
Submitted:
27 September 2023
Posted:
28 September 2023
You are already at the latest version
Abstract
In the present study, the efficiency of a biomimetic approach by coating demineralized bone matrix (DBM) amorphous calcium phosphate (DBM+CaP), including its combination with serum albumin (DBM+CaP+BSA), was investigated. The intact structure of DBM promotes the transformation of amorphous calcium phosphate (CaP) into DPCD with a characteristic plate shape and particle size of 5–35 µm. The inclusion of BSA in the coating resulted in a better and more uniform distribution of CaP on the surface of DBM trabeculae. MG63 cells showed that both the obtained forms of CaP and its complex with BSA did not exhibit cytotoxicity up to a concentration of 10 mg/ml in vitro. Ectopic (subcutaneous) implantation in rats revealed pronounced biocompatibility, as well as strong osteoconductive, osteoinductive, and osteogenic effects for both DBM+CaP and DBM+CaP+BSA, but more pronounced effects for DBM+CaP+BSA. In addition, for the DBM+CaP+BSA samples, a pronounced full physiological intrafibrillar biomineralization and proangiogenic effect with the formation of bone-morrow-like niches, accompanied by pronounced processes of intramedullary hematopoiesis, indicating a powerful osteogenic effect of this composite have been achieved.
Keywords:
bioactive materials
; bone grafts
; remineralization
; coatings
; bone tissue engineering
; demineralized bone matrix
; biomimetic scaffolds
; calcium phosphate-albumin composite
; bone marrow biomimetic
1. Introduction
Ideal bone substitutes are presented by highly porous materials with high biocompatibility, osteoinductivity, osteoconductivity, and osteogenic potential [1,2,3,4].
In current orthopaedic surgery, bone tissue autografts remain the gold standard, as this material already contains all the necessary components for full biontegration of the implanted tissue (living cells, signalling molecules, regulatory extracellular matrix (ECM), etc.). However, well-known complications and inevitable autograft deficiencies have stimulated the development of modern methods of bone tissue engineering, focusing on the creation of bone tissue biomimetics. Such mimetic constructs are required to contain ECM (or its full analog), which simultaneously provides haptotaxis (chemical cues on the surface), durotaxis (mechanical substrate compliance), and topotaxis (geometric features of the substrate) [5], as well as biological and chemical nature-like agents that provide chemotaxis, guided migration, and subsequent differentiation of the patient’s cells. Despite the abundance of synthetic scaffolds, in the above-mentioned context, the demineralized bone matrix has the most pronounced conformity for the provision of topotaxis, haptotaxis, and durotaxis because of its initial optimal geometry, the presence of all necessary signalling functional peptides on the surface of its collagen trabeculae, and because it is a physiological base for the precipitation of amorphous calcium phosphate, resulting in an optimal and specific hydroxyapatite for natural mechanisms of biomineralization. In addition, biological and chemical agents used for chemotaxis and differentiation of the recipient’s cells should also be natural, but remain affordable and controllable in terms of their activity (which is an issue, for instance, in the case of rhBMP-2 or stem cells) [6,7,8,9]. Thus, the utilized bioactive agents can be represented by calcium phosphates preceding the final form of hydroxyapatite (amorphous calcium phosphate, DCPD, TCP, OCP, etc.), as well as bioactive molecules, proteins, and lipids, which participate in the regeneration processes and normal osteogenesis [7,8,9,10].
Considering the deficiency of allogenic tissues in obtaining natural bone substitutes, it appears logical to use demineralized and antigen-free xenogeneic bone tissue [11,12,13]. The demineralized bone matrix consists almost entirely of type I collagen [14,15]. In addition to that, “hole zones” of bone collagen fibrils are not shielded by glycosaminoglycans leading to nearly non-energy-consuming adsorption of amorphous calcium phosphates with a formation of hydroxyapatite crystals being the terminal and bioinert structure, mechanically stabilizing the supporting bone tissue and presenting the main depot of calcium and phosphates in the organism [16,17]. The triple helix zone of collagen demonstrated a high degree of evolutionary stability, with variations in the amino acid content in different species of mammals not exceeding several percent [18]. It is worth noting that antigenicity and immunogenicity of collagen are caused in the first place by the release of antigenic determinants, which are normally hidden epitopes that interact with antibodies only after the triple helix is unfolded; that is, immunogenic factors can be only damaged and/or denatured collagen [19,20]. Another important consideration is the use of low-temperature calcium phosphates because of the inability of high-temperature synthesis in the body, which would be atypical for the organism and may mediate foreign body reactions and fibrous encapsulation [21,22]. The low-temperature synthesis of calcium phosphate imitates the physiological inorganic components of native bone tissue. At the same time, the use of hydroxyapatite precursors, but not hydroxyapatite itself, appears to be the most logical, as amorphous calcium phosphates are not only used to form tissue-specific hydroxyapatite but also directly drive the differentiation of migrating recipient’s cells [23,24].
Therefore, composite biomimetic bone tissue-like materials may be an effective osteoplastic material as a balanced complex of low-temperature calcium phosphates (precursors of hydroxyapatite) and additional bioactive natural proteins and lipids precipitated on the highly purified intact and non-immunogenic extracellular bone matrix with a preserved fibrillar structure. Such biomimetics of bone tissue should act as a kind of “primers” for the triggering of all processes necessary for bone tissue regeneration but performed directly by the body itself.
In our previous study, we took the first attempted to develop a material based on demineralized extracellular bone matrix with maximum preserved ultrastructure and the possibility of fine regulation of the physicochemical parameters of low-temperature DCPD deposition [25]. In the present work, the efficiency of the proposed approach was investigated in vitro and in vivo by exploiting precipitation of demineralized bone matrix amorphous calcium phosphate, including its combination with serum albumin.
2. Materials and Methods
2.1. Preparation of Demineralized bone matrixes
Initial reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used exactly as delivered. Except as noted below, all compounds and solvents used in this study were bought commercially and used without additional purification.
Using the author’s method, demineralized bone matrix (DBM) was produced (patent RU 2686309 C1, 04.25.2019). Adult bovine xenogeneic cancellous bone tissue underwent a multistage processing procedure that included complete decellularization and demineralization of the bone tissue while maintaining the greatest amount of microarchitectonics and structure of the fibrillar collagen matrix. Decellularized and demineralized three-dimensional porous bone-collagen blocks with axial dimensions of 1 × 1 × 0.5 cm were produced after carrying out all required operations.
For in vitro and in vivo experiments, the obtained blocks were sterilized by incubation in sterile phosphate-buffered saline (PBS) with the addition of antibiotics and antimycotics. The blocks were submerged in PBS containing antibiotics (gentamicin sulfate (0.02 mg/mL) (Sigma-Aldrich, Saint Louis, MO, USA), fluconazole (0.04 mg/mL) (Pfizer, Paris, France), ciprofloxacin (0.008 mg/mL) (Sigma-Aldrich, Saint Louis, MO, USA)) and incubated for 48 h at 37°C in a shaker incubator (Biosan, Riga, Latvia, USA) with continuous shaking. The blocks were washed three times in sterile PBS with a pH of 7.4 following a 24-hour period.
2.2. Quantification of Calcium and DNA Content
Using a Merck pestle microhomogenizer (Millipore-Sigma, USA), 10 mg tissue fragments (n = 5) were obtained from each group of samples before and after decellularization and homogenized in microtubes. The DNeasy Blood & Tissue Kit (QIAGEN, Limburg, Netherlands) was used to extract DNA from the tissue homogenate. Identical procedures were performed on an empty test tube used as a control, as they were on the experimental test tubes. The DNA in the resulting solutions was measured at 260 nm using a NanoVue Plus spectrophotometer (Biochrom, Holliston, USA).
Using absorption spectroscopy, the calcium concentrations of the control (native bone) and DBM samples both before and after implantation were identified. The material samples were dried in a hot-air sterilizer (Binder, Tuttlingen, Germany) for 12 h at 90°C before their dry weights were calculated. Subsequently, for 24 h at 20–25 °C, each sample was dissolved in 1 ml of 1M HCl. Calcium AS DiaS Arsenazo III kit (DiaSys, Holzheim, Germany) was used to measure the amount of dissolved calcium. The microplate reader Infinite F200 (Tecan, Männedorf, Switzerland) was used to quantify the optical density. Calcium mineralization values in the samples (µg calcium per mg of sample dry weight) were computed according to the manufacturer’s instructions.
2.3. Remineralization of DBM
The preparation of calcium phosphate coating was carried out under conditions that mimic physiological conditions as much as possible. The investigated samples were obtained as follows. In sterile conditions, samples (5 × 5 × 5 mm) were obtained by cutting bone blocks (DBM) with similar porosity and size. Separate solutions for remineralization were prepared, each with a volume of 30 ml. The components were added in the following order: distilled water, phosphate ion solution (0.588 M NH4H2PO4), bovine serum albumin (BSA)* (4% solution, *for DBM+CaP+BSA samples), the calcium ion solution (1 M Ca(NO3)24H2O). The resulting suspensions were rapidly mixed and used to fill fragments of DBM in 50 ml test tubes. The contents of the test tubes were subjected to vacuuming for 30 minutes at 20 mbar using a Millivac Maxi membrane pump (Millipore, USA). After vacuuming, the test tubes were sealed and placed in a shaking incubator at a temperature of 37°C and 60 RPM.
2.4. X-ray Diffraction Analysis and FTIR Spectroscopy
The phase compositions were investigated using a Shimadzu XRD-6000 diffractometer (Shimadzu, Tokyo, Japan) equipped with an automated imaging system that allowed data collection, graphical processing, and identification of the phases obtained from the JCPDS 2003 data bank. Powder-dried materials were subjected to X-ray phase analysis for general phase analysis using CuKα monochromatic radiation.
The normalized Chang technique was used to calculate the mass fractions of the samples. The Jana 2006 program was used to fully analyze the diffraction pattern for this purpose (wRp = 1.89%). The JCPDS 2003 data bank descriptions of the corundum number phases and nonoverlapping main peak intensities were used. Lattice constants were calculated in CelRef program using several peaks: (020), (021), (041), (-221), (151). Calculated results are in good agreement with values obtained in Jana2006.
The infrared (FT-IR) spectra of the tablets were recorded on an Avatar 330 FT-IR spectrometer (Thermo Nicolet Corporation, Madison, WI, USA) in the 4000–400 cm-1 wavelength region. Potassium bromide was combined with a small amount of powder (1 mg of powder in 50 mg of spectroscopic-grade KBr), and the mixture was pressed into a pellet. The pellets were examined at 20°C in the transmission mode of the main box.
2.5. MicroCT and Microstructure Analysis
In order to provide a detailed analysis of the morphological and density characteristics of porosity and thickness of materials in the Comprehensive TEX Archive Network (CT-an) program, microcomputer tomography (microCT) was performed on microtomography “SKYSCAN 1275” (Bruker micro-CT, Kontich, Belgium), with a resolution of 4.5 microns. The images were taken using a 13.76 m voxel size and 0.73° with NReconTM v.1.6.8.0, SkyScan, 2011 (Bruker, Kontich, Belgium). In the reconstruction, ring artifacts and beam-hardening corrections were made. The Data Viewer TM 1.4.4.0 program (Bruker, Kontich, Belgium) was then used to realign the reconstructed images. The quality of the specimens and their interior architectonics were investigated using this technique.
Scanning electron microscopy (Tescan VEGA III, Brno, Czech Republic) and energy-dispersive spectroscopy (EDS; INCA Energy Oxford Instruments, Abing-don, UK) were used to analyze the microstructure and morphology of the sample surfaces and slices. Before analysis, the samples were coated with gold using a Sputter Coater Q150R (Quorum Technologies, Lewes, UK). At pressures of 7.3 10-2 Pa in the column and 1.5 10-1 Pa in the chamber, surface pictures of the materials were produced. Calcium-to-phosphorus ratios were calculated using energy-dispersive X-ray spectroscopy and Oxford AZ-tecO 4.3 software (Oxford Instruments NanoAnalysis, High Wycombe, UK).
2.6. Cell Culture
Human osteoblast-like cells, MG-63, were obtained from the ATCC (Manassas, VA, USA). The cells were cultivated in the EMEM nutrient medium (Sigma-Aldrich, Milwaukee, WI, USA), supplemented with heat-inactivated fetal bovine serum (Gibco, Waltham, MA, USA) to a final concentration of 10% and 40 μg/mL gentamicin sulfate (Sigma-Aldrich, St. Louis, MO, USA), under conditions of 5% CO2 content in the air and at 37 °C. The cell cultures were tested for mycoplasma infection using the MycoFluor™Mycoplasma Detection Kit, and no mycoplasma was detected.
2.7. Cell Viability Assay
In vitro experiments with human osteoblast-like MG-63cells were seeded in an amount of 5 × 103 cells in 100 µL of complete growth medium into 96-well plates at different concentrations of calcium phosphate compounds (Corning Inc., Corning, NY, USA). After 24 h of cultivation, the medium was replaced with 100 μL of medium containing the above-mentioned СaP or CaP+BSA, at concentrations of 10, 3, 1, 0.3, and 0.1 mg/mL, and the cultivation was continued for 24 and 96 hours. The cells in the control conditions were cultured in the medium without the addition of CPC. СaP and CaP+BSA samples were pre-sterilized with 75% ethanol according to the indicated method [26].
Cell viability after incubation with CaP and CaP+BSA was evaluated by AlamarBlue (Invitrogen, Carlsbad, CA, USA). 100 μg/mL of AlamarBlue was added to the cells after 24 and 96 hours of incubation. The cells were then incubated for 4 hours at 37 C and 5% CO2 content in the air, then the fluorescence intensity was measured at an excitation wavelength of 560 nm and an emission wavelength of 595 nm using an Infinity F 200 plate reader (Tecan, Männedorf, Switzerland). Cell viability was assessed by the mean fluorescence intensity (MFI) of the resulting resofurin product. The viability of control cells not incubated with CaP and CaP+BSA was taken as 100%. Cell viability after incubation with CaP and CaP+BSA was calculated as a percentage relative to control by the formula: Cell viability% = (MFI cells after incubation with CaP and CaP+BSA/MFI control cells) * 100%. Evaluation of the effect of CaP and CaP+BSA on cells was conducted using a trypan blue exclusion assay [27].
2.8. Fluorescence microscopy
After 96 h of CPC incubation, the morphological state of the cells in culture was examined by staining the cells with the fluorescent dye Hoechst 33342 (blue nuclei of living and dead cells), Propidium Iodide (PI, red nuclei of dead cells), and Calcein AM (green cytoplasm of living cells). Hoechst 33342 1 μg/mL (Sigma-Aldrich, St. Louis, MO, USA), PI 1 μg/mL (Sigma-Aldrich, St. Louis, MO, USA), and Calcein AM 2 mM (Sigma-Aldrich, St. Louis, MO, USA) were added to the growth medium and incubated for 30 min at 37°C in a CO2 incubator (Binder GmbH, Tuttlingen, Germany). The cell-filled plate and investigated samples were moved into a microscope chamber at 37°C and 5% CO2 content. Microscopic analysis of the stained cell cultures and micro-images was performed using a Nikon Eclipse Ti-E microscope (Nikon, Tokyo, Japan).
2.9. Animals and Surgical Procedures
We used 18 Wistar male rats, aged two months and weighing 180-200 g. The animals were kept separately in temperature-controlled rooms (22°C) and fed a normal diet with unrestricted access to food and water. Experiments were conducted according to the Rules for Studies with Experimental Animals (Decree of the Russian Ministry of Health on August 12, 1997; No. 755). The Institute of Theoretical and Experimental Biophysics Commission on Biological Safety and Ethics approved this protocol in February 2018 (Protocol N15/2018). The rats were divided into three groups, with six rats in each group, and independent replicates were performed for each group.
This was performed using a widely known model of ectopic (subcutaneous) biomaterial implantation. This model most accurately captures the desired outcomes for confirming the osteoinductive and osteogenic capabilities of materials because it offers findings initiated by the substance alone rather than under the impact of the natural bone microenvironment [25,28,29,30].
The surgeries were performed under general anesthesia with Xylazine 13 μg/kg (Interchemie, Netherlands) and Zoletil 7 μg/kg (Virbac, Carros, France). To implant the samples, a 1.5-cm-wide transverse skin incision was made in the dorsal inter-scapular area and subcutaneous pockets were formed parallel to the skin using a smooth trocar, followed by implantation of the specimens at a depth of at least 2 cm from the incision line. Samples were implanted with full interstitial contact without restriction chambers or meshes. The first group of rats was implanted with DBM. The second group was implanted samples of DBM+CaP, and the third group was implanted samples of DBM+CaP+BSA. The animals were placed in front of a heating plate until they awakened for post-surgical recuperation.
Seven weeks (50 days) following the surgical treatment, the animals from each group were split at random and put to death using the carbon dioxide protocol. Immediately after humane euthanasia, to prevent autolysis, samples of implanted materials with surrounding recipient’s tissues were washed for 30 s with a cold (14 °C) isotonic solution and fixed for 48 h in neutral buffered formalin (NBF) at a tissue-fixator volume ratio of 1:30.
2.10. Histological Analysis
After the termination of fixation, the fragments of samples were washed with distilled water (3 × 3 min) to remove excessive phosphates and placed for no less than 12 h in medium Optimum Cutting Temperature (O.С.T.) Compound Tissue Tek (Sakura, Tokyo, Japan). Cross slices of DBM-samples (9 μm) were prepared by cryosectioning (MEV SLEE medical GmbH, Germany). The staining of samples was performed by a conventional method using H&E (Mayer’s Hematoxylin and Eosin Y), and differential staining for calcium deposits Alizarin red S (by the McGee-Russell method [31]) and collagen/non-collagen structures (by Lillie`s trichrome method) [31]. The micrographs of stained histological samples were obtained on a Nikon Eclipse Ti-E microscope station (Nikon, Tokyo, Japan) and processed using the software NIS Elements AR4.13.05 (Build 933).
2.11. Statistical Analysis
Results are presented as the mean ± standard deviation (M ± SD). Each of the in vitro experiments was carried out at least four times (n ≥ 4). The statistical significance of the difference was determined using Mann–Whitney U test.
The design of the experiment and related statistics (U test) were carried out using Python 3 (ver. 3.10.10) in development environment Spyder (v. 5.4.1) with libraries Pandas (v. 1.5.2), Numpy (v.1.24.2) and Scipy (v. 1.10.0). Plots were created using Python 3 (ver. 3.10.10) with libraries Seaborn (v. 0.12.2) and Matplotlib (v. 3.7.0).
3. Results
3.1. Results of efficiency evaluation of Bone matrix demineralization
After multistep treatment of xenogenic bone matrix (decellularization and de-mineralization of bone tissue protocol), it was found that residual calcium content in the samples did not exceed 0.83±0.41 µg/mg of tissue dry mass, with a residual donor cell DNA content of 2.38±0.76 ng/mg tissue mass, indicating a high degree of purification of the xenogenic collagen bone matrix.
3.2. Results of physico-chemical analysis and morphology
The phase compositions of the obtained samples were investigated by X-ray diffraction (XRD) and Fourier-transform infrared spectroscopy (FTIR). Full-profile analysis was performed using Jana2006 software, and refinement and indexing were carried out using WinXPow software. In both experiments, a single-phase crystalline DCPD was obtained, and the diffraction patterns corresponded to card № 9-0077 of the XRD base ICDD (Powder Diffraction File, Alphabetical Index Inorganic Compounds, Pennsylvania: JCPDS, 1997). The main peaks of DCPD ((020), (021), (041), (2 ̅21) etc.) are shown in Figure 1. The calculated lattice parameters are listed in Table 1. The lattice parameters for the BMD+CaP+BSA samples did not significantly differ from the tabulated values, indicating that BSA was absorbed on the surface of phosphates and no fragments were embedded in the structure [32].
The IR spectroscopy results were in good agreement with the XRD results. DCPD spectrum has characteristic modes of the HPO42- group at 875 and 987 cm-1 (ν1), 987, 1059, and 1135 cm-1 (ν3), and 525, 576, and 662 cm-1 (ν4). The stretching modes of the O-H bond of lattice water appear at 3550-3160 cm-1, bending modes at 1649 cm-1. H2O libration peak was observed at 791 cm-1. The peaks of absorbed water were observed at 2385 and 1718 cm-1 [33]. For the DBM+CaP+BSA samples, the modes characteristic of the Ca2+-BSA complex appear in the range of 1700-1200 cm-1. The peptide bonds of BSA exhibit three bands in the carbonyl absorption region, known as amides I, II, and III (indicated by blue lines in Figure 1) [34]. The band at 1648 belongs to the lattice water and amide I (C=O stretching), amide II appears as a mode at 1538 cm-1 (C–N stretching coupled to N–H bending), and peaks of amide III are observed at 1384 cm-1 (C–H in-plane bending vibration) and 1221 cm-1 (C–N stretching) [35,36]. The peak at 1221 cm-1 also corresponds to P-OH bending.
Scanning electron microscopy data confirmed the results obtained by spectral methods. Figure 2 shows that typical brushite particles are deposited in the DBM porous matrix. DCPD particles are represented by a lamellar form that is characteristic of calcium phosphate. The particle size varied from 5 µm to 35 µm. According to the MicroCT data, the inclusion of BSA in the coating composition led to a more uniform distribution on the surface of the sample pores. Violation of the matrix and destruction of the walls during the deposition process were not observed.
3.3. In vitro cytocompatibility evaluation results
The influence of CaP and its complex, CaP+BSA, on MG-63 cell viability was investigated. It was shown that the viability of cells incubated with CaP, as well as with CaP+BSA, did not differ from the control cells incubated without calcium phosphates in all the ranges of studied concentrations and experimental time points (24 and 96 h) (Figure 4a,b).
Morphological analysis of MG-63 cells cultivated with CaP and CaP+BSA confirmed these results. Figure 4c shows microscopic images of cells cultivated with CaP and CaP+BSA at maximum concentrations of 10, 3, and 1 mg/mL. The absence of dead cells stained with PI was observed in all concentrations. Moreover, cells cultivated with calcium phosphates had a normal fusiform-elongated morphology, which indicated the absence of calcium phosphate-related stress. Thus, the obtained results indicate the cytocompatibility of the studied calcium phosphates and their complexes with BSA.
3.4. Results of the assessment of biocompatibility, osteoconductive, and osteoinductive potential of sammples in vivo
3.4.1. DBM implantation results
The high biocompatibility of the implanted DBM and the absence of its osteoinductive and osteogenic potential were defined after 7-week ectopic implantation (Figure 5a-c). Preservation of the collagen matrix after decellularization and demineralization of bone tissue was indicated by the absence of both enzymatic and cell-mediated resorption of the implanted DBM (Figure 5a-c). No leukocyte or foreign-body giant cell (FBGC) invasion was observed in the implanted material, as well as degenerated mast cells in or around the specimens (Figure 5b). Signs of neocollagenesis were extremely poor, were observed only at the periphery of the sample, and were initiated by the remodeling processes of the peri-implant tissue (influence of the host tissue), but not by the DBM itself (Figure 5c). Additionally, there was weak growth of reactively altered connective tissue inside the specimen from the periphery of the peri-implant tissue. Complete involution of the fibrous capsule by the studied implantation period was identified, which evidences the biointegration of the implanted material. However, no signs of DBM mineralization were found, indicating the absence of both synthetic and mineralizing activities of osteoblasts (Figure 5a). Therefore, pure demineralized collagenous bone matrix is a highly biocompatible, non-immunogenic, osteoconductive, and biologically stable scaffold with no osteoinductive and osteogenic properties.
3.4.2. DBM+CaP implantation results
In contrast to the DBM specimens, in the implantation of DBM+CaP, a different pattern was observed (Figure 5d-f). In this group, a high degree of intratrabecular biomineralization without any signs of mineralized matrix degradation by osteoclasts was observed, that is, the nature of biomineralization was purely physiological (Figure 5d). Biovisualization of the obtained histological images showed that the grade of intratrabecular mineralization of the DBM was > 70%. In addition, there were no signs of synthetic activity of cells migrating in the matrix, and the cell response was steady and balanced (Figure 5f). At the same time, no negative adipocyte differentiation or positive osteoblast differentiation was observed (synthetic phenotype) (Figure 5e). It seems that in this pattern of remineralization, the matrix is accepted by the body as its own, and mineralization is carried out passively, with no mineralizing osteoblasts taking part. In addition, among the migrated cells, there were no histio-lymphocyte cells, mast cells, or FBGC; all cells observed in the matrix were mesenchymal cells (Figure 1e). Involution of the fibrous capsule by the observation period was also completed, which indicated the pronounced biocompatibility of the studied materials. Thus, remineralized by the proposed approach, DBM is an efficient material with a high degree of osteointegration and is a full-fledged osteogenic primer to initiate further regenerative processes in peri-implant tissue.
3.4.3. DBM+CaP+BSA implantation results
A remarkable pattern was found after seven weeks after implantation of the DBM + CaP +BSA specimens. The materials of this group were completely and physiologically intratrabecularly mineralized, and biovisualization of the obtained histological images showed that the degree of intramolecular mineralization of the bulk DBM collagen was almost 90% (Figure 5g).
The specimens of this group demonstrated extraordinarily active processes of chemo- and haptotaxis, migration, intense proliferation, and synthetic activity of cells inside the implanted materials (Figure 5g-i). Wherein, in the structure of the material, the formation of multicellular endosteal bone-marrow-like niches with a large number of myeloid precursors and erythroblastic islands was observed, accompanied by pronounced processes of intramedullary hematopoiesis (the samples were “sprinkled” of erythrocytes) with the formation of both a large number of venous sinuses and mature definitive vessels [37]. At the same time, there were no signs of proteolytic and cell-associated matrix degradation, or its destruction by aseptic utilization calcinosis, that is, this material, despite the active cell response, was indeed favored by the body. In contrast to our previous results, where we observed the indications of pronounced synthetic activity of osteoblasts and DBM remodelling in conformity with the host body, in this case, the matrix itself did not undergo remodelling and was accepted in full, and the synthetic cell activity was mostly connected to the formation of bone marrow-like structures and a complete vascular network. At the same time, given that the newly formed multicellular intertrabecular structures contained relatively large newly formed definitive blood vessels, it can be assumed that immature osteoblasts and M2 macrophages differentiated on the matrix actively produce angiogenic factors [38,39,40,41]. In addition, no negative signs of alterations in healthy tissues surrounding the implant or ectopic aseptic calcification of the peri-implant tissue were found, which indicates a strict material-associated localization of the identified effects.
In Figure 6, in the high magnification, it is visible that the formation of the multi-cellular endosteal bone marrow niches has been carried out (as in normal scenario) in the center of the intertrabecular space (Figure 6a,b, arrows indicate erythroblastic is-lands) surrounded by relatively “calm” mesenchymal cells, wherein the formation of neocollagenous matrix was observed mostly for the newly-formed complete blood vessels and sinuses (Figure 6с, arrows indicate formed definitive vessels).
Thus, remineralized with low-temperature CaP and BSA DBM possesses a high degree of biointegration and is an extremely bioactive osteogenic material that can fully integrate into the recipient’s bone tissue and initiate further regenerative processes, localizing its “on itself.”
In summary, we can assume that our proposed method of remineralization of highly purified demineralized bone matrix using low-temperature calcium phosphate compounds (HAp precursors), including the addition of serum albumin, is an effective and promising approach for obtaining osteoplastic materials with a pronounced osteogenic effect.
Such an active cellular response requires a separate and thorough study to determine whether this approach is acceptable for clinical use. For instance, these materials are hardly applicable in traumatology and maxillofacial surgery, where the formation of bone tissue with potential bone marrow is undesirable. It may be tentatively assumed that the use of such bioactive materials in spinal surgery or surgical correction of nonunions may be useful because such materials can simultaneously be both an effective osteogenic scaffold and a source of progenitor cells without additional costs for preliminary isolation, replication, and introduction of isogenic progenitor cells of the recipient in the scaffold.
On the other hand, as described by Torisawa et al. (2014), obtaining bone marrow mimetics for studying hematopoietic niche physiology in vitro [42] points to the fact that the approach used in our work may both decrease costs and increase the production efficiency of “bone marrow-on-a-chip” constructs in vitro, which also requires additional detailed investigations.
4. Discussion
The data obtained indicate that the biomimetic approach in the creation of osteoplastic materials is promising and should be based on three main “pillars”: (1) the use of a highly purified non-immunogenic extracellular bone matrix with preserved fibrillar structure and spatial architectonics, (2) the use of low-temperature calcium phosphate compounds as precursors of hydroxyapatite, and (3) the use of natural bioactive agents necessary for bone tissue regeneration.
The exploitation of intact demineralized bone collagen matrix is of utmost importance because this material can be completely purified and controlled for infections. At the same time, the intact extracellular bone matrix is non-immunogenic and can therefore undergo full remodelling and be incorporated into the body without the stage of intermediate resorption, which can significantly reduce the time of bone tissue regeneration.
Although various bone structures differentiate depending on the type of bone tissue (compact, spongy, etc.), the structure of the mineralized fibrils is conservative; thus, it acts as a universal elementary building block of bone [43]. Therefore, the proposed approach can be extended to other types of demineralized bone tissues to create a wider range of osteogenic materials.
Interesting results are presented by data on the implantation of a material remineralized with albumin, which makes us appreciate this simple protein as a promising tissue engineering material.
Albumin is the dominant plasma protein, constituting approximately 50% of the total protein concentration in blood plasma and up to 75% of colloidal activity. Albumin is a monomeric multidomain macromolecule that determines the oncotic pressure of plasma and the distribution of fluid between organs and tissues of the body, according to the classical Starling principle [44,45,46]. Under physiological conditions, there is a net movement of albumin from the intravascular to the interstitial space and back through lymphatic vessels [47]. Simultaneously, albumin itself can directly affect the integrity of blood vessels by binding to the interstitial matrix and subendothelium and changing the permeability of these layers for large molecules and solutes [48,49].
The concentration of albumin in the blood plasma is approximately 0.6 mM (4% wt/wt), but for clinical use, even 20% (wt/wt) solutions are utilized. Albumin is a classic pleiotropic protein that performs several important functions in the body. Under physiological conditions, HSA has the exceptional ability to bind ligands, providing a depot and carriage of endogenous and exogenous compounds such as metal ions, cholesterol [50], thyroxine [51], bilirubin and bile acids [52,53,54], nitric oxide [55], amino acids, and fatty acids [56]. In addition, almost 35 proteins and many peptides are associated with HSA (e.g., angiotensinogen, apolipoproteins, ceruloplasmin, clusterin, haemoglobin, plasminogen, prothrombin, and transferrin) [57], while the fraction of such peptides and proteins are collectively called “albumin” [58]. In addition, after acute hemolysis (due to trauma or post-ischemic reperfusion), albumin binds to the released heme and promotes its transfer to hemopexin, which provides receptor-mediated reuptake of heme by parenchymal liver cells [57,59] and which may be important for orthopaedic surgery and traumatology.
In addition, albumin can bind to almost all known drugs, as well as many nutraceuticals and toxic substances, largely determining their pharmacokinetics and toxicokinetics [60,61]. Albumin is known to provide the highest antioxidant capacity in human plasma. In addition, albumin is the main extracellular source of reduced sulfhydryl groups, which are present in the unprotected cysteine residue at position 31. These sulfhydryl groups, called thiols, are active scavengers of reactive oxygen species (ROS) and nitrogen species (RNS), especially superoxide hydroxyl and peroxynitrite radicals [45,62]. Albumin can also limit the production of these reactive compounds by binding to free Cu2+, an ion known to be particularly important for accelerating free radical production [66]. In addition, albumin is involved in the modulation of the immune response [45], neutralizes and removes potential toxins [57,61], and exhibits (pseudo)enzymatic properties and peroxidase activity [63] toward lipid hydroperoxides [44,64,65,66,67,68,69,70,71,72,73].
The role of albumin in calcium distribution is important. Normally, it is albumin that is an important carrier of Ca2+ in blood plasma, however, the affinity of albumin for Ca2+ binding is relatively weak (Kd 0,67 мМ, Ka = 1,5×103 M-1), and only about 45% of 2.4 mM circulating Ca2+ binds to albumin [74]. It has long been believed that albumin transports Ca2+ through carboxylate side chains on its surface [75,76]. In 1971, Pedersen showed that calcium can bind reversibly to serum albumin via 12 ± 1 independent binding sites with an association constant of 90–100 L/mol at 37°C in unbuffered solutions at pH 7.4, and an ionic strength of 0.15–0.16 [77]. Despite the work of Pedersen, only three selective Ca2+-binding sites in BSA were identified in 2012, all of which are located in domain I [78]. Only recently [79], almost 50 years after Pedersen’s work, 19 free and 11 stable bound Ca2+ docking sites (including the original three from the crystal structure) were additionally revealed, and a calcium-dependent change in albumin conformation was identified. This work clearly shows that the amount of Ca2+ ions binding to BSA increases with an increase in calcium concentration with a logarithmic-type dependence. However, in the proposed approach, the material worked not only as a calcium-binding and antioxidant protein, but also as a powerful osteogenic calcium-protein complex.
Based on the results of a literature analysis, it was revealed that albumin is physiologically present in the bone tissue and is the first protein secreted by bone cells in the case of bone damage. To obtain a graft that is very similar to the native tissue, it is necessary to replenish the albumin content in bone grafts [80]. It was also shown that during the healing of fractures in the femoral-diaphyseal tissues of rats, the expression of albumin significantly increased in the area of the fracture, while the content of albumin during cultivation significantly increased in the presence of parathyroid hormone (1-34), IGF-1, and zinc acexamate [81]. Albumin is also been shown to be expressed in osteoblasts and plays a role in regulating the expression of Runx2 or alpha1(I) collagen mRNA, which may be mediated by the intracellular mitogen-activated protein kinase (MAP-kinase) cascade in osteoblasts [82]. The number of osteoblast cells significantly increased when cultured in the presence of albumin (1.0 mg/ml) in vitro for 24-72 h, whereas this effect of albumin was completely eliminated in the presence of PD98049, staurosporine, or dibucaine, which are inhibitors of various protein kinases, and cycloheximide or 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole (DRB), which are inhibitors of transcriptional activity [83].
Many studies have also shown that the application of an albumin coating on the surface of hyaline cartilage xenografts causes a weakening of immune and inflammatory responses at the cellular, protein, and gene levels, and significantly fewer inflammatory cells (including neutrophils, macrophages, and lymphocytes) in coated xenogeneic materials, accompanied by significantly lower expression of genes encoding inflammation-associated cytokines, including MCP-1, IL-6, and IL-1β [84]. It has also been shown that albumin coatings improve bio- and immune compatibility, tissue formation, corrosion resistance, and antibacterial and anti-clotting properties of materials [85,86,87,88,89,90,91,92]. Albumin has been described in most studies as a protein that inhibits cell adhesion on inert surfaces; however, it is a potent cell adhesive in more physiological scaffolds such as mineralized bone allografts [93,94,95].
Kang et al. [96] showed that Ad-MSCs from adipose tissue showed a more homogeneous distribution and osteogenic differentiation on a porous albumin scaffold with collagen I gel than without collagen I gel. ALP activity and mineralization of the extracellular matrix in the construct with type I collagen were significantly higher than in the construct without type I collagen (p < 0.05). Thus, the combination of collagen I gel and serum albumin scaffolds has been shown to enhance osteogenic differentiation and homogeneous distribution of Ad-MSCs. Another study [89] showed that after in vitro incubation with MSCs, albumin-coated grafts recruited approximately twice as many cells as uncoated grafts.
Several studies have shown that albumin improves biointegration by improving the adhesion and proliferation of MSCs on mineralized human bone allografts and stimulating regeneration processes in peri-implant tissue [97,98,99,100,101]. In a double-blind randomized trial [102] it was demonstrated that albumin-coated grafts had the lowest level of postoperative pain, and after 6 and 12 weeks, there were signs of tissue remodelling, while uncoated xenografts were still separated from the host bone; after a year of implantation, complete remodelling and integration with the natural trabecular structure were revealed. Another study [97] focused on the co-cultivation of hADSCs and monocytes in vitro and showed that monocytes have the ability to degrade uncoated bovine bone pellets and that the albumin coating protects such grafts from degradation. Simultaneously, for samples coated with albumin, a significant decrease in the production of ROS and RNS and mitigation of gene expression of mitochondrial electron transport chain complexes was observed. Among the five complexes of the electron transport chain, the sites with the highest ability to produce ROS are complexes I and III [103]. Complex I produces ROS in the matrix, whereas complex III produces ROS both in the matrix and on the inner side of the membrane [103,104]. Moderate levels of mitochondrial ROS have recently been found to directly stimulate the production of proinflammatory cytokines, thus regulating the inflammatory response [105]. Cytokine analysis showed that the cultivation of stem cells and monocytes on albumin-coated grafts led to an increase in the levels of targeted cytokines (HGF, PGE-2, and IL-10) compared with uncoated xenografts, even under conditions that simulate inflammation [97]. Interestingly, the albumin coating was effective only on mineralized allogeneic human bone materials and demineralized bovine bone but not on hydroxyapatite scaffolds [106,107]. Another study [108] showed that albumin adsorbed on hydroxyapatite promotes significantly higher levels of cell adhesion than albumin adsorbed on control surfaces.
Several studies have shown the antimicrobial properties of albumin coating by preventing bacterial adhesion on the surface of implants coated with albumin [109,110,111]. Albumin also has antithrombotic and anticoagulant effects, possibly owing to its ability to bind to nitric oxide (NO) to form S-nitrosothiols [112], This inhibits the rapid inactivation of NO and prolongs its anti-aggregation effect on platelets [113,114]. Preliminary application of albumin to the implant surface (“albumin passivation”) effectively prevents platelet activation by creating a thin protein layer on the surface, which increases the hydrophilicity of the surface and prevents a biological reaction after contact with the blood of a hydrophobic material [92,115,116]. Materials with adsorbed native albumin have been shown to reduce the number of adherent platelets and their activation on surfaces. However, when the albumin structure was altered by crosslinking, the platelets were able to fully adhere and become activated to the modified albumin layer [88].
It has also been shown that the activity of the oxidative burst of neutrophils is noticeably lower when incubated with albumin but much higher when incubated with artificial colloids and crystalloids [117]. HSA has also been shown to suppress the respiratory release of neutrophils in response to exposure to cytokines relevant to the pathogenesis of critical illnesses (in particular, TNF) and complement components (e.g., C5A). Moreover, human serum albumin selectively and reversibly inhibits TNF-induced neutrophil spreading and the associated decrease in cAMP [118].
All the above studies show that albumin, along with all the previously mentioned properties, can also have pronounced positive effects, including (immunomodulatory functions) which are crucial for healing and tissue regeneration [63]. Thus, based on literature analysis and our data, we can assume that our proposed method of remineralizing highly purified DBM using low-temperature HAp precursors and serum albumin may be an effective and promising approach for obtaining osteoplastic materials with a pronounced osteogenic effect.
5. Conclusions
Composite bone tissue-like biomimetic constructs may be highly effective osteoplastic materials. These materials are required to contain ECM (or its full analog), which simultaneously provides haptotaxis, durotaxis, and topotaxis as well as biological and chemical nature-like agents that provide chemotaxis, guided migration, and subsequent differentiation of the patient’s cells. In addition, these biomimetic constructs must include low-temperature calcium phosphates (precursors of hydroxyapatite), bioactive natural proteins, and lipids that precipitate on the extracellular bone matrix. Such composite biomimetics should act as a kind of “primers” for triggering all processes necessary for bone tissue regeneration but should be performed directly by the organism itself. In the present study, the efficiency of a similar approach was investigated. The results showed pronounced biocompatibility and strong proangiogenic and osteogenic effects for both DBM+CaP and DBM+CaP+BSA, but more pronounced effects for DBM+CaP+BSA with the formation of bone-morrow-like niches in this composite. Thus, it can be stated that the use of a triad in the form of an intact demineralized bone matrix and low-temperature calcium phosphate compounds (HAp precursors) in combination with albumin is a promising and inspiring approach that can provide osteoinductive, osteoconductive, and angiogenic potentials, biological safety, easy market availability, long shelf life, and reasonable cost.
Author Contributions
Conceptualization, I.S.F., V.V.M. and A.Yu.T.; methodology, R.S.F., A.Yu.T., V.V.M. and K.A.M.; software, A.S.S. and K.S.K.; validation, R.S.F., A.S.S. and V.S.A.; formal analysis, O.A.K., M.A.S. and A.I.Z.; investigation, K.A.M., M.I.K., Y.V.L., I.V.S., P.V.S., K.V.P., P.S.S. and A.S.S.; resources, I.S.F., R.S.F. and V.S.K.; data curation, R.S.F., A.Yu.T., V.V.M., M.I.K., K.A.M. and V.S.A.; writing—original draft preparation, I.S.F., A.Yu.T., V.V.M., I.V.S. and K.A.M.; writing—review and editing, I.S.F., A.Yu.T. and R.S.F.; visualization, V.V.M., A.Yu.T., K.A.M., K.S.K. and I.S.F.; supervision, V.S.A. and V.S.K.; project administration, I.S.F. and A.S.S.; funding acquisition, V.S.A. and V.S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Science Foundation (RSF, № 22-73-00215).
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of Institute of Theoretical and Experimental Biophysics of Russian Academy of Sciences (protocol #N15/2018 from 01 February 2018).
Data Availability Statement
Data available on request from the authors.
Acknowledgments
We thank Ms. Kirsanova P.O. for performing part of the pre-analytical stage of histological analysis of pure DBM. The work was carried out using equipment from the Center for Collective Use of Research Equipment at the ITEB RAS and IMET RAS.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Busch, A.; Jäger, M.; Mayer, C.; Sowislok, A. Functionalization of Synthetic Bone Substitutes. Int J Mol Sci. 2021, 22, 4412. [Google Scholar] [CrossRef]
- Yang, Y.; Chu, C.; Xiao, W.; Liu, L.; Man, Y.; Lin, J.; Qu, Y. Strategies for advanced particulate bone substitutes regulating the osteo-immune microenvironment. Biomed Mater. 2022, 17, 022006. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Merchán, E.C. Bone Healing Materials in the Treatment of Recalcitrant Nonunions and Bone Defects. Int J Mol Sci. 2022, 23, 3352. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, M.L.; Dias, G.J.; Premadasa, K.M.G.P. A novel classification of bone graft materials. J Biomed Mater Res B Appl Biomater. 2022, 110, 1724–1749. [Google Scholar] [CrossRef]
- SenGupta, S.; Parent, C.A.; Bear, J.E. The principles of directed cell migration. Nat Rev Mol Cell Biol. 2021, 22, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Riederman, B.D.; Butler, B.A.; Lawton, C.D.; Rosenthal, B.D.; Balderama, E.S.; Bernstein, A.J. Recombinant human bone morphogenetic protein-2 versus iliac crest bone graft in anterior cervical discectomy and fusion: Dysphagia and dysphonia rates in the early postoperative period with review of the literature. J Clin Neurosci. 2017, 44, 180–183. [Google Scholar] [CrossRef]
- Crawford, C.H. 3rd; Carreon, L.Y.; McGinnis, M.D.; Campbell, M.J.; Glassman, S.D. 3rd; Carreon, L.Y.; McGinnis, M.D.; Campbell, M.J.; Glassman, S.D. Perioperative complications of recombinant human bone morphogenetic protein-2 on an absorbable collagen sponge versus iliac crest bone graft for posterior cervical arthrodesis. Spine (Phila Pa 1976) 2009, 34, 1390–1394. [Google Scholar] [CrossRef]
- Lu, D.C.; Tumialán, L.M.; Chou, D. Multilevel anterior cervical discectomy and fusion with and without rhBMP-2: a comparison of dysphagia rates and outcomes in 150 patients. J Neurosurg Spine 2013, 18, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Selph, S.; McDonagh, M.; Peterson, K.; Tiwari, A.; Chou, R.; Helfand, M. Effectiveness and harms of recombinant human bone morphogenetic protein-2 in spine fusion: a systematic review and meta-analysis. Ann Intern Med. 2013, 158, 890–902. [Google Scholar] [CrossRef]
- Riederman, B.D.; Butler, B.A.; Lawton, C.D.; Rosenthal, B.D.; Balderama, E.S.; Bernstein, A.J. Recombinant human bone morphogenetic protein-2 versus iliac crest bone graft in anterior cervical discectomy and fusion: Dysphagia and dysphonia rates in the early postoperative period with review of the literature. J Clin Neurosci. 2017, 44, 180–183. [Google Scholar] [CrossRef]
- Malagón-Escandón, A.; Hautefeuille, M.; Jimenez-Díaz, E.; Arenas-Alatorre, J.; Saniger, J.M.; Badillo-Ramírez, I.; Vazquez, N.; Piñón-Zarate, G.; Castell-Rodríguez, A. Three-Dimensional Porous Scaffolds Derived from Bovine Cancellous Bone Matrix Promote Osteoinduction, Osteoconduction, and Osteogenesis. Polymers (Basel) 2021, 13, 4390. [Google Scholar] [CrossRef] [PubMed]
- Al Qabbani, A.; Rani, K.G.A.; Syarif, J.; AlKawas, S.; Sheikh Abdul Hamid, S.; Samsudin, A.R.; Azlina, A. Evaluation of decellularization process for developing osteogenic bovine cancellous bone scaffolds in-vitro. PLoS One 2023, 18, e0283922. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ham, A.; Aguilar-Medina, M.; León-Félix, J.; Romero-Quintana, J.G.; Bermúdez, M.; López-Gutierrez, J.; Jiménez-Gastélum, G.; Avendaño-Félix, M.; Lizárraga-Verdugo, E.; Castillo-Ureta, H.; López-Camarillo, C.; Ramos-Payan, R. Extracellular matrix hydrogel derived from bovine bone is biocompatible in vitro and in vivo. Biomed Mater Eng. 2022, 33, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wei, M. Biomineralization of Collagen-Based Materials for Hard Tissue Repair. Int J Mol Sci. 2021, 22, 944. [Google Scholar] [CrossRef]
- Collagen. Available online: URL https://encyclopedia.pub/entry/2457 (accessed on 14 June 2023).
- Oosterlaken, B.M.; Vena, M.P.; de With, G. In Vitro Mineralization of Collagen. Adv Mater. 2021, 33, e2004418. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, W.; Wang, Z.; Yang, Y.; Sahai, N. Structure analysis of collagen fibril at atomic-level resolution and its implications for intra-fibrillar transport in bone biomineralization. Phys Chem Chem Phys. 2018, 20, 1513–1523. [Google Scholar] [CrossRef]
- Lynn, A.K.; Yannas, I.V.; Bonfield, W. Antigenicity and immunogenicity of collagen. J Biomed Mater Res B Appl Biomater. 2004, 71, 343–354. [Google Scholar] [CrossRef]
- Kasravi, M.; Ahmadi, A.; Babajani, A.; Mazloomnejad, R.; Hatamnejad, M.R.; Shariatzadeh, S.; Bahrami, S. , Niknejad, H. Immunogenicity of decellularized extracellular matrix scaffolds: a bottleneck in tissue engineering and regenerative medicine. Biomater Res. 2023, 27, 10. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Li, R.; Bai, H.; Zhu, Z.; Zhu, L.; Zhu, C.; Che, Z.; Liu, He.; Wang, J.; Huang, L. Collagen-based biomaterials for bone tissue engineering. Materials & Design 2021, 210, 110049. [Google Scholar]
- Trindade, R.; Albrektsson, T.; Tengvall, P.; Wennerberg, A. Foreign Body Reaction to Biomaterials: On Mechanisms for Buildup and Breakdown of Osseointegration. Clin Implant Dent Relat Res. 2016, 18, 192–203. [Google Scholar] [CrossRef]
- Brennan, M.Á.; Monahan, D.S.; Brulin, B.; Gallinetti, S.; Humbert, P.; Tringides, C.; Canal, C.; Ginebra, M.P.; Layrolle, P. Biomimetic versus sintered macroporous calcium phosphate scaffolds enhanced bone regeneration and human mesenchymal stromal cell engraftment in calvarial defects. Acta Biomater. 2021, 135, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, O.; Shiwaku, Y.; Hamai, R. Octacalcium phosphate bone substitute materials: Comparison between properties of biomaterials and other calcium phosphate materials. Dent. Mater. J. 2020, 39, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Almulhim, K.S.; Syed, M.R.; Alqahtani, N.; Alamoudi, M.; Khan, M.; Ahmed, S.Z.; Khan, A.S. Bioactive Inorganic Materials for Dental Applications: A Narrative Review. Materials (Basel). 2022, 15, 6864. [Google Scholar] [CrossRef] [PubMed]
- Fadeeva, I.S.; Teterina, A.Y.; Minaychev, V.V.; Senotov, A.S.; Smirnov, I.V.; Fadeev, R.S.; Smirnova, P.V.; Menukhov, V.O.; Lomovskaya, Y.V.; Akatov, V.S.; Barinov, S.M.; Komlev, V.S. Biomimetic Remineralized Three-Dimensional Collagen Bone Matrices with an Enhanced Osteostimulating Effect. Biomimetics (Basel). 2023, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Z.; Jiang, T.; Zou, X.; Lei, L.; Yan, W.; Yang, J.; Li, B. Strontium-substituted biphasic calcium phosphate microspheres promoted degradation performance and enhanced bone regeneration. J Biomed Mater Res A. 2020, 108, 895–905. [Google Scholar] [CrossRef]
- Ligasová, A.; Koberna, K. DNA Dyes-Highly Sensitive Reporters of Cell Quantification: Comparison with Other Cell Quantification Methods. Molecules. 2021, 26, 5515. [Google Scholar] [CrossRef]
- Teterina, A.Y.; Minaychev, V.V.; Smirnova, P.V.; Kobiakova, M.I.; Smirnov, I.V.; Fadeev, R.S.; Egorov, A.A.; Ashmarin, A.A.; Pyatina, K.V.; Senotov, A.S.; et al. Injectable Hydrated Calcium Phosphate Bone-like Paste: Synthesis, In Vitro, and In Vivo Biocompatibility Assessment. Technologies 2023, 11, 77. [Google Scholar] [CrossRef]
- Barradas, A.M.; Yuan, H.; van Blitterswijk, C.A.; Habibovic, P. Osteoinductive biomaterials: Current knowledge of properties, experimental models and biological mechanisms. Eur. Cell Mater 2011, 21, 407–429. [Google Scholar] [CrossRef]
- Habibovic, P.; de Groot, K. Osteoinductive biomaterials—Properties and relevance in bone repair. J. Tissue Eng. Regen. Med. 2007, 1, 25–32. [Google Scholar] [CrossRef]
- Lillie, R.D.; Fullmer, H.M. Histopathologic Technic and Practical Histochemistry; McGraw-Hill: New York, NA, USA, 1976; p. 942. [Google Scholar]
- Xiao, D.; Zhang, J.; Zhang, C.; Barbieri, D.; Yuan, H.; Moroni, L.; Feng, G. The role of calcium phosphate surface structure in osteogenesis and the mechanisms involved. Acta Biomater. 2020, 106, 22–33. [Google Scholar] [CrossRef]
- Rabadjieva, D.; Sezanova, K.; Gergulova, R.; Titorenkova, R.; Tepavitcharova, S. Precipitation and phase transformation of dicalcium phosphate dihydrate in electrolyte solutions of simulated body fluids: Thermodynamic modeling and kinetic studies. J Biomed Mater Res A 2020, 108, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Lenk, T.J.; Ratner, B.D.; Gendreau, R.M.; Chittur, K.K. IR spectral changes of bovine serum albumin upon surface adsorption. J Biomed Mater Res. 1989, 23, 549–569. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yang, X.; Ji, Z.; Zhu, L.; Ma, N.; Chen, D.; Jia, X.; Tang, J.; Cao, Y. DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N-Methylacetamide Fine Components. ACS Omega. 2020, 5, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Alhazmi, H.A. FT-IR spectroscopy for the identification of binding sites and measurements of the binding interactions of important metal ions with bovine serum albumin. Scientia Pharmaceutica 2019, 87, 5. [Google Scholar] [CrossRef]
- Figueiredo, J.F.; Mattix, M.E.; Papenfuss, T.L. Hematopoietic System. In Atlas of Histology of the Juvenile Rat, 1nd ed.; Picut, C.A., Ed.; Elsevier Inc.: San Diego, CA, U.S.A, 2016; pp. 349–371. [Google Scholar]
- Huang, J.-H.; He, H.; Chen, Y.-N.; Liu, Z.; Romani, M.D.; Xu, Z.-Y.; Xu, Y.; Lin, F.-Y. Exosomes derived from M2 Macrophages Improve Angiogenesis and Functional Recovery after Spinal Cord Injury through HIF-1α/VEGF Axis. Brain Sci. 2022, 12, 1322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, T.; Wu, T.; Xu, W.; Dou, G.; Wang, Y.; Guo, C. M2 macrophages promote vasculogenesis during retinal neovascularization by regulating bone marrow-derived cells via SDF-1/VEGF. Cell Tissue Res 2020, 380, 469–486. [Google Scholar] [CrossRef]
- Czekanska, E.; Stoddart, M.; Richards, R.; Hayes, J. In search of an osteoblast cell model for in vitro research. Eur. Cell. Mater. 2012, 24, 1–17. [Google Scholar] [CrossRef]
- Venkataiah, V.S.; Yahata, Y.; Kitagawa, A.; Inagaki, M.; Kakiuchi, Y.; Nakano, M.; Suzuki, S.; Handa, K.; Saito, M. Clinical Applications of Cell-Scaffold Constructs for Bone Regeneration Therapy. Cells 2021, 10, 2687. [Google Scholar] [CrossRef]
- Torisawa, Y.S.; Spina, C.S.; Mammoto, T.; Mammoto, A.; Weaver, J.C.; Tat, T.; Collins, J.J.; Ingber, D.E. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat Methods 2014, 11, 663–669. [Google Scholar] [CrossRef]
- Nudelman, F.; Lausch, A.J; Sommerdijk, N.A.; Sone, E.D. In vitro models of collagen biomineralization. J Struct Biol. 2013, 183, 258–269. [Google Scholar] [CrossRef]
- Peters, T. Jr. All about Albumin: Biochemistry, Genetics and Medical Applications. Academic Press: San Diego, USA, 1996; pp. 9–250.
- Evans, T.W. Review article: albumin as a drug – biological effects of albumin unrelated to oncotic pressure. Aliment. Pharmacol. Ther. 2002, 16 (Suppl. 5), 6–11. [Google Scholar] [CrossRef] [PubMed]
- Mendez, C.M.; McClain, C.J.; Marsano, L.S. Albumin therapy in clinical practice. Nutr. Clin. Pract. 2005, 20, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Gyntelberg, F. Transcapillary escape rate of albumin and plasma volume in essential hypertension. Circ. Res. 1973, 32, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Vick, J.; Vargas, F.F. Albumin modulation of paracellular permeability of pig vena caval endothelium shows specificity for pig albumin. Am J Physiol. 1993, 264, H1382–H1387. [Google Scholar] [CrossRef] [PubMed]
- Qiao, R.; Siflinger-Birnboim, A.; Lum, H.; Tiruppathi, C.; Malik, A.B. Albumin and Ricinus communis agglutinin decrease endothelial permeability via interactions with matrix. Am J Physiol. 1993, 265, C439–C446. [Google Scholar] [CrossRef]
- Sankaranarayanan, S.; Llera-Moya, M.; dela, Drazul-Schrader, D. ; Phillips, M.C.; Kellner-Weibel, G.; Rothblat, G.H. Serum albumin acts as a shuttle to enhance cholesterol efflux from cells. J. Lipid Res. 2013, 54, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Petitpas, I.; Petersen, C.E.; Ha, C.E.; Bhattacharya, A.A.; Zunszain, P.A.; Ghuman, J.; Bhagavan, N.V.; Curry, S. Structural basis of albumin-thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 6440–6445. [Google Scholar] [CrossRef]
- Ahlfors, C.E.; Wennberg, R.P. Bilirubin-albumin binding and neonatal jaundice. Semin Perinatol. 2004, 28, 334–339. [Google Scholar] [CrossRef]
- Pico, G.A.; Houssier, C. Bile salts-bovine serum albumin binding: spectroscopic and thermodynamic studies. Biochim Biophys Acta. 1989, 999, 128–134. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Shen, Y.; Zhu, Q.; Ding, F. Superior Dialytic Removal of Bilirubin and Bile Acids by Free Fatty Acid Displacement and Its Synergy With Albumin-Based Dialysis. ASAIO J. 2023, 69, 127–135. [Google Scholar] [CrossRef]
- Stamler, J.S.; Jaraki, O.; Osborne, J.; Simon, D.I.; Keaney, J.; Vita, J.; Singel, D.; Valeri, C.R.; Loscalzom, J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. U.S.A 1992, 89, 7674–7677. [Google Scholar] [CrossRef] [PubMed]
- van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Gundry, R.L.; Fu, Q.; Jelinek, C.A.; Van Eyk, J.E.; Cotter, R.J. Investigation of an albumin-enriched fraction of human serum and its albuminome. Proteomics Clin. Appl. 2007, 1, 73–88. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Bolli, A.; Fasano, M.; Notari, S.; Polticelli, F. (). Allosteric modulation of monomeric proteins. Biochemistry and Molecular Biology Education, 2005, 33, 169–176. [Google Scholar] [CrossRef]
- Roche, M.; Rondeau, P.; Singh, N.R.; Tarnus, E.; Bourdon, E. The antioxidant properties of serum albumin. FEBS Lett. 2008, 582, 1783–1787. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N.V. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int J Mol Sci 2021, 22, 10318. [Google Scholar] [CrossRef]
- Lee, H. , Cha M.K., Kim I.H. Activation of thiol-dependent antioxidant activity of human serum albumin by alkaline pH is due to the B-like conformational change. Arch. Biochem. Biophys. 2000, 380, 309–318. [Google Scholar] [CrossRef]
- Hurst, R.; Bao, Y.; Ridley, S.; Williamson, G. Phospholipid hydroperoxide cysteine peroxidase activity of human serum albumin. Biochem. J. 1999, 338, 723–728. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: from bench to bedside. Mol Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar] [PubMed]
- Carter, D.C.; Ho, J.X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. [Google Scholar]
- Bertucci, C.; Domenici, E. Reversible and covalent binding of drugs to human serum albumin: methodological approaches and physiological relevance. Curr. Med. Chem. 2002, 9, 1463–1481. [Google Scholar] [CrossRef]
- Curry, S. Beyond expansion: structural studies on the transport roles of human serum albumin. Vox Sang 2002, 83 (Suppl. 1), 315–319. [Google Scholar] [CrossRef] [PubMed]
- Curry, S. Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug. Metab. Pharmacokinet. 2009, 24, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U.; Chuang, V.T.; Otagiri, M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002, 25, 695–704. [Google Scholar] [CrossRef]
- Sakurai, Y.; Ma, S.F.; Watanabe, H.; Yamaotsu, N.; Hirono, S.; Kurono, Y.; Kragh-Hansen, U.; Otagiri, M. Esterase-like activity of serum albumin: characterization of its structural chemistry using p-nitrophenyl esters as substrates. Pharm. Res. 2004, 21, 285–292. [Google Scholar] [CrossRef]
- Sułkowska, A.; Bojko, B.; Równicka, J.; Sułkowski, W.W. Competition of cytarabine and aspirin in binding to serum albumin in multidrug therapy. Biopolymers 2006, 81, 464–472. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Kragh-Hansen, U. , Vorum H. Quantitative analyses of the interaction between calcium ions and human serum albumin. Clin. Chem. 1993, 39, 202–208. [Google Scholar] [CrossRef]
- Sokołowska, M.; Wszelaka-Rylik, M.; Poznański, J.; Bal, W. Spectroscopic and thermodynamic determination of three distinct binding sites for Co(II) ions in human serum albumin. J. Inorg. Biochem. 2009, 103, 1005–1013. [Google Scholar] [CrossRef]
- Eatough, D.J.; Jensen, T.E.; Hansen, L.D.; Loken, H.F.; Rehfeld, S.J. The binding of Ca2+ and Mg2+ to human serum albumin: a calorimetric study. Thermochim. Acta 1978, 25, 289–297. [Google Scholar] [CrossRef]
- Pedersen, K.O. Binding of calcium to serum albumin. I. Stoichiometry and intrinsic association constant at physiological pH, ionic strength, and temperature. Scand J Clin Lab Invest. 1971, 28, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol Immunol, 2012; 52(3-4), 174–182. [Google Scholar]
- Patel, D.; Haag, S.L.; Patel, J.S.; Ytreberg, F.M.; Bernards, M.T. Paired Simulations and Experimental Investigations into the Calcium-Dependent Conformation of Albumin. J Chem Inf Model. 2022, 62, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Márton, K.; Tamás, S.B.; Orsolya, N.; Béla, C.; Ferenc, D.; Péter, N.; Csaba, D.N.; Lajos, C.; Zsombor, L.; Eitan, M.; György. S. Materials (Basel), 2018, 11, E202. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Igarashi. A.; Misawa. H.; Tsurusaki. Y. Enhancement of albumin expression in bone tissues with healing rat fractures. J Cell Biochem. 2003, 89, 356–363. [Google Scholar] [CrossRef]
- Ishida, K.; Yamaguchi, M. Albumin regulates Runx2 and alpha1 (I) collagen mRNA expression in osteoblastic cells: comparison with insulin-like growth factor-I. Int J Mol Med. 2005, 16, 689–694. [Google Scholar]
- Ishida, K.; Yamaguchi, M. Role of albumin in osteoblastic cells: enhancement of cell proliferation and suppression of alkaline phosphatase activity. Int J Mol Med. 2004, 14, 1077–1081. [Google Scholar] [CrossRef]
- Tao, C.; Zhu, W.; Iqbal, J.; Xu, C.; Wang, D.A. Stabilized albumin coatings on engineered xenografts for attenuation of acute immune and inflammatory responses. J Mater Chem B. 2020, 8, 6080–6091. [Google Scholar] [CrossRef]
- Horváthy, D.B.; Simon, M.; Schwarz, C.M.; Masteling, M.; Vácz, G.; Hornyák, I.; Lacza, Z. Serum albumin as a local therapeutic agent in cell therapy and tissue engineering. BioFactors 2016, 43, 315–330. [Google Scholar] [CrossRef]
- Sanganeria, P.; Sachar, S.; Chandra, S.; Bahadur, D.; Ray, P.; Khanna, A. Cellular internalization and detailed toxicity analysis of protein-immobilized iron oxide nanoparticles. J. Biomed. Mater. Res. Part B Appl. Biomater, 2015; 103, 125–134. [Google Scholar]
- Ostroverkhov, P.; Semkina, A.; Nikitin, A.; Smirnov, A.; Vedenyapina, D.; Vlasova, K.; Kireev, I.; Grin, M.; Chekhonin, V.; Majouga, A.; Abakumov, M. Human serum albumin as an effective coating for hydrophobic photosensitizes immobilization on magnetic nanoparticles. J. Magn. Magn. Mater. 2019, 475, 108–114. [Google Scholar] [CrossRef]
- Amiji, M.; Park, H.; Park, K. Study on the prevention of surface-induced platelet activation by albumin coating. J. Biomater. Sci. Polym. Ed. 1992, 3, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Horváthy, D.B.; Schandl, K.; Schwarz, C.M.; Renner, K.; Hornyák, I.; Szabó, B.T.; Niculescu-Morzsa, E.; Nehrer, S.; Dobó-Nagy, C.; Doros, A.; Lacza, Z. Serum albumin–coated bone allograft (BoneAlbumin) results in faster bone formation and mechanically stronger bone in aging rats. J. Tissue Eng. Regen. Med. 2019, 13, 416–422. [Google Scholar] [CrossRef]
- Horváthy, D.B.; Vácz, G.; Cselenyák, A.; Weszl, M.; Kiss, L.; Lacza, Z. Albumin-coated bioactive suture for cell transplantation. Surg Innov. 2013, 20, 249–255. [Google Scholar] [CrossRef]
- Horváthy, D.B.; Vácz, G.; Szabó, T.; Szigyártó, I.C.; Toró, I.; Vámos, B.; Hornyák, I.; Renner, K.; Klára, T.; Szabó, B.T.; Dobó-Nagy, C.; Doros, A.; Lacza, Z. Serum albumin coating of demineralized bone matrix results in stronger new bone formation. J. Biomed. Mater. Res. Part B Appl. Biomater. 2016, 104, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cui, L.; Ren, Y.; Zou, Y.; Ma, J.; Wang, C.; Zheng, Z.; Chen, X.-B.; Zeng, R.; Zheng, Y. In vitro and in vivo biodegradation and biocompatibility of an MMT/BSA composite coating upon magnesium alloy AZ31. J. Mater. Sci. Technol. 2020, 47, 52–67. [Google Scholar] [CrossRef]
- Yamazoe, H.; Tanabe, T. Preparation of water-insoluble albumin film possessing nonadherent surface for cells and ligand binding ability. J. Biomed. Mater. Res. Part A. 2008, 86, 228–234. [Google Scholar] [CrossRef]
- Yamazoe, H.; Uemura, T.; Tanabe, T. Facile Cell Patterning on an Albumin-Coated Surface. Langmuir. 2008, 24, 8402–8404. [Google Scholar] [CrossRef]
- Wei, J.; Yoshinari, M.; Takemoto, S.; Hattori, M.; Kawada, E.; Liu, B.; Oda, Y. Adhesion of mouse fibroblasts on hexamethyldisiloxane surfaces with wide range of wettability. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 81B, 66–75. [Google Scholar] [CrossRef]
- Kang, B.J.; Kim, Y.; Lee, S.H.; Kim, W.H.; Woo, H.M.; Kweon, O.K. Collagen I gel promotes homogenous osteogenic differentiation of adipose tissue-derived mesenchymal stem cells in serum-derived albumin scaffold. J Biomater Sci Polym Ed. 2013, 24, 1233–1243. [Google Scholar] [CrossRef]
- Mijiritsky, E.; Gardin, C.; Ferroni, L.; Lacza, Z.; Zavan, B. Albumin-impregnated bone granules modulate the interactions between mesenchymal stem cells and monocytes under in vitro inflammatory conditions. Mater. Sci. Eng. C. 2020, 110, 110678. [Google Scholar] [CrossRef] [PubMed]
- Skaliczki, G.; Schandl, K.; Weszl, M.; Major, T.; Kovács, M.; Skaliczki, J.; Szendrői, M.; Dobó-Nagy, C.; Lacza, Z. Serum albumin enhances bone healing in a nonunion femoral defect model in rats: A computer tomography micromorphometry study. Int. Orthop. 2013, 37, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Weszl, M.; Skaliczki, G.; Cselenyák, A.; Kiss, L.; Major, T.; Schandl, K.; Bognar, E.; Stadler, G.; Peterbauer, A.; Csonge, L.; Lacza, Z. Freeze-dried human serum albumin improves the adherence and proliferation of mesenchymal stem cells on mineralized human bone allografts. J. Orthop. Res. 2012, 30, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Schandl, K.; Horváthy, D.B.; Doros, A.; Majzik, E.; Schwarz, C.M.; Csönge, L.; Abkarovits, G.; Bucsi, L.; Lacza, Z. Bone-Albumin filling decreases donor site morbidity and enhances bone formation after anterior cruciate ligament reconstruction with bone-patellar tendon-bone autografts. Int. Orthop. 2016, 40, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Klára, T.; Csönge, L.; Janositz, G.; Pap, K.; Lacza, Z. The use of structural proximal tibial allografts coated with human albumin in treating extensive periprosthetic knee-joint bone deficiency and averting late complications. Case Rep. Orv. Hetil. 2015, 156, 67–70. [Google Scholar] [CrossRef]
- Simonffy, L.; Minya, F.; Trimmel, B.; Lacza, Z.; Dobo-Nagy, C. Albumin-Impregnated Allograft Filling of Surgical Extraction Sockets Achieves Better Bone Remodeling Than Filling with Either Blood Clot or Bovine Xenograft. Int. J. Oral Maxillofac. Implant. 2020, 35, 297–304. [Google Scholar] [CrossRef]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp Gerontol, 2010; 45(7-8), 466–472. [Google Scholar]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Li, X. Fang, P. Mai, J. Choi, E.T. Wang, H. Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 2013, 6, 19. [Google Scholar] [CrossRef]
- Kuten Pella, O.; Hornyák, I.; Horváthy, D.; Fodor, E.; Nehrer, S.; Lacza, Z. Albumin as a Biomaterial and Therapeutic Agent in Regenerative Medicine. Int J Mol Sci. 2022, 23, 10557. [Google Scholar] [CrossRef]
- Klára, T.; Csönge, L.; Janositz, G.; Csernátony, Z.; Lacza, Z. Albumin-coated structural lyophilized bone allografts: A clinical report of 10 cases. Cell Tissue Bank. 2014, 15, 89–97. [Google Scholar] [CrossRef]
- Bernards, M.T.; Qin, C.; Jiang, S. MC3T3-E1 cell adhesion to hydroxyapatite with adsorbed bone sialoprotein, bone osteopontin, and bovine serum albumin. Colloids Surf. B 2008, 64, 236–247. [Google Scholar] [CrossRef]
- Costerton, J.W.; Cheng, K.J.; Geesey, G.G.; Ladd, T.I.; Nickel, J.C.; Dasgupta, M.; Marrie, T.J. Bacterial biofilms in nature and disease. Annu. Rev. Microbiol. 1987, 41, 435–464. [Google Scholar] [CrossRef]
- Kinnari, T.J.; I Peltonen, L.; Kuusela, P.; Kivilahti, J.; Könönen, M.; Jero, J. Bacterial Adherence to Titanium Surface Coated with Human Serum Albumin. Otol. Neurotol. 2005, 26, 380–384. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, W.; Zhu, G.; Lian, J.; Wang, J.; Hui, P.; He, S.; Chen, W.; Jiang, X. Albumin Broadens the Antibacterial Capabilities of Nonantibiotic Small Molecule-Capped Gold Nanoparticles. ACS Appl. Mater. Interfaces. 2019, 11, 45381–45389. [Google Scholar] [CrossRef]
- Marelli, D.; Paul, A.; Samson, R.; Edgell, D.; Angood, P.; Chiu, R.C. Does the addition of albumin to the prime solution in the cardiopulmonary bypass circuit affect clinical outcome? A randomized study. J Thorac Cardiovasc Surg. 1989, 98, 751–756. [Google Scholar] [CrossRef]
- Ishima, Y. Albumin-Based Nitric Oxide Traffic System for the Treatment of Intractable Cancers. Biol Pharm Bull, 2017; 40, 128–134. [Google Scholar]
- Stamler, J.S.; Jaraki, O.; Osborne, J.; Simon, D.I.; Keaney, J.; Vita, J.; Singel, D.; Valeri, C.R.; Loscalzo, J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci USA 1992, 89, 7674–7677. [Google Scholar] [CrossRef]
- Mulvihill, J.N.; Faradji, A.; Oberling, F.; Cazenave, J.-P. Surface passivation by human albumin of plasmapheresis circuits reduces platelet accumulation and thrombus formation. Experimental and clinical studies. J. Biomed. Mater. Res. 1990, 24, 155–163. [Google Scholar] [CrossRef]
- Maul, T.M.; Massicotte, P.; Wearden, P.D. ECMO Biocompatibility: Surface Coatings, Anticoagulation, and Coagulation Monitoring In Extracorporeal Membrane Oxygenation-Advances in Therapy; Firstenberg, M.S., Ed.; IntechOpen: London, UK, 2016; Volume 3, pp. 27–61. [Google Scholar]
- Rhee, P.; Wang, D.; Ruff, P.; Austin, B.; DeBraux, S.; Wolcott, K.; Burris, D.; Ling, G.; Sun, L. Human neutrophil activation and increased adhesion by various resuscitation fluids. Crit Care Med. 2000, 28, 74–78. [Google Scholar] [CrossRef]
- Nathan, C.; Xie, Q.W.; Halbwachs-Mecarelli, L.; Jin, W.W. Albumin inhibits neutrophil spreading and hydrogen peroxide release by blocking the shedding of CD43 (Sialophorin, Leukosialin). J Cell Biol. 1993, 122, 243–256. [Google Scholar] [CrossRef]
Figure 1.
XRD (A) and FTIR (B1, B2) data of samples DBM + CaP + BSA and DBM + CaP.
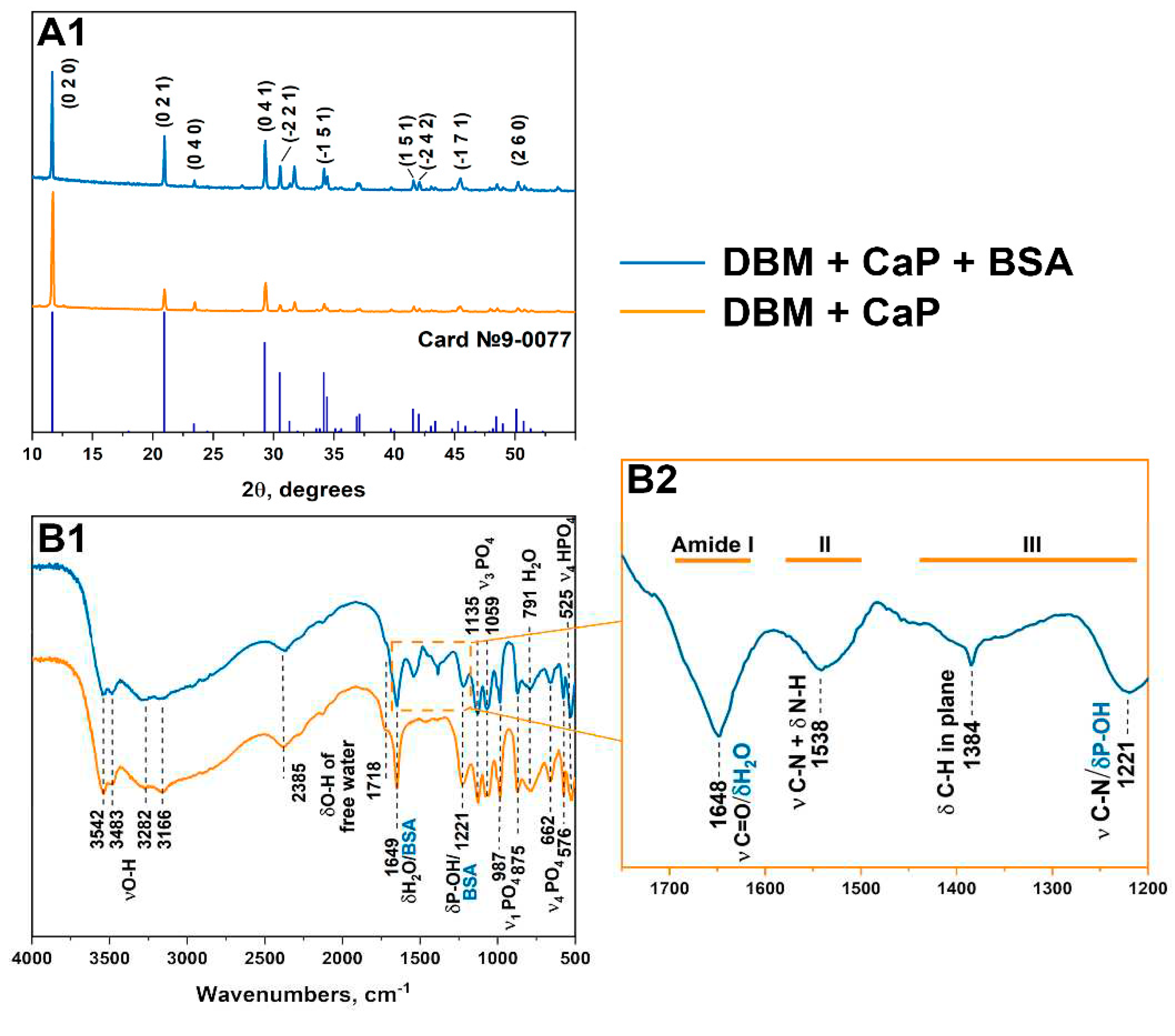
Figure 2.
MicroCT - images and SEM of the surface of the samples.
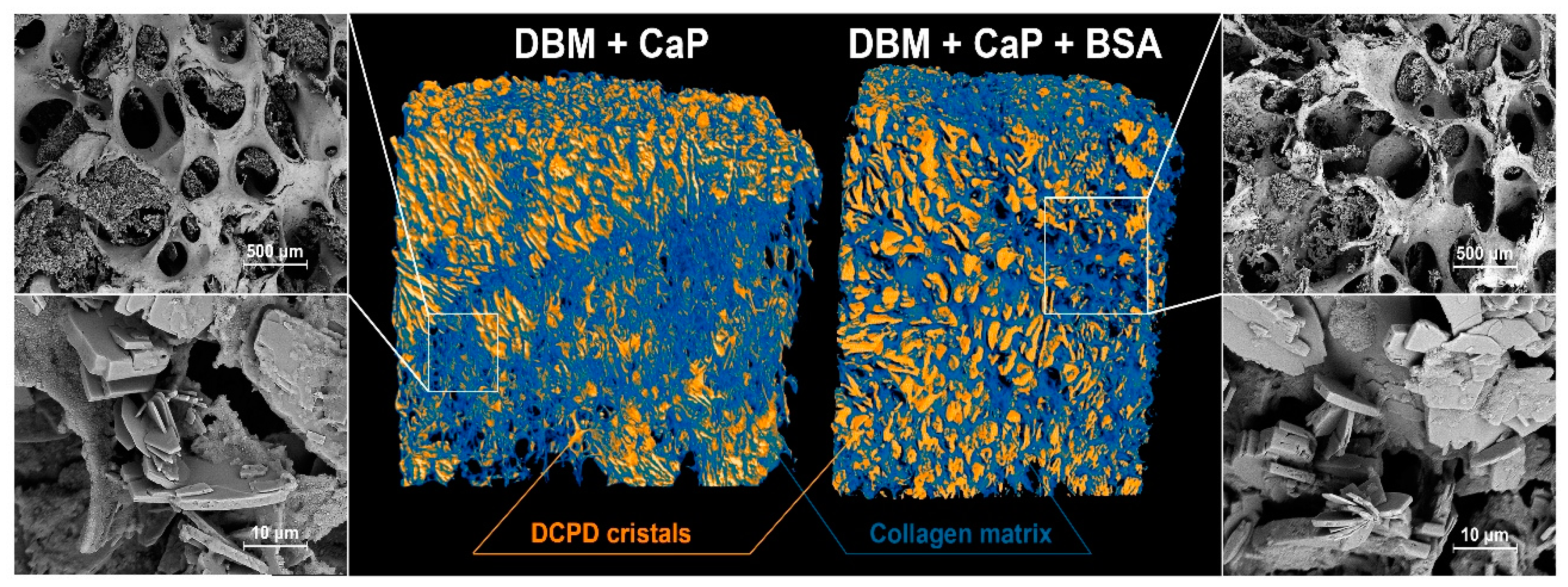
Figure 3.
Results of morphological analysis of DBM after demineralization and remineralization procedures; (A, C, E) SEM images of samples surface; (B, D, F) Alizarin red S Staining (McGee-Russell method: calcium deposits are colored red, ESM are colored light orange; light microscopy.
Figure 3.
Results of morphological analysis of DBM after demineralization and remineralization procedures; (A, C, E) SEM images of samples surface; (B, D, F) Alizarin red S Staining (McGee-Russell method: calcium deposits are colored red, ESM are colored light orange; light microscopy.
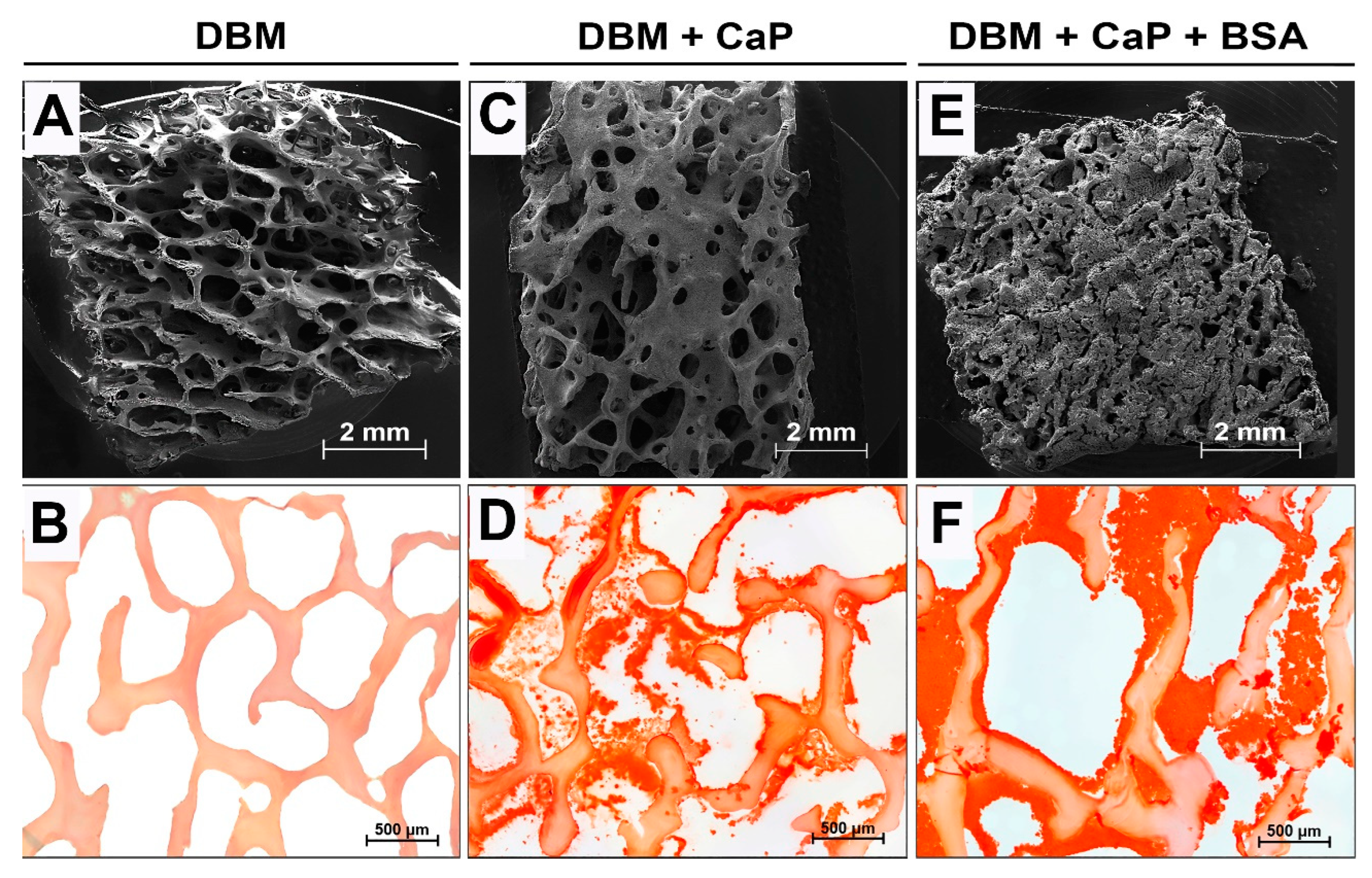
Figure 4.
Results of cell viability assays (а-b) and morphological analysis (c) of CaP and CaP+BSA samples. Cell viability of MG-63 cells after 24 h (a) and 96 h (b). Fluorescence micrographs of MG-63 cells after 96 h of cultivation with different concentrations of CaP and CaP+BSA (c); live cells are stained with Calcein AM (green), dead cells are stained with PI (red), nuclei of living and dead cells are stained with Hoechst 33342 (blue); fluorescent microscopy.
Figure 4.
Results of cell viability assays (а-b) and morphological analysis (c) of CaP and CaP+BSA samples. Cell viability of MG-63 cells after 24 h (a) and 96 h (b). Fluorescence micrographs of MG-63 cells after 96 h of cultivation with different concentrations of CaP and CaP+BSA (c); live cells are stained with Calcein AM (green), dead cells are stained with PI (red), nuclei of living and dead cells are stained with Hoechst 33342 (blue); fluorescent microscopy.
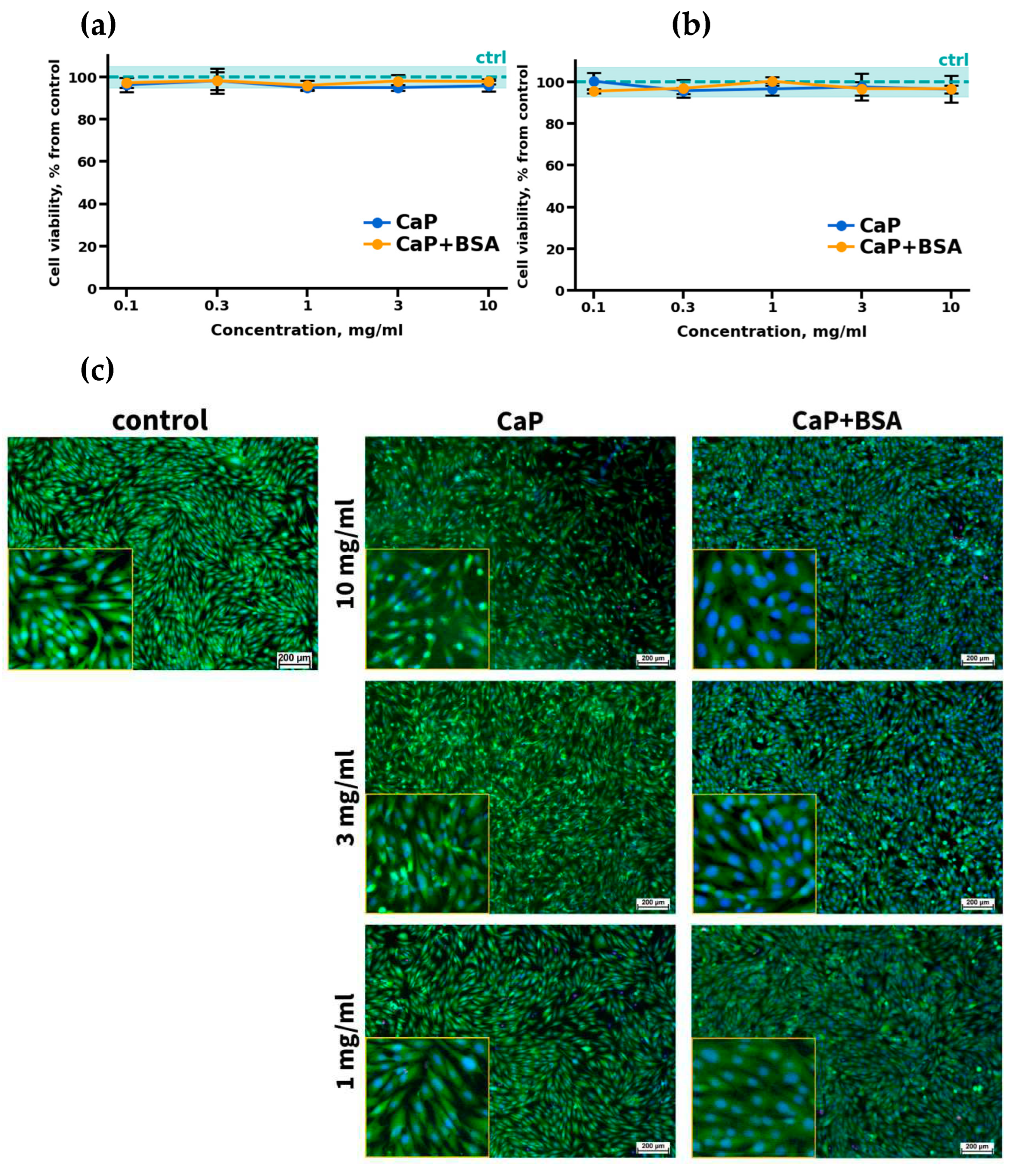
Figure 5.
Histological analysis of the implanted DBM specimens of control and experimental groups. (a-c) DBM without remineralization; (d-f) DBM+CaP specimens; (g-i) DBM+CaP+BSA specimens. (a, d, g) Alizarin red S staining (calcium depositions are stained orange-red), (a) due to the absence of the staining by Alizarin red S, the specimens were stained with Toluidine blue for visualization; (b, e, h) Haematoxylin & Eosin staining (H&E, cell nuclei are stained blue, erythrocytes are stained red, matrix components are stained pink); (c, f, i) Lillie’s trichrome staining (collagen is stained blue, venous sinuses and neocollagenous tissues are stained red, cell nuclei are stained violet-brown); light microscopy.
Figure 5.
Histological analysis of the implanted DBM specimens of control and experimental groups. (a-c) DBM without remineralization; (d-f) DBM+CaP specimens; (g-i) DBM+CaP+BSA specimens. (a, d, g) Alizarin red S staining (calcium depositions are stained orange-red), (a) due to the absence of the staining by Alizarin red S, the specimens were stained with Toluidine blue for visualization; (b, e, h) Haematoxylin & Eosin staining (H&E, cell nuclei are stained blue, erythrocytes are stained red, matrix components are stained pink); (c, f, i) Lillie’s trichrome staining (collagen is stained blue, venous sinuses and neocollagenous tissues are stained red, cell nuclei are stained violet-brown); light microscopy.
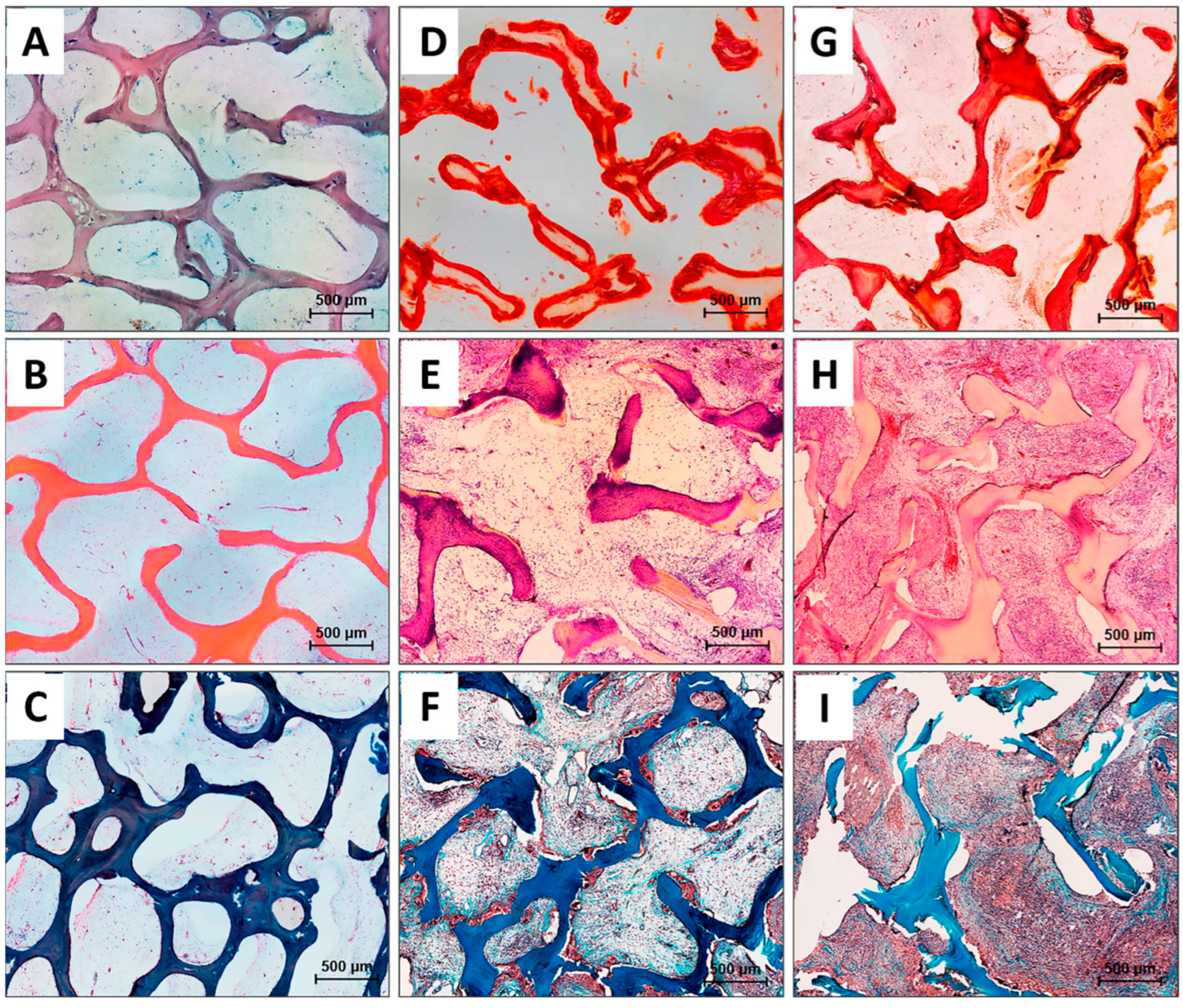
Figure 6.
Histological analysis of the implanted samples in the DBM+CaP+BSA group, magnified areas. (а-b) Hematoxylin & Eosin staining (H&E, cell nuclei are stained blue, erythrocytes are stained bright red, stroma components are pink, green arrows indicate erythroblastic islands); (с) Lillie’s trichrome (collagen is stained blue, aggregations of synthesizing cells are stained red, cell nuclei are violet-brown, yellow arrows indicate mature definitive vessels); light microscopy.
Figure 6.
Histological analysis of the implanted samples in the DBM+CaP+BSA group, magnified areas. (а-b) Hematoxylin & Eosin staining (H&E, cell nuclei are stained blue, erythrocytes are stained bright red, stroma components are pink, green arrows indicate erythroblastic islands); (с) Lillie’s trichrome (collagen is stained blue, aggregations of synthesizing cells are stained red, cell nuclei are violet-brown, yellow arrows indicate mature definitive vessels); light microscopy.
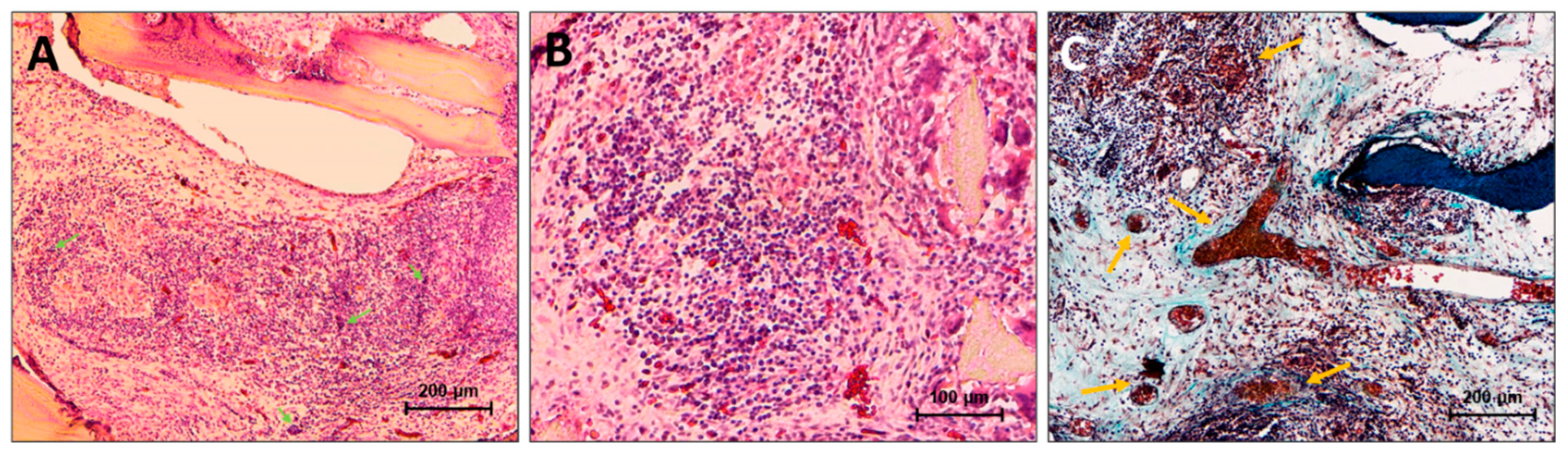

Table 1.
Calculated lattice constants of samples.
| a, Å | b, Å | c, Å | α, ° | β, ° | γ, ° | V, Å3 | |
|---|---|---|---|---|---|---|---|
| Card №9-0077 | 6.36 | 15.19 | 5.82 | 90.00 | 118.50 | 90.00 | 493.93 |
| CaP | 6.362(3) | 15.1609(24) | 5.8083(14) | 90.00 | 118.545(21) | 90.00 | 492.13(18) |
| CaP+BSA | 6.3610(20) | 15.168(3) | 5.8121(12) | 90.00 | 118.571(24) | 90.00 | 492.47(18) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



