
Submitted:
27 September 2023
Posted:
28 September 2023
You are already at the latest version
Abstract
: The interaction of the Activating Transcription Factor 6 (ATF6), a key effector of the unfolded protein response (UPR) in the endoplasmic reticulum, with the neuronal calcium sensor Downstream Regulatory Element Antagonist Modulator (DREAM) is a potential therapeutic target in neurodegeneration. Modulation of the interaction ATF6-DREAM with repaglinide (RP) induced neuroprotection in a model of Huntington´s disease. Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disorder with no cure, characterized by the progressive loss of motoneurons resulting in muscle denervation, atrophy, paralysis and death. The aim of this work was to investigate the potential therapeutic significance of DREAM as a target for intervention in ALS. We found that the expression of the DREAM protein was reduced in the spinal cord of SOD1G93A mice compared to wild type littermates. RP treatment improved motor strength and reduced the expression of the ALS progression marker collagen type XIX1 (Col191 mRNA) in the quadriceps muscle in SOD1G93A mice. Moreover, treated SOD1G93A mice showed reduced motoneuron loss and glial activation and increased ATF6 processing in the spinal cord. These results indicate that the modulation of the DREAM-ATF6 interaction ameliorates ALS symptoms in SOD1G93A mice.
Keywords:
ALS
; Repaglinide
; DREAM
; ATF6
; SOD1
; UPR
; motoneurons
; microglia
; astroglia
1. Introduction
The Downstream Regulatory Element Antagonist Modulator (DREAM) protein, also known as calsenilin or KChIP3, is a member of the potassium channel interacting protein (KChIP) family of neuronal Ca2+ sensors [1,2,3]. Encoded by four genes, KChIP1 to 4, KChIP proteins show distinct expression patterns in different organs as well as in different areas within the CNS [1,3,4] and have multiple functions [5,6,7], sometimes specific for the different family members (recently reviewed in [8,9]). In the case of DREAM/KChIP3, hereafter DREAM, specialized activities are achieved via specific protein-protein interactions (PPI) and the Ca2+-dependent binding to specific sites in the DNA [3,10]. Calcium binding to functional EF-hands in the DREAM protein triggers a conformational change that prevents binding to DRE sites in the DNA [3] and modifies its affinity for some interacting proteins e.g. CREM, CREB, GRK2, but not for others e.g. presenilins and Kv4 channels [10]. Importantly, changes in DREAM conformation are also induced upon binding of small molecules, like arachidonic acid [11], glinides [12] and some diaryl-urea derivatives [13], which affects the interacting properties of DREAM and modifies its protein-protein interactions [10]. In previous work, we reported that protein levels of DREAM were reduced in Huntington’s disease (HD) patients as well as in HD mice and HD knock-in cells [14]. We proposed that endogenous DREAM silencing in HD may be part of an early endogenous neuroprotective mechanism since i) induced DREAM haplodeficiency in R6/2 mice, a transgenic HD mouse model, delayed the onset of motor dysfunction, reduced striatal atrophy, and prolonged lifespan, ii) DREAM overexpression has the opposite effect in R6/2 mice and sensitized HD knock-in cells to mitochondrial stress and iii) block of DREAM activity with DREAM interacting molecules like repaglinide (RP) or PC330 reduced disease symptoms and cell viability. The process involves the interaction between DREAM and the activating transcription factor 6 (ATF6), one of the three endoplasmic reticulum (ER) sensors that activate the unfolded protein response (UPR) in response to ER stress [15]. Exposure to DREAM ligands reduces the DREAM-ATF6 interaction which increases ATF6 processing and improves the UPR and the neuronal survival [14]. Whether this mechanism applies to other neurodegenerative diseases is currently not known.
Amyotrophic lateral sclerosis (ALS) is the third most common neurodegenerative disease, after Alzheimer’s disease and Parkinson disease. ALS is characterized by a progressive loss of motoneurons accompanied by muscle atrophy and paralysis, triggering the death of the patient in an average period of 3 to 5 years after the onset of symptoms. Although a small percentage of ALS cases have a genetic origin with a known mutation, the majority of cases, 90-95%, are of unknown cause [16]. The first mutated gene associated with ALS was the gene of the superoxide dismutase 1 (SOD1) enzyme, which allowed to develop the SOD1G93A mouse model [17], one of the best characterized SOD1 transgenic mice. Numerous mechanisms involved in the pathogenesis of ALS have been described, including mitochondrial dysfunction, alterations in RNA function and abnormal protein processing, excitotoxicity, oxidative stress, neuroinflammation, and axonal transport damage. Despite this, there is currently no treatment capable of curing the disease, only palliative treatments that delay the progression of the disease by a few months. Therefore, there is a need to find a therapy capable of curing the disease or, at least, slowing its progression.
A previous study using a pan-KChIP antibody [18] indicated that changes in KChIP levels during disease progression in SOD1G93A mice may correlate with the astrocytic response and the excitotoxic death of spinal cord motoneurons. Here, using an antibody specific for DREAM/KChIP3, which does not cross react with other KChIP family members [19], we disclose a significant reduction of DREAM protein levels in the lumbar spinal cord. Moreover, we show that the administration of the DREAM interacting molecule RP to SOD1G93A mice improved motor strength, reduced the astrocytic and the microglial activation and partially rescued motoneurons from death. The mechanism involves an activation of ATF6 processing in the spinal cord of transgenic mice. The present study highlights the potential therapeutic relevance of DREAM as a target for intervention in the context of ALS associated neurodegeneration.
2. Results
2.1. Expression of DREAM is significantly reduced in the lumbar spinal cord of SOD1G93A mice.
To define a potential role for DREAM in ALS neurodegeneration, we assessed the level of DREAM protein in extracts of lumbar spinal cord from SOD1G93A mice and their wild type littermates. Using a specific DREAM antibody, we found that both monomeric and tetrameric forms of DREAM were significantly reduced in the lumbar spinal cord of symptomatic SOD1G93A mice (Figure 1A–D). The decrease in DREAM protein was reminiscent of that previously observed in the brain of R6/2 mice, a mouse model of Huntington´s disease (HD), where a further block of DREAM activity with RP ameliorated disease symptoms [14]. To investigate whether this endogenous silencing of DREAM expression in SOD1G93A mice has also a neuroprotective function, we seeked for a positive effect of the administration of the DREAM binding molecule, RP, in these mice.
2.2. Repaglinide treatment delayed the loss of motor strength in SOD1G93A mice.
Ten weeks-old SOD1G93A transgenic mice, corresponding to an early symptomatic stage of the disease, and WT littermates were exposed to RP in the drinking water for 8 weeks. Body weight, motor coordination and muscle strength were monitored weekly from week 10 to week 18 (Figure 2A–C).
As previously reported in SOD1G93A mice [18], disease progression was paralleled by a continuing reduction in body weight (Figure 2A). Chronic RP administration did not affect the normal growth in WT mice and did not modify the loss of body weight in SOD1G93A transgenic mice (Figure 2A). Similarly, chronic administration of RP did not improve the loss of motor coordination in the rotarod test and the latency to fall was similar in vehicle (DMSO) or RP-treated SOD1G93A mice (Figure 2B).
Notably, loss of muscle strength of SOD1G93A mice in the hanging-wire test was significantly reduced in those receiving RP compared to those treated with DMSO (Figure 2C). To further support the functional meaning of this result, we checked for changes in the expression of collagen type XIX alpha 1 gene (Col19a1), a biomarker associated with ALS progression, whose expression in the muscle of SOD1G93A mice negatively correlates with longevity [20]. Quantitative PCR (qPCR) analysis of the quadriceps muscle from SOD1-DMSO mice showed a 6.5-fold increase of Col19a1 mRNA expression compared to the WT-DMSO group (Figure 2D). Col19a1 mRNA levels in WT mice treated with RP were similar to those observed in the WT-DMSO group (Figure 2D). However, RP administration to SOD1G93A mice partially but significantly reversed the increase of Col19a1 mRNA (Figure 2D). This result indicates a positive effect of the RP treatment to delay disease progression in the quadriceps muscle of SOD1G93A mice.
2.3. Treatment with RP reduces motoneuron loss and improves gliosis in the ventral horn of SOD1G93A spinal cord.
To further substantiate a neuroprotective effect of RP in the SOD1G93A mouse model of ALS, we next checked a potential rescue of ventral horn motoneuron loss. Nissl staining of lumbar spinal cord sections of WT and SOD1G93A mice showed, as reported [21], a significant reduction of motoneurons in SOD1-DMSO compared to WT-DMSO mice (Figure 3A,B). Interestingly, RP treatment partially, but significantly, reduced motoneuron loss in SOD1G93A mice compared to the SOD1-DMSO group. RP did not exert any effect on motoneuron counts in the WT control groups (Figure 3A,B).
Moreover, we assessed whether RP was also affecting the glial reaction, a hallmark of neuroinflammation reported in SOD1G93A mice [22,23,24]. Immunohistochemical analysis of the ventral horn in the spinal cord showed that the expression of the microglial marker ionized calcium binding protein-1 (Iba1) was significantly higher in SOD1G93A mice than in WT mice (Figure 4A,B), suggesting the presence of activated microglia in the transgenic mice. Notably, RP-treated SOD1G93A mice presented a significant reduction of Iba1 staining compared to SOD1G93A mice treated with DMSO. The administration of repaglinide did not alter Iba1 expression in wild type mice (Figure 4A,B). We also stained spinal cord sections with an antibody for the glial fibrillary acidic protein (GFAP), an astroglial marker expressed by astrocytes that is up-regulated in association with inflammatory processes in neurodegenerative diseases [25,26]. As previously reported in these mice [23,24], GFAP immunoreactivity was increased in the ventral horn of SOD1G93A transgenic mice compared to WT littermates (Figure 4C,D). RP treatment did not change GFAP expression in the WT mice compared to the WT-DMSO group. However, RP administration reduced GFAP immunoreactivity in SOD1G93A mice compared to the SOD1-DMSO group (Figure 4C,D).
Taken together, these results indicate that RP treatment was able to reduce neuron loss and gliosis in SOD1G93A transgenic mice without exerting any deleterious effect in WT animals.
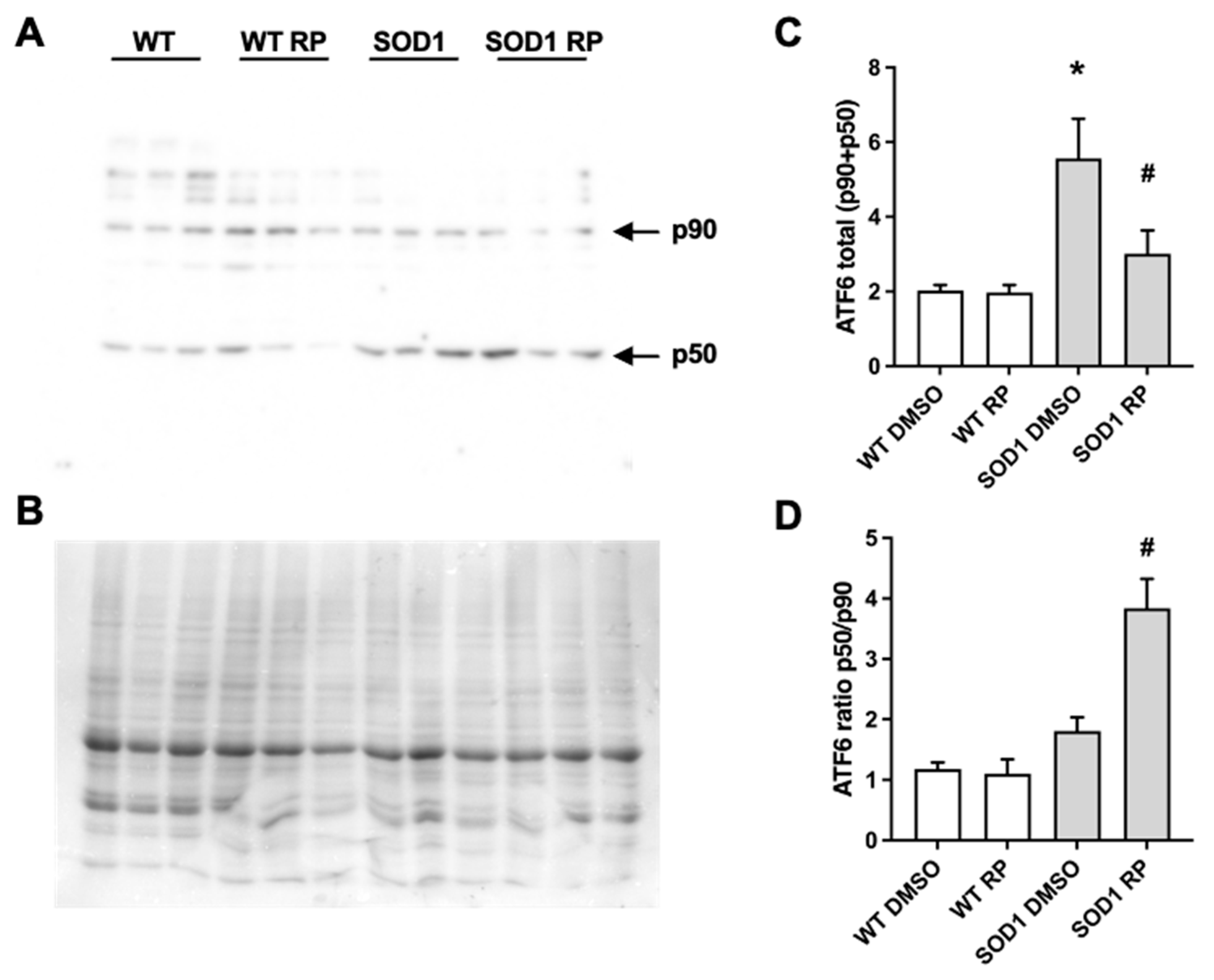
2.5. RP administration stimulates ATF6 processing in the spinal cord of SOD1G93A mice.
As mentioned, ATF6 is a key effector regulating the UPR in the endoplasmic reticulum (ER-UPR). In 2016, we reported that RP competes with the DREAM-ATF6 interaction increasing ATF6 processing and promoting neuroprotection in a mouse model of Huntington´s disease [14]. To test whether a similar mechanism is involved in the neuroprotective efects exerted by RP in the spinal cord of SOD1G93A mice, we studied the effect of RP administration on ATF6 levels and its processing. Western blot analysis revealed increased total levels of ATF6, full-length (p90) plus processed form (p50) in SOD1G93A mice treated with vehicle compared to WT mice (Figure 5). Moreover, ATF6 processing defined as the p50/p90 ratio, was not significantly different between SOD1G93A and WT DMSO-treated mice (Figure 5). Interestingly, in SOD1G93A mice RP administration increased the p50/p90 ratio and reduced total ATF6 (p90+p50) levels in compared to DMSO-treated SOD1G93A mice. These results linked the amelioration of disease symptoms following the administration of RP with the stimulation of ATF6 processing in the spinal cord of this ALS model. Finally, RP administration did not change total ATF6 levels nor ATF6 processing in WT mice (Figure 5).
3. Discussion
Accumulation of abnormal mutant SOD1 aggregates in the cytosol and the mitochondrial intermembrane space interferes with the assembly and maturation of cellular and mitochondrial proteins and triggers the unfolded protein response both at the endoplasmic reticulum (ER) and the mitochondria [15,27,28,29,30]. In addition, it has been suggested that mutant SOD1 protein interacts specifically with Derlin-1, a component of the ER-associated protein degradation (ERAD) machinery, and elicits ER stress-induced apoptosis in motoneurons through dysfunction of ERAD [31]. Several lines of evidence indicate that motoneurons in ALS are particularly sensitive to changes in ER stress [32,33]. Activation of the UPR at the ER (ER-UPR) depends on three ER transmembrane receptors that sense the ER stress signal; ATF6, inositol-requiring kinase 1 (IRE1) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) [15]. In this study we have shown that chronic RP administration to SOD1G93A transgenic mice induces neuroprotection and activation of ATF6 processing. Notably, we showed that in SOD1G93A spinal cord levels of total ATF6 are significantly increased compared to wild type mice, while the levels of activated ATF6 are similar in both genotypes. Previous in vitro studies have shown increased processing and nuclear translocation of ATF6 in N2a neuroblastoma cells overexpressing mutant SOD1G85R [34] and an increase in SOD1 aggregates in the motoneuronal cell line NSC-34 following ATF6 knockdown [35]. These data suggest a neuroprotective role of ATF6 activation in ALS. Our results support this notion, since block of the DREAM-ATF6 interaction after RP administration in SOD1G93A mice significantly increased the content of the transcriptionally active processed form of ATF6 and the following activation of the ER-UPR was associated with a decrease in motoneuron loss and gliosis and to a reduction in disease markers. Downstream effectors of ATF6 activation and whether the activation of ATF6 processing is the only mechanism responsible for the neuroprotective action of RP are currently under investigation. Furthermore, not only ATF6, but also IRE1 and PERK, the other two branches of the ER-UPR, have been shown to be activated in different ALS experimental models as well as in patient samples [31,36,37,38,39]. Genetic manipulation of these two UPR branches confirmed their importance in ALS. Thus, conditional deletion or knock-down of abnormally high levels of XBP1, the main downstream effector of the IRE1 pathway, in SOD1 mutant mice or patients motoneurons, respectively, delayed disease onset and increased lifespan or conferred significant neuroprotection in vitro [35,40]. Similarly, genetic ablation of elevated levels of ATF4, the main downstream effector of the PERK pathway, in mutant SOD1G93A mice prolonged lifespan and slowed disease progression [41]. The involvement of the PERK pathway in ALS, however, has been challenged by a more recent study [42] showing that PERK haploinsufficiency has no effect on disease progression in five different lines of mutant SOD1 transgenic mice. Moreover, genetic ablation of CHOP, another transcriptional effector downstream in the PERK pathway, or induced deficiency of GADD34, which potentiates the PERK pathway by stabilizing phospho-eIF2α, did not change disease progression in the same five mutant SOD1 transgenic mouse lines [42].
Taken together, the available experimental evidence supports the functional significance of ER-UPR in the fate of ALS motoneurons. This triggered the development of many small molecules to specifically target the different UPR components and their assessment as feasible candidates for therapeutic intervention in ALS. Ideally, besides to be avoid of unwanted side effects, a perfect candidate should potentiate the pro-survival function of the UPR, solving the protein aggregation problem and restoring protein homeostasis. At the same time, a perfect candidate should not over stimulate UPR activity leading to neuronal demise. Several strategies specific for each UPR branch were developed and the results recently reviewed [43,44,45,46]. In brief, i) to modulate the PERK pathway the strategies included the inhibition of PERK activation (GSK2606414, GSK2656157) [47,48], the block of downstream effects of elF2α phosphorylation (ISRIB, trazodone, dibenzoylmethane) [47,49,50,51,52] and the inhibition of elF2α phosphatases (guanabenz, salubrinal and sephin1) [33,53,54,55,56,57], ii) to modulate the IRE1 arm there are small molecules targeting the IRE1 RNase activity (4μ8C, salicylaldimines, SFT-083010, MKC-3946, toyocamycin) [58,59,60,61,62,63], to increase the XBP1 splicing levels (citrinins, Patulin, quercetin, apigenin) [64,65,66], to block kinase activity and/or interfere IRE1 oligomerization (sunitinid, APY29, KIRAs) [67,68,69,70] and iii) to modulate ATF6, increasing or decreasing its expression (different flavonoids including fisetin, apigenin, luteolin, baicalein, kaempferol) [65,71,72]. Moreover, several pharmacological chaperons have been assessed to restore defective protein homeostasis in ALS, extensively reviewed in Tao and Conn (2018) [73], that includes arimoclomol [74], tauroursodeoxycholic acid (TUDCA) and sodium 4-phenyl butyrate (4-PBA) [75,76] and geldamycin [77,78]. As a result, these studies identified potential molecules targeting the UPR that could be useful for ALS treatment and, some of them have been already enrolled in clinical trials of the disease e.g. guanabenz [54,79], a combination of the chemical chaperones TUDCA and 4-PBA, currently running a phase III trial with 600 patients.
Accumulation of abnormal mutant SOD1 aggregates in the mitochondrial intermembrane space interferes with the assembly and maturation of mitochondrial proteins, triggering mitochondrial dysfunction, which ultimately activates the UPR at the mitochondria (MT-UPR) [27,29,30]. Like the ER-UPR, the MT-UPR is a transcriptional program that controls the expression of mitochondrial proteases and chaperones designed to restore protein homeostasis at the mitochondria [80,81]. Interestingly, induction of the MT-UPR in SOD1G93A spinal cord precedes the onset of disease symptoms, follows disease progression and, in some cases, decays at disease end-stage [82,83]. DREAM is located is several subcellular compartments, including the mitochondria. The role of DREAM in mitochondrial function has not been addressed and whether RP administration has an effect on the mitochondrial response to SOD1G93A aggregation is currently unknown. The up-coming therapies to modulate the activation of mitochondrial UPR have been recently reviewed [84,85,86,87].
Chronic administration of RP has profound effects on motoneurons survival, the glial response and muscle strength. Moreover, it reduces the levels of Col19a1 mRNA, a marker of ALS associated with bad prognosis and fast disease progression [20]. Nevertheless, RP did not change the onset of disease, rate of body weight loss nor the deficits in motor coordination in the rotarod test. Using a similar dosing and pattern of administration, RP delayed onset and slowed progression, including the amelioration of motor deficits in the R6/1 mouse model of Huntington’s disease. Whether an increase in the dosage or a change in administration pattern of RP could render a significant effect on motor coordination or lifespan in ALS mice remains to be investigated. Moreover, the potential benefits of the new DREAM binding molecules of the PC-series, like PC330, PC260, [14,88,89,90,91], in SOD1G93A mice remain to be investigated. Noteworthy, the SOD1G93A mouse model is a quite aggressive model of familial ALS, showing a very early onset and very fast disease progression [92]. Whether RP or the new PC-molecules could offer improved results in other mouse models of familial or sporadic ALS is currently under investigation.
4. Materials and Methods
4.1. Animals and treatments.
4.1.1. Animals
Hemizygous B6SJL-Tg SOD1G93A males (stock number 002726) purchased from The Jackson Laboratory (Bar Harbor, ME, USA) were mated with C57BL/6J X SJL/J F1 hybrid females (B6SJLF1) purchased from Janvier Labs (Saint-Berthevin Cedex, France) to obtain hemizygous transgenic (SOD1G93A) and wild type mice used in this study. The mice were housed at the animal facilities in Centro de Investigación Biomédica de Aragón (CIBA) under a standard light-dark (12:12) cycle. Food and water were provided ad libitum.
All procedures were approved by the Ethic Committee for Animal Experiments from the University of Zaragoza and accordingly with the Spanish Policy for Animal Protection RD53/2013, which meets the European Union Directive 2010/63 on the protection of animals used for experimental and other scientific purposes. The approval codes are the PI45/22 and the approval date: 22 August 2022.
4.1.2. Treatments
RP or vehicle DMSO were administered orally in the drinking water at concentration of 2 μg/mL from week 10 to week 18 of age to SOD1G93A and wild type littermates. Drinking water was renewed weekly. Groups were sex-balanced.
4.2. Weight control and behavioral tests
Both WT and SOD1G93A mice were weekly weighted from the beginning of the treatment at week 10 of age. In addition, rotarod and hanging-wire tests were weekly performed in the SOD1G93A mice, although mice started to train on the rotarod one week before starting the treatment. Motor coordination was assessed using a rotarod (ROTA-ROD/RS, LE8200, LSILETICA, Scientific Instruments; Panlab, Barcelona, Spain). Animals were placed onto the cylinder at a constant speed of 14 rpm. The animals had three attempts to remain on the rotarod for a maximum of 180 s per trial, and the longest latency was recorded. Muscular strength was tested by the hanging-wire test. Each mouse was placed on a wire lid which was turned upside down. The latency from the beginning of the test until the mouse fell was timed. The animals had three attempts to remain for a maximum of 180 s per trial, and the longest latency was recorded. All behavioral experiments were carried out in blind conditions for treatment.
4.3. Western blot
Eighteen weeks-old WT and SOD1G93A mice treated with DMSO or RP were sacrificed under deep anesthesia and spinal cords were extracted. The spinal cord whole cell extracts preparation was performed as described [93]. In brief, tissue was homogenized on ice in RIPA buffer (9806, Cell Signaling Technology) supplemented with protease inhibitor (Complete EDTA-free, Roche) and 1 mM PMSF (phenylmethanesulfonyl fluoride). Extracts were cleared by centrifugation (14,000 g, 20 min. 4 °C). Samples (20–30 μg) were analyzed by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and immunoblot. PVDF (polyvinylidene difluoride) membranes were incubated (overnight, 4 °C) with specific antibodies to DREAM/KChIP3 (Ab730, [19]) and ATF6α (A303–719, Bethyl). Secondary antibodies used were HRP (horseradish peroxidase)-conjugated donkey anti-rabbit, IgG antibody (Jackson) and detection was with ECL Select (GE Healthcare). Equal protein loading was analyzed by Coomassie staining of the membrane after immunoblotting. Band and total lane (Coomassie) intensities were quantified with ImageLab software (BioRad). After densitometric analysis, band intensity ratio vs. Coomassie was normalized to DMSO-treated WT mice for each experiment.
4.4. Real-time qPCR.
RNA was isolated from the quadriceps muscle of 18 weeks-old WT and SOD1G93A mice treated with DMSO or RP using TRIzol, treated with DNase (Ambion), and reverse transcribed using hexamer primer and Moloney murine leukemia virus reverse transcriptase. To confirm the absence of genomic DNA, each sample was processed in parallel without reverse transcriptase. An assay from Applied Biosystems (Mm00483576_m1) was used for the collagen type XIX alpha 1 gene (Col19a1) and the relative expression was quantified by the 2-ΔΔCt method using hypoxanthine-phosphoribosyltransferase (Hprt) as a reference with primer 5′-TTGGATACAG GCCAGACTTTGTT-3′ and 5′-CTGAAGTACTCATTATAGTCAAGGGCATA-3′ and the MGB probe 5′-TTGAAATTCCAGACAAGTTT-3′.
4.5. Histology
4.5.1. Tissue Processing
Eighteen weeks-old WT and SOD1G93A mice treated with DMSO or RP were sacrificed under deep anesthesia and spinal cords were extracted. Spinal cords were fixed with 4% paraformaldehyde (pH 7.4) for 48 h at 4º C, cryoprotected in 30% sucrose (0.1 M PBS) and frozen, before sectioning into 20-µm-thick coronal sections on a cryostat.
4.5.2. Immunofluorescence, Nissl staining and Image Analysis.
Coronal spinal cord tissue sections were mounted on coated slides (Dako Flex IHC microscope slides), treated with sodium citrate 10 mM, pH 6.0, at 95º C for 3 min, and preincubated with 5% normal goat serum (NGS) in phosphate-buffered saline (0.1 M PBS, pH 7.4)/0.1% Triton-X 100 for 60 min. The primary antibodies rabbit polyclonal anti-Iba1 (1:500; Wako Chemicals) and mouse monoclonal anti-GFAP (1:250, Sigma-Aldrich) were diluted in 0.5% NGS in PBS and applied overnight at RT. The secondary antibodies Alexa Fluor-568 goat anti-mouse IgG and Alexa Fluor-488 goat anti-rabbit IgG (1:500; both from ThermoFisher) were diluted in 0.5% NGS in PBS and applied for 1h at RT. The slides were coverslipped in a medium containing p-phenylenediamine and bisbenzimide (Hoechst 33342; Sigma). For Nissl technique, the sections were dehydrated in 1:1 alcohol/chloroform overnight, rehydrated, incubated with 0.1% cresyl violet solution for 5 minutes, rinsed in distilled water, dehydrated, cleared with xylene and mounted with DPX (Fluka).
For quantification of fluorescence intensity, 8 or more high-resolution digital 200X widefield microphotographs from four consecutive sections per animal were taken at the same conditions of light and brightness/contrast, exposure time and gain parameters using a Leica DMI6000B epifluorescence microscope equipped with an OrcaR2 digital camera (Leica, Wetzlar, Germany). The microphotographs were used to measure the mean integrated density of labelling in the ventral horn of the spinal cord, using the ImageJ software (National Institutes of Health, Bethesda, MD). Data were normalized to DMSO-treated WT mice for each experiment.
A Leica DM4 B microscope coupled to a Leica DMC 5400 digital camera (Leica, Wetzlar, Germany) were used to study and photograph motoneurons (>20 μm diameter and polygonal-shaped) in the ventral horn of the spinal cords stained with the Nissl technique. High-resolution digital 100X widefield microphotographs (12 or more) from six consecutive sections per animal were taken at the same conditions of light and brightness/contrast, exposition time and gain parameters. Motor neurons were counted in the ventral horn area of the spinal cord using the cell counter plugin of the ImageJ software (National Institutes of Health, Bethesda, MD).
4.6. Statistical analyses
All data are shown as mean ± SEM. Differences were considered significant at p < 0.05. Parametric or nonparametric analysis was applied after normality test (Shapiro–Wilk). Student’s t-test was used to compare 2 groups. One-way or Two-way ANOVA followed by multiple comparison between selected groups were performed for more than 2 groups. Prism GraphPad Software 8.0 (La Jolla, CA, USA) was used to plot graphs and for statistical analysis.
5. Conclusions
In conclusion, to target the defective proteostasis in ALS modulating the ER-UPR seems like a plausible choice to develop new therapeutic venues. Importantly, this approach is transversal for different neurodegenerative diseases and successful molecules in the fine-tunning of the ER-UPR could be candidates, by themselves or in combination with other disease-specific therapies, for the treatment of these pathologies.
Author Contributions
Experimental Design, R.G-G., L.M-M., R.O. and J.R.N.; performed the experiments, R.G-G., L. M-M., P.G., X.M.D., A.C.C, I.P.-L., E.S. and R.D-M.; data Analysis: R.G-G., L. M-M., B.M. and P.G.; paper writing, R.G-G., L. M-M., R.O., B.M. and J.R.N.
Funding
This research was funded by Instituto de Salud Carlos III CIBERNED (RO and JRN) and PI21/00372 (RO); by Fondo Europeo de Desarrollo Regional (FEDER) “Una manera de hacer Europa” from the European Union (RO); by the Consolidated Groups Program from Gobierno de Aragón (RO) and by the Innovation Program from Asahi Kasei Pharma (JRN). R.G-G was supported by a post-doctoral contract from the Severo Ochoa Excellence Program (Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación).
Institutional Review Board Statement
The animal study protocol was approved by the Ethic Committee for Animal Experiments from the University of Zaragoza and accordingly with the Spanish Policy for Animal Protection RD53/2013, which meets the European Union Directive 2010/63 on the protection of animals used for experimental and other scientific purposes. The approval codes are the PI45/22 and the approval date: 22 August 2022.
Informed Consent Statement
Not applicable.
Data Availability Statement
All mentioned data are represented in the manuscript figures. Other additional data will be made available upon request.
Acknowledgments
We are grateful to Soledad Montalbán and Oscar Sánchez from the CNB Histology Facility and Jaime Fernández de Córdoba, Gianluca D'Agostino and Ana Oña from the Advanced Optical Microscopy Facility of the CNB for their technical help.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S., et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553-556. [CrossRef]
- Buxbaum, J.D.; Choi, E.K.; Luo, Y.; Lilliehook, C.; Crowley, A.C.; Merriam, D.E.; Wasco, W. Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat Med 1998, 4, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Carrion, A.M.; Link, W.A.; Ledo, F.; Mellstrom, B.; Naranjo, J.R. DREAM is a Ca2+-regulated transcriptional repressor. Nature 1999, 398, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Kovacs, I.; Zhang, Z. Differential distribution of KChIPs mRNAs in adult mouse brain. Brain Res Mol Brain Res 2004, 128, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Cantero-Recasens, G.; Butnaru, C.M.; Valverde, M.A.; Naranjo, J.R.; Brouwers, N.; Malhotra, V. KChIP3 coupled to Ca(2+) oscillations exerts a tonic brake on baseline mucin release in the colon. Elife 2018, 7. [Google Scholar] [CrossRef]
- Jerng, H.H.; Pfaffinger, P.J. Modulatory mechanisms and multiple functions of somatodendritic A-type K (+) channel auxiliary subunits. Front Cell Neurosci 2014, 8, 82. [Google Scholar] [CrossRef]
- Venn, N.; Haynes, L.P.; Burgoyne, R.D. Specific effects of KChIP3/calsenilin/DREAM, but not KChIPs 1, 2 and 4, on calcium signalling and regulated secretion in PC12 cells. Biochem J 2008, 413, 71–80. [Google Scholar] [CrossRef]
- Molinaro, P.; Sanguigno, L.; Casamassa, A.; Valsecchi, V.; Sirabella, R.; Pignataro, G.; Annunziato, L.; Formisano, L. Emerging Role of DREAM in Healthy Brain and Neurological Diseases. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Wu, L.Y.; Song, Y.J.; Zhang, C.L.; Liu, J. K(V) Channel-Interacting Proteins in the Neurological and Cardiovascular Systems: An Updated Review. Cells 2023, 12. [Google Scholar] [CrossRef]
- Rivas, M.; Villar, D.; Gonzalez, P.; Dopazo, X.M.; Mellstrom, B.; Naranjo, J.R. Building the DREAM interactome. Sci China Life Sci 2011, 54, 786–792. [Google Scholar] [CrossRef]
- Holmqvist, M.H.; Cao, J.; Knoppers, M.H.; Jurman, M.E.; Distefano, P.S.; Rhodes, K.J.; Xie, Y.; An, W.F. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J Neurosci 2001, 21, 4154–4161. [Google Scholar] [CrossRef]
- Okada, M.; Takezawa, D.; Tachibanaki, S.; Kawamura, S.; Tokumitsu, H.; Kobayashi, R. Neuronal calcium sensor proteins are direct targets of the insulinotropic agent repaglinide. Biochem J 2003, 375, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Bowlby, M.R.; Chanda, P.; Edris, W.; Hinson, J.; Jow, F.; Katz, A.H.; Kennedy, J.; Krishnamurthy, G.; Pitts, K.; Ryan, K., et al. Identification and characterization of small molecule modulators of KChIP/Kv4 function. Bioorg Med Chem 2005, 13, 6112-6119. [CrossRef]
- Naranjo, J.R.; Zhang, H.; Villar, D.; Gonzalez, P.; Dopazo, X.M.; Moron-Oset, J.; Higueras, E.; Oliveros, J.C.; Arrabal, M.D.; Prieto, A., et al. Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. J Clin Invest 2016, 126, 627-638. [CrossRef]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol Bioeng 2011, 108, 2777–2793. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat Rev Dis Primers 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X., et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772-1775. [CrossRef]
- Larrode, P.; Calvo, A.C.; Moreno-Martinez, L.; de la Torre, M.; Moreno-Garcia, L.; Molina, N.; Castiella, T.; Iniguez, C.; Pascual, L.F.; Mena, F.J.M., et al. DREAM-Dependent Activation of Astrocytes in Amyotrophic Lateral Sclerosis. Mol Neurobiol 2018, 55, 1-12. [CrossRef]
- Savignac, M.; Mellstrom, B.; Bebin, A.G.; Oliveros, J.C.; Delpy, L.; Pinaud, E.; Naranjo, J.R. Increased B cell proliferation and reduced Ig production in DREAM transgenic mice. J Immunol 2010, 185, 7527–7536. [Google Scholar] [CrossRef]
- Calvo, A.C.; Manzano, R.; Atencia-Cibreiro, G.; Olivan, S.; Munoz, M.J.; Zaragoza, P.; Cordero-Vazquez, P.; Esteban-Perez, J.; Garcia-Redondo, A.; Osta, R. Genetic biomarkers for ALS disease in transgenic SOD1(G93A) mice. PLoS One 2012, 7, e32632. [Google Scholar] [CrossRef]
- Lev, N.; Barhum, Y.; Lotan, I.; Steiner, I.; Offen, D. DJ-1 knockout augments disease severity and shortens survival in a mouse model of ALS. PLoS One 2015, 10, e0117190. [Google Scholar] [CrossRef]
- Lewis, K.E.; Rasmussen, A.L.; Bennett, W.; King, A.; West, A.K.; Chung, R.S.; Chuah, M.I. Microglia and motor neurons during disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis: changes in arginase1 and inducible nitric oxide synthase. J Neuroinflammation 2014, 11, 55. [Google Scholar] [CrossRef]
- Rando, A.; de la Torre, M.; Martinez-Muriana, A.; Zaragoza, P.; Musaro, A.; Hernandez, S.; Navarro, X.; Toivonen, J.M.; Osta, R. Chemotherapeutic agent 5-fluorouracil increases survival of SOD1 mouse model of ALS. PLoS One 2019, 14, e0210752. [Google Scholar] [CrossRef]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 2009, 187, 761–772. [Google Scholar] [CrossRef]
- Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Herranz, A.S.; Reimers, D.; Montero Vega, T.; Jimenez-Escrig, A.; Richart Lopez, L.A.; Bazan, E. Effects of intravenous administration of human umbilical cord blood stem cells in 3-acetylpyridine-lesioned rats. Stem Cells Int 2012, 2012, 135187. [Google Scholar] [CrossRef]
- Gonzalo-Gobernado, R.; Perucho, J.; Vallejo-Munoz, M.; Casarejos, M.J.; Reimers, D.; Jimenez-Escrig, A.; Gomez, A.; Ulzurrun de Asanza, G.M.; Bazan, E. Liver Growth Factor "LGF" as a Therapeutic Agent for Alzheimer's Disease. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [PubMed]
- Igoudjil, A.; Magrane, J.; Fischer, L.R.; Kim, H.J.; Hervias, I.; Dumont, M.; Cortez, C.; Glass, J.D.; Starkov, A.A.; Manfredi, G. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J Neurosci 2011, 31, 15826–15837. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum Mol Genet 2008, 17, 3303–3317. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergiya, C.; Beal, M.F.; Buck, J.; Manfredi, G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci 2005, 25, 2463–2470. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev 2008, 22, 1451–1464. [Google Scholar] [CrossRef]
- Filezac de L'Etang, A.; Maharjan, N.; Cordeiro Brana, M.; Ruegsegger, C.; Rehmann, R.; Goswami, A.; Roos, A.; Troost, D.; Schneider, B.L.; Weis, J., et al. Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat Neurosci 2015, 18, 227-238. [CrossRef]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Oh, Y.K.; Shin, K.S.; Yuan, J.; Kang, S.J. Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J Neurochem 2008, 104, 993–1005. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 2009, 23, 2294–2306. [Google Scholar] [CrossRef]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otin, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guegan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M., 2nd; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A 2006, 103, 6025–6030. [Google Scholar] [CrossRef] [PubMed]
- Montibeller, L.; de Belleroche, J. Amyotrophic lateral sclerosis (ALS) and Alzheimer's disease (AD) are characterised by differential activation of ER stress pathways: focus on UPR target genes. Cell Stress Chaperones 2018, 23, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 2010, 69, 346–355. [Google Scholar] [CrossRef]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S., et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781-795. [CrossRef]
- Matus, S.; Lopez, E.; Valenzuela, V.; Nassif, M.; Hetz, C. Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS One 2013, 8, e66672. [Google Scholar] [CrossRef] [PubMed]
- Dzhashiashvili, Y.; Monckton, C.P.; Shah, H.S.; Kunjamma, R.B.; Popko, B. The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol Dis 2019, 127, 527–544. [Google Scholar] [CrossRef]
- Charif, S.E.; Vassallu, M.F.; Salvanal, L.; Igaz, L.M. Protein synthesis modulation as a therapeutic approach for amyotrophic lateral sclerosis and frontotemporal dementia. Neural Regen Res 2022, 17, 1423–1430. [Google Scholar] [CrossRef]
- Hetz, C.; Axten, J.M.; Patterson, J.B. Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol 2019, 15, 764–775. [Google Scholar] [CrossRef]
- Medinas, D.B.; Gonzalez, J.V.; Falcon, P.; Hetz, C. Fine-Tuning ER Stress Signal Transducers to Treat Amyotrophic Lateral Sclerosis. Front Mol Neurosci 2017, 10, 216. [Google Scholar] [CrossRef]
- Zhao, C.; Liao, Y.; Rahaman, A.; Kumar, V. Towards Understanding the Relationship Between ER Stress and Unfolded Protein Response in Amyotrophic Lateral Sclerosis. Front Aging Neurosci 2022, 14, 892518. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M., et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 2015, 6, e1672. [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; Lopez, N.; Axten, J.M.; Sardi, S.P.; Hoozemans, J.J.M.; Hetz, C. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson's disease. Neurobiol Dis 2018, 112, 136–148. [Google Scholar] [CrossRef]
- Bugallo, R.; Marlin, E.; Baltanas, A.; Toledo, E.; Ferrero, R.; Vinueza-Gavilanes, R.; Larrea, L.; Arrasate, M.; Aragon, T. Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis 2020, 11, 397. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M., et al. Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768-1783. [CrossRef]
- Sidrauski, C.; McGeachy, A.M.; Ingolia, N.T.; Walter, P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Sidrauski, C.; Tsai, J.C.; Kampmann, M.; Hearn, B.R.; Vedantham, P.; Jaishankar, P.; Sokabe, M.; Mendez, A.S.; Newton, B.W.; Tang, E.L., et al. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife 2015, 4, e07314. [CrossRef]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D., et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935-939. [CrossRef]
- Dalla Bella, E.; Bersano, E.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chio, A.; Corbo, M.; Filosto, M.; Giannini, F., et al. The unfolded protein response in amyotrophic later sclerosis: results of a phase 2 trial. Brain 2021, 144, 2635-2647. [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D'Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Popko, B.; Tixier, E.; Roos, R.P. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis 2014, 71, 317–324. [Google Scholar] [CrossRef]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D., et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A 2012, 109, E869-878. [CrossRef]
- Kriss, C.L.; Pinilla-Ibarz, J.A.; Mailloux, A.W.; Powers, J.J.; Tang, C.H.; Kang, C.W.; Zanesi, N.; Epling-Burnette, P.K.; Sotomayor, E.M.; Croce, C.M., et al. Overexpression of TCL1 activates the endoplasmic reticulum stress response: a novel mechanism of leukemic progression in mice. Blood 2012, 120, 1027-1038. [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H., et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772-5781. [CrossRef]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M., et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311-1314. [CrossRef]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J., et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J 2012, 2, e79. [CrossRef]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C., et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem 2011, 286, 12743-12755. [CrossRef]
- Fribley, A.M.; Cruz, P.G.; Miller, J.R.; Callaghan, M.U.; Cai, P.; Narula, N.; Neubig, R.R.; Showalter, H.D.; Larsen, S.D.; Kirchhoff, P.D., et al. Complementary cell-based high-throughput screens identify novel modulators of the unfolded protein response. J Biomol Screen 2011, 16, 825-835. [CrossRef]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.L.; Zhang, Y.; Lee, K.P.; Harding, H.P.; Haynes, C.M.; Price, J.; Sicheri, F.; Ron, D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol Cell 2010, 38, 291–304. [Google Scholar] [CrossRef]
- Canosa, A.; Calvo, A.; Barberis, M.; Brunetti, M.; Restagno, G.; Cammarosano, S.; Ilardi, A.; Vigliani, M.C.; Chio, A.; Moglia, C. Amyotrophic lateral sclerosis onset after prolonged treatment with a VEGF receptors inhibitor. Amyotroph Lateral Scler Frontotemporal Degener 2015, 16, 129–130. [Google Scholar] [CrossRef]
- Fan, F.; Liu, F.; Shen, P.; Tao, L.; Zhang, H.; Wu, H. Salvianolic acid B, a new type I IRE1 kinase inhibitor, abrogates AngII-induced angiogenesis by interacting with IRE1 in its active conformation. Clin Exp Pharmacol Physiol 2023, 50, 82–95. [Google Scholar] [CrossRef]
- Feldman, H.C.; Vidadala, V.N.; Potter, Z.E.; Papa, F.R.; Backes, B.J.; Maly, D.J. Development of a Chemical Toolset for Studying the Paralog-Specific Function of IRE1. ACS Chem Biol 2019, 14, 2595–2605. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H., et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534-548. [CrossRef]
- Wang, Z.; Jiang, C.; Chen, W.; Zhang, G.; Luo, D.; Cao, Y.; Wu, J.; Ding, Y.; Liu, B. Baicalein induces apoptosis and autophagy via endoplasmic reticulum stress in hepatocellular carcinoma cells. Biomed Res Int 2014, 2014, 732516. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.S.; Yen, J.H.; Kou, M.C.; Wu, M.J. Luteolin and Apigenin Attenuate 4-Hydroxy-2-Nonenal-Mediated Cell Death through Modulation of UPR, Nrf2-ARE and MAPK Pathways in PC12 Cells. PLoS One 2015, 10, e0130599. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.X.; Conn, P.M. Pharmacoperones as Novel Therapeutics for Diverse Protein Conformational Diseases. Physiol Rev 2018, 98, 697–725. [Google Scholar] [CrossRef]
- Kieran, D.; Kalmar, B.; Dick, J.R.; Riddoch-Contreras, J.; Burnstock, G.; Greensmith, L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 2004, 10, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci Transl Med 2013, 5, 211ra156. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Batulan, Z.; Taylor, D.M.; Aarons, R.J.; Minotti, S.; Doroudchi, M.M.; Nalbantoglu, J.; Durham, H.D. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol Dis 2006, 24, 213–225. [Google Scholar] [CrossRef]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet 2001, 10, 1307–1315. [Google Scholar] [CrossRef]
- Clinicaltrialls.gov ID: NCT04516096. A Compassionate Use Protocol of AMX0035 for Treatment of Patients With Amyotrophic Lateral Sclerosis (ALS). Available online: https://www.clinicaltrials.gov/study/NCT04516096?term=Amylyx%20AMX0035&rank=1 (accessed on 13/09/2023).
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; Van Remmen, H. Metabolic and Stress Response Changes Precede Disease Onset in the Spinal Cord of Mutant SOD1 ALS Mice. Front Neurosci 2019, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Riar, A.K.; Burstein, S.R.; Palomo, G.M.; Arreguin, A.; Manfredi, G.; Germain, D. Sex specific activation of the ERalpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum Mol Genet 2017, 26, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Cilleros-Holgado, P.; Gomez-Fernandez, D.; Pinero-Perez, R.; Reche-Lopez, D.; Alvarez-Cordoba, M.; Munuera-Cabeza, M.; Talaveron-Rey, M.; Povea-Cabello, S.; Suarez-Carrillo, A.; Romero-Gonzalez, A., et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int J Mol Sci 2023, 24. [CrossRef]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Siegelin, M.D.; Dohi, T.; Raskett, C.M.; Orlowski, G.M.; Powers, C.M.; Gilbert, C.A.; Ross, A.H.; Plescia, J.; Altieri, D.C. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest 2011, 121, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P., et al. Activation of the Mitochondrial Unfolded Protein Response: A New Therapeutic Target? Biomedicines 2022, 10. [CrossRef]
- Lopez-Hurtado, A.; Burgos, D.F.; Gonzalez, P.; Dopazo, X.M.; Gonzalez, V.; Rabano, A.; Mellstrom, B.; Naranjo, J.R. Inhibition of DREAM-ATF6 interaction delays onset of cognition deficit in a mouse model of Huntington's disease. Mol Brain 2018, 11, 13. [Google Scholar] [CrossRef]
- Lopez-Hurtado, A.; Peraza, D.A.; Cercos, P.; Lagartera, L.; Gonzalez, P.; Dopazo, X.M.; Herranz, R.; Gonzalez, T.; Martin-Martinez, M.; Mellstrom, B., et al. Targeting the neuronal calcium sensor DREAM with small-molecules for Huntington's disease treatment. Sci Rep 2019, 9, 7260. [CrossRef]
- Peraza, D.A.; Cercos, P.; Miaja, P.; Merinero, Y.G.; Lagartera, L.; Socuellamos, P.G.; Izquierdo Garcia, C.; Sanchez, S.A.; Lopez-Hurtado, A.; Martin-Martinez, M., et al. Identification of IQM-266, a Novel DREAM Ligand That Modulates K(V)4 Currents. Front Mol Neurosci 2019, 12, 11. [CrossRef]
- Socuellamos, P.G.; Olivos-Ore, L.A.; Barahona, M.V.; Cercos, P.; Perez Pascual, M.; Arribas-Blazquez, M.; Naranjo, J.R.; Valenzuela, C.; Gutierrez-Rodriguez, M.; Artalejo, A.R. IQM-PC332, a Novel DREAM Ligand with Antinociceptive Effect on Peripheral Nerve Injury-Induced Pain. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Olivan, S.; Calvo, A.C.; Rando, A.; Munoz, M.J.; Zaragoza, P.; Osta, R. Comparative study of behavioural tests in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp Anim 2015, 64, 147–153. [Google Scholar] [CrossRef]
- Mellstrom, B.; Kastanauskaite, A.; Knafo, S.; Gonzalez, P.; Dopazo, X.M.; Ruiz-Nuno, A.; Jefferys, J.G.; Zhuo, M.; Bliss, T.V.; Naranjo, J.R., et al. Specific cytoarchitectureal changes in hippocampal subareas in daDREAM mice. Mol Brain 2016, 9, 22. [CrossRef]
Figure 1.
DREAM protein levels are reduced in the spinal cord of SOD1G93A mice. Western blot analysis of DREAM tetramer (A and C) and monomer (A and D) in spinal cord extracts from wild type (WT) or SOD1G93A (SOD1). Representative blot for DREAM monomer and tetramer expression (A) and Coomassie staining (loading control) (B). The results represent the mean ± SEM of 11-12 mice. ****p ≤ 0.001 vs WT (Student´s t-test).
Figure 1.
DREAM protein levels are reduced in the spinal cord of SOD1G93A mice. Western blot analysis of DREAM tetramer (A and C) and monomer (A and D) in spinal cord extracts from wild type (WT) or SOD1G93A (SOD1). Representative blot for DREAM monomer and tetramer expression (A) and Coomassie staining (loading control) (B). The results represent the mean ± SEM of 11-12 mice. ****p ≤ 0.001 vs WT (Student´s t-test).
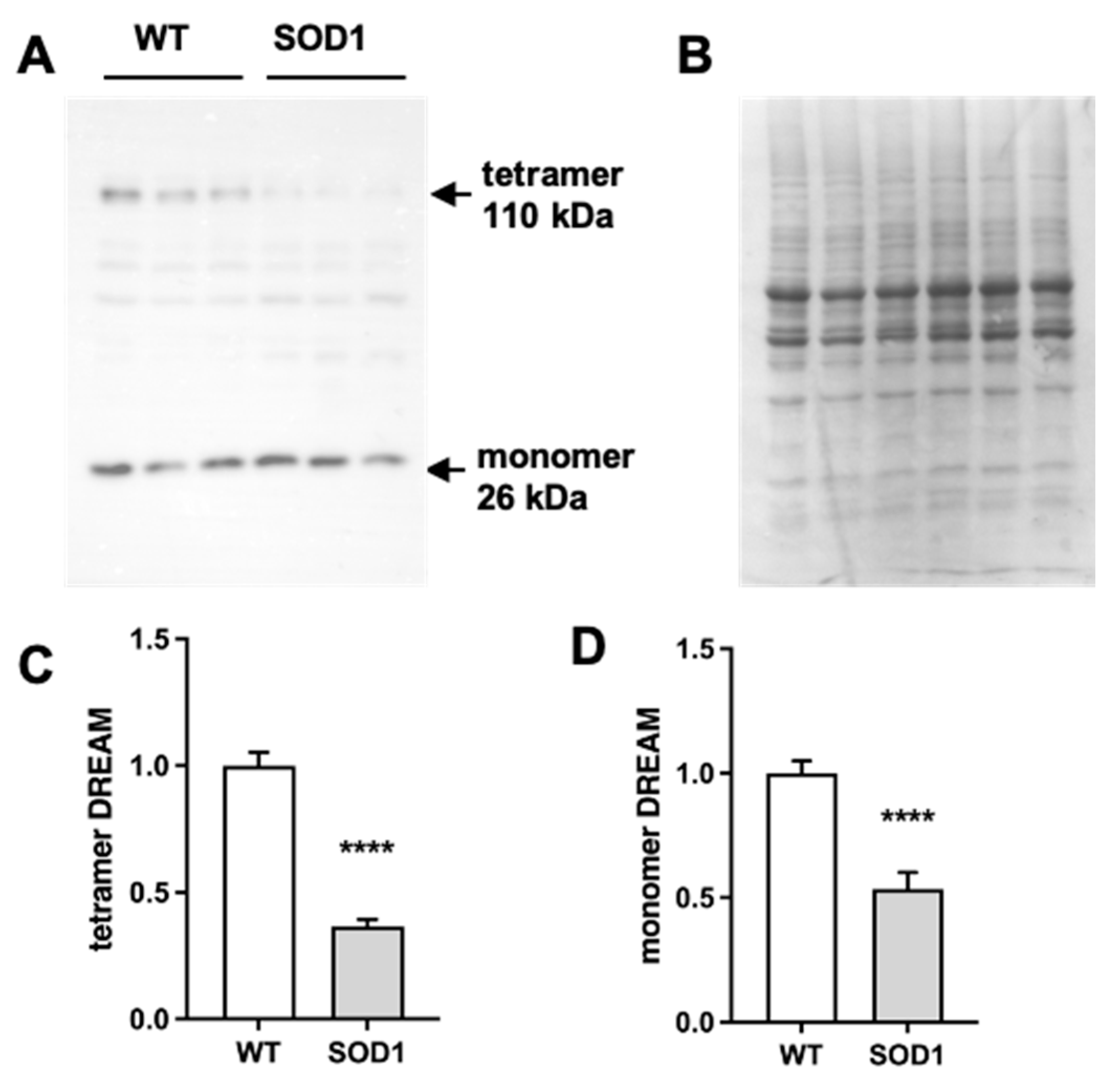
Figure 2.
Repaglinide ameliorates motor strength loss in SOD1G93A mice. Weight, hanging-wire and rotarod variation (%) was monitored from week 10 to 18 in wild type (WT) and SOD1G93A (SOD1) mice treated with DMSO or repaglinide (RP). Col19a1 expression was analyzed in the quadriceps muscle of WT and SOD1 mice treated with DMSO or RP. (A) Significant differences were found in the curves of weight variation between WT and SOD1 animals within each treatment group from week 14 onwards. Repaglinide did not protect from weight loss in SOD1 groups. (B) Similar values were obtained in the rotarod test between SOD1 DMSO and SOD1 RP groups. (C) Repaglinide improved latency in the hanging-wire test of SOD1 mice compared to SOD1 DMSO group. Significant differences were found between both curves. The results represent the mean ± SEM of 14-17 mice in A, 6 mice per group in B and 11 mice per group in C. +p ≤ 0.05 and +++p ≤ 0.001 vs SOD1 DMSO, #p ≤ 0.05 and ###p ≤ 0.001 vs SOD1 RP in A, *p ≤ 0.05 and ***p ≤ 0.001 vs SOD1 DMSO in B. (2-way ANOVA + Holm-Šídák’s test). (D) Real-time qPCR analysis of Col19a1 mRNA in the quadriceps muscle from WT and SOD1 mice receiving DMSO or RP. Values are normalized relative to Hprt mRNA levels. The results represent the mean ± SEM of 6-7 mice. **p ≤ 0.01 vs SOD1 DMSO, +p ≤ 0.05 and +++p ≤ 0.001 vs WT DMSO and #p ≤ 0.05 and ###p ≤ 0.001 vs WT RP (One-way ANOVA followed by the Holm-Šídák´s test).
Figure 2.
Repaglinide ameliorates motor strength loss in SOD1G93A mice. Weight, hanging-wire and rotarod variation (%) was monitored from week 10 to 18 in wild type (WT) and SOD1G93A (SOD1) mice treated with DMSO or repaglinide (RP). Col19a1 expression was analyzed in the quadriceps muscle of WT and SOD1 mice treated with DMSO or RP. (A) Significant differences were found in the curves of weight variation between WT and SOD1 animals within each treatment group from week 14 onwards. Repaglinide did not protect from weight loss in SOD1 groups. (B) Similar values were obtained in the rotarod test between SOD1 DMSO and SOD1 RP groups. (C) Repaglinide improved latency in the hanging-wire test of SOD1 mice compared to SOD1 DMSO group. Significant differences were found between both curves. The results represent the mean ± SEM of 14-17 mice in A, 6 mice per group in B and 11 mice per group in C. +p ≤ 0.05 and +++p ≤ 0.001 vs SOD1 DMSO, #p ≤ 0.05 and ###p ≤ 0.001 vs SOD1 RP in A, *p ≤ 0.05 and ***p ≤ 0.001 vs SOD1 DMSO in B. (2-way ANOVA + Holm-Šídák’s test). (D) Real-time qPCR analysis of Col19a1 mRNA in the quadriceps muscle from WT and SOD1 mice receiving DMSO or RP. Values are normalized relative to Hprt mRNA levels. The results represent the mean ± SEM of 6-7 mice. **p ≤ 0.01 vs SOD1 DMSO, +p ≤ 0.05 and +++p ≤ 0.001 vs WT DMSO and #p ≤ 0.05 and ###p ≤ 0.001 vs WT RP (One-way ANOVA followed by the Holm-Šídák´s test).
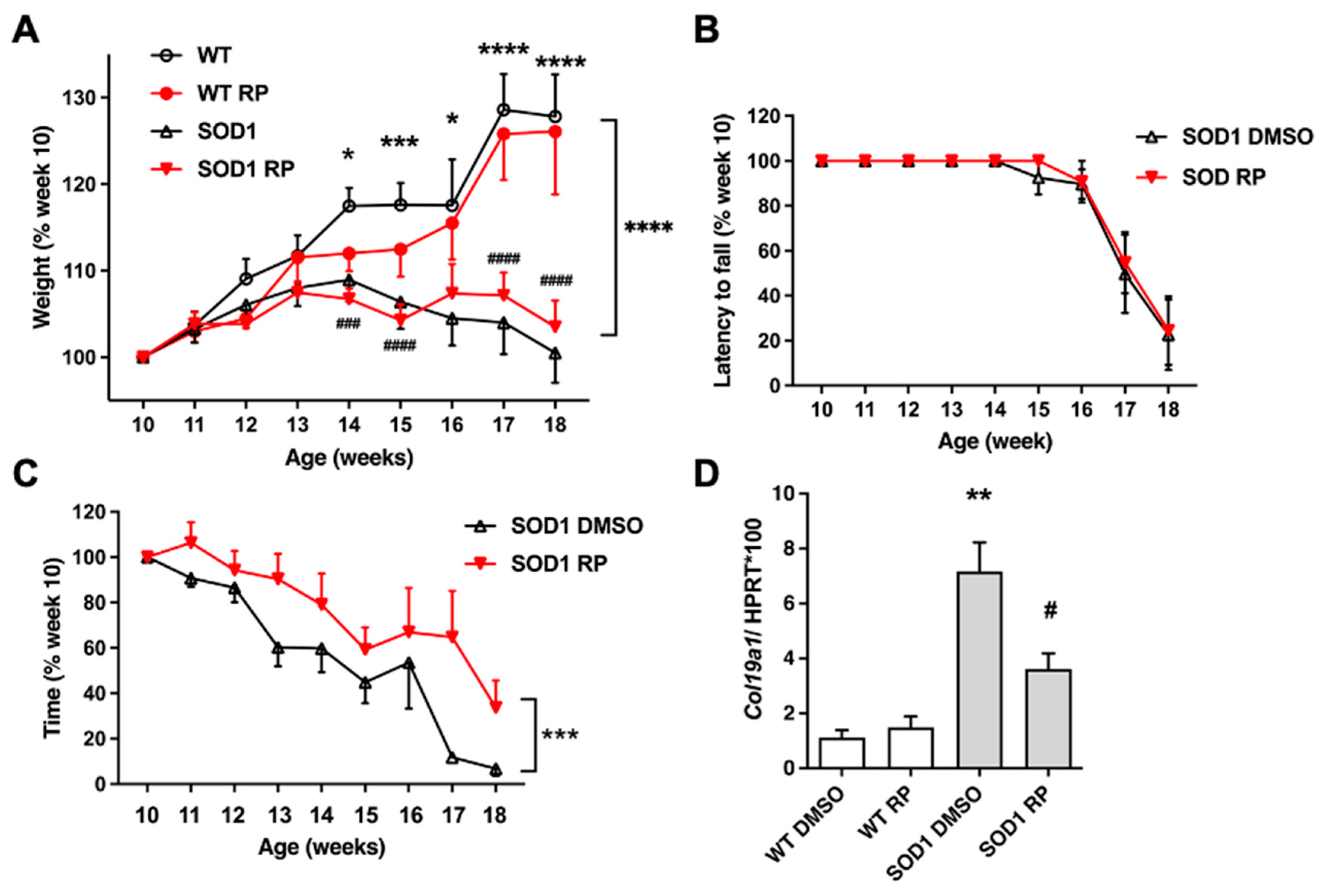
Figure 3.
Repaglinide reduces the loss of motor neurons in the spinal cord of SOD1G93A mice. (A) Representative images of Nissl-stained motor neurons in the ventral horn of the spinal cord. (B) Quantification of motor neurons number. The results represent the mean ± SEM of 5-6 mice. #p ≤ 0.05 vs SOD1 DMSO and ****p ≤ 0.001 vs WT DMSO (One-way ANOVA followed by the Holm-Šídák’s test). Scale bar 50 µm.
Figure 3.
Repaglinide reduces the loss of motor neurons in the spinal cord of SOD1G93A mice. (A) Representative images of Nissl-stained motor neurons in the ventral horn of the spinal cord. (B) Quantification of motor neurons number. The results represent the mean ± SEM of 5-6 mice. #p ≤ 0.05 vs SOD1 DMSO and ****p ≤ 0.001 vs WT DMSO (One-way ANOVA followed by the Holm-Šídák’s test). Scale bar 50 µm.
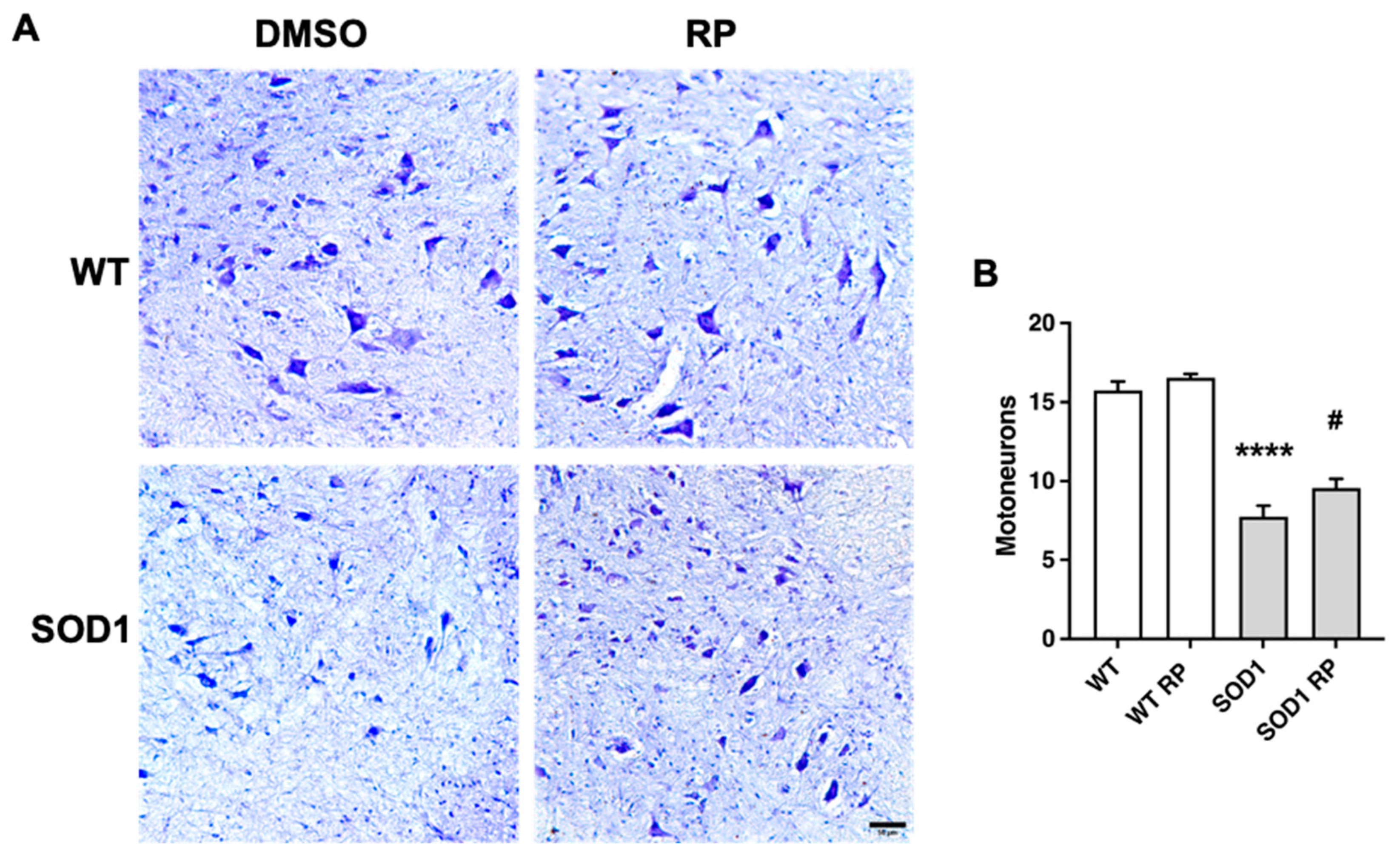
Figure 4.
Repaglinide improves gliosis in the spinal cord of SOD1G93A mice. Representative images of microglial Iba1+ cells (A) and astroglial GFAP+ cells (C) in the ventral horn of the spinal cord. Fluorescence intensity quantification of Iba-1 (B) and GFAP (D) immunoreactivity. The results represent the mean ± SEM of 5-6 mice. #p ≤ 0.05 vs SOD1 DMSO and ****p ≤ 0.001 vs WT DMSO (One-way ANOVA followed by the Holm-Šídák’s test). Scale bar 50 µm.
Figure 4.
Repaglinide improves gliosis in the spinal cord of SOD1G93A mice. Representative images of microglial Iba1+ cells (A) and astroglial GFAP+ cells (C) in the ventral horn of the spinal cord. Fluorescence intensity quantification of Iba-1 (B) and GFAP (D) immunoreactivity. The results represent the mean ± SEM of 5-6 mice. #p ≤ 0.05 vs SOD1 DMSO and ****p ≤ 0.001 vs WT DMSO (One-way ANOVA followed by the Holm-Šídák’s test). Scale bar 50 µm.
Figure 5.
Repaglinide activates ATF6 processing in the spinal cord of SOD1G93A mouse. Western blot analysis of total ATF6 levels (full-length p90 and processed N-terminal p50) (A and C) and ATF6 processing (p50/p90 ratio) (A and D) in spinal cord of WT and SOD1G93A mice receiving DMSO or repaglinide. A representative blot is shown (A). 90 kDa and 50 kDa bands corresponding to full-length ATF6 (p90) or processed N-terminal ATF6 (p50), respectively and Coomassie staining (loading control) is shown (B). The results represent the mean ± SEM of 18 mice analyzed by One-way ANOVA followed by Dunn’s test. * p ≤ 0.05 vs WT DMSO and #p ≤ 0.05 vs SOD1 DMSO.
Figure 5.
Repaglinide activates ATF6 processing in the spinal cord of SOD1G93A mouse. Western blot analysis of total ATF6 levels (full-length p90 and processed N-terminal p50) (A and C) and ATF6 processing (p50/p90 ratio) (A and D) in spinal cord of WT and SOD1G93A mice receiving DMSO or repaglinide. A representative blot is shown (A). 90 kDa and 50 kDa bands corresponding to full-length ATF6 (p90) or processed N-terminal ATF6 (p50), respectively and Coomassie staining (loading control) is shown (B). The results represent the mean ± SEM of 18 mice analyzed by One-way ANOVA followed by Dunn’s test. * p ≤ 0.05 vs WT DMSO and #p ≤ 0.05 vs SOD1 DMSO.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



