
Submitted:
25 September 2023
Posted:
27 September 2023
You are already at the latest version
Abstract
HIV-1 infection can activate the expression of human endogenous retroviruses (HERVs), particularly HERV-K (HML-2). HIV controllers (HICs) are rare people living with HIV (PLWH) who naturally control HIV-1 replication. The ability of HICs to control the expression of endogenous retroviruses has not been previously addressed. In this study, we measured ERVK-6 RNA expression in PBMCs of HICs (n = 23), antiretroviral (ART)-suppressed subjects (n = 8), and HIV-1-negative (NEG) individuals (n = 10) and correlated the transcript expression of ERVK-6 with multiple HIV-1 restriction factors. Our study reveals that ERVK-6 RNA expression in PBMCs from HICs was significantly downregulated compared with both ART and NEG control groups. Moreover, we detected that ERVK-6 RNA levels in PBMCs across all groups were negatively correlated with the expression levels of p21 and MCPIP1. In our previous study, these two cellular restriction factors were upregulated in HICs compared with control groups. Interestingly, p21 and MCPIP1 limit the activation of macrophages and T cells by downregulating the activity of NF-kB, a transcription factor that stimulates the LTR-driven transcription of HIV-1 and HERV-K proviruses. These findings support that HICs activate innate antiviral mechanisms that may simultaneously downregulate the transcription of both HIV-1 and endogenous retroviruses.
Keywords:
HIV-1
; HERV-K
; elite controllers
; viremic controllers
; restriction factors
1. Introduction
Human endogenous retroviruses (HERVs) are remnants of ancient viral infections integrated into the human genome throughout evolutionary history. These genetic elements are estimated to comprise nearly 8% of the human genome and are passed from generation to generation [1]. Although most HERVs are now functionally inactive due to mutations and deletions, some have been implicated in disease processes, such as autoimmune and neurodegenerative (such as amyotrophic lateral sclerosis – ALS) disorders, several types of cancer and immune dysregulation and coagulopathy in critical COVID-19 [2,3,4,5,6,7,8,9].
HERV-K represents the latest classified family, encompassing the most recently integrated HERV groups within the human genome [10]. HERV-Ks can be further divided into ten subfamilies (HML 1-10), with some retaining their structural integrity and ability to generate viral proteins [11]. Importantly, their high sequence similarity with contemporary retroviruses, such as HIV-1, has led to investigations into potential roles in HIV infection and pathogenesis. HIV-1 can activate HERV-K (HML-2) mRNA and protein expression in vitro [12,13,14,15]. Moreover, untreated PLWH display higher levels of HERV-K RNA in peripheral blood mononuclear cells (PBMCs) [4,13,16] or plasma [17,18,19] and higher titers of anti-HERV-K (HML-2) antibodies and HERV-specific CD8+ T-cell responses [19,20,21,22] compared to antiretroviral therapy (ART)-treated individuals and/or HIV-uninfected healthy subjects. However, the exact nature of the relationship between HERV-K and HIV-1 expression and the pathogenic impact of HERV-K activation in the setting of HIV-1 infection remains controversial.
Some findings support that expression of HERV-K during HIV-1 infection could directly affect immunopathogenesis. One study showed that the anti-HERV-K (HML-2) transmembrane (TM) antibody response was positively correlated with HIV plasma viremia and inversely correlated with CD4+ T-cell counts in viremic noncontrollers, suggesting that anti-TM antibodies could target HERV-K (HML-2) Env TM-expressing HIV-1-infected CD4+ cells and accelerate CD4+ T-cell depletion [13,17,22]. Interestingly, the HERV-K Env TM protein contains an immunosuppressive domain that modulates the release of different cytokines by PBMCs in vitro, including immunosuppressive IL-10, and may thus contribute to suppressing immune responses during HIV-1 infection [15]. Other studies propose that HERV-K expression could also exacerbate HIV-1 infection by stimulating the innate immune system and the expression of type I interferon in macrophages and probably other immune cells and, therefore, enhancing proinflammatory signaling in a harmful way [23].
Other findings, by contrast, support that increased expression of HERV-K and an enhanced immune response against HERV-K proteins may improve virologic control in HIV-1 infection. The induction of HERV-K antigen expression in HIV-1-infected cells could stimulate a HERV-K–specific cytotoxic CD8+ T-cell response, resulting in the targeted elimination of reservoir HIV-infected cells [15]. Some studies show an inverse correlation between HIV-1 plasma viral load and HERV-specific CD8+ T-cell responses or antibodies against HERV-K Gag and further observed that HIV controllers (HICs), a group of rare individuals who maintain undetectable viral loads without ART, have more robust cellular and antibody responses against HERV-K than ART-suppressed, viremic noncontrollers, and uninfected subjects [19,20,21,24]. Moreover, the level of HERV-K RNA expression in PBMCs was negatively associated with the level of T-cell activation in untreated PLWH, suggesting that HERV-K expression does not contribute to pathologic immune activation in chronic HIV-1 infection [16].
Previous studies revealed that LTR-directed transcription of both HERV-K (HML-2) and HIV-1 proviruses could be regulated by similar factors. One study demonstrated that HIV-1 Tat plays an important role in activating the expression of HERV-K (HML-2) in the setting of HIV-1 infection [25]. Activation of HERV-K (HML-2) transcription by Tat occurs in a partially different way than that of HIV-1 because it involves Tat’s interaction with cellular factors NF-kB and NF-AT but not with Cyclin T1 or P-TEFb. Another study with sorted PBMCs showed that increased HERV-K (HML-2) RNA expression in the setting of HIV-1 infection resulted from multiple cell sources, including those not infected by HIV-1 [4]. Thus, HIV-1 infection may also enhance HERV-K (HML-2) transcription in both infected and uninfected cells by indirect mechanisms that probably modulate the expression of key transcription factors, such as increased immune activation and inflammation [23] and/or mechanisms influencing the cellular epigenetic environment [26].
In a previous study conducted by our group, we discovered that the transcripts of two multifunctional cellular proteins, cyclin-dependent kinase (CDK) inhibitor 1A (CDKN1A/p21) and monocyte chemotactic protein-induced protein 1 (MCPIP1), were significantly elevated in HICs compared to ART-suppressed and HIV-1 negative control individuals [27]. The p21 can block HIV-1 by suppressing the biosynthesis of deoxyribonucleoside triphosphates (dNTPs), which are essential for viral reverse transcription, and by inhibiting the activity of CDK9, which is crucial for HIV-1 viral mRNA transcription [28,29,30,31,32]. MCPIP1 is critical in controlling inflammatory responses and maintaining immune homeostasis. It can block HIV-1 replication by promoting the degradation of several inflammatory cytokine mRNAs and viral mRNAs via its RNase activity [33,34]. Interestingly, p21 and MCPIP1 contribute to the limited activation of macrophages and T cells by downregulating NF-kB activity [34,35,36,37,38,39], which may also indirectly reduce the LTR-driven activation of HIV-1 and HERV-K proviruses.
We hypothesize that the upregulation of cellular restriction factors, such as p21 and MCPIP1, that negatively modulate HIV-1 transcription in HICs might also negatively affect the LTR-directed transcription of endogenous retroviruses. To test this hypothesis, we measured the ERVK-6 RNA expression levels (envelope protein fragment, also called HERV-K (HML-2) or HERV-K_7p22.1) in PBMCs of HIV controllers (n = 23), HIV-1 ART-suppressed subjects (n = 10), and HIV-1-negative individuals (n = 8) and correlated the transcript expression level of HERV-K with multiple HIV restriction factors.
2. Materials and Methods
2.1. Study Subjects
We analyzed a cohort of 23 HICs followed up at the Instituto Nacional de Infectologia Evandro Chagas (INI) in Rio de Janeiro, Brazil. All HICs maintained an RNA VL of < 2,000 copies/mL without antiretroviral therapy for at least five years and were subdivided into two subgroups: elite controllers (EC, n = 13) when most (≥ 70%) plasma VL determinations were below the limit of detection (LOD) and viremic controllers (VC, n = 10) when most (≥ 70%) VL determinations were > LOD and < 2,000 copies/mL. The limit of detection of plasma VL determinations varied over the follow-up period according to the Brazilian Ministry of Health guidelines, with methodologies being updated over time to improve sensitivity: Nuclisens HIV-1 RNA QT assay (Organon Teknika, Durham, NC, limit of detection: 80 copies/mL) from 1999 to 2007; the Versant HIV-1 3.0 RNA assay (bDNA 3.0, Siemens, Tarrytown, NY, limit of detection: 50 copies/mL) from 2007 to 2013; and the Abbott RealTime HIV-1 assay (Abbott Laboratories, Wiesbaden, Germany, the limit of detection: 40 copies/mL) from 2013 to today. Previous studies detailed these subjects' virological and immunological characteristics [27,40,41]. ART-suppressed subjects (ART, n = 8) and healthy HIV-1-uninfected subjects (NEG, n = 10) were used as controls.
2.2. mRNA gene-expression analysis
Total RNA was extracted from 1 × 107 PBMCs using an RNeasy mini kit (Qiagen, Hilden, North Rhine-Westphalia, Germany) in which buffer RLT was supplemented with β-mercaptoethanol and displaced on-column DNase treatment using a Qiagen RNase-Free DNase Set (Qiagen, Hilden, North Rhine-Westphalia, Germany) according to the manufacturer’s instructions. Total RNA yield and quality were determined using a NanoDrop® 8000 spectrophotometer and an Agilent® 2100 Bioanalyzer. Only samples with an RNA integrity number (RIN) greater than 8.0 were used for further analysis. Purified RNA (1 μg) was reverse transcribed to cDNA using an RT2 First Strand Kit (Qiagen, Hilden, North Rhine-Westphalia, Germany). The cDNA was mixed with RT2 SYBR Green/ROX qPCR Master Mix (Qiagen, Hilden, North Rhine-Westphalia, Germany), and the mixture was added to a customized RT2 RNA PCR Array (Qiagen, Hilden, North Rhine-Westphalia, Germany) to measure the mRNA expression of ERVK-6 envelope protein, also called HERV-K (HML-2) or HERV-K_7p22.1 (Gene RefSeq #PPH60565A-200/NM_001007236), 13 cellular restriction factors (CDKN1A/p21, ZC3H12A/MCPIP1, APOBEC3G, IFITM1, IFITM2, IFITM3, SAMHD1, Mx1, Mx2, SERINC3, SERINC5, SLFN11, and Tetherin/Bst2), and three housekeeping genes (GAPDH, β-actin, and RNase-P), according to the manufacturer’s instructions. Values of the crossing point at the maximum of the second derivative of the four-parameter fitted sigmoid curve second derivative, Cp, were determined for each sample. The efficiency of each amplification reaction was calculated as the ratio between the fluorescence of the cycle of quantification and the fluorescence of the cycle immediately preceding that. Genes used in the normalization among samples were selected by the geNorm method [42]. Data were expressed as fold changes in mRNA abundance calculated as the normalized gene expression in any test sample divided by the mean normalized gene expression in the control HIV-1 NEG group.
2.3. Data analyses
The comparisons of mean log(base 2)-fold changes (log-FC) in mRNA abundance were performed by either nonparametric t tests or one-way ANOVA permutation tests (B = 1000 permutations), followed by pairwise comparisons with Holm‒Bonferroni adjustment [43], for two or more groups, respectively. The Pearson coefficient was used for correlation analyses. Finally, a multivariate principal-component analysis (PCA) was performed for the log-transformed expression data to visualize the distribution of sample individuals according to either their group or their mRNA HERV-K 6 expression levels (divided into four different quartiles) in two-dimensional (2D) spaces. The proportion of explained variation was calculated by adding the successive proportions of variation explained to obtain the running total. The contributions (in percentage) of the variables to the principal components were calculated as (var. cos2 × 100)/(total cos2 of the component), where cos2 indicates square cosine or squared coordinates. Accordingly, the contributions (in percentage) of individuals to the principal components were calculated as (ind. cos2 × 100)/(total cos2 of the component). Ellipses of the quantiles 66% of the normal distribution adjusted to the individuals of the different interest groups in these new dimensional spaces are presented. A P value ≤ 0.05 was used as the significance level in the analysis. All analyses were performed using R software version 4.1.2 [44] and packages ‘base’ for descriptive and correlation analyses, ‘FactoMineR’ [45], and ‘factoextra’ [46] for PCA and its graphic representation.
3. Results
3.1. ERVK-6 mRNA levels are downregulated in HICs
We first evaluated the ERVK-6 transcript levels in the HIC and ART groups compared to the NEG group. We found a significant (P < 0.05) downregulation of ERVK-6 mRNA in the HIC (0.88 - mean fold-change), VC (0.84 - mean fold-change) and EC subgroups (0.86 - mean fold-change), while no significant differences in ERVK-6 expression levels were observed between the NEG and ART groups (0.94 - mean fold-change, P = 0.33) (Figure 1). In comparison to the ART group, we found a significant downregulation of ERVK-6 mRNA in VC (0.89-mean fold-change, P = 0.02) and a lower, but not significant, expression in EC (0.94-mean fold-change, P = 0.70) and HIC (0.92-mean fold-change, P = 0.08) (Figure 1). No significant differences in ERVK-6 expression levels were observed between the EC and VC subgroups (P = 0.08) (Figure 1).
3.2. ERVK-6 mRNA and cellular restriction factor levels are correlated
We next assessed whether reduced ERVK-6 transcripts in HICs could be associated with mRNA expression levels of 13 cellular restriction factors that act against HIV-1. Pearson's correlation of all groups combined showed that the mRNA expression of ERVK-6 was negatively correlated with both p21 (r = -0.48; P = 0.0013) and MCPIP1 (r = -0.35; P = 0.0248) (Figure 2A-B), positively correlated with SERINC3 (r = 0.58; P < 0.0001), SERINC5 (r = 0.60; P < 0.0001), APOBEC3G (r = 0.43; P = 0.005), IFITM2 (r = 0.43; P = 0.004), SAMHD1 (r = 0.43; P = 0.005), and SLFN11 (r = 0.40; P = 0.009) (Figure 2C-H) and was not significantly correlated (P > 0.05) with the other five restriction factors (Mx1, Mx2, IFITM1, IFITM3, and Tetherin) analyzed (Figure S1).
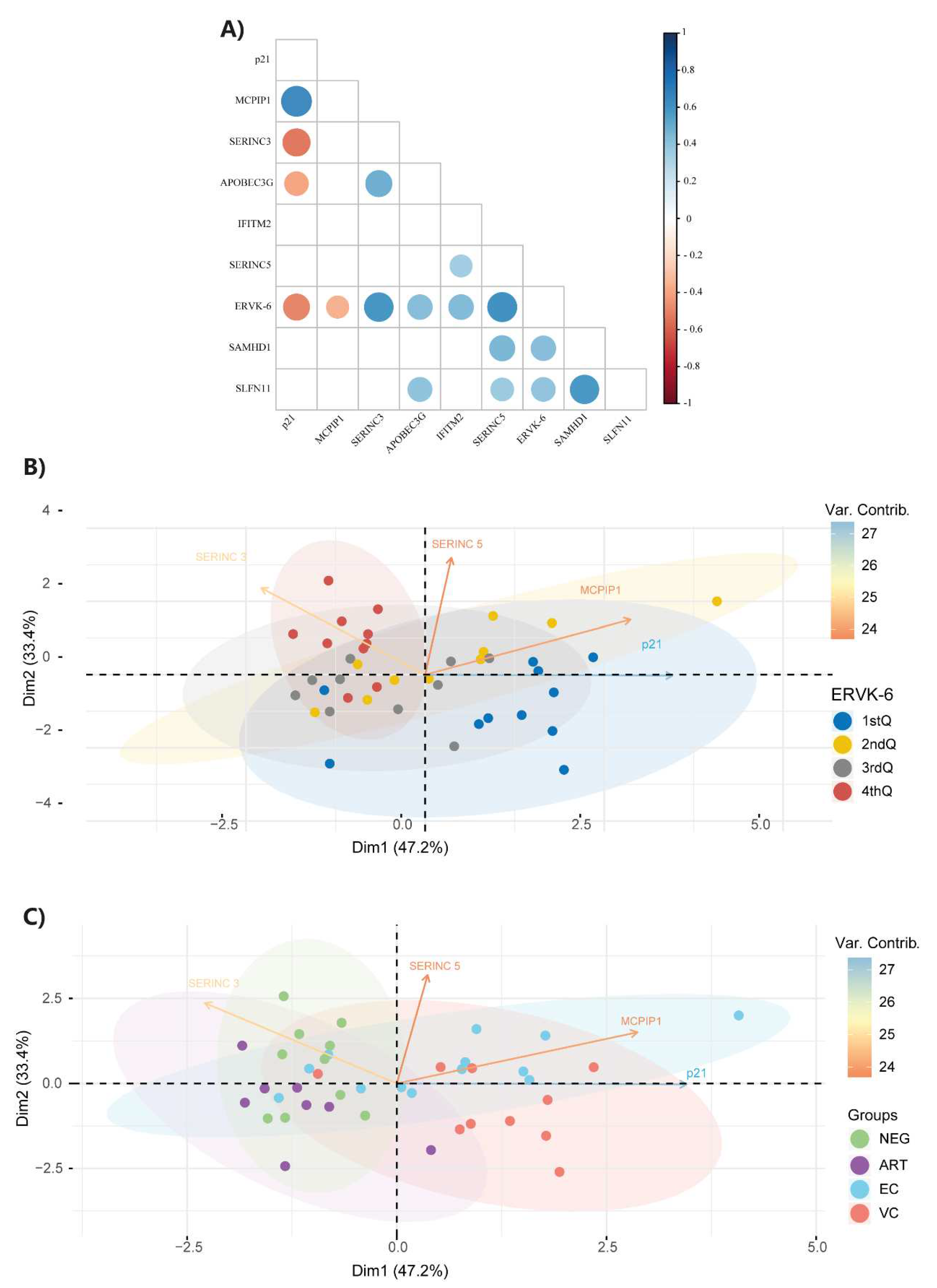
We also evaluated the correlation coefficient between restriction factors significantly correlated with ERVK-6 mRNA expression levels. We detected a significant negative correlation between p21 and SERINC3 (r = -0.53; P = 0.0004) and APOBEC3G (r = - 0.40; P = 0.009) (Figure 3A). Significant positive correlations were observed between SERINC3 and APOBEC3G (r = 0.49; P = 0.0013) and between SERINC5 and SLFN11 (r = 0.37; P = 0.018), SAMHD1 (r = 0.45; P = 0.003), and IFITM2 (r = 0.34; P = 0.028). Finally, we also observed positive correlations between SLFN11 and APOBEC3G (r = 0.41; P = 0.008) (Figure 3A).
Multivariate principal component analysis (PCA) comprising p21, MCPIP1, SERINC3, and SERINC5 variables is shown in Figure 3B and 3C according to either their mRNA ERVK-6 expression levels divided into four different quartiles (Figure 3C) or their group (Figure 3D). Our results revealed that 80.6% of the total variance in response to the four restriction factors was expressed by two principal components. The first component (Dim1) represented 47.2%, while the second (Dim2) represented 33.4% of the total variance. The variables p21 and MCPIP1 contributed most to the first component (Dim1), while SERINC5 and SERINC3 contributed most to the second component (Dim2).
In general, we observed a strong association between the ERVK-6 expression quartiles and the HIV-1 control groups (X2 = 32.293, df = 9, P = 0.0001772). This association was even more evident between the NEG control group and the quartiles with the highest ERVK-6 expression (i.e., 7 in the 4th quartile, 2 in the 3rd quartile, 1 in the 2nd quartile, and 0 in the 1st quartile) (X2 = 16.056, df = 3, P = 0.001104). The opposite situation, also highly associated, was observed between the VC group and ERVK-6 expression (7 in the 1st quartile, 1 in the 2nd quartile, 2 in the 3rd quartile, and 0 in the 4th quartile) (X2 = 13.641, df = 3, P = 0.003437). This association was confirmed when examining the HIC group (EC + VC), i.e., 10 in the 1st quartile, 8 in the 2nd quartile, 4 in the 3rd quartile, and only 1 in the 4th quartile (X2 = 17.413, df = 3, P = 0.0005812).
The separation between the HIC (EC + VC) and control (NEG + ART) groups occurred almost perfectly along the first component (Dim1), with the first group to the right of the origin (1st and 4th quadrants) and the second to the left of the origin (2nd and 3rd quadrants), except for two EC and one VC individuals, in the 3rd and 2nd quadrants, respectively; two of them had low ERVK-6 expression (2nd quartile), and another had very low ERVK-6 expression (1st quartile). The same observation regarding the first component (Dim1) was made with the levels of ERVK-6 expression, with the individuals with the lowest expression (1st and 2nd quartile) being further to the right of the origin (1st and 4th quadrants) and those with the highest expression to the left of the origin (2nd and 3rd quadrants). Notably, the individuals with the highest ERVK-6 expression (4th quartile) were concentrated in the 2nd quadrant of the PCA, coinciding with the predominance of healthy control individuals (NEG), while the individuals with the lowest ERVK-6 expression (1st quartile) were concentrated in the 4th quadrant of the PCA, coinciding with the predominance of VC.
4. Discussion
Previous studies revealed that HERV-K (HML-2) RNA expression in PBMCs from untreated viremic-PLWH was higher than that in ART-treated PLWH and/or HIV-uninfected healthy subjects [4,13,16] and that HICs have more robust cellular and antibody responses against HERV-K than ART-suppressed, viremic noncontrollers and uninfected subjects [20,21]. This led to the hypothesis that HERV-K expression in HICs could expose the immune system to HERV-K antigens that act as immutable targets for cytotoxic lysis, resulting in the virologic control of HIV-1. However, none of these studies analyzed HERV-K (HML-2) RNA expression in HICs. Our study reveals that HERV-K 6 (HML-2) RNA expression in PBMCs from HICs, particularly the VC subgroup, was significantly downregulated compared with both ART and NEG control groups.
We hypothesize that HICs may activate antiviral mechanisms that control the expression of both exogenous and endogenous retroviruses. Consistent with this notion, we detected that ERVK-6 RNA levels in PBMCs were negatively correlated with the expression levels of two cellular restriction factors, namely, p21 and MCPIP1. These proteins can block HIV-1 replication in macrophages and CD4+ T cells [29,30,33,47,48] and further limit aberrant immune activation [34,49,50]. Interestingly, p21 and MCPIP1 limit the activation of macrophages and T cells by downregulating the activity of NF-κB [34,35,36,37,38,39], a transcription factor that stimulates the LTR-driven transcription of HIV-1 and HERV-K proviruses [51,52]. In a previous study, we showed that MCPIP1 and p21 mRNA and protein expression levels are upregulated in PBMCs from our HIC cohort [27]. Therefore, we suggest that HICs can activate a homeostatic anti-inflammatory response that comprises antiviral factors, such as p21 and MCPIP1, to prevent excessive immune activation driven by residual HIV-1 replication. This negative homeostatic response inhibits the NF-κB pathway, which may reduce the efficiency of the LTR-driven transcription of HIV-1 and HERV-K proviruses.
Interestingly, we detected that ERVK-6 RNA expression in our cohort was positively correlated with two cellular restriction factors, SERINC3 and SERINC5, that may upregulate HERV-K (HML-2) RNA levels by activating the NF-κB pathway. SERINC3/5 were initially identified as cell restriction factors that can potently suppress HIV-1 infectivity by incorporating into budding viral particles and impairing subsequent virion fusion and infection of new target cells [53,54]. A recent study revealed that SERINC3/5 exhibited additional antiviral activities by forming a signaling complex with the mitochondrial antiviral signaling protein (MAVS) and TRAF6 at the mitochondria that increased the phosphorylation and activation of IRF3 and IκBα, thus cooperatively enhancing the NF-κB inflammatory pathway and type I IFN (IFN-I) production [55]. Moreover, we detected a significant negative correlation between the expression of p21 and SERINC3, suggesting that these restriction factors may display antagonist transcriptional regulation and may be part of the mechanism that controls the repression and activation of HERVs.
Our findings also revealed that the expression of some ISGs analyzed here (APOBEC3G, SLFN11, and SAMDH1) was positively correlated with the expression of both ERVK-6 and SERINC3/5. Cytosolic HERV-K dsRNA/cDNA may activate different nucleic acid sensors of innate immunity, such as retinoic acid-inducible gene-I-like receptors (RIG-I and MDA5) [56,57,58] and cGMPAMP synthase (cGAS) [59]. Activation of those innate immune sensors leads to phosphorylation and activation of TANK-binding kinase 1 (TBK1) and IκB kinase-ϵ (IKKϵ) kinases, which in turn activate the transcription factors IRF3 and NF-κB, ultimately leading to upregulation of type I IFNs, ISGs, and other proinflammatory cytokines. Interestingly, SAMHD1 and SLFN11 belong to a subset of ISGs that may be directly induced by IRF3 [60,61], and APOBEC3G may be positively modulated by the NF-κB pathway [62]. We propose that increased levels of SERINC3/5 and cytosolic HERV-K RNA/DNA may directly upregulate some ISGs by triggering the activation of IRF-3 and NF-κB, independent of the engagement of the type I IFN-JAK-STAT pathway.
The generation and maintenance of strong HERV-specific cellular and antibody responses in HICs seem inconsistent with the very low ERVK-6 mRNA expression detected here in PBMCs. Some findings, however, may explain these apparently conflicting results. Previous studies conducted in our and other cohorts of HICs showed that HIV-1 continues to replicate and evolve despite undetectable or extremely low levels of viremia [41,63,64,65,66,67,68]. Moreover, HIV-1 replication in HICs seems to mainly occur in the lymph nodes [68]. Thus, HERV-K expression in HICs may be mostly restricted to HIV-infected cells residing in lymph nodes, and expression of both HERV-K and HIV-1 antigens at those sites may stimulate the continuous generation of HERV-specific cellular and antibody responses. In contrast, most PBMCs and HIV-infected cells in the periphery display an antiviral transcriptional signature that limits the activation of both exogenous and endogenous retroviruses.
Persistent overexpression of IFN-α/β and ISGs is a hallmark of chronic HIV-1 infection and is associated with detrimental immune activation, bystander CD4+ T-cell apoptosis, and progressive disease [69,70]. Some data suggest that augmented HERV expression may play an active role in exacerbating and perpetuating chronic inflammation in some autoimmune diseases via type I IFN and MAVS positive signaling feedback loops [71]. On the other hand, HERV-K (HML-2) knockdown significantly downregulated genes containing interferon-stimulated response elements (ISREs) in their promoters in basal and IFN-γ-challenged macrophages and had implications for the paracrine activation of nearby cells following macrophage activation [23]. Moreover, repression of endogenous retrovirus in vivo alleviates tissue aging and, to some extent, organismal aging by attenuating innate immune responses [59]. These findings suggest that repression of HERV-K expression in HICs may contribute to attenuating the activation of innate immune responses, preventing chronic overexpression of IFN-α/β/ISGs, and preserving immune homeostasis in those subjects.
Previous studies from our cohort and other groups showed that HICs displayed elevated levels of some key plasma inflammatory markers, such as sCD14, IP-10, IL-18, and D-dimer, and higher CD8+ T-cell activation with respect to HIV-negative individuals [40,72,73,74,75,76,77,78]. Moreover, VCs displayed higher levels of immune activation and residual inflammation than ECs. The proinflammatory profile observed in HICs, however, is different from the aberrant inflammation observed in untreated noncontrollers [79,80]. Moreover, another study that performed gene expression analysis of PBMCs and purified CD4+ and CD8+ T cells from HICs showed that these individuals, compared to ART-treated individuals, have lower expression of several inflammatory genes, such as IL1A/B, IL-6, CXCL5, CXCL13, and CXCL1, but maintain upregulation of genes associated with cytotoxicity and the T-cell response [81]. We support that the low levels of HERV-K 6 (HML-2) detected here in HICs may be linked to the upregulation of an anti-inflammatory response that limits aberrant inflammation while preserving efficient antiviral T-cell responses. Such homeostatic anti-inflammatory mechanisms would be more critical for VCs due to the higher levels of chronic HIV-1 antigenic stimulation with respect to ECs.
5. Conclusions
Our findings revealed that ERVK-6 mRNA levels in PBMCs of HICs are even lower than those seen in aviremic ART-treated subjects and HIV-negative individuals. We found that ERVK-6 mRNA levels were negatively correlated with the expression of some restriction factors (p21 and MCPIP1) and positively correlated with the expression of others (SERINC3/5, APOBEC3G, IFITM2, SAMHD1, and SLFN11). We suggest that reduced expression of ERVK-6 in HICs may result from the upregulation of a distinctive antiviral and anti-inflammatory homoeostatic response that contributes to the control of HIV-1 replication and limits chronic immune activation and inflammation driven by persistent HIV-1 antigenic stimulation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Correlation between RF mRNA levels in PBMCs from HICs and ERVK-6 mRNA expression. The RF-normalized expression correlations were calculated in all groups. The point colors indicate the patient group, according to the legend. Correlation coefficients (Pearson’s ρ) are shown in each graph's upper right or left corner. P values < 0.05 were considered statistically significant.
Author Contributions
GB and TMLS conceived and designed the study and supervised the experiments. SSDA conducted the experiments and analyzed the data together with MR-A, ED, and GB. FH participated in sample processing and provided intellectual input. ED collaborated with the mRNA gene expression analysis. BH, BG, and VGV conducted patient recruitment and follow-up. F GB and TMLS conceived and designed the study and supervised the experiments. SSDA conducted the experiments and analyzed the data together with MR-A, ED, and GB. FH participated in sample processing and provided intellectual input. ED collaborated with the mRNA gene expression analysis. BH, BG, and VGV conducted patient recruitment and follow-up. FH, ED, and MGM provided intellectual input for results interpretations. SSDA, GB and MR-A wrote the first draft. All the authors have read and approved the final manuscript.
Funding
This work was supported by the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro–FAPERJ (Grant number E-26/110.123/2014) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico–CNPq (Grant Number 401220/2016-8). SSDA had her PhD funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro–FAPERJ. SSDA is financed by a Postdoctoral fellowship from the “Pós-Doutorado Nota 10 (PDR 10)” by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro–FAPERJ (Grant n° E-26/202.335/2019). GB is funded by CNPq (Grant nº 304883/2020-4) and FAPERJ (Grant nº E-26/202.896/2018).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Oswaldo Cruz Institute Ethics Committee (CAAE 03925112.0.0000.5248; 17 August 2018).
Informed Consent Statement
This study has written informed consent documents approved by the INI Institutional Review Board (Addendum 049/2010) and the Brazilian National Human Research Ethics Committee (CONEP 14430/2011). Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
To the patients who participated in the study, as well as all the INI and LabAIDS technical staff involved in the clinical follow-up and blood collection of these patients. We also thank the Plataforma de PCR em Tempo Real e Digital –RJ (RPT09A) –FIOCRUZ and Plataforma de Sequenciamento de Ácidos Nucléicos de Nova Geração–RJ (RPT01J) –FIOCRUZ.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jern, P.; Coffin, J.M. Effects of Retroviruses on Host Genome Function. Annu. Rev. Genet. 2008, 42, 709–732. https://doi.org/10.1146/annurev.genet.42.110807.091501. [CrossRef]
- Agoni, L.; Guha, C.; Lenz, J. Detection of Human Endogenous Retrovirus K (HERV-K) Transcripts in Human Prostate Cancer Cell Lines. Front. Oncol. 2013, 3. https://doi.org/10.3389/fonc.2013.00180. [CrossRef]
- Cegolon, L.; Salata, C.; Weiderpass, E.; Vineis, P.; Palù, G.; Mastrangelo, G. Human Endogenous Retroviruses and Cancer Prevention: Evidence and Prospects. BMC Cancer 2013, 13, 4. https://doi.org/10.1186/1471-2407-13-4. [CrossRef]
- Bhardwaj, N.; Maldarelli, F.; Mellors, J.; Coffin, J.M. HIV-1 Infection Leads to Increased Transcription of Human Endogenous Retrovirus HERV-K (HML-2) Proviruses In Vivo but Not to Increased Virion Production. J Virol 2014, 88, 11108–11120. https://doi.org/10.1128/JVI.01623-14. [CrossRef]
- Argaw-Denboba, A.; Balestrieri, E.; Serafino, A.; Cipriani, C.; Bucci, I.; Sorrentino, R.; Sciamanna, I.; Gambacurta, A.; Sinibaldi-Vallebona, P.; Matteucci, C. HERV-K Activation Is Strictly Required to Sustain CD133+ Melanoma Cells with Stemness Features. J Exp Clin Cancer Res 2017, 36, 20. https://doi.org/10.1186/s13046-016-0485-x. [CrossRef]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of Active Loci of a Human Endogenous Retrovirus in Neurons of Patients with Amyotrophic Lateral Sclerosis. Ann Neurol. 2011, 69, 141–151. https://doi.org/10.1002/ana.22149. [CrossRef]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; Von Geldern, G.; Johnson, K.; et al. Human Endogenous Retrovirus-K Contributes to Motor Neuron Disease. Sci. Transl. Med. 2015, 7. https://doi.org/10.1126/scitranslmed.aac8201. [CrossRef]
- Balada, E.; Vilardell-Tarrés, M.; Ordi-Ros, J. Implication of Human Endogenous Retroviruses in the Development of Autoimmune Diseases. International Reviews of Immunology 2010, 29, 351–370. https://doi.org/10.3109/08830185.2010.485333. [CrossRef]
- Temerozo, J.R.; Fintelman-Rodrigues, N.; Dos Santos, M.C.; Hottz, E.D.; Sacramento, C.Q.; De Paula Dias Da Silva, A.; Mandacaru, S.C.; Dos Santos Moraes, E.C.; Trugilho, M.R.O.; Gesto, J.S.M.; et al. Human Endogenous Retrovirus K in the Respiratory Tract Is Associated with COVID-19 Physiopathology. Microbiome 2022, 10, 65. https://doi.org/10.1186/s40168-022-01260-9. [CrossRef]
- Costas, J. Evolutionary Dynamics of the Human Endogenous Retrovirus Family HERV-K Inferred from Full-Length Proviral Genomes. Journal of Molecular Evolution 2001, 53, 237–243. https://doi.org/10.1007/s002390010213. [CrossRef]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, Characterization, and Comparative Genomic Distribution of the HERV-K (HML-2) Group of Human Endogenous Retroviruses. Retrovirology 2011, 8, 90. https://doi.org/10.1186/1742-4690-8-90. [CrossRef]
- Young, G.R.; Terry, S.N.; Manganaro, L.; Cuesta-Dominguez, A.; Deikus, G.; Bernal-Rubio, D.; Campisi, L.; Fernandez-Sesma, A.; Sebra, R.; Simon, V.; et al. HIV-1 Infection of Primary CD4 + T Cells Regulates the Expression of Specific Human Endogenous Retrovirus HERV-K (HML-2) Elements. J Virol 2018, 92, e01507-17. https://doi.org/10.1128/JVI.01507-17. [CrossRef]
- Contreras-Galindo, R.; López, P.; Vélez, R.; Yamamura, Y. HIV-1 Infection Increases the Expression of Human Endogenous Retroviruses Type K (HERV-K) in Vitro. AIDS Research and Human Retroviruses 2007, 23, 116–122. https://doi.org/10.1089/aid.2006.0117. [CrossRef]
- Vincendeau, M.; Göttesdorfer, I.; Schreml, J.M.H.; Wetie, A.G.N.; Mayer, J.; Greenwood, A.D.; Helfer, M.; Kramer, S.; Seifarth, W.; Hadian, K.; et al. Modulation of Human Endogenous Retrovirus (HERV) Transcription during Persistent and de Novo HIV-1 Infection. Retrovirology 2015, 12, 27. https://doi.org/10.1186/s12977-015-0156-6. [CrossRef]
- Jones, R.B.; Garrison, K.E.; Mujib, S.; Mihajlovic, V.; Aidarus, N.; Hunter, D.V.; Martin, E.; John, V.M.; Zhan, W.; Faruk, N.F.; et al. HERV-K–Specific T Cells Eliminate Diverse HIV-1/2 and SIV Primary Isolates. J. Clin. Invest. 2012, 122, 4473–4489. https://doi.org/10.1172/JCI64560. [CrossRef]
- Ormsby, C.E.; SenGupta, D.; Tandon, R.; Deeks, S.G.; Martin, J.N.; Jones, R.B.; Ostrowski, M.A.; Garrison, K.E.; Vázquez-Pérez, J.A.; Reyes-Terán, G.; et al. Human Endogenous Retrovirus Expression Is Inversely Associated with Chronic Immune Activation in HIV-1 Infection. PLoS ONE 2012, 7, e41021. https://doi.org/10.1371/journal.pone.0041021. [CrossRef]
- Contreras-Galindo, R.; Kaplan, M.H.; Markovitz, D.M.; Lorenzo, E.; Yamamura, Y. Detection of HERV-K(HML-2) Viral RNA in Plasma of HIV Type 1-Infected Individuals. AIDS Research and Human Retroviruses 2006, 22, 979–984. https://doi.org/10.1089/aid.2006.22.979. [CrossRef]
- Contreras-Galindo, R.; Almodóvar-Camacho, S.; González-Ramírez, S.; Lorenzo, E.; Yamamura, Y. Short Communication: Comparative Longitudinal Studies of HERV-K and HIV-1 RNA Titers in HIV-1-Infected Patients Receiving Successful versus Unsuccessful Highly Active Antiretroviral Therapy. AIDS Research and Human Retroviruses 2007, 23, 1083–1086. https://doi.org/10.1089/aid.2007.0054. [CrossRef]
- Garrison, K.E.; Jones, R.B.; Meiklejohn, D.A.; Anwar, N.; Ndhlovu, L.C.; Chapman, J.M.; Erickson, A.L.; Agrawal, A.; Spotts, G.; Hecht, F.M.; et al. T Cell Responses to Human Endogenous Retroviruses in HIV-1 Infection. PLoS Pathog 2007, 3, e165. https://doi.org/10.1371/journal.ppat.0030165. [CrossRef]
- De Mulder, M.; SenGupta, D.; Deeks, S.G.; Martin, J.N.; Pilcher, C.D.; Hecht, F.M.; Sacha, J.B.; Nixon, D.F.; Michaud, H.-A. Anti-HERV-K (HML-2) Capsid Antibody Responses in HIV Elite Controllers. Retrovirology 2017, 14, 41. https://doi.org/10.1186/s12977-017-0365-2. [CrossRef]
- SenGupta, D.; Tandon, R.; Vieira, R.G.S.; Ndhlovu, L.C.; Lown-Hecht, R.; Ormsby, C.E.; Loh, L.; Jones, R.B.; Garrison, K.E.; Martin, J.N.; et al. Strong Human Endogenous Retrovirus-Specific T Cell Responses Are Associated with Control of HIV-1 in Chronic Infection. J Virol 2011, 85, 6977–6985. https://doi.org/10.1128/JVI.00179-11. [CrossRef]
- Michaud, H.-A.; De Mulder, M.; SenGupta, D.; Deeks, S.G.; Martin, J.N.; Pilcher, C.D.; Hecht, F.M.; Sacha, J.B.; Nixon, D.F. Trans-Activation, Post-Transcriptional Maturation, and Induction of Antibodies to HERV-K (HML-2) Envelope Transmembrane Protein in HIV-1 Infection. Retrovirology 2014, 11, 10. https://doi.org/10.1186/1742-4690-11-10. [CrossRef]
- Russ, E.; Mikhalkevich, N.; Iordanskiy, S. Expression of Human Endogenous Retrovirus Group K (HERV-K) HML-2 Correlates with Immune Activation of Macrophages and Type I Interferon Response. Microbiol Spectr 2023, 11, e04438-22. https://doi.org/10.1128/spectrum.04438-22. [CrossRef]
- Tandon, R.; SenGupta, D.; Ndhlovu, L.C.; Vieira, R.G.S.; Jones, R.B.; York, V.A.; Vieira, V.A.; Sharp, E.R.; Wiznia, A.A.; Ostrowski, M.A.; et al. Identification of Human Endogenous Retrovirus-Specific T Cell Responses in Vertically HIV-1-Infected Subjects. J Virol 2011, 85, 11526–11531. https://doi.org/10.1128/JVI.05418-11. [CrossRef]
- Gonzalez-Hernandez, M.J.; Swanson, M.D.; Contreras-Galindo, R.; Cookinham, S.; King, S.R.; Noel, R.J.; Kaplan, M.H.; Markovitz, D.M. Expression of Human Endogenous Retrovirus Type K (HML-2) Is Activated by the Tat Protein of HIV-1. J Virol 2012, 86, 7790–7805. https://doi.org/10.1128/JVI.07215-11. [CrossRef]
- Hurst, T.; Magiorkinis, G. Epigenetic Control of Human Endogenous Retrovirus Expression: Focus on Regulation of Long-Terminal Repeats (LTRs). Viruses 2017, 9, 130. https://doi.org/10.3390/v9060130. [CrossRef]
- De Azevedo, S.S.D.; Ribeiro-Alves, M.; Côrtes, F.H.; Delatorre, E.; Spangenberg, L.; Naya, H.; Seito, L.N.; Hoagland, B.; Grinsztejn, B.; Veloso, V.G.; et al. Increased Expression of CDKN1A/P21 in HIV-1 Controllers Is Correlated with Upregulation of ZC3H12A/MCPIP1. Retrovirology 2020, 17, 18. https://doi.org/10.1186/s12977-020-00522-4. [CrossRef]
- Zhang, J.; Scadden, D.T.; Crumpacker, C.S. Primitive Hematopoietic Cells Resist HIV-1 Infection via p21Waf1/Cip1/Sdi1. J. Clin. Invest. 2007, 117, 473–481. https://doi.org/10.1172/JCI28971. [CrossRef]
- Bergamaschi, A.; David, A.; Le Rouzic, E.; Nisole, S.; Barré-Sinoussi, F.; Pancino, G. The CDK Inhibitor P21 Cip1/WAF1 Is Induced by FcγR Activation and Restricts the Replication of Human Immunodeficiency Virus Type 1 and Related Primate Lentiviruses in Human Macrophages. J Virol 2009, 83, 12253–12265. https://doi.org/10.1128/JVI.01395-09. [CrossRef]
- Valle-Casuso, J.C.; Allouch, A.; David, A.; Lenzi, G.M.; Studdard, L.; Barré-Sinoussi, F.; Müller-Trutwin, M.; Kim, B.; Pancino, G.; Sáez-Cirión, A. P21 Restricts HIV-1 in Monocyte-Derived Dendritic Cells through the Reduction of Deoxynucleoside Triphosphate Biosynthesis and Regulation of SAMHD1 Antiviral Activity. J Virol 2017, 91, e01324-17. https://doi.org/10.1128/JVI.01324-17. [CrossRef]
- Chen, H.; Li, C.; Huang, J.; Cung, T.; Seiss, K.; Beamon, J.; Carrington, M.F.; Porter, L.C.; Burke, P.S.; Yang, Y.; et al. CD4+ T Cells from Elite Controllers Resist HIV-1 Infection by Selective Upregulation of P21. J. Clin. Invest. 2011, 121, 1549–1560. https://doi.org/10.1172/JCI44539. [CrossRef]
- Elahi, S.; Weiss, R.H.; Merani, S. Atorvastatin Restricts HIV Replication in CD4+ T Cells by Upregulation of P21. AIDS 2016, 30, 171–183. https://doi.org/10.1097/QAD.0000000000000917. [CrossRef]
- Liu, S.; Qiu, C.; Miao, R.; Zhou, J.; Lee, A.; Liu, B.; Lester, S.N.; Fu, W.; Zhu, L.; Zhang, L.; et al. MCPIP1 Restricts HIV Infection and Is Rapidly Degraded in Activated CD4+ T Cells. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 19083–19088. https://doi.org/10.1073/pnas.1316208110. [CrossRef]
- Fu, M.; Blackshear, P.J. RNA-Binding Proteins in Immune Regulation: A Focus on CCCH Zinc Finger Proteins. Nat Rev Immunol 2017, 17, 130–143. https://doi.org/10.1038/nri.2016.129. [CrossRef]
- Sun, X.; Feng, W.; Guo, Y.; Wang, Q.; Dong, C.; Zhang, M.; Guan, Z.; Duan, M. MCPIP1 Attenuates the Innate Immune Response to Influenza A Virus by Suppressing RIG-I Expression in Lung Epithelial Cells. J Med Virol 2018, 90, 204–211. https://doi.org/10.1002/jmv.24944. [CrossRef]
- Chen, X.; Zhao, Q.; Xie, Q.; Xing, Y.; Chen, Z. MCPIP1 Negatively Regulate Cellular Antiviral Innate Immune Responses through DUB and Disruption of TRAF3-TBK1-IKKε Complex. Biochemical and Biophysical Research Communications 2018, 503, 830–836. https://doi.org/10.1016/j.bbrc.2018.06.083. [CrossRef]
- Lloberas, J.; Celada, A. P21 waf1/CIP1 , a CDK Inhibitor and a Negative Feedback System That Controls Macrophage Activation: HIGHLIGHTS. Eur. J. Immunol. 2009, 39, 691–694. https://doi.org/10.1002/eji.200939262. [CrossRef]
- Scatizzi, J.C.; Mavers, M.; Hutcheson, J.; Young, B.; Shi, B.; Pope, R.M.; Ruderman, E.M.; Samways, D.S.K.; Corbett, J.A.; Egan, T.M.; et al. The CDK Domain of P21 Is a Suppressor of IL-1β-Mediated Inflammation in Activated Macrophages: Innate Immunity. Eur. J. Immunol. 2009, 39, 820–825. https://doi.org/10.1002/eji.200838683. [CrossRef]
- Trakala, M.; Arias, C.F.; García, M.I.; Moreno-Ortiz, M.C.; Tsilingiri, K.; Fernández, P.J.; Mellado, M.; Díaz-Meco, M.T.; Moscat, J.; Serrano, M.; et al. Regulation of Macrophage Activation and Septic Shock Susceptibility via P21(WAF1/CIP1): Innate Immunity. Eur. J. Immunol. 2009, 39, 810–819. https://doi.org/10.1002/eji.200838676. [CrossRef]
- Côrtes, F.H.; De Paula, H.H.S.; Bello, G.; Ribeiro-Alves, M.; De Azevedo, S.S.D.; Caetano, D.G.; Teixeira, S.L.M.; Hoagland, B.; Grinsztejn, B.; Veloso, V.G.; et al. Plasmatic Levels of IL-18, IP-10, and Activated CD8+ T Cells Are Potential Biomarkers to Identify HIV-1 Elite Controllers With a True Functional Cure Profile. Front. Immunol. 2018, 9, 1576. https://doi.org/10.3389/fimmu.2018.01576. [CrossRef]
- De Azevedo, S.S.D.; Caetano, D.G.; Côrtes, F.H.; Teixeira, S.L.M.; Dos Santos Silva, K.; Hoagland, B.; Grinsztejn, B.; Veloso, V.G.; Morgado, M.G.; Bello, G. Highly Divergent Patterns of Genetic Diversity and Evolution in Proviral Quasispecies from HIV Controllers. Retrovirology 2017, 14, 29. https://doi.org/10.1186/s12977-017-0354-5. [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol 2002, 3, research0034.1. https://doi.org/10.1186/gb-2002-3-7-research0034. [CrossRef]
- Basso, D.; Pesarin, F.; Salmaso, L.; Solari, A. Nonparametric One-Way ANOVA. In Permutation Tests for Stochastic Ordering and ANOVA; Lecture Notes in Statistics; Springer New York: New York, NY, 2009; Vol. 194, pp. 105–132 ISBN 978-0-387-85955-2.
- R Core Team. R: A Language and Environment for Statistical Computing. 2021.
- Lê, S.; Josse, J.; Husson, F. FactoMineR : An R Package for Multivariate Analysis. J. Stat. Soft. 2008, 25. https://doi.org/10.18637/jss.v025.i01. [CrossRef]
- Kassambara, A. and Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 1.0.7.; 2020.
- Allouch, A.; David, A.; Amie, S.M.; Lahouassa, H.; Chartier, L.; Margottin-Goguet, F.; Barré-Sinoussi, F.; Kim, B.; Sáez-Cirión, A.; Pancino, G. P21-Mediated RNR2 Repression Restricts HIV-1 Replication in Macrophages by Inhibiting dNTP Biosynthesis Pathway. Proc. Natl. Acad. Sci. U.S.A. 2013, 110. https://doi.org/10.1073/pnas.1306719110. [CrossRef]
- Lin, R.-J.; Chien, H.-L.; Lin, S.-Y.; Chang, B.-L.; Yu, H.-P.; Tang, W.-C.; Lin, Y.-L. MCPIP1 Ribonuclease Exhibits Broad-Spectrum Antiviral Effects through Viral RNA Binding and Degradation. Nucleic Acids Research 2013, 41, 3314–3326. https://doi.org/10.1093/nar/gkt019. [CrossRef]
- Jura, J.; Skalniak, L.; Koj, A. Monocyte Chemotactic Protein-1-Induced Protein-1 (MCPIP1) Is a Novel Multifunctional Modulator of Inflammatory Reactions. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2012, 1823, 1905–1913. https://doi.org/10.1016/j.bbamcr.2012.06.029. [CrossRef]
- Uehata, T.; Iwasaki, H.; Vandenbon, A.; Matsushita, K.; Hernandez-Cuellar, E.; Kuniyoshi, K.; Satoh, T.; Mino, T.; Suzuki, Y.; Standley, D.M.; et al. Malt1-Induced Cleavage of Regnase-1 in CD4+ Helper T Cells Regulates Immune Activation. Cell 2013, 153, 1036–1049. https://doi.org/10.1016/j.cell.2013.04.034. [CrossRef]
- Chan, J.K.; Greene, W.C. Dynamic Roles for NF-κB in HTLV-I and HIV-1 Retroviral Pathogenesis: Pathogenic Human Retroviruses Subvert NF-κB. Immunological Reviews 2012, 246, 286–310. https://doi.org/10.1111/j.1600-065X.2012.01094.x. [CrossRef]
- Manghera, M.; Douville, R.N. Endogenous Retrovirus-K Promoter: A Landing Strip for Inflammatory Transcription Factors? Retrovirology 2013, 10, 16. https://doi.org/10.1186/1742-4690-10-16. [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef Promotes Infection by Excluding SERINC5 from Virion Incorporation. Nature 2015, 526, 212–217. https://doi.org/10.1038/nature15399. [CrossRef]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. https://doi.org/10.1038/nature15400. [CrossRef]
- Zeng, C.; Waheed, A.A.; Li, T.; Yu, J.; Zheng, Y.-M.; Yount, J.S.; Wen, H.; Freed, E.O.; Liu, S.-L. SERINC Proteins Potentiate Antiviral Type I IFN Production and Proinflammatory Signaling Pathways. Sci. Signal. 2021, 14, eabc7611. https://doi.org/10.1126/scisignal.abc7611. [CrossRef]
- Di Giorgio, E.; Xodo, L.E. Endogenous Retroviruses (ERVs): Does RLR (RIG-I-Like Receptors)-MAVS Pathway Directly Control Senescence and Aging as a Consequence of ERV De-Repression? Front. Immunol. 2022, 13, 917998. https://doi.org/10.3389/fimmu.2022.917998. [CrossRef]
- Min, X.; Zheng, M.; Yu, Y.; Wu, J.; Kuang, Q.; Hu, Z.; Ouyang, L.; Lu, S.; Zhao, M. Ultraviolet Light Induces HERV Expression to Activate RIG-I Signalling Pathway in Keratinocytes. Experimental Dermatology 2022, exd.14568. https://doi.org/10.1111/exd.14568. [CrossRef]
- Mikhalkevich, N.; O’Carroll, I.P.; Tkavc, R.; Lund, K.; Sukumar, G.; Dalgard, C.L.; Johnson, K.R.; Li, W.; Wang, T.; Nath, A.; et al. Response of Human Macrophages to Gamma Radiation Is Mediated via Expression of Endogenous Retroviruses. PLoS Pathog 2021, 17, e1009305. https://doi.org/10.1371/journal.ppat.1009305. [CrossRef]
- Liu, X.; Liu, Z.; Wu, Z.; Ren, J.; Fan, Y.; Sun, L.; Cao, G.; Niu, Y.; Zhang, B.; Ji, Q.; et al. Resurrection of Endogenous Retroviruses during Aging Reinforces Senescence. Cell 2023, 186, 287-304.e26. https://doi.org/10.1016/j.cell.2022.12.017. [CrossRef]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-Usage-Based Inhibition of HIV Protein Synthesis by Human Schlafen 11. Nature 2012, 491, 125–128. https://doi.org/10.1038/nature11433. [CrossRef]
- Yang, S.; Zhan, Y.; Zhou, Y.; Jiang, Y.; Zheng, X.; Yu, L.; Tong, W.; Gao, F.; Li, L.; Huang, Q.; et al. Interferon Regulatory Factor 3 Is a Key Regulation Factor for Inducing the Expression of SAMHD1 in Antiviral Innate Immunity. Sci Rep 2016, 6, 29665. https://doi.org/10.1038/srep29665. [CrossRef]
- Pauli, E.-K.; Schmolke, M.; Hofmann, H.; Ehrhardt, C.; Flory, E.; Münk, C.; Ludwig, S. High Level Expression of the Anti-Retroviral Protein APOBEC3G Is Induced by Influenza A Virus but Does Not Confer Antiviral Activity. Retrovirology 2009, 6, 38. https://doi.org/10.1186/1742-4690-6-38. [CrossRef]
- Bailey, J.R.; Williams, T.M.; Siliciano, R.F.; Blankson, J.N. Maintenance of Viral Suppression in HIV-1–Infected HLA-B*57+ Elite Suppressors despite CTL Escape Mutations. The Journal of Experimental Medicine 2006, 203, 1357–1369. https://doi.org/10.1084/jem.20052319. [CrossRef]
- Mens, H.; Kearney, M.; Wiegand, A.; Shao, W.; Schønning, K.; Gerstoft, J.; Obel, N.; Maldarelli, F.; Mellors, J.W.; Benfield, T.; et al. HIV-1 Continues To Replicate and Evolve in Patients with Natural Control of HIV Infection. J Virol 2010, 84, 12971–12981. https://doi.org/10.1128/JVI.00387-10. [CrossRef]
- Salgado, M.; Brennan, T.P.; O’Connell, K.A.; Bailey, J.R.; Ray, S.C.; Siliciano, R.F.; Blankson, J.N. Evolution of the HIV-1 Nefgene in HLA-B*57 Positive Elite Suppressors. Retrovirology 2010, 7, 94. https://doi.org/10.1186/1742-4690-7-94. [CrossRef]
- O’Connell, K.A.; Brennan, T.P.; Bailey, J.R.; Ray, S.C.; Siliciano, R.F.; Blankson, J.N. Control of HIV-1 in Elite Suppressors despite Ongoing Replication and Evolution in Plasma Virus. J Virol 2010, 84, 7018–7028. https://doi.org/10.1128/JVI.00548-10. [CrossRef]
- Pernas, M.; Tarancón-Diez, L.; Rodríguez-Gallego, E.; Gómez, J.; Prado, J.G.; Casado, C.; Dominguez-Molina, B.; Olivares, I.; Coiras, M.; León, A.; et al. Factors Leading to the Loss of Natural Elite Control of HIV-1 Infection. J Virol 2018, 92, e01805-17. https://doi.org/10.1128/JVI.01805-17. [CrossRef]
- Boritz, E.A.; Darko, S.; Swaszek, L.; Wolf, G.; Wells, D.; Wu, X.; Henry, A.R.; Laboune, F.; Hu, J.; Ambrozak, D.; et al. Multiple Origins of Virus Persistence during Natural Control of HIV Infection. Cell 2016, 166, 1004–1015. https://doi.org/10.1016/j.cell.2016.06.039. [CrossRef]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and Interferons: Who’s Interfering with Whom? Nat Rev Microbiol 2015, 13, 403–413. https://doi.org/10.1038/nrmicro3449. [CrossRef]
- Utay, N.S.; Douek, D.C. Interferons and HIV Infection: The Good, the Bad, and the Ugly. PAI 2016, 1, 107. https://doi.org/10.20411/pai.v1i1.125. [CrossRef]
- Russ, E.; Iordanskiy, S. Endogenous Retroviruses as Modulators of Innate Immunity. Pathogens 2023, 12, 162. https://doi.org/10.3390/pathogens12020162. [CrossRef]
- Côrtes, F.H.; Passaes, C.P.B.; Bello, G.; Teixeira, S.L.M.; Vorsatz, C.; Babic, D.; Sharkey, M.; Grinsztejn, B.; Veloso, V.; Stevenson, M.; et al. HIV Controllers With Different Viral Load Cutoff Levels Have Distinct Virologic and Immunologic Profiles. JAIDS Journal of Acquired Immune Deficiency Syndromes 2015, 68, 377–385. https://doi.org/10.1097/QAI.0000000000000500. [CrossRef]
- Caetano, D.G.; Ribeiro-Alves, M.; Hottz, E.D.; Vilela, L.M.; Cardoso, S.W.; Hoagland, B.; Grinsztejn, B.; Veloso, V.G.; Morgado, M.G.; Bozza, P.T.; et al. Increased Biomarkers of Cardiovascular Risk in HIV-1 Viremic Controllers and Low Persistent Inflammation in Elite Controllers and Art-Suppressed Individuals. Sci Rep 2022, 12, 6569. https://doi.org/10.1038/s41598-022-10330-9. [CrossRef]
- Hunt, P.W.; Brenchley, J.; Sinclair, E.; McCune, J.M.; Roland, M.; Page-Shafer, K.; Hsue, P.; Emu, B.; Krone, M.; Lampiris, H.; et al. Relationship between T Cell Activation and CD4 + T Cell Count in HIV-Seropositive Individuals with Undetectable Plasma HIV RNA Levels in the Absence of Therapy. J INFECT DIS 2008, 197, 126–133. https://doi.org/10.1086/524143. [CrossRef]
- Krishnan, S.; Wilson, E.M.P.; Sheikh, V.; Rupert, A.; Mendoza, D.; Yang, J.; Lempicki, R.; Migueles, S.A.; Sereti, I. Evidence for Innate Immune System Activation in HIV Type 1–Infected Elite Controllers. The Journal of Infectious Diseases 2014, 209, 931–939. https://doi.org/10.1093/infdis/jit581. [CrossRef]
- Pereyra, F.; Lo, J.; Triant, V.A.; Wei, J.; Buzon, M.J.; Fitch, K.V.; Hwang, J.; Campbell, J.H.; Burdo, T.H.; Williams, K.C.; et al. Increased Coronary Atherosclerosis and Immune Activation in HIV-1 Elite Controllers. AIDS 2012, 26, 2409–2412. https://doi.org/10.1097/QAD.0b013e32835a9950. [CrossRef]
- Noel, N.; Boufassa, F.; Lécuroux, C.; Saez-Cirion, A.; Bourgeois, C.; Dunyach-Remy, C.; Goujard, C.; Rouzioux, C.; Meyer, L.; Pancino, G.; et al. Elevated IP10 Levels Are Associated with Immune Activation and Low CD4+ T-Cell Counts in HIV Controller Patients. AIDS 2014, 28, 467–476. https://doi.org/10.1097/QAD.0000000000000174. [CrossRef]
- Platten, M.; Jung, N.; Trapp, S.; Flossdorf, P.; Meyer-Olson, D.; Schulze Zur Wiesch, J.; Stephan, C.; Mauss, S.; Weiss, V.; Von Bergwelt-Baildon, M.; et al. Cytokine and Chemokine Signature in Elite Versus Viremic Controllers Infected with HIV. AIDS Research and Human Retroviruses 2016, 32, 579–587. https://doi.org/10.1089/aid.2015.0226. [CrossRef]
- Neuhaus, J.; Jacobs, Jr, D.R.; Baker, J.V.; Calmy, A.; Duprez, D.; La Rosa, A.; Kuller, L.H.; Pett, S.L.; Ristola, M.; Ross, M.J.; et al. Markers of Inflammation, Coagulation, and Renal Function Are Elevated in Adults with HIV Infection. J INFECT DIS 2010, 201, 1788–1795. https://doi.org/10.1086/652749. [CrossRef]
- Deeks, S.G. HIV Infection, Inflammation, Immunosenescence, and Aging. Annu. Rev. Med. 2011, 62, 141–155. https://doi.org/10.1146/annurev-med-042909-093756. [CrossRef]
- Hocini, H.; Bonnabau, H.; Lacabaratz, C.; Lefebvre, C.; Tisserand, P.; Foucat, E.; Lelièvre, J.-D.; Lambotte, O.; Saez–Cirion, A.; Versmisse, P.; et al. HIV Controllers Have Low Inflammation Associated with a Strong HIV-Specific Immune Response in Blood. J Virol 2019, 93, e01690-18. https://doi.org/10.1128/JVI.01690-18. [CrossRef]
Figure 1.
The ERVK-6 mRNA levels are downregulated in HIC. Boxplots represent the interquartile and sample median (central solid black line) of the relative changes (fold-change values relative to the mean of HIV-1-uninfected (NEG) subjects) of ERVK-6 in HIC (A) and VC and EC subgroups (B) comparing with NEG and ART-suppressed subjects (ART) ERVK-6's expression. P values < 0.05 were considered statistically significant.
Figure 1.
The ERVK-6 mRNA levels are downregulated in HIC. Boxplots represent the interquartile and sample median (central solid black line) of the relative changes (fold-change values relative to the mean of HIV-1-uninfected (NEG) subjects) of ERVK-6 in HIC (A) and VC and EC subgroups (B) comparing with NEG and ART-suppressed subjects (ART) ERVK-6's expression. P values < 0.05 were considered statistically significant.
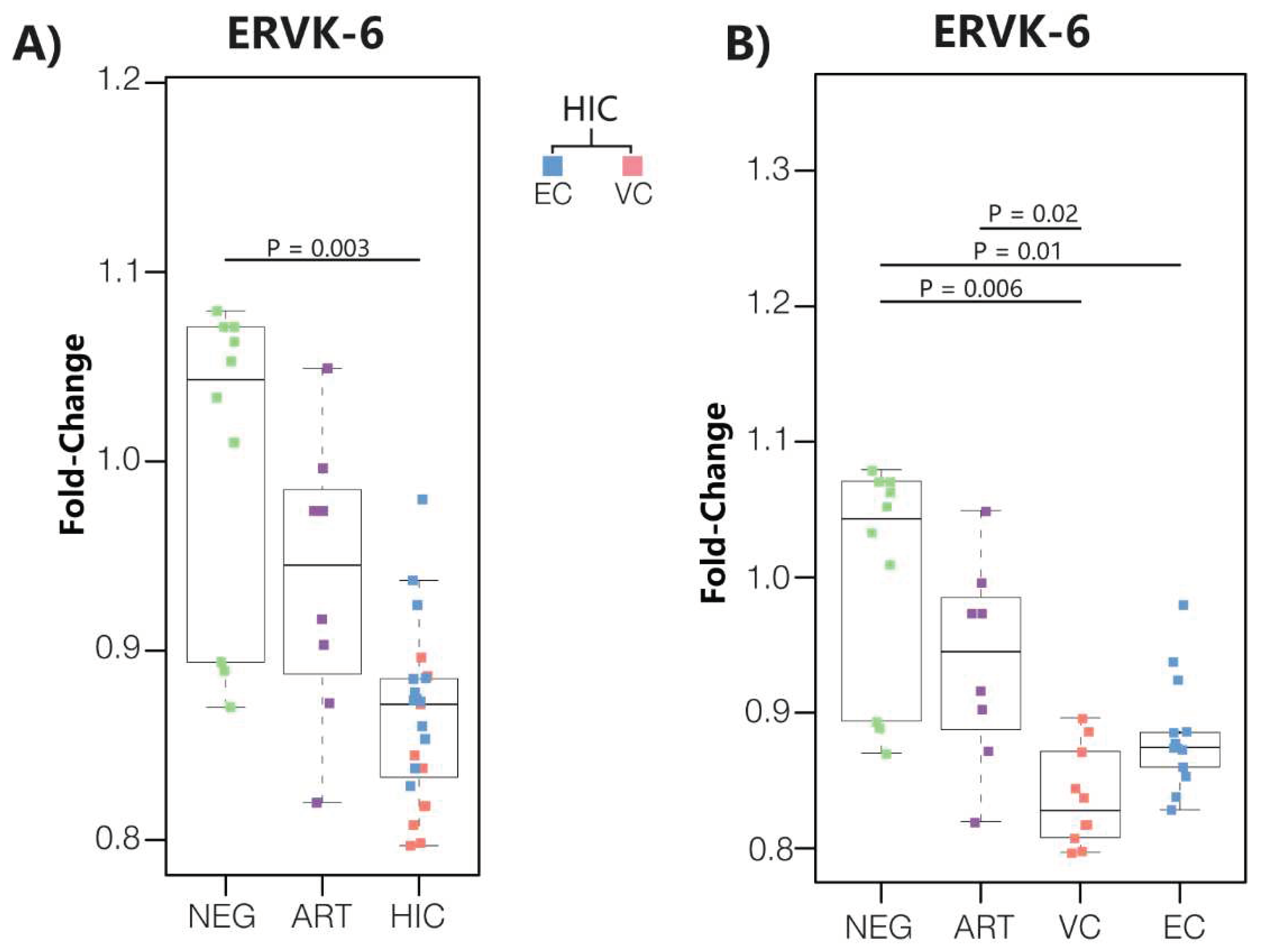
Figure 2.
Correlation between mRNA levels of ERVK-6 and RF (p21, MCPIP1, SERINC3, SERINC5, APOBEC3G, IFITM2, SAMHD1 and SLFN11) in PBMC from HIC (VC and EC) and control groups (NEG and ART). The points’ colors indicate the patient group, according to the legend. Correlation coefficients (Pearson’s ρ) are shown in each graph's upper right or left corner. P values < 0.05 were considered statistically significant.
Figure 2.
Correlation between mRNA levels of ERVK-6 and RF (p21, MCPIP1, SERINC3, SERINC5, APOBEC3G, IFITM2, SAMHD1 and SLFN11) in PBMC from HIC (VC and EC) and control groups (NEG and ART). The points’ colors indicate the patient group, according to the legend. Correlation coefficients (Pearson’s ρ) are shown in each graph's upper right or left corner. P values < 0.05 were considered statistically significant.
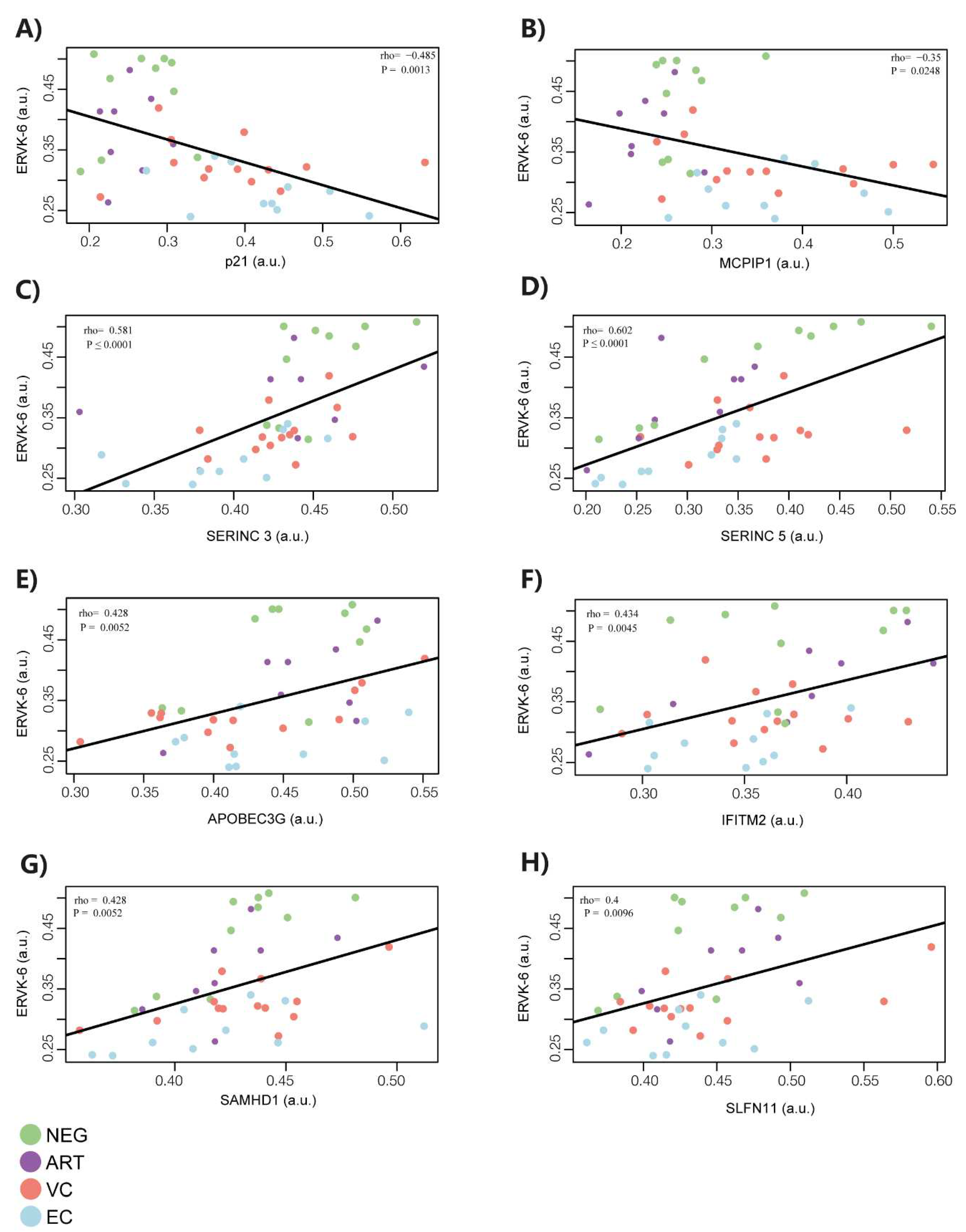
Figure 3.
(A) Correlogram representing the matrices of Pearson's rank order correlation coefficient (ρ) between RFs and ERVK-6 mRNA expression levels. (B,C) Principal Component Analysis showing the four principal components of the dataset. In (B), each circle represents one sample, and the circle's color indicates the mRNA ERVK-6 expression levels (divided into four different quartiles), according to the legend in the right corner. In (C), the circles representing each sample were colored according to groups of the study (NEG, ART, EC, and VC), as shown in the legend in the right corner.
Figure 3.
(A) Correlogram representing the matrices of Pearson's rank order correlation coefficient (ρ) between RFs and ERVK-6 mRNA expression levels. (B,C) Principal Component Analysis showing the four principal components of the dataset. In (B), each circle represents one sample, and the circle's color indicates the mRNA ERVK-6 expression levels (divided into four different quartiles), according to the legend in the right corner. In (C), the circles representing each sample were colored according to groups of the study (NEG, ART, EC, and VC), as shown in the legend in the right corner.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



