
Submitted:
25 September 2023
Posted:
25 September 2023
You are already at the latest version
Abstract
Marek’s disease (MD) is an important neoplastic disease caused by serotype 1 Marek's disease virus (MDV-1), which results in severe economic losses worldwide. Despite vaccination practices that have controlled the MD epidemic, current increasing MD-suspected cases indicate the persistent viral infections circulating among vaccinated chicken farms in many countries. However, the lack of available information about phylogeny and molecular characterization of circulating MDV-1 field strains in Taiwan reveals a potential risk in MD outbreaks. This study investigated the genetic characteristics of 18 MDV-1 isolates obtained from 17 vaccinated chicken flocks in Taiwan between 2018 and 2020. Based on the sequences of the meq oncogene, phylogenetic analysis demonstrated that the circulating Taiwanese MDV-1 field strains were predominantly in a single cluster that showed high similarity with strains/isolates from countries of East Asian region. Because the isolated strains were obtained from CVI988/Rispens vaccinated chicken flocks and the molecular characteristics of the Meq oncoprotein showed the features like vvMDV and vv+MDV strains, the circulating Taiwanese MDV-1 field strains may have higher virulence compared with vvMDV pathotype. In conclusion, the presented data demonstrate the circulation of hypervirulent MDV-1 strains in Taiwan and highlight the importance of routine surveillance and precaution strategies in response to the emergence of enhanced virulent MDV-1.
Keywords:
Marek’s disease virus
; vaccinated chicken flocks
; meq oncogene
; genetic characteristics
; virulence
1. Introduction
Marek’s disease (MD), caused by Gallid alphaherpesvirus 2 (GaHV-2), is a critical, highly contagious avian viral disease that induces serial clinical manifestations including systemic visceral lymphoma, neurological disorders, paralysis, and immunosuppression in infected chickens, resulting in considerable economic losses in poultry industry [1,2]. The etiological agent GaHV-2, also commonly known as serotype 1 of Marek’s disease virus (MDV-1), belongs to a member of the genus Mardivirus in the subfamily Alphaherpesvirinae of the family Herpesviridae which also consists of other nononcogenic MDV species: Gallid alphaherpesvirus 3 (GaHV-3) is serotype 2 of MDV (MDV-2), and Meleagrid alphaherpesvirus 1, also known as turkey herpesvirus (HVT), is serotype 3 [3]. Nononcogenic MDVs were developed as first-generation vaccines and were soon after introduced to many countries for MD prevention [4]. Based on the pathotyping protocol referring to the virulent properties in surmounting specific vaccinal protection, the population of MDV-1 can be classified into various pathotypes including mild (m), virulent (v), very virulent (vv), and very virulent plus (vv+) [5,6]. The increasing emergence of MD cases has been revealed in current reports among vaccinated chicken flocks in many countries, which suggests a probable rise in evolved MDV-1 field strains associated with enhanced virulence [7,8].
The MDV-1 genome encodes more than 200 genes, some of which are unique oncogenes that are mostly involved in viral pathogenesis [9]. The Meq oncoprotein encoded by the meq oncogene was the first discovered oncoprotein whose N-terminal basic- leucine- zipper (bZIP) domain and C-terminal proline-rich transactivation domain were identified as major functional factors associated with MDV-1 virulence and oncogenicity [10,11]. Recent studies have reported that specific amino acid mutations, along with proline contents and the number of 4-proline-repeat stretches (PPPPs) within Meq oncoprotein, are correlated with MDV-1 virulence [12,13]. Therefore, in addition to the laborious in vivo pathotyping assay, alternative methods based on the molecular characteristics of meq oncogene sequences and the corresponding encoded Meq oncoprotein have been commonly used for phylogenetic analysis and virulence prediction of novel MDV-1 isolates and have been published in numerous studies from various countries [14,15,16,17,18].
Despite the widely and routine application of vaccination, outbreaks of MD still occasional occurred in vaccinated chicken farms in numerous Asian countries, including China [19], India [20], Iran [21], Japan [22] and Thailand [23]. During the past 20 years, MD-related cases have frequently been found in chicken populations in Taiwan; however, the nearest published report of very virulence MDV-1 appearing and circulating among local chickens or layers in poultry flocks in Taiwan was before the 21st century [24]. The constant lack of continuous monitoring of the genotypes and virulence of the circulating MDV-1 strains in Taiwan has lead MD prevention become a thorny issue, which may result in inadequate responses to the sudden MD epidemic. In this study, we present the phylogenetic and virulence characteristics of current circulating MDV-1 isolates in Taiwan through sequence analysis of the meq oncogene obtained from vaccinated chicken flocks from 2018 to 2020.
2. Materials and Methods
2.1. Samples
During January 2018 to December 2020, the chicken cases which pathologically diagnosed with MD suspect from Animal Disease Diagnostic Center of National Pingtung University of Science and Technology (NPUST) were included in this study. The submitted chickens, including layers and native chickens, had been vaccinated with commercial univalent or bivalent MDV vaccines. The gross lesion tissue from these MD suspect chickens were examined by PCR assay for MDV-1 detection [25,26] and then stored at -80 ℃ for further gene analysis.
2.2. Nucleic Acid Extraction and Field Virus Detection
A total of 17 cases were randomly selected among the MDV detected cases (Table 1). The nucleic acid was extracted from collected tissue samples by using TANBead® Nucleic Acid Extraction Kit (TANBEAD, Taiwan) following the manufacturer’s instructions and stored at -20℃. All extracted nucleic acid samples were further examined for avian leukosis virus (ALV) infection [27]. In addition, each case was also detected for the positivity of suspected common avian pathogens, such as Newcastle disease virus (NDV) [28], infectious bursa disease virus (IBDV) [29], infectious bronchitis virus (IBV) [30], chicken anemia virus (CAV) [29], Fowl adenovirus (FAV) [31], Mycoplasma synoviae (MS) [32], Fowl poxvirus (FPV) [33], etc.
2.3. PCR for Meq Oncogene
The meq oncogene was amplified with primers EcoR-Q-for: GGTGATATAAAGACGATAGTCATG and EcoR-Q-rev: CTCATACTTCGGAACTCCTGGAG by conventional PCR to produce 1,625-bp DNA fragment as described previously [14].
2.4. Cloning and Sequencing
The amplified meq oncogene products were purified by the FavorPrepTM Gel purification Mini Kit (FAVORGEN® BIOTECH CORP., Taiwan) according to the manufacturer’s instructions and were cloned into T-vector using the T&A Cloning Vector Kit (Yeastern Biotech Co., Ltd., Taiwan). After blue- white screening, the plasmid-transformed colony was picked and cultured to acquire meq gene-carried plasmids for sequencing. Consensus sequences of the meq oncogene, which were confirmed by Sanger sequencing, were further verified and assembled using BLAST alignment analysis. The obtained nucleotide sequences of meq oncogenes of Taiwanese MDV-1 isolates were submitted to the GenBank database with the accession numbers: OQ576796-OQ576813.
2.5. Genetic Analysis
A total of 37 selected meq oncogene sequences were retrieved from the GenBank database as references (Table S1) for comparison with obtained sequences of Taiwanese isolates in this study. Nucleotide and amino acid identifications were conducted by alignment of Taiwanese isolates and references using Clustal W software [34]. The phylogenetic tree was constructed by MEGA version X [35] software using neighbor-joining (NJ) algorithms under the Tamura-Nei model with 1,000 bootstrap replicates. To characterize the specific substitution of deduced amino acids, the sequences of Meq oncoprotein of Taiwanese isolates were compared with selected references. Additionally, the proline content and the number of PPPP motifs within the Meq oncoprotein of Taiwanese isolates were evaluated.
3. Results
3.1. Profiles of Collected Samples
During 2018 to 2022, MDV-1 detection rates of the submitted chicken cases were 7.6%, 4.6%, 3.24%, 2.2%, and 2.2%, respectively. The information and the status of coinfection with other avian diseases of the randomly selected 17 cases were shown in Table 1. The presence of other poultry pathogens in the examined samples along with MDV indicates that pathogen coinfections in chicken flocks occur frequently nowadays in Taiwan. The chicken cases in this study mainly showed lymphoma lesions in a variety of organs and tissues, such as the ovary, lung, heart, mesentery, kidney, liver, spleen, thymus, pancreas, proventriculus, intestine, and skeletal muscle, and few of them had neuronal lesions, indicating that the visceral lymphoma of MD was of a major epidemic form (Figure 1).
3.2. Phylogenetic Analysis of Meq Oncogenes of Taiwanese MDV-1 Isolates
A total of 18 MDV-1 isolates were obtained from 17 vaccinated chicken flocks in Taiwan between 2018 and 2020. The genetic features of these 18 obtained meq oncogenes were characterized through phylogenetic analysis with 37 selected reference genes of identified isolates or strains, which were collected from various locations and available in the GenBank database. Among the selected reference strains, 25 of the strains were pathotyped [5,6]. The phylogenetic tree demonstrated that the analyzed meq oncogenes in this study could be separated into 4 clusters (Figure 2). Cluster 1 involved all of the Chinese strains, Thai isolates, Taiwanese isolates, and some of the Japanese isolates. The vaccine strains, mild virulent strain, and Australian strains were all included in Cluster 2. Most classic USA strains representing pathotypes of very virulent and very virulent plus were grouped into Cluster 3, whereas part of USA strains were divided into Cluster 4 with isolates from India and Japan. The homology range among the members of Cluster 1 was 99.3-100% nucleotide identity and 98.5-100% amino acid identity, respectively. Notably, five of 18 Taiwanese isolates showed the closest relationship with the very virulent strain LS of China and 4 recently identified isolates of Thailand (100% nucleotide identity and 100% amino acid identity, respectively). In addition, the frequent appearance of branches from 13 Taiwanese isolates in Cluster 1 indicated a high probability of individual evolution of MDV-1. These results suggested the circulation of MDV-1 for a certain duration among chicken flocks in Taiwan, which brought about geographical genetic polymorphism.
3.3. Molecular Characterization of Meq Oncoproteins of Taiwanese MDV-1 Isolates
The deduced amino acid sequences were compared with known pathotyping strains from different countries to investigate the characterization of Meq oncoproteins of Taiwanese isolations (Table 2). Specific amino acid substitutions, including positions 71, 77, 80, 88, 93, 110, 115, and 119 in the basic leucine zipper domain and 139, 153, 176, 180, 217, 218, 277, 283, and 326 in the transactivation domain of the Meq oncoprotein, were verified previously to be correlated with MDV-1 virulence [13]. All of the Meq oncoproteins of Taiwanese isolates shared identical substitutions at positions 71(A), 77(E), 80(Y), 115(A), and 176(R) with the vvMDV strains LS and GX0101 [36,37], and the vv+MDV strains LTS and SD2012-1 [38,39] of China. Although genetic analysis demonstrated the existence of high virulence MDV-1 isolates in Taiwan, four unique substitution positions 119(R), 153(Q), 176(A) and 277(P) were not present in Taiwanese isolates while comparing with the classic vv+MDV strains N and 648A of USA. Moreover, the proline content and the number of PPPP repeats within the Meq oncoprotein were also used as virulence predictors for Taiwanese isolates, as in previous reports [13,40]. Compared with vaccine and mild MDV-1 strains (Table 3), the Taiwanese isolates were lack of insertions and showed related lower proline contents as well as PPPP motif numbers, which supported the high virulence prediction.
4. Discussion
This is the first report of MDV-1 virulence by molecular analyses during the nearest 20 years after the study on the polymorphism of MDV-1 isolates and the presence of vvMDV in Taiwan [24]. The present study revealed the occurrence and genetic properties of the MDV-1 field strains circulating in Taiwan based on the sequence analysis of 18 virulence-associated meq oncogenes obtained from 17 vaccinated chicken flocks collected during 2018-2020. Therefore, understanding the genetic characterization of Taiwan MDV-1 has become a primary concern for disease prevention and control.
Phylogenetic analysis demonstrated all Taiwanese isolates were grouped into the same cluster involving predominantly highly virulent MDV-1 strains from China and field isolates from Thailand and Japan. Some Taiwanese isolates showed complete genetic identity to the LS strain, which was isolated from the Sichuan province of China and classified as the vvMDV pathotype [36]. High similarity features are also represented in Thai field isolates, which were recently published as being in close phylogenetic relationship with MDV-1 strains from China [23], indicating that these field MDV-1 strains may share a common ancestor and evolutionary direction. Interestingly, Guangxi Province is geographically closer to Taiwan and Thailand than to Sichuan Province; however, based on phylogenetic analysis, the strains from Guangxi, GX070060 and GX070079, showed less phylogenetic relationships with Taiwanese and Thai isolates. The reasons of these findings are still unknown, but the possibility of pathogen transmission by wild birds could be taken into consideration [41]. In the present study, the “LS-like” MDV-1 field isolates, including TW/011/18, TW/123/19, TW/141B/19, TW/029/20 and TW/116/20, were obtained from different flocks in various collecting years, indicating that these isolates were dominant stains persistently circulating in chicken farms in Taiwan. The persistent detection of such strains from vaccinated flocks might due to the genetic adaptation in the chicken flocks and farms and the immune escape from the vaccine protection [23,42].
Taiwanese MDV-1 isolates were all clustered together in the Cluster 1 of the phylogenetic tree and spread in several different branches, which revealed not only geographically restricted evolution, but also the genetic diversity as in previous investigations [17,43]. Notably, the isolates from Southern Japan were grouped into the cluster with Taiwanese MDV-1 and Chinese strains, whereas the Northern Japanese isolates were clustered into another group with USA and Indian strains, suggesting a possible independent construction of geographical phylogeny in East Asia.
The spontaneous mutations of oncogenes, especially the meq oncogene, on the MDV-1 genome have been regarded as important roles corresponding to increasing virulence [44]. The Meq oncoprotein, which is known to play a critical role in MDV-1 pathogenicity, has shown unexpectedly higher mutation rates than general DNA viruses and even resembles RNA viruses [45]. Although the causes for such high mutation frequency of MDV-1 have not been fully clarified, most investigations have demonstrated that the improper use of vaccines can lead to the induction of positive selection from the field viruses, eventually resulting in viral diversity [46,47]. With the annually found MD clinical cases and the genetic diversity of meq oncogenes in our results (Figure 2, Cluster 1), the positive selection of the viruses in vaccinated chicken flocks of Taiwan may drive the viral evolved direction toward enhanced virulence of MDV-1.
Specific sequence characterization of the Meq oncoprotein has been reported as a predictor for MDV-1 pathotype and can be applied on the virulence prediction for novel isolated MDV strains instead of in vivo classification [20,23,40]. It has been reported previously that amino acid mutations at positions 71 (Ala), 77 (Glu), 80 (Tyr), 115 (Ala), and 176 (Arg) were the main feature of highly virulent MDV-1 of Chinese strains [18,19,22]. The results of sequence analyses in our study showed that all obtained Taiwanese MDV-1 field strains represented the molecular characteristics of the mutations as the previous report of China strains, supporting the high virulent potential of these Taiwanese MDV-1 strains. In addition, the mutations at positions 77, 80, 115, and, 176 of Meq oncoproteins seem to be common features of Chinese, Thai, Japanese and Taiwanese MDV-1 field strains, and could be considered as the accessible markers for molecular identification of East and Southeast Asian MDV-1 isolates.
Insertions appearing in meq oncogenes of mild and attenuated strains, such as CU-2 and CVI988/Rispens, cause the expression of longer Meq oncoproteins, resulting in the presence of higher proline contents and more PPPP motifs than those of virulent MDV-1 strains which were correlated with low virulence characteristics of MDV strains [13]. In contrary, no insertions in meq oncogenes of N and 648A strains of USA have lower proline contents and fewer PPPP motifs, leading to high virulence MDV strains [13,40]. Our findings in the present study showed the related lower proline contents and fewer PPPP motifs, and the values were between those of vvMDV and vv+MDV USA strains. In addition, the related lower proline contents and fewer PPPP motifs of Taiwanese strains were similar to the values of vvMDV and vv+MDV Chinese strains. These results indicated that the circulating MDV-1 field strains in Taiwan were potentially hypervirulent, but their exact pathotypes still required further classification by in vivo pathotyping experiments.
At present, the use of vaccines in flocks is still a vital and effective way to control MDV epidemics [4]. In Taiwan, vaccination programs for young chickens via bivalent vaccines of two commercial live strains CVI988/Rispens of MDV-1 and FC126 of HVT have been common practiced across the whole poultry industry. To the best of our knowledge, bivalent vaccination is available for producing a protective immune response against most virulent MDVs, including vvMDV and vv+MDV pathotypes, but the occurrence of clinical MD cases due to immune failure in chicken flocks around the world, including in Taiwan, which raising high attention to the problems regarding the vaccine application. Current commercial MD vaccines are all cell-associated types, which come with more difficulties in transportation, storage, and administration than other live vaccines. Vaccination efficiency can be affected by the reconstituted conditions, performance, dose uniformity of vaccines, etc. [48,49]. In Taiwan, we have examined the immune status by detecting MDV from feather tips at 14-21 post-vaccination day after the pullets were applied the CVI988 and/or HVT-FC126 on 1 day of age. Only 48% (16/33) of chicken flocks were vaccinated successfully (achieving 70% immunization coverage) [50]. After monitoring the 7 flocks from a layer breeding farm, in which the pullets were vaccinated by applying the same patch of CVI988 vaccine, and the same injection machine and procedure were used, various detection rates of the vaccinated virus in the 7 flocks were found (30-90%) [50]. Ununiformed vaccine doses received by pullets were considered be the possible reason for the uneven vaccination efficacy, and applying the well-mixed vaccines was important to prevent immune failure.
Coinfection of avian viruses, such as MDV, IBDV, NDV, CAV, reovirus, and reticuloendotheliosis virus, can induce immunosuppression in infected hosts, resulting in reduced vaccination efficiency [51,52]. The coexistence of poultry immunosuppressive disease virus together with MDV has been detected in the cases of the present study, suggesting that the chicken flocks in Taiwan may also suffer under the immune suppression and cannot have proper protection after vaccination.
In conclusion, the phylogenetic findings on geographical diversity of meq oncogenes suggested an ongoing evolution in circulating Taiwanese MDV-1 strains, which already adapted to the chicken farms in Taiwan. The circulation of field MDV-1 strains in Taiwan were dominated in a single cluster with potentially hypervirulent characterization. Routine surveillance of field MDV-1 strains and monitoring of immune status on poultry farms will be needed for development of effective vaccines and control strategies in response to the emergence of enhanced virulent Taiwanese MDV-1 strains.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Profile of MDV-1 isolates/strains used in this study.” can be found online.
Author Contributions
Conceptualization, Y-Y Lien and M-C Cheng; methodology, G-H Lai and M-C Cheng; formal analysis, Y-L Tsai, G-H Lai and M-C Cheng; resources, M-C Cheng and Y-Y Lien; writing—original draft preparation, G-H Lai and M-C Cheng; writing—review and editing, G-H Lai, Y-Y Lien, Y-L Tsai and M-C Cheng; funding acquisition, M-C Cheng. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Ethics Statement
Not applicable.
Data Availability Statement
The novel meq oncogene sequences generated in this study were submitted to the GenBank database.
Acknowledgments
We appreciate the support from the Animal Disease Diagnostic Center of National Pingtung University of Science and Technology for providing the clinical samples and the co-operation of pathogenic diagnosis and virus detection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Calnek, B.W. Pathogenesis of Marek’s disease virus infection. Curr. Top. Microbiol. Immunol. 2001, 255, 25-55;. [CrossRef]
- Kennedy, D. A.; Cairns, C.; Jones, M. J.; Bell, A.; Salathé, R. M.; Baigent, S. J.; Nair, V. K.; Dunn, P. A.; Read, A. F. Industry-wide surveillance of Marek’s disease virus on commercial poultry farms. Avian Dis. 2017, 61, 153-164;. [CrossRef]
- Bulow, V. V.; Biggs, P. M. Differentiation between strains of Marek’s disease virus and turkey herpesvirus by immunofluorescence assay. Avian Pathol. 1975, 4, 133-146;. [CrossRef]
- Jarosinski, K. W.; Tischer, B. K.; Trapp, S.; Osterrieder, N. Marek’s disease virus: lytic replication, oncogenesis and control. Expert Rev. Vaccines. 2006, 5, 761-772;. [CrossRef]
- Witter, R. L. Increased virulence of Marek’s disease virus field isolates. Avian Dis. 1997, 41, 149-163.
- Witter, R. L.; Calnek, B. W.; Buscaglia, C.; Gimeno, I. M.; Schat, K. A. Classification of Marek’s disease viruses according to pathotype: philosophy and methodology. Avian Pathol. 2005, 34: 75-90;. [CrossRef]
- Bertzbach, L. D.; Conradie, A. M.; You, Y.; Kaufer, B. B. Latest insights into Marek’s disease virus pathogenesis and tumorigenesis. Cancers (Basel). 2020, 12, 647;. [CrossRef]
- Yehia, N.; El-Sayed, H. S.; Omar, S. E.; Erfan, A.; Amer, F. Genetic evolution of Marek’s disease virus in vaccinated poultry farms. Vet. World. 2021, 14, 1342-1353;. [CrossRef]
- Ongor, H.; Timurkaan, N.; Abayli, H.; Karabulut, B.; Kalender, H.; Tonbak, S.; Eroksuz, H.; Cetinkaya, B. First report of serotype-1 Marek’s disease virus (MDV-1) with oncogenic form in backyard turkeys in Turkey: a molecular analysis study. BMC Vet Res. 2022, 18: 30;. [CrossRef]
- Jones, D.; Lee, L.; Liu, J. L.; Kung, H. J.; Tillotson, J. K. Marek’s disease virus encodes a basic-leucine zipper gene resembling the fos/jun oncogenes that is highly expressed in lymphoblastoid tumors. Proc. Natl. Acad. Sci. 1992, 89, 4042-4046;. [CrossRef]
- Deng, X.; Li, X.; Shen, Y.; Qiu, Y.; Shi, Z.; Shao, D.; Jin, Y.; Chen, H.; Ding, C.; Li, L.; Chen, P.; Ma, Z. The Meq oncoproteins of Marek’s disease virus interacts with p53 and inhibits its transcriptional and apoptotic activities. Virol. J. 2010, 7, 348;. [CrossRef]
- Shamblin, C. E.; Greene, N.; Arumugaswami, V.; Dienglewicz, R. L.; Parcells, M. S. Comparative analysis of Marek’s disease virus (MDV) glycoprotein-, lytic antigen pp38- and transformation antigen Meq-encoding genes: association of meq mutations with MDVs of high virulence. Vet. Microbiol. 2004, 102, 147-167;. [CrossRef]
- Renz, K. G.; Cooke, J.; Clarke, N.; Cheetham, B. F.; Hussain, Z.; Islam, A. F. M. F.; Tannock, G. A.; Walkden-Brown, S. W. Pathotyping of Australian isolates of Marek’s disease virus and association of pathogenicity with meq gene polymorphism. Avian Pathol. 2012, 41, 161-176;. [CrossRef]
- Mescolini, G.; Lupini, C.; Felice, V.; Guerrini, A.; Silveira, F.; Cecchinato, M.; Catelli, E. Molecular characterization of the meq gene of Marek’s disease viruses detected in unvaccinated backyard chickens reveals the circulation of low- and high-virulence strains. Poult. Sci. 2019, 98, 3130-3137;. [CrossRef]
- Dunn, J. R.; Pyrkosz, A. B.; Steep, A.; Cheng, H. H. Identification of Marek’s disease virus genes associated with virulence of US strains. J. Gen. Virol. 2019, 100, 1132-1139;. [CrossRef]
- Bayoumi, M.; El-Saied, M.; Ahmed, B.; El-Mahdy, M.; Amer, H. Gallid Alphaherpesvirus 2 in the Egyptian turkeys: Molecular characterization and establishment of a universal system for phylogenetic classification. Intervirology. 2021, 64, 156-164;. [CrossRef]
- Murata, S.; Machida, Y.; Isezaki, M.; Maekawa, N.; Okagawa, T.; Konnai, S.; Ohashi, K. Genetic characterization of a Marek’s disease virus strain isolated in Japan. Virol. J. 2020, 17, 186;. [CrossRef]
- Song, B.; Zeb, J.; Hussain, S.; Aziz, M. U.; Circella, E.; Casalino, G.; Camarda, A.; Yang, G.; Buchon, N.; Sparagano, O. A review on the Marek’s disease outbreak and its virulence-related meq genovariation in Asia between 2011-2021. Animals (Basel). 2022, 12, 540;. [CrossRef]
- Yu, Z-H.; Teng, M.; Luo, J.; Wang, X-W.; Ding, K.; Yu, L-L.; Su, J-W.; Chi, J-Q.; Zhao, P.; Hu, B.; Zhang, G-P.; Liu, J-X. Molecular characteristics and evolutionary analysis of field Marek’s disease virus prevalent in vaccinated chick flocks in recent years in China. Virus Genes. 2013, 47, 282-291;. [CrossRef]
- Kannaki, T. R.; Priyanka, E.; Nishitha, Y.; Krishna, S. V.; Haunshi, S.; Subbiah, M. Molecular detection and phylogenetic analysis of Marek’s disease virus virulence-associated genes from vaccinated flocks in southern India reveals circulation of virulent MDV genotype. Transbound. Emerg. Dis. 2022, 69, 244-253;. [CrossRef]
- Ghalyanhilangeroudi, A.; Hosseini, H.; Nazarpak, H. H.; Molouki, A.; Dezfoulian, O.; Morshed, R. Molecular characterization and phylogenetic analysis of Marek’s disease virus in Iran. Avian Dis. 2022, 66, 1-5;. [CrossRef]
- Abd-Ellatieff, H. A.; Rawash, A. A. A.; Ellakany, H.; Goda, W. M.; Suzuki, T.; Yanai, T. Molecular characterization and phylogenetic analysis of a virulent Marek’s disease virus field strain in broiler chickens in Japan. Avian Pathol. 2018, 47, 47-57;. [CrossRef]
- Wannaratana, S.; Tunterak, W.; Prakairungnamthip, D.; Sasipreeyajan, J.; Thontiravong, A. Genetic characterization of Marek’s disease virus in chickens in Thailand reveals a high genetic diversity of circulating strains. Transbound. Emerg. Dis. 2022, 69, 3771-3779;. [CrossRef]
- Lin, J. A.; Chen, C. P. First isolation and characterization of very virulent Marek’s disease virus in Taiwan. J. Vet. Med. Sci. 1996, 58, 1011-1015;. [CrossRef]
- World Organization for Animal Health (OIE). Chapter 3.3.13: Marek’s disease. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals; OIE, 2018, pp. 952-963; URL: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/3.03.13_MAREK_DIS.pdf.
- Murata, S.; Chang, K-S.; Lee, S-I.; Konnai, S.; Onuma, M.; Ohashi, K. Development of a nested polymerase chain reaction method to detect oncogenic Marek’s disease virus from feather tips. J. Vet. Diagn. Invest. 2007, 19, 471-478;. [CrossRef]
- Abdel-Latif, M. M.; Khalafalla, A. I. Detection by PCR of multiple subgroups of Avian leukosis virus (ALV) in broilers in the Sudan. J. Anim. Vet. Adv. 2005, 4, 407-413; URL: http://medwelljournals.com/abstract/?doi=javaa.2005.407.413.
- Lin, M. Y.; Liu, H. J.; Ke, G. M. Genetic and antigenic analysis of Newcastle disease viruses from recent outbreaks in Taiwan. Avian Pathol. 2003, 32, 345-350;. [CrossRef]
- Caterina, K. M.; Frasca Jr, S.; Girshick, T.; Khan, M. I. Development of a multiplex PCR for detection of avian adenovirus, avian reovirus, infectious bursal disease virus, and chicken anemia virus. Mol. Cell. Probes. 2004, 18, 293-298;. [CrossRef]
- Handberg, K. J.; Nielsen, O.; Pedersen, M. W.; Jørgensen, P. H. Detection and strain differentiation of infectious bronchitis virus in tracheal tissues from experimentally infected chickens by reverse transcription-polymerase chain reaction. Comparison with an immunohistochemical technique. Avian Pathol. 1999, 28, 327-335;. [CrossRef]
- Meulemans, G.; Boschmans, M.; Berg, T. P.; Decaesstecker, M. Polymerase chain reaction combined with restriction enzyme analysis for detection and differentiation of fowl adenoviruses. Avian Pathol. 2001, 30, 655-660;. [CrossRef]
- Lauerman, L. H.; Hoerr, F. J.; Sharpton, A. R.; Shah, S. M.; van Santen, V. L. Development and application of a polymerase chain reaction assay for Mycroplasma synoviae. Avian Dis. 1993, 37, 829-834.
- Lee, L. H.; Lee, K. H. Application of the polymerase chain reaction for the diagnosis of fowl poxvirus infection. J. Virol. Methods. 1997, 63, 113-119;. [CrossRef]
- Thompson, J. D.; Higgins, D. G.; Gibson, T. J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673-4680;. [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547-1549;. [CrossRef]
- Tian, M.; Zhao, Y.; Lin, Y.; Zou, N.; Liu, C.; Liu, P.; Cao, S.; Wen, X.; Huang, Y. Comparative analysis of oncogenic genes revealed unique evolutionary features of field Marek’s disease virus prevalent in recent years in China. Virol. J. 2011, 8, 121;. [CrossRef]
- Cui, Z.; Zhuang, G.; Xu, X.; Sun, A.; Su, S. Molecular and biological characterization of a Marek’s disease virus field strain with reticuloendotheliosis virus LTR insert. Virus Genes. 2010, 40, 236-243;. [CrossRef]
- Zhang, Y-P.; Li, Z-J.; Bao, K-Y.; Lv, H-C.; Gao, Y-L.; Gao, H-L.; Qi, X-L.; Cui, H-Y.; Wang, Y-Q.; Ren, X-G.; Wang, X-M. Pathogenic characteristics of Marek’s disease virus field strains prevalent in China and the effectiveness of existing vaccines against them. Vet Microbiol. 2015, 177: 62-8;. [CrossRef]
- Gong, Z.; Zhang, K.; Li, L.; Wang, H.; Qiu, Y.; Li, I.; Hou, G.; Yu, J.; Wang, J.; Shan, Hu. Effect of vaccination with different types and dosages against a very virulent Marek’s disease virus strain. J. Mol. Genet. Med. 2014, 8:4.
- Lachheb, J.; Mastour, H.; Nsiri, J.; Kaboudi, K.; Choura, I.; Ammouna, F.; Amara, A.; Ghram, A. Newly detected mutations in the Meq oncogene and molecular pathotyping of very virulent Marek’s disease herpesvirus in Tunisia. Arch Virol. 2020, 165, 2589-2597;. [CrossRef]
- Murata, S.; Hayashi, Y.; Kato, A.; Isezaki, M.; Takasaki, S.; Onuma, M.; Osa, Y.; Asakawa, M.; Konnai, S.; Ohashi, K. Surveillance of Marek’s disease virus in migratory and sedentary birds in Hokkaido, Japan. Vet. J. 2012, 192, 538-540;. [CrossRef]
- Mescolini, G.; Lupini, C.; Davidson, I.; Massi, P.; Tosi, G.; Catelli, E. Marek’s disease viruses circulating in commercial poultry in Italy in the years 2015-2018 are closely related by their meq gene phylogeny. Transbound. Emerg. Dis. 2020, 67, 98-107;. [CrossRef]
- Lv, H.; Zhang, Y.; Sun, G.; Bao, K.; Gao, Y.; Qi, X.; Cui, H.; Wang, Y.; Li, K.; Gao, L.; Pan, Q.; Wang, X.; Liu, C. Genetic evolution of Gallid herpesvirus 2 isolated in China. Infect. Genet. Evol. 2017, 51, 263-274;. [CrossRef]
- Conradie, A. M.; Bertzbach, L. D.; Trimpert, J.; Patria, J. N.; Murata, S.; Parcells, M. S.; Kaufer, B. B. District polymorphisms in a single herpesvirus gene are capable of enhancing virulence and mediating vaccinal resistance. PLoS Pathog. 2020, 16, e1009104;. [CrossRef]
- Padhi, A.; Parcells, M. S. Positive selection derives rapid evolution of the meq oncogene of Marek’s disease virus. PLoS One. 2016, 11, e0162180;. [CrossRef]
- Davison, F.; Nair, V. Use of Marek’s disease vaccines: Could they be driving the virus to increasing virulence? Expert Rev. Vaccines. 2005, 4, 77-88;. [CrossRef]
- Read, A. F.; Baigent, S. J.; Powers, C.; Kgosana, L. B.; Blackwell, L.; Smith, L. P.; Kennedy, D. A.; Walkden-Brown, S. W.; Nair, V. K. Imperfect vaccination can enhance the transmission of highly virulent pathogens. PLoS Biol. 2015, 13, e1002198. [CrossRef]
- Landman, W. J. M.; Verschuren, S. B. E. Titration of Marek’s disease cell-associated vaccine virus (CVI 988) of reconstituted vaccine and vaccine ampoules from Dutch hatcheries. Avian Dis. 2003, 47, 1458-65;. [CrossRef]
- López de Juan Abad, B. A.; Cortes, A. L.; Correa, M.; Gimeno, I. M. Evaluation of factors that influence dose variability of Marek’s disease vaccines. Avian Dis. 2019, 63, 591-598;. [CrossRef]
- Tsai, Y. W.; Kang, C. Y.; Chiao, K. H.; Tsai, Y. L.; Lien, Y. Y.; Cheng, M. C. Detection of vaccine strain virus (serotype 1) in layer chicks feather tips after immunization against Marek’s disease. Presented at the 3rd joint meeting of veterinary science in east Asia, Pingtung, Taiwan, 1st May 2023.
- Sun, G-R.; Zhang, Y-P.; Zhou, L-Y.; Lv, H-C.; Zhang, F.; Li, K.; Gao, Y-L; Qi, X-L.; Cui, H-Y.; Wang, Y-Q.; Gao, L.; Pan, Q.; Wang, X-M.; Liu, C-J. Co-infection with Marek’s disease virus and reticuloendotheliosis virus increases illness severity and reduces Marek’s disease vaccine efficacy. Viruses. 2017, 9, 158;. [CrossRef]
- Zhang, Y.; Cui, N.; Han, N.; Wu, J.; Cui, Z.; Su, S. Depression of vaccinal immunity to Marek’s disease by infection with Chicken infectious anemia virus. Front. Microbiol. 2017, 8, 1863;. [CrossRef]
Figure 1.
The gross lesions of MDV-1 clinical cases. The gross lesions in MDV-1 infected chickens in this study include: Enlarged liver (A) with white neoplastic nodules; the variable size of multifocal grayish-white nodules in the heart (B) and spleen (C); numerous white nodules throughout the intestinal serosa surface (D); thickened proventricular wall with multiple white prostrusions (E); multifocal white nodules in kidney (F); neoplastic mass occupied ovary (F); enlarged left sacrum nerve plexus (G).
Figure 1.
The gross lesions of MDV-1 clinical cases. The gross lesions in MDV-1 infected chickens in this study include: Enlarged liver (A) with white neoplastic nodules; the variable size of multifocal grayish-white nodules in the heart (B) and spleen (C); numerous white nodules throughout the intestinal serosa surface (D); thickened proventricular wall with multiple white prostrusions (E); multifocal white nodules in kidney (F); neoplastic mass occupied ovary (F); enlarged left sacrum nerve plexus (G).
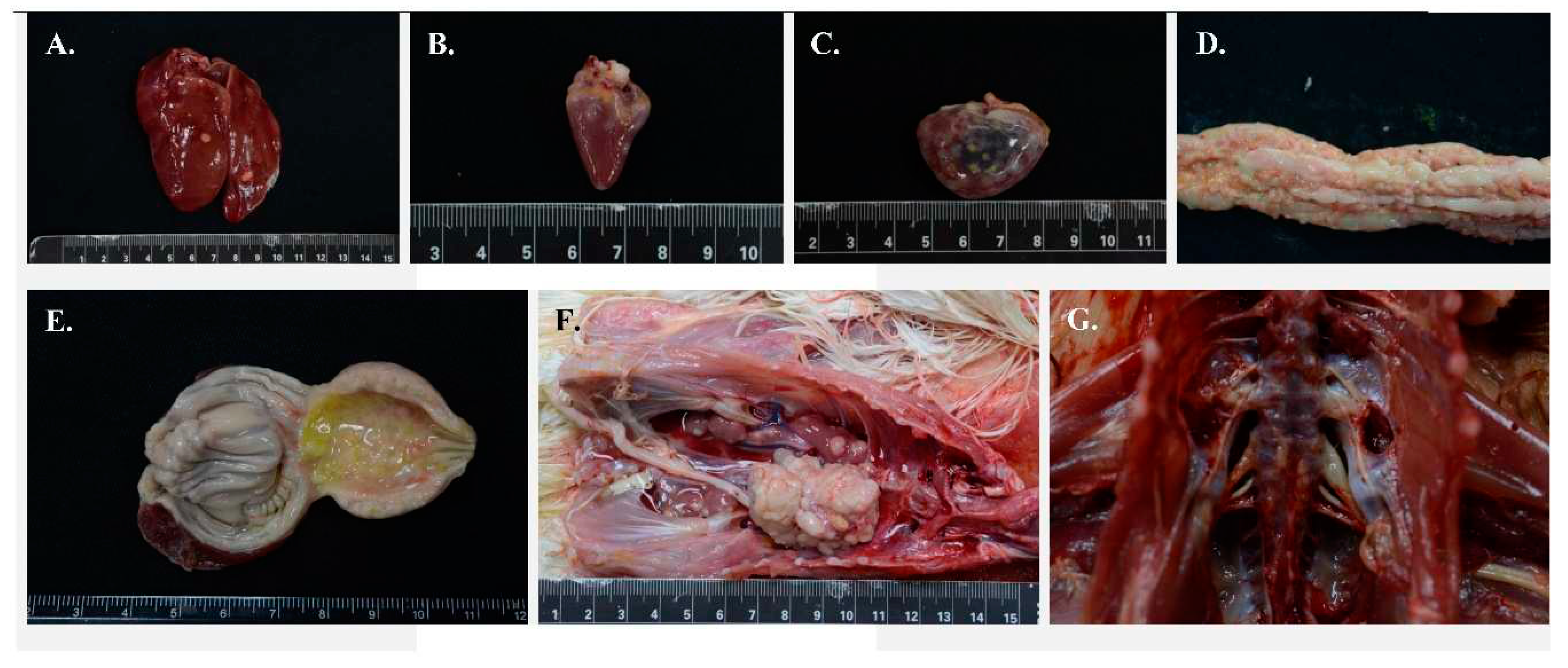
Figure 2.
Phylogenetic tree of MDV-1 isolates. The phylogenetic tree was built by using neighbor-joining (NJ) based on the complete nucleotide sequences of meq oncogene obtained from reference MDV-1 isolates/strains and Taiwan field isolates. All reference isolate/strain names are labeled with the corresponding abbreviation of countries. The colored dots indicate respective field MDV-1 strains in different countries and the attenuated/mild strains.
Figure 2.
Phylogenetic tree of MDV-1 isolates. The phylogenetic tree was built by using neighbor-joining (NJ) based on the complete nucleotide sequences of meq oncogene obtained from reference MDV-1 isolates/strains and Taiwan field isolates. All reference isolate/strain names are labeled with the corresponding abbreviation of countries. The colored dots indicate respective field MDV-1 strains in different countries and the attenuated/mild strains.
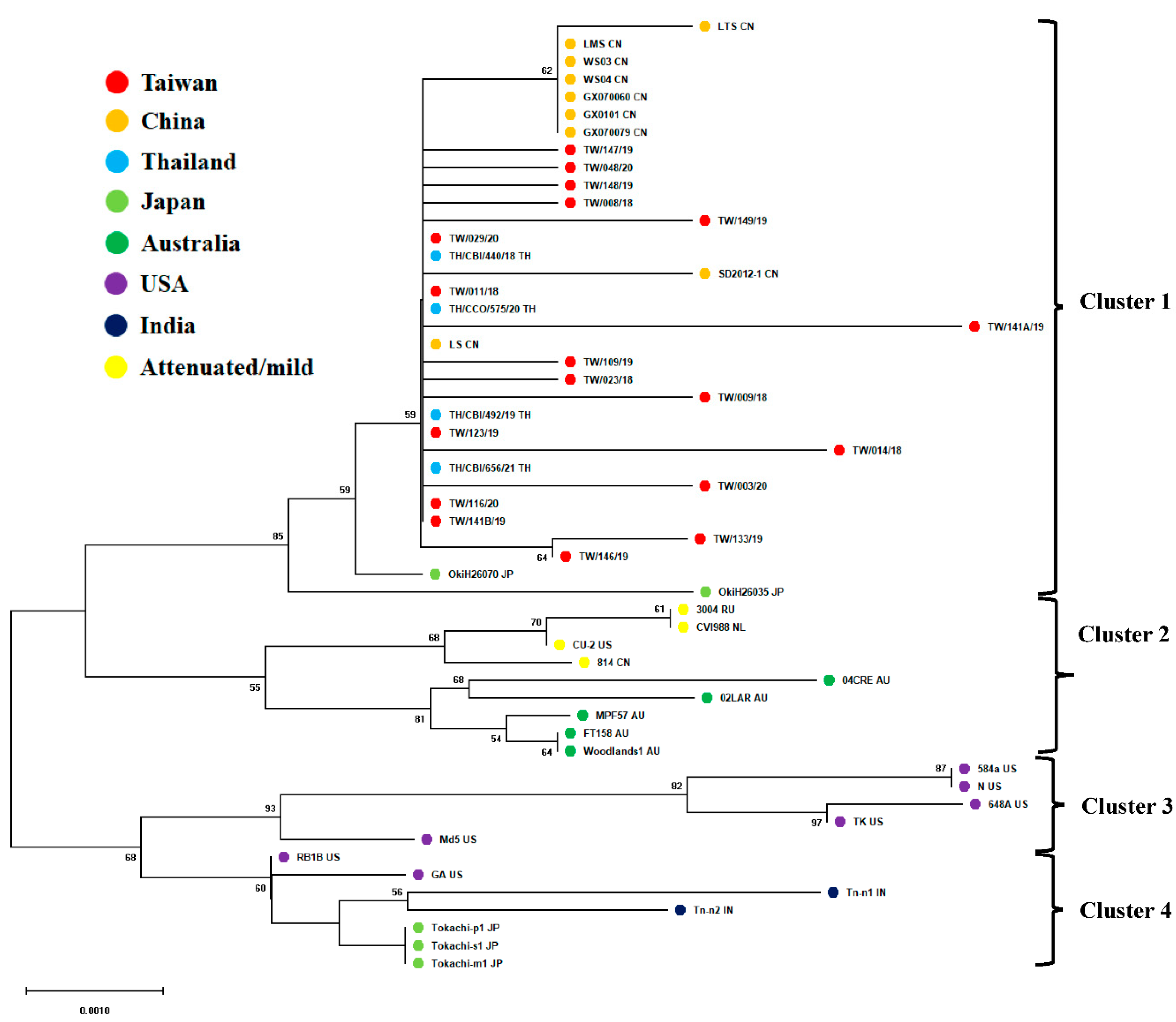

Table 1.
Information of 17 confirmed MDV-1 cases obtained from chicken flocks in Taiwan.
| Flock ID | Location | Year | Age (weeks) | Genetic line | Vaccine types | pathogen | Isolate |
|---|---|---|---|---|---|---|---|
| P107-008 | Changhua | 2018 | 20 | Hy-Line | CVI988 | MDV, CAV | TW/008/18 |
| P107-009 | Pingtung | 2018 | 17 | Hy-Line | CVI988 | MDV, CAV | TW/009/18 |
| P107-011 | Pingtung | 2018 | 19 | Hy-Line | CVI988 | MDV, CAV | TW/011/18 |
| P107-014 | NA | 2018 | 26 | Hy-Line | CVI988 | MDV | TW/014/18 |
| P107-023 | Pingtung | 2018 | 35 | Hy-Line | CVI988 | MDV, CAV | TW/023/18 |
| P108-109 | Tainan | 2019 | 31 | Native Chicken | CVI988 + HVT | MDV, CAV | TW/109/19 |
| P108-123 | Pingtung | 2019 | NA | HENDRIX | CVI988 + HVT | MDV | TW/123/19 |
| P108-133 | Pingtung | 2019 | 10 | Layer, NA | CVI988 + HVT | MDV | TW/133/19 |
| P108-141 | Chiayi | 2019 | 14, 18 | HENDRIX | CVI988 + HVT | MDV, IBV, CAV, IBDV | TW/141A/19TW/141B/19 |
| P108-146 | Chiayi | 2019 | 19 | Layer, NA | CVI988 + HVT | MDV, CAV | TW/146/19 |
| P108-147 | Tainan | 2019 | 23 | Layer, NA | CVI988 + HVT | MDV | TW/147/19 |
| P108-148 | Kaohsiung | 2019 | 19 | Layer, NA | CVI988 + HVT | MDV, CAV | TW/148/19 |
| P108-149 | Chiayi | 2019 | 23 | Layer, NA | CVI988 + HVT | MDV, CAV | TW/149/19 |
| P109-003 | Taitung | 2020 | 27 | HENDRIX | CVI988 + HVT | MDV | TW/003/20 |
| P109-029 | Chiayi | 2020 | 8 | HENDRIX | CVI988 + HVT | MDV, NDV | TW/029/20 |
| P109-048 | Pingtung | 2020 | 18 | Hy-Line | CVI988 | MDV, FAV | TW/048/20 |
| P109-116 | Chiayi | 2020 | 29 | HENDRIX | CVI988 + HVT | MDV, IBV, CAV, MS, FPV | TW/116/20 |
NA: Not available. MDV: Marek’s disease virus; CAV: Chicken anemia virus; IBV: Infectious bronchitis virus; IBDV: Infectious bursal disease virus; NDV: Newcastle disease virus; FAV: Fowl adenovirus; MS: Mycoplasma synoviae; FPV: Fowl poxvirus.
Table 2.
Specific amino acid substitutes in Meq oncoprotein of presented Taiwan MDV-1 field isolates compared with the reference strains.
Table 2.
Specific amino acid substitutes in Meq oncoprotein of presented Taiwan MDV-1 field isolates compared with the reference strains.
| Strain/Isolate | Pathotype | 71 | 77 | 80 | 88 | 93 | 110 | 115 | 119 | 139 | 153 (PPPP) |
176 (PPPP) |
180 | 217/276b (PPPP) |
218/277b (PPPP) |
277/336b | 283/342b | 326/385b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 648A (USA) | vv+MDV | A | K | D | A | Q | C | V | R | T | Q | A | A | A | P | P | A | T |
| N (USA) | vv+MDV | A | K | D | A | Q | C | V | R | T | Q | A | A | A | P | P | A | T |
| Md5 (USA) | vvMDV | A | K | D | A | Q | C | V | C | T | P | P | T | A | P | L | V | T |
| RB1B (USA) | vvMDV | A | K | D | A | Q | C | V | C | T | P | P | T | P | P | L | A | T |
| GA (USA) | vMDV | A | K | D | A | Q | C | V | C | T | P | P | T | P | P | L | A | T |
| 571 (USA) | vMDV | A | E | D | A | Q | C | V | C | T | P | H | T | P | P | L | A | T |
| CU-2 (USA) | mMDV | S | E | D | A | Q | S | V | C | T | P | P | T | P | P | L | A | I |
| CVI988/Rispens (NL) | attMDV | S | E | D | A | Q | S | V | C | T | P | P | T | P | P | L | A | I |
| SD2012-1 (CN) | vv+MDV | A | E | Y | T | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| LTS | vv+MDV | A | E | Y | A | Q | C | A | C | A | P | R | T | A | P | L | A | T |
| GX0101 (CN) | vvMDV | A | E | Y | A | Q | C | A | C | A | P | R | T | A | P | L | A | T |
| LS (CN) | vvMDV | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TH/CBI/656/21 (THA) | NAa | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| OkiH26035 (JP) | NA | A | E | Y | A | Q | C | A | C | T | P | P | T | A | P | L | A | T |
| TW/008/18 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/009/18 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/011/18 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/014/18 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/023/18 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/109/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/123/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/133/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/141A/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/141B/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/146/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/147/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/148/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/149/19 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/003/20 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/029/20 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/048/20 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
| TW/116/20 | NA | A | E | Y | A | Q | C | A | C | T | P | R | T | A | P | L | A | T |
a NA: not available b Amino acid position based on 59-a.a. insertion-containing Meq oncoprotein which is predominantly in lower virulence strain.
Table 3.
The proline content and the number of 4-proline-repeat (PPPP) within Meq oncoproteins in the presented Taiwan MDV-1 field isolates and reference strains.
Table 3.
The proline content and the number of 4-proline-repeat (PPPP) within Meq oncoproteins in the presented Taiwan MDV-1 field isolates and reference strains.
| Strain/Isolate | Pathotype | Size of Meq (a. a.) |
Insertion size (a. a.) | Proline contents (%) | Number of PPPPs |
|---|---|---|---|---|---|
| CVI988/Rispens (NL) | attMDV | 398 | 59 | 23.1 | 7 |
| CU-2 (USA) | mMDV | 398 | 59 | 23.1 | 7 |
| 648A (USA) | vv+MDV | 339 | Nilb | 20.9 | 2 |
| N (USA) | vv+MDV | 339 | Nil | 20.9 | 2 |
| Md5 (USA) | vvMDV | 339 | Nil | 21.3 | 4 |
| RB1B (USA) | vvMDV | 339 | Nil | 21.5 | 5 |
| GA (USA) | vMDV | 339 | Nil | 21.5 | 5 |
| 571 (USA) | vMDV | 339 | Nil | 21.2 | 4 |
| SD2012-1 (CN) | vv+MDV | 339 | Nil | 20.9 | 3 |
| LTS (CN) | vv+MDV | 339 | Nil | 20.9 | 3 |
| GX0101 (CN) | vvMDV | 339 | Nil | 20.9 | 3 |
| LS (CN) | vvMDV | 339 | Nil | 20.9 | 3 |
| TH/CBI/656/21 (THA) | NAa | 339 | Nil | 20.9 | 3 |
| OkiH26035 (JP) | NA | 339 | Nil | 21.2 | 4 |
| TW/008/18 | NA | 339 | Nil | 20.9 | 3 |
| TW/009/18 | NA | 339 | Nil | 20.9 | 3 |
| TW/011/18 | NA | 339 | Nil | 20.9 | 3 |
| TW/014/18 | NA | 339 | Nil | 20.6 | 3 |
| TW/023/18 | NA | 339 | Nil | 20.9 | 3 |
| TW/109/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/123/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/133/19 | NA | 339 | Nil | 20.6 | 3 |
| TW/141A/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/141B/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/146/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/147/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/148/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/149/19 | NA | 339 | Nil | 20.9 | 3 |
| TW/003/20 | NA | 339 | Nil | 21.3 | 3 |
| TW/029/20 | NA | 339 | Nil | 20.9 | 3 |
| TW/048/20 | NA | 339 | Nil | 20.9 | 3 |
| TW/116/20 | NA | 339 | Nil | 20.9 | 3 |
a NA: not available b Nil: nothing.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



