
Submitted:
29 September 2023
Posted:
30 September 2023
You are already at the latest version
Abstract
Hepatitis C virus (HCV) infection is a worldwide public health problem. Chronic infection by HCV can lead to liver cirrhosis or cancer. Although some immune-competent individuals can clear the virus, others develop chronic HCV disease due to viral mutations or an impaired immune response. IFNs type I and III and the signal transduction induced by them are essential for a proper antiviral effect. Research on the viral cycle and immune escape mechanisms have generated the basis of therapeutic strategies to achieve a sustained virological response (SVR). The first therapies were based on IFN-, then IFN-α plus ribavirin (IFN-RBV); then, pegylated-IFN--RBV (PEGIFNα-RIV) to improve cytokine pharmacokinetics. However, the maximum SVR was 60%, and several significant side effects were observed, decreasing the patients' treatment adherence. The development of direct-acting antivirals (DAAs) significantly enhanced SVR (> 90%); the compounds were able to inhibit HCV replication without significant side effects, even in pediatric populations. The management of coinfected patients HBV-HCV and HCV-HIV has also improved based on DAA and PEG-IFNα-RBV (HBV-HCV). CD4 cells are crucial for an effective antiviral response. IFNλ3, IL28B, TNF-α, IL-10, TLR-3, and TLR-9 gene polymorphisms are involved in viral clearance, therapeutic responses, and hepatic pathologies. Future research focuses on searching for strategies to circumvent resistance-associated substitution (RAS) to DAAs, develop new therapeutic schemes for different medical conditions, including organ transplant, and develop vaccines for long-lasting cellular and humoral responses with cross-protection to different HCV genotypes. The goal is to minimise the probability of HCV infection, HCV chronicity and hepatic carcinoma.
Keywords:
Hepatitis C virus
; chronic HCV
; antivirals
; Sustained virological response
; IFN therapy
; Vaccines
1. Introduction
Hepatitis C virus (HCV) was discovered by Harvey J. Alter, Michael Houghton and Charles M. Rice (Nobel Prize in Medicine and Physiology in 2020). Their research was crucial to identifying and characterising the viral genome, developing serological and molecular diagnostic methodologies, and providing fundamental bases to study the virus pathophysiology [1,2].
Structurally, HCV is a positive-stranded RNA virus with a lipoprotein envelope that presents a spherical structure of approximately 55 nm in diameter and is taxonomically located within the genus Hepacivirus of the family Flaviviridae [1,2,3]. The genome is approximately 9.6 kb long and codes for a polyprotein of about 3,010 amino acids that is proteolytically processed by viral and cellular enzymes to generate at least ten proteins (Figure 1). These proteins include three "structural" polypeptides: 1) the nucleocapsid or "core" protein (C) and two envelope proteins (E1 and E2); 2) two proteins that are essential for virion production (p7 and NS2); and five nonstructural proteins that are essential in viral replication (NS3, NS4A, NS4B, NS5A and NS5B) (Figure 1) [1,2,3].
HCV RNA has been successfully detected in blood (including serum and plasma), saliva, tears, seminal fluid, ascitic fluid and cerebrospinal fluid [2,3,4,5,6]. Around 185 million people are infected with HCV [4,5]. In particular, HCV infections have been linked to intravenous drug abuse or poor medical practices in hemodialysis, transfusion of hemo components and blood products in resource-limited areas of the world. The frequency of perinatal and sexual transmission is low [2,3,4,5,6]. According to estimates by the Center for Disease Control (CDC) for 2013, HCV infection accounted for the highest number of deaths and mortality rate (5.0 deaths/100,000 population) among hepatitis of viral origin [7,8]. The number of deaths from HCV infection in 2012 compared to 2015 increased from 18,650 to 19,629 in the US [7,8].
Viral transmission requires that infectious virions come into contact with susceptible host cells through specific and co-receptors or non-specific receptors. The key receptors for HCV entry are CD81 [9], the scavenger receptor class B member 1 (SCARB1) [10], the proteins of the tight junction, Claudin-1 (CLDN1) and occludin (OCLN) [11], EGFR (epidermal growth factor receptor) and EPH receptor A2 (ephrin type-A receptor 2), involved in HCV virus entry and probably in oncogenic transformation [12]. Other receptors which are not highly specific but are involved in virus entry and escape are the very low-density lipoprotein receptor (VLDL-R), the LDL receptor (LDL-R) [13,14], Niemann-Pick C1-Like 1 receptor (NPC1L1) [15], Heparan sulfate proteoglycan (HSPG) [16] and the Fc receptors by immune complex [17]. Due to the variety of receptors and co-receptors, non-hepatic cells, including leukocytes, are infected, or their function is affected by the viral infection [18,19,20].
HCV has an enormous replicative capacity, reaching 105-107 IU/ml titers in the first days of infection [3,6]. Only a minority of those infected particles are spontaneously eliminated by the immune response [22]. The efficiency depends on the virus's heterogeneity, mechanisms of immune response evasion, and host-specific factors such as age, race, sex and genetic markers [5,22,23,24]. During the initial response, the host immune system reacts similarly to other viral infections by inducing the transcription and secretion of type I and III interferons to restrict viral replication [5,6,21]. IFN signalling cascade includes Janus kinases (JAK), the STAT (signal transducers and activators of transcriptions family) transcription factors that dimerise and translocate from the cytosol, the IRF (IFN-stimulated gene factor), which binds to IFN-stimulated response elements 3 and 9 (ISREs) are gene promoters, leading to the transcription of numerous IFN-stimulated genes (ISGs) [25]. The HCV viral core protein interferes with STAT signalling by decreasing STAT1 accumulation and promoting its proteosome-dependent degradation, affecting the IFN-λ signalling pathway favouring HCV replication [26,27]. IFNα activates STAT2, which also affects HCV replication [28]. However, IL-8, induced upon the viral infection, can facilitate viral escape by suppressing the IFNα signalling (antagonism) [29].
Other critical players in the cellular immune response against HCV are 1) the blockade of Protein Kinase R (PKR) phosphorylation and dimerisation by HCV proteins [30,31]; 2) The expression of human Myxovirus resistance protein A (MxA) that inhibits HCV replication by activating the JAK-STAT pathway independently of IFNα [32]; 3) IFN signalling is enhanced in females, especially at a young age, compared to males; the effect seems dependent on estradiol levels [33,34]. In the early stages, HCV clearance occurs predominantly by non-cytolytic effector mechanisms induced by the IFN-stimulated genes (ISG) in hepatocytes [5,6,22,23]. Nevertheless, these signals may be insufficient to decrease viral burden at the initial stage.
After 4 to 8 weeks, HCV-specific T lymphocytes are recruited to the liver in the second or late phase of acute hepatitis; this phase lasts 4 to 10 weeks and is the best opportunity for the immune system to clear the virus [5,6,22,23]. However, MHC antigen presentation can be suppressed by NS4A/B virus proteins in infected cells [35,36], and HCV core proteins NS3, NS5A, and NS5B can activate apoptosis of mature dendritic cells [35,36], inhibiting innate response and decreasing antigen presentation [22,35,36]. The absence of antigens results in the lack of recognition of infected cells [22,35,36]. Peripheral NK cell cytotoxic response in HCV patients is usually impaired [20], suggesting that tissue NK cells could not eliminate the virus; peripheral T-cell responses were unaffected [35,36,37]. As shown in the coinfected HIV-HCV patients, the CD3CD4 cell population may be crucial in virus clearance [37,38]. Viral clearance is achieved in 1 of 3 infected patients and requires a sustained and prolonged specific CD4+ and CD8+ T lymphocyte response against different HCV proteins [21,22,38,39]. Moreover, the gene polymorphisms of the host, mainly IFNλ3, IFNλ4, IL28B, inflammatory cytokines (IL-12, TNFα), IL-10, and Toll-like receptors, especially in their role in coinfection with HBV and HIV, are crucial in the efficiency of viral clearance [36]. Viral escape and chronicity can be detected by the lack of expression of proteins induced by ISGs; the virus effectively blocked the IFN signal pathway [4,22,23,24,40]. Subjects with chronic hepatitis have a 25% higher risk of developing liver cirrhosis and hepatocarcinoma after 10-40 years of infection due to the continuous necrosis and inflammation of the liver [4,6]. It is the second cause of liver cancer worldwide [6,24].
The virus gene variability is mainly due to the low corrective activity in the RNA polymerase (NS5B), responsible for viral genome replication, generating multiple variants [6,23,41,42,43]. According to the current classification, seven HCV genotypes are identified based on nucleotide variability in the genome sequences analysed from various geographic regions [6,23,41,42,43,44,45]. Genotypes 1 to 4 vary in distribution and prevalence depending on the geographic area [6,23,41,42,43,44,45]. Genotype 1 is the most common in the United States, Latin America and Europe, accounting for 46% of all infections, followed by genotypes 3 (22%) and 2 and 4 (13% each). Around 40% of all infections in Asia are genotype 3, and genotype 4 is the most common (71%) in North Africa. Patients infected with genotype 1 have a lower therapeutic response than those infected with other genotypes [6,41,42,43,44,45].
HCV coinfection with HBV and/or HIV has also been described to affect the immune response, viral burden and chronicity [6,46,47,48]. HBV-HCV coinfection is common in some endemic regions, and it is often difficult to establish since patients have undetectable levels of antibodies against HBV surface proteins but have detectable levels of HBV DNA (a consistent marker of active replication) [48]. HCV infection can activate IFN type I and III synthesis, decreasing HBV replication; however, if HCV downregulates IFN I and III signalling, it facilitates liver infection by both viruses [47,48]. The severity of coinfection also depends on the genotype of the HCV virus and the host's immune [47,48]. Viral replication is very high in immunosuppressed or immune-deficient patients and requires special attention [48]. The coinfected individuals have a higher incidence of liver disease and hepatocellular carcinoma [46,47,48].
Since the modes of transmission of blood-borne viruses HIV, HBV and HCV, coinfection of two or three viruses is highly probable in endemic areas [46,47,48]. The risk of multimorbidity is higher in injection drug users; these patients have a three-fold higher risk of developing hepatic disease than those infected only with HCV [46,47,48]. The viral load of all the viruses involved may be enhanced in coinfection, impairing viral clearance due to an inefficient immune response [46,47,48]. In the three viral infections, effective CD4 cells besides IFNs type I and III are crucial for virus clearance. Lower numbers of circulating CD4 facilitate the viral escape of these viruses [46,47,48]. Even though CD8 cells are critical to eliminating viral-infected cells, recent reports reveal that CD8 is involved in liver damage [46,47,48,49,50]. The increase in primed CD8 cells is probably responsible for the augmented liver damage in coinfected individuals [46,47,48,49,50].
The treatment of HCV infection has represented a real challenge in therapeutics to develop treatments capable of generating a sustained virological response (SVR) (Figure 1). SVR is achieved when HCV RNA is no longer detectable in the blood after 12 weeks of therapy, with decreased antibody titers and improved liver pathology. HCV reinfection rarely occurs; nonetheless, the infection of non-hepatic cells may facilitate viral escape [4,5,6]. The development of preventive HCV vaccines remains another primary strategy to eliminate the disease globally. The extreme genetic diversity of HCV represents a well-known obstacle in developing an effective vaccine [41].
2. Interferon α and Ribavirin
Interferons do not have a unique mechanism of action. The intracellular signals, second messengers and proteins induced by IFNs are responsible for the antiviral activity [36,40,51,52,53]; virus resistance, as described before, refers to inhibiting those signalling events directly or indirectly by the virus [51,52,53].
IFNα therapy was first tested by Hoofnagle et al. [54] in a study showing decreased levels of aminotransferases in 8 of 10 chronically HCV-infected patients. Another investigation of 44 German patients infected with HCV and treated with IFN α 2b showed undetectable serum HCV RNA levels and normal alanine aminotransferase levels in 98 % of patients after 24 weeks of treatment [54].
To improve standard IFNα 2b therapy, the drug ribavirin (RBV), a guanosine nucleoside analogue with antiviral activity, was introduced in the late 1990s [55,56]. This drug was adjusted to body mass for 24 or 48 weeks, ranging from 1,000 to 1,200 mg orally daily. The SVR was much higher with the combined IFN-RBV therapy compared to the standard treatment consisting of IFN alone [57,58].
The mechanisms of action of RBV have not yet been fully elucidated. Nevertheless, four possible mechanisms have been proposed: 1) antiviral effect against HCV RNA polymerase-dependent RNA, 2) depletion of the intracellular pool of guanosine triphosphate (GTP), 3) induction of misincorporation of nucleotides by viral RNA polymerase, and 4) alteration in the cytokine balance from a Th2-type to a Th1-type profile with antiviral properties [59].
In the early 2000s, the structure of IFN was modified by adding polyethene glycol (PEG) chains [60,61,62,63,64,65,66]. Pegylation of IFN (PEGIFN) confers constant absorption, longer half-life in serum and lower systemic clearance. These changes allow sustained serum concentrations and improve SVR when combined with RBV [60,61,62,63,64,65,66]. PEGIFN has two forms of presentation in HCV treatment: pegylated IFNα2a (PEG IFNα2a) and pegylated IFNα2b (PEG IFNα2b). PEG IFN α2a produces a higher SVR than IFN PEG α2b when combined with RBV, achieving a cure rate between 56-54% and fewer secondary effects [65,66]. A randomised study by Manns et al. reported the advantage of PEGIFN treatment with RBV compared to IFNα2b monotherapy [66].
The standard treatment for many years for HCV infection was PEGIFN/ RBV; nonetheless, the side effects led to a decrease in adherence to the treatment in the early stages [67]. The most relevant side effects described were 1) anaemia; around 54% of the treated patients reported a reduction in haemoglobin of ≥3 g/dL; 2) neutropenia and thrombocytopenia Compensatory drugs such as granulocyte colony-stimulating factor (G-CSF) and thrombopoietin receptor agonists (eltrombopag) were used in these patients [67,68,69,70]. 3) Chronic fatigue syndrome and psychiatric symptoms such as depression [67,68,69,70]. Given the poor adherence to treatment with PEGIFN/RBV and the low percentage of SVR, particularly against HCV genotype 1, other therapeutic tools have been introduced after IFN, Figure 2.
3. Direct-Acting Antivirals (DAA)
Viral proteases are considered important targets for antiviral therapy [71,72,73]. NS3/4 proteases, the multifunctional protein NS5A, and the viral polymerase NS5B have been the most studied potential therapeutic targets in HCV [71,72,73]. A range of drugs, direct-acting antivirals (DAAs), have been developed against HCV proteases. The future aim is to find more specific and compelling compounds with few side effects [71,72,73]. A summary of the current DAA is presented in Table 1.
3.1. NS3/4-Protease Inhibitors
Proteolytic cleavages at the hepatitis C virus (HCV) polyprotein generating NS3-NS4A, NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B are produced by the virus-encoded serine protease in NS3 [74,75]. The enzyme is specific; it uses an extended polydentate binding cleft with several recognition subsites [74,75]. This polypeptide forms a heterodimeric complex with the NS4A protein, an essential cofactor that induces proteolytic activity and helps anchor the heterodimer to the endoplasmic reticulum [72,73,74,75].
The first DAAs were designed based on enzyme active sites [75]. However, HCV proteases require a large peptide substrate, with which the enzyme establishes multiple weak interactions distributed over an extended surface area. The requirement for large substrates was a significant concern in developing orally bioavailable small-molecule drugs [72,75].
Ciluprevir was the first NS3 protease inhibitor developed against HCV [72,75]. It demonstrated early viral load reduction in humans and established the proof of concept for future HCV protease inhibitors. The development of ciluprevir was discontinued due to cardiac toxicity observed in monkeys at high doses [72,75].
3.1.1. Telaprevir and Boceprevir
In 2011, telaprevir was the first DAA approved by the US Food and Drug Administration (FDA). As an NS3/4A protease inhibitor, it was designed to treat the viral infection by genotype 1 or patients previously treated with IFN-RBV [76]. The telaprevir/IFN/RBV combination offered an SVR between 60-75% [76]. The recommended dose of this drug was 750mg three times a day for 12 weeks. Nonetheless, this schedule generated considerable side effects, including pruritus, rash, anaemia, nausea, vomiting and anal discomfort. Skin erythema led to discontinuation of treatment in 4% of patients. Severe skin reactions of the Stevens-Johnson syndrome type have also been reported and were the most critical limitation in the use of telaprevir [76].
In 2011, boceprevir, another NS3/4A protease inhibitor approved by the European Medicines Agency for HCV treatment, was introduced [77]. This drug was co-administered with PEGIFN-RBV and was only effective in patients infected with viral genotype 1, with an SVR of 67-68% [77]. The dose of boceprevir was 800 mg every 8 hours, and the duration of treatment depended on response, ranging from 24 to 48 weeks [78]. Therapy was discontinued in all patients with HCV RNA levels above 100 IU at week 12 or if viral RNA was present at week 24, as SVR is rarely achieved under these circumstances and prevents the development of resistance to boceprevir. The main adverse effect attributed to this DAA is anaemia, so these patients were prescribed erythropoietin during the treatment [73].
Both boceprevir and telaprevir have been classified as category B drugs in pregnancy, i.e. with no evidence of risk in humans. However, both drugs are currently discontinued due to the emergence of more effective and pangenotypic DAAs [73].
3.1.2. Asunaprevir
It is an efficient N3/4A protease inhibitor with favoured liver distribution used as a co-treatment with IFNPRG-RBV or other DAA of patients infected with HCV genotypes 1 to 4 [79]. However, the drug's pharmacodynamics is markedly affected by using drugs that inhibit the organic anion-transporting polypeptide, like rifampin, mild hepatic impairment and Asiatic ethnic group [73]. A new scheme asunaprevir is used in China, Japan and part of Europe using daclastavir and beclabuvir (an indolobenzapine-like allosteric inhibitor of HCV polymerase). This combination, administered for 12 weeks, achieves an SVR ≥ 90%, regardless of cirrhosis stage, previous use of RBV or IFN failure [80]. Despite its high SVR rate, asunaprevir is limited to a twice-daily dosing schedule, safety concerns involving patients with advanced liver and kidney disease, and minimal potency against HCV genotypes outside of genotype 1. Its use should be an additional option for specific patient subgroups. However, resistance-associated mutations were observed in the coinfected HCV/HIV patients and changing its use to other DAA has been suggested [81].
3.1.3. Simeprevir
In 2013, simeprevir, the first single daily dose DAA (50 to 200mg), was approved for treating HCV genotype 1 in the US and genotypes 1 and 4 in the European Union [82]. Simeprevir was approved for use with PEGIFN+RBV but was subsequently approved as combination therapy with sofosbuvir with SVR rates of 79-100% reported after 12-24 weeks of treatment. Combining simeprevir and sofosbuvir represented the first completely IFN-free oral combination therapy for treating HCV genotype 1 [83]. Adverse reactions with simeprevir are less common than first-generation DAAs. The most frequent was rash (photosensitivity), followed by hyperbilirubinaemia, pruritus and nausea. Like other drugs, simeprevir is a DAA that has been discontinued because it has shown efficacy in only two HCV genotypes [73].
3.1.4. Paritaprevir
In 2014, paritaprevir was approved by the FDA in a fixed-dose co-formulated combination (FDC) with ritonavir (an HIV protease inhibitor) and ombitasvir (an NS5A inhibitor) for treating HCV for genotype 4 [84]. For HCV genotype 1, the combination of paritaprevir/ritonavir/ombitasvir with dasabuvir (a non-nucleoside polymerase inhibitor) is known as PrOD [85]. The combination does not require dose adjustment for patients with renal impairment but is contraindicated in patients with severe hepatic dysfunction due to the risk of potential toxicity. Side effects of PrOD with or without RBV are rare but include fatigue, nausea, pruritus, other skin reactions, insomnia and asthenia. In phase 3 trials of PrOD with or without RBV, less than 1% of subjects discontinued treatment due to a side effect. In the latest treatment guidelines of the American Association for the Study of Liver Diseases (AASLD) and the Infectious Diseases Society of America (IDSA), PrOD is no longer included in the treatment guidelines [73].
3.1.5. Grazoprevir
Over the years, better DAAs have been developed that exhibit fewer side effects. In 2016, the HCV NS3/4A protease inhibitor grazoprevir was approved by the FDA because studies showed that the kidneys excrete less than 1% of the drug, so no dose adjustments were needed in patients with stage 4 and 5 chronic kidney disease [86]. Grazoprevir was combined with elbasvir (NS5A inhibitor) for a combination therapy administered once daily for 12 weeks for patients infected with HCV genotypes 1b and 4 [86]. An SVR > 95 % has been reported in patients treated with the combination of grazoprevir and elbasvir [87].
3.1.6. Glecaprevir and Voxileprevir
Glecaprevir, an NS3/4A protease inhibitor, which is used in combination with pibrentasvir for individuals infected with all genotypes (1 to 6) of HCV, without cirrhosis or compensated cirrhosis, and for those patients infected with HCV genotype 1 who have previously received an NS3/4A or NS5A DAA [88]. The reported adverse effects are mild, and the SVR is around 90 %, depending on the genotype and previous treatment.
For treatment failure with first-generation DAAs, voxileprevir, an HCV NS3/4a protease inhibitor, is used alongside two previously approved drugs, sofosbuvir and velpatasvir, in a fixed-dose combination [89]. It demonstrated an SVR of 96-97% at 12 weeks of treatment in patients infected with genotypes 1-6 [89].
3.2. NS5B Polymerase Inhibitors
3.2.1. Nucleotide: Sofosbuvir
Another class of DAAs developed for treating hepatitis C are the nucleotide analogues that inhibit the viral polymerase. These drugs bind with high affinity to the active site of the RNA-dependent RNA polymerase, the product of the NS5B gene, causing premature termination of the RNA strand and inhibiting HCV replication. Sofosbuvir was initially used in combination with ribavirin for the management of patients infected with genotype 2 (12 weeks), genotype 3 (24 weeks), genotype 1 (24 weeks) and in combination therapy with IFN - PEG +RBV (12 weeks) for genotype 1. These regimens were associated with higher SVR rates than PEGIFN +RBV-only treatments, with no significant side effects reported by patients [90]. In 2014, the FDA approved using sofosbuvir and ledipasvir (NS5BA inhibitor) for treating HCV genotype 1, 4, 5 or 6 infections [91]. Finally, in 2017, the combined use of sofosbuvir, voxilelprevir and velpatasvir was approved in those patients who showed treatment failure with other DAAs [91]. Additionally, sofosbuvir has been used with RBV to treat patients coinfected with HIV and HCV genotypes 1, 2 and 3, reporting high SVR rates [92].
3.2.2. Non-nucleotide: Dasabuvir
Non-nucleotide analogue polymerase inhibitors bind outside the active site of NS5B RNA polymerase and cause allosteric inhibition of the enzyme. They generally have a lower barrier to resistance than nucleotide-analogue polymerase inhibitors. Dasabuvir is the first non-nucleotide analogue type DAA approved in 2014 for treating HCV genotype 1 [93]. It is usually administered alongside paritaprevir/ritonavir/ombitasvir; it was effective in a group of patients [94]. However, dasabuvir is limited as monotherapy due to its low genotypic coverage and the possibility of adding to other therapies [95].
3.3. NS5A Polymerase Inhibitors
NS5A inhibitors target the NS5A protein, which is essential for HCV RNA replication and viral assembly. The DAAs discussed below have antiviral activity primarily against genotype 1, with activity varying against other genotypes. Combining a pan-genotypic NS5A inhibitor with pan-genotypic nucleotide analogue polymerase inhibitors currently represents the most widely used treatment against HCV [96].
3.3.1. Ledipasvir (LDV)
Ledipasvir, the first NS5A inhibitor approved by the FDA in 2014 for treating HCV genotype 1, is available only as FDC with sofosbuvir [97]. The treatment is administered for 12 weeks in patients without previous treatment, without cirrhosis or compensated cirrhosis, reporting SVRs greater than 90% [97]. In HIV-coinfected patients, treatment schemes for 12 weeks with sofosbuvir/ledipasvir provided an SVR of 96%. Similarly, this therapeutic regimen was approved as initial treatment in pediatric patients, as sofosbuvir/ledipasvir is well tolerated and highly effective in children between 3 and 6 years of age with chronic HCV infection. Phase 3 studies showed that less than 1% of subjects discontinued treatment due to side effects [98].
3.3.2. Daclatasvir (DCV).
Another NS5A inhibitor approved in 2014 by the European Union and later in 2015 by the FDA was daclastavir [99]. Its use was indicated in conjunction with sofosbuvir for many years because of its synergistic antiviral activity for HCV genotypes 1 and 3 [100]. Cytochrome P450 3A4 metabolises the compound, and dose adjustment was required when used with drugs that significantly affect these enzymes, such as certain antiretrovirals [101].
3.3.3. Ombitasvir
By 2014, ombitasvir, an NS5A protein inhibitor, was approved to treat HCV genotype 1 and in 2015 for genotype 4 as part of the paritaprevir/ritonavir/ombitasvir FDC [102]. It was used as therapy for HCV-infected patients who did not respond to the first protease inhibitors [102]. Several studies have demonstrated its high efficacy in patients infected with genotype 1 who received previous treatment and those infected with HIV. Ombistavir is discontinued and has been replaced by pangenotypic antivirals [73].
3.3.4. Elbasvir
Approved by the FDA for 2016 was elbasvir, an NS5A replication complex inhibitor with broad genotypic activity for use in combination with grazoprevir in infections with HCV genotypes 1 and 4, as well as in patients who have failed to achieve SVR with previous DAA regimens [103]. The combination of grazoprevir and elbasvir was active against NS3/4A protease inhibitor resistance in vitro and in vivo for genotype 1b [104]. No dosing modifications are required for renal or mild to moderate hepatic impairment patients. Side effects are similar to those reported for grazoprevir [73].
3.3.5. Velpatasvir and Pibrentasvir
Velpatasvir was approved by the FDA in 2016 for use alongside sofosbuvir for treating patients infected with genotypes 1 to 6 [105]. High SVRs upon veltaspavir treatment were observed for all genotypes, except in patients infected with genotype 3, where RBV was incorporated into the treatment. Currently, the ASDL/IDSA guidelines recommend 12 weeks of treatment with sofosbuvir (400 mg)/velpatasvir (100 mg) for previously untreated adult patients and pediatric patients older than 3 years of age infected with HCV [106]. Similarly, velpatasvir is used in conjunction with sofosbuvir and voxileprevir in cases of treatment failure, specifically with sofosbuvir and glecaprevir/pibrentasvir regimens [107]. The combination glecaprevir/pibrentasvir has been highly effective in drug users with an SVR of 93 %. In other patients, the SVR was around 95 % [108].
Figure 2 represents a historical overview of the treatments used in HCV.
4. Treatment in Coinfection (HCV/HIV, HBV/HCV, HBV/HCV/HIV).
There are approximately 2 million people worldwide who have HIV/HCV coinfections; most of them (1.3 million) are parenteral drug users. The risk of HCV infection in HIV patients is six times higher than in the general population. It is recommended to routinely test for HCV infection in persons with recent HIV infection due to the similarities in infection routes and the increased risk of liver pathology compared to patients without HIV infection [46,47].
Current IDSA guideline recommendations suggest that patients living with treatment-naïve HIV and HCV coinfection, regardless of liver involvement (with or without cirrhosis), are good candidates for DAAs, based on the results of the phase 4 MINMON (minimal monitoring approach) clinical trial conducted by Solomon et al. 2022 [109]. The treatment scheme involved sofosbuvir and velpatasvir with an SVR of 95%. DAA treatment benefits patients with HIV infection while promoting HCV clearance [110].
In HBV/HCV coinfection, defining the genotype of HCV causing the infection is essential since the coinfection can be HCV-dominant, in which DAA drugs will provide a clear rationale [111]. It is vital to monitor circulating HBV DNA even in the nonexistence of antibodies and circulating HBV core protein [111]. Immunocompromised individuals should be closely monitored to avoid viral escape and hepatic disease [111]. PEGIFN-RBV can be added to prevent HBV replication to decrease chronic infection and liver damage. There is a risk of HBV reactivation after DAA treatment in the coinfected patients [112,113]. HBV core antigen and DNA should be closely monitored, and PEG IFN-RBV therapy should be used if required to avoid cirrhosis and the occurrence of hepatocarcinoma [113].
There is little information in the literature about the coinfection with the three viruses, probably because of the lack of detectable antibodies against HBV proteins in coinfections [46]. HIV patients treated with triple therapy for an extended period may be partially protected from new HCV or HBV infections [46,47]. In naïve patients, active HCV and HBV replication may induce early liver damage. In HBV/HIV coinfection, tenofovir and entecavir are recommended to block HBV and HIV replication [46,47]. These drugs may benefit patients with triple infection after DAA treatment has concluded. Recently, Cairoli et al. [114] and Mirzaei et al. [115] have reported the importance of miRNA in coinfected individuals. Future research should focus on potential biomarkers to facilitate infection screening and response to therapy.
5. Treatment of Pregnant Women, Vertical Transmission. Pediatric Care.
Vertical transmission of HCV occurs in 5.8% of infants from HCV-infected women and up to 12 % of HIV/HCV coinfected women [116]. Reports published by the CDC in 2016 revealed the increasing risk of perinatal HCV transmission in specific high-risk areas of the United States. HCV incidence has increased among young adults and women of childbearing age in these areas [116,117,118,119]. In the absence of an HCV vaccine, there is an immediate need to improve the availability of HCV screening among at-risk individuals, including children born to HCV-infected mothers [117,118,119].
The USPSTF (United States Preventive Services Task Force) and the CDC issued recommendations in 2020 concerning the importance of HCV screening pregnant women at the start of prenatal and during pregnancy and those individuals undergoing fertility treatment [117]. It is also suggested to request HCV tests in sperm and ovule donors. Antiviral therapy is recommended before pregnancy is considered. Cesarean delivery is not recommended to prevent perinatal transmission [117]. Breastfeeding is not contraindicated except in the context of an HIV-coinfected mother [117,118,119]. No large-scale studies have been conducted to evaluate the safety of DAAs during pregnancy, and some groups suggest using DAA during pregnancy on a case-by-case basis [120,121]. However, the Maternal-Fetal Society of ACOG (American College of Obstetrics and Gynecology) recommends that AAD regimens should only be used in the context of a clinical trial or that antiviral treatment should be deferred to the postpartum period, as they are not currently approved for use in pregnancy [122]. They also suggest measures to reduce the risk of transmission during delivery by recommending avoiding internal fetal monitoring, prolonged rupture of membranes and episiotomy [122]. An open-label phase 1 clinical trial evaluated the use of ledipasvir/sofosbuvir in pregnant women between the second and third trimester of gestation through pharmacokinetic studies, concluding that the treatment was safe and effective [123]. The study included only 29 pregnant women, a limited population to extrapolate the results on a large scale [123]. There is a need for increased research on antiviral therapies in pregnant women.
HCV infection in children and adolescents is a critical problem in underdeveloped countries. It has been estimated worldwide that 3.5 million children and adolescents are chronically infected with HCV [124,125]. Treatment of the pediatric population based on adult pharmacokinetics has been rationalised by adjusting the adult successful treatment schemes [124,125,126,127]. It is important to note that parental HBV and HCV infection may be risk factors for hepatic and non-hepatic cancers in children [128]; therefore, ensuring that infected children are treated as soon as possible is critical. In HCV-HIV coinfection, there is a high risk of vertical transmission and a high possibility of chronic liver disease due to an immature immune response. Research in pediatric coinfection is urgently needed.
6. Host Genetics, Infection and Response to HCV Treatments
Several studies have found an association between the host genetic factor, spontaneous clearance of the virus, the treatment response and the risk of fibrosis and/or hepatocarcinoma. Most study single nucleotide polymorphisms (SNP) are from the IL28B gene; however, several other SNP in different genes have been involved: interferon-λ3, interferon-λ4, IL-12, TNF, IL-10 and Toll-like receptors 3 and 9 [129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144]. Moreover, a relationship has been recently shown between increased mortality and epigenetic changes related to age in a group of intravenous drug users coinfected with HCV-HIV [145]. These results suggest that epigenetic modifications may also be relevant to the infection and may jeopardise the effectiveness of the treatment.
Several SNP near the IL28B gene have been related to the spontaneous resolution of HCV infection [129]. Also, the SNPs rs12979860 and rs8099917 are strongly associated with response to treatment with PEGIFN/RBV [130,131,132]. Patients with the homozygous CC genotype achieve a higher SVR with PEGIFN/RBV treatment than patients with TT or CT genotypes [132]. A significantly higher SVR was reported for TT homozygous allele of IL28B gene rs8099917 compared to the other genotypes [133].
Four SNPs, rs12979860 and rs8099917 for IL28B, and rs368234815 and rs117648444 for IFN-λ4 have been associated with HCV susceptibility to infection and response to the treatment [133,134,135]. Ge et al. [130] showed that in rs12979860 SNP, the favourable allele (CC genotype) was reported in Caucasians who responded positively to IFN treatment. In contrast, the unfavourable TT genotype was more common in African Americans [130]. The African Americans with the CC genotype of the SNP rs12979860 responded better to treatment, with higher SVR, than Caucasians with the TT genotype [130].
Patients with the SNP rs12979860 CC polymorphism are more likely to achieve SVR at 12 weeks of treatment with sofosbuvir/daclastavir in genotype 4 infected Egyptian patients [134]. This gene has been studied both as a predictor of response and as a factor of resistance to treatment in patients treated with sofosbuvir, raising the possibility of performing genetic analysis for SNP in IL-28B before initiating antiviral treatment [137,138].
There is a significant association between HCV infection and coinfection with genetic polymorphisms of Toll-like receptors [146,147,148]. The allele of TLR3 rs13126816 decreased the odds of a virologic response to HCV therapy in HCV/HIV coinfected patients [147], while TLR9 rs352140 (G/A variant) may be necessary in HCV and HBV/HCV coinfection [148].
The polymorphisms of Interferon Regulatory Factors have not been studied in detail. Talaat and coworkers [149], in a small group of Egyptian patients, have shown that the IRF3 SNP polymorphism rs2304204 (-925A/G) is a protective genotype for liver cirrhosis. Based on the antiviral effect of IFN pathways, it is crucial to analyse the genetic polymorphism, which may be more prevalent in the infected population and a risk factor in endemic areas.
The miRNAs can also be considered key players in the antiviral response. New reports on miRNA have provided insights into single and multiple infections and possible targets for therapy [114,115,150,151]. However, large-scale studies are required to validate miRNA signatures in HCV-infected and coinfected individuals and their response to treatment.
7. Resistance Associated Substitutions
The main challenge with DAA treatment is resistance-associated substitution (RAS), the leading cause of DAA resistance [152,153,154]. This treatment resistance is an inevitable and intrinsic problem, as HCV is highly adaptable [152,153,154]. These mutations are due to the lack of correction of HCV polymerase, leading to base mutations for each viral replication [152,153,154]. SARs occur because of different treatment regimens, the various HCV genotypes and subtypes, and geographic distribution. SARs in other regions of the NS3/4 protease (F53S, Q80K/R, S122R, R155K, A156T/V, D168) are important causes of treatment failure with protease inhibitors, such as simeprevir, grazoprevir, asunaprevir, and paritaprevir [155].
On the other hand, NS5A inhibitors are an indispensable component of all first-line DAA regimens, making them the class of HCV drugs where resistance is most relevant. RAS found after NS5A inhibitors (M28A/G/T, Q30E/H/R, L31F/M/V, P32L/S, H58D, Y93H) are more frequent in HCV genotypes 1b and genotypes 3. The presence of these RAS in NS5A was demonstrated to show a decrease in SVR in the first week of antiviral treatment with sofosbuvir/daclastavir, especially in HCV genotypes 3 [155,156].
The spectrum of mutations associated with NS5B inhibitors is similarly broad. Because all nucleoside inhibitors target the highly conserved active sites of polymerases, these inhibitors tend to be pan-generic. However, RAS such as S282T have been identified to decrease the efficacy of sofosbuvir and the C316N mutation that reduces dasubuvir's effectiveness [153,155,156].
Even though the number of patients with RAS has not been reported as highly significant, and the consensus is that the change of therapy if SVR is not achieved, there is a need for good epidemiological studies involving patients infected only with HCV and coinfected to determine the actual extent of RAS. These studies should not limit the generation of more specific and effective compounds for treatment.
8. Other strategies
The World Health Organization (WHO) aims to achieve a ~90% reduction in new HCV infections by 2030 [157]. Treatment efficacy has improved since the introduction of DAAs, achieving up to 95% pangenotypic cure; however, there are still challenges, especially in high endemic areas. There is a need to develop preventive and therapeutic HCV vaccines; candidate vaccines studied have shown limited efficacy due to low immunogenicity [158,159,160,161]. There is a debate about whether B-lymphocyte and T-lymphocyte responses are necessary to develop an effective vaccine [162]; however, new strategies to develop a specific and long-lasting vaccine have improved after the experiences encountered with the SARS-CoV-2 virus [163,164]. A protein-based HCV vaccine could induce cell-mediated and humoral immunity; nonstructural proteins and HCV E1/E2 proteins [165,166] seem promising. Other strategies based on the spontaneous clearance of the virus may be used. Recent studies have shown that combining several adjuvants can be useful in increasing the efficacy of vaccines by induction of several receptors of innate immunity. An example of this is the study on HCV-related immunoadjuvants, where an emulsion (MF59), lipid-based nanoparticles (archaeosomes) and a combination delivery immunostimulating system: Alhydrogel-MPL, using recombinant HCV E1E2 glycoproteins, were compared. All formulations with adjuvants showed enhanced immunogenicity with significant neutralisation activity compared to antigen alone; however, no cellular response was detected for the formulation with MF59 adjuvant [166].
On the other hand, a study by Lin et al. [167] evaluated the use of recombinant HCV polypeptides combined with various Th1-type adjuvants and replication-defective alphaviral particles encoding HCV proteins in various priming/boosting modalities in BALB/c mice. Chimeric alphaviral-defective particles derived from Venezuelan equine encephalitis virus and Sindbis encoding the gpE1/gpE2 heterodimer of HCV envelope glycoprotein (E1E2) or nonstructural proteins 3, 4, and 5 (NS345) elicited a robust CD8+ T cell response but a low CD4+ helper T cell response to these HCV gene products. In contrast, recombinant E1E2 glycoproteins with MF59 adjuvant containing a CpG oligonucleotide elicited strong CD4+ helper T cell responses but no CD8+ T cell responses. A recombinant NS345 polyprotein also stimulated strong CD4+ T lymphocyte responses but no CD8+ T cell responses when used with ISCOMATRIX™ containing CpG. Obtaining optimal CD4+ and CD8+ T cell responses against E1E2 and NS345 was achieved by sensitising with Th1 adjuvants and then boosting with defective chimeric alphaviruses expressing these HCV genes. Therefore, these authors concluded that the formulation of this vaccine and the regimen used may be effective for treating HCV. Therefore, these authors concluded that the formulation of this vaccine and the regimen used might be effective in humans for protection against this highly heterogeneous virus [167].
The failure of a T-cell vaccine based on the use of viral vectors expressing HCV nonstructural protein sequences to prevent chronic hepatitis C has indicated that the induction of neutralising antibodies (NAb) should be essential in future vaccines. Finally, the generation of Nab by vaccines should contain the main target of this type of antibodies, which are structural proteins, including HCV envelope glycoproteins (E1 and E2) [168].
Another strategy of interest in HCV therapeutics has been the use of checkpoint inhibitors previously used in cancer treatments. This approach has begun to be used as a treatment in patients with hepatocarcinoma and untreated HCV infection, showing no toxicity in using checkpoint inhibitors in these patients and thus opening up a range of possibilities for untreated and cured HCV oncology patients [169].
Treatment of acute HCV infection in patients undergoing organ transplantation has not been adequately explored [170]. It is clear that also there is a risk of infection from the donor, which may jeopardise the recipient's response. Also, there is a lack of data regarding using DAAs in managing chronic HCV-infected patients with HCV superinfection. In HCV superinfection, there is a potential genotype switch and mixed viral strain infection in patients with HCV superinfection; pan-genotypic DAA regimens are the preferred treatment choices to secure satisfactory viral eradication. It is essential to clarify that the risk of reinfection after successful DAA treatment is shallow; however, due to the possibility of the virus infecting non-hepatic cells, the chance of tissue niches should not be discarded.
In patients undergoing solid transplants, monoclonal therapy blocking the possible entry of the virus into the cells has been explored [170,171,172]. Since the monoclonal antibody against CD81 can be used to treat colorectal, liver and gastric cancers [172], it can be used in transplanted patients.
HCV can infect hepatic and non-hepatic cells by the lipid receptors, the scavenger receptor B type I, the LDL receptor, the ApoE receptor and glycosaminoglycan or heparan sulfate proteoglycan [173]. Lipid-lowering drugs, including Ezetimibe have been suggested to inhibit the infection of the virus to the cell and, in conjunction with other therapies, have been tested in chronic patients and patients at risk [173]. It is assumed that no well-designed clinical trial has been posted, that the inhibition of cholesterol synthesis will decrease the possibility of internalisation of the complex, and it will also affect the lipid moiety of the cell capsid [173]. Exetimibe seems to be more effective along with DAA in this objective [173,174]. Several clinical trials are posted concerning the use of 1) fluvastatin and simvastatin to improve IFN or PEGIFN sensitivity (NCT01377909). 2) Statins to potentiate the effect of the DAA combination Sofosbuvir/Daclatasvir (Egypt) NCT03490097 3) The trial NCT00487318, a randomised control study, including genotypes 1 and 3, in which statin is added to the combination Sofosbuvir/Daclatasvir/Ribavirin to test if statin potentiates the antiviral effect. 4) The trial NCT00926614 (San Antonio, Texas, USA) aimed to study the effect of an insulin-sensitising thiazolidinedione plus a atorvastatin improves sustained virologic response rates in patients who have previously not responded or relapsed on standard PEGIFN and ribavirin therapy. 5) use of different doses of rosuvastatin and atorvastatin to determine if these compounds affect HCV viral load and liver parameters (NCT00446940). However, there is no single and reliable publication on these trials.
Recently, it has been shown that heparanse-1 its upregulated by HCV infection and favours its replication [175]. Heparanase-1 has been involved in tumour growth [176] and it may be related to hepatocarcinoma induced HCV infection.
A recent trial has shown that Erlotinib treatment is safe in noncirrhotic CHC patients. An antiviral activity at 100 mg/d confirms a functional role of EGFR as an HCV host factor in patients [177]. The inhibition of viral entry with dasatinib was also described, the role of EphA2 was then confirmed [173]. In similar fashion the inhibition of claudin-1 by monoclonal antibodies has been studied [173] and just recently a new humanised version has been described [178] and no results of trials have been published.
Finally, RNA interference (RNAi) gene silencing and antisense oligonucleotide suppressive
Functions have been described. The most promising therapy involves miRNA-122 and blocking miR-155 to prevent hepatocellular carcinoma after HCV infection [179].
9. Conclusions
According to WHO, there are about 1.5 million new HCV infections annually and an estimated 8 million chronically infected people worldwide. Even though most individuals clear the virus, many individuals require appropriate therapy. Therapies have evolved from IFN, pegylated-IFN, and RBV to the DAAs, significantly decreasing the viral burden in infected patients. However, there are significant limitations. Simple and economical diagnostic tests are required in health centres with high incidences of infections, generally underdeveloped countries. Early detection reduces the epidemiological transmission of the virus and decreases the risk of developing chronic hepatitis.
The emergence of RASs puts pressure on developing new antiviral treatments with high efficacy, SVR, and low rates of adverse events. There is a need for an effective vaccine to minimise infection and protect vulnerable populations, especially in underdeveloped countries. There is also a need for an epidemiological follow-up of the treated populations to analyse the effectiveness of treatments and medical attention. Any effort to cure and protect individuals from this infectious disease is worthwhile. Figure 3 summarises of all the therapies discussed in the text.
Several areas require research. The difficulties in eradicating HCV and HBV infection in endemic areas are complicated; new genotypes may be developed, complicating the treatment and medical care, the management of transplanted patients and chronic patients, especially elders, with low therapeutic options and finally, the development of protective vaccines which will be a breakthrough, especially for underdeveloped countries.
Author Contributions
Conceptualisation, C.M., A.H.G., F.C., and S.M.; investigation, C.M., F.C., J.B.D.S..; writing—original draft preparation, C.M., F.C., S.M.; writing—review and editing, A.H.G, J.B.D.S.; visualisation, A.H.G., F.C., J.B.D.S..; supervision, J.B.D.S..; project administration, A.H.G..; funding acquisition, A.H.G. All authors have read and agreed to the published version of the manuscript.”
Funding
This work was financed by the National Fund for Science, Technology, and Innovation Of Venezuela (FONACIT): an entity attached to the Ministry of Popular Power for Science and Technology of the Bolivarian Republic of Venezuela (MINCYT). C.M. and F.C. are fellows of the program. AHG is the responsible person. JBDS is partially financed by the National Institute of Virology and Bacteriology [Programme EXCE LES, ID Project No. LX22NPO5103] - - Funded by the European Union - Next Generation EU from the Ministry of Education, Youth and Sports of the Czech Republic (MEYS)]
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable
Acknowledgments
The authors would like to thank Dr Mercedes Zabaleta, Director of the Institute of Immunology, Universidad Central de Venezuela, and Dr Leopoldo Deibis for their support.
Conflicts of Interest
The authors declare no conflict of interest
References
- Ghany, M.G.; Lok, A.S.F.; Dienstag, J.L.; Feinstone, S.M.; Hoofnagle, J.H.; Jake Liang, T.; Seeff, L.B.; Cohen, D.E.; Bezerra, J.A.; Chung, R.T. The 2020 Nobel Prize for Medicine or Physiology for the Discovery of Hepatitis C Virus: A Triumph of Curiosity and Persistence. Hepatology 2021, 74, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Kao, J.-H. Acute hepatitis C virus infection: clinical update and remaining challenges. Clin. Mol. Hepatol. 2023, 29, 623–642. [Google Scholar] [CrossRef]
- Dubuisson, J. ; Cosset FL Virology and cell biology of the hepatitis C virus life cycle An update. J Hepatol. 2014;61(1):S3-13.
- Lee, M.-H.; Yang, H.-I.; Yuan, Y.; L'Italien, G.; Chen, C.-J. Epidemiology and natural history of hepatitis C virus infection. . 2014, 20, 9270–9280. [Google Scholar] [PubMed]
- Alazard-Dany, N.; Denolly, S.; Boson, B.; Cosset, F.-L. Overview of HCV Life Cycle with a Special Focus on Current and Possible Future Antiviral Targets. Viruses 2019, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Basit, H.; Tyagi, I.; Koirala, J. Hepatitis C. In: StatPearls [Internet]. Treasure Island (FL) 2023 Mar 26.: StatPearls Publishing. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430897/.
- Hofmeister, M.G.; Rosenthal, E.M.; Barker, L.K.; Rosenberg, E.S.; Barranco, M.A.; Hall, E.W.; Edlin, B.R.; Mermin, J.; Ward, J.W.; Ryerson, A.B. Estimating Prevalence of Hepatitis C Virus Infection in the United States, 2013-2016. Hepatology 2019, 69, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Stasi, C.; Silvestri, C.; Voller, F. Update on Hepatitis C Epidemiology: Unaware and Untreated Infected Population Could Be the Key to Elimination. SN Compr. Clin. Med. 2020, 2, 2808–2815. [Google Scholar] [CrossRef] [PubMed]
- Fénéant, L.; Levy, S.; Cocquerel, L. CD81 and Hepatitis C Virus (HCV) Infection. Viruses 2014, 6, 535–572. [Google Scholar] [CrossRef]
- Westhaus, S.; Deest, M.; Nguyen, A.T.; Stanke, F.; Heckl, D.; Costa, R.; Schambach, A.; Manns, M.P.; Berg, T.; Vondran, F.W.; et al. Scavenger receptor class B member 1 ( SCARB1 ) variants modulate hepatitis C virus replication cycle and viral load. J. Hepatol. 2017, 67, 237–245. [Google Scholar] [CrossRef]
- Liu, S.; Yang, W.; Shen, L.; Turner, J.R.; Coyne, C.B.; Wang, T. Tight Junction Proteins Claudin-1 and Occludin Control Hepatitis C Virus Entry and Are Downregulated during Infection To Prevent Superinfection. J. Virol. 2009, 83, 2011–2014. [Google Scholar] [CrossRef]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef]
- Mazumdar, B.; Banerjee, A.; Meyer, K.; Ray, R. Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 2011, 54, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Lavie, M.; Dubuisson, J. Interplay between hepatitis C virus and lipid metabolism during virus entry and assembly. Biochimie 2017, 141, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ray, K. NPC1L1 identified as a novel HCV entry factor. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 124–124. [Google Scholar] [CrossRef]
- Xu, Y.; Martinez, P.; Séron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of Hepatitis C Virus Interaction with Heparan Sulfate Proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [PubMed]
- Marino, R.; Deibis, L.; De Sanctis, J.B.; Bianco, N.E.; Toro, F. Interaction of immune complexes isolated from hepatitis C virus-infected individuals with human cell lines. Med Microbiol. Immunol. 2005, 194, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, A.; Shimada, M.; Obata, T. Leukocytes are the major target of hepatitis C virus infection: Possible mechanism of multiorgan involvement including the heart. Glob. Hear. 2010, 5, 51–58. [Google Scholar] [CrossRef]
- Toro, F.; Conesa, A.; Garcia, A.; Deibis, L.; Bianco, N.E. ; De Sanctis, JB HCV RNA sequences in eosinophils of chronic HCV-infected patients. J Med. 1999; 30(3-4):279-288.
- Corado, J.; Toro, F.; Rivera, H.; Bianco, N.E.; Deibis, L. ; De Sanctis, JB Impairment of natural killer (NK) cytotoxic activity in hepatitis C virus (HCV) infection. Clin Exp Immunol. 1997;109(3):451-457.
- Dustin, L.; Bartolini, B.; Capobianchi, M.; Pistello, M. Hepatitis C virus: life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin. Microbiol. Infect. 2016, 22, 826–832. [Google Scholar] [CrossRef]
- Janiak M, Caraballo Cortes K, Demkow U, Radkowski M. Spontaneous Elimination of Hepatitis C Virus Infection. Adv Exp Med Biol. 2018;1039:45-54.
- Chigbu, D.I.; Loonawat, R.; Sehgal, M.; Patel, D.; Jain, P. Hepatitis C Virus Infection: Host–Virus Interaction and Mechanisms of Viral Persistence. Cells 2019, 8, 376. [Google Scholar] [CrossRef]
- Axley, P.; Ahmed, Z.; Ravi, S.; Singal, A.K. Hepatitis C Virus and Hepatocellular Carcinoma: A Narrative Review. J. Clin. Transl. Hepatol. 2018, 6, 1–6. [Google Scholar] [CrossRef]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef]
- Lin, W.; Kim, S.S.; Yeung, E.; Kamegaya, Y.; Blackard, J.T.; Kim, K.A.; Holtzman, M.J.; Chung, R.T. Hepatitis C Virus Core Protein Blocks Interferon Signaling by Interaction with the STAT1 SH2 Domain. J. Virol. 2006, 80, 9226–9235. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J. ; He, SF; Liu, Y.; Zhao, P.; Bian, ZQ; Qi, ZT Inhibition of STAT Pathway Impairs Anti-Hepatitis C Virus Effect of Interferon Alpha. Cell Physiol Biochem. 2016; 40(1-2):77-90.
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Honjoh, C.; Kato, Y.; Yoshiki, H.; Hotta, H.; Sada, K. STAT1 is essential for the inhibition of hepatitis C virus replication by interferon-λ but not by interferon-α. Sci. Rep. 2016, 6, 38336. [Google Scholar] [CrossRef] [PubMed]
- Polyak, S.J.; Khabar, K.S.A.; Rezeiq, M.; Gretch, D.R. Elevated Levels of Interleukin-8 in Serum Are Associated with Hepatitis C Virus Infection and Resistance to Interferon Therapy. J. Virol. 2007, 75, 6209–6211. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Shi, S.T.; Romano, P.R.; Barber, G.N.; Lai, M.M.C. Inhibition of the Interferon- Inducible Protein Kinase PKR by HCV E2 Protein. Science 1999, 285, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xin, X.; Wang, M.; Han, L.; Li, J.; Hao, Y.; Zheng, C.; Shen, C. Myxovirus resistance protein A inhibits hepatitis C virus replication through JAK-STAT pathway activation. Arch. Virol. 2018, 163, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Barbaglia, M.N.; Harris, J.M.; Smirnov, A.; Burlone, M.E.; Rigamonti, C.; Pirisi, M.; Minisini, R.; Magri, A. 17β-Oestradiol Protects from Hepatitis C Virus Infection through Induction of Type I Interferon. Viruses 2022, 14, 1806. [Google Scholar] [CrossRef] [PubMed]
- Marchi, E.; Ramamurthy, N.; Ansari, M.A.; E Harrer, C.; Barnes, E.; Klenerman, P.; Hcv, S. Defining the key intrahepatic gene networks in HCV infection driven by sex. Gut 2023, 72, 984–994. [Google Scholar] [CrossRef]
- Stuart, J.D.; Salinas, E.; Grakoui, A. Immune system control of hepatitis C virus infection. Curr. Opin. Virol. 2021, 46, 36–44. [Google Scholar] [CrossRef]
- Dustin, L.B. Innate and Adaptive Immune Responses in Chronic HCV Infection. Curr. Drug Targets 2017, 18, 826–843. [Google Scholar] [CrossRef]
- Corado, J.A.; Toro, F.I.; Baroja, M.L.; Bianco, N.E.; Machado, I.V.; Shapiro; Gershtein; Elias; Zuckerman; Salman; et al. CD3- and CD28-Activating Pathways in HCV Infection. Viral Immunol. 1994, 7, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Auma, A.W.N.; Shive, C.L. ; Kostadinova, L:; Anthony, D.D. Variable Normalisation of Naïve CD4+ Lymphopenia and Markers of Monocyte and T Cell Activation over the Course of Direct-Acting Antiviral Treatment of Chronic Hepatitis C Virus Infection. Viruses. 2021;14(1):50.
- Ferrufino, R.Q.; Rodrigues, C.; Figueiredo, G.M.; Gleison, D.; Yapura, S.; de Matos, M.L.M.; Witkin, S.S.; Mendes-Correa, M.C. Factors Associated with Spontaneous Clearance of Recently Acquired Hepatitis C Virus among HIV-Positive Men in Brazil. Viruses 2023, 15, 314. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-C.; Sung, P.S.; Park, S.-H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Marascio, N.; Torti, C.; Liberto, M.C.; Focá, A. Update on different aspects of HCV variability: focus on NS5B polymerase. BMC Infect Dis 2014;14 (Suppl 5), S1.
- Echeverría, N.; Moratorio, G.; Cristina, J.; Moreno, P. Hepatitis C virus genetic variability and evolution. World J Hepatol. 2015;7(6):831-845.
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–40. [Google Scholar] [CrossRef] [PubMed]
- Gobran, S.T.; Ancuta, P.; Shoukry, N.H. A Tale of Two Viruses: Immunological Insights Into HCV/HIV Coinfection. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Shahriar, S.; Araf, Y.; Ahmad, R.; Kattel, P.; Sah, G.S.; Rahaman, T.I.; Sadiea, R.Z.; Sultana, S.; Islam, S.; Zheng, C.; et al. Insights Into the Coinfections of Human Immunodeficiency Virus-Hepatitis B Virus, Human Immunodeficiency Virus-Hepatitis C Virus, and Hepatitis B Virus-Hepatitis C Virus: Prevalence, Risk Factors, Pathogenesis, Diagnosis, and Treatment. Front. Microbiol. 2022, 12, 780887. [Google Scholar] [CrossRef]
- Konstantinou, D.; Deutsch, M. The spectrum of HBV/HCV coinfection: epidemiology, clinical characteristics, viral interactions and management. Ann gastroenterol. 2015; 28(2), 221–228.
- Wieland, S.; Makowska, Z.; Campana, B.; Calabrese, D.; Dill, M.T.; Chung, J.; Chisari, F.V.; Heim, M.H. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 2013, 59, 2121–2130. [Google Scholar] [CrossRef]
- Nkongolo, S.; Mahamed, D.; Kuipery, A.; Vasquez, J.D.S.; Kim, S.C.; Mehrotra, A.; Patel, A.; Hu, C.; McGilvray, I.; Feld, J.J.; et al. Longitudinal liver sampling in patients with chronic hepatitis B starting antiviral therapy reveals hepatotoxic CD8+ T cells. J. Clin. Investig. 2023, 133, e158903. [Google Scholar] [CrossRef]
- Perales, C.; Beach, N.M.; Gallego, I.; Soria, M.E.; Quer, J.; Esteban, J.I.; Rice, C.; Domingo, E.; Sheldon, J. Response of Hepatitis C Virus to Long-Term Passage in the Presence of Alpha Interferon: Multiple Mutations and a Common Phenotype. J. Virol. 2013, 87, 7593–7607. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J. M.; Alejo, A.; Martín, V.; Sevilla, N. Viral pathogen-induced mechanisms to antagonise mammalian interferon (IFN) signalling pathway. CMLS 2021, 78(4), 1423–1444. [Google Scholar] [CrossRef] [PubMed]
- Mertowska, P.; Smolak, K.; Mertowski, S.; Grywalska, E. Immunomodulatory Role of Interferons in Viral and Bacterial Infections. Int. J. Mol. Sci. 2023, 24, 10115. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; Mullen, K.D.; Jones, D.B.; Rustgi, V.; Di Bisceglie, A.; Peters, M.; Waggoner, J.G.; Park, Y.; Jones, E.A. Treatment of Chronic Non-A, Non-B Hepatitis with Recombinant Human Alpha Interferon. New Engl. J. Med. 1986, 315, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Jaeckel, E.; Cornberg, M.; Wedemeyer, H.; Santantonio, T.; Mayer, J.; Zankel, M.; Pastore, G.; Dietrich, M.; Trautwein, C.; Manns, M.P. Treatment of Acute Hepatitis C with Interferon Alfa-2b. New Engl. J. Med. 2001, 345, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Aghemo, A. , Rumi, M. G., & Colombo, M. (2010). Pegylated interferons alpha2a and alpha2b in the treatment of chronic hepatitis C. Nature reviews. Gastroenterology & hepatology, 7(9), 485–494.
- McHutchison, J.G.; Gordon, S.C.; Schiff, E.R.; Shiffman, M.L.; Lee, W.M.; Rustgi, V.K.; Goodman, Z.D.; Ling, M.-H.; Cort, S.; Albrecht, J.K. Interferon Alfa-2b Alone or in Combination with Ribavirin as Initial Treatment for Chronic Hepatitis C. New Engl. J. Med. 1998, 339, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; Marcellin, P.; Lee, S.S.; Niederau, C.; Minuk, G.S.; Ideo, G.; Bain, V.; Heathcote, J.; Zeuzem, S.; Trepo, C.; et al. Randomised trial of interferon α2b plus ribavirin for 48 weeks or for 24 weeks versus interferon α2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. Lancet 1998, 352, 1426–1432. [Google Scholar] [CrossRef]
- Thomas, E.; Ghany, M.G.; Liang, T.J. The Application and Mechanism of Action of Ribavirin in Therapy of Hepatitis C. Antivir. Chem. Chemother. 2012, 23, 1–12. [Google Scholar] [CrossRef]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Gonçales, F.L.J.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon Alfa-2a plus Ribavirin for Chronic Hepatitis C Virus Infection. New Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef]
- Hadziyannis, S. J. , Sette, H., Jr, Morgan, T. R., Balan, V., Diago, M., Marcellin, P., Ramadori, G., Bodenheimer, H., Jr, Bernstein, D., Rizzetto, M., Zeuzem, S., Pockros, P. J., Lin, A., Ackrill, A. M., & PEGASYS International Study Group. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomised study of treatment duration and ribavirin dose. Ann Intern Med 2004; 140(5), 346–355.
- Ascione, A.; De Luca, M.; Tartaglione, M.T.; Lampasi, F.; Di Costanzo, G.G.; Lanza, A.G.; Picciotto, F.P.; Marino–Marsilia, G.; Fontanella, L.; Leandro, G. Peginterferon Alfa-2a Plus Ribavirin Is More Effective Than Peginterferon Alfa-2b Plus Ribavirin for Treating Chronic Hepatitis C Virus Infection. Gastroenterology 2010, 138, 116–122. [Google Scholar] [CrossRef]
- Rumi, M.G.; Aghemo, A.; Prati, G.M.; D’Ambrosio, R.; Donato, M.F. , Soffredini, R., et al. Randomised Study of Peginterferon-α2a Plus Ribavirin vs Peginterferon-α2b Plus Ribavirin in Chronic Hepatitis C. Gastroenterology. 2010;138(1):108-15.
- Alavian, S.M.; Behnava, B.; Tabatabaei, S.V. The Comparative Efficacy and Safety of Peginterferon Alpha-2a vs. 2b for the Treatment of Chronic HCV Infection: A Meta-Analysis. 2010, 10, 121–131. [Google Scholar]
- Nakamura, M.; Kanda, T.; Miyamura, T.; Wu, S.; Nakamoto, S.; Yokosuka, O. Alanine Aminotransferase Elevation during Peginterferon Alpha-2a or Alpha-2b plus Ribavirin Treatment. Int. J. Med Sci. 2013, 10, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.-H.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.-H.; Chang, M.-L.; Huang, T.-J.; Yeh, C.-T.; Chiu, W.-N.; Chiang, M.-S.; Chen, M.-Y. Comparison of Compliance and Efficacy of Pegylated Interferon α-2a and α-2b in Adults with Chronic Hepatitis C. J. Interf. Cytokine Res. 2019, 39, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Afdhal, N.H.; Dieterich, D.T.; Pockros, P.J.; Schiff, E.R.; Shiffman, M.L.; Sulkowski, M.S.; Wright, T.; Younossi, Z.; Goon, B.L.; Tang, K.; et al. Epoetin alfa maintains ribavirin dose in HCV-infected patients: a prospective, double-blind, randomized controlled study. Gastroenterology 2004, 126, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Van Thiel, D.H.; Faruki, H.; Friedlander, L.; Fagiuoli, S.; Caraceni, P.; Molloy, P.J.; Kania, R.J.; I Wright, H. Combination treatment of advanced HCV associated liver disease with interferon and G-CSF. . 1995, 42, 907–12. [Google Scholar] [PubMed]
- Afdhal, N.H.; Dusheiko, G.M.; Giannini, E.G.; Chen, P.; Han, K.; Mohsin, A.; Rodriguez–Torres, M.; Rugina, S.; Bakulin, I.; Lawitz, E.; et al. Eltrombopag Increases Platelet Numbers in Thrombocytopenic Patients With HCV Infection and Cirrhosis, Allowing for Effective Antiviral Therapy. Gastroenterology 2014, 146, 442–452.e1. [Google Scholar] [CrossRef] [PubMed]
- O'Leary, J.G.; Davis, G.L. Review: Hepatitis C virus replication and potential targets for direct-acting agents. Ther. Adv. Gastroenterol. 2010, 3, 43–53. [Google Scholar] [CrossRef]
- Milani, A.; Basimi, P.; Agi, E.; Bolhassani, A. Pharmaceutical Approaches for Treatment of Hepatitis C virus. Curr. Pharm. Des. 2020, 26, 4304–4314. [Google Scholar] [CrossRef]
- A Bernal, L.; Soti, V. Hepatitis C Virus: Insights Into Its History, Treatment, Challenges, and Future Directions. Cureus 2023, 15, e43924. [Google Scholar] [CrossRef]
- Failla, C.; Tomei, L.; De Francesco, R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol. 1994, 68, 3753–3760. [Google Scholar] [CrossRef] [PubMed]
- Tabata K, Neufeldt CJ, Bartenschlager R. Hepatitis C Virus Replication. Cold Spring Harb Perspect Med. a: 2020;10(3), 2020.
- Kiang, T.K.L.; Wilby, K.J.; Ensom, M.H.H. Telaprevir: Clinical Pharmacokinetics, Pharmacodynamics, and Drug–Drug Interactions. Clin. Pharmacokinet. 2013, 52, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Gordon, L.A.; Fung, H.B. Boceprevir: A Protease Inhibitor for the Treatment of Hepatitis C. Clin. Ther. 2012, 34, 2021–2038. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. C. , Reddy, K. R., Jacobson, I. M., Poordad, F., Bronowicki, J. P., Bacon, B., Buti, M., Hu, K. Q., Pedicone, L. D., Burroughs, M., Brass, C. A., Albrecht, J. K., & Lawitz, E. J. (2014). Boceprevir plus peginterferon α-2b/ribavirin in chronic hepatitis C genotype 1: impact of baseline viral load on sustained virologic response. Journal of clinical gastroenterology, 48(5), 435–443.
- Akamatsu, N.; Sugawara, Y.; Kokudo, N. Asunaprevir (BMS-650032) for the treatment of hepatitis C virus. Expert Rev. Anti-infective Ther. 2015, 13, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M. , Ueno, T., Shiozaki, T., Li, H., & Garimella, T. (2019). Safety Exposure-Response Analysis for Daclatasvir, Asunaprevir, and Beclabuvir Combinations in HCV-Infected Subjects. Journal of clinical pharmacology, 59(4), 557–565.
- Cao, Y.; Bao, Y.; Xia, W.; Wu, H.; Wei, F.; Zhang, Y.; Zhang, R.; Xu, X. Resistance-associated mutations to HCV protease inhibitors naturally pre-existed in HIV/HCV coinfected, treatment-naïve patients. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Childs-Kean, L.M.; Hand, E.O. Simeprevir and Sofosbuvir for Treatment of Chronic Hepatitis C Infection. Clin. Ther. 2015, 37, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, M.S.; Vargas, H.E.; Di Bisceglie, A.M.; Kuo, A.; Reddy, K.R.; Lim, J.K.; Morelli, G.; Darling, J.M.; Feld, J.J.; Brown, R.S.; et al. Effectiveness of Simeprevir Plus Sofosbuvir, With or Without Ribavirin, in Real-World Patients With HCV Genotype 1 Infection. Gastroenterology 2016, 150, 419–429. [Google Scholar] [CrossRef]
- Klibanov, O.M.; Gale, S.E.; Santevecchi, B. Ombitasvir/Paritaprevir/Ritonavir and Dasabuvir Tablets for Hepatitis C Virus Genotype 1 Infection. Ann. Pharmacother. 2015, 49, 566–581. [Google Scholar] [CrossRef]
- Deeks, E.D. Ombitasvir/Paritaprevir/Ritonavir Plus Dasabuvir: A Review in Chronic HCV Genotype 1 Infection. Drugs 2015, 75, 1027–1038. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Deeks, E.D. Elbasvir/Grazoprevir: A Review in Chronic HCV Genotypes 1 and 4. Drugs 2017, 77, 911–921. [Google Scholar] [CrossRef]
- Ahmed, H.; Abushouk, A.I.; Menshawy, A.; Attia, A.; Mohamed, A.; Negida, A.; Abdel-Daim, M.M. Meta-Analysis of Grazoprevir plus Elbasvir for Treatment of Hepatitis C Virus Genotype 1 Infection. Ann. Hepatol. 2018, 17, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Lamb Y., N. (2017). Glecaprevir/Pibrentasvir: First Global Approval. Drugs, 77(16), 1797–1804.
- Chahine, E.B.; Kelley, D.; Childs-Kean, L.M. Sofosbuvir/Velpatasvir/Voxilaprevir: A Pan-Genotypic Direct-Acting Antiviral Combination for Hepatitis C. Ann. Pharmacother. 2018, 52, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Puoti, M. , Panzeri, C., Rossotti, R., & Baiguera, C. (2014). Efficacy of sofosbuvir-based therapies in HIV/HCV infected patients and persons who inject drugs. Digestive and liver disease : official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver, 46 Suppl 5, S206–S211.
- Smith, M. A. , Chan, J., & Mohammad, R. A. (2015). Ledipasvir-sofosbuvir: interferon-/ribavirin-free regimen for chronic hepatitis C virus infection. The Annals of pharmacotherapy, 49(3), 343–350.
- Devan, P.; Tiong, K.L.A.; Neo, J.E.; Mohan, B.P.; Wijarnpreecha, K.; Tam, Y.C.S.; Coppola, N.; Preda, C.M.; Wong, Y.J. Treatment Outcomes of Sofosbuvir/Velpatasvir/Voxilaprevir in Direct-Acting Antiviral-Experienced Hepatitis C Virus Patients: A Systematic Review and Meta-Analysis. Viruses 2023, 15, 1489. [Google Scholar] [CrossRef] [PubMed]
- Gentile, I.; Buonomo, A.R.; Borgia, G. Dasabuvir: A Non-Nucleoside Inhibitor of NS5B for the Treatment of Hepatitis C Virus Infection. Rev. Recent Clin. Trials 2014, 9, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Gamal, N.; Andreone, P. Working together to tackle HCV infection: ombitasvir/paritaprevir/ritonavir and dasabuvir combination. Drugs Today 2015, 51, 303–14. [Google Scholar] [CrossRef] [PubMed]
- King, J.R.; Zha, J.; Khatri, A.; Dutta, S.; Menon, R.M. Clinical Pharmacokinetics of Dasabuvir. Clin. Pharmacokinet. 2017, 56, 1115–1124. [Google Scholar] [CrossRef]
- Gitto, S.; Gamal, N.; Andreone, P. NS5A inhibitors for the treatment of hepatitis C infection. J. Viral Hepat. 2016, 24, 180–186. [Google Scholar] [CrossRef]
- Bourlière, M.; Adhoute, X.; Ansaldi, C.; Oules, V.; Benali, S.; Portal, I.; Castellani, P.; Halfon, P. Sofosbuvir plus ledipasvir in combination for the treatment of hepatitis C infection. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1483–1494. [Google Scholar] [CrossRef]
- Belperio, P.S.; Shahoumian, T.A.; Loomis, T.P.; Mole, L.A.; Backus, L.I. Real-world effectiveness of daclatasvir plus sofosbuvir and velpatasvir/sofosbuvir in hepatitis C genotype 2 and 3. J. Hepatol. 2019, 70, 15–23. [Google Scholar] [CrossRef]
- Sabry, N.; Kamel, A.M.; Cordie, A.; Esmat, G. Daclatasvir as a hepatitis C infection treatment option: an up-to-date evaluation. Expert Opin. Pharmacother. 2023, 24, 1–12. [Google Scholar] [CrossRef]
- Lim, S.G.; Aghemo, A.; Chen, P.-J.; Dan, Y.Y.; Gane, E.; Gani, R.; Gish, R.G.; Guan, R.; Jia, J.D.; Lim, K.; et al. Management of hepatitis C virus infection in the Asia-Pacific region: an update. Lancet Gastroenterol. Hepatol. 2017, 2, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, Y.; Eley, T.; Fura, A.; Li, W.; Bertz, R.J.; Garimella, T. Daclatasvir: A Review of Preclinical and Clinical Pharmacokinetics. Clin. Pharmacokinet. 2018, 57, 911–928. [Google Scholar] [CrossRef] [PubMed]
- Badri, P.S.; Shuster, D.L.; Dutta, S.; Menon, R.M. Clinical Pharmacokinetics of Ombitasvir. Clin. Pharmacokinet. 2017, 56, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-J.; Huang, C.-F.; Yu, M.-L. Elbasvir and grazoprevir for the treatment of hepatitis C. Expert Rev. Anti-infective Ther. 2021, 19, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Lahser, F.C.; Bystol, K.; Curry, S.; McMonagle, P.; Xia, E.; Ingravallo, P.; Chase, R.; Liu, R.; Black, T.; Hazuda, D.; et al. The Combination of Grazoprevir, a Hepatitis C Virus (HCV) NS3/4A Protease Inhibitor, and Elbasvir, an HCV NS5A Inhibitor, Demonstrates a High Genetic Barrier to Resistance in HCV Genotype 1a Replicons. Antimicrob. Agents Chemother. 2016, 60, 2954–2964. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.-D.; Fu, X.; He, Y.-Q.; Li, C.-Y.; Guo, M.; Qiao, M. Safety and efficacy of sofosbuvir-velpatasvir: A meta-analysis. Medicine 2022, 101, e31183. [Google Scholar] [CrossRef] [PubMed]
- Ghany MG, Morgan TR; AASLD-IDSA Hepatitis C Guidance Panel. Hepatitis C Guidance 2019 Update: American Association for the Study of Liver Diseases-Infectious Diseases Society of America Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Hepatology. 2020 Feb;71(2):686-721.
- Pearlman, B.; Perrys, M.; Hinds, A. Sofosbuvir/Velpatasvir/Voxilaprevir for Previous Treatment Failures With Glecaprevir/Pibrentasvir in Chronic Hepatitis C Infection. Am. J. Gastroenterol. 2019, 114, 1550–1552. [Google Scholar] [CrossRef]
- Reau, N. , Cheng, W. H., Shao, Q., Marx, S. E., Brooks, H., & Martinez, A. (2023). Real-World Effectiveness of 8-Week Glecaprevir/Pibrentasvir in Treatment-Naïve, Compensated Cirrhotic HCV Patients. Infectious diseases and therapy, 12(7), 1849–1860.
- Solomon, S.S.; Wagner-Cardoso, S.; Smeaton, L.; A Sowah, L.; Wimbish, C.; Robbins, G.; Brates, I.; Scello, C.; Son, A.; Avihingsanon, A.; et al. A minimal monitoring approach for the treatment of hepatitis C virus infection (ACTG A5360 [MINMON]): a phase 4, open-label, single-arm trial. Lancet Gastroenterol. Hepatol. 2022, 7, 307–317. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Aronsohn, A.; Price, J.; Re, V.L.; Heald, J.; Demisashi, G.; Durzy, E.; Davis-Owino, A.; Tynes, S. ; Aasld-Idsa Hcv Guidance the American Association for the Study of Liver Diseases–Infectious Diseases Society of America HCV Guidance Panel Hepatitis C Guidance 2023 Update: American Association for the Study of Liver Diseases– Infectious Diseases Society of America Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Clin. Infect. Dis. 2023. [Google Scholar] [CrossRef]
- Mavilia, M.G.; Wu, G.Y. HBV-HCV Coinfection: Viral Interactions, Management, and Viral Reactivation. J. Clin. Transl. Hepatol. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Pisaturo, M.; Macera, M.; Alessio, L.; Calò, F.; Coppola, N. Hepatitis B Virus (HBV) Reactivation Following Pharmacological Eradication of Hepatitis C Virus (HCV). Viruses 2019, 11, 850. [Google Scholar] [CrossRef] [PubMed]
- Maqsood, Q.; Sumrin, A.; Iqbal, M.; Younas, S.; Hussain, N.; Mahnoor, M.; Wajid, A. Hepatitis C virus/Hepatitis B virus coinfection: Current prospectives. Antivir. Ther. 2023, 28. [Google Scholar] [CrossRef] [PubMed]
- Cairoli, V.; Valle-Millares, D.; Terrón-Orellano, M.C.; Luque, D.; Ryan, P.; Dominguez, L.; Martín-Carbonero, L.; Santos, I.D.L.; De Matteo, E.; Ameigeiras, B.; et al. MicroRNA signature from extracellular vesicles of HCV/HIV co-infected individuals differs from HCV mono-infected. J. Mol. Med. 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, R.; Karampoor, S.; Korotkova, N.L. The emerging role of miRNA-122 in infectious diseases: Mechanisms and potential biomarkers. Pathol. - Res. Pr. 2023, 249, 154725. [Google Scholar] [CrossRef]
- Benova L, Mohamoud YA, Calvert C, Abu-Raddad LJ. Vertical Transmission of Hepatitis C Virus: Systematic Review and Meta-analysis. Clin Infect Dis. 2014;59(6):765-73.
- US Preventive Services Task Force, Owens DK, Davidson KW, Krist AH, Barry MJ, Cabana M, et al. Screening for Hepatitis C Virus Infection in Adolescents and Adults: US Preventive Services Task Force Recommendation Statement. JAMA. 10 de marzo de 2020;323(10):970.
- Schillie, S.; Wester, C.; Osborne, M.; Wesolowski, L.; Ryerson, A.B. CDC Recommendations for Hepatitis C Screening Among Adults — United States, 2020. MMWR. Recomm. Rep. 2020, 69, 1–17. [Google Scholar] [CrossRef] [PubMed]
- AASLD-IDSA HCV Guidance Panel. Hepatitis C Guidance 2018 Update: AASLD-IDSA Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. Clin. Infect. Dis. 2018, 67, 1477–1492. [Google Scholar] [CrossRef] [PubMed]
- Kushner, T.; Lange, M.; Sperling, R.; Dieterich, D. Treatment of Women With Hepatitis C Diagnosed in Pregnancy: a Co-Located Treatment Approach. Gastroenterology 2022, 163, 1454–1456.e1. [Google Scholar] [CrossRef] [PubMed]
- AbdAllah M, Alboraie M, Abdel-Razek W, et al. Pregnancy outcome of anti-HCV direct-acting antivirals: real-life data from an Egyptian cohort. Liver Int 2021; 41:1494–7.
- Hughes, B.L.; Page, C.M.; Kuller, J.A. Hepatitis C in pregnancy: screening, treatment, and management. Am. J. Obstet. Gynecol. 2017, 217, B2–B12. [Google Scholar] [CrossRef]
- A Chappell, C.; Scarsi, K.K.; Kirby, B.J.; Suri, V.; Gaggar, A.; Bogen, D.L.; Macio, I.S.; A Meyn, L.; E Bunge, K.; E Krans, E.; et al. Ledipasvir plus sofosbuvir in pregnant women with hepatitis C virus infection: a phase 1 pharmacokinetic study. Lancet Microbe 2020, 1, e200–e208. [Google Scholar] [CrossRef]
- Bitnun, A. The management of infants, children, and youth at risk for hepatitis C virus infection. Paediatr. Child Heal. 2021, 26, 440–440. [Google Scholar] [CrossRef]
- Schwarz, K.B.; Rosenthal, P.; Murray, K.F.; Honegger, J.R.; Hardikar, W.; Hague, R.; Mittal, N.; Massetto, B.; Brainard, D.M.; Hsueh, C.; et al. Ledipasvir-Sofosbuvir for 12 Weeks in Children 3 to <6 Years Old With Chronic Hepatitis C. Hepatology 2020, 71, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.; Seetharaman, K.; Anushree, N. Treatment of hepatitis C in children and adolescents: how far have we reached? World J. Pediatr. 2023, 19, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Peralta, R.P.; Wirth, S.; Squires, R.H.; Mutschler, F.; Lang, T.; Pawlowska, M.; Sluzewski, W.; Majda-Stanislawska, E.; Fischler, B.; Balistreri, W.F.; et al. Elbasvir/grazoprevir in children aged 3–18 years with chronic HCV genotype 1 or 4 infection: a pharmacokinetic modeling study. Hepatol. Commun. 2023, 7, e0031–e0031. [Google Scholar] [CrossRef] [PubMed]
- E Heck, J.; Wu, C.-K.; Huang, X.; Chew, K.W.; Tong, M.; Federman, N.; Ritz, B.; A Arah, O.; Li, C.-Y.; Yu, F.; et al. Cohort study of familial viral hepatitis and risks of paediatric cancers. Leuk. Res. 2021, 51, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Miri, H.H.; Fazeli, P.; Ali-Hassanzadeh, M.; Bemani, P.; Kabelitz, D.; Kalantar, K. Correlation between IL-28 polymorphism and spontaneous clearance in HCV patients: systematic review and meta-analysis. Arch. Virol. 2021, 166, 2469–2478. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, K.; Watanabe, T.; Tanaka, Y. Role of IL28B for chronic hepatitis C treatment toward personalised medicine. J Gastroenterol Hepatol. 2014 ;29:241-9.
- Hayes, C.N.; Imamura, M.; Aikata, H.; Chayama, K. Genetics of IL28B and HCV—response to infection and treatment. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 406–417. [Google Scholar] [CrossRef]
- Bhushan, A.; Ghosh, S.; Bhattacharjee, S.; Chinnaswamy, S. Confounding by Single Nucleotide Polymorphism rs117648444 (P70S) Affects the Association of Interferon Lambda Locus Variants with Response to Interferon-α-Ribavirin Therapy in Patients with Chronic Genotype 3 Hepatitis C Virus Infection. J. Interf. Cytokine Res. 2017, 37, 369–382. [Google Scholar] [CrossRef]
- Ito, K.; Higami, K.; Masaki, N.; Sugiyama, M.; Mukaide, M.; Saito, H.; Aoki, Y.; Sato, Y.; Imamura, M.; Murata, K.; et al. The rs8099917 Polymorphism, When Determined by a Suitable Genotyping Method, Is a Better Predictor for Response to Pegylated Alpha Interferon/Ribavirin Therapy in Japanese Patients than Other Single Nucleotide Polymorphisms Associated with Interleukin-28B. J. Clin. Microbiol. 2011, 49, 1853–1860. [Google Scholar] [CrossRef]
- Riva E, Scagnolari C, Turriziani O, Antonelli G. Hepatitis C virus and interferon type III (interferon-λ3/interleukin-28B and interferon-λ4): genetic basis of susceptibility to infection and response to antiviral treatment. Clin Microbiol Infect. 2014;20(12):1237-45.
- Ibrahim MK, AbdElrahman M, Bader El Din NG, Tawfik S, Abd-Elsalam S, Omran D, Barakat AZ, Farouk S, Elbatae H, El Awady MK. The impact of genetic variations in sofosbuvir metabolising enzymes and innate immunity mediators on treatment outcome in HCV-infected patients. Microb Pathog. 2022 Jan;162:105311.
- Loucks CM, Lin JJ, Trueman JN, Drögemöller BI, Wright GEB, Chang WC, Li KH, Yoshida EM, Ford JA, Lee SS, Crotty P, Kim RB, Al-Judaibi B, Schwarz UI, Ramji A, Farivar JF, Tam E, Walston LL, Ross CJD, Carleton BC. Patient-specific genetic factors predict treatment failure in sofosbuvir-treated patients with chronic hepatitis C. Liver Int. 2022 Apr;42(4):796-808.
- Abdelnajid, D.M.; Elmowafy, A.Y.; Rostaing, L.; Elrakaiby, M.T. Prediction of response to sofosbuvir-based therapy using serum interleukin-12 and single nucleotide polymorphism of the interleukin 28B gene as predictive factors in HCV positive genotype-4 patients. Medicine 2023, 102, e34125. [Google Scholar] [CrossRef]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; et al. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nishida, N.; Sugiyama, M.; Kurosaki, M.; Matsuura, K.; Sakamoto, N.; Nakagawa, M.; Korenaga, M.; Hino, K.; Hige, S.; et al. Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009, 41, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Bochud, P.-Y.; George, J. Host – hepatitis C viral interactions: The role of genetics. J. Hepatol. 2016, 65, S22–S32. [Google Scholar] [CrossRef] [PubMed]
- Asthana, M.; Sahu, S.K.; Kumar, A.; Mohanty, S.; Chakrabarti, S.; Das, P.; Chattopadhya, N.R.; Chatterjee, K.; Singh, S.P.; Rajasubramaniam, S.; et al. Role of Interleukin 28B Polymorphisms in Response to Interferon Based Therapy for Hepatitis C Virus Clearance. Curr. Drug Metab. 2018, 19, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M. A. , Aranday-Cortes, E., Ip, C. L., da Silva Filipe, A., Lau, S. H., Bamford, C., Bonsall, D., Trebes, A., Piazza, P., Sreenu, V., Cowton, V. M., STOP-HCV Consortium, et al. (2019). Interferon lambda 4 impacts the genetic diversity of hepatitis C virus. eLife, 8, e42463.
- Ferreira, J.; Oliveira, M.; Bicho, M.; Serejo, F. Role of Inflammatory/Immune Response and Cytokine Polymorphisms in the Severity of Chronic Hepatitis C (CHC) before and after Direct Acting Antiviral (DAAs) Treatment. Int. J. Mol. Sci. 2023, 24, 1380. [Google Scholar] [CrossRef]
- Liang, X.; Justice, A.C.; Marconi, V.C.; Aouizerat, B.E.; Xu, K. Co-occurrence of injection drug use and hepatitis C increases epigenetic age acceleration that contributes to all-cause mortality among people living with HIV. Epigenetics 2023, 18, 2212235. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y. , Nepal, N., & Jin, S. Z. (2021). Toll-like receptors and hepatitis C virus infection. Hepatobiliary & pancreatic diseases international : HBPD INT, 20(6), 521–529.
- Jiménez-Sousa et al. (2015) Jiménez-Sousa MA. TLR3 polymorphisms are associated with virologic response to hepatitis C virus (HCV) treatment in HIV/HCV coinfected patients. Journal of Clinical Virology. 6: 2015;66, 2015.
- Xu, Y.; Xue, W.; Gao, H.; Cui, J.; Zhao, L.; You, C. Association of toll-like receptors single nucleotide polymorphisms with HBV and HCV infection: research status. PeerJ 2022, 10, e13335. [Google Scholar] [CrossRef]
- Talaat, R.M.; Elsayed, S.S.; Abdel-Hakem, N.E.; El-Shenawy, S.Z. Genetic Polymorphism in Toll-Like Receptor 3 and Interferon Regulatory Factor 3 in Hepatitis C Virus-Infected Patients: Correlation with Liver Cirrhosis. Viral Immunol. 2022, 35, 609–615. [Google Scholar] [CrossRef]
- Öksüz, Z.; Gragnani, L.; Lorini, S.; Temel, G. .; Serin, M.S.; Zignego, A.L. Evaluation of Plasma miR-17-5p, miR-24-3p and miRNA-223-3p Profile of Hepatitis C Virus-Infected Patients after Treatment with Direct-Acting Antivirals. J. Pers. Med. 2023, 13, 1188. [Google Scholar] [CrossRef]
- Kulkarni, V. , Jayakumar, S., Mohan, M., & Kulkarni, S. (2023). Aid or Antagonise: Nuclear Long Noncoding RNAs Regulate Host Responses and Outcomes of Viral Infections. Cells, 12(7), 987.
- Mushtaq S, Hashmi AH, Khan A, Asad Raza Kazmi SM and Manzoor S (2022) Emergence and Persistence of Resistance-Associated Substitutions in HCV GT3 Patients Failing Direct- Acting Antivirals. Front. Pharmacol. 13:8944heng60.
- Sølund C, Pedersen MS, Fahnøe U, Filskov J, Jenssen H, Weis N, Schønning K, Bukh J. Pre-existing, treatment-specific resistance-associated substitutions in hepatitis C virus genotype 1 and 3 and viral RNA titers during treatment with direct-acting antivirals. APMIS. 2023 Aug;131(8):426-433.
- Devan, P.; Tiong, K.L.A.; Neo, J.E.; Mohan, B.P.; Wijarnpreecha, K.; Tam, Y.C.S.; Coppola, N.; Preda, C.M.; Wong, Y.J. Treatment Outcomes of Sofosbuvir/Velpatasvir/Voxilaprevir in Direct-Acting Antiviral-Experienced Hepatitis C Virus Patients: A Systematic Review and Meta-Analysis. Viruses 2023, 15, 1489. [Google Scholar] [CrossRef]
- Zeng, H.; Li, L.; Hou, Z.; Zhang, Y.; Tang, Z.; Liu, S. Direct-acting Antiviral in the Treatment of Chronic Hepatitis C: Bonuses and Challenges. Int. J. Med Sci. 2020, 17, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Malandris, K. , Kalopitas, G., Theocharidou, E., & Germanidis, G. (2021). The Role of RASs /RVs in the Current Management of HCV. 2096. [Google Scholar]
- Thomas, D.L. Global Elimination of Chronic Hepatitis. New Engl. J. Med. 2019, 380, 2041–2050. [Google Scholar] [CrossRef] [PubMed]
- Garcia A, Fernandez S, Toro F, Sanctis J. An Overview of Hepatitis C Vaccines. Recent Pat Inflamm Allergy Drug Discov. 21 de julio de 2014;8(2):85-91.
- Bailey JR, Barnes E, Cox AL. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology. enero de 2019;156(2):418-30.
- Duncan, J.D.; Urbanowicz, R.A.; Tarr, A.W.; Ball, J.K. Hepatitis C Virus Vaccine: Challenges and Prospects. Vaccines 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Kardani, K.; Sadat, S.M.; Kardani, M.; Bolhassani, A. The next generation of HCV vaccines: a focus on novel adjuvant development. Expert Rev. Vaccines 2021, 20, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.-L.; Wrensch, F.; Pierce, B.G.; Baumert, T.F.; Foung, S.K.H. Mapping Determinants of Virus Neutralization and Viral Escape for Rational Design of a Hepatitis C Virus Vaccine. Front. Immunol. 2018, 9, 1194. [Google Scholar] [CrossRef] [PubMed]
- Elbahrawy, A.; Atalla, H.; Alboraie, M.; Alwassief, A.; Madian, A.; El Fayoumie, M.; Tabll, A.A.; Aly, H.H. Recent Advances in Protective Vaccines against Hepatitis Viruses: A Narrative Review. Viruses 2023, 15, 214. [Google Scholar] [CrossRef] [PubMed]
- Adugna, A. Therapeutic strategies and promising vaccine for hepatitis C virus infection. Immun Inflamm Dis. 2023;11(8):e977.
- Fauvelle C, Colpitts CC, Keck Z yong, Pierce BG, Foung SKH, Baumert TF. Hepatitis C virus vaccine candidates inducing protective neutralising antibodies. Expert Rev Vaccines. 2016;15(12):1535-44.
- Landi, A.; Law, J.; Hockman, D.; Logan, M.; Crawford, K.; Chen, C.; Kundu, J.; Ebensen, T.; Guzman, C.; Deschatelets, L.; et al. Superior immunogenicity of HCV envelope glycoproteins when adjuvanted with cyclic-di-AMP, a STING activator or archaeosomes. Vaccine 2017, 35, 6949–6956. [Google Scholar] [CrossRef]
- Lin, Y.; Kwon, T.; Polo, J.; Zhu, Y.-F.; Coates, S.; Crawford, K.; Dong, C.; Wininger, M.; Hall, J.; Selby, M.; et al. Induction of Broad CD4 + and CD8 + T-Cell Responses and Cross- Neutralizing Antibodies against Hepatitis C Virus by Vaccination with Th1-Adjuvanted Polypeptides Followed by Defective Alphaviral Particles Expressing Envelope Glycoproteins gpE1 and gpE2 and Nonstructural Proteins 3, 4, and 5. J. Virol. 2008, 82, 7492–7503. [Google Scholar] [CrossRef]
- Gomez-Escobar, E.; Roingeard, P.; Beaumont, E. Current Hepatitis C Vaccine Candidates Based on the Induction of Neutralizing Antibodies. Viruses 2023, 15, 1151. [Google Scholar] [CrossRef]
- Alkrekshi, A.; Tamaskar, I. Safety of Immune Checkpoint Inhibitors in Patients with Cancer and Hepatitis C Virus Infection. Oncol. 2021, 26, e827–e830. [Google Scholar] [CrossRef]
- Patnaik, R.; Tsai, E. Hepatitis C Virus Treatment and Solid Organ Transplantation. . 2022, 18, 85–94. [Google Scholar] [PubMed]
- Desombere I, Mesalam AA, Urbanowicz RA, Van Houtte F, Verhoye L, Keck ZY, Farhoudi A, Vercauteren K, Weening KE, Baumert TF, Patel AH, Foung SKH, Ball J, Leroux-Roels G, Meuleman P. A novel neutralising human monoclonal antibody broadly abrogates hepatitis C virus infection in vitro and in vivo. Antiviral Res. 2017 Dec;148:53-64.
- Bailly, C.; Thuru, X. Targeting of Tetraspanin CD81 with Monoclonal Antibodies and Small Molecules to Combat Cancers and Viral Diseases. Cancers 2023, 15, 2186. [Google Scholar] [CrossRef] [PubMed]
- Carriquí-Madroñal, B.; Lasswitz, L.; von Hahn, T.; Gerold, G. Genetic and pharmacological perturbation of hepatitis-C virus entry. Curr. Opin. Virol. 2023, 62, 101362. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Cypel, M.; Kumar, D.; Dahari, H.; Ribeiro, R.V.P.; Marks, N.; Kamkar, N.; Bahinskaya, I.; Onofrio, F.Q.; A Zahoor, M.; et al. Short-course, direct-acting antivirals and ezetimibe to prevent HCV infection in recipients of organs from HCV-infected donors: a phase 3, single-centre, open-label study. Lancet Gastroenterol. Hepatol. 2020, 5, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Gallard, C.; Lebsir, N.; Khursheed, H.; Reungoat, E.; Plissonnier, M.-L.; Bré, J.; Michelet, M.; Chouik, Y.; Zoulim, F.; Pécheur, E.-I.; et al. Heparanase-1 is upregulated by hepatitis C virus and favors its replication. J. Hepatol. 2022, 77, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yuan, F.; Zhou, H.; Quan, J.; Liu, C.; Wang, Y.; Xiao, F.; Liu, Q.; Liu, J.; Zhang, Y.; et al. Potential roles of heparanase in cancer therapy: Current trends and future direction. J. Cell. Physiol. 2023, 238, 896–917. [Google Scholar] [CrossRef] [PubMed]
- Saviano, A.; Habersetzer, F.; Lupberger, J.; Simo-Noumbissie, P.; Schuster, C.; Doffoël, M.; Schmidt-Mutter, C.; Baumert, T.F. Safety and Antiviral Activity of EGFR Inhibition by Erlotinib in Chronic Hepatitis C Patients: A Phase Ib Randomized Controlled Trial. Clin. Transl. Gastroenterol. 2022, 13, e00492. [Google Scholar] [CrossRef]
- Colpitts, C.C.; Tawar, R.G.; Mailly, L.; Thumann, C.; Heydmann, L.; Durand, S.C.; Xiao, F.; Robinet, E.; Pessaux, P.; Zeisel, M.B.; et al. Humanisation of a claudin-1-specific monoclonal antibody for clinical prevention and cure of HCV infection without escape. Gut 2018, 67, 736–745. [Google Scholar] [CrossRef]
- Pascut, D.; Hoang, M.; Nguyen, N.N.Q.; Pratama, M.Y.; Tiribelli, C. HCV Proteins Modulate the Host Cell miRNA Expression Contributing to Hepatitis C Pathogenesis and Hepatocellular Carcinoma Development. Cancers 2021, 13, 2485. [Google Scholar] [CrossRef]
Figure 1.
HCV genomic virus structure and viral proteins.

Figure 2.
History of advances in treatment against chronic HCV infection and evolution of sustained viral response. NANBH: non-A, non-B hepatitis; IFN-α: interferon α; Peg-IFN: pegylated interferon-α. Over the years, there has been a marked improvement in HCV clearance efficiency.
Figure 2.
History of advances in treatment against chronic HCV infection and evolution of sustained viral response. NANBH: non-A, non-B hepatitis; IFN-α: interferon α; Peg-IFN: pegylated interferon-α. Over the years, there has been a marked improvement in HCV clearance efficiency.
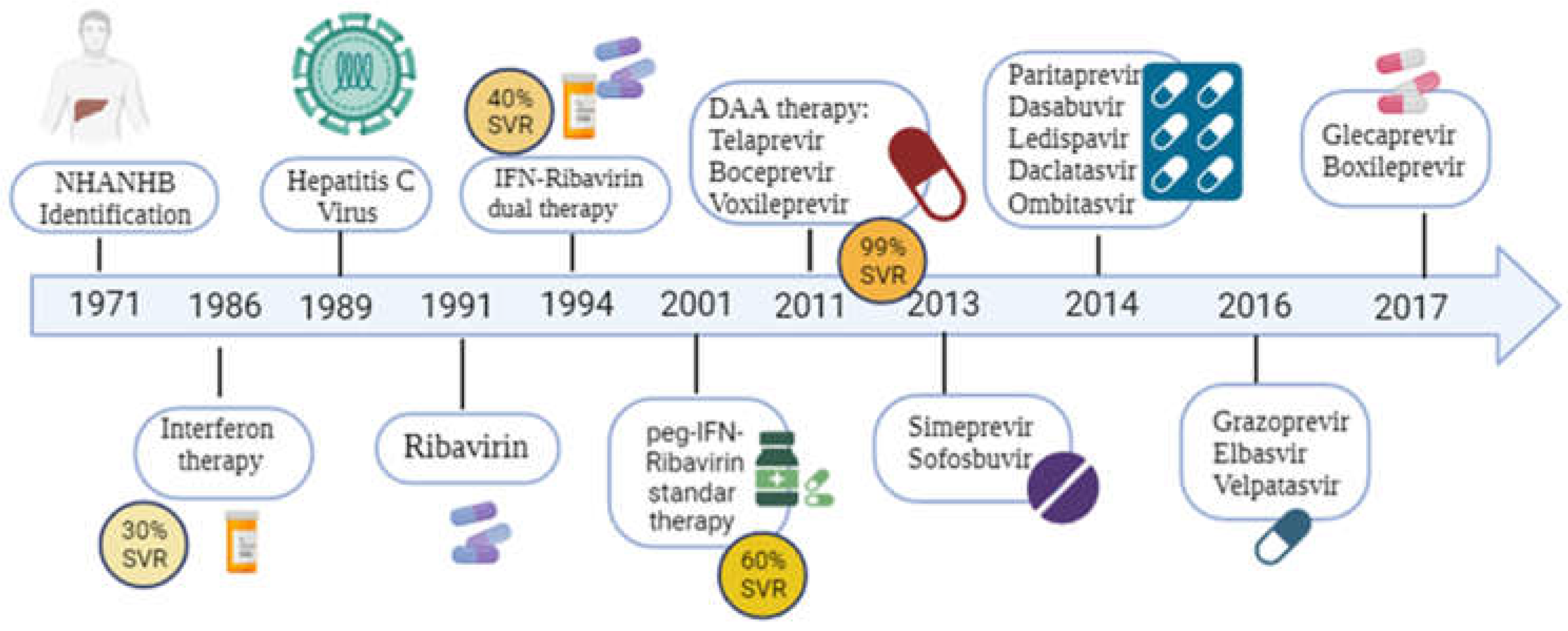
Figure 3.
New and old therapeutic strategies against HCV infection. The figure represents therapeutic targets, from cell binding /infection to general antiviral drugs such as IFNα. The old and new inhibitors of NS3/NS4, NS5A and NS5B are represented. Several vaccine strategies are being tested. Statins, Ezetimibe and other lipid-lowering drugs have been tested in clinical trials; however, no results have been posted. ITX-5061 (antagonist of scavenger receptor B1) was evaluated for safety (clinical trial phase I NCT01165359 in HCV naïve patients and NCT01560468 in liver transplant patients). Ezetimibe has been used in transplant patients from HCV-positive donors (clinical trial NCT04017338). Other inhibitors are either been tested in clinical trials or new schemes have been developed to undergo clinical trials. .
Figure 3.
New and old therapeutic strategies against HCV infection. The figure represents therapeutic targets, from cell binding /infection to general antiviral drugs such as IFNα. The old and new inhibitors of NS3/NS4, NS5A and NS5B are represented. Several vaccine strategies are being tested. Statins, Ezetimibe and other lipid-lowering drugs have been tested in clinical trials; however, no results have been posted. ITX-5061 (antagonist of scavenger receptor B1) was evaluated for safety (clinical trial phase I NCT01165359 in HCV naïve patients and NCT01560468 in liver transplant patients). Ezetimibe has been used in transplant patients from HCV-positive donors (clinical trial NCT04017338). Other inhibitors are either been tested in clinical trials or new schemes have been developed to undergo clinical trials. .
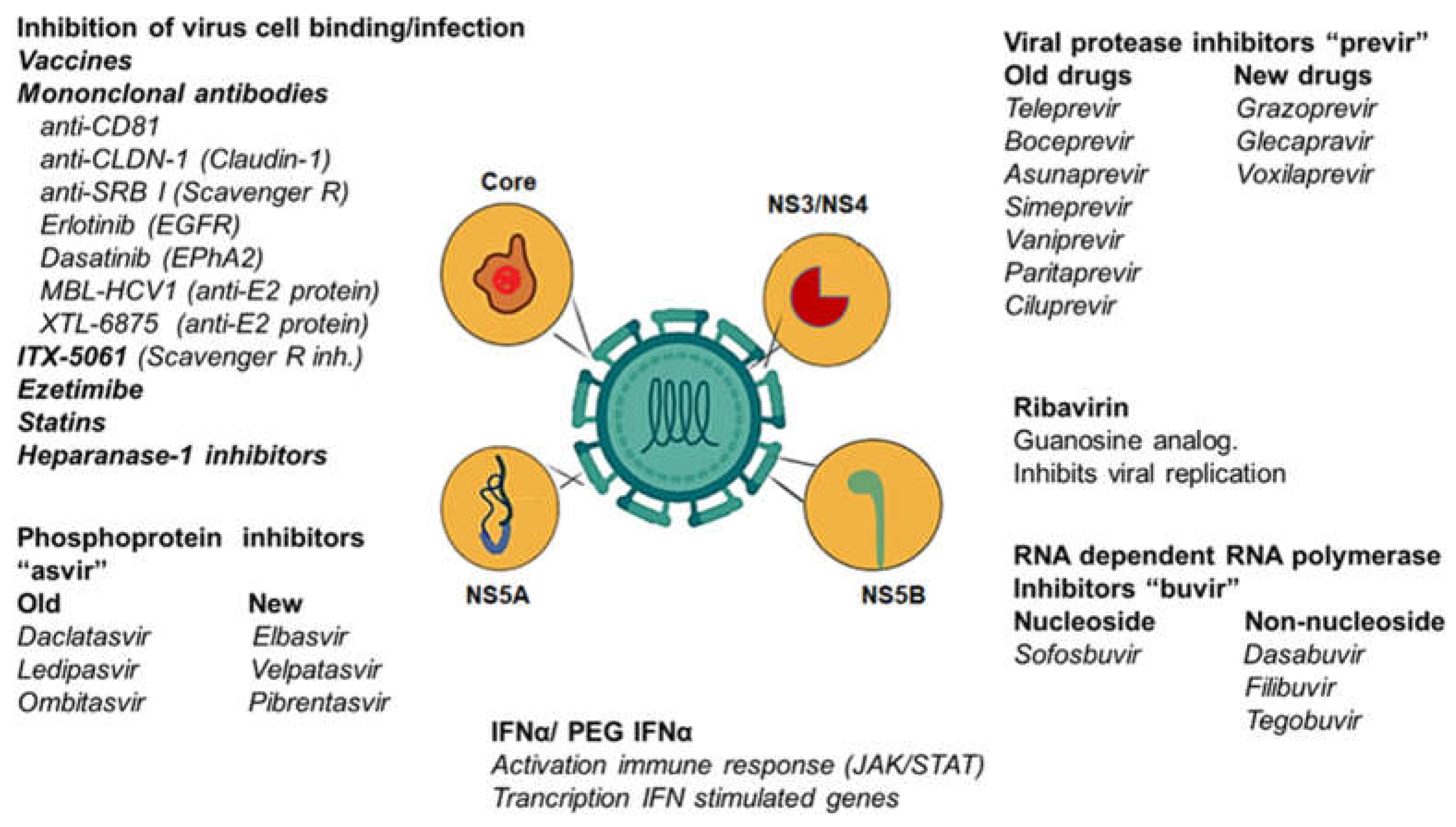
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



