genes.
DS-related ALL (ALL-DS) differs from non-DS ALL in terms of cytogenetic subgroups and genetic driver events and aberrant expression of CRLF2, JAK2 mutations, and RAS pathway activating mutations are frequent in ALL-DS.
Recent advancements in single-cell multi-omics technologies have provided unprecedented insights into the cellular and molecular heterogeneity of DS-associated hematologic neoplasms. Single-cell RNA sequencing and digital spatial profiling enable the identification of rare cell subpopulations, characterization of clonal evolution dynamics, and exploration of the tumor microenvironment's role. These approaches may help identify new druggable targets and tailor therapeutic interventions based on distinct molecular profiles, ultimately improving patient outcomes with the potential to guide personalized medicine approaches and the development of targeted therapies.
Keywords:
acute myeloid leukemia
; acute lymphoblastic leukemia
; single-cell RNA sequencing
; Down syndrome
; trisomy 21
; personalized medicine
; multi-omics approach
1. Trisomy 21 and leukemia
Down syndrome (DS) is the most common chromosomal disorder in humans1. It results from a full trisomy of chromosome 21 (T21) in 90% of cases, with remaining patients harboring other chromosome 21 abnormalities or mosaicisms. Notably, while the risk of solid tumors is reduced throughout life, DS is associated with an increased risk of developing leukemia, especially during the first years of life. In particular, DS children have a 150-fold and 7-20-fold increased risk of developing acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), respectively.
Although recent scientific progress, the molecular basis of DS predisposition to leukemia remains elusive. Patently, the perturbation of hematopoiesis in DS individuals is driven by chromosome 21. Besides T21-related mechanisms, several additional drivers have been described to be involved in DS-associated leukemogenesis, including concomitant oncogenic mutations, epigenetic and transcriptional alterations, and changes in selection dynamics within the fetal liver niche. Recently, single-cell multi-omics technologies has led to unbiased investigation of cellular profiles at unprecedented resolution in all hematological areas, including DS-associated hematologic neoplasms2,3.
Overall, DS represents the human phenotype model of genomic gain dosage imbalances, and the implementation of emerging single-cell analyses constitute an unprecedented opportunity to decipher the molecular consequences of genome dosage imbalance with potential groundbreaking consequences for non-DS leukemogenesis.
2. Myeloid proliferations related to Down syndrome
Myeloid proliferations in DS constitute a unique model of step-wise leukemogenesis that occurs during both pre- and post-natal life. Myeloid leukemia associated with DS (ML-DS) phenotypically recapitulates the features of acute megakaryoblastic leukemia (AMKL). Virtually all cases of ML-DS are preceded by a preleukemic phase termed transient abnormal myelopoiesis (TAM), which occurs in 5-30% of all DS neonates and is driven by somatic mutations in the GATA1 gene4. Mutations in GATA1 (mostly in exon 2) arise in utero from the 21st week of gestation and result in the expression of a shorter, N-terminal deleted protein (GATA1s). Although TAM spontaneously resolves within the first 3 months after birth in the majority of newborns, 20% of patients subsequently develop ML-DS before the age of five years. Leukemic progression occur from the initial GATA1-mutated clone through the acquisition of additional mutations in cohesin genes, CTCF, or genes of the RAS or JAK/STAT pathway5.
Using multi-omic assessment of mRNA and multiplexed protein epitope expression, Jardine and collegues2 uncovered an intrinsic bias of fetal HSPCs in DS that is underpinned by genome-wide transcriptional changes. In detail, MKs from DS fetuses expressed higher levels of regulons for GABPA (encoded on chromosome 21) and lower levels of FLI1 (a driver of MK differentiation), in line with previous data. Most myeloid lineages overexpressed TNF and TNF signaling pathway gene, consistent with the higher levels of circulating TNF in DS. Moreover, the expression of NOTCH1 and NOTCH ligands NOV (CCN3) and DLK1 was significantly higher in endothelium and HSPCs from fetuses with DS, respectively. These findings add novel insights into the role of cell-intrinsic and extrinsic regulation of differentiation in the development of ML-DS.
Recently, Wagenblast and colleagues6 developed a humanized model that faithfully recapitulates the full spectrum of DS premalignant and malignant leukemia using CRISPR/Cas9-mediated gene editing of primary human disomic and trisomic fetal HSPCs followed by xenotransplantation. In detail, the introduction GATA1 mutations caused preleukemia only in trisomic long-term HSCs. Simultaneous overexpression of a subset of chromosome 21 microRNAs (miR-99a, miR-125b-2, and miR-155) contributed to preleukemia initiation, while their removal inhibited GATA1s-induced preleukemia development. Furthermore, leukemic progression was independent of T21 and occurred in multiple HSPCs through additional mutations in cohesin genes. Interestingly, CD117+/KIT was identified as a driver of propagation of preleukemia and leukemia cells, and pharmacological KIT inhibition targeted both GATA1-induced preleukemic and primary TAM patient cells.
Aimed to investigate the cooperation between GATA1s and secondary genetic abnormalities, Arkoun and collegues7 used CRISPR/Cas9 technology to sequentially introduce GATA1s, MPLW515K, and haploinsufficiency of SMC3 (Cohesin complex subunit) in human disomic and trisomic induced pluripotent stem cells (iPSCs). GATA1s profoundly reshaped iPSC-derived hematopoiesis and cooperated with SMC3 haploinsufficiency to induce an even more profound failure of the GATA1-dependent MK differentiation program, including NFE2 downregulation. While T21 enhanced the proliferative phenotype, impairment of MK differentiation was independent of T21, consistent with previous findings6, thus suggesting that leukemic progression is independent of T21 and can be induced by the synergistic interaction between GATA1s and cohesin gene mutations.
Unlike non-DS AMKL, ML-DS harbors a generally favorable prognosis with excellent overall survival (OS). Notwithstanding, outcomes are dismal for patients with refractory/relapsed disease, with an OS rate <20%. Discernibly, prevention of leukemic development by the targeting of preleukemic clones represents an attractive therapeutic strategy.
3. Acute lymphoblastic leukemia related to Down syndrome
DS-related ALL (ALL-DS) is a distinct entity displaying clinical and biological features that differ from non-DS ALL. The age of diagnosis extends into adolescents and young adulthood, with a peak age that is slightly higher compared to non-DS ALL. The increased risk of ALL in DS is almost exclusively limited to the B-cell precursor (BCP) phenotype, with few reported cases of T-cell ALL.
Common cytogenetic subgroups of childhood non-DS-ALL are less represented in DS patients, including both favorable and unfavorable abnormalities8. Among genetic driver events, aberrant expression of CRLF2 has been well characterized and can be found in 5-15% of sporadic B-ALL versus 60% of ALL-DS. Other studies uncovered a high frequency of JAK2 mutations in ALL-DS, particularly the R683 mutation (18-28% of ALL-DS). Interestingly, several studies reported tight association between aberrant CRLF2 expression and JAK2 mutations, suggesting a cooperation between the two alterations. In addition to –and mutually exclusively with- JAK mutations, some reports identified RAS pathway activating mutations in up to 30% of ALL-DS patients.
Recently, Turati and colleagues9 showed that chemotherapy, while having little impact on genetic heterogeneity, exerts an extensive action on the transcriptional and epigenetic profile, resulting in a bottleneck selection of a genetically polyclonal but phenotypically uniform population.
In a more recent work10, the same study group presented compelling data suggesting that specific genotype-phenotype relationships have functional relevance in terms of leukemic progression and treatment resistance. Using serial transplantation assays and SNP-array, the authors showed that individual genetic lesions are restricted to well-defined cell immunophenotypes, corresponding to different stages of the leukemic differentiation hierarchy. Reconstruction of the leukemia phylogenetic tree demonstrated that the dominant population at relapse originated from a rare, highly quiescent, and developmentally primitive clone, representing a reservoir for relapse. More interestingly, genotypes and phenotypes with functional relevance were shown to co-segregate within the disease, in contrast with previous findings in non-DS ALL9. Overall, these findings depict ALL-DS as a complex matrix of cells exhibiting extensive genetic and epigenetic heterogeneity with dynamic clonal evolution and competition, and reinforce the idea that chemotherapy can act as a critical selective force for the preferential expansion of selected leukemic compartments.
In remarkable contrast to ML-DS, patients with ALL-DS typically have poor outcomes, with lower OS and higher relapse rate. Moreover, ALL-DS patients have a higher susceptibility to chemotherapy-related toxicities, resulting in increased risk of treatment-related morbidities and mortalities. On that basis, treatment of ALL-DS patients can be challenging, and the identification of specific molecular features aimed at reducing both relapse risk and treatment-related toxicities represents a major goal for the future.
4. Single-cell analysis: extending the frontiers of ML/ALL-DS
In recent years, next generation sequencing (NGS) provided a paramount contribution in the understanding of cancer biology. Neverthless, bulk genomic profiling methods fail to accurately resolve the clonal architecture of tumor populations and are limited in the identification of relapse-driving clones.
The development of single-cell analysis techniques, including single-cell RNA sequencing (scRNA-seq) and spatial single-cell imaging, offers a promising opportunity to gain insights into the biology of cancer development and progression. The use of scRNA-seq may allow the identification of rare cell subpopulations and the characterization of clonal evolution dynamics in order to decipher the mechanisms supporting treatment resistance and disease relapse. Moreover, scRNA-seq and spatial single-cell imaging have the potential to investigate the role of tumor microenvironment (TME) in supporting leukemia cell growth and survival. Ultimately, such depth of exploration may allow for the development of novel targeted therapies aimed at improving the patient outcomes.
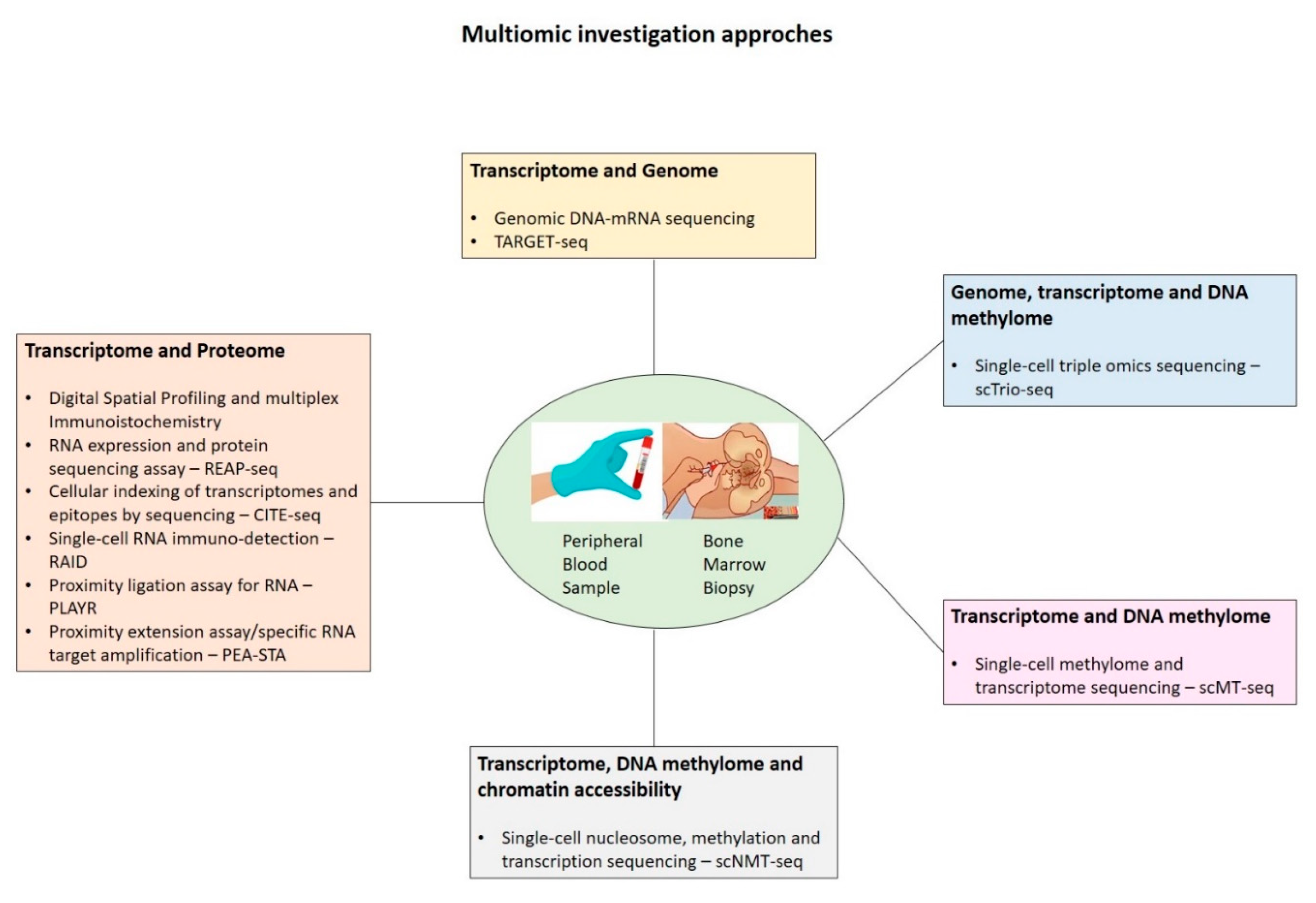
Finally, the integration of single-cell techniques within multi-omics (Figure 1), by allowing the joint analysis of genome, transcriptome, epigenome, and proteome at the single-cell level, may enable pivotal new insights into the complex interplay between intracellular and intercellular molecular mechanisms driving disease pathogenesis, evolution, and recurrence.
In conclusion, DS leukemogenesis represents a unique disease setting to study human preleukemia and the evolutionary steps that lead to fully transformed leukemia. The increasingly widespread access to platforms based on cell-by-cell technologies has allowed to overcome the limitations of conventional NGS, while shedding light on the complexity of tumor composition and clonal evolution In the era of personalized medicine, single-cell research has the potential to provide a more detailed understanding of the biology of DS leukemogenesis in order to identify new druggable targets and tailor the therapeutic intervention according to distinct molecular profiles at risk.
Author Contributions
EP and GC wrote the manuscript, MLR, AR, GM and LDA revised and provided important intellectual content.
Funding
This research received “Ricerca Corrente” funding from the Italian Ministry of Health to cover publication costs.
Consent for publication
All authors contributed to the research and approved the final manuscript.
Availability of data and material
Not applicable.
Competing interests
The authors declare no conflict of interest.
Ethics approval and consent to participate
Not applicable.
References
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; et al. Down syndrome. Nature Reviews Disease Primers. 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Jardine, L.; Webb, S.; Goh, I.; et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature. 2021, 598, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Potter, N.; Jones, L.; Blair, H.; et al. Single-cell analysis identifies CRLF2 rearrangements as both early and late events in Down syndrome and non-Down syndrome acute lymphoblastic leukaemia. Leukemia. 2019, 33, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, J.; Greene, M.; McDevitt, M.A.; et al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nature genetics. 2002, 32, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Labuhn, M.; Perkins, K.; Matzk, S.; et al. Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with Down syndrome. Cancer cell. 2019, 36, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Wagenblast, E.; Araújo, J.; Gan, O.I.; et al. Mapping the cellular origin and early evolution of leukemia in Down syndrome. Science. 2021, 373, eabf6202. [Google Scholar] [CrossRef] [PubMed]
- Arkoun, B.; Robert, E.; Boudia, F.; et al. Stepwise GATA1 and SMC3 mutations alter megakaryocyte differentiation in a Down syndrome leukemia model. The Journal of Clinical Investigation 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Bhansali, R.; Izraeli, S.; Hijiya, N.; Crispino, J.D. The biology, pathogenesis and clinical aspects of acute lymphoblastic leukemia in children with Down syndrome. Leukemia. 2016, 30, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Turati, V.A.; Guerra-Assunção, J.A.; Potter, N.E.; et al. Chemotherapy induces canalization of cell state in childhood B-cell precursor acute lymphoblastic leukemia. Nature cancer. 2021, 2, 835–852. [Google Scholar] [CrossRef] [PubMed]
- Lutz, C.; Turati, V.A.; Clifford, R.; et al. Complex genotype-phenotype relationships shape the response to treatment of Down Syndrome Childhood Acute Lymphoblastic Leukaemia. bioRxiv 2022:2022.2002. 2006.479302. 2022. [Google Scholar]
Figure 1.
Several multiomics approaches combining genomic, transcrisptomic, proteomic and spatial imaging data from bone marrow biopsy and/or peripheral blood sample.
Figure 1.
Several multiomics approaches combining genomic, transcrisptomic, proteomic and spatial imaging data from bone marrow biopsy and/or peripheral blood sample.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



