
Submitted:
21 September 2023
Posted:
25 September 2023
You are already at the latest version
Abstract
Rationale: Chronic Granulomatous Disease (CGD) is a rare primary immunodeficiency. Although most mutations are linked to the X chromosome, therefore affecting males, there are autosomal recessive forms that therefore affect both sexes. Clinically it is characterized by recurrent and severe bacterial and fungal infections, with formation of granulomas, due to the inability of phagocytes to generate reactive oxygen compounds, necessary for the intracellular death of phagocytosed microorganisms. Patient concerns: We reported a 12-year-old boy with pneumonia with pneumatocele of possible staphylococcal etiology at one year of age, bilateral orchitis secondary to Salmonella with hematogenous spread, osteomyelitis due to Salmonella, gastroenteritis due to Salmonella enteritidis, liver abscesses due to Staphycococcus aureus and lymphomatoid papulosis and pyodermititis. Diagnoses: In the biochemical diagnosis, the presence of yellow dye (NBT) and absence of dark bluish black dye (formazan) was evidenced. Regarding the genetic diagnosis, the presence of the mutation in exon 10 of CYBB (OMIM 306400) (Xp21.1) that encodes the gp91phox protein was demonstrated. Interventions and outcomes: Treatment was started with trimethoprim/sulfamethoxazole 400/80 mg every 24 hours via oral, and antifungal prophylaxis with itraconazole 200 mg every 24 hours orally is added, after which he does not present serious bacterial or fungal infections, not being able to administer immunomodulatory treatment with recombinant human Interferon gamma-1b. Lessons: The clinical finding of frequent severe infections associated with granulomas, CGD should be suspected. Treatment of this primary immunodeficiency is trimethoprim/sulfamethoxazole and itraconazole. Importantly, it indicated that the mutation in exon 10 of CYBB (Xp21.1) that encodes the gp91phox might influence the pathogenesis of CGD.
Keywords:
Chronic-granulomatous disease
; NADPH oxidase
; human neutrophils
; superoxide
; immune deficiciency
; recurrent infections
1. Introduction
The multi-subunit phagocyte NADPH oxidase generates reactive oxygen species and is crucial for host defense [1]. Deficiencies in individual subunits (gp91phox, p22phox, p47phox, p67phox, and p40phox) of phagocytic NADPH oxidase cause chronic granulomatous disease (CGD), a serious inborn error of immunity characterized by severe infections with catalase-positive organisms. Life-threatening infections with Staphylococcus, Salmonella, Burkholdheria, and nocardia are well documented. Fungal infections due to species such as Aspergillus are also common and are associated with severe morbidity. Although improved prophylaxis and antimicrobial treatment, accompanied by the advent of bone marrow transplantation for CGD, have improved outcomes, it remains a serious disease that requires rapid identification and treatment. We previously demonstrated that EROS/CYBC1 is essential for the generation of reactive oxygen species because it chaperones the membrane-bound gp91phox-p22phox heterodimer in both mice and humans [2,3,4]. Some authors have shown that mutations in EROS/CYBC1 can cause CGD5 [3,5]. The small number of cases described so far appear to have some typical features of CGD, such as opportunistic infections and inflammatory bowel disease, but also some features not normally associated with CGD, including autoimmune hemolytic anemia, chronic glomerulonephritis and even some viral infections. So far, only three pathogenic mutations in EROS/CYBC1 have been identified. The case we described in 2018 was a private mutation restricted to one family [3] (as a result of a consanguineous marriage) and Arnadottir et al. described eight patients with a mutation that is probably limited to the Icelandic population [5]. Very recently, a group from Rome described another isolated case in which a homozygous mutation caused complete EROS deficiency. This was mainly characterized by a sarcoid-like syndrome and also by probable infectious pneumonia. Interestingly, the authors showed that the loss of gp91phox was not complete in all cell types [6]. This is consistent with our previous studies in mice where there was minor preservation of the respiratory burst in response to some stimuli in neutrophils, not macrophages [2]. Similarly, Arnadottir et al. showed some residual gp91phox in neutrophils from a patient with EROS deficiency but absence of the protein in monocytes [5].
CGD is a rare primary immunodeficiency, with an incidence of 1 in 200,000 to 250,000 live births [7]. Although most mutations are linked to the X chromosome, therefore affecting males, there are autosomal recessive forms that therefore affect both sexes, which occur more frequently in communities with a greater number of consanguineous marriages [8]. Clinically it is characterized by recurrent and severe bacterial and fungal infections, with formation of granulomas, due to the inability of phagocytes to generate reactive oxygen compounds, necessary for the intracellular death of phagocytosed microorganisms. Biochemically, it is characterized by mutations that generate loss or functional inactivation of one of the subunits of the NADPH oxidase complex.
Belzer et al. [9] review psychosocial considerations in children with rare diseases. Rare diseases affect children and their families infrequently, but with a significant high impact. The diagnostic odyssey that is undertaken as part of having a child with a rare disease is immense and involves practical, emotional, relational, and contextual issues that are not well understood. Children with rare diseases have chronic and complex medical conditions that require a complicated environment of care from numerous clinical caregivers. Both children and their families may feel isolated and stigmatized in education, employment and the workplace, or lack social support or understanding [9,10]. Different studies have analyzed the quality of life in primary immunodeficiencies such as severe combined immunodeficiency due to RAG1 and RAG2 deficiency [10], common variable immunodeficiency [11] or X-linked immunodeficiency (Bruton’s agammaglobulinemia) [12]. Among the immunodeficiencies by default in phagocytosis is chronic granulomatous disease (CGD). Pulvirenti et al. [13], Varni et al. [16] and Cole et al. [15] analyze the quality of life and emotional health of children with CGD.
According to the classification of primary immunodeficiencies compiled by the Primary Immunodeficiency Expert Committee (PID EC) of the International Union of Immunological Societies (IUIS), the CGD is congenital defect of macrophage function [16]. More recently, X-linked CGD, gp91phox, by falling superoxide production, is classified into defects of respiratory burst [17].
Interesting findings have been found in these authors that show that both the health-related quality of life (HRQoL), as well as the emotional, social and cognitive aspects are affected in children suffering from this pathology. Thus, Pulvirenti et al. [13] revealed that children with CGD showed greater difficulties in the social and school area, having more difficulty relating to peers and behavioral and emotional problems. Findings that are in line with the study by Varni et al. [14,18], which reveals that in general psychosocial health and emotional functioning in children with chronic pathologies, as well as in their parents, is lower compared to healthy children, affecting the HRQoL of these children, patients and their parents. Cole et al. [15,18] evaluated the quality of life of children with CGD who received a hematopoietic stem cell transplant (HSCT) and revealed that their HRQoL is good after HSCT, resulting in better emotional, social and school functioning.
2. Clinical Case
Male patient first seen at age 12 years with pneumonia with pneumatocele of possible staphylococcal etiology at one year of age, bilateral orchitis secondary to Salmonella with hematogenous spread, osteomyelitis due to Salmonella, gastroenteritis due to Salmonella enteritidis, liver abscesses due to Staphycococcus aureus and lymphomatoid papulosis and pyodermititis.
Previously, between 3 and 10 years of life he developed multiple recurrent bacterial infections: pneumonia, acute otitis media, sinusitis, and peritonsillar abscesses. At 5, 7, and 8 years of age, he presented acute community pneumonia that required intensive therapy in the intensive care unit. In the basic immunological study, hypergammaglobulinemia and normal lymphocyte populations were observed. The allergen-specific serum IgG values against fungi are: Alternaria alternata 27.10 mg/L (normal range: 0-9.33 mg/L); Aspergillus fumigatus 64 mg/L (normal range: 0-37.86 mg/L); Aspergillus flavus 49.50 mg/L (normal range: 2.02-20.78 mg/L); Cladosporium herbarum 43.20 mg/L (normal range: 2.02-20.78 mg/L) and Candida albicans 106 mg/L (normal range: 1-29.14 mg/L).
In the biochemical diagnosis, the presence of yellow dye (NBT) and absence of dark bluish black dye (formazan) was evidenced. Regarding the genetic diagnosis, the presence of the mutation in exon 10 of CYBB (OMIM 306400) (Xp21.1) that encodes the gp91phox protein was demonstrated. The absence of male brothers together with the presence of a sister with no higher infections than those of the general population, make us think of a type of inheritance linked to the X chromosome.
3. Discussion
3.1. Pathophysiology
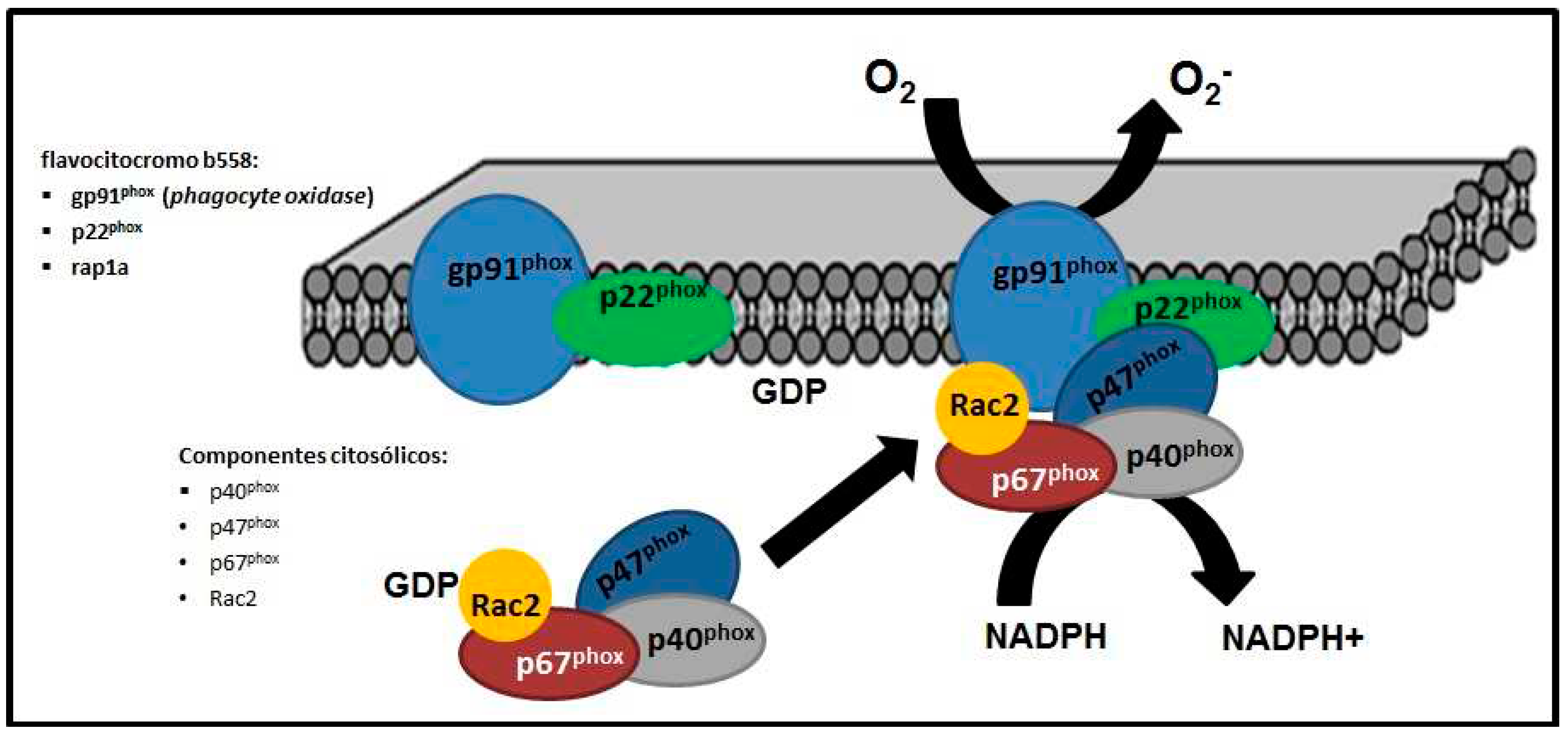
The assembled NADPH (nicotinamide adenine dinucleotide phosphate) oxidase complex is located in the cytoplasmic membrane of phagocytes, made up of membrane elements such as flavocytochrome b558 [formed in turn by the proteins gp91phox (phagocyte oxidase) and p22phox and rap1a] and four cytosolic components, the proteins p40phox, p47phox, p67phox and Rac2 [16,17] (Figure 1).
3.2. Diagnostic
The presence of bacterial and fungal infections and granulomas are mainly characteristic of EGC [18], with the response to viral infections being normal. Bacterial infections are usually symptomatic, accompanied by fever and leukocytosis; while fungal infections present with less fever and less leukocytosis. The microorganisms responsible for most infections are Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Nocardia, and Aspergillus [19,20].
Given the clinical finding of frequent severe infections associated with granulomas, CGD should be suspected. There are two tests available that estimate the formation of reactive oxygen species (respiratory burst) that assess the function of neutrophils [21,22]:
• Nitrobluetetrazolium (NBT) test, which allows colorimetric evaluation of the ability of leukocytes to reduce a yellow dye (NBT), where normal phagocytes transform it into formazan (bluish-black pigment). EGC-carrying mothers would have a mixed population of NBT positive and negative (+/-) cells.
• Test for conversion of dihydrorhodamine-123 (DTH) to rhodamine-123 measured by flow cytometry, which uses the same principle as the NBT test, but with a different stain, which can also detect X-carrier status.
Depending on the chromosomal abnormality, CGD can be classified as X-linked or autosomal recessive. Of the five proteins that make up the enzyme complex, gp91phox is encoded by the CYBB gene of the X chromosome (OMIM 306400), its mutation is responsible for 65% of CGDs, and originates the X-linked form. More have been described of 300 mutations described in the CYBB gene being registered in the XCGDbase (http://bioinf.uta.fi/CYBBbase), of which almost 200 are unique. Most are distributed in the 13 exons and intron-exon junctions. RNA processing site mutations represent about 10% of them [19,20].
The genes of the other components of the NADPH oxidase complex, p47, p40 and p67, are encoded respectively by the genes NCF1 (7q11.23), NCF4 (22q13.1) and NCF2 (1q25), corresponding to autosomal recessive forms, affecting both sexes [16,21]. The NCF1 gene mutation is responsible for 25% of all CGD, while the NCF4 and NCF2 mutations are responsible for the remaining 10% (5% each) of all CGD.
3.3. Treatment
Treatment was started with trimethoprim/sulfamethoxazole 400/80 mg every 24 hours via oral, and antifungal prophylaxis with itraconazole 200 mg every 24 hours orally is added, after which he does not present serious bacterial or fungal infections, not being able to administer immunomodulatory treatment with recombinant human Interferon gamma-1b.
Although immunity against viral infections is not affected, influenza vaccination is recommended due to possible bacterial complications that may arise. Magorlis et al. [24] demonstrated the effectiveness of itraconazole as antifungal prophylaxis. In the randomized study, 39 patients were assigned to receive placebo or itraconazole for one year. All patients received antibacterial prophylaxis and most received interferon gamma (IFN-γ) prophylaxis. In the placebo group, 7 invasive fungal infections were reported compared to one case in the group that received itraconazole 5 mg/kg orally once a day [25,26].
Recombinant human interferon gamma-1b (IFN-γ) is produced in an E. coli expression system. Gatlin et al. [28] conducted a randomized multicenter study with IFN-γ including 128 patients with CGD, who received IFN-γ 50 μg/m2 or placebo subcutaneously three times a week for an average of 8.9 months. 46% of patients in the placebo group developed at least one serious infection, while only 30% of patients in the placebo group were free of serious infections. IFN-γ was shown to reduce the frequency of serious infections over the 12-month trial period by 77% in IFN-γ-treated patients, compared to 30% in the placebo-treated group (p=0.0006) twenty-one.
It has been pointed out that HSCT is the definitive cure for CGD, obtaining better results in young patients and with fewer sequelae, although it is also effective in patients with recurrent serious infections despite prophylaxis, difficult-to-treat infections, or inflammatory diseases [29,30,31]. Gene therapy with retroviral and lentiviral vectors can reconstitute NADPH oxidase activity in deficient cells.
Although studies on HRQoL are scarce in this field, the findings cited in this regard [13,14,15] highlight that HRQoL is a variable to be taken into account in the clinical picture of CGD, which must be evaluated and addressed in order to guide more effective treatments that can cover not only the clinical picture but also the psychosocial picture, through a multidisciplinary approach, which allows evaluating and treating these patients and their families, in an integral way, responding to the needs that they present in all their areas and dimensions.
4. Conclusion
In conclusion, the clinical finding of frequent severe infections associated with granulomas, CGD should be suspected. Treatment of this primary immunodeficiency is trimethoprim/sulfamethoxazole and itraconazole. Importantly, it indicated that the mutation in exon 10 of CYBB (Xp21.1) that encodes the gp91phox might influence the pathogenesis of CGD. In the resolution of this clinical case, the importance of the multidisciplinary approach to immunodeficiencies was discussed.
Abbreviations
HRQoL health-related quality of life
DTH dihydrorhodamine-123 (DTH)
CGD chronic granulomatous disease
IFN-γ interferon gamma-1b
NADPH nicotinamide adenine dinucleotide phosphate
NBT nitrobluetetrazolium test
HSCT hematopoietic stem cell transplantation
References
- P.M. Mortimer, S.A. Mc Intyre, D.C. Thomas. Beyond the extra respiration of phagocytosis: NADPH oxidase 2 in adaptive immunity and inflammation. Front. Immunol., 12 (2021), . [CrossRef]
- D.C. Thomas, S. Clare, J.M. Sowerby, M. Pardo, J.K. Juss, D.A.Goulding, et al. Eros is a novel transmembrane protein that controls the phagocyte respiratory burst and is essential for innate immunity. J. Exp. Med., 214 (2017), pp. 1111-1128.
- D.C. Thomas, L.-M. Charbonnier, A. Schejtman, H. Aldhekri, E.L.Coomber, E.R. Dufficy, et al. EROS/CYBC1 mutations: decreased NADPH oxidase function and chronic granulomatous disease. J. Allergy Clin. Immunol., 143 (2019), pp. 782-785. Available from http://www.jacionline.org/article/S0091674918314234/fulltext. [CrossRef]
- L.O. Randzavola, P.M. Mortimer, E. Garside, E.R. Dufficy, A.Schejtman, G. Roumelioti, et al. Title EROS-mediated control of NOX2 and P2X7 biosynthesis. (2021), 10.1101/2021.09.14.460103.
- G.A. Arnadottir, G.L. Norddahl, S. Gudmundsdottir, A.B.Agustsdottir, S. Sigurdsson, B.O. Jensson, et al. A homozygous loss-of-function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat. Commun. (2018), p. 9. [CrossRef]
- M. Chiriaco, A. De Matteis, C. Cifaldi, G. Di Matteo, B. Rivalta, C.Passarelli, et al. Characterization of AR-CGD female patient with a novel homozygous deletion in CYBC1 gene presenting with unusual clinical phenotype. Clin. Immunol., 251 (2023), Article 109316. [CrossRef]
- Heyworth P, Cross A, Curnutte J. Chronic granulomatous disease. Curr Opin Immunol 2003; 15: 578-84.
- Köker MY, Camcýoðlu Y, van Leeuwen K, Kýlýç SÞ, Barlan I, Yýlmaz M, et al. Clinical, functional, and genetic characterization of Chronic Granulomatous Disease in 89 turkish patients. J Allergy Clin Immunol 2013;132:1156-63.
- Belzer LT, Wright SM, Goodwin EJ, Singh MN, Carter BS. Psychosocial Considerations for the Child with Rare Disease: A Review with Recommendations and Calls to Action. Children (Basel). 2022 Jun 21;9(7):933. [CrossRef]
- Abd Hamid IJ, Slatter MA, McKendrick F, Pearce MS, Gennery AR. Long-Term Health Outcome and Quality of Life Post-HSCT for IL7Rα-, Artemis-, RAG1- and RAG2-Deficient Severe Combined Immunodeficiency: a Single Center Report. J Clin Immunol. 2018 Aug;38(6):727-732. [CrossRef]
- Andersen JB, Midttun K, Feragen KJB. Measuring quality of life of primary antibody deficiency patients using a disease-specific health-related quality of life questionnaire for common variable immunodeficiency (CVID_QoL). J Patient Rep Outcomes. 2019 Feb 26;3(1):15. [CrossRef]
- Gao S, Hu S, Duan H, Wang L, Kong X. Clinical characteristics and prenatal diagnosis for 22 families in Henan Province of China with X-linked agammaglobulinemia (XLA) related to Bruton’s tyrosine kinase (BTK) gene mutations. BMC Med Genet. 2020 Jun 17;21(1):131. [CrossRef]
- Pulvirenti F, Sangerardi M, Plebani A, Soresina A, Finocchi A, Pignata C et al. Health-Related Quality of Life and Emotional Difficulties in Chronic Granulomatous Disease: Data on Adult and Pediatric Patients from Italian Network for Primary Immunodeficiency (IPINet). J Clin Immunol. 2020 Feb;40(2):289-298. [CrossRef]
- Varni JW, Limbers CA, Burwinkle TM. Impaired health-related quality of life in children and adolescents with chronic conditions: a comparative analysis of 10 disease clusters and 33 disease categories/severities utilizing the PedsQL 4.0 Generic Core Scales. Health Qual Life Outcomes. 2007 Jul 16;5:43. doi:10.1186/1477-7525-5-43.
- Cole T, McKendrick F, Titman P, Cant AJ, Pearce MS, Cale CM et al. Health related quality of life and emotional health in children with chronic granulomatous disease: a comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol. 2013 Jan;33(1):8-13. [CrossRef]
- Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME et al. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015 Nov;35(8):696-726.
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022 Oct;42(7):1473-1507.
- Agudelo-Flórez P, Navarro S, Luttges P, López JA, Norambuena X, Navarrete S CL, et al. Identificación de mutaciones en el gen CYBB que llevan al fenotipo de la enfermedad granulomatosa crónica ligada al cromosoma X: reporte de una nueva mutación. Rev Med Chile 2006;134:965-72.
- Leiding JW, Holland SM. Chronic Granulomatous Disease. In: Pagon RA, Adam MP, Ardinger HH et al., editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014.
- Holland SM. Chronic Granulomatous Disease. Clin Rev Allergy Immunol 2010; 38: 3-10.
- Van den Berg J, Van Koppen E, Ahlin A, Belohradsky B, Bernatowska E, Corbeel L. Chronic Granulomatous Disease: The European Experience. PLoS ONE. 2009;4:e5234. [CrossRef]
- Cale CM, Morton L, Goldblatt D. Cutaneous and other lupus-like symptoms in carriers of X-linked chronic granulomatous disease: incidence and autoimmune serology. Clin Exp Immunol. 2007;148:79-84. [CrossRef]
- Roos D, de Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol 2013; 175: 139-49. [CrossRef]
- Margolis DM, Melnick DA, Alling DW, Gallin JI: Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis 1990;162:723-6.
- Gallin JI, Alling DW, Malech HL, Wesley R, Koziol D, Marciano B, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med 2003;348:2416-22.
- Mouy R, Veber F, Blanche S, Donadieu J, Brauner R, Levron JC, et al. Long-term itraconazole prophylaxis against Aspergillus infections in thirty-two patients with chronic granulomatous disease. J Pediatr 1994;125:998-1003. [CrossRef]
- Petropoulou T, Liese J, Tintelnot K, Gahr M, Belohradsky BH. Long-term treatment of patients with itraconazole for the prevention of Aspergillus infections in patients with chronic granulomatous disease (CGD). Mycoses 1994;37:64-9.
- Gatlin JI, Malech HL, Weening RS, Curnutte JT, Quie PG, Jaffe HS, Ezekowitz RAB: A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group. N Engl J Med 1991;324:509-16.
- Seger RA. Hematopoietic stemcell transplantation for chronic granulomatous disease. Immunol Allergy Clin North Am. 2010; 30: 195-208.
- Kang EM, Marciano BE, DeRavin S, Zarember KA, Holland SM, Malech HL. Chronic granulomatous disease: overview and hematopoietic stemcell transplantation versus conventional treatment. Acta Paediatrica 2013;102:1087-94.
- Åhlin A, Fugeläng J, de Boer M, Ringden O, Fasth A, Winiarski J. Chronic granulomatous disease – haematopoietic stemcell transplantation versus conventional treatment. Acta Pædiatrica 2013; 102: 1087-94.
Figure 1.
Schematic representation of the assembly of the NADPH (nicotinamide adenine dinucleotide phosphate) oxidase enzyme complex, which generates superoxide anion (O2-), hydrogen peroxide (H2O2), hypochlorous acid (HOCl).
Figure 1.
Schematic representation of the assembly of the NADPH (nicotinamide adenine dinucleotide phosphate) oxidase enzyme complex, which generates superoxide anion (O2-), hydrogen peroxide (H2O2), hypochlorous acid (HOCl).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



