
Submitted:
29 September 2023
Posted:
30 September 2023
You are already at the latest version
Abstract
Therapeutic phages are primarily chosen based on their in vitro bacteriolytic activity. Although anti-phage antibodies are known to inhibit phage infection, the influence of other immune system components is less well known. An important anti-bacterial and anti-viral innate immune system that may interact with phages is the complement system, a cascade of proteases that recognizes and targets invading microorganisms. In this study, we aimed to study the effects of serum components such as complement on the infectivity of different phages targeting Pseudomonas aeruginosa. We used a fluorescence-based assay to monitor the killing of P. aeruginosa by phages of different morphotypes in presence of human serum. Our results reveal that several myophages are inhibited by serum in a concentration-dependent way, while the activity of four podophages and one siphophage tested in this study is not affected by serum. By using specific nanobodies blocking different components of the complement cascade, we show that activation of the classical complement pathway is a driver of phage inhibition. To determine the mechanism of inhibition, we produced bioorthogonally labeled fluorescent phages to study their binding by means of microscopy and flow cytometry. We show that phage adsorption is hampered in presence of active complement. Our results indicate that interactions with complement may affect in vivo activity of therapeutically administered phages. A better understanding of this phenomenon is essential to optimize the design and application of therapeutic phage cocktails.
Keywords:
phage therapy
; phage inhibition
; complement system
; Pseudomonas aeruginosa
; myophages
1. Introduction
Over the last decades, the increase in multi-drug resistant bacteria has spurred a renewed interest in bacteriophages (phages) as an alternative to antibiotics[1]. Phages used in therapy predominantly follow a lytic cycle, which means that they can rapidly kill infected bacteria and propagate by spreading to neighboring cells[2]. These phages belong in the majority of cases to the order Caudovirales, and are morphologically characterized by an icosahedral capsid, or head, which contains the genetic material, and a tail. The tail can further determine the morphotype of the phage. Phages with contractile tails are named myophages, phages with a short tail are named podophages, and phages with a long, non-contractile tail are named siphophages. In contrast to most antibiotics, phages have the advantage of being very specific in the bacteria that they can target. However, this also means that phage therapy often has to be highly tailored to the needs of each patient. Therapeutic success may therefore depend on using the most optimal phages to treat each infection[3].
A pathogen for which phage therapy is particularly relevant is Pseudomonas aeruginosa. This species of gram-negative bacteria is part of the ESKAPE list, a group of pathogens that are the leading cause of nosocomial infections worldwide. It is an opportunistic pathogen that causes infections ranging from burn wound complications to pneumonia and severe sepsis. It is particularly worrisome for immunocompromised patients with underlying conditions, such as cystic fibrosis or hematological malignancies. Antibiotic resistance in P. aeruginosa is rising rapidly, making it a prime candidate for treatment with phage therapy[4]. The potential of phage therapy against P. aeruginosa infections is showcased in recent case reports[5,6,7,8]. Several clinical studies have also been carried out to evaluate the safety of phage therapy in humans against this pathogen, with positive outcomes[9,10]. Efficacy is also assessed in some of these studies, although the results can be difficult to interpret because of the challenges in study design[11,12]. Nonetheless, most of the evidence comes from clinical cases and case series, with the number of randomized clinical trials being still limited.
There are several reasons why the efficacy of phage therapy is still disputed. Bacteria can evade phage infection via several defense mechanisms, and they can rapidly mutate to develop resistance to phages[13,14]. To prevent this, mixes of different phages, or cocktails, are often administered. However, finding several phages to target a pathogenic strain can be challenging, and the optimization of phage cocktail compositions is not always straightforward[15,16]. Another major caveat is that the patient’s immune system may recognize and target phages as exogenous organisms. This has been shown to occur in different ways, such as induction of neutralizing antibodies[17] or phagocytosis of phage virions by immune cells[18]. Depending on the patient and the route of administration, the immune response could lead to inactivation of the phages before they can reach their goal.
A major part of the immune system that phages encounter when entering the blood and tissues is the complement system. The complement system mediates innate immunity to exogenous entities, including pathogens. It consists of a cascade of proteases that can activate via different pathways: the classical pathway is activated through the action of antibodies, the alternative pathway creates an amplification loop that increases activation, and the lectin pathway is triggered by recognition of molecular patterns[19]. Following activation, the pathways converge at the formation of a C3 convertase, which cleaves C3 causing C3b to deposit on the bacterial surface. C3b can act as an opsonin, triggering and promoting phagocytosis by immune cells. The presence of C3b also causes the C3 convertase to switch specificity to recognize C5. Eventually, this leads to formation of the membrane attack complex (MAC), which can directly lyse gram-negative bacteria[20]. Although complement could in some instances act synergistically with phage therapy[21], there is also evidence that complement can recognize viral particles[22], and even phages[23].
Our goal is to study the effect of human serum, and in particular of the complement system, on lytic phage activity. To address this, we have assessed different P. aeruginosa phages in presence of serum. Our results reveal that serum inhibits the lytic activity of certain phages, predominantly myophages. To study the involvement of the complement system in this phenomenon, we have used nanobodies inhibiting specific complement proteins. In this way, we demonstrate that recognition of phages by early components of the complement cascade can impair their binding to bacteria. The fact that different phages perform differently when challenged by the innate immune system could have important repercussions in the way that we select phages for therapy.
2. Materials and Methods
2.1. Phages and Strains
Phage PBJ was isolated in our lab after two rounds of amplification from a mixed stock using P. aeruginosa strain PAO1 as host. Original stocks of Pseudomonas phages 14-1, LKD16 and LUZ19 were kindly provided by Rob Lavigne (KU Leuven). Pseudomonas phages (Pa collection) and phage K were obtained from the Fagenbank (Delft, Netherlands). Phage amplification was carried out by infecting the host strain PAO1 overnight at 37°C in Lysogeny Broth (LB). Bacterial debris was removed by centrifugation at 11,000 RCF for 40 minutes at 4°C. Phages were then incubated for 2 hours on ice with a solution of 10% PEG-8000 and 0.5 M NaCl and precipitated by centrifugation at 11,000 RCF for 40 minutes at 4°C. The preparation was mixed with chloroform, after which the aqueous phase was sterilized using a 0.2 μm filter and incubated with DNase and RNase (5 μg/mL each) for 30 minutes at room temperature. Final purity was achieved by filtration through a Zeba Spin desalting column (40K MWCO, Thermo Fisher Scientific). Phages were recovered and stored in SM buffer (100 mM NaCl, 8 mM MgSO4·7H2O, 50 mM Tris-Cl).
Phages labeled with an azido-handle were produced in a PAO1 metZ knockout strain, obtained from the University of Washington collection[24]. Phage amplification was performed by infecting the bacteria overnight in M9 minimal medium supplemented with methionine (5 mg/L) and the methionine homolog azido-homo-alanine (40 mg/L). After removal of bacterial remains, phages were pelleted using PEG-8000/NaCl as described above, filter-sterilized, and concentrated using an Amicon filter (100kDa cut-off). To achieve fluorescent labeling, phages were incubated with DBCO-AF488 (Jena Bioscience) for 1 hour at 37°C. Excess dye was washed off by washing with SM buffer in an Amicon filter (100kDa cut-off).
PAO1 expressing sf-Cherry were obtained by transforming PAO1 with the plasmid pUCP30T modified to express sfCherry[25]. Bacteria were made competent by washing in 300 mM sucrose, after which the plasmid was introduced by electroporation. Selection was performed on plates containing gentamycin (30 μg/mL). S. aureus strain ATCC 19,685 was kindly provided by Ana Rita Costa (Fagenbank; TU Delft).
2.2. Genome Sequencing and Bioinformatics Analysis
The genomic DNA library for phage PBJ was prepared with the Nextera Flex kit (Illumina, USA) and sequenced using the Illumina MiniSeq using a paired-end approach (2*150 bp). The reads were then controlled for quality using FastQC[26] and we used Trimmomatic[27] to remove adapter sequences, filter by length (>50 bp), and trim lower quality regions (Trimmomatic options: ILLUMINACLIP:NexteraPE-PE.fa:2:30:10LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50). The genomes were assembled de novo using the SPAdes assembler with default options, including --careful[28]. The quality of the assembly were inspected with Bandage[29] to allow us to extract the relevant circular phage contigs. The genome of phage PBJ was annotated using Pharokka[30], and compared with the closely related phage 14-1 genome (NC_011703.1).
2.3. Bacterial Growth
For all experiments, bacteria were cultured overnight in LB from a single colony at 37°C with shaking. On the day of the experiment, bacteria diluted 1:31 in LB and sub-cultured to mid-log phase (OD600nm ~ 0.5). Then bacteria were washed in RPMI buffer (RPMI 1640, ThermoFisher, supplemented with 0.05% human serum albumin, Sanquin), pelleted and resuspended to an OD600nm of 1.0 (~ 8 × 108 bacteria/mL) in RPMI buffer. Bacteria were further diluted as specified for the different assays.
2.4. Serum Preparation and Reagents
Human pooled serum (HPS) was prepared as previously described[31]. Briefly, blood was drawn from healthy volunteers, allowed to clot, and centrifuged to isolate the serum. Serum of 15–20 donors was pooled, aliquoted, and stored at −80 °C. Heat inactivated HPS (HI HPS) was prepared by treating HPS at 56 °C for 30 min to selectively inactivate complement proteins. Pooled IgG/IgM was isolated from HPS as described before[32]. Compstatin was kindly provided by John Lambris (University of Philadelphia). OMCI was produced and purified as previously described[33]. Complement-targeting nanobodies were produced as described by their developers[34,35,36,37]. Monoclonal mouse antibody bH6 recognizing human activated C3 (Hycult Biotech, The Netherlands) was produced as previously described[20].
2.5. Microplate Reader Assays
Bacteria (PAO1), phages and Sytox green Nucleic Acid stain (ThermoFisher) were mixed together to a final concentration of ~ 2 x 107 bacteria/mL bacteria and 5 μM Sytox green. Concentration of phages is dependent on the MOI, which is indicated for each experiment. Where indicated, human pooled serum, heat-inactivated human pooled serum, purified IgG/IgM from pooled serum or serum with complement inhibitors was added in the concentrations specified in each of the figures. For experiments with pre-opsonized bacteria, prior to addition of phages, bacteria were incubated with serum at 37°C for 30 min, centrifuged at 3500 rpm for 8 minutes, and resuspended in fresh medium. Fluorescence measurements were performed in a microplate reader (FLUOstar Omega, BMG Labtech) at 37°C, in a clear, flat-bottom 96-well plate (Corning), with the following settings: λexcitation = 490 nm, bandwidth = 14 nm; λemission = 537 nm, bandwidth 30 nm; gain = 1300.
2.6. Determination of Bacterial Viability
Bacteria (S. aureus strain ATCC 19685) were mixed with phage K to a concentration of 2 x 107 bacteria/mL and a phage concentration of 2 x 108 bacteria/mL (MOI of 10). The mixture was incubated at 37°C with shaking for 120 minutes. Samples were taken and serial dilutions were performed in RPMI buffer. 5 µL of each sample were plated on LB agar and incubated overnight at 37°C. Colonies were counted, and the cfu/mL was calculated.
2.7. Flow Cytometry
Bacteria (PAO1-sfCherry) at a final concentration of ~ 2 x 107 bacteria/mL were mixed with PBJ-AF488 in various MOIs and, in some cases, 10% HPS. Nanobodies (anti-C1q and anti-C3) were when indicated at 1 μM. Heat inactivated PBJ-AF488 (heat treatment: 85°C for 50 minutes) was used as a control at an MOI of 200. Samples were incubated for 5 minutes at 37°C with shaking, after which bacteria were washed in cold buffer and pelleted at 4000 rpm at 4°C for 8 minutes. The supernatant was removed, and bacteria were resuspended in a solution of bH6-AF405 antibody against C3b at 3 μg/mL in RPMI 1640 with 0.05% HAS. After incubation on ice for 10 minutes, samples were fixed using cold PFA to a final concentration of 1%. Samples were finally pelleted at 3500 rpm for 8 minutes, resuspended in fresh buffer to a concentration between 106 and 107 bacteria/mL and analyzed via flow cytometry (MACS Quant, Miltenyi Biotech). Per condition, 10 μl were measured. Bacteria were gated using sfCherry signal height.
2.8. Widefield Fluorescence Microscopy
Bacteria (PAO1-sfCherry) were grown to mid-log phase (OD600nm ~ 0.5) in LB, pelleted and resuspended to an OD600nm of 1.0 (~ 8 x 108 bacteria/mL) in RPMI 1640 with 0.05% HSA. Bacteria at a final concentration of 108 bacteria/mL were mixed with phages at an MOI of 50. HPS was added to some conditions in a concentration of 10%. The mixture was incubated for 5 minutes at 37°C with shaking. Then, samples were fixed using cold PFA to a final concentration of 1%. After a 15-minute incubation on ice, the samples were centrifuged at 3500 rpm for 8 minutes. The supernatant was removed, and the samples were resuspended in fresh RPMI to an estimated concentration of 4 × 108 bacteria/mL. Then, the samples were immobilized on microscope slides prepared with agarose pads. To make the agarose pads, 50 μL 1% agarose gel in PBS were added to an objective slide, after which a siliconized cover slip was applied on top of the agar gel to flatten it. Once the agarose was solid, the cover slips were removed from the agar and 10 μL of the sample were added on the pad. After the samples were dry, a cover slip was placed on the pad. The samples were imaged using a Leica TCS SP5 II microscope with an HC PL APO CS 100x/1.4 OIL objective (Leica Microsystems, The Netherlands). The images were obtained by overlaying the modes phase contrast and widefield fluorescence with cube filters for RFP (N21, dichroic mirror: 580 nm) and GFP (dichroic mirror: 500 nm) respectively.
2.9. Data Analysis and Statistical Testing
Data visualization and statistical analyses were performed in GraphPad Prism 9 and are further specified in the figure legends. Flow cytometry data was analyzed using FlowJo™ v10.8.1 Software. Widefield fluorescence microscopy images were processed using Fiji.
3. Results
3.1. Human Serum Impairs Activity of Myophages PBJ and 14-1
We studied the effect of human serum on the activity of phages against the serum-resistant P. aeruginosa strain PAO1. We evaluated four different virulent phages: myophages PBJ and 14-1 and podophages LKD16 and LUZ19. PBJ is a Pbunavirus that belongs to the Pbunavirus pv141 species with 96% sequence identity over 96% of the genome of phage 14-1. Phages 14-1, LKD16 and LUZ19 have been extensively characterized in literature[38,39,40,41]. The phages were combined in different multiplicities of infection (MOIs) with PAO1 in presence of different concentrations of human pooled serum (HPS). Phage-mediated killing was monitored using the fluorescent DNA dye assay as previously described[42]. Briefly, we used the membrane-impermeant DNA dye Sytox green, which can only stain bacterial DNA once the cells are damaged or lysed. This assay allows us to monitor phage-mediated damage in real time, where an increase in fluorescence correlates with bacterial killing. When comparing all four phages, we found that the addition of HPS inhibited damage induction for the myophages, resulting in a much slower increase of the fluorescence signal (Figure 1a,b). In contrast, no differences in activity were observed for the podophages in presence or absence of HPS. Based on these results, we used time of phage-mediated damage induction as a readout for phage activity to compare all conditions more easily. In this way, a shorter time indicates a more rapid or efficient phage infection. We confirmed that damage induction by phage PBJ was delayed with the addition of HPS. The effect is concentration-dependent, where a higher concentration of HPS was needed to inhibit a higher phage MOI (Figure 1c). Phage 14-1 presented a similar pattern, where the lower MOIs (0.1 and 1) showed clear inhibition by HPS (Figure 1d). However, this phage seemed slightly less sensitive to HPS than PBJ, as its activity was largely unaffected at the highest MOI. Strikingly, the activity of podophages LKD16 and LUZ19 did not change with the addition of HPS (Figure 1e,f). These observations indicate that serum can selectively inhibit certain phages in a concentration-dependent manner, while other phages are not sensitive to inhibition by serum.
3.2. Serum-Mediated Inhibition Is Found for a Broader Set of Myophages
To further determine whether the effect is specific to the phage morphotype, we evaluated the activity of 9 more myophages, 2 podophages and 1 siphophage in presence and absence of human serum. These were uncharacterized phages initially isolated from sewage water and classified according to tail morphology[43]. Phage activity was assessed using the fluorescent DNA dye assay. In absence of serum, we observed differences in the killing kinetics of the different phages, with latent periods ranging from around 30 to over 100 minutes (Figure S1). When serum was added, time of damage induction was greatly increased for 7 out of the 9 myophages (Figure 2a, Figure S1). In contrast, the activity of myophages Pa33 and Pa36, podophages Pa18 and Pa29 and siphophage Pa28 was not affected by the addition of serum. Of note, siphophage Pa28 and podophage Pa29 were not very active in absence of serum, which could be due to PAO1 not being the optimal host for this phage. These observations suggest that some myophages, if not all, could be more sensitive to serum-mediated inhibition. However, given that these phages and their receptors have not been characterized, it could be that the effect is not dependent on the morphotype, but rather due to the phages being closely related or targeting the same receptor. To rule this out, we tested whether serum can also inhibit phage K, a myophage targeting Staphylococcus aureus which uses wall teichoic acid (WTA) as a receptor. To do so, we incubated bacteria with the phage in presence or absence of serum and examined bacterial viability through colony counting. Indeed, no reduction in colony-forming units (CFU) was detected when phages were added in presence of serum (Figure 2b). As phage K targets a receptor absent on P. aeruginosa, it seems that inhibition by serum is not dependent on the phage receptor. These results further suggest that myophages are more sensitive to inhibition by human serum.
3.3. Phage Inhibition in Serum Is Mediated by the Early Stages of the Complement Cascade
Next, we investigated the components in serum responsible for the inhibitory effect. We first assessed whether this effect is due to complement or exclusively mediated by neutralizing antibodies. For this, we combined phages with HPS, HPS with heat-inactivated complement (HI HPS), or IgG and IgM purified from HPS, and studied killing kinetics with the fluorescent DNA dye assay (Figure 3a). Phages performed equally well in presence of antibodies and heat inactivated serum as they do in the control condition. The lack of an effect of purified antibodies and heat inactivated serum on phage activity suggests that the complement system drives phage inhibition. In all complement activation pathways, cleavage of C3 and subsequent deposition of the cleavage product C3b is necessary for the downstream conversion of C5. To further look into this, we examined phage activity in presence of serum with the addition of specific complement inhibitors: compstatin, which inhibits C3 cleavage[44,45], and OmCI, which inhibits C5 cleavage[46] (Figure 3b). We observed that compstatin partially rescued phage activity in presence of serum, while OmCI did not prevent inhibition by serum at all. Therefore, our results point to the involvement of the early stages of the complement system in the inhibitory effect of serum on phage PBJ. Of note, there was no difference in activity of phages in presence of heat inactivated serum or heat inactivated serum with complement inhibitors (Figure S2). However, as addition of compstatin only partially recovered phage activity we tested an additional panel of complement inhibitors, in this case nanobodies. Nanobodies are single-domain antibody fragments that bind antigens, with the advantage of having a much smaller size than an antibody[47]. We used nanobody C1qNb75, which inhibits activation of the complement classical pathway by binding C1q, the first step of the cascade[34]. Next to this, we used nanobodies hC3Nb1 and hC3Nb2, which inhibit cleavage of C3 in respectively the alternative pathway or all activation pathways[35,36]. Finally, we used sdAb_E4, a C5 binder, to inhibit the terminal stage of the complement cascade[37]. We assessed phage activity in serum in presence of the nanobodies in a range of concentrations. To compare their effects, we analyzed influx of fluorescent DNA dye after 90 minutes of infection (Figure 3c,d). The C1q-targeting nanobody was able to recover phage activity in a concentration-dependent manner to the level of the control. Both of the C3-targeting nanobodies exhibited an intermediate effect, comparable to that of compstatin. As expected, the C5-targeting nanobody barely improved phage activity even at the highest concentration, which is in line with our observations with OmCI. Taken together, our results indicate that phage inhibition is likely caused by complement and more specifically by activation of the classical pathway.
3.4. Inhibition of Phages by Complement Occurs at the Stage of Phage Adsorption
One of the stages of phage infection which the complement system could be hampering is binding of phages to their host. Phage adsorption can be measured with assays where bacteria are incubated with phages and the remaining free plaque forming units are quantified. However, in such an assay both phage adsorption and phage inactivation would translate to a decrease in free plaque forming units. To circumvent this, we assessed phage adsorption by using fluorescently tagged phages.
Producing fluorescent phages poses a challenge. Random labeling strategies may compromise the activity of the phage as the dye has to react with the capsid and tail proteins[48]. We circumvented this by producing phages tagged with a chemical handle that can be functionalized through click chemistry[49]. To do so, we used a methionine-auxotrophic strain of P. aeruginosa, which requires exogenous methionine to grow. Methionine can also be partially substituted by its structural homologue L-azidohomoalanine, a non-canonical amino acid that contains an azide group. In a situation of methionine scarcity, bacteria will incorporate L-azidohomoalanine into their proteins. If phages are propagated with these bacteria as hosts, their proteins will also be azide-tagged (Figure 4a)[50]. The resulting virions are then available for biorthogonal labeling via click chemistry, as the azide groups can react efficiently with alkyne groups attached to a fluorophore to create a stable covalent bond. We amplified PBJ in this way and used the molecule DBCO functionalized with an Alexa-488 dye to fluorescently label our azido-tagged phages, obtaining PBJ-AF488.
We confirmed the labeling of PBJ-AF488 by means of widefield fluorescence microscopy. To do so, we incubated fluorescently labeled phage with PAO1 expressing cytoplasmic sfCherry (PAO1-sfCherry) to allow the phages to bind. We successfully detected bacteria with bound phages on their surface, as well as isolated phage clusters (Figure 4b). Addition of serum in this setting resulted in a decrease in the number of bacteria with attached phage particles (Figure 4c). Next, we used flow cytometry to confirm whether complement indeed hampers phage binding with a more quantitative approach. In this assay, we again used PAO1-sfCherry to facilitate gating (Figure S3). PBJ-AF488 or heat-inactivated PBJ-AF488 was incubated with bacteria for 5 minutes to allow for phage adsorption in presence of serum or serum with inhibitors. Analysis of phage binding in these conditions revealed that phage fluorescent signal increases when increasing the MOI in absence of serum (Figure 4d). Although a relatively high MOI was necessary to obtain a sufficiently high phage binding signal, it seems that phage binding was not yet saturated even at the highest MOI. Heat-inactivated phage did not bind to bacteria, supporting that the signal was specific for binding of active phages. When serum was added, phage binding showed a sharp decrease (Figure 4d,e). As seen before in the phage lysis assays, the inhibitory effect became less pronounced as the MOI increased. In accordance with our previous results, we could recover phage binding almost fully by inhibiting serum with the C1q-targeting nanobody (C1qNb75), while the nanobody targeting all forms of C3 (hC3Nb2) only partially recovered phage binding. In all of the conditions analyzed, the shift in phage fluorescent signal occurred homogeneously, meaning that phage binding was evenly distributed throughout the bacterial population (Figure 4e). Next to phage binding, we also stained the samples for bound C3b to confirm that there was complement activation on the bacterial surface. Very high levels of C3b were detected on bacteria incubated with active serum (Figure 4f,g). Interestingly, both nanobodies could completely inhibit C3b deposition in the concentrations tested. In the conditions treated with the C3 nanobody, phage biding was still inhibited even in absence of C3b deposition. In conclusion, these results show that active complement, and in particular C1q, hinders the ability of phages to bind to their host.
3.5. Complement Inhibitory Effect Is Not Mediated by Receptor Blockage
Our results from phage binding assays performed with fluorescent phages reveal that complement blocks phage binding to bacteria. This could be due to complement components shielding receptors on the bacterial surface. However, it could also be caused by a direct interaction of complement with the viral particles. To discriminate which of these processes is hampering phage binding, we first studied the infectivity of phages on bacteria pre-opsonized with HPS. Opsonization was achieved by incubating bacteria with HPS for 30 minutes at 37oC and washing away the unbound serum components. We confirmed that bacteria treated in this way were indeed covered with complement components C3b (Figure 5a). We then used the fluorescent DNA dye assay to assess whether phages induce lysis of pre-opsonized bacteria (Figure 5b). When phages were added to the bacteria together with fresh buffer (RPMI), we could still observe an increase in fluorescence signal. In the case where phages were added together with fresh serum phage infection was still inhibited, consistent with our previous findings. Our results indicate that phage inhibition is not mediated by complement blocking phage adsorption to receptors on the bacterial surface.
4. Discussion
The past decade has seen an unprecedented increase in interest on bacteriophage therapy. New efforts are being made to investigate its clinical potential, while physicians strive to work around the stringent regulations attached to its use. At the same time, numerous case reports have been published in the past few years highlighting the potential of phages as therapeutics, both to substitute and complement antibiotics. In this context, it is important to keep advancing our fundamental knowledge on how phages may behave inside the body of a patient.
The purpose of our study was to explore the interactions of phages with the human immune system, and in particular with the complement system. To tackle this issue, we used human serum as a source of complement and characterized the performance of phages in this medium using different assays. Our results revealed that some phages are inhibited by serum in a concentration-dependent way, while others remain unaffected. When looking at a broader panel of phages, it was only the myophages that were challenged by this phenomenon. However, due to the limited number of podo- and siphophages available for us to test, it is difficult to fully relate the observation to the phage morphotype. Furthermore, we do not know the receptors of all the phages we tested, what their optimal host is or how closely related they are to each other. Nonetheless, when we tested the activity of S. aureus myophage K, we saw that it was also inhibited by serum, further hinting at the importance of the morphotype. In any case, examination of a more complete and well characterized set of phages, including more phages targeting other bacterial species, would be valuable in finding patterns to potentially predict sensitivity to serum.
We were also able to show that phage inhibition is mediated by the early stages of the classical complement pathway, as infective activity could be rescued by blocking C1q or C3. The action of the classical pathway, initiated by the deposition of C1q, can kickstart complement activation, while the alternative pathway can later on contribute to mounting a bigger reaction. This is reflected by our results with the nanobodies targeting C1q and C3. While both C3-targeting nanobodies could partially counteract the inhibitory effect of serum, it was only when C1q was also targeted that we observed a full recovery of the phage infection.
In this study, we also demonstrate a novel technique for producing fluorescently labeled phages in P. aeruginosa, based on incorporation of the non-natural amino acid L-azidohomolalanine and biorthogonal labeling by means of click chemistry. This method is more labor-intensive than random labeling approaches, but we found that it better preserved the activity of the phages. Furthermore, azido-chains incorporated into the phage capsid could potentially be functionalized with other molecules for a variety of applications. Using this technique, we were able to produce fluorescently labeled PBJ in high titers and study its binding to PAO1 through microscopy and flow cytometry. A high MOI of phage was needed in these experiments to produce a detectable fluorescent signal. This constitutes a limitation, as the experimental conditions were not directly comparable to those of the other assays performed in this study. Nonetheless, the experiments described in section 4 revealed that complement hampers phages by interfering with their ability to bind to the surface of the host. An intuitive explanation for this could be that C1q or C3b could shield phage receptors. However, when we assessed phage activity on bacteria opsonized with complement components using the fluorescent DNA dye assay, we observed that phages were still able to induce lysis. We hypothesize that complement activation may have a direct neutralizing effect on phages. The ability of complement to recognize and opsonize viral particles has been shown in studies with eukaryotic viruses[51], and the same could hold true for phages. Attachment of heavy proteins such as C1q or C3b to the viral particles could interfere with their infectivity. This could be particularly relevant for myophages, as they rely on the contraction of their tail to introduce their genetic material into the host[52]. Addressing these questions would be an interesting perspective for future studies.
Although we found that phage inhibition was not mediated by neutralizing antibodies in our assays, specific anti-phage antibodies in our serum pool could also be important in activating the classical complement pathway. Antibodies against head proteins of Escherichia coli (E. coli) myophage T4 have been found in individuals who have never been subjected to phage therapy[53]. The same proteins could induce production of neutralizing antibodies and complement activation when used to immunize mice. Similarly, in a study performed on an E. coli podophage T7 peptide display library, recognition of certain peptides by IgM was seen to initiate a neutralizing complement response[54]. Nonetheless, there is also evidence that activation of the classical pathway on viruses can occur independently of antibodies, for example through the interaction of C1q with serum C reactive protein[55]. Additionally, direct binding of C1q to viruses such as dengue, influenza A and SARS-Cov2 seems to be sufficient in some cases to reduce infectivity[56,57,58]. Inactivation of phages by complement components independent of antibodies could explain the observations reported in several studies where phage activity was compromised in presence of human serum or plasma[59,60,61].
Taken together, our results provide evidence that certain phages may be less competent than others in targeting bacteria in certain physiological situations, like in blood or tissues where complement is present. Although it remains unclear how these results would translate to an in vivo situation, our observations could still have implications on how we select phages for therapy. Researchers are working on ways to optimize this process and make it faster and more efficient. To this effect, phage biobanks are currently being set up in different countries, and sharing phages between different institutions is gradually becoming a more usual practice. Several phages against P. aeruginosa have been characterized and are being used as medicinal products in the context of magistral phage preparations[62,63]. Efforts are also being made in engineering phages to increase their host range, as well as in optimizing phage cocktails to avoid the problem of phage resistance. Additionally, bioinformatic pipelines have been developed that may allow to predict which phages will be more effective against a certain clinical isolate[64]. However, in spite of all these important advances, interactions of phages with the patient’s innate immune system are still largely ignored in the process of therapeutic phage selection. In light of our findings, we suggest that performing screenings in presence of serum would be valuable when designing phage cocktails.
In conclusion, this study shows that complement activation via the classical pathway can inhibit anti-bacterial activity of certain phages, with myophages being more susceptible to this effect. We show that inhibition is not mediated by deposition on the bacterial surface but is rather caused by a direct effect on phages themselves. Our findings highlight the need to better understand how phages behave in physiological conditions when administered to a patient as a therapeutic. Selection of phages according to their expected performance in human blood or tissues could contribute to improving the efficacy of phage therapy.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Human serum inhibits a variety of myophages targeting PAO1; Figure S2: Inhibition of phages by serum is mediated by the complement system; Figure S3. Example of gating strategy of PAO1-sfCherry for flow cytometry experiments.
Author Contributions
Conceptualization: J.E.E., S.H.M.R., B.W.B. and P.J.H.; methodology: J.E.E. and S.O.D.; validation: J.E.E.; formal analysis: J.E.E., C.T.B. and C.L.; investigation: J.E.E., S.O.D. and C.T.B.; writing—original draft preparation: J.E.E.; writing—review and editing: J.E.E., C.L., B.W.B. and P.J.H.; visualization: J.E.E.; supervision: S.H.M.R., B.W.B. and P.J.H.; project administration: S.H.M.R.; funding acquisition: S.H.M.R. and P.J.H. All authors have read and agreed to the published version of the manuscript.
Funding
C.L. is supported by a junior postdoctoral fellowship from the FWO (12D8623N).
Informed Consent Statement
Human blood from healthy volunteers was collected with informed consent in accordance with the Declaration of Helsinki. Approval was obtained from the medical ethics committee of the UMC Utrecht, The Netherlands.
Data Availability Statement
All the datasets generated and analyzed during this study are presented in the main text or the Supplementary Materials. Genomic sequence of phage PBJ is available from Genbank, accession number: OR611941.
Acknowledgments
The present work was part of the research program of the Netherlands Centre for One Health (www.ncoh.nl). The authors would like to thank Rob Lavigne and Ana Rita Costa for supplying phage stocks and host strains. We thank Sayali Gorivale (KU Leuven) for her help in sequencing the genome of phage PBJ. We thank John Lambris for providing compstatin. We thank Carla Gosselaar-de Haas (UMCU) for producing the complement-targeting nanobodies, Loutje van der Sman (UMCU) for producing PBJ-AF488, and Dani Heesterbeek (UMCU) for producing the AF405-tagged antibody bH6. We thank Jos van Strijp (UMCU) for the critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin Microbiol Rev 2019, 32, e00066-18. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, S.A.; Hatfull, G.F.; Mutalik, V.K.; Schooley, R.T. Phage Therapy: From Biological Mechanisms to Future Directions. Cell 2023, 186, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annual Review of Medicine 2022, 73, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Vilas Boas, D.; Sillankorva, S.; Azeredo, J. Phage Therapy: A Step Forward in the Treatment of Pseudomonas Aeruginosa Infections. Journal of Virology 2015, 89, 7449–7456. [Google Scholar] [CrossRef] [PubMed]
- Ferry, T.; Kolenda, C.; Laurent, F.; Leboucher, G.; Merabischvilli, M.; Djebara, S.; Gustave, C.-A.; Perpoint, T.; Barrey, C.; Pirnay, J.-P.; et al. Personalized Bacteriophage Therapy to Treat Pandrug-Resistant Spinal Pseudomonas Aeruginosa Infection. Nat Commun 2022, 13, 4239. [Google Scholar] [CrossRef]
- Simner, P.J.; Cherian, J.; Suh, G.A.; Bergman, Y.; Beisken, S.; Fackler, J.; Lee, M.; Hopkins, R.J.; Tamma, P.D. Combination of Phage Therapy and Cefiderocol to Successfully Treat Pseudomonas Aeruginosa Cranial Osteomyelitis. JAC-Antimicrobial Resistance 2022, 4, dlac046. [Google Scholar] [CrossRef] [PubMed]
- Cesta, N.; Pini, M.; Mulas, T.; Materazzi, A.; Ippolito, E.; Wagemans, J.; Kutateladze, M.; Fontana, C.; Sarmati, L.; Tavanti, A.; et al. Application of Phage Therapy in a Case of a Chronic Hip-Prosthetic Joint Infection Due to Pseudomonas Aeruginosa: An Italian Real-Life Experience and In Vitro Analysis. Open Forum Infectious Diseases 2023, 10, ofad051. [Google Scholar] [CrossRef]
- Van Nieuwenhuyse, B.; Van der Linden, D.; Chatzis, O.; Lood, C.; Wagemans, J.; Lavigne, R.; Schroven, K.; Paeshuyse, J.; de Magnée, C.; Sokal, E.; et al. Bacteriophage-Antibiotic Combination Therapy against Extensively Drug-Resistant Pseudomonas Aeruginosa Infection to Allow Liver Transplantation in a Toddler. Nat Commun 2022, 13, 5725. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Floch, R.L.; et al. Efficacy and Tolerability of a Cocktail of Bacteriophages to Treat Burn Wounds Infected by Pseudomonas Aeruginosa (PhagoBurn): A Randomised, Controlled, Double-Blind Phase 1/2 Trial. The Lancet Infectious Diseases 2019, 19, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Uyttebroek, S.; Chen, B.; Onsea, J.; Ruythooren, F.; Debaveye, Y.; Devolder, D.; Spriet, I.; Depypere, M.; Wagemans, J.; Lavigne, R.; et al. Safety and Efficacy of Phage Therapy in Difficult-to-Treat Infections: A Systematic Review. The Lancet Infectious Diseases 2022, 22, e208–e220. [Google Scholar] [CrossRef]
- Stacey, H.J.; De Soir, S.; Jones, J.D. The Safety and Efficacy of Phage Therapy: A Systematic Review of Clinical and Safety Trials. Antibiotics 2022, 11, 1340. [Google Scholar] [CrossRef] [PubMed]
- Pirnay, J.-P.; Djebara, S.; Steurs, G.; Griselain, J.; Cochez, C.; Soir, S.D.; Glonti, T.; Spiessens, A.; Berghe, E.V.; Green, S.; et al. Retrospective, Observational Analysis of the First One Hundred Consecutive Cases of Personalized Bacteriophage Therapy of Difficult-to-Treat Infections Facilitated by a Belgian Consortium 2023, 2023.08.28.23294728.
- Egido, J.E.; Costa, A.R.; Aparicio-Maldonado, C.; Haas, P.-J.; Brouns, S.J.J. Mechanisms and Clinical Importance of Bacteriophage Resistance. FEMS Microbiol Rev 2022, 46, fuab048. [Google Scholar] [CrossRef] [PubMed]
- Markwitz, P.; Lood, C.; Olszak, T.; van Noort, V.; Lavigne, R.; Drulis-Kawa, Z. Genome-Driven Elucidation of Phage-Host Interplay and Impact of Phage Resistance Evolution on Bacterial Fitness. ISME J 2022, 16, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Bustamante, C.A.; Dedrick, R.M.; Garlena, R.A.; Russell, D.A.; Hatfull, G.F. Toward a Phage Cocktail for Tuberculosis: Susceptibility and Tuberculocidal Action of Mycobacteriophages against Diverse Mycobacterium Tuberculosis Strains. mBio 2021, 12, e00973-21. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Haas, P.-J.; van Noort, V.; Lavigne, R. Shopping for Phages? Unpacking Design Rules for Therapeutic Phage Cocktails. Current Opinion in Virology 2022, 52, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Łusiak-Szelachowska, M.; Żaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Kłak, M.; Fortuna, W.; Letkiewicz, S.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; et al. Phage Neutralization by Sera of Patients Receiving Phage Therapy. Viral Immunol 2014, 27, 295–304. [Google Scholar] [CrossRef]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapała, J.; Drab, M.; Jończyk-Matysiak, E.; Lecion, D.; Kaźmierczak, Z.; Beta, W.; Majewska, J.; Harhala, M.; et al. Mammalian Host-Versus-Phage Immune Response Determines Phage Fate in Vivo. Sci Rep 2015, 5, 14802. [Google Scholar] [CrossRef]
- Heesterbeek, D.A.C.; Angelier, M.L.; Harrison, R.A.; Rooijakkers, S.H.M. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J Innate Immun 2018, 10, 455–464. [Google Scholar] [CrossRef]
- Heesterbeek, D.A.; Bardoel, B.W.; Parsons, E.S.; Bennett, I.; Ruyken, M.; Doorduijn, D.J.; Gorham, R.D.; Berends, E.T.; Pyne, A.L.; Hoogenboom, B.W.; et al. Bacterial Killing by Complement Requires Membrane Attack Complex Formation via Surface-Bound C5 Convertases. EMBO J 2019, 38, e99852. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, A.M.; Elgaml, A.; Ali, Y.M. Bacteriophage Therapy Increases Complement-Mediated Lysis of Bacteria and Enhances Bacterial Clearance After Acute Lung Infection With Multidrug-Resistant Pseudomonas Aeruginosa. J Infect Dis 2019, 219, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Ostrycharz, E.; Hukowska-Szematowicz, B. New Insights into the Role of the Complement System in Human Viral Diseases. Biomolecules 2022, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Hodyra-Stefaniak, K.; Lahutta, K.; Majewska, J.; Kaźmierczak, Z.; Lecion, D.; Harhala, M.; Kęska, W.; Owczarek, B.; Jończyk-Matysiak, E.; Kłopot, A.; et al. Bacteriophages Engineered to Display Foreign Peptides May Become Short-circulating Phages. Microb Biotechnol 2019, 12, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.A.; Alwood, A.; Thaipisuttikul, I.; Spencer, D.; Haugen, E.; Ernst, S.; Will, O.; Kaul, R.; Raymond, C.; Levy, R.; et al. Comprehensive Transposon Mutant Library of Pseudomonas Aeruginosa. Proceedings of the National Academy of Sciences 2003, 100, 14339–14344. [Google Scholar] [CrossRef]
- Barbier, M.; Damron, F.H. Rainbow Vectors for Broad-Range Bacterial Fluorescence Labeling. PLOS ONE 2016, 11, e0146827. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Current Protocols in Bioinformatics 2020, 70, e102. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive Visualization of de Novo Genome Assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Bouras, G.; Nepal, R.; Houtak, G.; Psaltis, A.J.; Wormald, P.-J.; Vreugde, S. Pharokka: A Fast Scalable Bacteriophage Annotation Tool. Bioinformatics 2023, 39, btac776. [Google Scholar] [CrossRef]
- Berends, E.T.M.; Dekkers, J.F.; Nijland, R.; Kuipers, A.; Soppe, J.A.; van Strijp, J.A.G.; Rooijakkers, S.H.M. Distinct Localization of the Complement C5b-9 Complex on Gram-Positive Bacteria. Cell Microbiol 2013, 15, 1955–1968. [Google Scholar] [CrossRef]
- Zwarthoff, S.A.; Magnoni, S.; Aerts, P.C.; van Kessel, K.P.M.; Rooijakkers, S.H.M. Method for Depletion of IgG and IgM from Human Serum as Naive Complement Source. In The Complement System: Innovative Diagnostic and Research Protocols; Roumenina, L.T., Ed.; Methods in Molecular Biology; Springer US: New York, NY, 2021; pp. 21–32. ISBN 978-1-07-161016-9. [Google Scholar]
- Nunn, M.A.; Sharma, A.; Paesen, G.C.; Adamson, S.; Lissina, O.; Willis, A.C.; Nuttall, P.A. Complement Inhibitor of C5 Activation from the Soft Tick Ornithodoros Moubata. J Immunol 2005, 174, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Laursen, N.S.; Pedersen, D.V.; Gytz, H.; Zarantonello, A.; Bernth Jensen, J.M.; Hansen, A.G.; Thiel, S.; Andersen, G.R. Functional and Structural Characterization of a Potent C1q Inhibitor Targeting the Classical Pathway of the Complement System. Frontiers in Immunology 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.K.; Pihl, R.; Gadeberg, T.A.F.; Jensen, J.K.; Andersen, K.R.; Thiel, S.; Laursen, N.S.; Andersen, G.R. A Potent Complement Factor C3–Specific Nanobody Inhibiting Multiple Functions in the Alternative Pathway of Human and Murine Complement. J Biol Chem 2018, 293, 6269–6281. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.; Jensen, R.K.; Hansen, A.G.; Gadeberg, T.A.F.; Thiel, S.; Laursen, N.S.; Andersen, G.R. A C3-Specific Nanobody That Blocks All Three Activation Pathways in the Human and Murine Complement System. J Biol Chem 2020, 295, 8746–8758. [Google Scholar] [CrossRef] [PubMed]
- Yatime, L.; Merle, N.S.; Hansen, A.G.; Friis, N.A.; Østergaard, J.A.; Bjerre, M.; Roumenina, L.T.; Thiel, S.; Kristensen, P.; Andersen, G.R. A Single-Domain Antibody Targeting Complement Component C5 Acts as a Selective Inhibitor of the Terminal Pathway of the Complement System and Thus Functionally Mimicks the C-Terminal Domain of the Staphylococcus Aureus SSL7 Protein. Frontiers in Immunology 2018, 9. [Google Scholar] [CrossRef]
- Ceyssens, P.-J.; Miroshnikov, K.; Mattheus, W.; Krylov, V.; Robben, J.; Noben, J.-P.; Vanderschraeghe, S.; Sykilinda, N.; Kropinski, A.M.; Volckaert, G.; et al. Comparative Analysis of the Widespread and Conserved PB1-like Viruses Infecting Pseudomonas Aeruginosa. Environmental Microbiology 2009, 11, 2874–2883. [Google Scholar] [CrossRef]
- Watkins, S.C.; Sible, E.; Putonti, C. Pseudomonas PB1-Like Phages: Whole Genomes from Metagenomes Offer Insight into an Abundant Group of Bacteriophages. Viruses 2018, 10, 331. [Google Scholar] [CrossRef]
- Ceyssens, P.-J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic Analysis of Pseudomonas Aeruginosa Phages LKD16 and LKA1: Establishment of the phiKMV Subgroup within the T7 Supergroup. J Bacteriol 2006, 188, 6924–6931. [Google Scholar] [CrossRef]
- Lavigne, R.; Lecoutere, E.; Wagemans, J.; Cenens, W.; Aertsen, A.; Schoofs, L.; Landuyt, B.; Paeshuyse, J.; Scheer, M.; Schobert, M.; et al. A Multifaceted Study of Pseudomonas Aeruginosa Shutdown by Virulent Podovirus LUZ19. mBio 2013, 4, e00061-00013. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.E.; Toner-Bartelds, C.; Costa, A.R.; Brouns, S.J.J.; Rooijakkers, S.H.M.; Bardoel, B.W.; Haas, P.-J. Monitoring Phage-Induced Lysis of Gram-Negatives in Real Time Using a Fluorescent DNA Dye. Sci Rep 2023, 13, 856. [Google Scholar] [CrossRef]
- Costa, A.R.; van den Berg, D.F.; Esser, J.Q.; Muralidharan, A.; van den Bossche, H.; Bonilla, B.E.; van der Steen, B.A.; Haagsma, A.C.; Nobrega, F.L.; Haas, P.-J.; et al. Accumulation of Defense Systems Drives Panphage Resistance in Pseudomonas Aeruginosa. Microbiology 2022. [Google Scholar]
- Ricklin, D.; Lambris, J.D. Compstatin: A Complement Inhibitor on Its Way to Clinical Application. Adv Exp Med Biol 2008, 632, 273–292. [Google Scholar] [PubMed]
- Lamers, C.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Compstatins: The Dawn of Clinical C3-Targeted Complement Inhibition. Trends in Pharmacological Sciences 2022, 43, 629–640. [Google Scholar] [CrossRef]
- Nunn, M.A.; Sharma, A.; Paesen, G.C.; Adamson, S.; Lissina, O.; Willis, A.C.; Nuttall, P.A. Complement Inhibitor of C5 Activation from the Soft Tick Ornithodoros Moubata. The Journal of Immunology 2005, 174, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.-K.; Odongo, S.; Radwanska, M.; Magez, S. Nanobodies: A Review of Generation, Diagnostics and Therapeutics. Int J Mol Sci 2023, 24, 5994. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, D.R.; Jiang, H.; Kalin, J.H.; Chen, Z.; Cole, P.A. Site-Specific Protein Labeling with NHS-Esters and the Analysis of Ubiquitin Ligase Mechanisms. J Am Chem Soc 2018, 140, 9374–9378. [Google Scholar] [CrossRef]
- Bird, R.E.; Lemmel, S.A.; Yu, X.; Zhou, Q.A. Bioorthogonal Chemistry and Its Applications. Bioconjugate Chem. 2021, 32, 2457–2479. [Google Scholar] [CrossRef]
- Strable, E.; Prasuhn, D.E.Jr.; Udit, A.K.; Brown, S.; Link, A.J.; Ngo, J.T.; Lander, G.; Quispe, J.; Potter, C.S.; Carragher, B.; et al. Unnatural Amino Acid Incorporation into Virus-Like Particles. Bioconjugate Chem. 2008, 19, 866–875. [Google Scholar] [CrossRef]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement Evasion Strategies of Viruses: An Overview. Front Microbiol 2017, 8, 1117. [Google Scholar] [CrossRef]
- Maghsoodi, A.; Chatterjee, A.; Andricioaei, I.; Perkins, N.C. How the Phage T4 Injection Machinery Works Including Energetics, Forces, and Dynamic Pathway. Proceedings of the National Academy of Sciences 2019, 116, 25097–25105. [Google Scholar] [CrossRef]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kaźmierczak, Z.; Letarov, A.; Górski, A. Immunogenicity Studies of Proteins Forming the T4 Phage Head Surface. J Virol 2014, 88, 12551–12557. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, A.V.; Zhang, G.; Sebestyén, M.G.; Wolff, J.A.; Bock, I. The Interactions of Peptides with the Innate Immune System Studied with Use of T7 Phage Peptide Display. Molecular Therapy 2000, 2, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Bíró, A.; Rovó, Z.; Papp, D.; Cervenak, L.; Varga, L.; Füst, G.; Thielens, N.M.; Arlaud, G.J.; Prohászka, Z. Studies on the Interactions between C-Reactive Protein and Complement Proteins. Immunology 2007, 121, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Douradinha, B.; McBurney, S.P.; de Melo, K.M.S.; Smith, A.P.; Krishna, N.K.; Barratt-Boyes, S.M.; Evans, J.D.; Nascimento, E.J.M.; Marques, E.T.A. C1q Binding to Dengue Virus Inhibits Infection of THP-1 and Cellular Inflammatory Responses. Virus Res 2014, 179, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Varghese, P.M.; Kishore, U.; Rajkumari, R. Human C1q Regulates Influenza A Virus Infection and Inflammatory Response via Its Globular Domain. International Journal of Molecular Sciences 2022, 23, 3045. [Google Scholar] [CrossRef]
- Beirag, N.; Varghese, P.M.; Neto, M.M.; Al Aiyan, A.; Khan, H.A.; Qablan, M.; Shamji, M.H.; Sim, R.B.; Temperton, N.; Kishore, U. Complement Activation-Independent Attenuation of SARS-CoV-2 Infection by C1q and C4b-Binding Protein. Viruses 2023, 15, 1269. [Google Scholar] [CrossRef]
- Gu Liu, C.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.C.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-Antibiotic Synergy Is Driven by a Unique Combination of Antibacterial Mechanism of Action and Stoichiometry. mBio 2020, 11. [Google Scholar] [CrossRef]
- Shinde, P.; Stamatos, N.; Doub, J.B. Human Plasma Significantly Reduces Bacteriophage Infectivity Against Staphylococcus Aureus Clinical Isolates. Cureus 14, e23777. [CrossRef]
- Molendijk, M.M.; Phan, M.V.T.; Bode, L.G.M.; Strepis, N.; Prasad, D.K.; Worp, N.; Nieuwenhuijse, D.F.; Schapendonk, C.M.E.; Boekema, B.K.H.L.; Verbon, A.; et al. Microcalorimetry: A Novel Application to Measure In Vitro Phage Susceptibility of Staphylococcus Aureus in Human Serum. Viruses 2023, 15, 14. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.-P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-Controlled Small-Scale Production of a Well-Defined Bacteriophage Cocktail for Use in Human Clinical Trials. PLoS One 2009, 4, e4944. [Google Scholar] [CrossRef]
- Duyvejonck, H.; Merabishvili, M.; Vaneechoutte, M.; de Soir, S.; Wright, R.; Friman, V.-P.; Verbeken, G.; De Vos, D.; Pirnay, J.-P.; Van Mechelen, E.; et al. Evaluation of the Stability of Bacteriophages in Different Solutions Suitable for the Production of Magistral Preparations in Belgium. Viruses 2021, 13, 865. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Boeckaerts, D.; Stock, M.; De Baets, B.; Lavigne, R.; van Noort, V.; Briers, Y. Digital Phagograms: Predicting Phage Infectivity through a Multilayer Machine Learning Approach. Current Opinion in Virology 2022, 52, 174–181. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Human serum inhibits lytic activity of myophages. P. aeruginosa strain PAO1 was incubated with phages PBJ, 14-1, LKD16 or LUZ19 at 37oC in presence of the DNA dye Sytox green. Phage-mediated damage is signaled by an increase in fluorescence intensity of Sytox green. a) Fluorescence intensity (relative fluorescence units, RFU) over time of PAO1 infected with different phages at an MOI of 1 in absence of serum. Uninfected bacteria were used as control. b) Fluorescence intensity (RFU) over time of PAO1 infected with different phages at an MOI of 1 in presence of 10% human pooled serum (HPS). Control are uninfected bacteria incubated with 10% HPS. c-f) Time of damage induction (min) by phage c) PBJ, d) 14-1, e) LDK16 and f) LUZ19, at a range of MOIs (0.1, 1, 10) combined with HPS in different concentrations (0 – 30%). Time of damage induction is determined as the time after addition of phages at which the fluorescence curve experiences a sharp and steady increase (1000 RFU over the previous measurement). a-f) Data represent mean ± SD of three independent experiments.
Figure 1.
Human serum inhibits lytic activity of myophages. P. aeruginosa strain PAO1 was incubated with phages PBJ, 14-1, LKD16 or LUZ19 at 37oC in presence of the DNA dye Sytox green. Phage-mediated damage is signaled by an increase in fluorescence intensity of Sytox green. a) Fluorescence intensity (relative fluorescence units, RFU) over time of PAO1 infected with different phages at an MOI of 1 in absence of serum. Uninfected bacteria were used as control. b) Fluorescence intensity (RFU) over time of PAO1 infected with different phages at an MOI of 1 in presence of 10% human pooled serum (HPS). Control are uninfected bacteria incubated with 10% HPS. c-f) Time of damage induction (min) by phage c) PBJ, d) 14-1, e) LDK16 and f) LUZ19, at a range of MOIs (0.1, 1, 10) combined with HPS in different concentrations (0 – 30%). Time of damage induction is determined as the time after addition of phages at which the fluorescence curve experiences a sharp and steady increase (1000 RFU over the previous measurement). a-f) Data represent mean ± SD of three independent experiments.
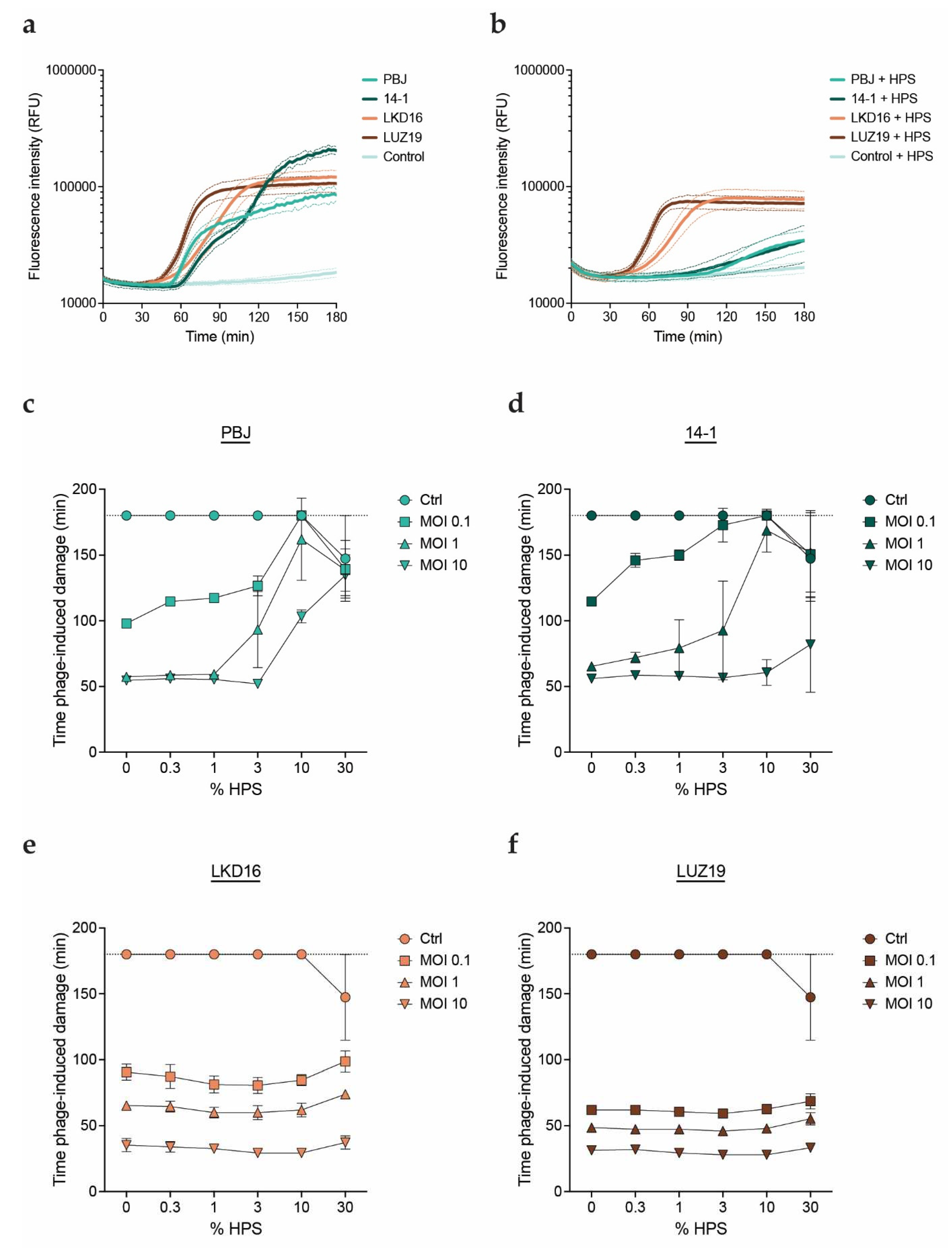
Figure 2.
Human serum inhibits a variety of myophages targeting PAO1. a) Time of damage induction (min) of the different phages as determined by the fluorescent DNA dye assay. Time of damage induction is determined here as the time after addition of phages at which the fluorescence curve experiences a sharp and steady increase (700 RFU over the previous measurement). Phages were added at an MOI of 1 and incubated with PAO1 at 37oC in absence of serum (control) or in presence of 10% HPS. b) S. aureus strain ATCC 19,685 was incubated with phage K at an MOI of 10 in absence (control) or presence of 10% HPS for 2 hours at 37oC, after which bacteria were plated and incubated overnight. Number of recovered colonies per plated volume is expressed as colony forming units per mL (CFU/mL). Black dotted line represents detection limit. a, b) Data represent mean ± SD of three independent experiments.
Figure 2.
Human serum inhibits a variety of myophages targeting PAO1. a) Time of damage induction (min) of the different phages as determined by the fluorescent DNA dye assay. Time of damage induction is determined here as the time after addition of phages at which the fluorescence curve experiences a sharp and steady increase (700 RFU over the previous measurement). Phages were added at an MOI of 1 and incubated with PAO1 at 37oC in absence of serum (control) or in presence of 10% HPS. b) S. aureus strain ATCC 19,685 was incubated with phage K at an MOI of 10 in absence (control) or presence of 10% HPS for 2 hours at 37oC, after which bacteria were plated and incubated overnight. Number of recovered colonies per plated volume is expressed as colony forming units per mL (CFU/mL). Black dotted line represents detection limit. a, b) Data represent mean ± SD of three independent experiments.
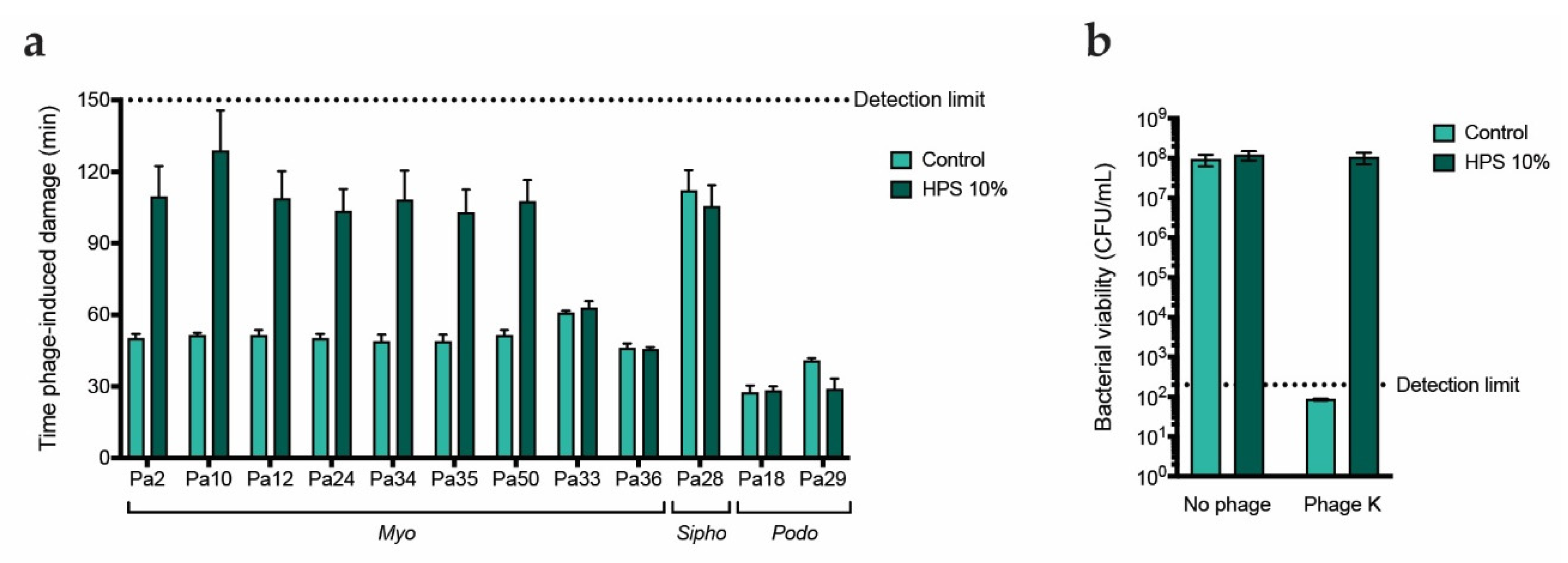
Figure 3.
Inhibition of phages by serum is mediated by the complement system. PAO1 was incubated with phage PBJ at 37oC in presence of the DNA dye Sytox green. a) Fluorescence intensity (RFU) over time (min) of bacteria infected with PBJ at an MOI of 10 (control), in presence of 10% HPS, 10% HPS with heat-inactivated complement (HI HPS), or IgG and IgM purified from HPS at a concentration equivalent to 10%. b) Fluorescence intensity (RFU) over time (min) of bacteria infected with PBJ at an MOI of 10 (control), in presence of 10% HPS, 10% HPS with 50 μM compstatin, or 10% HPS with 20 μg/mL OmCI. c) Fluorescence intensity (RFU) of bacteria after 90 minutes of infection with PBJ at an MOI of 10 in presence of 10% HPS and nanobodies in a range of concentrations. Value for bacteria infected with phage in absence of serum is shown as a control. d) Fluorescence intensity (RFU) over time (min) of bacteria infected with PBJ at an MOI of 10 in absence of serum (control) or in presence of 10% HPS and nanobodies at a concentration of 3 μM. a-d) Data represent mean ± SD of three independent experiments.
Figure 3.
Inhibition of phages by serum is mediated by the complement system. PAO1 was incubated with phage PBJ at 37oC in presence of the DNA dye Sytox green. a) Fluorescence intensity (RFU) over time (min) of bacteria infected with PBJ at an MOI of 10 (control), in presence of 10% HPS, 10% HPS with heat-inactivated complement (HI HPS), or IgG and IgM purified from HPS at a concentration equivalent to 10%. b) Fluorescence intensity (RFU) over time (min) of bacteria infected with PBJ at an MOI of 10 (control), in presence of 10% HPS, 10% HPS with 50 μM compstatin, or 10% HPS with 20 μg/mL OmCI. c) Fluorescence intensity (RFU) of bacteria after 90 minutes of infection with PBJ at an MOI of 10 in presence of 10% HPS and nanobodies in a range of concentrations. Value for bacteria infected with phage in absence of serum is shown as a control. d) Fluorescence intensity (RFU) over time (min) of bacteria infected with PBJ at an MOI of 10 in absence of serum (control) or in presence of 10% HPS and nanobodies at a concentration of 3 μM. a-d) Data represent mean ± SD of three independent experiments.
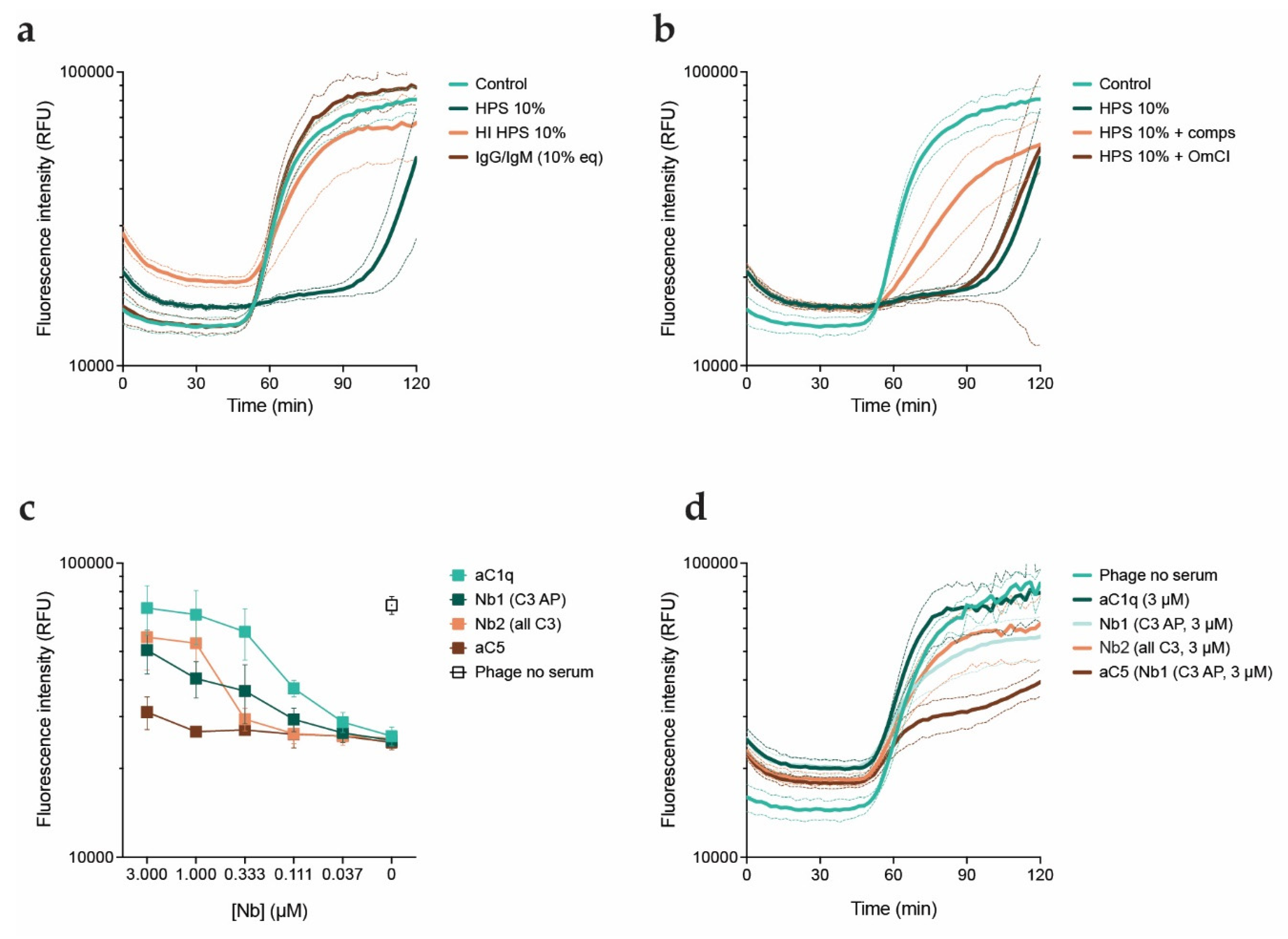
Figure 4.
Phages tagged with an azide group and labeled with a fluorescent marker were used to study their binding to bacteria. a) Schematic view of the technique used to produce azido-tagged phages. A methionine auxotroph PAO1 mutant was cultured in minimal medium supplemented with L-azidohomoalanine. Phages were amplified overnight at 37oC using methionine auxotroph PAO1 as host. The resulting phage progeny incorporates the non-canonical amino acid L-azidohomoalanine in place of methionine in its proteins, making them available for biorthogonal labeling via click chemistry. b, c) PAO1-sfCherry (red) were incubated with PBJ-AF488 (green, MOI 50) for 5 minutes at 37oC in b) buffer or c) 10% HPS, fixed in 1% PFA, washed and immobilized on agar pads. Images were acquired with a 100X immersion objective and are overlays of the modes phase contrast and widefield fluorescence with cube filters. Images are representative of three independent experiments. d-g) Flow cytometry was performed on PAO1-sfCherry incubated with PBJ-AF488 at different MOIs. When indicated, 10% HPS, anti-C1q nanobody (1 μM) or anti-C3 nanobody (1 μM) was added for 5 minutes at 37oC. Samples were stained with a monoclonal anti-C3-AF405 antibody and fixed in 1% PFA before measuring. Bacteria were gated on sfCherry-height and forward scatter-height signals. d) Geometric mean of the signal corresponding to bound phages. Heat inactivated (HI) phage is used as a negative control. e) Histograms of phage fluorescent signal at an MOI of 200 or HI phage at an MOI of 400 in the various serum conditions. f) Histograms of fluorescent signal corresponding to C3b deposition (AF405) on bacteria treated with the various serum conditions. g) Geometric mean of the signal corresponding to C3b (AF405) for the conditions with PBJ-AF488 at an MOI of 200, or HI phage at an MOI of 400. d, g) Data represent mean ± SD of three independent experiments. e, f) Signal was normalized by number of events. A representative graph of three independent experiments is shown.
Figure 4.
Phages tagged with an azide group and labeled with a fluorescent marker were used to study their binding to bacteria. a) Schematic view of the technique used to produce azido-tagged phages. A methionine auxotroph PAO1 mutant was cultured in minimal medium supplemented with L-azidohomoalanine. Phages were amplified overnight at 37oC using methionine auxotroph PAO1 as host. The resulting phage progeny incorporates the non-canonical amino acid L-azidohomoalanine in place of methionine in its proteins, making them available for biorthogonal labeling via click chemistry. b, c) PAO1-sfCherry (red) were incubated with PBJ-AF488 (green, MOI 50) for 5 minutes at 37oC in b) buffer or c) 10% HPS, fixed in 1% PFA, washed and immobilized on agar pads. Images were acquired with a 100X immersion objective and are overlays of the modes phase contrast and widefield fluorescence with cube filters. Images are representative of three independent experiments. d-g) Flow cytometry was performed on PAO1-sfCherry incubated with PBJ-AF488 at different MOIs. When indicated, 10% HPS, anti-C1q nanobody (1 μM) or anti-C3 nanobody (1 μM) was added for 5 minutes at 37oC. Samples were stained with a monoclonal anti-C3-AF405 antibody and fixed in 1% PFA before measuring. Bacteria were gated on sfCherry-height and forward scatter-height signals. d) Geometric mean of the signal corresponding to bound phages. Heat inactivated (HI) phage is used as a negative control. e) Histograms of phage fluorescent signal at an MOI of 200 or HI phage at an MOI of 400 in the various serum conditions. f) Histograms of fluorescent signal corresponding to C3b deposition (AF405) on bacteria treated with the various serum conditions. g) Geometric mean of the signal corresponding to C3b (AF405) for the conditions with PBJ-AF488 at an MOI of 200, or HI phage at an MOI of 400. d, g) Data represent mean ± SD of three independent experiments. e, f) Signal was normalized by number of events. A representative graph of three independent experiments is shown.
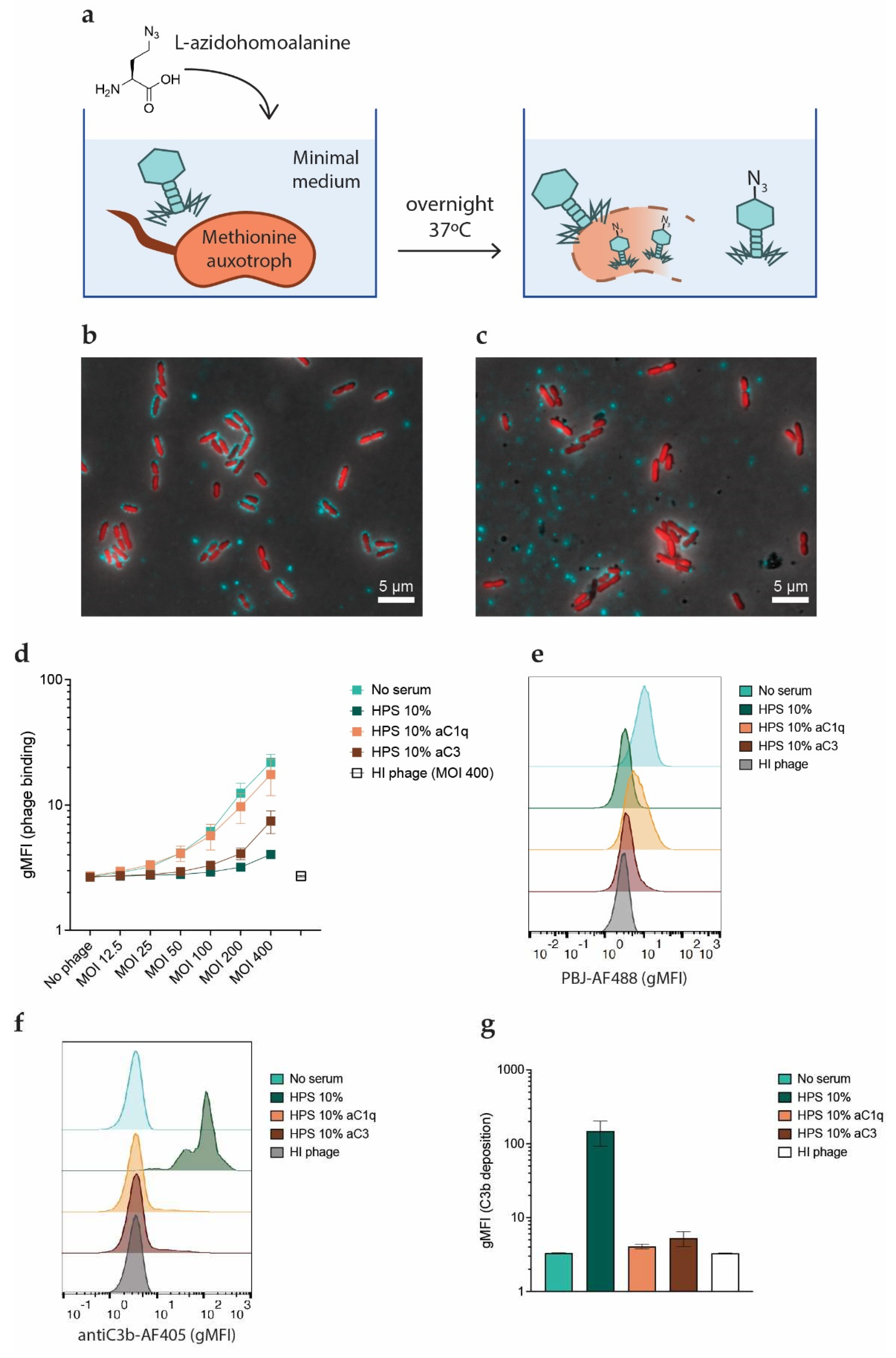
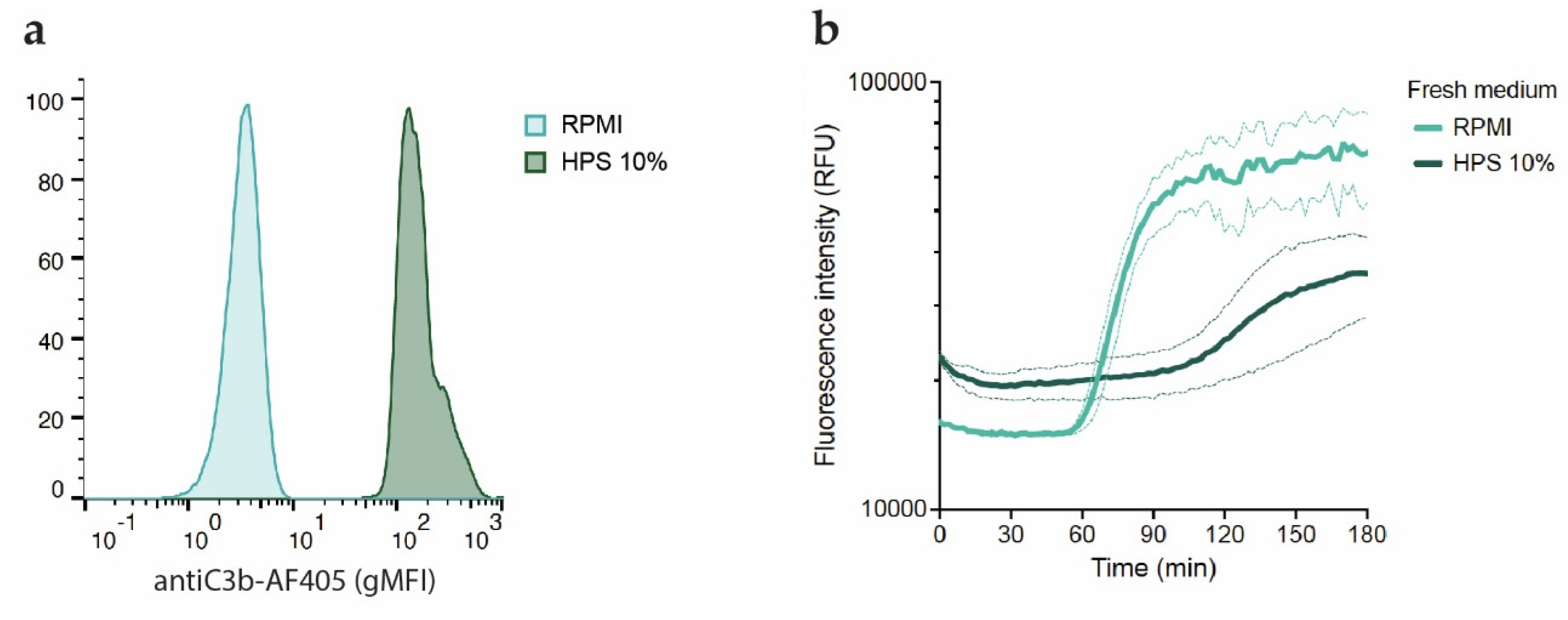
Figure 5.
Inhibition of phages by the complement system is not caused by blockage of phage receptors. a) Flow cytometry histograms showing C3b deposition (AF405) on bacteria incubated with 10% HPS for 30 minutes at 37oC after removal of unbound serum components and staining with aC3b-AF405 nanobody. Signal was normalized by number of events. A representative graph of three independent experiments is shown. b) Fluorescence intensity (RFU) over time of complement pre-opsonized PAO1 incubated at 37oC with phage PBJ (MOI 10) added either in buffer (RPMI) or in 10% HPS in presence of Sytox green. Data represent mean ± SD of three independent experiments.
Figure 5.
Inhibition of phages by the complement system is not caused by blockage of phage receptors. a) Flow cytometry histograms showing C3b deposition (AF405) on bacteria incubated with 10% HPS for 30 minutes at 37oC after removal of unbound serum components and staining with aC3b-AF405 nanobody. Signal was normalized by number of events. A representative graph of three independent experiments is shown. b) Fluorescence intensity (RFU) over time of complement pre-opsonized PAO1 incubated at 37oC with phage PBJ (MOI 10) added either in buffer (RPMI) or in 10% HPS in presence of Sytox green. Data represent mean ± SD of three independent experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



