
Submitted:
27 September 2023
Posted:
29 September 2023
You are already at the latest version
Abstract
Pregnancy entails bidirectional interactions between the developing fetus, the maternal tissues, and the organ systems. To this end, the phenomenon of migration of fetal cells (FCs) into the maternal circulation is poorly understood. Here, we review literature underlying the migration of FCs from the placenta to the maternal circulation, which is likely a dynamic process, including trophoblast invasion, placental angiogenesis, modulation of maternal immune responses, and enlargement of maternal organs. As placental neovascularization fosters direct connections between fetal and maternal circulatory systems, the trophoblast, a pivotal to placental development, adeptly deploys an array of invasive strategies to breach maternal tissue barriers, facilitating FC escapade into the maternal circulation. The intricate balance struck by the maternal immune system, which both acts as a guardian against potential foreign cell threats and orchestrates a niche conducive to FC survival and differentiation, is facilitated by finely tuned interactions among regulatory T cells, cytokines, and inhibitory receptors. FC presence in mothers’ circulation may be clinically relevant and unveil novel molecular participants like lncRNA, exosomes, and intricate signaling pathways that drive innovative clinical approaches for diagnostics and therapeutics. Ongoing research should reshape our knowledge of pregnancy and maternal-fetal health by improving our understanding of fetal-maternal interactions.
Keywords:
Angiogenesis
; blood vessels
; endothelial cells
; fetal cells
; Flk1
; migration
; placenta
; pregnancy
; trophoblast
; vasculogenesis
; VEGF
; VEGFR2
1. Introduction
Pregnancy is a fundamental physiological process, characterized by an array of hormonal and physiological adaptations within the maternal organs geared towards accommodating the burgeoning fetus. Following fertilization, the egg differentiates into the human blastocyst composed of two primary cell lineages: a) the inner cell mass, responsible for developing into the embryo, and b) the outer layer known as the trophectoderm, which eventually forms the placenta. The process of implantation begins when trophoblast cells breach the surface epithelial lining of the decidua, attaching and invading it, ultimately leading to placental formation. The success of a pregnancy relies on the reciprocal interplay between the invading trophoblast and a receptive maternal decidua. In addition to decidual cells and endothelial cells (ECs), immune cells that infiltrate the maternal decidua constitute a significant cellular component. One fascinating aspect of this reciprocal interplay is the migration of fetal cells (FCs) into the maternal circulation [1,2,3]. This phenomenon entails the migration of FCs, including those of trophoblastic origin, into the maternal bloodstream [1,2,3]. FCs found in the maternal system are likely to trigger a series of genomic and non-genomic responses that facilitate maternal-fetal communication and influence the overall course of pregnancy. This exchange is governed by a complex interplay of hormones, cytokines, and cellular interactions, each playing a vital role in shaping the maternal body's adaptive response to the presence of the developing fetus. Understanding the mechanisms and consequences of FC migration is crucial, not only for understanding the dynamics of pregnancy, but also for shedding light on potential implications for maternal health, disease, and treatment strategies. This review will explore both what is known and what remains unknown about this remarkable biological phenomenon.
The first documented evidence of fetal-to-maternal cell migration was reported in 1893 by George Schmorl [1,2] who identified the presence of trophoblast cells (TBs) in the pulmonary vasculature of deceased women whose pregnancies were affected by eclampsia [1,2]. Recent studies demonstrate that in both human and murine pregnancies, FCs migrate through the placenta and enter the maternal circulation where they engraft into various maternal tissues. This can begin as early as 6 weeks of gestation and will peak just prior to term. Interestingly, circulating FCs can persist in a woman’s tissues for decades after pregnancy where they have been shown to integrate into injured maternal tissues. The most recent technique used to examine the presence of FCs in maternal tissues is the use of in situ hybridization which demonstrated male DNA (Y-chromosome) in women who had sons and was not found in women with daughters or no pregnancies at all [3,4,5]. In one woman, they detected the presence of male DNA 27 years after her last pregnancy, alluding to the longevity of these cells within the maternal tissues [6]. While there is a plethora of data indicating the presence of FCs in maternal organs, there is no clear consensus on the molecular or phenotypic traits they possess. Recent reports hint at their heterogenous nature and suggest these circulating FCs may possess the potential to differentiate into one or more cell types and/or secrete paracrine factors involved in subsequent physiological and pathological mechanisms [2].
Genetically engineered mouse experiments utilizing flow cytometry detected eGFP+ FC populations present within various maternal organs comprised of both progenitor and differentiated cell phenotypes [2]. These FC populations express immature progenitor CD34 and hematopoietic markers PTPRC, SLAMF1, and CXCR4 proteins [2], endothelial cell (EC) marker platelet and endothelial cell adhesion molecule 1 (PECAM1, also known as CD31), as well as several mesenchymal cell markers [2]. Distribution of these cell surface markers varied significantly by maternal organ. For example, PTPRC+ cells were more predominant in maternal bone marrow, whereas ITGB1+ cells were found in higher proportions in the blood [3]. This pattern of distribution may suggest there are specific recruitment mechanisms orchestrating FC migration. FCs might respond to maternal organ microenvironmental cues, leading to the acquisition of a favorable cellular phenotype. These interactions are likely to also include cell-cell or cell-matrix adhesion events [2]. Notably, the maternal lung contained the highest number of eGFP+ FCs among all the maternal organs examined [2]. As a highly vascularized organ, lung is likely to be the preferred reservoir of migratory FCs (as opposed to other maternal organs) with nuanced molecular mechanisms governing the migration pattern [Table 1] [2,7].
2. What is known about Blood Exchange Across the Placental Barrier and Fetal Cell Migration?
The mechanism of FC migration is not well understood, however, the placenta likely plays an integral role in the cellular exchange that occurs between the mother and growing fetus [8]. The placenta is considered a fetomaternal organ, comprised of both zygote-derived and maternal cells [8]. Placentation in both humans and murine model systems is hemochorial, and interaction between fetal and maternal circulations involve chorionic, zygote-derived TBs [2]. Upon implantation, there is rapid proliferation of the trophectoderm of the blastocyst, resulting in the unique signature cell type of the placenta (trophoblast) [Figure 1] [2]. The proliferative villous cytotrophoblast cells (VCT) can differentiate into two main sub-populations, the syncytiotrophoblasts (SCT) and extravillous trophoblasts (EVT). The multinucleated SCTs, located in the epithelium of the villi, are the main trophoblast cell type responsible for the exchange of nutrients between the mother and fetus and contribute to hormone production [9,10]. The EVTs, however, also help anchor the placenta to the maternal decidua and assist in the remodeling of the maternal spiral arteries which are essential for proper placental vascular development. Proper development of these placental vascular networks is central to its functions and critical to the overall well-being of both the mother and baby. Abnormal development of this vascular network can lead to numerous undesirable outcomes and pregnancy related complications, many of which can follow a woman well into post-partum [11,12].

Table 1.
Fetal cells in various maternal tissue during congenic pregnancies. Fetal microchimeric cells in various maternal tissues during congenic pregnancies.
Table 1.
Fetal cells in various maternal tissue during congenic pregnancies. Fetal microchimeric cells in various maternal tissues during congenic pregnancies.
| Mouse | Gestation age (week) | GFP+ fetuses/total | Blood | Bone marrow | Spleen | Liver | Heart | Lung | Kidney | Brain |
|---|---|---|---|---|---|---|---|---|---|---|
| P1 | First | Unknown | 0 | 0 | 2 | 0 | 0 | 332 | 2 | np |
| P2 | Second | 1/3 | 0 | 13 | 7 | 7 | 431 | 45 | 0 | 60 |
| P3 | Second | 5/9 | 0 | 0 | 13 | 0 | 85 | 317 | 0 | np |
| P4 | Third | Unknown | 0 | nd | 12 | 2 | 77 | 129 | 0 | np |
| P5 | Third | 2/7 | 12 | 0 | 2 | 1 | 185 | 833 | 405 | np |
| P6 | Third | 3/6 | 0 | 3 | 2 | 2 | 5 | 734 | 1 | np |
| P7 | Third | 4/9 | 0 | 0 | 8 | 3 | 0 | 365 | 1 | np |
| P8 | Third | 6/9 | 0 | 0 | 2 | 5 | 12 | 1200 | 13 | np |
| P9 | Third | 3/6 | 0 | 0 | 19 | 0 | 197 | 347 | 45 | 0 |
Table 1. Number of eGFP+ FCs in maternal tissues. Groups of C57BL/6J female mice were crossed with C57BL/6J males expressing the eGFP sequence and were sacrificed at various times during pregnancy (P1-P9) [2,7]. Numbers indicate the quantity of GFP sequences/million maternal genome equivalents. nd: not determined; in this case the total number of nucleated cells isolated was less than 10,000. np: not performed. Among all the maternal organs profiled, the lung displayed the highest percentage of eGFP+ FCs. Consequently, it also exhibited the most varied distribution of cellular markers [2,7].
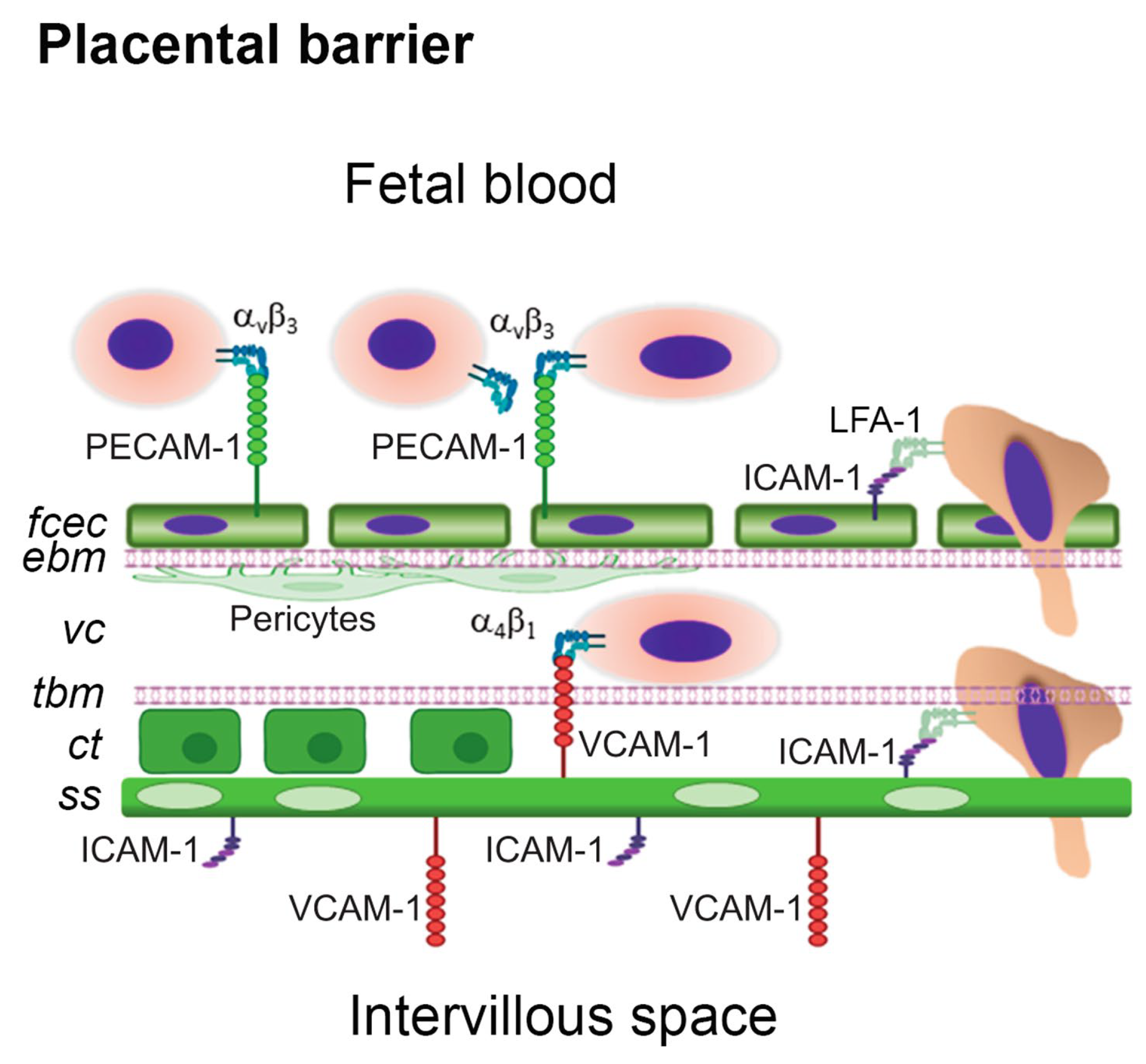
While there is no consensus on how these cells cross into the maternal circulation, one proposed method of FC migration is by integrin-mediated transmigration and involves the use of cell adhesion molecules. The fetal capillary EC layer expresses adhesion molecules CD31/PECAM-1 and ICAM-1 which have been implicated in neutrophil adhesion and transmigration across EC monolayers [Figure 3]. While the functional ligand for PECAM1 in this model system is unknown, recent studies suggest αvβ3 integrin may play play an important role in this cellular exchange. In this proposed mechanism of FC migration, FCs may adhere and transmigrate into maternal circulation across the ternal circulation across the placental barrier in a similar manner by which lymphocytes cross the blood brain barrier [8].
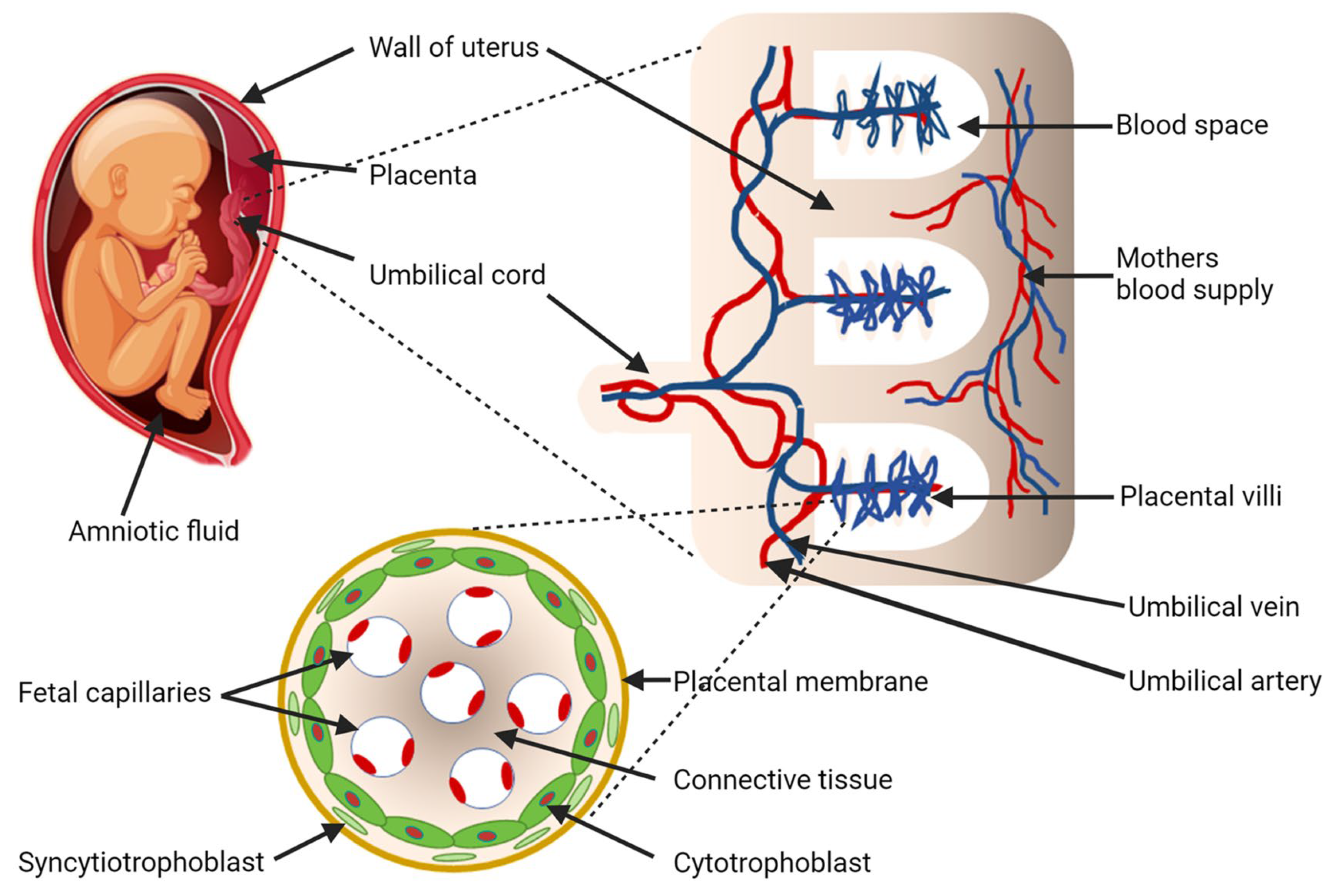
Figure 1.
Fetomaternal interaction, blood exchange between mother and baby, and putative mechanism of fetal cell migration into maternal circulation. Maternal spiral arteries facilitate blood exchange with the fetus via the intervillous space. Fetal blood flow begins at the umbilical cord and travels to the fetal capillaries within the villous trees [7]. The layer of trophoblast cells lining the surface of villous trees are at the center of the interface between the fetal and maternal circulation [8,9,10,18]. Image adapted using Biorender.
Figure 1.
Fetomaternal interaction, blood exchange between mother and baby, and putative mechanism of fetal cell migration into maternal circulation. Maternal spiral arteries facilitate blood exchange with the fetus via the intervillous space. Fetal blood flow begins at the umbilical cord and travels to the fetal capillaries within the villous trees [7]. The layer of trophoblast cells lining the surface of villous trees are at the center of the interface between the fetal and maternal circulation [8,9,10,18]. Image adapted using Biorender.
It has been a long-held view that the presence of these FCs in the maternal circulation contribute to maternal pathology [2,3,13], however, recent reports demonstrate these cells play a reparative role in response to maternal injury. In previous animal experiments, FCs were reported to be recruited to sites of maternal lung and heart injury [13,14]. Characterization of FCs with Y-chromosome markers suggested the presence of trophoblast-derived immature ECs and progenitor cells [2,3,4,5,7]. However, it remains unknown whether these potential immature FCs are recruited to the maternal lung and play a role in repair of vascular structures in response to vascular injury.
Figure 2.
Placental vasculogenesis and angiogenesis. Placental vasculogenesis begins with the recruitment of hemangioblastic progenitors followed by the formation of cords. After endothelial tubes are formed, pericytes stabilize this process. As vasculogenesis stops, new vessels emerge from a pre-existing vascular network via the process of angiogenesis. The key drivers of these processes are the VEGF family of growth factors and their transmembrane receptors, VEGFR1 (flt1) and VEGFR-2 (Flk1). Hypoxia can stabilize HIF-1α thereby inducing the expression of VEGF to stimulate placental angiogenesis [38,49]. Image adapted using Biorender.
Figure 2.
Placental vasculogenesis and angiogenesis. Placental vasculogenesis begins with the recruitment of hemangioblastic progenitors followed by the formation of cords. After endothelial tubes are formed, pericytes stabilize this process. As vasculogenesis stops, new vessels emerge from a pre-existing vascular network via the process of angiogenesis. The key drivers of these processes are the VEGF family of growth factors and their transmembrane receptors, VEGFR1 (flt1) and VEGFR-2 (Flk1). Hypoxia can stabilize HIF-1α thereby inducing the expression of VEGF to stimulate placental angiogenesis [38,49]. Image adapted using Biorender.

3. Is pregnancy a precursor to cardiovascular disease in mothers?
During and after pregnancy, the mothers experience extensive hemodynamic, metabolic, and hormonal changes that place an incredible burden on a woman's cardiovascular system. In fact, it is estimated that roughly 1-4% of all pregnancies worldwide are complicated by cardiovascular disease [11,12], and this percentage increases when the mother has predisposing risk factors such as hypertension, obesity, diabetes, and other hypertensive disorders. In developed countries, pregnancies complicated by cardiovascular disease is the leading cause of maternal mortality.
Figure 3.
A simplified representation of the human placental barrier showing a hypothetical mechanism of fetal cell capture, adhesion and transmigration. The placental barrier is made of fetal capillary endothelial cells (fcec), an endothelial basement membrane (ebm), the villous core (vc) with pericytes (p) and extracellular matrix, a trophoblastic basement membrane (tbm), a layer of proliferative cytotrophoblasts (ct) in the first trimester, and a multinucleated syncytium of syncytiotrophoblasts (ss) [8]. In the mouse, the trophoblastic layers differ in that there are two syncytiotrophoblastic layers and the cytotrophoblastic layer is outermost facing the intervillous interface [28,29,30]. The prevailing hypothesis is that FCs may adhere and transmigrate across the placental barrier in a manner analogous to diapedesis. Image adapted using Biorender.
Figure 3.
A simplified representation of the human placental barrier showing a hypothetical mechanism of fetal cell capture, adhesion and transmigration. The placental barrier is made of fetal capillary endothelial cells (fcec), an endothelial basement membrane (ebm), the villous core (vc) with pericytes (p) and extracellular matrix, a trophoblastic basement membrane (tbm), a layer of proliferative cytotrophoblasts (ct) in the first trimester, and a multinucleated syncytium of syncytiotrophoblasts (ss) [8]. In the mouse, the trophoblastic layers differ in that there are two syncytiotrophoblastic layers and the cytotrophoblastic layer is outermost facing the intervillous interface [28,29,30]. The prevailing hypothesis is that FCs may adhere and transmigrate across the placental barrier in a manner analogous to diapedesis. Image adapted using Biorender.
It is important to address how these physiological changes and critical adaptations in a woman's body occur to allow for the normal development of the fetus, as well as why and how cardiovascular complications develop during pregnancy. Among these adaptations, hemodynamic shifts, such as changes in blood composition and plasma volume, can arise within the first few weeks of pregnancy, often modulating a woman's cardiovascular system before the first fetal heartbeat. Starting in the first trimester, vascular resistance decreases, resulting in a decrease in mean arterial pressure [11,12]. This is partly due to an overall reduction in response to vasoconstrictive agents such as angiotensin II as well as an increase in levels of relaxing agents such as nitric oxide [11,12]. This decrease in vascular resistance activates the renin-aldosterone system leading to water and sodium retention and allows for volume expansion to compensate. It is during this time that a low resistance uteroplacental circulation is created. As systemic vascular resistance slowly increases (towards late gestation), there is a corresponding increase in blood flow to the placenta to maintain the growing needs of the developing fetus [Figure 4].
Figure 4.
Role of Etv2/Er71 in the emergence of mesoderm and vascular endothelial cells. Etv2 is crucially required for vascular mesoderm formation (VSP), whereas cells double positive for Flk-1+PDGFRα+ diverge into Flk-1+PDGFRα- [45]. VEGF-Flk1 signaling further promotes this action by up-regulating Etv2 expression [45,46,47,48,49]. Expression of Etv2 leads to VSP induction and EC/HPC generation with expression of additional hemato-endothelial genes. Image adapted using Biorender.
Figure 4.
Role of Etv2/Er71 in the emergence of mesoderm and vascular endothelial cells. Etv2 is crucially required for vascular mesoderm formation (VSP), whereas cells double positive for Flk-1+PDGFRα+ diverge into Flk-1+PDGFRα- [45]. VEGF-Flk1 signaling further promotes this action by up-regulating Etv2 expression [45,46,47,48,49]. Expression of Etv2 leads to VSP induction and EC/HPC generation with expression of additional hemato-endothelial genes. Image adapted using Biorender.

Other major cardiovascular changes during pregnancy include an increase in heart rate due to the low afterload activation of baroreceptors in the cardiopulmonary and renal systems, further stimulating the sympathetic nervous system [11,12]. During pregnancy, catecholamine release increases heart rate and contractibility by 15-20 % and stroke volume is additionally increased by 20-30% as well [11,12]. Pregnancy can also lead to significant changes in the aortic vessel wall including the loss of elastic fibers, fragmentation of the reticulum fibers and lower levels of acid mucopolysaccharides [12]. Furthermore, hormonal shifts such as an increase in progesterone, can lead to non-collagenous protein depositions within the vasculature, thereby raising the risk of aortic dissection. During pregnancy, a woman's body also shifts to a hypercoagulable state with high levels of clotting factors and a corresponding decrease in serum anti-coagulant levels. This is presumably in preparation for the significant blood loss that occurs during childbirth [11,12]. Interestingly, the pattern of adverse cardiovascular events (that arise during pregnancy) has been hypothesized to be linked to the timing of these major physiological and hemodynamic changes. According to the worldwide registry of pregnancies affected by cardiovascular disease, incidence of maternal heart failure seems to correlate with the timing of major changes in cardiac output [11,12]. This finding was further confirmed as peaks in incidence of maternal heart failure happened around the same time as changes in cardiac output with the first peak (in incidence of maternal heart failure) occurring as cardiac output plateaus (between week 26-30), and the second peak (for both cardiac output and incidence of maternal heart failure) arising shortly after delivery, when the uteroplacental circulation is in an overload state [11,12].
4. What is the Relationship between Placental Hormones and the Development of Peripartum Cardiomyopathy (PPCM)?
PPCM is a type of cardiomyopathy that is characterized by left ventricular dysfunction (systolic heart failure) and usually occurs within the last month of pregnancy or in the first few months after delivery [14,15]. While there have been many proposed factors contributing to the pathophysiology of this disease, recent literature suggests maternal hormones play a crucial role in the development of PPCM [14]. Whereas most adverse cardiovascular events that occur during pregnancy happen around the time of major hemodynamic changes, onset of PPCM tend to only happen in peripartum when hormonal changes are at their peak [14].
The proposal that PPCM is a vascular disease triggered by hormonal shifts in peripartum was first developed by researchers who generated a mouse model of PPCM by deleting the STAT3 gene in cardiomyocytes [14]. In this model, the authors observed an overall decrease in STAT3 levels in the LV of patients with end-stage heart failure due to PPCM. They also demonstrated that the loss of STAT3 in murine hearts leads to a decrease in expression of genes such as manganese superoxide dismutase (MnSOD) which helps shield the heart from harmful reactive oxygen species (ROS) [14]. A rise in levels of ROS results in the secretion of cathepsin D and subsequent cleavage of prolactin into a 16 kDa fragment. Prolactin is a hormone commonly expressed in late gestation and cleavage of prolactin (by cathepsin D) triggers EC apoptosis [14]. As a result, cardiac muscle-specific STAT3 knockout mice showed significant vascular dropout during late gestation and pregnancy-induced DCM, a classic phenotype of PPCM [14]. This phenotype was able to be reversed by treating these mice with bromocriptine, essentially blocking expression of prolactin from the pituitary and proving the importance of maternal hormones in the development of PPCM [14].
A similar model demonstrating the involvement of maternal hormones in the development of PPCM was developed by a research group who deleted proliferator-activated receptor-gamma coactivator-1α (PGC-1α) in mouse cardiomyocytes. (PGC-1α) drives expression of MnSOD in a manner similar to the STAT3 model, thereby suppressing ROS [14]. Additionally, (PGC-1α) also drives expression of VEGF so cardiac deletion of PGC-1α not only resulted in EC apoptosis, but also the loss of VEGF/VEGFR2-mediated angiogenesis [14]. In this model, only treatment with both bromocriptine and VEGF could rescue the PPCM phenotype [14]. Not only did this confirm the theory that changes in maternal hormones can trigger PPCM in peripartum, but it also led to critical insights as to how mis-regulated hormones (secreted by the placenta) can impact VEGF-mediated signaling resulting in maternal vasculopathy [14].
5. What is the role of sFlt1 in the Development of PPCM?
In an investigation into the factor responsible for inhibiting VEGF in the PGC-1α mouse model, the authors addressed the hypothesis that a key contributing factor may be a soluble variant of VEGF receptor 1 known as soluble Fms-like tyrosine kinase 1 (sFlt1) [14]. During late gestation, the placenta secretes various factors into the maternal circulation including sFlt1. During late gestation, most of the free VEGF in the maternal circulation is neutralized by sFlt1 [14]. The heart and other vascular organs can defend themselves from circulating anti-angiogenic factors by secretion of local VEGF. This action, however, is compromised in the PGC-1α model of PPCM whereas local VEGF is inhibited by sFlt1 [14]. In this PGC-1α model of PPCM, only treatment with both VEGF and bromocriptine could rescue this phenotype [14]. Interestingly, when administering sFlt1 to non-pregnant mice (with this cardiac-specific deletion of PGC-1α), they still displayed symptoms of cardiomyopathy [14]. Moreover, patients pregnant with twin babies and those presenting with pre-eclampsia have been shown to have increased circulating levels of sFLT1. In addition, both conditions have been previously identified as predisposing risk factors for the development of PPCM [14]. Altogether these observations indicate that hormones produced by the placenta during pregnancy (such as sFLT1) can impact VEGF signaling leading to the development of cardiomyopathies by posing a toxic challenge to the maternal vasculature [14]. Further studies elucidating the mechanisms by which these hormones (secreted by the placenta) impact VEGF signaling could lead to critical insights into how diseases such as PPCM develop during gestation as well as help identify targets for therapeutic intervention [14].
It is important to note that out of all the varying forms of cardiomyopathy, PPCM has the best overall rate of recovery [15]. According to a study reported by the New England Journal of Medicine, in which 1230 patients with cardiomyopathy were enrolled and grouped by underlying cause of cardiomyopathy, patients with PPCM displayed the best long-term survival outcome [15]. Although elegant experiments have implicated the secretion of anti-angiogenic hormones (from the placenta) to be significant contributing factors in the development of this PPCM, it is still unclear why the rate of recovery for these patients is so much higher than that of other forms of cardiomyopathy [15]. Could fetal stem/progenitor cells that migrate to the mother during pregnancy assist in the repair and recovery of the damaged heart and lung endothelium? We know that FCs can migrate to maternal vascular organs during pregnancy, however, there is no clear consensus on their overall role and or how they contribute to maternal health and disease. It is also unclear whether their migration is in response to circulating hormones (secreted by the placenta) and what impact they have (if any) on downstream VEGF angiogenic signaling.
6. How does the Placenta Develop and Establish its Vascular Networks?
The placenta is a highly dynamic and vascularized organ that is responsible for nutrient uptake and gas exchange between the mother and fetus [16,17,18]. It is derived from extraembryonic tissue and develops within the first few days after implantation. Proper placentation is critical to maintaining a successful pregnancy and failure in its development can lead to numerous pregnancy-related complications such as pre-eclampsia, pre-term labor, and intrauterine growth restriction (IUGR) [19,20,21,22,23,24,25,26].
The main cell type and building block of the placenta is the trophoblast. TBs arise from the trophectoderm (TE) which develops approximately 5 days after fertilization [27,28,29,30,31,32,33]. The formation of the TE, and subsequent interaction with the uterine luminal epithelium, marks the first cellular fate decision and catalyzes the start of placenta development. Shortly after implantation, TE stem cells give rise to early mononuclear cytotrophoblasts (CTBs) and the primitive syncytium (PS) [8]. The latter then expands into the maternal decidua, while at the same time, the inner cell mast gives rise to the embryo and primitive yolk sac. Formation of the placental villus tree structures begin around day 10 post-conception and proliferative CTBs, which form the primary villi (PV), extend into the maternal decidua [8,10]. CTBs at the tips of anchoring villi undergo extravillous trophoblast (EVT) differentiation, a process critical to placenta vascular development and needed to meet the growing demands of the baby [Figure 3]. These cells are known for their migratory and invasive capabilities and play a major role in the remodeling of maternal spiral arteries [28,29,30]. Shortly thereafter, the PV are transformed into secondary villi while the epithelial surface expands by the continuous proliferation and fusion of the developing villous cytotrophoblast (vCTB). This process helps give rise to the synctiotrophoblast layer (STB) which is a multinucleated cell layer spanning the entire outside surface of the villous placenta and is in direct contact with the maternal blood supply [28,29,30]. A key function of this layer is to assist in nutrient uptake and gas exchange between the mother and developing baby as well as synthesize signaling hormones such as hCG, progesterone, and lactogen. Additionally, this layer acts as a barrier by preventing the passage of large molecules and is essentially hidden from the maternal immune system by their lack of HLA molecule expression [29].
By approximately the 17th day after conception, vasculogenesis peaks as hemangiogenic clusters initiate the formation of EC tubes [8,10,12]. Pericytes are then recruited to stabilize this process as the vascular network expands and connects with the fetal vasculature. Interestingly, the formation of the placenta vascular network occurs before the formation of the embryonic vascular network as the first blood vessels visibly evident within the placenta arise while the embryo still exists in three germ layers [8,10,12,35,36,37]. Additionally, the connection between the placenta vascular network and the fetal umbilical vascular network does not occur until at least 32 days post-conception [37]. It is important to note that while both placenta and fetal vasculogenesis occur through similar mechanisms (and are highly coordinated), the development of their individual vascular networks are completed separately [37]. Formation and subsequent expansion of this vascular network is an integral part of placental development and necessary to support the growing fetus.
The two main cellular processes responsible for the generation and maintenance of the placental vascular network are vasculogenesis and angiogenesis. Vasculogenesis is a process in which blood vessels develop de novo from pluripotent mesenchymal stem cells and usually occurs between day 18 and 35 post conception [37]. Angiogenesis is essentially the formation of new vessels from an already existing vascular network and requires the highly coordinated actions of invading trophoblasts and ECs [37]. This process begins around day 32 post conception and continues throughout gestation in two sub-phases. The first phase of placental angiogenesis is the branching phase which occurs around day 32 post-conception and lasts until approximately 24 weeks into gestation [35,37]. During angiogenesis, pre-vascular networks form in response to growth factors derived from cytotrophoblasts, and the endothelial tube segments (formed during vasculogenesis) that extend into the primitive capillary network. Around 24 weeks, as pregnancy progresses, this branching phase is followed by an elongation phase in which there is a marked decrease in trophoblast proliferation and subsequent rise in EC proliferation [37]. Key mediators of placental vasculogenesis and angiogenesis are the VEGF family of growth factors and their transmembrane receptors VEGFR1 (flt1) and VEGFR-2 (Flk1). During placenta development, VEGF-A binds to both VEGFR-1 and VEGFR-2, whereas PlGF only binds to VEGFR-1 [Figure 2] [19]. The soluble form of VEGFR1 (sFlt1) has been implicated as a marker of many pregnancy-related complications such as preeclampsia where alterations in the development of placental vascular networks occur. Inhibition of downstream VEGF and PIGF signaling by sFlt1 can result in a diminished placental vascular network and lead to decreased EC proliferation, migration, and tube formation [37,38,39].
In early gestation, VEGF-A expression is high in the cytotrophoblast cells and known to aid in the formation of the hemangioblastic cords from precursor cells [38]. Along with regulating placenta angiogenesis, VEGF plays an important role in EVT invasion and remodeling of the maternal spiral arteries [38]. By the 6th week of gestation, endovascular cytotrophoblasts invade and break down the smooth muscle cell layer, essentially modifying the maternal spiral arteries leading to high-capacitance and low-resistance vessels. This process is usually completed by 20 weeks of pregnancy and helps meet the demands of the developing fetus by increasing uteroplacental perfusion [11,12,30,35,36,38].
Hypoxia is a key signaling factor in placental angiogenesis and maternal artery remodeling. A low oxygen environment in early gestation allows for adequate EVT outgrowth and expansion [11,12,21,22]. As pregnancy progresses, oxygen levels rise allowing for increased blood flow toward the intravillous space. Insufficient EVT invasion can lead to impaired spiral artery remodeling and ultimately diminished placental angiogenesis [11,12,21]. Recent studies have demonstrated the importance of dynamic hypoxia signaling and its effect on VEGF-mediated placenta angiogenesis [25,40]. Hypoxia inducible factor 1-α (HIF-1α) has been known to mediate cellular adaptation in response to hypoxia. When stabilized, HIF-1α translocates into the nucleus where it binds to hypoxia response elements on target genes, many of which are involved in angiogenesis [40]. The HIF-1α/VEGF signaling pathway has already been shown to play a pivotal role in tumor angiogenesis. Given the mechanistic similarities between tumor and placental angiogenesis, it is hypothesized that HIF-1α/VEGF signaling (under hypoxia stimulation) may also be critical to placenta villous development [40].
To address this, placental tissues from elective and missed abortions were isolated and evaluated for the expression of HIF-1α and VEGF as well as the microvessel density in these early placenta tissues. Next, they compared expression of HIF-1α and VEGF to that of normal (healthy) controls [Figure 4]. Missed abortion is a common pregnancy complication in which the development of the fetus is abruptly halted and may stem from abnormal development of the placenta vasculature [40]. Immunohistochemistry staining for HIF-1α and VEGF showed a decrease in both HIF-1α and VEGF expression in tissue sections isolated from patients with missed abortions as compared to those of normal controls or elective abortions. Microvessel density was also decreased in placenta sections from missed abortions as opposed to those from elective abortions [40].
Using a human villous trophoblast cell line (HTR8/SVneo) to evaluate the expression of HIF-1α and VEGF under hypoxic and normoxic conditions [40], studies showed that under hypoxic conditions, qRT-PCR analysis confirmed an increase in both HIF-1α and VEGF mRNA expression. In addition, siRNA knockdown of HIF-1α led to a decrease in VEGF mRNA expression within human trophoblasts [40]. This effect was followed by a notable increase in tube formation under hypoxic conditions, but diminished after siRNA knockdown of HIF-1α, suggesting that hypoxia modulates total HIF-1α/VEGF mRNA [40].
This data provides evidence to support the notion that HIF-1α acts upstream of VEGF, and that lack of HIF-1α expression leads to diminished development of the placenta vascular network [40]. Hypoxia and dynamic HIF-1α signaling in human villous trophoblasts is just one of many mediators that can affect VEGF-driven placental angiogenesis. Investigations into the causality of disruptions in this delicate angiogenic process and identifying key signaling factors leading to its failure could help us better understand the mechanisms underlying pregnancy complications caused by placental vascular insufficiency such as with missed abortions.
7. What are the Proximal Factors that Regulate Vascular EC Fate Determination?
Cardiovascular disease is a significant cause of mortality in the United States and many patients suffering from vascular deficiencies have very limited treatment options available to them. Accordingly, there is a need for alternative sources to promote therapeutic angiogenesis and aid in tissue repair processes. Angiogenesis is a highly coordinated process where new blood-vessels are formed from a pre-existing vascular network. This occurs during growth and development or in response to tissue injury to help restore blood supply and promote wound healing [6]. As in placental angiogenesis, ECs form the inner lining of blood vessels and play a pivotal role in regulating sprouting angiogenesis and vascular function.
In the adult, ECs remain in a quiescent state until a physiological stimulus such as hypoxia or tissue damage occurs. Quiescent ECs respond to this stimulus by dividing rapidly and initiating sprouting angiogenesis. This process is mediated by EC tip cell migration and stalk cell proliferation [41]. The key angiogenic factor that drives vessel branching is vascular endothelial growth factor (VEGF). VEGF promotes angiogenesis by binding to VEGF receptor (VEGFR2) causing the activation of EC tip cells followed by stalk cell proliferation. Delta-like ligand (DLL4), expressed on tip cells, binds to notch receptors on the stalk cell initiating notch-mediated lateral inhibition and contributing to the specification of tip versus stalk cell phenotype [42,43,44,45,46]. As stalk cells proliferate, they extend the sprout toward the VEGF gradient. This process ends with a new lumenized vessel and the organization of ECs into a continuous monolayer forming a tight barrier that allows for conduction of blood flow [43,50]. The resultant onset of blood flow through the neovasculature leads to an increase in tissue oxygenation. Angiogenesis is a fundamental biological process required for tissue homeostasis and organ function [43,47]. EC dysfunction leading to perturbations in this process underlies various vascular pathologies resulting in vessel malformation or regression.
Fetal liver kinase (Flk)1, also known as Vascular endothelial growth factor receptor 2 (VEGFR2), is a cell surface receptor and key mediator of VEGF-driven angiogenesis. VEGF/Flk1 signaling can activate many downstream signaling cascades (including p38 MAPK) leading to cell proliferation and survival [34,45,46,47,48]. Flk1 deficient mice die in early embryogenesis due to their inability to form proper blood and vascular networks, thus indicating it plays a critical role in vascular development [45,46,47,48]. Flk1 along with transcription factor Etv2 is essential for vasculogenesis during embryonic development and the promotion of an EC fate [47,48,49]. Etv2 is a member of the ETS family of transcription factors and although the genetic hierarchy of Flk1 and Etv2 has been debated, several studies allude to their ability to bidirectionally activate each other depending on the stage of mesodermal development [45,46,47]. Experiments performed in mouse ECs (mESCs), have shown that Etv2 can induce de novo generation of Flk1+ progenitors [45,46,47]. In addition, Etv2 can regulate Flk1 signaling by binding directly to promoter or enhancer regions on target genes and driving EC progenitors toward a mature EC lineage [44,45,46,49]. Recent advances in ChIP sequencing technology have led to the identification of various downstream targets of Etv2 that are critical to vascular development. Most recently, Etv2 was revealed to regulate VEGF-Flk1 signaling via binding to selective miRNA targets [46].
Several studies have also emphasized the importance of the Flk1-MAPK signaling axis in correlation with Etv2 activation [Figure 4]. Further investigation into this connection demonstrated that overexpression of Etv2 can lead to the activation of MAPK downstream signaling while inhibition of MAPK led to a decrease in Etv2-mediated Flk1+ cell generation [45,46]. Additionally, MAPK can upregulate Flk1 expression via JUN/FOS binding on the Flk1 enhancer [45,46]. These studies also provided evidence to suggest that both Etv2 and Flk1 can bidirectionally activate each other and modulate subsequent downstream VEGF signaling [45]. One example of this was reported by a group of investigators that demonstrated VEGF can signal through Flk1 in a p38 MAPK dependent pathway to activate expression of Creb. Activation of Creb can then lead to upregulation of Etv2 [45]. Together, the above-described studies demonstrated the crucial roles that both Etv2 and Flk1 have in VEGF-mediated angiogenesis and EC fate decisions. Given that EC dysfunction is a key defining feature in many vascular disorders, identifying signaling intermediaries with the capacity to drive progenitor populations toward an EC fate could lead to novel approaches for stimulating angiogenesis and tissue repair in vascular disorders such as CVD. Given that many pregnancy complications are additionally marked by vascular insufficiencies, these insights could then be translated into an applicable pregnancy model.
8. What is known about the migration of FCs to the mother during pregnancy?
The presence of FCs within maternal organs was first described by George Schmorl, a physician who observed placenta-derived TBs within the lung vasculature of women who had suffered from eclampsia [8]. FCs are seen in the maternal circulation as early as 6 weeks gestation and increase with gestational age. By the second trimester, it is estimated that between 1-6 FCs/mL are present in maternal venous blood [52,53,54]. Within a murine model system, FCs can be detected in the maternal blood as early as 10-12 days post-conception [52,53,54]. This data is consistent with the establishment of the uteroplacental circulation. Formation of the fetal capillary network usually occurs by day 12 post-conception and coincides with the onset of the fetal circulation and the subsequent completion of organogenesis [28]. The migration of FCs into the maternal circulation reaches a peak just prior to delivery and quickly declines in the weeks following. Some migratory FCs, however, have been shown to persist in maternal organs for decades post-partum, although researchers have yet to uncover their exact cellular identity and role during pregnancy [2,8,52,53].
The detection of FCs in maternal tissues was made possible by the use of fluorescence in situ hybridization (FISH) methods [6,54,55]. Using the FISH technique to detect male DNA (Y-chromosome), they were able to uniformly detect the presence of FCs within maternal tissues in women who conceived sons, but not in women with daughters or no pregnancies at all [6,54]. To date, there is no consensus on what triggers the migration of FCs to maternal organs, or the precise mechanism by which they migrate [8]. Although many questions regarding their phenotypic traits and mechanistic actions remain unanswered, a strong correlation exists between developmental placental abnormalities and increased FC migration into the maternal circulation. Other common factors such as gestational age can affect their migration as well.
A widely held view was that migratory FCs played a role in maternal pathology, however, recent literature suggests they may play a more reparative role in response to maternal injury [5,6,7,8,52,55]. Investigators have detected more FCs in tissue sections of women suffering from diseases vs those from healthy controls [5,6,7,8,52,55]. Additionally, the lineage potential of these migratory FCs varied widely by maternal tissue type [2,7].
The migratory pattern displayed by FCs in response to maternal injury was observed using FISH hybridization methods where the presence of male DNA (Y-chromosome) was detected in the liver of a woman diagnosed with hepatitis who had been pregnant with a son 17 years prior [56]. After performing a liver biopsy, sections were probed for detection of male DNA (Y-chromosome) which displayed an average of 400 male nuclei per cm2 of tissue [56]. This result was then confirmed by PCR amplification of polymorphic STR sequences [56]. It is important to note that apart from these FCs possessing the Y-chromosome marker, they were indistinguishable (morphologically) from other cells in the liver. This data provided evidence to suggest that upon migration to the maternal liver, they then differentiate into functional hepatocytes and persist within the maternal liver for years after giving birth [52,53,56]. This also gave rise to the hypothesis that these migratory FCs may in fact be pluripotent in nature with the capacity to differentiate into multiple cellular lineages [2,5,55]. A similar result was found in thyroid biopsy sections of women with thyroid adenomas, reinforcing this hypothesis of FC migration occurring in response to maternal injury [5]. Similarly, these cells were found to differentiate into multiple cellular lineages within the maternal thyroid although research on whether they can behave as functional cells (interacting with the maternal microenvironment) to aid in tissue repair processes has not been conclusive [52].
To test this hypothesis in vivo, experiments used transgenic mouse models where virgin C57BL/6 females were mated to males transgenic for the enhanced green fluorescent protein (eGFP). This breeding scheme was used in order to detect the presence of eGFP+ FCs within the black background of the WT mother [2,5]. Using FACS analysis, they could then isolate eGFP+ FCs directly from the maternal tissues and assess their cellular identity by evaluating expression of specific cellular markers. In addition, after sorting out all the eGFP+ FCs in their tissue of interest, they examined the role of CCL2/CCR2 signaling in wound healing [5,57].
To address if FC migration occurs in response to maternal injury, experiments utilized a transgenic mouse model, and excisional skin wounds were introduced on the dorsal side of the pregnant females at embryonic day 15.5. Quantification by FACS analysis detected an increase in eGFP+ FCs in the wound bed of pregnant females one day after inducing injury [57]. This increase in eGFP+ FCs was also observed in the blood and bone marrow of pregnant females as well, indicating not only an increase in FC migration within the maternal blood circulation, but also at the site of injury, proving these FCs do in fact migrate in response to maternal injury [57].
An elegant study evaluated the ability of FC migration to the maternal heart in response to cardiac injury [5]. To this end, a myocardial infarction and ischemia/reperfusion injury was introduced in the mother at approximately day 12 gestation [5]. DNA from injured hearts was then collected and examined for the presence of eGFP+ FCs; thereafter, quantitative PCR analysis demonstrated greater expression of cells positive for eGFP in injured maternal hearts than those of sham and non-infarcted controls [5]. Further examination by confocal microscopy of ventricular heart sections from pregnant females showed a high percentage of eGFP+ cells in or around the infarct zones confirming these eGFP+ FCs homed to site of maternal injury [5]. Additionally, the eGFP+ cells found in the injured myocardium of pregnant females also expressed markers for varying cardiac lineages including cardiomyocytes, smooth muscle cells, and ECs [5]. Interestingly, when plating these cells on a feeder layer derived from isolated cardiomyocytes of neonatal cyclin A2 transgenic mice, the isolated eGFP+ FCs differentiated into beating cardiomyocytes and expressed cardiac troponin [5]. After culturing for 5 weeks in chamber slides, they also expressed gap junction marker connexin 43, suggesting an electromechanical connection between the eGFP+ FC-derived cardiomyocytes and the feeder cardiomyocytes [5]. This finding was significant in that these cells not only were able to express a variety of cellular cardiac lineage markers, but also demonstrated their ability to adopt functional phenotypic traits of neighboring cells within the maternal heart. When investigating the cellular identity of total eGFP+ FCs isolated from injured maternal hearts, they also demonstrated their ability to express a variety of cardiac stem/progenitors markers including NKX2.5, Sca-1, nanog, and sox2 [5]. Trophoblast stem cell marker cdx2 was also found to be expressed in 38% of eGFP+ FCs isolated from injured maternal hearts [5]. In previous studies, Cdx2 has been shown to regulate trophoblast stem (TS) cell development and proliferation. This raised the question of whether TS cells (originating from the placenta) can migrate to sites of injury where they then differentiate into multiple cardiac cell lineages [5]. Collectively, these data suggest that fetal stem/progenitor cells can migrate to the mother's tissues in response to injury, where they likely differentiate into multiple cellular lineages. The cellular fates these FCs adopt, as well as their functional capabilities, seem to vary by maternal organ. The signaling mechanisms behind this migration pattern as well as how they interact with neighboring cells within the maternal microenvironment, however, are still unclear and need to be further explored.
9. What are the Cellular Fates of Migratory FCs?
There is no consensus on the exact cellular identity and origin of migratory FCs that enter the maternal circulation during pregnancy. Given the lack of reliable human trophoblast markers and limitations with human tissue samples, this task has been extremely difficult to perform. Early studies evaluating pregnancy-related progenitors (PRPCs) have provided new insights into the heterogeneous nature of these cells and found that overall expression and distribution of cellular markers seems to vary depending on the maternal tissue they are found in [54]. FCs possessing male DNA, isolated from peripheral blood samples, have been shown to express both hematopoietic and mesenchymal markers whereas FCs within maternal epithelial tissues display higher expression of epithelial markers. Another study highlighted this correlation further by assessing the lineage potential of FCs isolated from maternal epithelial tissues vs those isolated from maternal lymph and blood [52]. According to their results, between 20%-50% of FCs found in maternal cervical epithelium specimens expressed epithelial marker cytokeratin whereas 90% of FCs isolated from maternal lymph node and spleen specimens expressed hematopoietic marker CD45 [52].
To identify and characterize the FCs within a murine model, C57BL/6 WT virgin females were mated to males transgenic for the eGFP sequence. Various maternal organs were isolated at embryonic day 18 when FC migration into the mother is at its peak, and flow cytometry was used to detect the number of eGFP+ cells that were also positive for expression of each antigen marker [2]. According to their results, the maternal lung displayed the highest number of eGFP+ FCs and exhibited the most varied distribution of cellular expression with significant expression of CD31, CD34, CD44, ITGAM, and ITGB1. Conversely, other maternal tissues such as the spleen and bone marrow (isolated at embryonic day 18 gestation) displayed higher specificity with one cell marker being predominately expressed as opposed to the varied distribution pattern observed within the maternal lung [2]. In the maternal blood, expression of ITGB1 was predominantly expressed suggesting a likely MSC origin. Within the maternal bone marrow and spleen, there was significant PTPRC expression by eGFP+ FCs whereas within the maternal kidney, eGFP+ FCs favored expression of EC marker CD31 and ITGB1 [2]. Therefore, it is likely that fetal-derived progenitors migrate to a wide range of maternal organs during pregnancy, and their cellular fate seems to vary depending on the maternal organ, where the microenvironment allows for adhesion, differentiation and acquisition of phenotypic markers. It would be a rewarding research question to address if these migratory FCs can aid in tissue repair and angiogenic processes. In principle, it could lead to the development of therapeutics for pregnancy-related complications where placental angiogenesis is impaired. It is important to note that due to the low levels of eGFP+ FCs found within many maternal organs, it would be presumptuous to draw a complete conclusion from the cellular expression data. Further research underlying the molecular mechanism behind what triggers the migration of FCs into the mother needs to be completed in order to increase recruitment of endogenous fetal progenitors to maternal injury sites, yield better reproducibility, and gain a better understanding of their cellular identity.
To elucidate the mechanism behind what triggers the migration of FCs to sites of maternal injury, experiments were designed to evaluate a population of fetal-derived myeloid progenitors cells defined as CD11b+CD34+CD31+ that had been recruited to sites of maternal injury during pregnancy [57]. This study found that C-C chemokine receptor 2 (a chemokine receptor expressed on bone marrow progenitors and macrophages) was overexpressed on fetal progenitors at sites of maternal skin injury [57]. Additionally, treatment with Ccl2 (its main ligand) increased the migration of fetal progenitors and improved wound healing time exclusively in pregnant and postpartum females [57]. Utilization of Ccr2KO/KO mice confirmed this pattern of recruitment and led to one of the first mechanistic breakthroughs in this area of research. In their study, the authors addressed the hypothesis that during pregnancy, monocytes and ECs from wounds secrete Ccl2 leading to the recruitment of CD11b+CD34+CD31+ FCs from the bone marrow of the mother [57]. This recruitment pattern was largely due to the overexpression of Ccr2 on FCs which once at the site of skin injury, transdifferentiate into mural and ECs aiding wound healing cellular processes [57]. They also found that this population of fetal-derived MPCs displayed more pluripotent and proliferative properties than adult progenitors of the same phenotype. This novel strategy could prove useful when searching for an alternative to induce pluripotent stem cell or embryonic stem cell therapies. Mobilization of endogenous fetal stem cells could be beneficial in developing therapies for mothers with genetic or acquired diseases as they are semi-allogeneic and well tolerated by the maternal immune system [57].
10. What are the advantages and limitations of pregnancy research models?
Due to regulatory limitations, developing a reliable research model for studying pregnancy and pregnancy-related disorders has been difficult. Even with access to human biopsy tissue samples, proving statistical significance can be challenging as tissue sample availability can be minimal and vary from patient-to-patient, resulting in low reproducibility. Utilization of a murine model system has helped bridge this gap by providing a robust platform for studying the migration of fetal progenitors (to the mother) during pregnancy [31]. Additionally, recent advances in isolating mouse and human-derived trophoblast stem cells have provided the unique opportunity to examine placental development in vitro [33,58]. New research strategies and methodologies are needed to further our knowledge on the role and cellular identity of migratory FCs, and it is equally important to consider the advantages and limitations of each model system and how we can ultimately address clinically relevant questions of human pregnancy associated vascular diseases.
There are known differences between human and rodent placentation, however, the mouse has been a useful model system to investigate the development of placenta due to its genetic homogeneity and ease of genetic manipulation [26,30,32,59]. Both human and rodent placentation is hemochorial, meaning the fetal epithelium is bathed in maternal blood [29,30,31,32]. There are several functional similarities between mice and humans in the vascular villous units of the placenta (responsible for gas exchange and nutrient uptake) that make the mouse a suitable animal model for studying placentation [27,30,32,59]. In fact, most key events of placenta development in mice are similar to human placentation and more highly conserved than that of larger animals such as sheep [29,30,31,58] Interestingly, the mouse was the first animal from which trophoblast stems cells were isolated and cultured in vitro, and studies utilizing mTSCs have led to a deeper understanding of the molecular and regulatory events underlying trophoblast differentiation and lineage development [28].
There are, however, several key anatomical differences worth mentioning between murine and human placentation. One significant difference being in the layers of trophoblasts. In the mouse labyrinth, there are three layers of trophoblasts whereas in the chorionic villi of human placentas there are two in early gestation and as pregnancy progresses, that shifts to one functional layer in late gestation [29,30]. Another key difference is in the process of EVT invasion and remodeling of the maternal spiral arteries. As mentioned earlier, EVT invasion and subsequent remodeling of the maternal spiral arteries is a critical component of early placental development as the woman's body adapts to meet the needs of the growing baby [29,30,31]. Alterations in this process can lead to pregnancy-related complications such as fetal growth restriction and pre-eclampsia. In mice, however, the parietal giant cells and glycogen trophoblasts are only minimally invasive as opposed to the highly invasive EVTs in human placentas [29,30,31]. This is an important distinction, especially when examining pregnancy disorders with underlying vascular deficiencies, as is the case with pre-eclampsia [11,12].
In both mice and humans, placental development begins with trophectoderm formation [28,29,30,31,32,33,38,46]. Gene knockout studies have identified key transcriptional regulators in placental development, governing trophoblast growth and specialization, which are common processes in both humans and mice [29,30]. For example, Oct4 and homeobox protein CDX2 are important for initiation of the first cellular fate decision [30]. Transcriptional coactivator YAP1, along with transcription factor TEAD4, induces expression of Cdx2 marking the first cellular fate decision whereas cells of the outer morula become biased towards a TE fate [60]. In contrast, Oct4 transcripts are largely enriched in the ICM of human blastocytes which go on to later form the embryo. In the first trimester of human pregnancy, Cdx2 is strongly expressed in vCTBs but is rapidly downregulated as pregnancy progresses [60]. The role of TEAD4 in placentation is highly conserved between mice and humans [30,60]. In mice, TEAD4 and CDX2 co-operate with transcription factors such as: ELF5, ETS2, ESRRB, EOMES, GATA3, SOX2, and TFAP2C to form a functional compartment in early trophoblast development [60]. Thus, coordination between these transcription factors drives trophoblast maintenance and differentiation. Another important signaling factor needed for mouse trophoblast development is fibroblast growth factor (FGF). Esrrb and Sox2 are dependent on fibroblast growth factor signaling which is also needed for early trophoblast maintenance. Mouse-derived trophoblast stem cell lines have been established using a combination of mouse embryonic fibroblasts (MEFs) as a feeder layer and FGF4. Both FGF4 and TGF are required for mouse TS cells to maintain their undifferentiated state in vitro [29,30,58]. In human, as Esrrb and Sox2 expression is low in CTBs, and thus their role in trophoblast maintenance remains unknown. Additionally, EOMES expression in human TEs is absent, whereas expression of this transcription factor is essential for maintenance and self-renewal in mTSCs [29,30]. ELF5 is another transcription factor that plays an important role in regulating trophoblast maintenance and differentiation in both mice and humans [21,30]. ELF5 is essential for both human and mouse trophoblast stem cell maintenance. vCTBs that express CDX2 and ELF5 in the first trimester are suggested to be good candidates for cells that retain stem cell potential [30].
One of the major limitations in both human and mouse models of early trophoblast development is the lack of specific trophoblast markers [30]. In mice, many of the trophoblast lineage markers are also expressed either concurrently or at later stages in the embryo, therefore, there is no one unequivocal marker of trophoblast identity [30]. Despite the lack of specific trophoblast lineage markers, there is functional diversity between expression of these markers in the embryo and trophoblast due to formation of divergent protein complexes within each compartment [30]. Although similar markers of identity are expressed in both the embryo and trophoblast, they each form unique protein complexes that lead to non-overlapping sets of genomic elements. These unique combinations of signaling factors help differentiate trophoblast cellular identity from embryonic identity [30]. A good example of this is in mTSCs as ELF5 preferentially interacts with EOMES. The formation of this EOMES-ELF5 complex helps drive trophoblast expression by binding to additional trophoblast self-renewal genes. This complex is disbanded, however, when TFAP2C levels exceed those of EOMES and self-renewal shifts to differentiation as the formation of a new protein complex consisting of TFAP2C-ELF5 promotes the up-regulation of trophoblast differentiation [30]. This is an example of unique protein complexes that can help distinguish trophoblastic from embryonic lineage cells.
In recent years, the development of mouse pregnancy models has yielded exciting findings on how placental villous development affects embryonic development. This was demonstrated by the knockout mouse models of retinoblastoma (Rb) tumor suppressor gene and the myc proto-oncogene which led to intra-uterine lethality. Recent experiments using conditional knockouts and tetraploid complementation assays showed gene function was returned in the trophoblast lineages [30]. This led to the hypothesis that many embryonic defects could be rescued by providing the mutant embryo with a functional placenta. Interestingly, from approximately 100 different mouse mutants surveyed, researchers have discovered the placenta is morphologically abnormal in roughly two-thirds of them [30]. This realization prompted further investigation into the genotype-phenotype axis between the embryo and placenta during gestation in hopes that we can better understand how placenta defects can lead to specific fetal pathologies [30]. Clearly, defective placentation can lead to fetal defects such as vascular deficiencies. For example, in Pparg and Mapk14 mutants, defective placentation was identified as a main contributor to embryonic heart defects. While the specific causal and mechanistic reasons behind this correlation in embryonic vascular defects and altered placentation remain unknown, this area of research is fertile with novelty and innovation [30].
Significant progress has been made in developing human-derived trophoblast cell (hTSC) lines and organoid models to address these research topics in vitro more effectively. Previously, primary trophoblasts could only be propagated in vitro from transformed trophoblastic tumors. Human embryonic stem cell (hESC)-derived TBs lines have been useful but do not faithfully replicate the earliest steps in trophoblast differentiation and expansion, which is critical for predicting placental function [30,33]. The development of hTSCs have allowed researchers to better understand transcriptional regulation in early human trophoblast differentiation and is likely to lead to the identification of key signaling mediators involved in early trophoblast differentiation and propagation as well as the development of better therapeutic interventions for complications effecting both mother and baby [29,30,31,32,33].
11. Can animal models of pregnancy-related disease be used for drug screening?
The animal models of pregnancy-related disease can be used as a platform for pharmacological drug screening. For example, the inhibition or anomaly of vascular development has been associated with fetal loss, congenital human abnormalities, and cognitive deficits. Animal research on pregnancy and related disease could be used as an in vivo tool for the rigorous exploration of high-throughput drug screening and comprehensive toxicity assessments. For instance, the detection and quantification of a) fetal cells, b) genomic/non-genomic factors, c) exosomes, d) cytokines, e) hormones, in both the circulatory milieu and bronchoalveolar lavage (BAL) fluid bear the potential to create novel diagnostic, prognostic, and therapeutic paradigms [61,62,63,64]. Importantly, the platform might afford the opportunity to systematically assay pharmacological agents, whether agonists or antagonists, capable of modulating angiogenic processes associated with gestation. Such agents stand poised to exert differential influences on EC proliferation, survival, apoptosis, and thereby choreograph the formation of endothelial cell capillary branching networks and the course of pregnancy. Furthermore, this platform could also offer substantial utility in the evaluation of anticancer compounds, specifically targeting those with an enhanced cardiovascular safety index. Given the continuous exposure of ECs to hemodynamic shear stress within the circulatory system and their intricate interplay with local cell populations, we envisage harnessing this model to substantially expedite the preclinical drug development trajectory for an array of vascular diseases associated with pregnancy and women's health.
12. Summary
FCs found in maternal circulation are likely heterogenous but have intrigued investigators due to their potential implications in maternal health. Initially seen as potential contributors to maternal pathology, these cells are likely to have multifaceted roles, including immune modulation and tissue repair. However, their precise role remains a subject of ongoing investigation and further research is needed to fully understand their functions and potential applications in diagnosing and treating pregnancy-related disorders. FCs found in mothers circulation represent a promising and an understudied area of research that could shed light on the complex interaction between maternal and fetal systems and revolutionize maternal health, as well as the therapeutic landscape.
Author Contributions
Original Draft Preparation, C.A.; Original Graphics Preparation, C.A.; Review & Editing, C.A., V.M., R.D.M., K.W.; Visualization, K.W.; Supervision, K.W.; Funding Acquisition, C.A., V.M., K.W.
Funding
C.A. was supported by NIH-T32HL007829 and NIH-T32HL144459. K.W. was supported, in part, by the American Heart Association (AHA) Grants GRNT33700162 and TPA34910205. V.M. was supported by the NIH-T32 HL007829 and AHA Pre-doctoral Fellowship 19PRE34450173 (National Affiliate).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Herzenberg, L.A.; Bianchi, D.W.; Schröder, J.; Cann, H.M.; Iverson, G.M. Fetal cells in the blood of pregnant women: detection and enrichment by fluorescence-activated cell sorting. Proc Natl Acad Sci USA 1979, 76, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, Y.; Johnson, K.L.; Peter, I.; Tighiouart, H.; Bianchi, D.W. Fetal Cells in the Pregnant Mouse Are Diverse and Express a Variety of Progenitor and Differentiated Cell Markers. Biol. Reprod. 2009, 81, 26–32. [Google Scholar] [CrossRef]
- Campagnoli, C.; Roberts, I.A.G.; Kumar, S.; Bennett, P.R.; Bellantuono, I.; Fisk, N.M. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood 2001, 98, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Huu, S.N.; Oster, M.; Uzan, S.; Chareyre, F.; Aractingi, S.; Khosrotehrani, K. Maternal neoangiogenesis during pregnancy partly derives from fetal endothelial progenitor cells. Proc Natl Acad Sci USA 2007, 104, 1871–1876. [Google Scholar] [CrossRef]
- Kara, R.J.; Bolli, P.; Karakikes, I.; Matsunaga, I.; Tripodi, J.; Tanweer, O.; Altman, P.; Shachter, N.S.; Nakano, A.; Najfeld, V.; et al. Fetal Cells Traffic to Injured Maternal Myocardium and Undergo Cardiac Differentiation. Circ. Res. 2012, 110, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, D.W.; Zickwolf, G.K.; Weil, G.J.; Sylvester, S.; DeMaria, M.A. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc. Natl. Acad. Sci. USA 1996, 93, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Khosrotehrani, K.; Johnson, K.L.; Guégan, S.; Stroh, H.; Bianchi, D.W. Natural history of fetal cell microchimerism during and following murine pregnancy. J. Reprod. Immunol. 2005, 66, 1–12. [Google Scholar] [CrossRef]
- Turco, M.; Moffett, A. Development of the human placenta. Development. 2019, 146, dev163428. [Google Scholar] [CrossRef] [PubMed]
- James, J.L.; Lissaman, A.; Nursalim, Y.N.S.; Chamley, L.W. Modelling human placental villous development: designing cultures that reflect anatomy. Cell. Mol. Life Sci. 2022, 79, 384. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2019, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Ramlakhan, K.P.; Johnson, M.R.; Roos-Hesselink, J.W. Pregnancy and cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Aplin, J.D.; Myers, J.E.; Timms, K.; Westwood, M. Tracking placental development in health and disease. Nat. Rev. Endocrinol. 2020, 16, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Dawe, G.S.; Tan, X.W.; Xiao, Z. Cell migration from baby to mother. Cell adhesion migration. 2007, 1, 19–27. [Google Scholar] [CrossRef]
- Davis, M.B.; Arany, Z.; McNamara, D.M.; Goland, S.; Elkayam, U. Peripartum Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020, 75, 207–221. [Google Scholar] [CrossRef]
- Felker, G.M.; Thompson, R.E.; Hare, J.M.; et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000, 342, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Maltepe, E.; Fisher, S.J. Placenta: The Forgotten Organ. Annu. Rev. Cell Dev. Biol. 2015, 31, 523–552. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, S.; Song, T.; Yin, Y.; Tan, C. Placental Angiogenesis in Mammals: A Review of the Regulatory Effects of Signaling Pathways and Functional Nutrients. Adv. Nutr. Int. Rev. J. 2021, 12, 2415–2434. [Google Scholar] [CrossRef]
- O'Brien, K.; Wang, Y. The Placenta: A Maternofetal Interface. Annu. Rev. Nutr. 2023, 43, 301–325. [Google Scholar] [CrossRef]
- Paauw, N.D.; Lely, A.T. Cardiovascular Sequels During and After Preeclampsia. Adv Exp Med Biol. 2018, 1065, 455–470. [Google Scholar]
- Soncin, F.; Natale, D.; Parast, M.M. Signaling pathways in mouse and human trophoblast differentiation: a comparative review. Cell. Mol. Life Sci. 2014, 72, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.D.; De Long, N.E.; Wang, R.C.; Yazdi, F.T.; Holloway, A.C.; Raha, S. Angiogenesis in the Placenta: The Role of Reactive Oxygen Species Signaling. BioMed Res. Int. 2015, 2015, 814543. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth—New Insights from Mouse Models. Front. Endocrinol. 2018, 9, 570. [Google Scholar] [CrossRef] [PubMed]
- Kirollos, S.; Skilton, M.; Patel, S.; Arnott, C. A Systematic Review of Vascular Structure and Function in Pre-eclampsia: Non-invasive Assessment and Mechanistic Links. Front. Cardiovasc. Med. 2019, 6, 166. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.; Puttemans, P.; Benagiano, G. Placental bed research: I. The placental bed: from spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 2019, 221, 437–456. [Google Scholar] [CrossRef]
- Cheng, S.B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef] [PubMed]
- Opichka, M.A.; Rappelt, M.W.; Gutterman, D.D.; Grobe, J.L.; McIntosh, J.J. Vascular Dysfunction in Preeclampsia. Cells 2021, 10, 3055. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hu, Y.; Ma, J. Animal models of the placenta accreta spectrum: current status and further perspectives. Front. Endocrinol. 2023, 14, 1118168. [Google Scholar] [CrossRef] [PubMed]
- Gauster, M.; Moser, G.; Wernitznig, S.; Kupper, N.; Huppertz, B. Early human trophoblast development: from morphology to function. Cell. Mol. Life Sci. 2022, 79, 345. [Google Scholar] [CrossRef]
- James, J.L.; Lissaman, A.; Nursalim, Y.N.S.; Chamley, L.W. Modelling human placental villous development: designing cultures that reflect anatomy. Cell. Mol. Life Sci. 2022, 79, 384. [Google Scholar] [CrossRef] [PubMed]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2019, 21, 27–43. [Google Scholar] [CrossRef]
- Knöfler, M.; Haider, S.; Saleh, L.; Pollheimer, J.; Gamage, T.K.J.B.; James, J. Human placenta and trophoblast development: key molecular mechanisms and model systems. Cell Mol Life Sci. 2019, 76, 3479–3496. [Google Scholar] [CrossRef] [PubMed]
- Karvas, R.M.; Khan, S.A.; Verma, S.; Yin, Y.; Kulkarni, D.; Dong, C.; Park, K.-M.; Chew, B.; Sane, E.; Fischer, L.A.; et al. Stem-cell-derived trophoblast organoids model human placental development and susceptibility to emerging pathogens. Cell Stem Cell 2022, 29, 810–825. [Google Scholar] [CrossRef]
- Io, S.; Kabata, M.; Iemura, Y.; Semi, K.; Morone, N.; Minagawa, A.; Wang, B.; Okamoto, I.; Nakamura, T.; Kojima, Y.; et al. Capturing human trophoblast development with naive pluripotent stem cells in vitro. Cell Stem Cell 2021, 28, 1023–1039. [Google Scholar] [CrossRef] [PubMed]
- Koyano-Nakagawa, N.; Garry, D.J. Etv2 as an essential regulator of mesodermal lineage development. Cardiovasc. Res. 2017, 113, 1294–1306. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.D.; De Long, N.E.; Wang, R.C.; Yazdi, F.T.; Holloway, A.C.; Raha, S. Angiogenesis in the Placenta: The Role of Reactive Oxygen Species Signaling. BioMed Res. Int. 2015, 2015, 814543. [Google Scholar] [CrossRef] [PubMed]
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; Epstein, F.H.; Sukhatme, V.P.; Karumanchi, S.A. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Charnock-Jones, D.S.; Kaufmann, P.; Mayhew, T.M. Aspects of human fetoplacental vasculogenesis and angiogenesis. I. molecular regulation. Placenta. 2004, 25, 103–113. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef]
- Zhi, Z.; Yang, W.; Liu, L.; Jiang, X.; Pang, L. Early missed abortion is associated with villous angiogenesis via the HIF-1α/VEGF signaling pathway. Arch. Gynecol. Obstet. 2018, 298, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Norton, K.-A.; Popel, A.S. Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis. Sci. Rep. 2016, 6, 36992. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Naiche, L.A.; Villa, S.R.; Kitajewski, J.K. Endothelial Cell Fate Determination: A Top Notch Job in Vascular Decision-Making. Cold Spring Harb Perspect Med 2022, 12, a041183. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, D.; Yu, Y.Y.; Kang, I.; Cha, M.J.; Kim, J.Y.; Park, C.; Watson, D.K.; Wang, T.; Choi, K. Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep. 2015, 16, 654–669. [Google Scholar] [CrossRef]
- Kataoka, H.; Hayashi, M.; Nakagawa, R.; Tanaka, Y.; Izumi, N.; Nishikawa, S.; Jakt, M.L.; Tarui, H.; Nishikawa, S. Etv2/ER71 induces vascular mesoderm from Flk1+PDGFRa+ primitive mesoderm. Blood. 2011, 118, 6975–6986. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, D.H.; Kim, J.K.; Choi, H.S.; Dwivedi, B.; Rupji, M.; Kowalski, J.; Green, S.J.; Song, H.; Park, W.J.; Chang, J.Y.; Kim, T.M.; Park, C. ETV2/ER71 regulates the generation of FLK1+ cells from mouse embryonic stem cells through miR-126-MAPK signaling. Stem Cell Res Ther. 2019, 10, 328. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Itoh, H.; Hirashima, M.; Ogawa, M.; Nishikawa, S.; Yurugi, T.; Naito, M.; Nakao, K.; Nishikawa, S.-I. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 2000, 408, 92–96. [Google Scholar] [CrossRef]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; De Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Kim, T.M.; Malik, A.B. Transcriptional Regulation of Endothelial Cell and Vascular Development. Circ. Res. 2013, 112, 1380–1400. [Google Scholar] [CrossRef]
- Lui, K.; Zangi, L.; A Silva, E.; Bu, L.; Sahara, M.; A Li, R.; Mooney, D.J.; Chien, K.R. Driving vascular endothelial cell fate of human multipotent Isl1+ heart progenitors with VEGF modified mRNA. Cell Res. 2013, 23, 1172–1186. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Khosrotehrani, K.; Johnson, K.L.; Cha, D.H.; Salomon, R.N.; Bianchi, D.W. Transfer of Fetal Cells With Multilineage Potential to Maternal Tissue. JAMA 2004, 292, 75–80. [Google Scholar] [CrossRef]
- Khosrotehrani, K.; Bianchi, D.W. Multi-lineage potential of fetal cells in maternal tissue: a legacy in reverse. J. Cell Sci. 2005, 118, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, S.; Hoffman, A.M.; Johnson, K.L.; Bianchi, D.W. Pregnancy-associated progenitor cells: an under-recognized potential source of stem cells in maternal lung. Placenta. 2011, 32 (Suppl. 4), S298–S303. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Smith, J.B.; Jimenez, S.A. Identification of Fetal DNA and Cells in Skin Lesions from Women with Systemic Sclerosis. New Engl. J. Med. 1998, 338, 1186–1191. [Google Scholar] [CrossRef]
- Johnson, K.L.; Samura, O.; Nelson, J.L.; McDonnell, W.M.; Bianchi, D.W. Significant fetal cell microchimerism in a nontransfused woman with hepatitis C: Evidence of long-term survival and expansion. Hepatology 2002, 36, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Castela, M.; Nassar, D.; Sbeih, M.; Jachiet, M.; Wang, Z.; Aractingi, S. Ccl2/Ccr2 signalling recruits a distinct fetal microchimeric population that rescues delayed maternal wound healing. Nat Commun. 2017, 8, 15463. [Google Scholar] [CrossRef] [PubMed]
- Red-Horse, K.; Zhou, Y.; Genbacev, O.; Prakobphol, A.; Foulk, R.; McMaster, M.; Fisher, S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004, 114, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Dassanayake, M.; Langen, E.; Davis, M.B. Pregnancy Complications as a Window to Future Cardiovascular Disease. Cardiol. Rev. 2020, 28, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Arutyunyan, A.; Roberts, K.; Troulé, K.; Wong, F.C.K.; Sheridan, M.A.; Kats, I.; Garcia-Alonso, L.; Velten, B.; Hoo, R.; Ruiz-Morales, E.R.; et al. Spatial multiomics map of trophoblast development in early pregnancy. Nature 2023, 616, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ma, Y.; Wu, H.; Wang, Y.-L. Placenta-Derived MicroRNAs in the Pathophysiology of Human Pregnancy. Front. Cell Dev. Biol. 2021, 9, 646326. [Google Scholar] [CrossRef]
- Ogoyama, M.; Takahashi, H.; Suzuki, H.; Ohkuchi, A.; Fujiwara, H.; Takizawa, T. Non-Coding RNAs and Prediction of Preeclampsia in the First Trimester of Pregnancy. Cells. 2022, 11, 2428. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, H.; Hong, W.; Wei, Y.; Tian, H.; Huang, X.; Jia, L.; Zheng, J.; Duan, T.; He, Q.; Wang, K. Human Placental Endothelial Cell and Trophoblast Heterogeneity and Differentiation Revealed by Single-Cell RNA Sequencing. Cells. 2022, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Apicella, C.; Ruano, C.S.M.; Thilaganathan, B.; Khalil, A.; Giorgione, V.; Gascoin, G.; Marcellin, L.; Gaspar, C.; Jacques, S.; Murdoch, C.E.; et al. Pan-Genomic Regulation of Gene Expression in Normal and Pathological Human Placentas. Cells 2023, 12, 578. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



