
Submitted:
25 September 2023
Posted:
27 September 2023
You are already at the latest version
Abstract
In the present work, we report on a 3D MOF of {[Pb5(3-OH)(3-NO3)3(6-pmdc)3·H2O}n formula (pmdc = pyrimidine-4,6-dicarboxylate) synthesized by an oven-heated solvent-free procedure. The large connectivity afforded by the three ligands in their coordination to lead(II) ions grows cubic building units characterized by a central Pb atom with an unusual coordination index of 12 and six pmdc ligands occupying the faces. These cubic units are linked one another giving rise to a quite condensed structure that represents an unprecedented topology showing the (4.62)6(43)2(45.610)3(45.68.82)6(46.69)6(612.83) point symbol. The crystalline material has been characterized by routine physico-chemical techniques to confirm its purity and its thermal behaviour has been also studied by thermogravimetric and thermodiffractometric analyses. The solid presents greenish blue photoluminescent emission based on pmdc ligands, as revealed time-dependent density-functional theory (TD-DFT) calculations, by that is substantially more intense than in free H2pmdc ligand according to its improved quantum yield. The emissive capacity of the material is further analysed according to decreasing temperature of the polycrystalline sample, finding that sizeable long-lasting phosphorescence is present.
Keywords:
lead(II)
; metal-organic framework
; pyrimidine-4
; 6-dicarboxylate
; new topology
; photoluminescence properties
; time-dependent density-functional theory
1. Introduction
During the last two decades, the preparation and characterization of metal-organic frameworks (MOFs) [1] has received an increasing attention because these materials possess outstanding microporosity consisting of specific surface areas higher than 7000 m2 g- 1 but also pore volumes larger than 4 cm3 g–1 [2,3]. That structural feature is also accompanied by a wide topological diversity [4] that is not only capable for modulating the pore volume and size but also the surface functionalities according to the selection of the basic constituents of the framework: metal ions and organic ligands selection [5,6,7]. Nowadays, in spite of all the steps taken on structural prediction from the coordination characteristics between most of metal ions (mainly transition metal ions) and several chemical functions of ligands [8], controlling the final framework is still challenging particularly for representative elements given their usually large coordination environments [9], a fact that prevents the simplification of the networks on the basis of nodes and spacers (foundations of reticular chemistry) [10,11,12,13]. In fact, synthetic efforts in designing novel coordination polymers have primarily focused on the use of s-, d- or f-block metal ions [14,15,16,17,18], while less attention has been paid to p-block ions despite their significance in working fields such as electroluminescence, photovoltaic conversion, fluorescent sensors, and organic light-emitting diodes [19,20,21]. As a heavy p-block metal ion, lead(II) presents a large ion radius, flexible coordination environment, and variable stereochemical activity, which provides unique opportunities for achieving unusual architectures with uncommon properties [22,23].
On another level, the presence of both ligands and metal ions disposed in an ordered manner along the framework imbues these materials with other relevant properties such as magnetism,[24,25,26], catalysis [27], electronics [28], spintronics [29,30] and photoluminescence (PL) [19,31]. In particular, luminescent CPs are currently an important branch of these sorts of compounds and are known to provide tunable emission derived from not only their framework but also their interplay with surrounding medium (solvents, guest molecules, composites) [32]. As a consequence, the number of applications deriving from PL properties of MOFs has been multiplied in such a way that today it serves as a basis for many purposes, such as the chemical sensing of ions, biomolecules, the pH of a solution, temperature and pressure [33,34,35,36]. Focusing on the inherent luminescence arising from the framework, the emission may be derived from metal ions and/or organic ligands. Particularizing to the latter, ligand-centered emissions are often originated from π-systems or charge transfer complexes formed between neighboring ligands in the excited state, so their presence plays an essential role in the PL properties. That emission capacity is often improved in CPs because the coordination affords rigidity to the ligand that reduces the vibrational motions leading to the quenching of PL. Moreover, the presence of metal ions lacking quenching derived from intraionic electronic transitions (such as d-d transitions occurring for most of transition elements), and/or heavy ions affording spin-orbit coupling effect stabilizing excited states may also help in promoting PL in MOFs [37].
Based on the previous ideas and persuaded by our propose to crystallize coordination polymers and MOFs with interesting PL properties [35,38,39,40,41], in the present work we are describing the preparation and characterization of a new 3D MOF based on pyrimidine-4,6-dicarboxylate (pmdc) ligand and a lead(II). Such a combination can be considered as a logical step in view of our previous success of pmdc to give rise to luminescent compounds with zinc(II) and lanthanide(III) ions [42,43,44,45]. On the present case, we employ lead(II) with the aim of generating an unusual topological framework on the basis of its great coordination capacity to carboxylate groups and the premise suggesting that such a heavy ion might afford good PL properties to the material. A complete computational study is also performed to shed light on those properties.
2. Results and Discussion
2.1. Solvent-Free Synthetic Approach
The present lead-based MOF has been obtained through a less habitual synthetic procedure, coined by us and others as solvent-free, which has been largely used in the preparation of organic molecules [46,47] but scarcely employed in the case of metal-organic compounds [48,49]. Nonetheless, this alternative procedure to the main solvothermal method, which consists of the mechano-chemical mixture of the reagents followed by thermal treatment, was successfully employed from 2012 on in the preparation of metal-azolate MOFs starting from metal oxide or hydroxides [50,51]. As observed in those works, solvent-free procedure has shown to render pure and crystalline materials in a very high yield, in such a way that it may consider as a cheaper and environmentally-friendly approach compared to those methods (evaporation or solvothermal methods) that require an organic solvent. When we first employed this methodology, we observed that its success is mainly due exclusively to the reagents used, particularly with regard to their ease to form a melt during the acid-base reaction taking place. As an example, diazole/triazole-like ligands are characterized for low melting points that favour the formation of melts in which metal oxides and hydroxides are reacted. The latter metal sources count on the advantage of consisting of anions that cannot remain in the mixture as impurities [52]. Accordingly, we focused on other reagents to reproduce the formation of the melt by selecting metal salts that are melted upon relatively low temperature to extend the use of the solvent-free procedure to ligands possessing high melting point such as pmdc [45,53]. For instance, the use of hygroscopic metal salts (often hydrated salts) in combination with organic carboxylic acids may lead, after being ground, sealed and heated, to the complete mixture of the reagents and the presence of enough water resulting from the acid-base reaction to allow for the crystallization of a MOF. Moreover, some anions such as nitrates and acetates acknowledge the capacity to be decomposed during the heating process to avoid their presence in the final product [54].
On that basis, the synthesis followed in the present case has been carefully selected to meet all the previous keypoints. by mixing Lead(II) nitrate and pyrimidine-4,6-dicarboxylic acid are mixed in a mortar and, once smoothly hand-ground, the resulting mixture is placed into an airtight glass vessel that is oven-heated for 3 days at 150 °C to allow the crystallization of a MOF while the nitric acid formed during the acid-base reaction is decomposed and released when the vessel is open at room temperature.
2.2. Structural Description of {[Pb5(μ3-OH)(μ3-NO3)3(μ6-pmdc)3]·H2O}n (1)
Compound 1 crystallises in the Rc space group and consists of a highly condensed 3D framework where the Pb(II) atoms are linked through μ6-pmdc ligands in addition to the μ3-hydroxide and μ3-nitrate anions. The asymmetric unit contains three different lead atoms, each one showing a different coordination environment. The Pb1 atom shows a heptacoordinated NO6Pb chromophore that best resembles a capped trigonal prism though it is also close to a capped octahedron [S(ctpr) = 2.16 / S(coc) = 2.87] according to the continuous shape measurements calculated with SHAPE program [55]. The environment is formed by one chelating and three monodentate pmdc ligands, a nitrate anion, and the hydroxide anion. The Pb2 atom coordinates to two N,O-chelating pmdc ligands, two nitrate anions, and to two monodentate pmdc ligands with slightly larger bond distances (Table 1), thus rendering an octacoordinated polyhedron resembling a slightly distorted square antiprism [S(sapr) = 1.77]. At last, the coordination sphere of the Pb3 atom, meanwhile, is fully occupied by twelve nitrate oxygen atoms leading to a distorted cuboctahedron [S(coc) = 1.83] (Figure 1). Despite the astonishing coordination environment with that high coordination number, it must be remarked that such a geometry has been previously reported for other lead(II)-based complexes [56,57,58] deposited in the CCDC [59], although this is the first example in which such a fragment pertains to an extended network of a MOF.
On its part, the crystallographically unique pmdc ligand exhibits a previously non-reported κ2N11,O171:κ2N13,O181:κO171:κO172:κO182:κO182 coordination mode in which it is coordinated to six Pb centres. In detail, pmdc acts as a bridge between equivalent Pb1 and Pb2 atoms imposing Pb···Pb distances ranging from 4.04 to 7.22 Å depending on the ligand group involved (only the carboxylate group or the carboxylate and also the pyrimidine ring). As a consequence of such tangle of links, cubic building units are formed, whose faces are occupied by the pmdc ligands and metal ions decorate the vertexes and edges. The Pb3 atom is placed in the centre of the cube linking the Pb1 and Pb2 atoms through the nitrato bridging ligands. It is also worth mentioning that Pb1 atoms exhibit a large mutual connectivity being connected themselves by means of the hydroxide anion that dispose them in a triangular shape (Figure 2).
The cubic building units are self assembled by sharing the edges in such a way that each one is surrounded by twelve surrounding units, giving rise to a highly condensed 3D framework that consists of a six-nodal network [(3-c)2(3-c)6(6-c)6(6-c)6(6-c)3(6-c) stoichiometry] possessing a (4.62)6(43)2(45.610)3(45.68.82)6(46.69)6(612.83) point symbol, which is a new topology that has been registered as jcr4 in the TOPOS database [60]. The crystallisation water molecules are occluded within isolated small voids that are generated in the framework between the pmdc ligands of two adjacent cubes, being highly disordered without any remarkable interaction with the 3D crystal building (Figure 3).
2.3. Thermal Behaviour
The thermogravimetric measurement was performed in polycrystalline sample of compound 1 as an additional characterization to verify the chemical formula (Figure 4). Starting from room temperature, the compound does not exhibit any mass loss upon heating up to 100 °C despite the presence of a crystallisation water molecule. However, the TG curve experiments a very slight decrease in the 150–300 °C range whose mass loss corresponds to a water molecule. Therefore, it can be confirmed that compound 1 possesses a complete lattice water molecule per formula unit (calc. 1.0, exp. 1.2% of mass loss), which finds it difficult to be released from the framework since it is occluded within the isolated pores. In the thermodiffractometric study conducted over sample from the same batch, it is observed that the compound preserves its crystal structure because the diffractograms are practically unchanged up to 210 °C. From this temperature on, the PXRD patterns show a significant shift of most of their maxima to higher 2θ angles, which is consistent with the gradual loss of the lattice molecules and subsequent compression of the framework. The loss of water, which presents a weak endothermic character in view of the DTA curve, is followed by the structural collapse in which pmdc ligand and nitrate and hydroxide anions are decomposed. This step leads to PbO as final residue of the combustion (cald. 36.4, exp. 37.0% of mass remained in the experiment) above 400 °C after two successive exothermic processes.
To end up with the thermal characterization, just to mention that the dehydrated amorphous compound obtained at ca. 300 °C cannot revert back into the pristine material neither by gas diffusion nor by water soaking procedures. In the first one, the anhydrous powder was placed into a closed vessel containing an isolated open vessel of water, in such a way that water saturated atmosphere is generated. In the second one, the product is soaked in water for 6 h and filtered. Amorphous products were obtained in both experiments, confirming the irreversibility of the thermal dehydration, which is in agreement with the possible structural collapse occurring to the compound.
2.4. Photoluminescence Properties
We explored the photoluminescence properties of compound 1 in view of the good emissive properties previously shown by CPs based on pmdc ligand [44,45,61]. To start with, the emission spectrum recorded at room temperature on polycrystalline sample (Figure 5a) presents a wide band covering the whole visible spectrum that is composed of two main contributions with the maxima peaking at ca. 460 and 550 nm. Focusing on the wavelength of the emission maximum, the excitation spectrum reveals the presence of a very weak band in the 300-360 nm range followed by the main band (peaking at 450 nm) that cannot be completely observed due to its overlap with the emission. The first excitation band is concordant with the diffuse reflectance spectrum (Figure S12) in which the occurrence of at least two bands are observed in the 220-400 nm range. Compared to the spectroscopic data of the free H2pmdc ligand (in which λex,max = 335 and λem,max = 435 nm), compound 1 shows a strong red shift of 115 nm (Figure S5), meaning that the photoluminescence properties of this compound cannot be fully explained on the basis of the usual electronic transitions of the π-π* electronic of pmdc. The emission lifetime was also measured for both emission maxima (Figure S6). The signal was very weak for the first shoulder (λem,max = 460 nm) and the lifetime very short so it had to be estimated by deconvolution with an appropriate instrument refinement file (τ = 2.8(1) ns). The maxima of λem,max = 550 nm presented a larger lifetime of 72(3) µs (averaged over two components, see ESI) estimated from the general multiexponential expression (It = A0 + A1exp(t/τ1) + A2exp(t/τ2)). Moreover, the emission quantum yield (QY) measured for compound 1 almost doubles that of the H2pmdc ligand recorded under equivalent experimental setup, being of 1.7(2)% vs 0.8(2)%, respectively.
In view of the observed spectra, TDDFT calculations were performed on a suitable model of compound 1 consisting of a monomeric anionic fragment containing all ligands, which was previously optimized at DFT level (see Computational details and Figure S7). According to the results obtained with Gaussian software (Figure 5b), the main excitations occurring in the compound are related with two theoretical electronic transitions: i) LUMO ← HOMO-5 occurring at 395 nm and ii) LUMO ← HOMO-7 at 335 nm, which would give rise to the seventh (S7) and tenth (S10) excited states. Although both excitations possess high oscillator strengths, it must be highlighted that the former is much higher than the latter (see Table S3). Moreover, both HOMO-5 and HOMO-7 orbitals are mainly located over the coordinated hydroxide and carboxylate groups, while HOMO-7 also possesses some participation of the non-bonding electrons of Pb(II). Considering that LUMO is assigned as a π* orbital of one of the pmdc ligands, the PL excitation of this compound may have both a mixed ligand-to-metal charge transfer (LMCT) and ligand-to-ligand charge transfer (LLCT) origin. These results mean that, under the employed experimental setup with the λex = 325 nm laser excitation, and though the absorbed energy could be enough as to excite the S10 state, the S7 state could more probably be involved in the PL excitation process. In any case, given that these states are unlikely to be directly involved in the emission process according to Kasha’s rule [62,63], which anticipates the lowest-lying state to be the emission donor, we focused on the optimization of the first excited state (S1) to elucidate the theoretical luminescence energy. According to this calculation, the S1 lies 18868 cm-1 (530 nm) above the ground state, meaning that the PL emission occurs from that state and that there must be an energy transfer from S7 and S10 during the structure relaxation.
To confirm the validity of the previous calculations, we also computed the excitation and emission spectra by means of the ESD module in ORCA using the same mononuclear model. The calculated excitation spectrum shows a symmetric band peaking at 350 nm, suggesting that there is a slight blue shift of the calculated band with respect to the experimentally measured one (Figure 6), whose maximum seems to be over 400 nm although the band might be somewhat mixed with the emission. Nonetheless, the excitation band for the main emission (λem = 550 nm) may be well observed experimentally at 15 K, in which the excitation band is centred at 390 nm, thus confirming the slight blue shift for the calculated spectrum. On its part, the calculated emission spectrum reproduces very well the band maximum of the experimental measurement with a small blue shifted of ∆ = 15 nm. However, the shape is not that accurate although the calculated spectrum presents a distinguishable shoulder in the region of 460 nm.
The PL characterization of the material was also conducted at low temperature by placing the sample into a close cryostat linked to a helium pump. The emission spectrum recorded at the same conditions (λex = 325 nm) resembles that of RT although the first shoulder (λem = 460 nm) becomes a well-defined and somewhat structured band (with higher relative intensity compared to RT, Figure S8). Moreover, the main band, at λem,max = 525 nm at 15 K, is somewhat split in such a way that the resulting wide maxima is broadened and covers the region of 500-600 nm. The occurrence of the band at 600 nm as well as the increasing intensity of the band at 460 nm seems to be a consequence of the decreasing non-radiative vibrational quenching due to the low temperature, because the excitation spectra collected at all emission wavelengths show the same pattern (Figure S9). This fact means that there is no significant change in the radiative pathways by the effect of the temperature. The experimental lifetimes were also estimated for the two main bands of the emission spectrum for comparative purposes. Surprisingly, the first band (λem = 460 nm) consists of a slightly shorter fluorescent process (τ = 0.8(2) ns), an opposite result to the usual trend by which lowering the temperature brings an enlargement of the lifetimes due to a decrease of the vibrational quenching (Figure S10). However, this unexpected behaviour may be understood with the analysis of the second and dominant band of λem,max = 525 nm, for which the average lifetime is substantially lengthened (2337(56) µs at 15 K vs 72(3) µs at RT). Such a large lifetime, in the range of milliseconds, may be classified within the low part of long-lived phosphorescence [19], an emission that leads to an afterglow that is traced by human eye once the excitation source is turned off. The large difference in the emission lifetime between the first and second bands is clearly observed in the time-resolved emission spectra (TRES) experiment conducted at low temperature (Figure S11). Immediately after the absorption of the pulsed light, the first band fully disappears and the emission spectrum consists of a unique and symmetrical band centred at 525 nm, which is maintained along the whole experiment. In this regard, it is worth noticing that the second band (at low temperature) contains at least two bands of different lifetimes given that the high intensity around 550-600 nm is rapidly vanished after 1 ms in the TRES.
3. Materials and Methods
3.1. Synthesis of {[Pb5(μ3-OH)(μ3-NO3)3(μ6-pmdc)3]·H2O}n (1)
Single crystals of compound 1 were obtained by following a solvent-free synthesis in which stoichiometric amounts of H2pmdc (0.0306 g, 0.15 mmol) and Pb(NO3)2 (0.0497 g, 0.15 mmol) were mixed and hand-grinded carefully to avoid the formation of a melt at room temperature. Then, the solid mixture was rapidly transferred into a glass tube which, once sealed, led to the hermetically closed reaction vessel. The vessel was placed in an autoclave and heated up to 150 °C for 48 h, after which it was slowly cooled down (5 °C/min) to room temperature. Finally, the product was gently washed with a water/ethanol solution to remove rests of unreacted reagent. Yield: 90-95% (based on metal). Anal. Calc. for C18H11N9O24Pb5 (%): C, 12.32; H, 0.52; N, 7.18; Pb, 59.02. Found: C, 12.68; H, 0.63; N, 7.12; Pb, 59.15.
It is worth noting that performing the synthesis in the 150-210 °C range also leads to pure sample of the compound but in the form of polycrystalline powder.
3.2. Physical Measurements
Elemental analysis (C, H, N) was performed on the polycrystalline sample of compound 1 by using a Euro EA Elemental Analyser while the lead content of the sample was determined by means of ICP-AES measurements performed on a Horiba Yobin Yvon Activa spectrometer. FTIR spectrum was collected in the 4000-400 cm-1 spectral region and measured on a FTIR 8400S Shimadzu spectrometer using KBr pellets. The study of the thermal analysis (TG/DTA) was performed on a TA Instruments SDT 2960 instrument under synthetic air (79% N2/21% O2), applying a heating rate of 5 °C/min.
3.3. X-ray Diffraction Data Collection and Structure Determination
Single crystal X-ray diffraction (SCXRD) data collection was acquired at 100(2) K on an Agilent Technologies Supernova diffractometer (λCu-κα = 1.5418 Å) and the data reduction was performed by means of CrysAlisPro program [64]. The crystal structure was solved by direct methods using SHELXT program [65] and refined with the Olex2 (1.5 version) software [66] including all reflections by means of full-matrix least-squares. Hydrogen atoms were located in the difference Fourier map but their position was fixed using riding models involving isotropic thermal displacement parameters of 120% those of their parent atoms, except for the hydroxide anion, in which thermal ellipsoids of 150% were applied. Moreover, the lattice water molecule could not be properly refined because it showed an extremely large displacement parameter owing to its partial absence in the crystal as confirmed by thermal gravimetric analysis (see ESI). CCDC 2295990contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).

Table 2.
Single crystal X-ray diffraction data and structure refinement details of compound 1.
| Compound | 1 |
|---|---|
| Empirical formula | C18H9N9O23Pb5 |
| Formula weight (g mol-1) | 1755.31 |
| Crystal system | Trigonal |
| Space group | Rc |
| a (Å) | 14.3004(2) |
| c (Å) | 53.369(1) |
| V (Å3) | 9451.8(3) |
| Z | 6 |
| Reflections collected | 1944 |
| Unique data/parameters | 1926/164 |
| Rint | 0.0549 |
| GoF (S)a | 1.260 |
| R1b/wR2 [I > 2σ(I)]c | 0.0521/0.1169 |
| R1b/wR2 [all]c | 0.0524/0.1170 |
a S = [∑w(F02 – Fc2)2 / (Nobs – Nparam)]1/2. b R1 = ∑||F0|–|Fc|| / ∑|F0|; c wR2 = [∑w(F02 – Fc2)2 / ∑wF02]1/2; w = 1/[σ2(F02) + (aP)2 + bP] where P = (max(F02,0) + 2Fc2)/3 with a = 0.0001 and b = 2663.0154.
Powder X-ray diffraction (PXRD) patterns were measured on a Philips X´PERT diffractometer using monochromatic Cu-Kα radiation (λ = 1.5418 Å) in the 5 < 2θ < 50° (2θ) range using a fixed slit, a step size of 0.02° and an acquisition time of 2.5 s per step. Pattern matching analysis of the full diffractogram was performed with FULLPROF program [67] using the cell parameters and space group from SCXRD experiment. The thermodiffractometry was conducted at room temperature and acquiring a diffractogram in the 5 < 2θ < 38° range every 20 °C (following a heating rate of 5 °C/min) in the 30-410 °C range.
3.4. Photoluminescence Measurements
Photoluminescence excitation and emission spectra were acquired on an Edinburgh Instruments FLS920 spectrometer equipped with a close cycle helium cryostat. All measurements were done under high vacuum (of ca. 10–9 mbar) to avoid the presence of oxygen or water in the sample holder. An IK3552R-G HeCd continuous laser (325 nm) was employed and a Müller-Elektronik-Optik SVX1450 Xe lamp were employed as excitation source for the steady-state measurements, whereas a pulsed led (λ = 340 nm) or laser diode LDH-P-C-375 (λ = 375 nm) was used for the decay curves. The fluorescence counts were detected on a photomultiplier tube (PMT) coupled to the spectrometer. The overall quantum yield (%) was measured in solid state and at room temperature by means of a Horiba Quanta–φ integrating sphere using an Oriel Instruments MS257 lamp as excitation source and an iHR550 spectrometer from Horiba to analyze the emission. Photographs taken on irradiated polycrystalline samples of compound 1 were performed with an Olympus optical microscope illuminated with a Hg lamp and equipped with a CCD detector.
3.5. Computational Calculations
Computational calculations were carried out on an excerpt cut from X-ray coordinates of compound 1 consisting of a central Pb(II) ion containing all the complete ligand molecules. The model, possessing the [Pb(pmdc)4(OH)(NO3)]8- formula, was optimized with Gaussian 16 package [68] keeping some constraints for the bonds and angles of the coordination shell as well as some dihedral angles of the pmdc ligand and performing a frequency calculation to confirm that the geometry corresponds to an energy minimum. These calculations were conducted with DFT with the hybrid B3LYP functional [69,70] and the 6-31G+(d) basis set [71] for all atoms except for Pb(II), for which the LANL2DZ [72] basis set along, and the corresponding effective core potential (ECP) was employed. PL spectra were calculated with two different strategies. On the one hand, the 70 lowest excitation states were calculated by the TD-DFT method implemented in Gaussian 16 package to identify the main excitations during the absorption process. The results were analysed with GaussSum [73] and molecular orbitals plotted with GaussView 6 [74]. On the other hand, the ground and first excited states were also optimized with ORCA 5.0.3 program [75,76] at DFT level of theory with B3LYP and employing a ZORA Hamiltonian in order to account for relativistic effects [77] as well as the atom-pairwise dispersion correction with the Becke-Johnson damping scheme (D3BJ) [78,79]. Several constraints were added in order to freeze most of free rotations of the non-coordinating pmdc ligands in the employed model, in order to mimic the real situation of the 3D crystal. The appropriate ZORA-def2-TZVP basis set was selected for C, H, N, and O atoms, whereas a SARC-ZORA-TZVP basis set for Pb, including coulomb and exchange contributions [80,81]. Then, excitation and emission spectra were computed with the modules using the hessians of the optimized geometries of the S0 and S1 states and the spin–orbit coupling (SOC) matrix elements.
4. Conclusions
A 3D metal-organic framework, with {[Pb5(μ3-OH)(μ3-NO3)3(μ6-pmdc)3]·H2O}n (1) formula, has been crystallized by reacting pyrimidine-4,6-dicarboxylic acid with lead(II) nitrate with a mechano-thermal procedure in the absence of a solvent. Compound 1 is obtained as a pure product because both the main byproduct (nitric acid) and rests of unreacted reagents are dissolved during the washing procedure. In addition to pmdc ligands showing a hexadentate coordination mode, nitrate and hydroxide ions present in the reaction medium also act as ligands of the MOF, which presents an intricate structure possessing an unprecedented topology with the (4.62)6(43)2(45.610)3(45.68.82)6(46.69)6(612.83) point symbol. Owing to the coordination of pmdc to Pb(II) ions in the framework, PL properties of the MOF are improved with respect to the free H2pmdc ligand, because luminescence brightness is increased and quantum yield is almost doubled up to 1.7% for compound 1. The overall panchromatic emission of the compound at room temperature is due to the occurrence of two bands of different nature in terms of emission durability, being composed of a blue fluorescent component and a yellowish green phosphorescence in view of their lifetimes. Moreover, TDDFT calculations also point out to the participation of other ligands or even the metal ion (LLCT and/or LMCT) as possible reason for the PL observed in the material. Interestingly, the MOF acquires long-lived phosphorescence at temperatures below liquid nitrogen reaching a lifetime of around 2 ms at 15 K that makes of this compound the unique pmdc-based example showing luminescent emission lasting in the range of milliseconds.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1. Packing of compound 1 along the c axis viewing direction; Figure S2. Packing of compound 1 along the a axis viewing direction; Table S1. Continuous Shape Measurements for the coordination environments for compound 1. The lowest SHAPE values are shown in bold blue, indicating best fits; Figure S3. Pattern-matching analysis of polycrystalline sample of compound 1; Table S2. Main IR absorption bands (cm–1) for the compound 1; Figure S4. Comparison of the FTIR spectra of compound 1 and the free H2pmdc ligand; Figure S5. Excitation (λem = 435 nm) and emission (λex = 335 nm) spectra of H2pmdc ligand recorded at room temperature; Figure S6. Decay curves of the emission of compound 1 at room temperature for the main emission maxima; Figure S7. Monomeric model taking from the X-ray coordinates used for the calculations of the PL properties of compound 1; Table S3. Calculated main excitation and emission energies (nm), singlet electronic transitions and associated oscillator strengths of model of compound 1 in gas phase; Figure S8. Emission spectrum of compound 1 collected at 15 K under monochromatic laser light (λex = 325 nm); Figure S9. Comparison of the low temperature excitation spectra collected over the two maxima observed in the emission of compound 1; Figure S10. Decay curves of the emission of compound 1 at low temperature for the main emission maxima; Figure S11. TRES recorded at 15 K for compound 1 showing a selection of spectra.
Author Contributions
Conceptualization, J.C.; methodology, J.C.; software, J.M.M., I.V.Y. and L.R.B.; validation, J.M.M., J.A.G., A.Z.L.; formal analysis, J.A.G. and A.R.D.; investigation, L.R.B., J.C.; resources, J.C.; data curation, O.P.C., J.A.G. and J.C.; writing—original draft preparation, L.R.B. and J.C.; writing—review and editing, A.R.D and J.C.; visualization, A.Z.L., O.P.C. and I.V.Y.; supervision, J.C.; project administration, A.R.D., J.C.; funding acquisition, A.R.D., J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Gobierno Vasco/Eusko Jaurlaritza (IT1755-22, IT1722-22 and IT1500-22) and Junta de Andalucía (ProyExcel_00386 and FQM-394). This publication is also part of the I+D+i projects of PGC2018-102052-A-C22 and PGC2018-102052-B-C21 codes, funded by MCIN/ AEI/10.13039/501100011033/ and “FEDER Una manera de hacer Europa”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
Acknowledgments
The authors are thankful for the technical and human support provided by SGIker of UPV/EHU and European funding (ERDF and ESF), and wish to acknowledge the terrific help of all reviewers of the present manuscript whose comments helped to improve the quality of the work. L.R.B. is grateful to the UPV/EHU for her predoctoral fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compound reported in this work is available from the authors.
References
- Batten, S.R.; Champness, N.R.; Chen, X.M.; Garcia-Martinez, J.; Kitagawa, S.; Öhrström, L.; O’Keeffe, M.; Suh, M.P.; Reedijk, J. Terminology of metal-organic frameworks and coordination polymers (IUPAC recommendations 2013). Pure Appl. Chem. 2013, 85, 1715–1724. [Google Scholar] [CrossRef]
- Farha, O.K.; Eryazici, I.; Jeong, N.C.; Hauser, B.G.; Wilmer, C.E.; Sarjeant, A.A.; Snurr, R.Q.; Nguyen, S.T.; Yazaydın, A.Ö.; Hupp, J.T. Metal–Organic Framework Materials with Ultrahigh Surface Areas: Is the Sky the Limit? J. Am. Chem. Soc. 2012, 134, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, L.; Wang, J. Design strategies for MOF-derived porous functional materials: Preserving surfaces and nurturing pores. J. Mater. 2021, 7, 440–459. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Yaghi, O.M. Deconstructing the Crystal Structures of Metal–Organic Frameworks and Related Materials into Their Underlying Nets. Chem. Rev. 2012, 112, 675–702. [Google Scholar] [CrossRef]
- Feng, L.; Wang, K.-Y.; Lv, X.-L.; Yan, T.-H.; Zhou, H.-C. Hierarchically porous metal–organic frameworks: synthetic strategies and applications. Natl. Sci. Rev. 2019, 7, 1743–1758. [Google Scholar] [CrossRef]
- Lu, W.; Wei, Z.; Gu, Z.Y.; Liu, T.F.; Park, J.; Park, J.; Tian, J.; Zhang, M.; Zhang, Q.; Gentle, T.; et al. Tuning the structure and function of metal-organic frameworks via linker design. Chem. Soc. Rev. 2014, 43, 5561–5593. [Google Scholar] [CrossRef]
- Allendorf, M.D.; Stavila, V. Crystal engineering, structure-function relationships, and the future of metal-organic frameworks. CrystEngComm 2015, 17, 229–246. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of metal-organic frameworks (MOFs): Routes to various MOF topologies, morphologies, and composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef] [PubMed]
- Morsali, A.; Hashemi, L. Main Group Metal Coordination Chemistry. In Main Group Metal Coordination Polymers; John Wiley & Sons, Ltd, 2017; pp. 183–204 ISBN 9781119370772.
- Yaghi, O.M. Reticular Chemistry in All Dimensions. ACS Cent. Sci. 2019, 5, 1295–1300. [Google Scholar] [CrossRef]
- Taddei, M.; Howarth, A.J.; Uemura, T. Introduction to molecular engineering in MOFs: beyond reticular chemistry. Mol. Syst. Des. Eng. 2023, 8, 700. [Google Scholar] [CrossRef]
- Seco, J.M.; Pérez-Yáñez, S.; Briones, D.; García, J.Á.; Cepeda, J.; Rodríguez-Diéguez, A. Combining Polycarboxylate and Bipyridyl-like Ligands in the Design of Luminescent Zinc and Cadmium Based Metal–Organic Frameworks. Cryst. Growth Des. 2017, 17, 3893–3906. [Google Scholar] [CrossRef]
- Fernandez, B.; Beobide, G.; Sanchez, I.; Carrasco-Marin, F.; Seco, J.M.; Calahorro, A.J.; Cepeda, J.; Rodriguez-Dieguez, A. Controlling interpenetration for tuning porosity and luminescence properties of flexible MOFs based on biphenyl-4,4 ’-dicarboxylic acid. CrystEngComm 2016, 18, 1282–1294. [Google Scholar] [CrossRef]
- García-Couceiro, U.; Castillo, O.; Cepeda, J.; Lanchas, M.; Luque, A.; Pérez-Yáñez, S.; Román, P.; Vallejo-Sánchez, D.; Garcia-Couceiro, U.; Castillo, O.; et al. Influence of the Synthetic Conditions on the Structural Diversity of Extended Manganese-Oxalato-1,2-bis(4-pyridyl)ethylene Systems. Inorg. Chem. 2010, 49, 11346–11361. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gao, Y.; Li, N.; Ge, L.; Bu, X.; Feng, P. Transition metal-based bimetallic MOFs and MOF-derived catalysts for electrochemical oxygen evolution reaction. Energy Environ. Sci. 2021, 14, 1897–1927. [Google Scholar] [CrossRef]
- García-García, A.; Toral, V.; Del Moral, J.F.Q.; Pretel, A.G.; Morales, D.P.; Salinas-Castillo, A.; Cepeda, J.; Choquesillo-Lazarte, D.; Bobinger, M.; Salmerón, J.F.; et al. Selectivity of Relative Humidity Using a CP Based on S-Block Metal Ions. Sensors 2022, 22, 1664. [Google Scholar] [CrossRef]
- Sun, S.; Zhao, Y.; Wang, J.; Pei, R. Lanthanide-based MOFs: synthesis approaches and applications in cancer diagnosis and therapy. J. Mater. Chem. B 2022, 10, 9535–9564. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, S.; Zhao, Z.-S.; Wang, Z.; Zhang, R.; Liu, L.; Han, Z.-B. Recent progress in lanthanide metal–organic frameworks and their derivatives in catalytic applications. Inorg. Chem. Front. 2021, 8, 590–619. [Google Scholar] [CrossRef]
- San Sebastian, E.; Rodríguez-Diéguez, A.; Seco, J.M.; Cepeda, J. Coordination Polymers with Intriguing Photoluminescence Behavior: The Promising Avenue for Greatest Long-Lasting Phosphors. Eur. J. Inorg. Chem. 2018, 2155–2174. [Google Scholar] [CrossRef]
- Gutiérrez, M.; Martín, C.; Souza, B.E.; Van der Auweraer, M.; Hofkens, J.; Tan, J.-C. Highly luminescent silver-based MOFs: Scalable eco-friendly synthesis paving the way for photonics sensors and electroluminescent devices. Appl. Mater. Today 2020, 21, 100817. [Google Scholar] [CrossRef]
- Karmakar, A.; Li, J. Luminescent MOFs (LMOFs): recent advancement towards a greener WLED technology. Chem. Commun. 2022, 58, 10768–10788. [Google Scholar] [CrossRef] [PubMed]
- Buston, J.E.H.; Claridge, T.D.W.; Heyes, S.J.; Leech, M.A.; Moloney, M.G.; Prout, K.; Stevenson, M. Structural investigations of a lead(iv) tetraacetate–pyridine complex. Dalton Trans. 2005, 3195–3203. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yuan, D.; Han, L.; Ma, E.; Wu, M.; Lin, Z.; Hong, M. Mono- and Bilayered Lead(II)–bpno Polymers with Unusual Low Energy Emission Properties (bpno = 4,4’-Bipyridine N,N’-Dioxide). Eur. J. Inorg. Chem. 2005, 2005, 2054–2059. [Google Scholar] [CrossRef]
- Nadar, S.S.; Rathod, V.K. Magnetic-metal organic framework (magnetic-MOF): A novel platform for enzyme immobilization and nanozyme applications. Int. J. Biol. Macromol. 2018, 120, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Mínguez Espallargas, G.; Coronado, E. Magnetic functionalities in MOFs: from the framework to the pore. Chem. Soc. Rev. 2018, 47, 533–557. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Long, J.R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2011, 2, 2078–2085. [Google Scholar] [CrossRef]
- Yang, D.; Gates, B.C. Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research. ACS Catal. 2019, 9, 1779–1798. [Google Scholar] [CrossRef]
- Mancuso, J.L.; Mroz, A.M.; Le, K.N.; Hendon, C.H. Electronic Structure Modeling of Metal–Organic Frameworks. Chem. Rev. 2020, 120, 8641–8715. [Google Scholar] [CrossRef]
- Karnieli, A.; Tsesses, S.; Bartal, G.; Arie, A. Emulating spin transport with nonlinear optics, from high-order skyrmions to the topological Hall effect. Nat. Commun. 2021, 12, 1092. [Google Scholar] [CrossRef]
- Huizi-Rayo, U.; Gutierrez, J.; Seco, J.M.; Mujica, V.; Diez-Perez, I.; Ugalde, J.M.; Tercjak, A.; Cepeda, J.; San Sebastian, E. An Ideal Spin Filter: Long-Range, High-Spin Selectivity in Chiral Helicoidal 3-Dimensional Metal Organic Frameworks. Nano Lett. 2020, 20, 8476–8482. [Google Scholar] [CrossRef]
- Cui, Y.; Yue, Y.; Qian, G.; Chen, B. Luminescent Functional Metal–Organic Frameworks. Chem. Rev. 2012, 112, 1126–1162. [Google Scholar] [CrossRef]
- Liu, J.Q.; Luo, Z.D.; Pan, Y.; Kumar Singh, A.; Trivedi, M.; Kumar, A. Recent developments in luminescent coordination polymers: Designing strategies, sensing application and theoretical evidences. Coord. Chem. Rev. 2020, 406, 213145. [Google Scholar] [CrossRef]
- Wang, S.; Sun, B.; Su, Z.; Hong, G.; Li, X.; Liu, Y.; Pan, Q.; Sun, J. Lanthanide-MOFs as multifunctional luminescent sensors. Inorg. Chem. Front. 2022, 9, 3259–3266. [Google Scholar] [CrossRef]
- Leo, P.; Briones, D.; García, J.A.; Cepeda, J.; Orcajo, G.; Calleja, G.; Rodríguez-Diéguez, A.; Martínez, F. Strontium-Based MOFs Showing Dual Emission: Luminescence Thermometers and Toluene Sensors. Inorg. Chem. 2020, 59, 18432–18443. [Google Scholar] [CrossRef] [PubMed]
- Seco, J.M.; Sebastián, E.S.; Cepeda, J.; Biel, B.; Salinas-Castillo, A.; Fernández, B.; Morales, D.P.; Bobinger, M.; Gómez-Ruiz, S.; Loghin, F.C.; et al. A Potassium Metal-Organic Framework based on Perylene-3,4,9,10-tetracarboxylate as Sensing Layer for Humidity Actuators. Sci. Rep. 2018, 8, 14414. [Google Scholar] [CrossRef]
- Pajuelo-Corral, O.; Pérez-Yáñez, S.; Vitorica-Yrezabal, I.J.; Beobide, G.; Zabala-Lekuona, A.; Rodríguez-Diéguez, A.; Seco, J.M.; Cepeda, J. A metal-organic framework based on Co(II) and 3-aminoisonicotinate showing specific and reversible colourimetric response to solvent exchange with variable magnet behaviour. Mater. Today Chem. 2022, 24, 100794. [Google Scholar] [CrossRef]
- Cepeda, J.; Rodríguez-Diéguez, A. Tuning the luminescence performance of metal-organic frameworks based on d10metal ions: From an inherent versatile behaviour to their response to external stimuli. CrystEngComm 2016, 18, 8556–8573. [Google Scholar] [CrossRef]
- Zabala-Lekuona, A.; Cepeda, J.; Oyarzabal, I.; Rodríguez-Diéguez, A.; García, J.A.; Seco, J.M.; Colacio, E. Rational design of triple-bridged dinuclear ZnIILnIII-based complexes: a structural, magnetic and luminescence study. CrystEngComm 2017, 19, 256–264. [Google Scholar] [CrossRef]
- Seco, J.M.; Rodríguez-Diéguez, A.; Padro, D.; García, J.A.; Ugalde, J.M.; San Sebastian, E.; Cepeda, J. Experimental and Theoretical Study of a Cadmium Coordination Polymer Based on Aminonicotinate with Second-Timescale Blue/Green Photoluminescent Emission. Inorg. Chem. 2017, 56, 3149–3152. [Google Scholar] [CrossRef]
- Pajuelo-Corral, O.; Razquin-Bobillo, L.; Rojas, S.; García, J.A.; Choquesillo-Lazarte, D.; Salinas-Castillo, A.; Hernández, R.; Rodríguez-Diéguez, A.; Cepeda, J.; Ochoa, S. Lanthanide(III) Ions and 5-Methylisophthalate Ligand Based Coordination Polymers: An Insight into Their Photoluminescence Emission and Chemosensing for Nitroaromatic Molecules. Nanomater. 2022, Vol. 12, Page 3977 2022, 12, 3977. [Google Scholar] [CrossRef]
- Briones, D.; Leo, P.; Cepeda, J.; Orcajo, G.; Calleja, G.; Sanz, R.; Rodríguez-Diéguez, A.; Martínez, F. Alkaline-earth metal based MOFs with second scale long-lasting phosphor behavior. CrystEngComm 2018, 20, 4793–4803. [Google Scholar] [CrossRef]
- Pajuelo-Corral, O.; García, J.A.; Castillo, O.; Luque, A.; Mendicute-Fierro, C.; Rodríguez-Diéguez, A.; Cepeda, J. A Lamellar Zn-Based Coordination Polymer Showing Increasing Photoluminescence upon Dehydration. Molecules 2023, 28, 5643. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, J.; Beobide, G.; Castillo, O.; Luque, A.; Pérez-Yáñez, S. Structural diversity of coordination compounds derived from double-chelating and planar diazinedicarboxylate ligands. Coord. Chem. Rev. 2017, 352, 83–107. [Google Scholar] [CrossRef]
- Cepeda, J.; Balda, R.; Beobide, G.; Castillo, O.; Fernández, J.; Luque, A.; Pérez-Yáñez, S.; Román, P.; Vallejo-Sánchez, D. Lanthanide(III)/Pyrimidine-4,6-dicarboxylate/Oxalate Extended Frameworks: A Detailed Study Based on the Lanthanide Contraction and Temperature Effects. Inorg. Chem. 2011, 50, 8437–8451. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, J.; Pérez-Yáñez, S.; Beobide, G.; Castillo, O.; García, J.Á.; Lanchas, M.; Luque, A. Enhancing luminescence properties of lanthanide(iii)/pyrimidine-4,6-dicarboxylato system by solvent-free approach. Dalton Trans. 2015, 44, 6972–6986. [Google Scholar] [CrossRef] [PubMed]
- Zangade, S.; Patil, P. A Review on Solvent-free Methods in Organic Synthesis. Curr. Org. Chem. 2019, 23, 2295–2318. [Google Scholar] [CrossRef]
- Tanaka, K.; Toda, F. Solvent-Free Organic Synthesis. Chem. Rev. 2000, 100, 1025–1074. [Google Scholar] [CrossRef]
- Mei, J.; Duan, A.; Wang, X. A Brief Review on Solvent-Free Synthesis of Zeolites. Mater. (Basel, Switzerland) 2021, 14, 788. [Google Scholar] [CrossRef]
- López-Cabrelles, J.; Romero, J.; Abellán, G.; Giménez-Marqués, M.; Palomino, M.; Valencia, S.; Rey, F.; Mínguez Espallargas, G. Solvent-Free Synthesis of ZIFs: A Route toward the Elusive Fe(II) Analogue of ZIF-8. J. Am. Chem. Soc. 2019, 141, 7173–7180. [Google Scholar] [CrossRef]
- Lin, J.-B.; Lin, R.-B.; Cheng, X.-N.; Zhang, J.-P.; Chen, X.-M. Solvent/additive-free synthesis of porous/zeolitic metal azolate frameworks from metal oxide/hydroxide. Chem. Commun. 2011, 47, 9185–9187. [Google Scholar] [CrossRef]
- Pichon, A.; Lazuen-Garay, A.; James, S.L. Solvent-free synthesis of a microporous metal-organic framework. CrystEngComm 2006, 8, 211–214. [Google Scholar] [CrossRef]
- Lanchas, M.; Arcediano, S.; Aguayo, A.T.; Beobide, G.; Castillo, O.; Cepeda, J.; Vallejo-Sánchez, D.; Luque, A. Two appealing alternatives for MOFs synthesis: Solvent-free oven heating vs. microwave heating. RSC Adv. 2014, 4. [Google Scholar] [CrossRef]
- Cepeda, J.; Pérez-Yáñez, S.; Beobide, G.; Castillo, O.; García, J.Á.; Luque, A. Photoluminescence Modulation in Lanthanide(III)/Pyrazine-2,5-dicarboxylato/Nitrato Frameworks. Eur. J. Inorg. Chem. 2015, 4318–4328. [Google Scholar] [CrossRef]
- Yuvaraj, S.; Fan-Yuan, L.; Tsong-Huei, C.; Chuin-Tih, Y. Thermal Decomposition of Metal Nitrates in Air and Hydrogen Environments. J. Phys. Chem. B 2003, 107, 1044–1047. [Google Scholar] [CrossRef]
- Alvarez, S.; Avnir, D.; Llunell, M.; Pinsky, M. Continuous symmetry maps and shape classification. The case of six-coordinated metal compounds. New J. Chem. 2002, 26, 996–1009. [Google Scholar] [CrossRef]
- Cramer, R.E.; Mitchell, K.A.; Hirazumi, A.Y.; Smith, S.L. Crystal structures of [Pb(NO3)6]4– and [Ba(NO3)6]4– salts of 24-pyrimidinium crown 6 {5,12,19,26,33,40-hexaamino-3,10,17,24,31,38-hexamethyl [1.6](1,5)pyrimidiniophane}. J. Chem. Soc. Dalt. Trans. 1994, 563–569. [Google Scholar] [CrossRef]
- Litecká, M.; Gyepes, R.; Vargová, Z.; Vilková, M.; Almáši, M.; Walko, M.; Imrich, J. Toxic metal complexes of macrocyclic cyclen molecule – synthesis, structure and complexing properties. J. Coord. Chem. 2017, 70, 1698–1712. [Google Scholar] [CrossRef]
- Adhikari, B.B.; Ohto, K.; Schramm, M.P. p-tert-Butylcalix[6]arene hexacarboxylic acid conformational switching and octahedral coordination with Pb(ii) and Sr(ii). Chem. Commun. 2014, 50, 1903–1905. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package topospro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Pajuelo-Corral, O.; García, J.A.; Castillo, O.; Luque, A.; Rodríguez-Diéguez, A.; Cepeda, J. Single-ion magnet and photoluminescence properties of lanthanide(III) coordination polymers based on pyrimidine-4,6-dicarboxylate. Magnetochemistry 2021, 7, 1–20. [Google Scholar] [CrossRef]
- Khan, A.U.; Kasha, M. Mechanism of four-level laser action in solution excimer and excited-state proton-transfer cases. Proc. Natl. Acad. Sci. 1983, 80, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- del Valle, J.C.; Catalán, J. Kasha{’}s rule: a reappraisal. Phys. Chem. Chem. Phys. 2019, 21, 10061–10069. [Google Scholar] [CrossRef]
- Agilent CrysAlisPro Data Collection and Processing Software for Agilent X-ray Diffractometers. Technol. UK Ltd, Yarnton, Oxford, UK 2014, 44, 1–53.
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O. V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.K.; Puschmann, H. {\it OLEX2}: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Condens. Matter 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 {R}evision {C}. 01 2016.
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 720–723. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView {V}ersion {6} 2019.
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Snijders, J.G.; Baerends, E.J. The zero-order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules. J. Chem. Phys. 1996, 105, 6505–6516. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence{,} triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Coordination shells of the crystallographically independent metal ions: (a) Pb1, (b) Pb2 and (c) Pb3. Colour codes: nitrogen, blue; oxygen, red; lead, green.
Figure 1.
Coordination shells of the crystallographically independent metal ions: (a) Pb1, (b) Pb2 and (c) Pb3. Colour codes: nitrogen, blue; oxygen, red; lead, green.
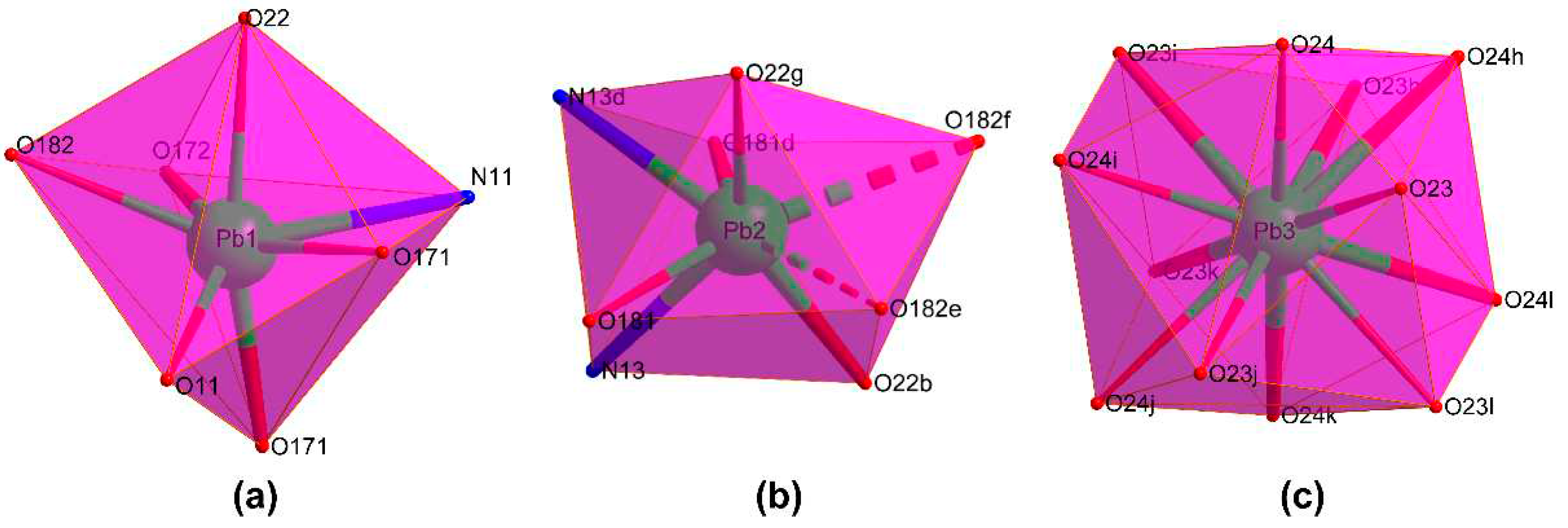
Figure 2.
Coordination modes of the ligands within cubic building unit of compound 1. Colour codes: carbon, grey; hydrogen, white; nitrogen, blue; oxygen, red; lead, green.
Figure 2.
Coordination modes of the ligands within cubic building unit of compound 1. Colour codes: carbon, grey; hydrogen, white; nitrogen, blue; oxygen, red; lead, green.
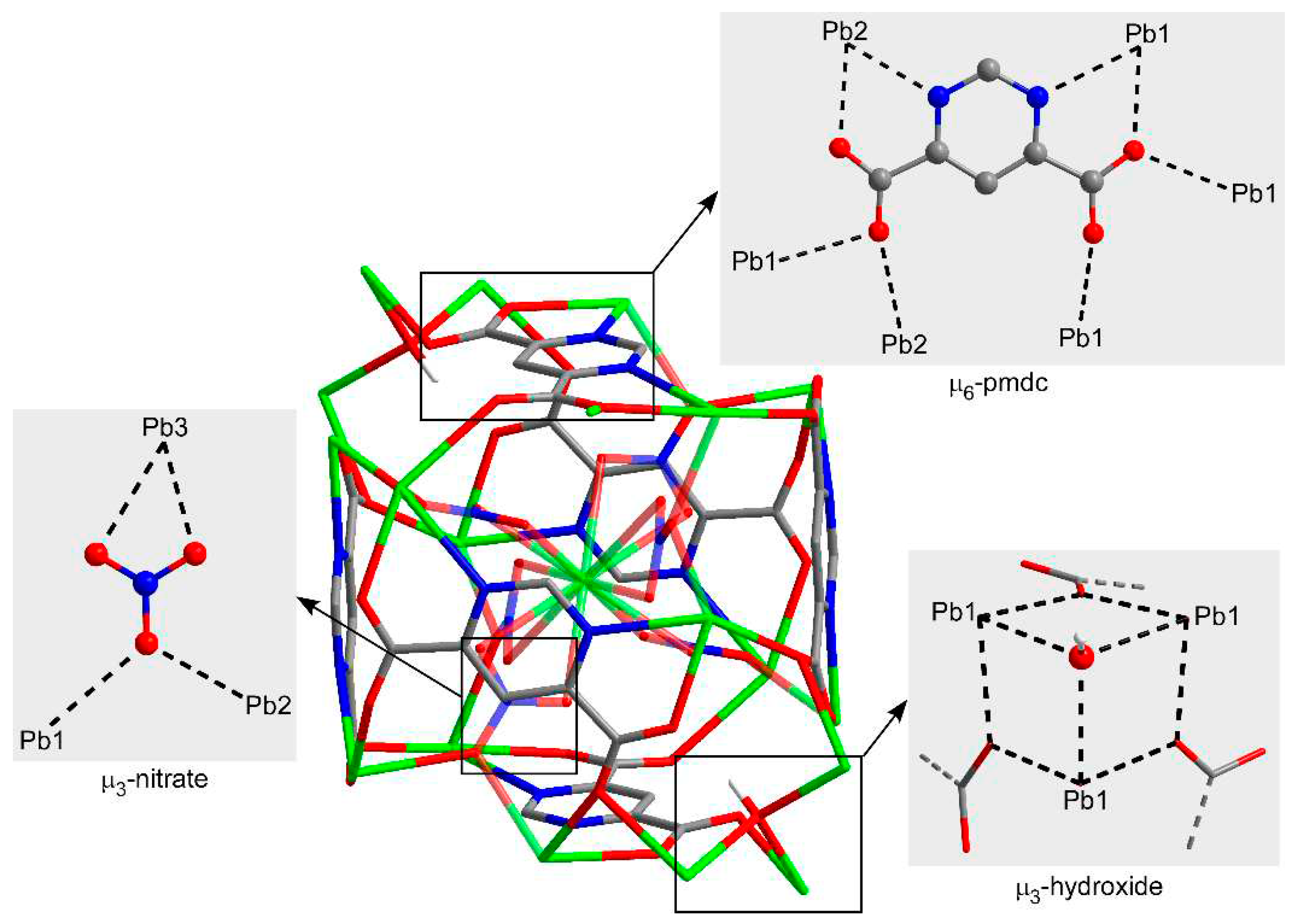
Figure 3.
Crystal packing of compound 1 showing the isolated voids (golden solid) containing water molecules.
Figure 3.
Crystal packing of compound 1 showing the isolated voids (golden solid) containing water molecules.
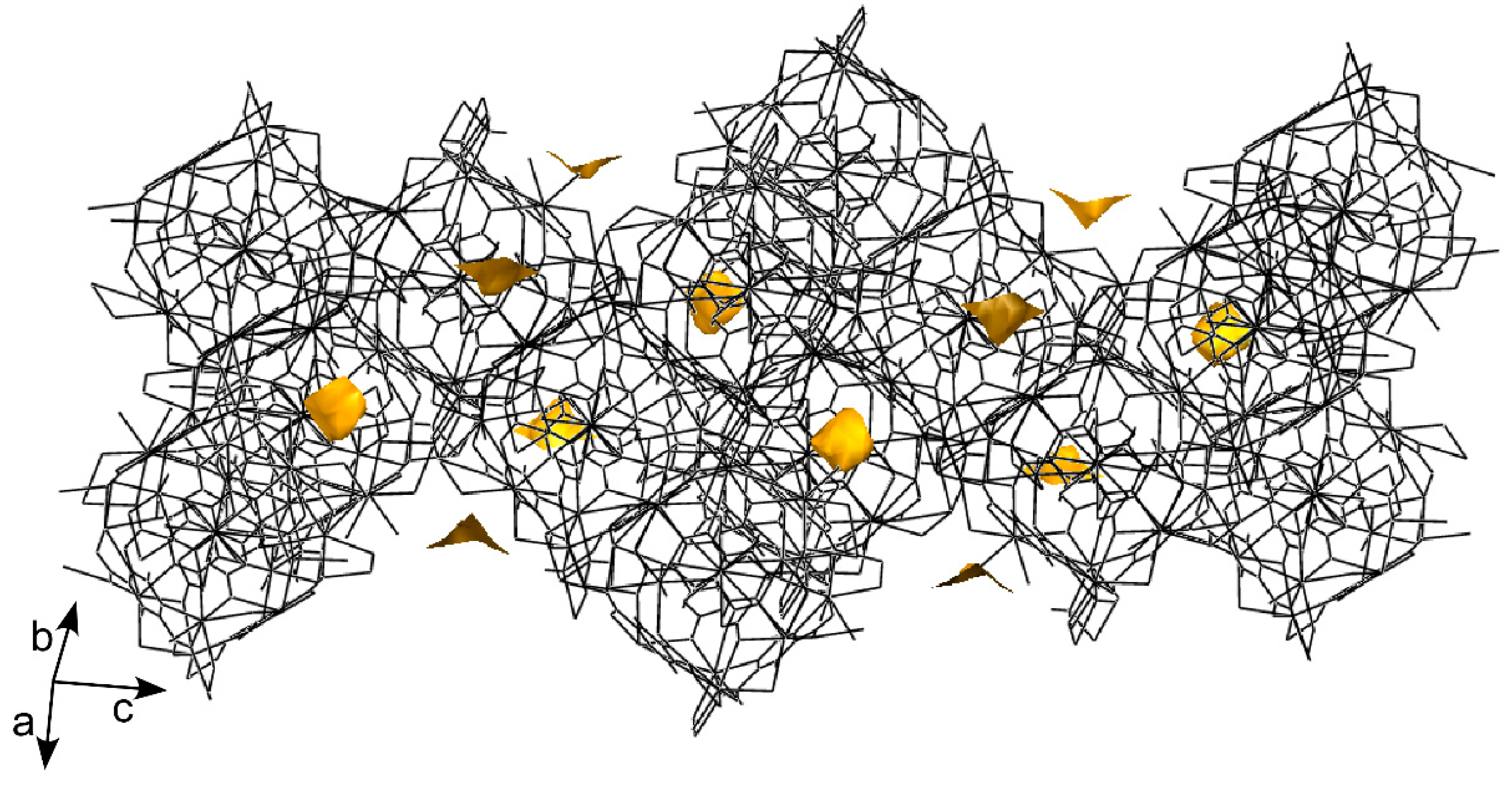
Figure 4.
Thermo-structural behaviour of compound 1 analysed by means of a (a) thermodiffractometric study in the 30-410 °C temperature range and (b) thermogravimetry.
Figure 4.
Thermo-structural behaviour of compound 1 analysed by means of a (a) thermodiffractometric study in the 30-410 °C temperature range and (b) thermogravimetry.
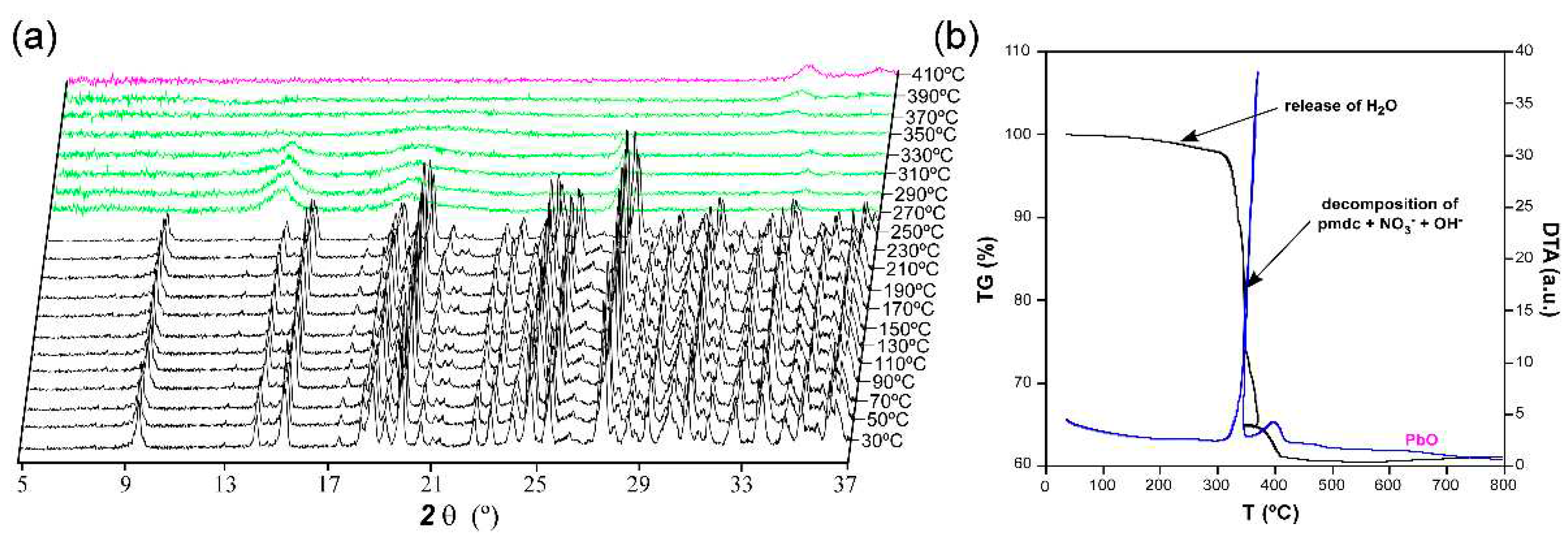
Figure 5.
(a) Experimental excitation (blue) and emission (red) spectra of 1 at RT. (b) Schematic representation of the most relevant calculated excitations.
Figure 5.
(a) Experimental excitation (blue) and emission (red) spectra of 1 at RT. (b) Schematic representation of the most relevant calculated excitations.
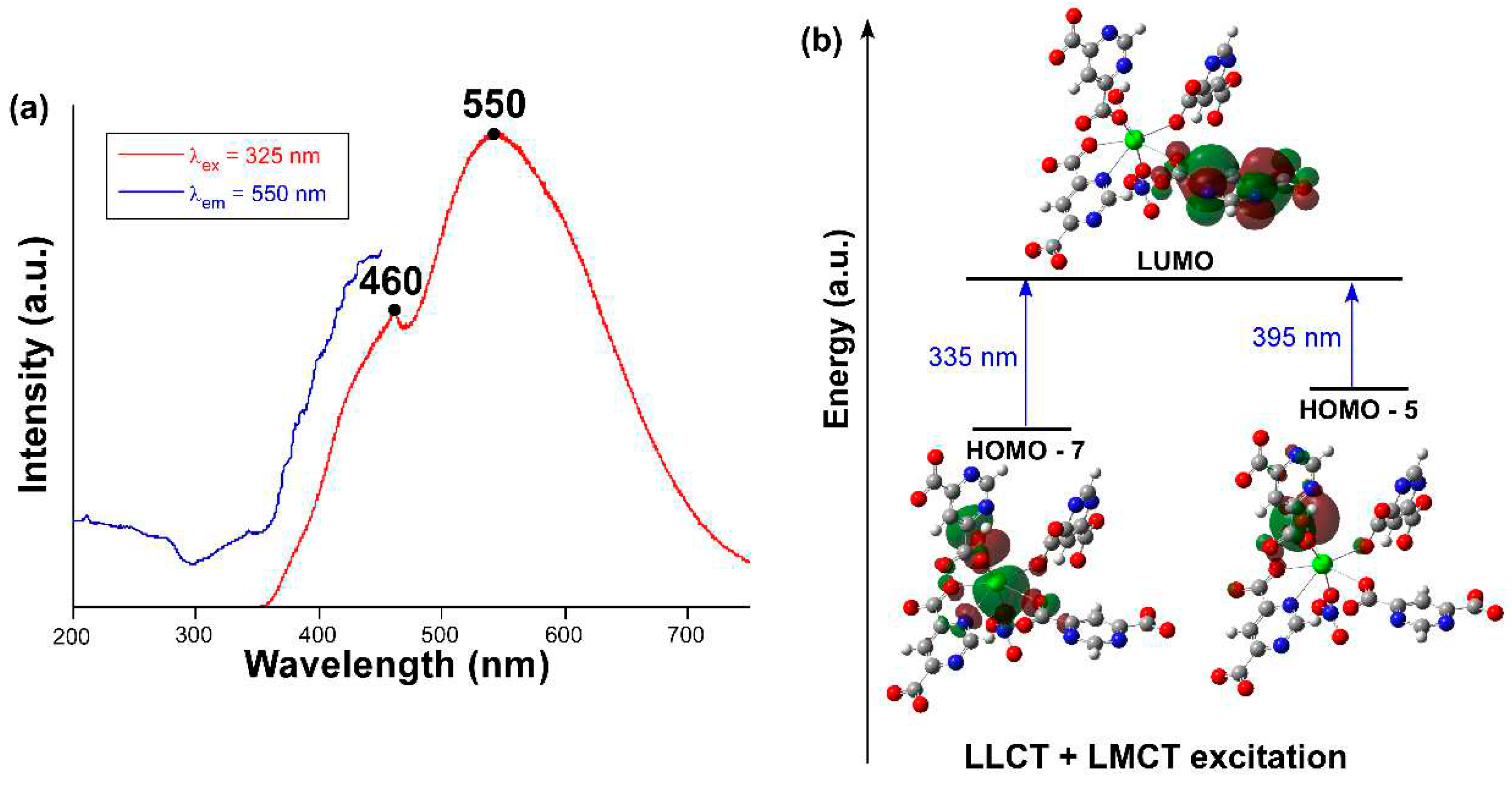
Figure 6.
(a) Excitation and (b) emission spectra showing the measured (solid line) and calculated (dotted line) bands.
Figure 6.
(a) Excitation and (b) emission spectra showing the measured (solid line) and calculated (dotted line) bands.
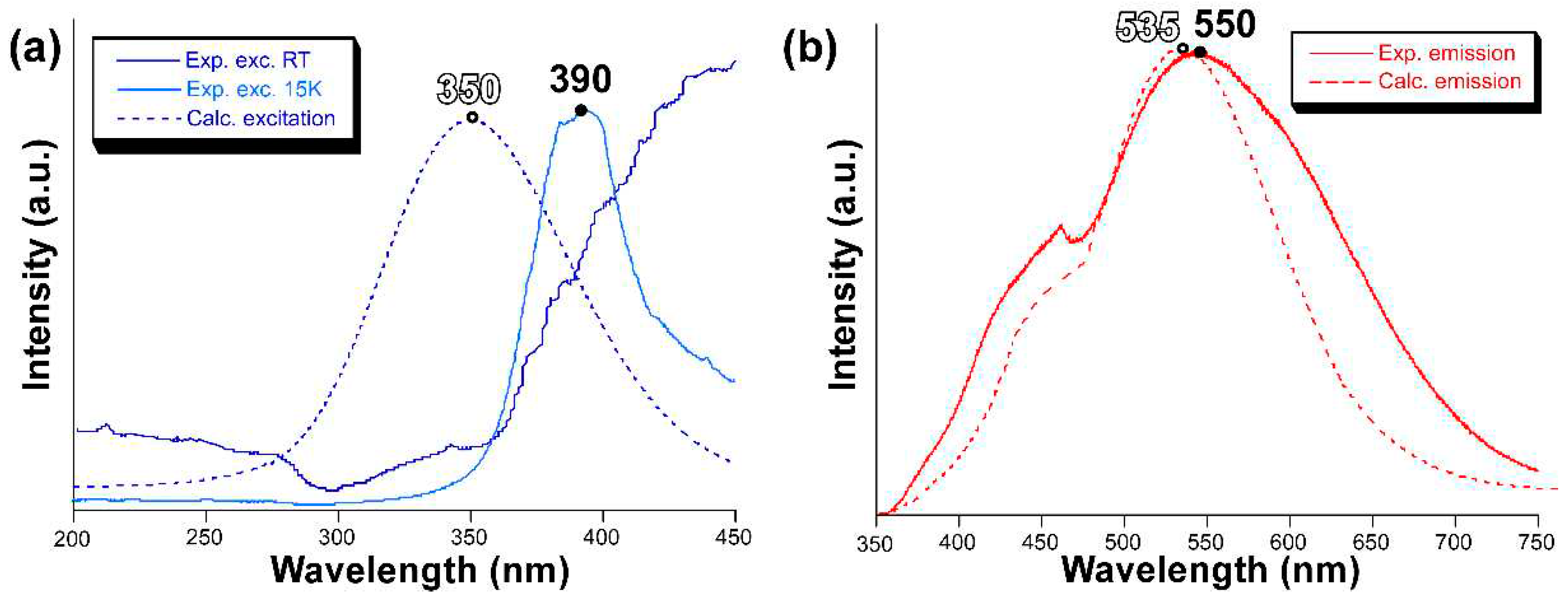
Table 1.
Selected coordination bond distances for the Pb(II) ions of compound 1 (Å).1.
| Pb1–N11 | 2.767(12) | Pb2–N13 | 2.548(12) | Pb3–O23 | 2.744(12) |
| Pb1–O171 | 2.510(12) | Pb2–N13d | 2.548(12) | Pb3–O23h | 2.744(12) |
| Pb1–O171a | 2.432(11) | Pb2–O181 | 2.506(11) | Pb3–O23i | 2.744(12) |
| Pb1–O172b | 2.753(12) | Pb2–O181d | 2.506(11) | Pb3–O23j | 2.744(12) |
| Pb1–O182c | 2.707(10) | Pb2–O182e | 2.914(11) | Pb3–O23k | 2.744(12) |
| Pb1–O11 | 2.478(11) | Pb2–O182f | 2.914(11) | Pb3–O23l | 2.744(12) |
| Pb1–O22 | 2.679(13) | Pb2–O22b | 2.686(12) | Pb3–O24 | 2.865(16) |
| Pb2–O22g | 2.686(12) | Pb3–O24h | 2.865(16) | ||
| Pb3–O24i | 2.865(16) | ||||
| Pb3–O24j | 2.865(16) | ||||
| Pb3–O24k | 2.865(16) | ||||
| Pb3–O24l | 2.865(16) |
1 Symmetry codes: (a) –x + y + 1, –x + 1, z; (b) x – y – 1/3, x – 2/3, –z + 1/3; (c) –y + 1/3, –x + 2/3, z + 1/6; (d) y + 1/3, x – 1/3, –z + 1/6; (e) –y, x – y – 1, z; (f) x – y – 2/3, –y – 1/3, –z + 1/6; (g) x – 1/3, x – y – 2/3, z – 1/6; (h) y + 2/3, –x + y + 1/3, –z + 1/3; (i) x – y – 1/3, x – 2/3, –z + 1/3; (j) –y, x – y – 1, z; (k) –x + 2/3, –y – 2/3, –z + 1/3; (l) –x + y +1, –x, z.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



