
Submitted:
28 September 2023
Posted:
29 September 2023
You are already at the latest version
Abstract
Background: Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are the two most common subtypes within rare genetic photodermatoses, which can lead to incapacitating pain episodes. Methods: We reviewed previous and current treatment options for EPP/XLP, notably with a focus on metabolism, pharmacokinetics, mechanism of action, key clinical trial results, and limitations of Dersimelagon [MT-7117]. Results: In review of treatment modalities for EPP/XLP, Dersimelagon has demonstrated an acceptable safety profile with the ease of once-daily oral dosage. Conclusions: Dersimelagon should be a considered as a potential treatment for EPP/XLP. Ongoing studies will provide additional insights into expanding the treatment options for those suffering from EPP/XLP.
Keywords:
Erythropoietic Protoporphyria
; X-linked protoporphyria
; oral
; dersimelagon
Introduction
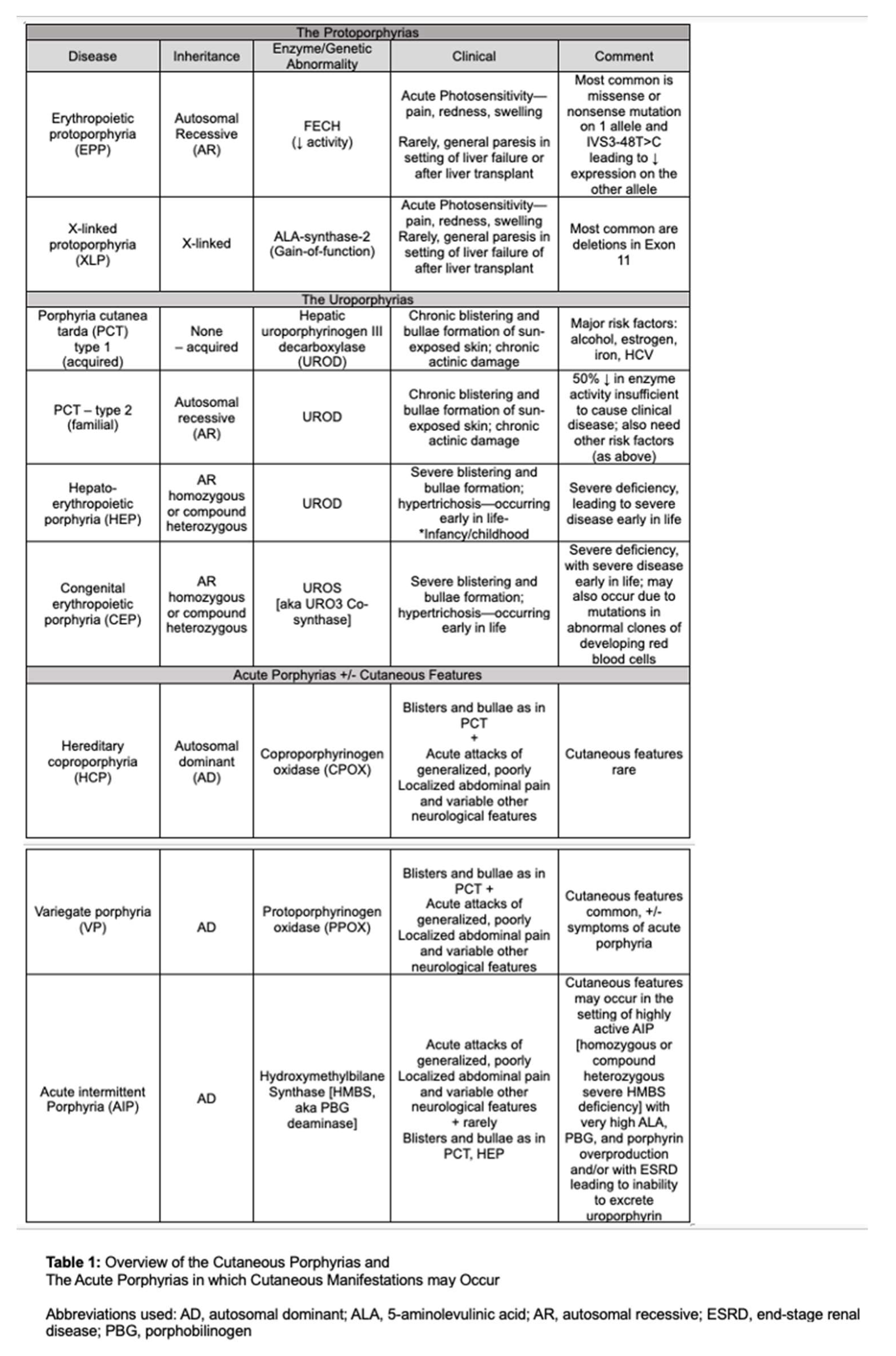
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are rare genetic photodermatoses with an estimated prevalence between 1 in 75,000 and 1 in 200,000 among the Caucasian population; however, the frequency of pathogenic ferrochelatase (FECH) mutations in the UK Biobank database suggest the potential for a higher prevalence of 184/200,000 [1,2,3]. While rare, EPP/XLP is one of the two most common cutaneous porphyrias [the other being porphyria cutanea tarda, either acquired or inherited] and the most common porphyria overall in the pediatric population [2,3,11]. EPP and XLP are inborn errors of heme biosynthesis resulting in abnormal accumulation of erythrocyte protoporphyrin IX (ePPIX), which can lead to debilitating pain episodes [4,5,6,7,8,9,10]. The cutaneous porphyrias, as well as acute hepatic porphyrias that may present with cutaneous manifestations, are summarized in Table 1.
EPP is due to biallelic mutations in the FECH gene, which encodes for the enzyme responsible for the last step in heme biosynthesis, wherein PPIX is chelated with iron to form heme [8]. Most commonly, either a missense or nonsense mutation in one FECH allele occurs in trans to a non-coding splice variant [IVS3-48T>C] in the other allele [12,13]. The missense or nonsense mutation contributes to reduced expression of the normal protein, by nearly halving its synthesis. When coupled with the other mutated hypomorphic allele, this leads to a 75% or greater decrease in FECH activity – causing an accumulation of PPIX in late erythroblasts with clinically evident disease [13]. Patients with a higher accumulation of ePPIX report a faster onset of pain attacks [14,15,16]. Beyond the classic cutaneous manifestations, up to 47% of patients with EPP are found to have iron deficiency anemia. Though less common, progressive cholestatic liver disease may result in 2-5% of patients ultimately resulting in liver failure.
The enzyme ALA-synthase 2 (ALAS2), responsible for the initial step in erythroid heme biosynthesis is located on the X-chromosome. This gene was found to be associated with a variety of gain-of-function mutations that result in increased ALAS2 activity, causing an overproduction of protoporphyrin IX [12,13]. XLP was previously described as an X-linked dominant trait with essentially 100% penetrance, but there is recent evidence of heterozygous females who show varying phenotypic and biochemical heterogeneity, reflecting the random varying degree of X-chromosomal inactivation of the X-chromosome that carries the mutant gene [12]. In EPP/XLP, blue light, [peak effect at 410 nm, the Soret band], is absorbed by protoporphyrin which is excited to a singlet electron state; as it returns to the lower energy triplet state, it emits energy that leads to increased oxidative stress and pro-inflammatory cascades and cytokines. These, in turn, lead to pain, edema, and inflammatory responses [14].
However, the typical attacks of severe, diffuse abdominal pain or other neuro-visceral features associated with acute hepatic porphyrias are only rarely observed in patients with EPP/XLP, in the setting of liver failure, before or after liver transplantation [15,16]. When EPP or XLP is suspected, testing first involves the measurement of total erythrocyte protoporphyrin levels. Elevated levels of fractionated erythrocyte metal-free and zinc protoporphyrin help distinguish between EPP and XLP. In XLP, a significant portion (>25%) of protoporphyrin in red blood cells is bound to zinc. Confirming the specific diagnosis is supported by genetic testing through sequencing FECH or ALAS2 genes [17,45].
Dersimelagon [MT-7117] was designed specifically for the treatment of phototoxicity in EPP. This review serves to provide increased awareness and understanding of this novel oral agent that has shown clinically meaningful and statistically significant benefits in phase 2 clinical trials demonstrating its safety and efficacy in the treatment of EPP/XLP.
Previous therapies
Behavioral modification is typically advised as first-line treatment, such as sun avoidance, use of zinc oxide sunblock, and protective clothing. However, this is insufficient due to the infeasibility of sun avoidance and indoor exposure to damaging wavelengths of sunlight passing through windows or arising from artificial light sources. Additionally, most sunscreens are more protective against lower wavelength UV radiation and not the frequencies that cause EPP symptoms [18].
Narrowband ultraviolet B phototherapy has been attempted as a prophylactic measure in EPP. There were proposals for patients with EPP/XLP to purposely experience controlled and gradually increasing UVB radiation during the springtime to produce skin thickening with increased melanin production. This controlled form of UVB radiation requires administration by a trained professional and may be inconvenient for patients as there, it involves exposure at least three times weekly for at least five weeks during the spring. A small European study with 12 patients noted it was ineffective for 5 of these patients; not meeting any statistical or clinical significance [22].
Subsequently, there has been a treatment goal to induce eumelanogenesis without exposure to UV radiation. Early studies of beta-carotene as a treatment option necessitated daily high doses [a minimum of 180 mg/day for adults] for at least three months to reach carotenoid blood levels of at least 800 ug/dL [20,35,36]. Evaluation of these studies found data supporting the use of beta-carotene treatment efficacy were contradictory and efficacy inversely correlated with study quality [19]; i.e Krook, et al. were unable to find a significant difference between beta-carotene and placebo for photosensitivity in patients with EPP [21].
Treatment with cimetidine has been purported to inhibit delta-aminolevulinic acid [ALA] synthase, proposed thereby to reduce PPIX levels in patients with EPP. [But note that such inhibition has been proposed for ALA synthase-1, the hepatic form of the enzyme, rather than ALA synthase-2, the erythroid form of the enzyme.] Though data are limited to case reports or small series, without placebo controls or double-blind designs, there is some suggestion that photosensitivity may be decreased with cimetidine treatment [5]. An ongoing Phase-2, double-blind, placebo-controlled cross-over trial of cimetidine is currently underway in the United States (NCT05020184).
Additionally, there has not been clinical success with the use of vitamin C, or N-acetyl cysteine in the treatment of EPP [47,48,49]. A systemic review by Minder, et al. in 2009 concluded: “The available data are insufficient to prove the efficacy of any treatments studied so far in EPP.” With the advent of newer therapeutics, these previous treatments outlined above have now largely been abandoned due to inefficacy [19].
Currently, the only approved therapy for EPP and XLP is afamelanotide [Scenesse, Clinuvel Pharma], which focuses on increasing eumelanin production by melanocytes, with an expected reduction in the severity of phototoxic reactions to sunlight or other strong light. Afamelanotide has little if any, effect on the ongoing overproduction of PPIX [46]. It was approved in Europe by the European Medicines Agency and in the US by the FDA in 2019, as the first effective medical treatment for EPP [23,46]. Afamelanotide, [Nle4, D-Phe7]-a-MSH], also known as Melanotan 1, is an analog of a-MSH, which stimulates the production of eumelanin as a non-selective agonist of melanocortin receptors. It has higher potency and stability than the natural MSH peptide. A recent observational animal study from Switzerland suggests a dose-dependent protective effect from liver damage related to EPP [51]. Afamelanotide is administered as a subcutaneous implant, which is injected every two months and gradually releases the active peptide. This treatment was found to reduce severe phototoxic reactions and improve quality of life; however, a trained professional is needed to administer the treatment. Nausea and loss of appetite are the most notable side effects, perhaps, due to the molecule's ability to activate other MCRs non-selectively [24,25]. Other adverse events related to afamelanotide include headache, nasopharyngitis, hyperpigmentation, and back pain [26]. In May 2023, Swetlitz outlined challenges with obtaining afamelanotide for EPP or XLP [27]; notably concerns regarding patient accessibility [Clinuvel Pharma is currently supplying afamelanotide to only few selected centers] and costs [currently prices in the US are ~ $55,000/ implant] [27]. Thus, its use has been restricted to relatively few patients with EPP/XLP.
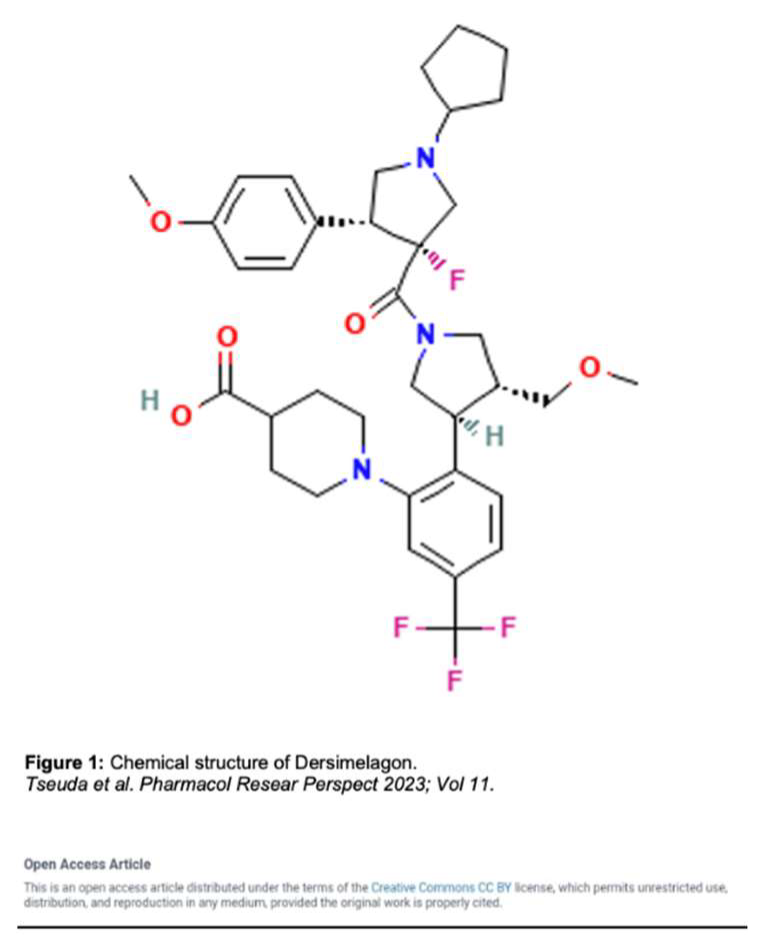
Dersimelagon (Figure 1) is an orally active small molecule selective melanocortin-1 receptor agonist, which increases the production of eumelanin resulting in increased skin pigmentation and anti-inflammatory effects resulting in increased duration of symptom-free sunlight exposure.
Metabolism and Pharmacokinetics of Dersimelagon
A key component of the appeal of Dersimelagon is its oral bioavailability. Mitsubishi Tanabe Pharma Corporation has developed modification methods to improve its stability as a non-peptide [28]. Its molecular structure (Figure 1 [31]) has extensive terminal modification to strengthen and protect the molecule from rapid degradation before reaching its target receptors. In contrast, afamelanotide is unsuitable for oral administration due to liability of its breakdown in the small intestine by proteases and peptidases [28,29].
The molecular stability of Dersimelagon was examined by Ogawa et al., who assessed oral bioavailability and pharmacokinetics of Dersimelagon tablets under a variety of gastric conditions such as fed-state, fasting-state, acidic beverage consumption, high-fat meals, and with a proton pump inhibitor (PPI) [30]. The 50-participant study was an open-label, multicenter, randomized, 2-cohort, sequential, and crossover study in which participants were randomized into two cohorts to receive 300 mg or 100 mg tablets under three gastric conditions: fasted (10+ hours), fed (high-fat breakfast with and without esomeprazole 40 mg 2 hours before breakfast) with water or a caffeine-free acidic carbonated beverage (355 mL of Canada Dry© ginger ale at a pH of 2.8). No effect was observed on overall exposure following consumption of a high-fat meal, and Cmax was higher (22%, 90% confidence interval [CI] 1.05–1.42) in a fed state compared with fasted conditions. Similarly, overall exposure AUC of Dersimelagon was comparable following administration alone or in combination with esomeprazole; however, coadministration of esomeprazole led to a slight decrease in Cmax (fasted: 9%, 90%CI 0.77–1.07; fed: 24%, 90%CI 0.66–0.88) compared with administration of Dersimelagon alone. In general, the consumption of an acidic beverage increased time to Cmax regardless of fed or fasted status and decreased overall exposure to AUC and Cmax of Dersimelagon [30].
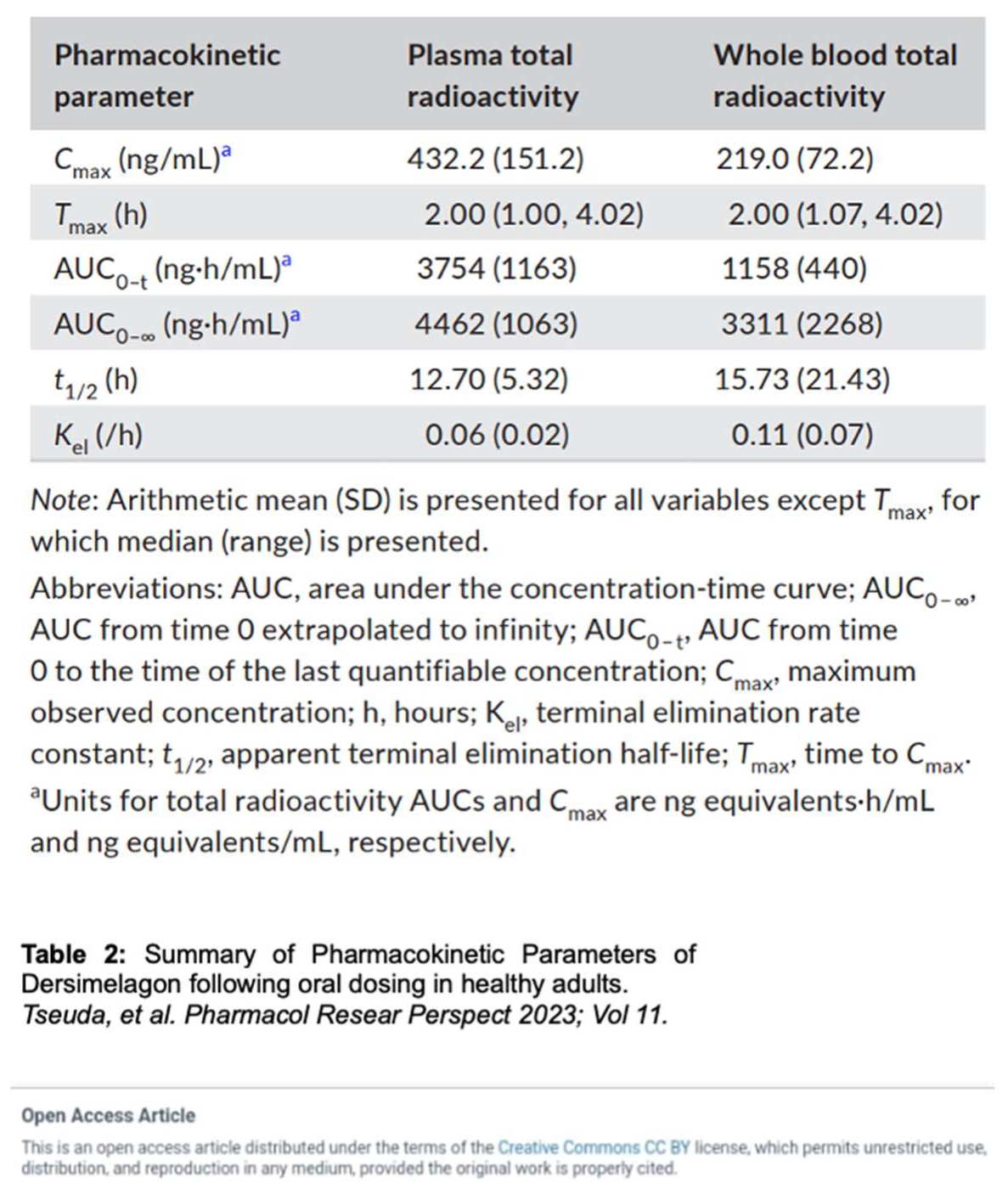
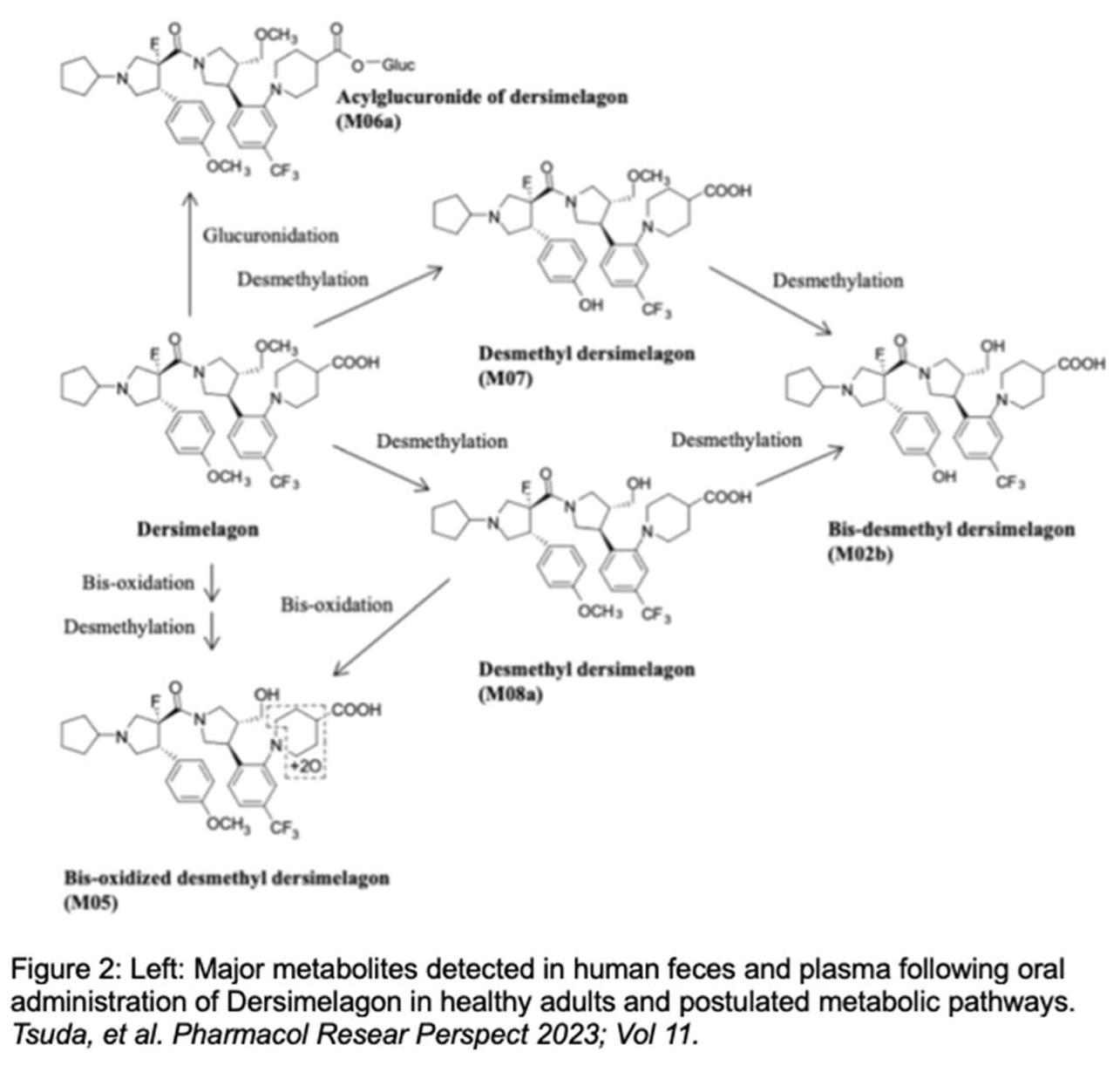
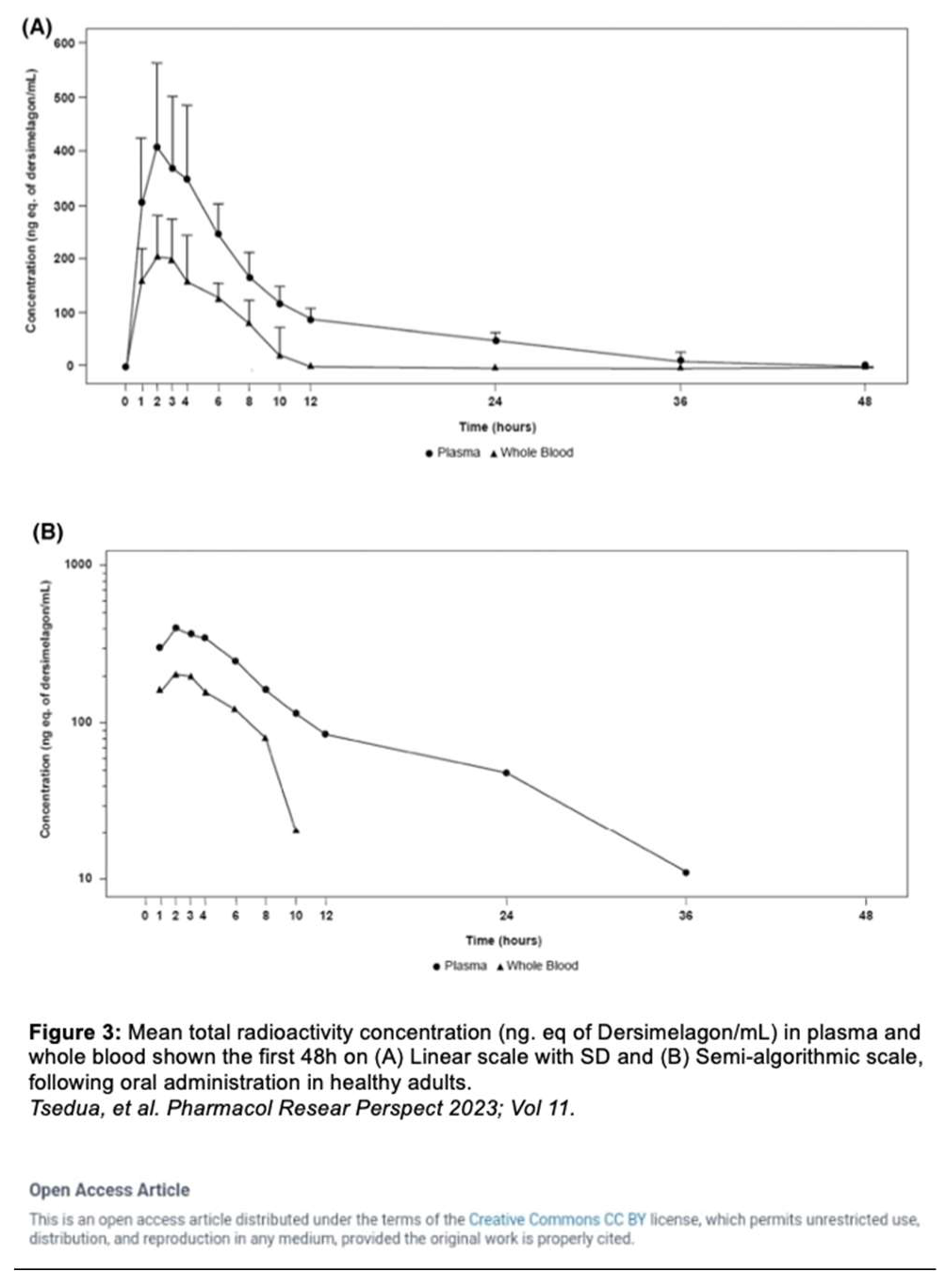
Tsuda et al. evaluated the absorption, metabolism, and excretion of Dersimelagon in rats, monkeys, and 6 humans (white males aged 30-65 years, weights 60-110 kg, and with BMI of 18 – 32 kg/m2, who were overall healthy and free from illness or disease). Participants received a single oral 100 mg dose of radioactive [14C] Dersimelagon. Blood, urine, and fecal samples were collected periodically to evaluate Cmax, Tmax, AUC, apparent t1/2, and terminal elimination rate constant (Kel). The median Tmax was 2 hours in humans (Table 2) [31]. As shown in Figure 2 [31], Dersimelagon is extensively metabolized to the glucuronide in the liver, which is eliminated in bile. In the intestine, some of the glucuronide is metabolized back to the parent drug. In humans, elimination of radioactivity in urine was negligible (excretion of radioactivity into the urine: 0.31% of dose), and the primary route of excretion was feces, with more than 90% of the radioactivity recovered through 5 days post-dose. Based on these findings, it was concluded a significant amount of radioactivity from [14C] Dersimelagon was not retained in the human body (Figure 3) [31]
Mechanism of Action
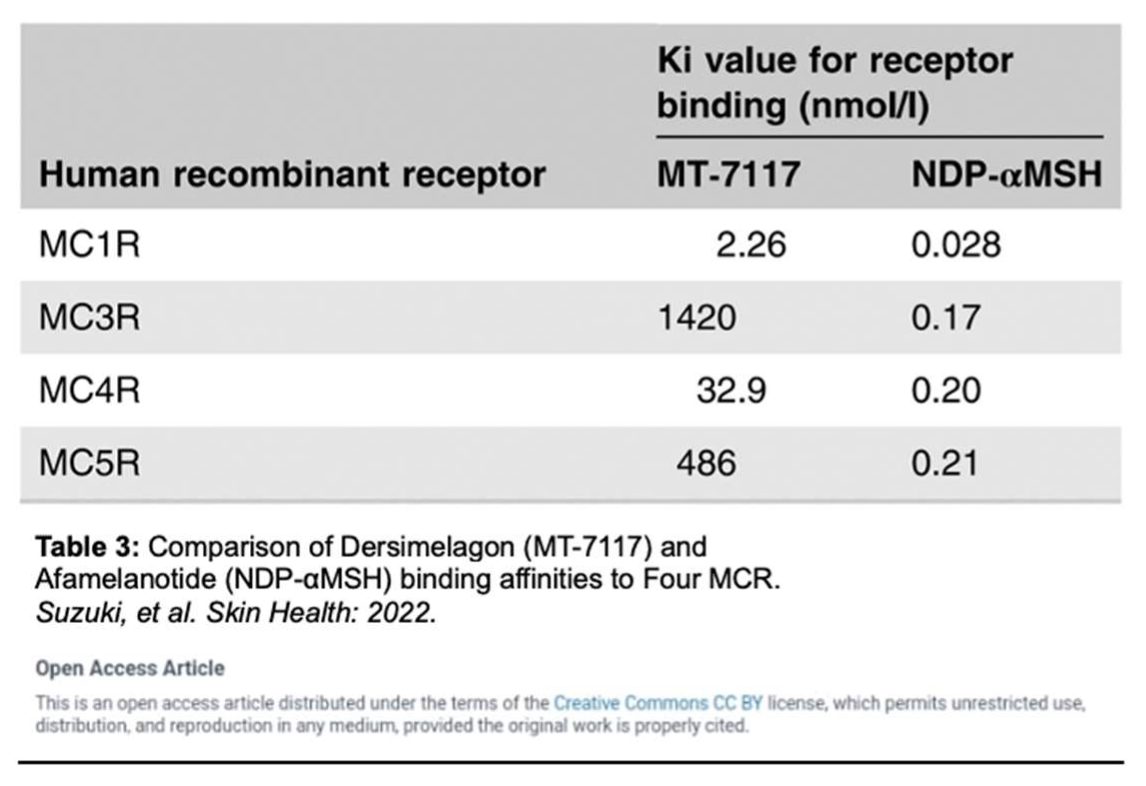
A mechanism of photoprotection is provided through the increased production of eumelanin, a black-brown pigment generated by melanocytes, keratinocytes, monocytes, endothelial cells, and fibroblasts. Eumelanin produced by melanocytes is influenced by five known distinct melanocortin receptors. Dersimelagon has the highest affinity for the melanocortin-1 receptor (MC1R), a G-protein coupled receptor on melanocytes that binds to melanocortin to regulate skin and hair color (Table 3) [33,34,37].
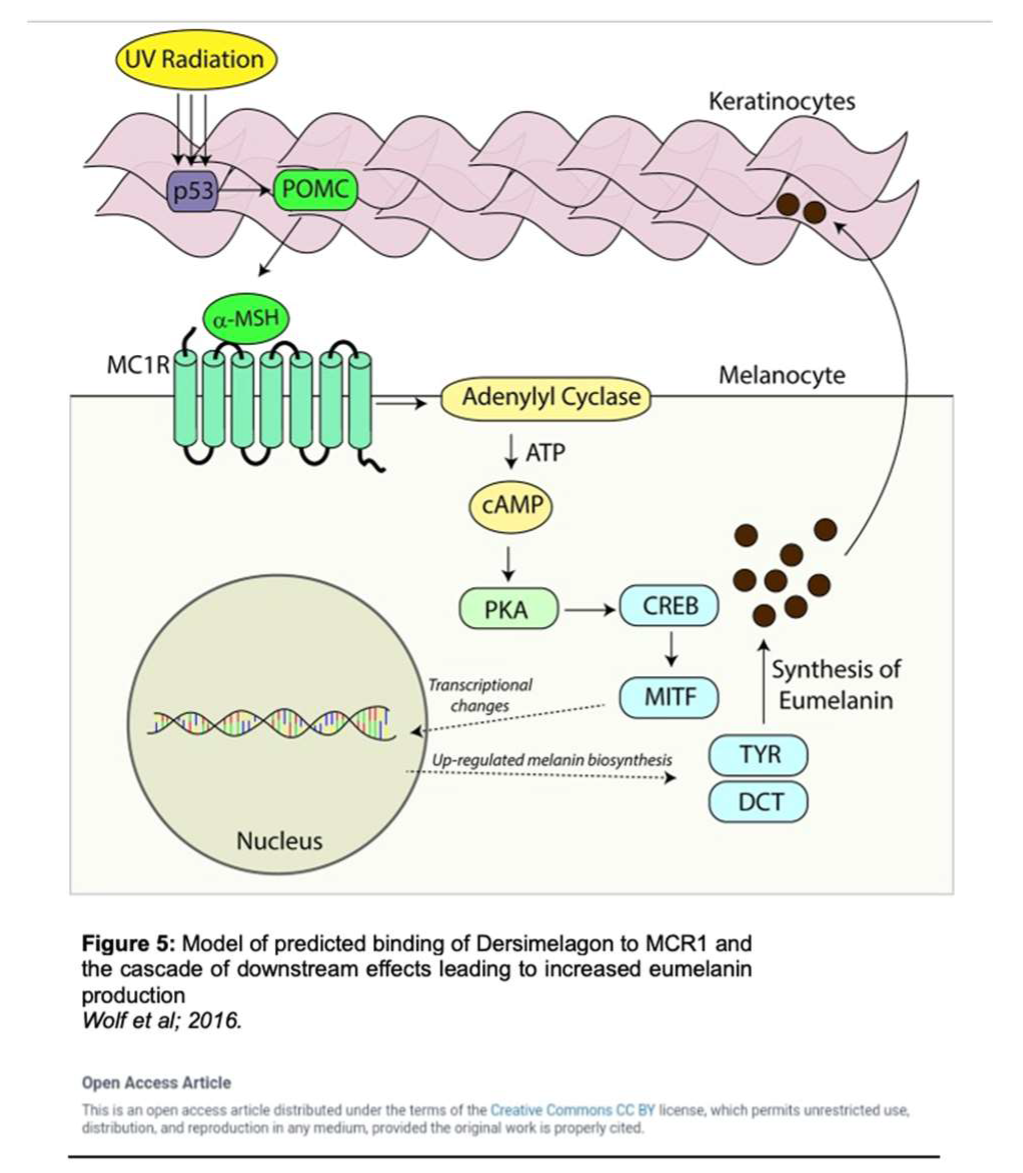
MC1R is established as the main driver for human pigmentation [25]. Potent peptide analogs of α-melanocyte-stimulating hormone (α-MSH) (which binds to MC1R as seen in Figure 4) have been developed and extensively tested and demonstrated the effect of enhancing the repair of DNA photoproducts and reducing reactive oxygen species (ROS) generation and apoptosis in ultraviolet radiation-irradiated melanocytes [25]. Activation of MC1R has been shown to produce eumelanin through an intracellular cascade reaction as outlined in Figure 5 [34]. In this cascade, the agonist α-MSH binds to MC1R in the setting of solar irradiation, which then induces dissociation of the G α-subunit. Adenyl cyclase is activated, leading to an accumulation of intracellular cAMP. Protein kinase A is then activated by this cAMP, thereby phosphorylating a variety of downstream effector pathways including induction of the CREB and Mitf transcription factor networks, which ultimately leads to the expression of tyrosinase, and other enzymes involved in melanin synthesis [34].
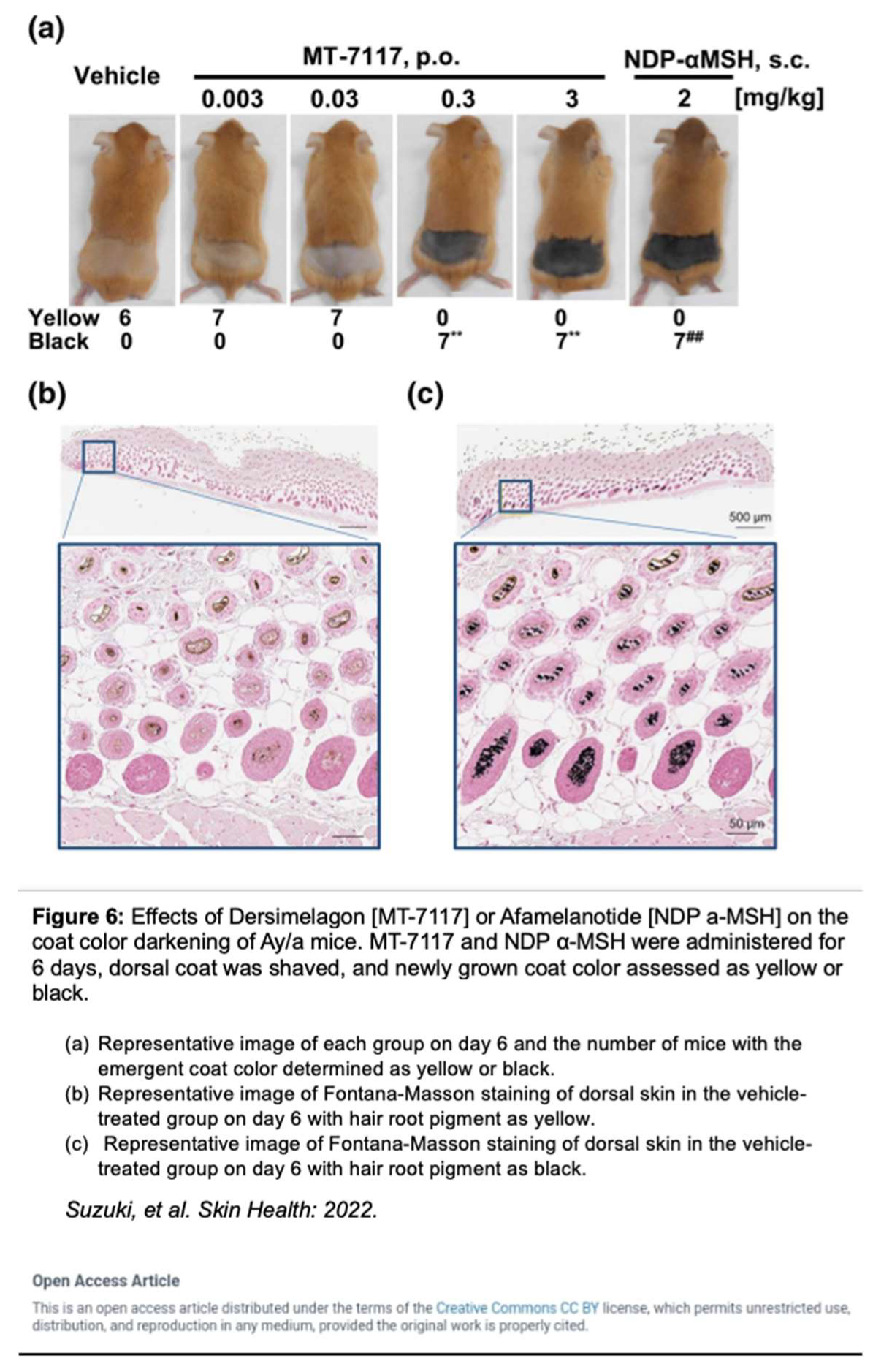
Dersimelagon increases cAMP levels in a dose-dependent manner. Pharmacokinetic studies by Suzuki et al. revealed that Dersimelagon was able to produce Emax values similar to that of αMSH suggesting that they have similar agonistic activity on variants of MC1R [29]. Plasma concentrations of the drug were analyzed and revealed a greater than dose-proportional increase in serum levels of Dersimelagon with increased oral dosing in animal studies. Oral administration of Dersimelagon produced the MC1R agonistic activity levels necessary to induce skin pigmentation in murine models (Figure 6). While Dersimelagon is selective for MC1R, Afamelanotide non-selectively binds MC1R, MC3R, and other melanocortin receptors [19,29]. Specifically, nausea often produced by afamelanotide is primarily attributed to its binding to MC3R and downstream effects. It has been noted that Dersimelagon, compared to afamelanotide, induced similar melanin production with 20-fold reduced potency in cAMP production. [29].
Key clinical trial results for Dersimelagon
The first-in-human phase 1 trial conducted by Ogasawara et al. for Dersimelagon, enrolled 144 healthy participants (143 completed the trial) and demonstrated an acceptable safety profile in these patients. Thirty-four of these patients received a placebo, with the remaining patients receiving at least 1 dose of Dersimelagon (doses ranged from 1 to 600 mg). Additionally, 36 of the Dersimelagon patients received multiple doses ranging from 30 to 450 mg. There were few treatment-emergent adverse events (TEAEs) with no death or serious AE reported. Most symptoms reported were mild to moderate in severity, though, overall, patients receiving multiple doses of Dersimelagon had more TEAEs. Most common TEAEs reported were related to skin pigmentation, including lentigo, skin hyperpigmentation, and melanocytic nevi (two cases were severe but non-malignant). Additionally, there were no statistically significant effects of age or race on the pharmacokinetics of Dersimelagon; however, increased exposure was observed in females compared to males, which was attributed to differences in the body weight generally observed in male versus female participants [38].
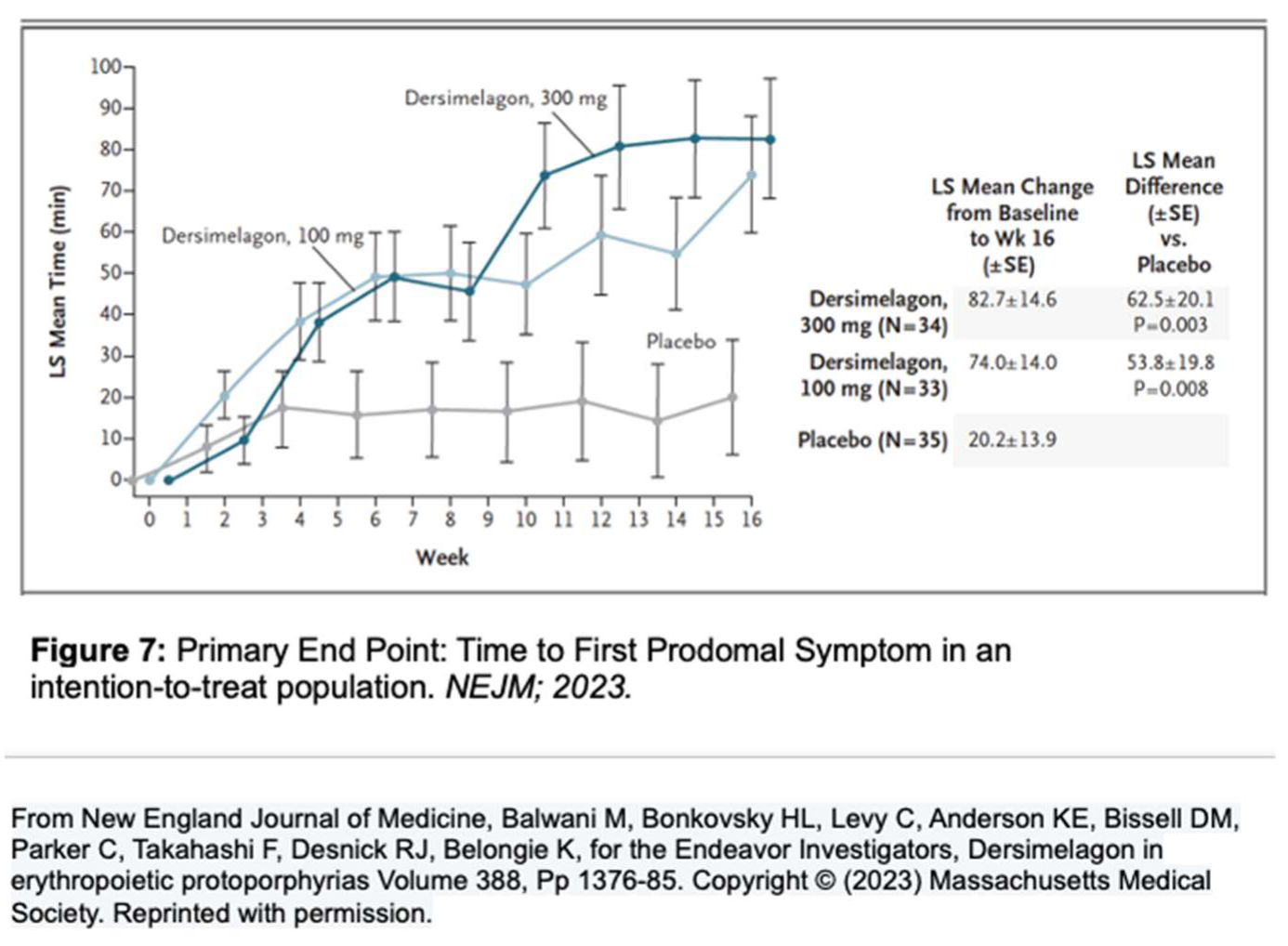
A phase 2 randomized, double-blind, placebo-controlled study by Balwani et al. set out to evaluate the safety and efficacy of Dersimelagon concerning the time to prodromal symptoms (TTP) and severity of symptoms associated with sunlight exposure for patients with EPP or XLP (Figure 7) [1]. The population included both males and females with confirmed diagnoses of EPP between the ages of 18-75 who were assigned in a 1:1:1 ratio to receive a placebo, Dersimelagon 100 mg, or Dersimelagon 300 mg once daily for 16 weeks. Exclusion criteria included those with acute or chronic renal disease, pregnancy or lactation, clinically significant hepatobiliary disease; history of non-EPP photodermatoses, excessive alcohol intake, melanoma, psychiatric disease, treatment with phototherapy, antioxidant agents, or afamelanotide within 3 months before trial. Of the 102 randomized patients (93 with EPP and 9 with XLP), a statistically significant increase in the least squares mean change in TTP from baseline to week 16 was noted, with the 100 mg group being 74+14minutes (p = 0.008) and the 300 mg group being 82.7+14.6 minutes (p = 0.003), v placebo (20.2+13.9 minutes). Secondary endpoints, including incidence rate and total number of phototoxic pain events, and total duration of sunlight exposure without prodromal symptoms, all demonstrated improvement, regardless of Dersimelagon dose [39].
Dersimelagon was well tolerated overall. Most adverse events were mild to moderate in severity and resolved within the trial period. The groups evaluated were placebo, Dersimelagon 100 mg, and Dersimelagon 300 mg, respectively. The most frequently reported adverse effect was nausea (12%, 15%, and 46% respectively), followed by headache (18%, 18%, and 29%), freckles (0%, 15%, and 31%), and skin hyperpigmentation (0%, 9%, and 31%). The one serious adverse event that led to discontinuation by a patient was an anaphylactic reaction that was deemed by the investigators to be unrelated to Dersimelagon [39].
Post-hoc analysis of this phase 2 trial evaluated patients’ perspectives of their quality of life and the role of geographic location within different seasons (given anticipated changes in UV light intensity). Two subgroups (Subgroup 1: Spring/Summer; Subgroup 2: Fall/Winter) were evaluated in both the northern and southern United States. Patients taking Dersimelagon were found to have a statistically significant increase in time to symptoms development with sun exposure in both subgroups and geographical locations. For example, in Subgroup 1, when compared to placebo, time to symptoms increased by 38.0 minutes with Dersimelagon 100 mg and 39.7 minutes with Dersimelagon 300 mg. By assessing this metric across all groups, there was a significant increase when compared with placebo by 21.9 minutes with Dersimelagon 100 mg and by 22.1 minutes with Dersimelagon 300 mg (P=.032; P=.004, respectively). Additionally, an online questionnaire to assess the quality of life was completed by 75 of the 102 patients enrolled. Of those taking Dersimelagon 100 mg, 57.6% rated their EPP as “very much better” and 69.7% rated they were “much more often” able to be outside at the end of the study. Likewise, among those who used Dersimelagon 300 mg, positive ratings were given by 31.4% and 48.6%, respectively. [40,41].
Limitations
As outlined above, most adverse events with the use of Dersimelagon were mild to moderate in severity and resolved within the trial period. Participation in the trial was discontinued by 4 patients in the placebo group (voluntary withdrawal), 2 patients in the 100-mg Dersimelagon group (1 for nonadherence and 1 for a serious adverse event), and 4 patients in the 300-mg Dersimelagon group (1 voluntary withdrawal, 2 withdrawals for adverse events, and 1 withdrawal for clinically significant lichen planus found during nevi assessment). The serious adverse event that led to discontinuation by 1 patient in the 100-mg Dersimelagon group was an anaphylactic reaction that was deemed by the investigators to be unrelated to Dersimelagon [39]. Additionally, there is incomplete blinding in this trial due to increases in skin pigmentation when taking Dersimelagon, which allowed many subjects to correctly guess if they were assigned to the active drug group.
In the phase 2 trial by Balwani et al., limitations included a lack of formal statistical power analysis, although such limitations are common in studies of rare diseases such as EPP/XLP.
Continuing concerns surrounding the chronic use of Dersimelagon primarily involve the long-term safety and tolerability of the drug. A concern arises regarding the possible stimulation of abnormal melanocytes in the human body, given that melanoma tumors comprise a mixture of cells, with only a subset expressing MC1R. Some experiments demonstrated that MC1R increases its expression as the melanoma cell becomes more differentiated [42,43]. There is some association with elevated α-MSH in a subset of melanoma without any indication that it initiates melanoma induction [42]. It should be noted that a literature review conducted in 2013 on afamelanotide evaluated concerns about its potential to contribute to the malignant transformation of melanocytes. In this review, no documented cases of malignancy due to afamelanotide were noted, and instead, the authors suggested that it may inhibit melanoma cell proliferation. Suzuki et al. studied the effects of Dersimelagon in their preclinical, in vitro studies on 5 separate human melanoma cell lines, and found that Dersimelagon did not affect the proliferation of these cell lines [29]. Additionally, Murata et al. showed that α-MSH did not stimulate increased proliferation or invasion of malignant cells in a nonclinical, in vitro study [43].
To further evaluate the potential for drug-induced liver injury, more subjects observed for longer times of treatment are needed. In cross-sectional studies of patients with EPP/XLP, up to 25% had a reported elevation in liver enzymes. A rare, yet severe and rapidly progressive form of liver involvement that can lead to acute liver failure, known as protoporphyric hepatopathy, occurs in a small proportion (2-5%) of patients with protoporphyria. It is currently not clear if this rare but serious complication of EPP/XLP is due to the natural history of the underlying genetic disease or superimposed etiologies (biliary tract disease, viral hepatitis, autoimmune conditions, alcohol, etc.) [32]. Drugs or chemicals that lead to liver injury, especially with cholestasis, are of special concern in EPP/XLP [1,8,9,15,16,32].
Conclusions
Strides are being made to gain insight into the true prevalence of EPP/XLP, which is likely under-recognized. As such, there may be an underappreciation for the devastating effects on patients with these conditions. The only current EMA- or FDA-approved therapy, afamelanotide, is subcutaneously injected. Additional uncertainty of receiving this therapeutic has been posed, including only a few centers that have been provided with afamelanotide, the expertise required for administration, and significant cost concerns. Dersimelagon offers hope for an alternative agent to improve patient quality of life. Dersimelagon thus far has demonstrated an acceptable safety profile with the ease of once-daily oral dosage. Ongoing studies will provide additional insights into expanding the treatment options for those suffering from EPP/XLP.
Author Contributions
Conceptualization, HB, SR, MA, KM.; methodology, KM, MA, HB, SR; resources, KM, HB, SR, MA, NU; data curation, KM, HB, SR, MA, NU; writing—original draft preparation, KM, SR, MA, HB; writing—review and editing, KM, HB, SR, MA, NU; visualization, HB, KM, SR, MA; supervision, HB, SR, KM; project administration, HB, SR.; funding acquisition, HB, SR, KM. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by funds provided to Atrium Wake Forest Baptist Health from the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health. The funding organization had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Institutional Review Board Statement
As consistent with federal regulations, an Institutional Review Board waiver of consent in the review of a pharmacologic agent does not directly involve patients. There is more than minimal risk to participants with a prospect of direct benefit to participants.
Informed Consent Statement
Not applicable as this review did not directly involve human subjects.
Data Availability Statement
Research data was not collected in this review.
Acknowledgments
We acknowledge the support and contributions awarded to Atrium Health Wake Forest Baptist in the ongoing research and study of porphyrias.
Conflicts of Interest
In the past 3 years, Dr. Bonkovsky has served as a paid consultant to Alnylam Pharma, Disc Medicine, Mitsubishi-Tanabe Pharma North America, and Recordati Rare Chemicals. He has been the site PI of clinical research grants awarded to Atrium Health Wake Forest Baptist for the study of porphyrias from Disc Medicine, Mitsubishi-Tanabe Pharma, NA, and Alnylam Pharma. Dr. Bonkovsky has been the site PI of clinical research grants awarded to Atrium Health Wake Forest Baptist for the study of primary sclerosing cholangitis from Gilead Sciences and for the study of primary biliary cholangitis from Calliditas, SA. In the past 3 years, Dr. Rudnick has served as a paid speaker and consultant to Alnylam Pharmaceuticals, and consultant to Alexion Pharmaceuticals and Recordati Rare Chemicals. Drs. Agnew, Madigan, and Urooj report no potential conflicts of interest.
References
- Balwani, M.; Bonkovsky, H.; Levy, C.; et al. Dersimelagon in Erythropoietic Protoporphyrias. New England J Med. 2023, 388, 1376–1385. [Google Scholar] [CrossRef]
- Dickey, A.K.; Quick, C.; Ducamp, S.; Zhu, Z.; Feng, Y.-C.A.; Naik, H.; Balwani, M.; Anderson, K.E.; Lin, X.; Phillips, J.E.; et al. Evidence in the UK Biobank for the underdiagnosis of erythropoietic protoporphyria. Genet Med. 2020, 23, 140–148. [Google Scholar] [CrossRef]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 2012, 36, 849–857. [Google Scholar] [CrossRef]
- Heerfordt, I.M.; Philipsen, P.A.; Wulf, H.C. Bringing the gentle properties of daylight photodynamic therapy indoors: A systematic review of efficacy and safety. Photodiagnosis Photodyn. Ther. 2022, 39, 102858. [Google Scholar] [CrossRef]
- Heerfordt, I.M.; Lerche, C.M.; Wulf, H.C. Cimetidine for erythropoietic protoporphyria. Photodiagnosis Photodyn. Ther. 2022, 38, 102793. [Google Scholar] [CrossRef]
- Lim, H.W. Mechanisms of phototoxicity in porphyria cutanea tarda and erythropoietic protoporphyria. Immunology Series 1989, 46, 671–85. [Google Scholar]
- Bottomley, S.S.; Tanaka, M.; A Everett, M. Diminished erythroid ferrochelatase activity in protoporphyria. J Lab Clin Med. 1975, 86, 126–31. [Google Scholar]
- Bonkowsky, H.L.; Bloomer, J.R.; Ebert, P.S.; Mahoney, M.J. Heme synthetase deficiency in human protoporphyria. Demonstration of the defect in liver and cultured skin fibroblasts. J. Clin. Investig. 1975, 56, 1139–1148. [Google Scholar] [CrossRef]
- Levy, C. Overview of Liver Involvement in Patients With Erythropoietic Protoporphyria. Gastroenterol Hepatol. 2023, 19, 104–107. [Google Scholar]
- Todd, D. Erythropoietic protoporphyria. British J Dermatol. 1994, 131, 751–766. [Google Scholar]
- Schulenburg-Brand, D.; Katugampola, R.; Anstey, A.; et al. The cutaneous porphyrias. Dermatol Clin. 2014, 32, 369–84. [Google Scholar] [CrossRef]
- Brancaleoni, V.; Balwani, M.; Granata, F.; Graziadei, G.; Missineo, P.; Fiorentino, V.; Fustinoni, S.; Cappellini, M.D.; Naik, H.; Desnick, R.; et al. X-chromosomal inactivation directly influences the phenotypic manifestation of X-linked protoporphyria. Clin. Genet. 2015, 89, 20–26. [Google Scholar] [CrossRef]
- Balwani, M.; Doheny, D.; Bishop, D.F.; et al. Porphyrias Consortium of the National Institutes of Health Rare Diseases Clinical Research Network: Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med. 2013, 19, 26–35. [Google Scholar]
- Di Pierro, E.; Granata, F.; De Canio, M.; Rossi, M.; Ricci, A.; Marcacci, M.; De Luca, G.; Sarno, L.; Barbieri, L.; Ventura, P.; et al. Recognized and Emerging Features of Erythropoietic and X-Linked Protoporphyria. Diagnostics 2022, 12, 151. [Google Scholar] [CrossRef]
- Singal, A.K.; Parker, C.; Bowden, C.; Thapar, M.; Liu, L.; McGuire, B.M. Liver transplantation in the management of porphyria. Hepatology 2014, 60, 1082–1089. [Google Scholar] [CrossRef]
- McGuire, B.M.; Bonkovsky, H.L.; Carithers, R.L.; Chung, R.T.; Goldstein, L.I.; Lake, J.R.; Lok, A.S.; Potter, C.J.; Rand, E.; Voigt, M.D.; et al. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transplant. 2005, 11, 1590–1596. [Google Scholar] [CrossRef]
- Balwani, M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: pathophysiology, genetics, clinical manifestations, and management. Mol. Genet. Metab. 2019, 128, 298–303. [Google Scholar] [CrossRef]
- Kaye, E.T.; Levin, J.A.; Blank, I.H.; Arndt, K.A.; Anderson, R.R. Efficiency of Opaque Photoprotective Agents in the Visible Light Range. Arch. Dermatol. 1991, 127, 351–355. [Google Scholar] [CrossRef]
- Minder, E. Afamelanotide, an antagonistic analog of alpha-melanocyte-stimulating hormone, dermal phototoxicity of erythropoietic protoporphyria. Expert Opin Invest Drugs 2010, 19, 1591–602. [Google Scholar] [CrossRef]
- Mathews-Roth, M.M.; Pathak, M.A.; Fitzpatrick, T.B.; Harber, L.C.; Kass, E.H. β-Carotene as an Oral Photoprotective Agent in Erythropoietic Protoporphyria. JAMA 1974, 228, 1004. [Google Scholar] [CrossRef]
- Krook, G.; Haeger-Aronsen, B. beta-Carotene in the treatment of erythropoietic protoporphyria. A short review. Acta Derm Venereol Suppl. 1982, 100, 125–9. [Google Scholar]
- Sivaramakrishnan, M.; Woods, J.; Dawe, R. Narrowband ultraviolet B phototherapy in erythropoietic protoporphyria: case series. Br. J. Dermatol. 2014, 170, 987–988. [Google Scholar] [CrossRef]
- Wensink, D.; Wagenmakers, M.A.; Langendonk, J.G. Afamelanotide for prevention of phototoxicity in erythropoietic protoporphyria. Expert Rev. Clin. Pharmacol. 2021, 14, 151–160. [Google Scholar] [CrossRef]
- Levine, N.; Sheftel, S.N.; Eytan, T.; Dorr, R.T.; Hadley, M.E.; Weinrach, J.C.; Ertl, G.A.; Toth, K.; McGee, D.L.; Hruby, V. Induction of Skin Tanning by Subcutaneous Administration of a Potent Synthetic Melanotropin. JAMA 1991, 266, 2730–2736. [Google Scholar] [CrossRef]
- García-Borrón, J.C.; Abdel-Malek, Z.; Jiménez-Cervantes, C. MC1R, the cAMP pathway, and the response to solar UV: extending the horizon beyond pigmentation. Pigment. Cell Melanoma Res. 2014, 27, 699–720. [Google Scholar] [CrossRef]
- Lane, A.M.; McKay, J.T.; Bonkovsky, H.L. Advances in the management of erythropoietic protoporphyria – role of afamelanotide. Appl. Clin. Genet. 2016, 9, 179–189. [Google Scholar] [CrossRef]
- Swetlitz, I. Clinuvel limits the availability of Scenesse for rare genetic disease. Bloomberg.com. 2023. https://www.bloomberg.com/news/articles/2023-05-01/clinuvel-limits-availability-of-scenesse-for-rare-genetic-disease#xj4y7vzkg.
- Yao, J.-F.; Yang, H.; Zhao, Y.-Z.; Xue, M. Metabolism of Peptide Drugs and Strategies to Improve their Metabolic Stability. Curr. Drug Metab. 2018, 19, 892–901. [Google Scholar] [CrossRef]
- Suzuki, T.; Kawano, Y.; Matsumoto, G.; et al. Melanogenic effect of dersimelagon (MT-7117), a novel oral melanocortin 1 receptor agonist. Skin Health Dis. 2022, 2, e78. [Google Scholar] [CrossRef]
- Ogawa, K.; Ide, R.; Belongie, K.; Tsuda, M.; Kawanishi, H.; Teng, R.; Ogasawara, A. The Oral Bioavailability and Effect of Various Gastric Conditions on the Pharmacokinetics of Dersimelagon in Healthy Adult Volunteers. Clin. Pharmacol. Drug Dev. 2023, 12, 493–501. [Google Scholar] [CrossRef]
- Tsuda, M.; Ogawa, K.; Endou, T.; Goto, T.; Ogasawara, Y.; Ogasawara, A. Absorption, metabolism, and excretion of [14C]dersimelagon, an investigational oral selective melanocortin 1 receptor agonist, in preclinical species and healthy volunteers. Pharmacol. Res. Perspect. 2023, 11, e01084. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Schned, A.R. Fatal liver failure in protoporphyria. Synergism between ethanol excess and the genetic defect. Gastroenterol 1986, 90, 191–201. [Google Scholar] [CrossRef]
- Mun, Y.; Woo, K.; Dongyun, S. Melanocortin 1 Receptor (MC1R): Potentials as therapeutic targets. Pre-prints 2023, 2023051558.
- Horrell, E.M.W.; Boulanger, M.C.; D’orazio, J.A. Melanocortin 1 Receptor: Structure, Function, and Regulation. Front. Genet. 2016, 7, 95. [Google Scholar] [CrossRef]
- Mathews-Roth, M.M. Treatment of erythropoietic protoporphyria with beta-carotene. Photodermatol 1984, 1, 318–21. [Google Scholar] [CrossRef]
- Mathews-Roth, M.M.; Pathak, M.A.; Fitzpatrick, T.B.; Harber, L.H.; Kass, E.H. Beta Carotene Therapy for Erythropoietic Protoporphyria and Other Photosensitivity Diseases. Arch. Dermatol. 1977, 113, 1229–1232. [Google Scholar] [CrossRef]
- Dorr, R.T.; Ertl, G.; Levine, N.; Brooks, C.; Bangert, J.L.; Powell, M.B.; Humphrey, S.; Alberts, D.S. Effects of a Superpotent Melanotropic Peptide in Combination With Solar UV Radiation on Tanning of the Skin in Human Volunteers. Arch. Dermatol. 2004, 140, 827–835. [Google Scholar] [CrossRef]
- Ogasawara, A.; Ogawa, K.; Ide, R.; Ikenaga, Y.; Fukunaga, C.; Nakayama, S.; Tsuda, M. Results from a first-in-human study of dersimelagon, an investigational oral selective MC1R agonist. Eur. J. Clin. Pharmacol. 2023, 79, 801–813. [Google Scholar] [CrossRef]
- Belongie, K.; Takahashi, F.; Martin, S. 34420 Perspective of patients with erythropoietic protoporphyria treated with dersimelagon, a selective melanocortin-1 receptor agonist: Results of the ENDEAVOR study exit questionnaire. J. Am. Acad. Dermatol. 2022, 87, AB92. [Google Scholar] [CrossRef]
- Belongie, K.; Takahashi, F.; Mukai, S.; Endou, M.; Desnick, R. 33323 Dersimelagon, a melanocortin-1 receptor agonist, maintains efficacy regardless of seasons in subjects with erythropoietic protoporphyria: Results from a post hoc analysis of phase 2 clinical trial data. J. Am. Acad. Dermatol. 2022, 87, AB156. [Google Scholar] [CrossRef]
- Fabrikant, J.; Touloei, K.; Brown, S. A review and update on melanocyte stimulating hormone therapy: afamelanotide. J Drugs Dermatol. 2013, 12, 775–9. [Google Scholar]
- Murata, J.; Ayukawa, K.; Ogasawara, M.; Fujii, H.; Saiki, I. Alpha-melanocyte-stimulating hormone blocks invasion of reconstituted basement membrane (Matrigel) by murine B16 melanoma cells. Invasion Metastasis 1997, 17, 82–93. [Google Scholar]
- Kondo, M.; Suzuki, T.; Kawano, Y.; Kojima, S.; Miyashiro, M.; Matsumoto, A.; Kania, G.; Błyszczuk, P.; Ross, R.L.; Mulipa, P.; et al. Dersimelagon, a novel oral melanocortin 1 receptor agonist, demonstrates disease-modifying effects in preclinical models of systemic sclerosis. Arthritis Res. Ther. 2022, 24, 1–17. [Google Scholar] [CrossRef]
- Sandberg, S.; Talstad, I.; Høvding, G.; Bjelland, N. Light-induced release of protoporphyrin, but not of zinc protoporphyrin, from erythrocytes in a patient with greatly elevated erythrocyte protoporphyrin. Blood 1983, 62, 846–51. [Google Scholar] [CrossRef]
- Langendonk, J.G.; Balwani, M.; Anderson, K.E.; Bonkovsky, H.L.; Anstey, A.V.; Bissell, D.M.; Bloomer, J.; Edwards, C.; Neumann, N.J.; Parker, C.; et al. Afamelanotide for Erythropoietic Protoporphyria. New Engl. J. Med. 2015, 373, 48–59. [Google Scholar] [CrossRef]
- Corbett, M.; Herxheimer, A.; Magnus, I.; et al. The long-term treatment with beta-carotene in erythropoietic protoporphyria: a controlled trial. Br J Dermatol. 1977, 97, 655–62. [Google Scholar] [CrossRef]
- Collins, P.; Ferguson, J. Narrow-band UVB (TL-01) phototherapy: an effective preventative treatment for the photodermatoses. Br. J. Dermatol. 2010, 132, 956–963. [Google Scholar] [CrossRef]
- Bijlmer-Iest, J.C.; De La Faille, H.B.; Van Asbeck, B.S.; Van Hattum, J.; Van Weelden, H.; Marx, J.J.; Koningsberger, J.C. Protoporphyrin photosensitivity cannot be attenuated by oral N-acetylcysteine. Photodermatol Photoimmunol Photomed. 1992, 9, 245–9. [Google Scholar]
- McGuire, B.M.; Bonkovsky, H.L.; Carithers, R.L.; Chung, R.T.; Goldstein, L.I.; Lake, J.R.; Lok, A.S.; Potter, C.J.; Rand, E.; Voigt, M.D.; et al. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transplant. 2005, 11, 1590–1596. [Google Scholar] [CrossRef]
- https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-22-2585.
- Minder, A.-E.; Schneider-Yin, X.; Zulewski, H.; Minder, C.E.; Minder, E.I. Afamelanotide Is Associated with Dose-Dependent Protective Effect from Liver Damage Related to Erythropoietic Protoporphyria. Life 2023, 13, 1066. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



