
Submitted:
29 September 2023
Posted:
30 September 2023
You are already at the latest version
Abstract
Ewing sarcoma (EWS) is an aggressive pediatric malignancy of the bone and soft tissues in need of novel therapeutic options. To identify potential therapeutic targets, we focused on essential biological pathways that are upregulated by EWS-FLI1, the primary oncogenic driver of EWS, including mitotic proteins such as Aurora Kinase A (AURKA), Kinesin Family Member 15 (KIF15) and its binding partner, targeting protein for Xklp2 (TPX2). KIF15/TPX2 cooperates with KIF11, a key mitotic kinesin essential for mitotic spindle orientation. Given the lack of clinical grade KIF15/TPX2 inhibitors, we chose to target KIF11 (using SB-743921) in combination with AURKA (using VIC-1911), given phosphorylation of KIF15S1169 by Aurora A is required for its targeting to the spindle. In vitro, the drug combination demonstrated strong synergy (Bliss score ≥10) at nanomolar doses. Colony formation assay revealed significant reduction in plating efficiency (1-3%) and increased percent accumulation of cells in G2/M phase with the combination treatment (45-52%) upon cell-cycle analysis indicating mitotic arrest. In vivo studies in EWS xenograft mouse models showed significant tumor reduction and overall effectiveness: drug combination vs vehicle control (p≤0.01), SB-743921 (p≤0.01) and VIC-1911 (p≤0.05). Kaplan-Meier curves demonstrated superior overall survival with the combination compared to vehicle or monotherapy arms (p≤0.0001).
Keywords:
Ewing sarcoma
; drug synergy
; Kinesin Family Member 11
; Aurora Kinase A
; SB-743921
; and VIC-1911
1. Introduction
Ewing sarcoma (EWS) is the second most common pediatric bone sarcoma after osteosarcoma affecting kids and young adults. EWS is a rare cancer with an annual incidence of 2.93 cases per million per year 1. The pathogenic event in EWS is a somatic chromosomal translocation resulting in a fusion oncogene, EWS-ETS, and efforts to target it therapeutically have been unsuccessful. Chemotherapy has improved the 5-year overall survival rate for patients with localized disease (to approximately 60-70%) but unfortunately is less effective on metastatic (30%), refractory and recurrent disease (<15%) indicating that there is still a great unmet need for effective therapies 2. To identify new therapies for EWS, we previously conducted a multi-pronged approach using in silico predictions of drug activity via integrated bioinformatics approach in parallel with an in vitro screen of FDA-approved drugs. We uncovered drug targets 3,4 essential for mitotic spindle formation and cell cycle progression such as Kinesin Family Member 15 (KIF15) and its binding partner TPX25, and Aurora Kinase A (AURKA), which are upregulated by EWS-FLI1,which is the most predominant EWS-ETS fusion and the primary oncogenic driver of EWS.
KIF15 is a plus-end directed kinesin that localizes to spindle microtubules and chromosomes and plays a role in maintaining spindle bipolarity during mitosis 6. Though KIF15 is not essential for bipolar spindle formation during normal cell division, KIF15 compensates when the function of key motor Kinesin Family Member 11 (KIF11/Eg5) is inhibited 7. There are several KIF11 inhibitors that can disrupt the mitotic spindle bipolarity function mediated by KIF11; however, in most studies, resistance is observed as KIF15 replaces the functions of KIF11 in a TPX-2 dependent manner 7,8. Importantly, we and others have shown that KIF15 cooperates with KIF11 to promote bipolar spindle assembly and formation 9, which is essential for proper sister chromatid segregation and when KIF15 is genetically silenced, the efficacy of KIF11 inhibition is significantly enhanced. In previous studies we designed an RNAi-based screen of the “druggable genome” to identify putative points of molecular vulnerability across a diverse panel of ovarian cancer cell lines 10. These screens identified KIF11 as an essential protein in maintaining tumor cell viability. KIF11 is a tetrameric crosslinker and mitotic motor protein which facilitates mitotic progression through metaphase and anaphase by binding and pushing apart microtubules in the bipolar spindle. Hence, dual inhibition of KIF11 and KIF15/TPX2 axis is essential to disrupt mitotic activity of a cancer cell.
AURKA is a serine/threonine kinase with crucial functions in mitosis and has aberrant expression in most cancer types 11-13. AURKA-mediated phosphorylation regulates the functions of a diverse set of AURKA substrates, some of which are mitosis regulators, including KIF15 14. Studies have shown that dual inhibition of KIF11 and AURKA can overcome KIF15 dependent drug resistance and in KIF11 inhibitor-resistant HeLa cells, dual inhibition of KIF11 and AURKA led to the formation of monopolar spindles indicating the potency of combined targeting of these proteins 15. Due to the lack of clinically relevant inhibitors to target KIF15 directly, we sought to indirectly target KIF15 using a new clinically relevant AURKA inhibitor, VIC-1911. Previous studies have shown that AURKA directly regulates KIF15 and phosphorylation of KIF15S1169 by Aurora A is required for its targeting to the spindle 16. VIC-1911 (developed by VITRAC Therapeutics, LLC) or formerly known as TAS-119 is a novel, selective, and orally active small molecule inhibitor of AURKA developed for the treatment of solid tumors and hematologic malignancies. In preclinical studies VIC-1911 demonstrated anti-tumor activity 17,18 and is currently being studied in phase1 clinical trials for advanced solid tumors 19.
KIF11 has also been identified by the pharmaceutical industry as a viable target to develop anti-cancer drugs 20-22. Although these KIF11 inhibitors are generally well tolerated by patients 23, the clinical response rates as monotherapies in patients with advanced adult cancers are typically less than 10% 24,25. SB-743921 (also known as Kinesin Spindle Protein Inhibitor) is a second-generation small molecule ATPase inhibitor of KIF11. It has been reported to have greater than 40,000-fold sensitivity for KIF11 over other kinesins 26. SB-743921 has been used in several preclinical studies and has demonstrated significant anti-tumor activity and is being evaluated in clinical trials for multiple cancers 26,27. Both KIF11 and AURKA inhibitors have been employed in clinical trials as single agent or in combination with chemotherapy but have not been efficacious, indicating the need to reevaluate their mode of action and clinical limitations 12,28,29. Based on these clinical outcomes, we sought to evaluate these single-drug agents by the dual targeting of key mitotic regulators. To our knowledge, the following is the first study to report the in vitro synergistic activities and in vivo anti-tumor efficacy of this drug combination in EWS.
2. Materials and Methods
2.1. DepMap Portal Gene Expression Analysis
RNA expression levels of KIF11, KIF15, AURKA, and TPX2 across the Cancer Cell Line Encyclopedia were obtained from the DepMap Portal (RRID:SCR_017655) published by The Broad Institute 30. Expression levels were separated by cancer type and sorted by median value. Cancer types with two or fewer representatives or of hematological origin were excluded from analysis. This yielded 903 samples across 26 different tumor types. Data were plotted using GraphPad Prism software (RRID:SCR_002798).
2.2. Cell Lines and Cell Culture
We selected a panel of confirmed cell lines which are representative of the most common EWS-ETS fusion types; namely EWS-FLI1 type I (CHLA-9, CHLA-10, CHLA-32, TC-32, & TC-71), EWS-FLI1 type II (SK-ES-1), EWS-FLI1 type III (CHLA-258) fusion, and EWS-ERG fusion (COG-E-352) 31. For non-EWS controls, rhabdomyosarcoma cell line; RMS-13, osteosarcoma cell lines; U2-OS and bone marrow derived mesenchymal stem cells (BMD-MSCs) were utilized. BMD-MSCs (ATCC #PCS-500-012, (RRID:CVCL_A0YN), SK-ES-1 (ATCC #HTB-86, RRID:CVCL_0627), U2-OS (ATCC # HTB-96, RRID:CVCL_0042), and RMS-13 (ATCC #CRL-2061,RRID:CVCL_0041) were purchased from the American Type Culture Collection (ATCC). In addition, TC-71 (RRID: CVCL_2213), TC-32 (RRID: CVCL_7151), CHLA-32 (RRID: CVCL_M151), CHLA-9 (RRID:CVCL_M150), CHLA-10 (RRID:CVCL_6583), COG-E-352 (RRID:CVCL_M153), and CHLA-258 (RRID:CVCL_A058) cell lines were obtained from the Children’s Oncology Group (COG). All cell lines mentioned above have been authenticated by STR profiling. TC-71, CHLA-9, CHLA-10, CHLA-32, CHLA-258 and COG-E-352 were cultured in Iscove's Modified Dulbecco's Medium (Gibco #12440-053) supplemented with 20% FBS, 2 mM L-glutamine and 1X Insulin-Transferrin-Selenium (Gibco #41400-045). SK-ES-1 was cultured in McCoy's 5A medium (ATCC #30-2007) with 15% FBS, TC-32, CRL-2061 and U2-OS were cultured in RPMI-1640 (HyClone #SH30027.01) medium with 10% FBS. BMD MSC cells were cultured in MSC basal media (ATCC# PCS-500-030) supplemented with 7% FBS, rhIGF-1 (15 ng/mL, R&D systems #233-FB-025), rhFGF-b (125 pg/mL, R&D systems #291-G1-200) and L-Alanyl-L-Glutamine (2.4mM, Fisher scientific # AAJ6699606).
2.3. Drugs
Ispinesib (MedChem Express, HY-50759), Filanesib (MedChem Express, #HY-15187), SB-743921 (MedChem Express, #HY-12069) and VIC-1911 was obtained from VITRAC Therapeutics. Drugs were re-suspended in DMSO for invitro studies.
2.4. Cell Viability and Drug Synergy Assay
500 cells/well (80 µL) were plated in triplicates in 96-well plates in 8x8 matrix. Drugs (20 µL) were added the next day (18-24 hrs) after seeding cells and incubated for 72 hours before cell viability readings were measured. CellTiter-Glo reagent (Promega # G7572) mixed with Glo Lysis buffer (Promega # E2661) in 1:2 ratio was used for analysis. Equal volume of reagent to cell culture media was added. After adding the reagent, plates were mixed and incubated for 10 mins and readings were measured using TECAN Infinite 200 PRO plate reader (RRID:SCR_019033). Percentage inhibition of cell viability was calculated and synergy scores were assessed using SynergyFinder (RRID:SCR_019318) 32. Dose-response curves, heat-maps and 2D synergy plots are generated by the software, Bliss scoring algorithm was used to calculate synergy. Synergy scores less than -10 indicate that the interaction between the two drugs is likely to be antagonistic, scores from -10 to 10 indicate an additive effect, and scores higher than 10 indicate a synergistic effect.
2.5. Colony Formation Assay
500 cells/well (TC-71 and SK-ES-1) were plated in triplicates in a 6-well plate. Cells were treated with drugs the following day after which the plates were incubated and observed for 7 days. After the 7-day period, cells were washed twice with 2mL of 1X PBS and fixed with 500 µL methanol for 30 mins. Cells were then stained with 1 mL of crystal violet solution (0.1% w/v, Sigma Aldrich, Cat# C0775-25G) at room temperature for 30-40 mins. Stained colonies were imaged and quantified using Celigo Imaging Cytometer (Nexcelom Biosciences, RRID:SCR_018808). Data acquisition and imaging was done in a blinded manner. Plating efficiency is calculated by the following formula: Number of colonies formed/ Number of colonies plated x 100%.
2.6. Cell Cycle Analysis
TC-71 and CHLA-10 cells (1,50,000 per well) were plated in triplicates in a 6-well plate for each condition. Drugs were added the next day and incubated for 24 hrs. After drug treatment, cells were centrifuged at 300g for 5 minutes and washed twice with 1X PBS solution. Cells were then fixed with 70% ethanol and then stained with 500µL of FxCycle™ PI/RNase Staining Solution (Thermo Fisher Scientific, Cat# F10797). DNA content was measured using the Attune™ NxT flow cytometer (Invitrogen, RRID:SCR_019590). Cells were gated based on vehicle treatment of each cell line. Data was analyzed using FlowJo software v10 (BD Biosciences, RRID:SCR_008520). Data acquisition was done in a blinded manner.
2.7. Capillary Western Blot (Wes) Analysis
Capillary Western analyses were performed using the ProteinSimple Wes System. Final concentration of 0.4 µg/µl of protein samples were used for analysis and samples were diluted with 0.1 × Sample Buffer. Then 4 parts of diluted sample were combined with 1-part 5x Fluorescent Master Mix (containing 5x sample buffer, 5x fluorescent standard, and 200 mM DTT) and heated at 95 °C for 5 min. The Fluorescent Master Mix contains three fluorescent proteins that act as a ‘ruler’ to normalize the distance for each capillary because the molecular weight ladder is only on the first capillary and each capillary is independent. After this denaturation step, the prepared samples, blocking reagent, 1:50 diluted primary antibodies; KIF11 or Eg5 (Cell Signaling Technology Cat# 4203, RRID:AB_10545760), p-KIF11 (Thermo Fisher Scientific Cat# PA5-105186, RRID:AB_2816659), KIF15 (Proteintech Cat# 55407-1-AP, RRID:AB_11182836) , AURKA (Novus Cat# NBP1-51843SS, RRID:AB_11018019), p-AURKA (Novus Cat# NBP3-05434) PARP(46D11) (Cell Signaling Technology Cat# 9532, RRID:AB_659884), and β-actin (Cell Signaling Technology Cat# 12262, RRID:AB_2566811), HRP-conjugated secondary antibodies (Anti-rabbit (Biotechne, #DM-001) and anti-mouse (Biotechne, #DM-002) and chemiluminescent substrate were dispensed into designated wells in an assay plate. A biotinylated ladder provided molecular weight standards for each assay. After plate loading, the separation electrophoresis and immunodetection steps take place in the fully automated capillary system. Compass for Simple Western (RRID:SCR_022930) software was used to analyze data and process results. For the quantification of the Wes blots, area of the bands was used and was determined by the integrated analysis tool in Wes Compass software. Lane normalization factor was calculated using the following formula: Observed signal of housekeeping protein (β-Actin) for each lane/Highest observed signal of housekeeping protein on the blot. Protein expression or normalized experimental signal for each protein/sample was calculated as Observed experimental signal/Lane normalization factor .
2.8. Tumor Xenograft and Drug Treatment Studies
Six-week-old female NSG mice were inoculated subcutaneously in the right flanks with a 200 µL suspension of TC-71 (2 × 106 cells/site) mixed with an equal volume of ice-cold Matrigel (Corning #354234). Appropriate amount of SB-743921 drug was re-suspended in 10% DMSO, 40% PEG300, 5% Tween-80 and 45% PBS. VIC-1911 drug was re-suspended in 0.5% Hydroxypropyl methylcellulose solution. After tumors reached approximately 500 mm3, mice were randomized into four treatment groups (n=10 mice per group as per power analysis) and treated as follows: For study 1 (Supplementary Figure S4) groups included, 1) Vehicle control; equivalent dose of SB-743921 vehicle (intraperitoneal) and VIC-1911 vehicle (oral gavage), 2) SB-743921 only (2.5 mg/kg), 3) VIC-1911 only (37.5 mg/kg), and 4) combination of SB-743921 (2.5 mg/kg) and VIC-1911(37.5 mg/kg). Mice were treated with the vehicle and drugs, every other day for 20 days. For study 2, (Figure 7) 1) Vehicle control; equivalent dose of SB-743921 vehicle (intraperitoneal) and VIC-1911 vehicle (oral gavage), 2) SB-743921 only (1.25 mg/kg), 3) VIC-1911 only (37.5 mg/kg), and 4) combination of SB-743921 (1.25 mg/kg) and VIC-1911(37.5 mg/kg). Mice were treated with the vehicle and drugs, 3 times a week for 42 days. Tumor volume and body weight were measured 3 times per week. Tumor volumes were measured with calipers and calculated using the following formula: volume (mm3) = length × (width)2/2. Mice were humanely euthanized, and gross necropsies were performed when tumor volumes exceeded 4,000 mm3. The researchers were not blinded during drug treatment, data collection or analysis.
2.9. Data Analysis and Statistics
In vitro data were reported as mean ± SD of 3 independent experiments. In the statistical analyses for the in vivo xenograft study, Kaplan-Meier survival curves were used to determine the difference in survival among the treatment groups. A p-value less than 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 9.0 software (RRID:SCR_002798).
3. Results
3.1. In Silico Bioinformatics Screen Identifies Mitotic Proteins Essential for EWS Growth
Using integrated bioinformatics and high-throughput screening, we have previously identified and validated several drugs, which were predicted to reverse the EWS disease signatures and/or EWS-ETS-dependent signatures 3. We then computed and sorted the expression changes of individual genes associated with the EWS signatures upon treatment with the validated drugs. We identified the top fifteen genes (Table 1) whose transcript levels were significantly (p< 0.05) downregulated by these drugs. Among these genes, KIF15 (encoding for a motor kinesin) was ranked first and its potential binding partner, TPX2 (encoding for a microtubule-associated protein that mediates bipolar spindle assembly and mitosis) was ranked fourteenth on the list. Notably, in separate but parallel investigations conducted by our team, we observed significant vulnerability of the spindle assembly motor proteins KIF11, KIF15, and their binding partner TPX2 in epithelial ovarian cancer (unpublished data). Furthermore, we found that elevated levels of KIF15 and TPX2 contribute to resistance against the KIF11 inhibitor (KIF11i), as demonstrated in both in vitro and in vivo studies. We also identified another key mitotic kinase Aurora kinase A (AURKA) to be ranked sixth in our screen. Furthermore, bioinformatic analysis of RNA expression data from solid cancers within DepMap portal indicated that KIF15 expression levels were the highest in Ewing sarcoma and that KIF11 expression levels were the second highest in Ewing Sarcoma, only behind synovial sarcomas (Figure 1A). Interestingly, our analyses found that both KIF11 and KIF15 are highly expressed across all the various sarcomas included in the portal.
STRING protein-protein interaction analysis of the top-fifteen candidates indicates association between the proteins involved in cell cycle progression (Supplementary Figure S1A). Gene enrichment analysis indicated the biological pathways such as AURKA signaling (Supplementary Figure S1B) and biological processes such as spindle assembly (Supplementary Figure S1C) to be the key pathways mediated by the top-fifteen genes. KIF11, KIF15, TPX2, and AURKA are abundantly expressed in several cell line models of EWS at the protein level (Figure 1B,C, Supplementary Figure S2). We next investigated publicly available databases to check for the RNA expression of these proteins in patient tumor samples using BioGPS (E-GEOD-12102) 33 (Supplementary Figure S1D). These proteins are expressed in all EWS patients indicating the feasibility of targeting them. Small molecule inhibitors to KIF15 such as KIF15-IN-1 have been tested in preclinical studies 34,35 but none of them have been advanced to clinical trials, hence we chose to target the mitotic proteins upstream, KIF11 and AURKA with clinically available inhibitors for this study.
3.2. Synergistic Inhibition of EWS Growth In Vitro by KIF11 and AURKA Inhibitors
We performed drug synergy assays to identify the most synergistic KIF11 inhibitor in combination with AURKA inhibitor VIC-1911. Given there are several KIF11 inhibitors that have been tested in clinical trials for different types of cancers, we tested the three most widely used inhibitors, e.g., Ispinesib, Filanesib and SB-743921 (also known as kinesin spindle protein inhibitor). We initially tested these drug combinations in TC-71, an EWS-FLI1 Type I fusion containing cell line. An 8 x 8 matrix was used to test different combinations of both inhibitors. Cell inhibition was measured using the CellTiter-Glo viability assay and drug synergy was calculated via Synergy Finder 36. Bliss algorithm was used to measure synergy, based on which scores greater than 10 indicate significant synergistic interaction between the two drugs. We observed synergistic interaction with all the three different KIF11 inhibitors used. Synergy scores were observed to be the greatest with the SB-743921 and VIC-1911 combination (24.62) (Figure 2A) when compared to synergy scores of Ispinesib (19.54) (Figure 2B) and Filanesib (16.81) (Figure 2C) in combination with VIC-1911.Hence, we decided to use the combination of SB-743921+ VIC-1911 for this study.
3.3. Drug Synergy Is Observed in Different EWS Fusion Type Cell Lines
We next assessed if the drug combination was sensitive to different EWS-ETS fusion types and tested the combination of SB-743921 and VIC-1911 across multiple EWS cell lines bearing different EWS fusions. EWS-FLI1 Type II (SKES-1) (Figure 3A), EWS-FLI1 Type III (CHLA-258) (Figure 3B), and EWS-ERG (COG-E-352) (Figure 3C) cell lines yielded strong synergistic drug activity with the combination treatment. This finding indicates that this drug combination is efficient at reducing EWS cell viability irrespective of fusion type. We also tested treatment naïve EWS-FLI1 Type I fusion bearing cell lines CHLA-9, CHLA-32 and TC-32 with the combination (Supplementary Figure S3A-C) and found that these cells have greater susceptibility and synergy compared to the other cells lines established post-treatment. Again, these results suggest that dual inhibition is effective irrespective of the treatment status of EWS cells. Synergy was also observed in other cancer types such as osteosarcoma (U2OS) and rhabdomyosarcoma (RMS-13) (Supplementary Figure S4A,B). However, in control BMD-MSCs (Supplementary Figure S4C), the presumptive progenitor cells to EWS 37, the combination was not effective in inhibiting cell growth indicating enhanced sensitivity of these drugs for more rapidly proliferating cancer cells and a possible therapeutic window for this drug combination.
3.4. Combination Treatment with SB-743921 and VIC-1911 Reduces Colony Formation In Vitro
We next performed colony formation assay to measure clonogenicity of the drug combination. Different drug combinations were tested alone and in combination. We used the drug dosages that were in the synergistic range and assessed the ability of single cells to form colonies. We seeded very low number (500 cells/well) of SKES-1 cells and TC-71 cells in a 6-well plate and drugs were added the next day after seeding. The cells were incubated for 7 days and were fixed and stained using crystal violet. The plates were imaged, and colonies were quantified using the Celigo Imaging Cytometer. We observed significantly fewer colonies in the wells treated with the drug combination as compared to single drug treatment and the control group (Figure 4A) and significant reduction in plating efficiency with the combination treatment. Mean plating efficiencies for SK-ES-1 cells are control (100.5%), 0.625 nM SB-743921 (103.3%), 0.312 nM SB-743921 (104.9%), 25 nM VIC-1911 (59.6%), 12.5 nM VIC-1911 (84%), 0.625 nM SB-743921 + 12.5 nM VIC-1911 (2%) and 0.625 nM SB-743921 + 25 nM VIC-1911 (0.8%). For TC-71 cells, control (113.1.%), 0.625 nM SB-743921 (20%), 0.312 nM SB-743921 (48.9%), 25 nM VIC-1911 (60.7%), 12.5 nM VIC-1911 (38.6%), 0.625 nM SB-743921 + 12.5 nM VIC-1911 (1.6%) and 0.625 nM SB-743921 + 25 nM VIC-1911 (1.8%). (Figure 4B). This result indicated that the combination treatment has a significant effect on EWS cancer cell survival in vitro.
3.5. Cell Cycle Analysis Indicates Combination Treatment Arrests the Cells in G2/M Phase
KIF11 38 and AURKA 39 inhibitors have known roles in mediating G2/M arrest and in turn halting cell division. We performed cell-cycle analyses in TC-71 and CHLA-10 cells to test if the drug combination was effective at arresting the cells in G2/M phase compared to single drugs alone. We observed that there was an increase in the number of cells in G2/M and sub G1 phase (cell fragments) indicating cell cycle arrest and apoptosis, respectively with the combination treatment (Figure 5A). Quantification of the percentage of cells in G2/M and subG1 phase indicates significant effects on cell growth and viability with the combination treatment (Figure 5B).
3.6. Protein Expression Post-Combination Treatment Indicates an Increase in Expression of KIF11 and AURKA
We performed a capillary electrophoresis based western blot assay (Wes) to determine protein expression levels following drug treatment. We first checked the protein levels of KIF11 and AURKA upon treatment with their specific inhibitors’ SB-743921 and VIC-1911. We observed that compared to control and single-drug treatment groups, there was an increase in accumulation of KIF11 and AURKA proteins in the combination treatment group (Figure 6A,B, & Supplementary Figures S5). This finding could be due to increased accumulation of cells in G2/M phase as observed previously (Figure 5B). We next tested the status of AURKA and KIF11 phosphorylation following drug treatment and found enhanced phosphorylation of AURKA at Thr288 and of KIF11 at Thr926 and corresponding decrease in KIF15 protein levels with the drug combination group. We also observed an increase in expression of cleaved-poly (ADP-ribose) polymerase (c-PARP) upon SB-743921 drug treatment, which was enhanced in combination with VIC-1911 indicating induction of apoptosis (Figure 6A,B & Supplementary Figures S5). These finding continue to support drug targeting of KIF11 and AURKA and subsequent down regulation of KIF15 as a potential new therapeutic approach in children and young adults diagnosed with recurrent EWS. Taken together, these protein expression data indicate that combination treatment inhibits EWS growth via perturbation of the KIF11/15 pathways, contributing to G2/M arrest and apoptosis.
3.7. Combination Treatment Synergistically Leads to Tumor Regression in EWS Xenograft Mouse Model
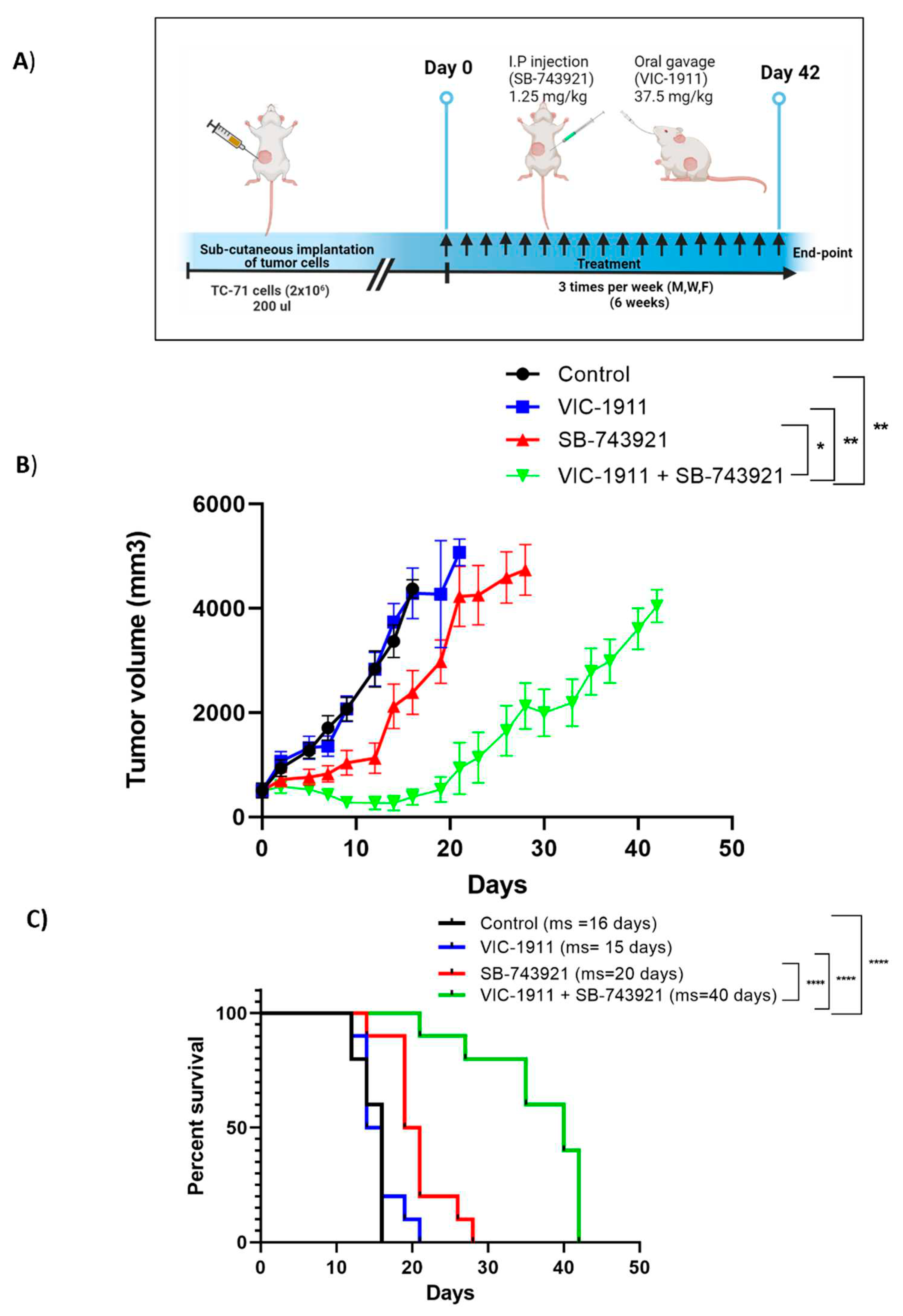
To test the efficacy of the drug combination in vivo, we employed a EWS mouse xenograft model. Female NSG mice (6-8 weeks) were implanted sub-cutaneously with 2x106 TC-71 cells, once tumors reached approximately 500 mm3, mice were grouped into 4 treatment arms. In efficacy Study 1, mice were grouped into controls, SB-743921 (2.5 mg/kg), VIC-1911 (37.5 mg/kg) or their combination (Supplementary Figure 6A) and were treated every other day for 20 days. We observed significant tumor regression with the drug combination compared to control or the VIC-1911 monotherapy arm (Supplementary Figure 6B). At day 32 (12 days after ending treatment), the animals treated with the drug combination demonstrated no evidence of measurable disease based on palpation. We followed survival (based on regulations by our IUCUC) and observed that the tumor bearing animals treated with the combination survived significantly longer (70 days) than the vehicle and monotherapy treatment arms (Supplementary Figure 6C). Despite the desired and significant efficacy observed, the animals displayed a significant loss in body weight when treated with 2.5 mg/kg of SB-743921 alone or combination (Supplementary Figure S7A). Although the animals’ body weights recovered when treatment was discontinued, we designed a second efficacy study in which the dosage of SB-743921 was reduced to 1.25 mg/kg/treatment and the treatment regimen altered. In efficacy Study 2 tumor bearing animals were treated with vehicle (control), SB-743921 (1.25 mg/kg), VIC-1911(37.5 mg/kg) or their combination 3-times a week (Monday, Wednesday and Friday) for up to 6 weeks (42 days). Mice were monitored until defined end-point symptoms were observed (Figure 7A). Based on the approve IUCUC protocol, animals were euthanized when the tumor reached 4,000 mm3. As shown in Figure 7B the combination group continued to show the greatest level of efficacy as compared to the vehicle control or the monotherapy arms. However, when using a lower levels of SB-743921 in combination with VIC-1911, the tumors recurred more quickly. The combination treatment also had a significant effect on improving the overall survival time of the mice compared to control and single drug treatment groups (Figure 7C). Compared to the animals in efficacy Study1, their body weights were more stable throughout the treatments (Supplementary Figure S7B). Combined, these data indicate that synergistic targeting of KIF11 and AURKA with specific inhibitors causes significant delay in tumor growth and improved overall survival compared to single drugs alone.
Figure 7.
Tumor efficacy studies in a TC-71 xenograft model. A) Timeline of in vivo study is represented. SB-743921 (1.25 mg/kg) and VIC-1911 (37.5 mg/kg) were dosed intraperitoneally and orally respectively. Mice were treated with these drugs 3 times a week, for 6 weeks (18 treatments) until end-point was reached. We observed significant B) tumor regression (control vs combination, SB-743921 vs combination and VIC-1911 vs combination p≤0.01**, p≤0.01**, p≤0.05 as assessed by one-way ANOVA) at day 28 C) Kaplan-Meier curves indicate overall survival with the combination treatment (VIC-1911 and SB-743921) compared to monotherapy and vehicle control groups (control vs combination (p≤0.0001****) , VIC-1911 only vs combination, (p≤0.0001****) and SB-743921 only vs combination (p≤0.0001****) as measured by Log-rank (Mantel-Cox) test). Median survival for each group is represented on the survival-curve.
Figure 7.
Tumor efficacy studies in a TC-71 xenograft model. A) Timeline of in vivo study is represented. SB-743921 (1.25 mg/kg) and VIC-1911 (37.5 mg/kg) were dosed intraperitoneally and orally respectively. Mice were treated with these drugs 3 times a week, for 6 weeks (18 treatments) until end-point was reached. We observed significant B) tumor regression (control vs combination, SB-743921 vs combination and VIC-1911 vs combination p≤0.01**, p≤0.01**, p≤0.05 as assessed by one-way ANOVA) at day 28 C) Kaplan-Meier curves indicate overall survival with the combination treatment (VIC-1911 and SB-743921) compared to monotherapy and vehicle control groups (control vs combination (p≤0.0001****) , VIC-1911 only vs combination, (p≤0.0001****) and SB-743921 only vs combination (p≤0.0001****) as measured by Log-rank (Mantel-Cox) test). Median survival for each group is represented on the survival-curve.
4. Discussion
Antimitotic drugs are among the most important chemotherapeutic agents available to oncologists and continue to be a clinical staple in the treatment of most solid tumors, including EWS 40,41. Newly diagnosed EWS patients are treated with an aggressive chemotherapeutic regimen which consists of 14 cycles with vincristine, doxorubicin, cyclophosphamide, alternating with ifosfamide and etoposide (VDC/IE) 42. These drugs have both short-term and long-term adverse effects in patients such as myelosuppression, cardiotoxicity, neuropathy, and secondary malignancies 43,44. Despite extensive therapy utilized commonly in new diagnoses, at least one-fourth of patients with localized disease will relapse after completion of therapy. Meanwhile for newly diagnosed patients with metastatic disease, recurrence rates are even higher with treatment failure seen in 50%–80% depending on the location of metastases 45. For patients with relapse or for those who are refractory to initial therapy, the odds of long-term survival are low. In addition, there is currently no standard management for this group of patients, raising many questions about how best to proceed. Hence, there is an unmet need for novel therapies, especially targeted agents for EWS for this patient population. Therefore, more specific inhibitors of mitosis could avoid the side effects of anti-microtubule agents (e.g., vincristine) such as neuropathy 46.
Current therapies for EWS that are under development mainly focus on targeting the EWS/FLI1 fusion protein, DNA-damage repair pathways, tyrosine kinase inhibition, immunotherapy, and cell therapies 47. Although multiple randomized trials are being conducted involving many international groups, the outcomes have been disappointing indicating the need for novel therapies 48. We have previously used multi-pronged approach using in silico predictions of drug activity via an integrated bioinformatics approach in parallel with an in vitro screen of FDA-approved drugs and identified key molecules, some of which include mitotic proteins that essential for EWS progression 3. Aberrant cell cycle progression is the key hallmark for cancers, and a variety of cell cycle agents have been used for treatment of cancers in the clinic, including EWS 49.
Among these cell cycle inhibitors, mitotic inhibitors specifically prevent formation of bipolar spindles and normal assembly of chromosomes leading to mitotic arrest and apoptosis 50. Mitotic inhibitors have been used in clinical trials for several different cancer types, but major limitations associated with the use of these inhibitors include off-target toxicity affecting non-neoplastic cells and tumor recurrence associated with monotherapies 51. Mitotic inhibitors can cause mitotic slippage leading to aneuploidy and chromosomal instability causing drug resistance 52. Therefore, it is of critical importance to a use combination of drugs that target multiple pathways and synergistically inhibit tumor growth. Synergistic drug combinations have greater potency at lower and physiologically relevant doses compared to monotherapies. In this study, we have employed a synergistic combination of mitotic inhibitors targeting KIF11 and AURKA to halt EWS tumor growth in vitro and in vivo as assessed in a mouse xenograft model.
KIF11 53 and AURKA 12 are overexpressed in many different cancer types indicating the importance of this mitotic machinery to facilitate aggressive tumor growth (Figure 1A). We leveraged the publicly available datasets to assess the RNA expression of these proteins in EWS patient tumor samples and found uniform expression in patient tumors irrespective of the disease state indicating the feasibility of targeting these proteins in EWS patients (Supplementary Figure S1). We tested different KIF11 inhibitors including Ispinesib, Filanesib and SB-743921 in combination with VIC-1911 (Figure 2). Ispinesib, SB-743921 and Filanesib have been tested in multiple clinical trials for several malignancies alone and in combination with other chemotherapies 29. Ispinesib (SB-715992) was tested in a pediatric phase I clinical trial on several different cancer types and was found to be well tolerated in pediatric patients 23. This finding suggests that KIF11 inhibitors are highly clinically relevant and since they have never been tested for EWS patients before, they have the potential to be developed as a new treatment option for these patients. SB-743921 was synthesized to be more selective and potent by replacing quinazoline ring in Ispinesib with the chromen-4-one ring and resulted in a five-fold increase in its potency over the parent compound 54. SB-743921 was used for the remainder of the study as we observed it to be very potent and had the highest synergy with VIC-1911 in comparison with other KIF11 inhibitors.
One of the main mechanisms of action mediated by the drug combination we found was by enhancing G2/M mediated cell cycle arrest and subG1 cells (Figure 5), ultimately resulting in enhanced apoptosis mediated cell death (Figure 6). Further studies need to be done to identify additional mechanisms of action and pathways affected by this combination. We also tested other cancer cell lines such as osteosarcoma and rhabdomyosarcoma and observed synergy with this drug combination in our preliminary studies (Supplementary Figure S4), which is consistent with elevated KIF11 and KIF15 levels (Figure 1A), indicating the potential for this drug combination in targeting other tumor types, especially sarcomas. Finally, mouse xenograft studies showed that the combination treatment of SB-743921 and VIC-1911 led to a significant delay in tumor growth compared to the single drugs alone, even though the tumors eventually developed therapy resistance (Supplementary Figures S6 and S7). Although the experiments with the higher levels of SB-743921 (2.5 mg/kg) showed sustained tumor regression and increased overall survival in combination with VIC-1911 (Supplementary Figure S6B,C), we repeated the experiments with lower levels of SB-743921 due to significant body weight loss in mice (Supplementary Figure S7A). Nevertheless, together these in vivo studies warranted additional studies to evaluate different dosing schemes along with assessing other classes of KIF11 inhibitors in combination with VIC-1911 to provide support for potential clinical translation.
Systemic cytotoxicity is often one of the major limiting factors for many chemotherapeutic regimens, hence developing an effective drug delivery approach will largely benefit the field of precision medicine in general and specifically the area of targeted therapy. Future studies will be focused on employing targeted drug delivery approaches given the effectiveness of the drug combination that we observe in the current study. Overall, we have identified a novel combination of mitotic inhibitors targeting KIF11 and AURKA that is highly synergistic in inhibiting the growth of an aggressive tumor such as Ewing sarcoma. Our findings are highly relevant, timely and clinically translatable given the lack of proper therapies for this rare and orphaned pediatric disease.
5. Conclusions
This study identified a synergistic combination of inhibitors for KIF11 (SB-743921) and AURKA (VIC-1911) that demonstrated significant pre-clinical activity in vitro and efficacy in vivo in Ewing sarcoma. Further studies will be required to define whether these are the best in class drugs to use and the dosing schedule to administer in order to minimize drug toxicity. These drugs have been tested in early phase clinical trials either individually or in combination with other drugs for several adult tumor types and have shown acceptable safety profiles. Our studies support considering dual targeting of these biological pathways for future clinical trial design to address an unmet need in children and young adults diagnosed with EWS.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Figure S1. A) STRING protein-protein interaction analysis for the top-fifteen genes is represented. Gene enrichment analysis of the top-fifteen genes by FunRich (Functional Enrichment Analysis Tool) software indicating the key B) biological pathways, and C) biological processes mediated by these genes. D) RNA expression of KIF11, AURKA, KIF15 and TPX2 from EWS patients were accessed from BioGPS. Supplementary Figure S2 (related to Figure 1B & C). A) Quantification of Wes blots data are shown. Supplementary Figure S3. Dose-response curves, dose-response matrix, and heat map indicating synergy scores in different EWS cell lines. Combination treatment resulted in the following synergy scores in A) CHLA-9 (27.45), B) CHLA-32 (38.97), and C) TC-32 (31.28). Supplementary Figure S4. Dose-response curves, dose-response matrix, and heat map indicating synergy scores in different cancer cell lines and control. Combination treatment resulted in the following synergy scores in A) U2-OS (17.91), B) CRL-2061 (13.16), and C) BMD-MSCs - control (13.04). Supplementary Figure S5 (related To Figure 6). Quantification of Wes blot data for Figure 6 is shown. Supplementary Figure S6. A) Timeline of in vivo study is represented. SB-743921 (2.5 mg/kg) and VIC-1911 (37.5 mg/kg) were dosed intraperitoneally and orally respectively. Mice were treated with these drugs every other day for 20 days (11 treatments) and monitored until end-point criteria were met. We observed significant B) tumor regression (control vs combination, VIC-1911 vs combination, p≤0.001*** as assessed by one-way ANOVA) and C) overall survival. p≤0.001***, p≤0.001*** and p-value** ≤0.01 is determined by logrank (Mantel-cox) test and indicated on the graph. Supplementary Figure S7. The body weights of mice are represented for the tumor efficacy A) efficacy Study 1 and B) efficacy Study 2. p≤0.0001****, p≤0.001***, p≤0.01** and p≤0.05 as measured by one-way ANOVA.
Author Contributions
S.M. Turaga: Investigation, visualization, methodology, writing-original draft. V. Vishwakarma: Investigation, methodology, writing-review, and editing. S.L. Hembruff: Investigation. B.K. Gibbs: Investigation, writing-review, and editing. P. Sabu: Investigation. R.V. Puri: Investigation. H.B. Pathak: Investigation, methodology, writing-review and editing G. Samuel: Conceptualization, supervision, writing-review, editing and funding acquisition A.K. Godwin: Conceptualization, supervision, funding acquisition, writing–original draft, writing-review, and editing.
Funding
This work is supported in part by a grant from Noah’s Bandage awarded to GS and AKG, Braden’s Hope for Childhood Cancer to AKG, a KUMC Biomedical Training Research Fellowship awarded to SMT, and the Kansas Institute for Precision Medicine (P20 GM130423, AKG PI).
Institutional Review Board Statement
All in vivo efficacy studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Kansas Medical Center (KUMC) (IACUC# 2020–2549).
Informed Consent Statement
“Not applicable.”
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
We acknowledge VITRAC Therapeutics LLC for providing the drug VIC-1911 for this study. We acknowledge the Flow Cytometry Core Laboratory, which is sponsored, in part, by the NIH/NIGMS COBRE grant P30 GM103326 and the NIH/NCI Cancer Center grant P30 CA168524.
Conflicts of Interest
AKG is a co-founder of Sinochips Diagnostics, serves as a scientific advisory board member to Biovica, Clara Biotech, EXOKĒRYX, VITRAC Therapeutics, and Sinochips Diagnostics, and receives research funding from Predicine and VITRAC Therapeutics. The remaining authors declare no potential conflicts of interest.
References
- Esiashvili, N.; Goodman, M.; Marcus, R.B., Jr. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol. 2008, 30, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Balamuth, N.J.; Womer, R.B. Ewing's sarcoma. Lancet Oncol. 2010, 11, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Pessetto, Z.Y.; Chen, B.; Alturkmani, H.; et al. In silico and in vitro drug screening identifies new therapeutic approaches for Ewing sarcoma. Oncotarget. 2017, 8, 4079–4095. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Baltezor, M.; Rajewski, L.; et al. Targeted inhibition of histone deacetylase leads to suppression of Ewing sarcoma tumor growth through an unappreciated EWS-FLI1/HDAC3/HSP90 signaling axis. J Mol Med (Berl). 2019, 97, 957–972. [Google Scholar] [CrossRef]
- Mann, B.J.; Balchand, S.K.; Wadsworth, P. Regulation of Kif15 localization and motility by the C-terminus of TPX2 and microtubule dynamics. Mol Biol Cell. 2017, 28, 65–75. [Google Scholar] [CrossRef]
- Brouwers, N.; Mallol Martinez, N.; Vernos, I. Role of Kif15 and its novel mitotic partner KBP in K-fiber dynamics and chromosome alignment. PLoS One. 2017, 12, e0174819. [Google Scholar] [CrossRef]
- Raaijmakers, J.A.; van Heesbeen, R.G.; Meaders, J.L.; et al. Nuclear envelope-associated dynein drives prophase centrosome separation and enables Eg5-independent bipolar spindle formation. Embo j. 2012, 31, 4179–4190. [Google Scholar] [CrossRef]
- Vanneste, D.; Takagi, M.; Imamoto, N.; Vernos, I. The role of Hklp2 in the stabilization and maintenance of spindle bipolarity. Curr Biol. 2009, 19, 1712–1717. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Macurek, L.; Janssen, A.; Geers, E.F.; Alvarez-Fernandez, M.; Medema, R.H. Kif15 cooperates with eg5 to promote bipolar spindle assembly. Curr Biol. 2009, 19, 1703–1711. [Google Scholar] [CrossRef]
- Sethi, G.; Pathak, H.B.; Zhang, H.; et al. An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. PLoS One. 2012, 7, e47086. [Google Scholar] [CrossRef]
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: novel therapy targets in cancers. Oncotarget. 2017, 8, 23937–23954. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: molecular mechanisms and opportunities for Cancer therapy. Mol Cancer. 2021, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp Mol Med. 2021, 53, 835–847. [Google Scholar] [CrossRef] [PubMed]
- van Heesbeen, R.; Raaijmakers, JA.; Tanenbaum, M.E.; et al. Aurora A, MCAK, and Kif18b promote Eg5-independent spindle formation. Chromosoma 2017, 126, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.T.; Erdal, S.; Huang, S.; Poon, R.Y. Synergism between inhibitors of Aurora A and KIF11 overcomes KIF15-dependent drug resistance. Mol Oncol. 2014, 8, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- van Heesbeen, R.G.H.P.; Raaijmakers, J.A.; Tanenbaum, M.E.; et al. Aurora A, MCAK, and Kif18b promote Eg5-independent spindle formation. Chromosoma. 2017, 126, 473–486. [Google Scholar] [CrossRef]
- Sootome, H.; Miura, A.; Masuko, N.; Suzuki, T.; Uto, Y.; Hirai, H. Aurora A Inhibitor TAS-119 Enhances Antitumor Efficacy of Taxanes In Vitro and In Vivo: Preclinical Studies as Guidance for Clinical Development and Trial Design. Mol Cancer Ther. 2020, 19, 1981–1991. [Google Scholar] [CrossRef]
- Miura, A.; Sootome, H.; Fujita, N.; et al. TAS-119, a novel selective Aurora A and TRK inhibitor, exhibits antitumor efficacy in preclinical models with deregulated activation of the Myc, β-Catenin, and TRK pathways. Invest New Drugs. 2021, 39, 724–735. [Google Scholar] [CrossRef]
- Robbrecht, D.G.J.; Lopez, J.; Calvo, E.; et al. A first-in-human phase 1 and pharmacological study of TAS-119, a novel selective Aurora A kinase inhibitor in patients with advanced solid tumours. Br J Cancer. 2021, 124, 391–398. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat Rev Cancer. 2012, 12, 527–539. [Google Scholar] [CrossRef]
- Carol, H.; Lock, R.; Houghton, P.J.; et al. Initial testing (stage 1) of the kinesin spindle protein inhibitor ispinesib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2009, 53, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Lad, L.; Luo, L.; Carson, J.D.; et al. Mechanism of inhibition of human KSP by ispinesib. Biochemistry. 2008, 47, 3576–3585. [Google Scholar] [CrossRef] [PubMed]
- Souid, A.K.; Dubowy, R.L.; Ingle, A.M.; et al. A pediatric phase I trial and pharmacokinetic study of ispinesib: a Children's Oncology Group phase I consortium study. Pediatr Blood Cancer. 2010, 55, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Saez, I.; Skoufias, D.A. Eg5 targeting agents: From new anti-mitotic based inhibitor discovery to cancer therapy and resistance. Biochem Pharmacol. 2021, 184, 114364. [Google Scholar] [CrossRef]
- Lee, C.W.; Bélanger, K.; Rao, S.C.; et al. A phase II study of ispinesib (SB-715992) in patients with metastatic or recurrent malignant melanoma: a National Cancer Institute of Canada Clinical Trials Group trial. Invest New Drugs. 2008, 26, 249–255. [Google Scholar] [CrossRef]
- Holen, K.D.; Belani, C.P.; Wilding, G.; et al. A first in human study of SB-743921, a kinesin spindle protein inhibitor, to determine pharmacokinetics, biologic effects and establish a recommended phase II dose. Cancer Chemother Pharmacol. 2011, 67, 447–454. [Google Scholar] [CrossRef]
- Zhu, L.; Xiao, F.; Yu, Y.; et al. KSP inhibitor SB743921 inhibits growth and induces apoptosis of breast cancer cells by regulating p53, Bcl-2, and DTL. Anticancer Drugs. 2016, 27, 863–872. [Google Scholar] [CrossRef]
- Falchook, G.S.; Bastida, C.C.; Kurzrock, R. Aurora Kinase Inhibitors in Oncology Clinical Trials: Current State of the Progress. Semin Oncol. 2015, 42, 832–848. [Google Scholar] [CrossRef]
- Shahin, R.; Aljamal, S. Kinesin spindle protein inhibitors in cancer: from high throughput screening to novel therapeutic strategies. Future Sci OA. 2022, 8, Fso778. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Samuel, G.; Crow, J.; Klein, J.B.; et al. Ewing sarcoma family of tumors-derived small extracellular vesicle proteomics identify potential clinical biomarkers. Oncotarget. 2020, 11, 2995–3012. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: an interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jin, X.; Tsueng, G.; Afrasiabi, C.; Su, A.I. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 2016, 44, D313–D316. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.E.; Chen, G.Y.; Kendrick, N.D.; et al. Dual inhibition of Kif15 by oxindole and quinazolinedione chemical probes. Bioorg Med Chem Lett. 2019, 29, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Milic, B.; Chakraborty, A.; Han, K.; Bassik, MC.; Block, S.M. KIF15 nanomechanics and kinesin inhibitors, with implications for cancer chemotherapeutics. Proc Natl Acad Sci U S A. 2018, 115, E4613–e4622. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: an interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–w743. [Google Scholar] [CrossRef]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007, 11, 421–429. [Google Scholar] [CrossRef]
- Daigo, K.; Takano, A.; Thang, P.M.; et al. Characterization of KIF11 as a novel prognostic biomarker and therapeutic target for oral cancer. Int J Oncol. 2018, 52, 155–165. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, L.; Zhou, Q.; et al. Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells. Cancer Cell Int. 2020, 20, 9. [Google Scholar] [CrossRef]
- van Vuuren RJ, Visagie MH, Theron AE, Joubert AM. Antimitotic drugs in the treatment of cancer. Cancer Chemother Pharmacol. 2015, 76, 1101–1112. [Google Scholar] [CrossRef]
- Paier, C.R.K.; Maranhão, S.S.; Carneiro, T.R.; et al. Natural products as new antimitotic compounds for anticancer drug development. Clinics (Sao Paulo) 2018, 73 (suppl 1), e813s. [Google Scholar] [CrossRef] [PubMed]
- Womer, R.B.; West, D.C.; Krailo, M.D.; et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol. 2012, 30, 4148–4154. [Google Scholar] [CrossRef] [PubMed]
- Marina, N.M.; Liu, Q.; Donaldson, S.S.; et al. Longitudinal follow-up of adult survivors of Ewing sarcoma: A report from the Childhood Cancer Survivor Study. Cancer. 2017, 123, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.N.; Chastain, K.; Chou, J.F.; et al. Morbidity and mortality after treatment of Ewing sarcoma: A single-institution experience. Pediatr Blood Cancer. 2017, 64. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef]
- Was, H.; Borkowska, A.; Bagues, A.; et al. Mechanisms of Chemotherapy-Induced Neurotoxicity. Front Pharmacol. 2022, 13, 750507. [Google Scholar] [CrossRef]
- Gartrell, J.; Rodriguez-Galindo, C. Ewing sarcoma: investigational mono- and combination therapies in clinical trials. Expert Opin Investig Drugs. 2021, 30, 653–663. [Google Scholar] [CrossRef]
- Felix, A.; Berlanga, P.; Toulmonde, M.; et al. Systematic review of phase-I/II trials enrolling refractory and recurrent Ewing sarcoma: Actual knowledge and future directions to optimize the research. Cancer Med. 2021, 10, 1589–1604. [Google Scholar] [CrossRef]
- Bailey, K.; Cost, C.; Davis, I.; et al. Emerging novel agents for patients with advanced Ewing sarcoma: a report from the Children's Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Res. 2019, 8. [Google Scholar] [CrossRef]
- Deep, G.; Agarwal, R. New combination therapies with cell-cycle agents. Curr Opin Investig Drugs. 2008, 9, 591–604. [Google Scholar]
- Yan, V.C.; Butterfield, H.E.; Poral, A.H.; et al. Why Great Mitotic Inhibitors Make Poor Cancer Drugs. Trends Cancer. 2020, 6, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic slippage: an old tale with a new twist. Cell Cycle. 2019, 18, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, L.; Wu, Y.; Li, J. KIF11 As a Potential Pan-Cancer Immunological Biomarker Encompassing the Disease Staging, Prognoses, Tumor Microenvironment, and Therapeutic Responses. Oxid Med Cell Longev. 2022, 2022, 2764940. [Google Scholar] [CrossRef] [PubMed]
- Novais, P.; Silva, P.M.A.; Amorim, I.; Bousbaa, H. Second-Generation Antimitotics in Cancer Clinical Trials. Pharmaceutics. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
In silico bioinformatics screen identifies mitotic proteins essential for EWS growth. A) DepMap portal was used to access the RNA-expression data across different cancer cell lines. Capillary based analysis of protein lysates from EWS cell lines indicating expression of B) KIF11 and AURKA, and C) KIF15 and TPX2 protein levels.
Figure 1.
In silico bioinformatics screen identifies mitotic proteins essential for EWS growth. A) DepMap portal was used to access the RNA-expression data across different cancer cell lines. Capillary based analysis of protein lysates from EWS cell lines indicating expression of B) KIF11 and AURKA, and C) KIF15 and TPX2 protein levels.
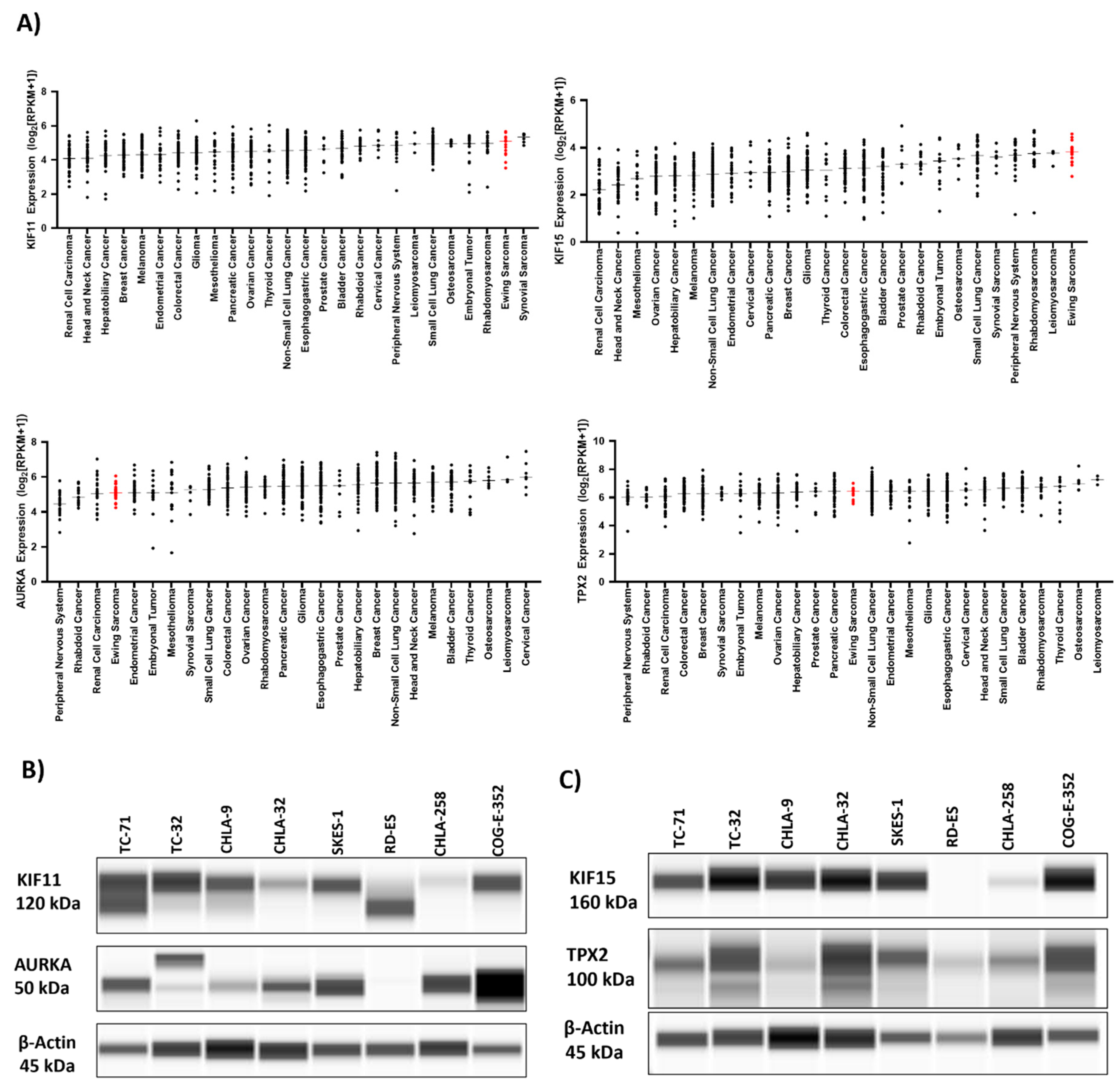
Figure 2.
Synergistic inhibition of EWS growth by VIC-1911 and different KIF11 inhibitors. Dose-response curves, dose-response matrix, and heat map indicating synergy scores in TC-71 EWS cell line. A) SB-743921 and VIC-1911 combination resulted in Bliss synergy score of 24.62 for the highlighted drug combination, B) Ispinesib and VIC-1911 combination resulted in a Bliss synergy score of 19.54 for the highlighted drug combination, and C) Filanesib and VIC-1911 combination resulted in a Bliss synergy score of 16.81 for the highlighted drug combination. Biological triplicates (mean ± SEM, n=3) were used for all the drug combinations tested in the synergy assays.
Figure 2.
Synergistic inhibition of EWS growth by VIC-1911 and different KIF11 inhibitors. Dose-response curves, dose-response matrix, and heat map indicating synergy scores in TC-71 EWS cell line. A) SB-743921 and VIC-1911 combination resulted in Bliss synergy score of 24.62 for the highlighted drug combination, B) Ispinesib and VIC-1911 combination resulted in a Bliss synergy score of 19.54 for the highlighted drug combination, and C) Filanesib and VIC-1911 combination resulted in a Bliss synergy score of 16.81 for the highlighted drug combination. Biological triplicates (mean ± SEM, n=3) were used for all the drug combinations tested in the synergy assays.
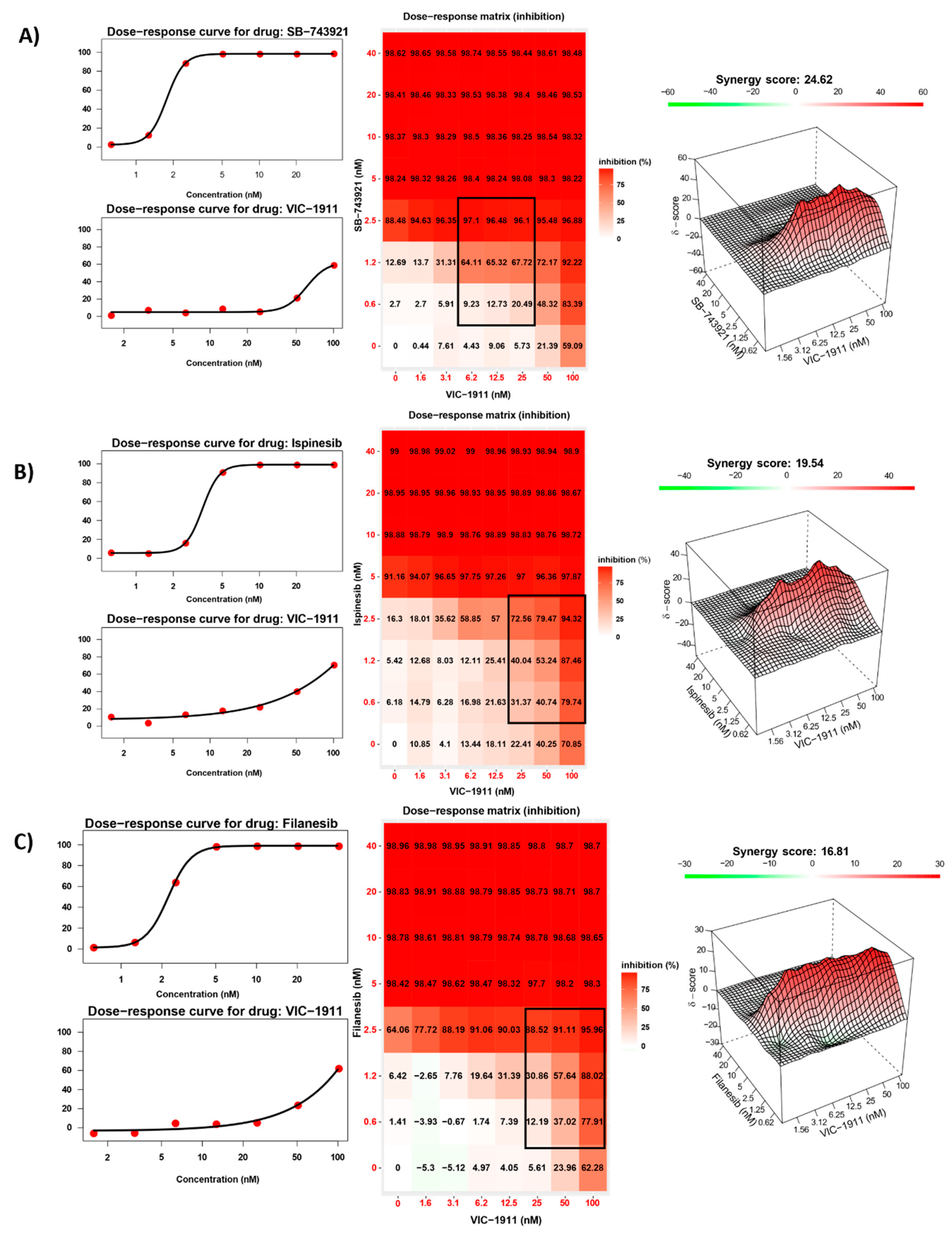
Figure 3.
Synergy is observed in different EWS-ETS fusion bearing cell lines. Dose-response curves, dose-response matrix, and heat map indicating synergy scores in different EWS cell lines. Combination treatment with SB-743921 and VIC-1911 resulted in following synergy scores in A) SK-ES-1 (26.22), B) CHLA-258 (23.29), and C) COG-E-352 (30.13). Biological triplicates (mean ± SEM, n=3) were used for all the drug combinations tested in the synergy assays.
Figure 3.
Synergy is observed in different EWS-ETS fusion bearing cell lines. Dose-response curves, dose-response matrix, and heat map indicating synergy scores in different EWS cell lines. Combination treatment with SB-743921 and VIC-1911 resulted in following synergy scores in A) SK-ES-1 (26.22), B) CHLA-258 (23.29), and C) COG-E-352 (30.13). Biological triplicates (mean ± SEM, n=3) were used for all the drug combinations tested in the synergy assays.
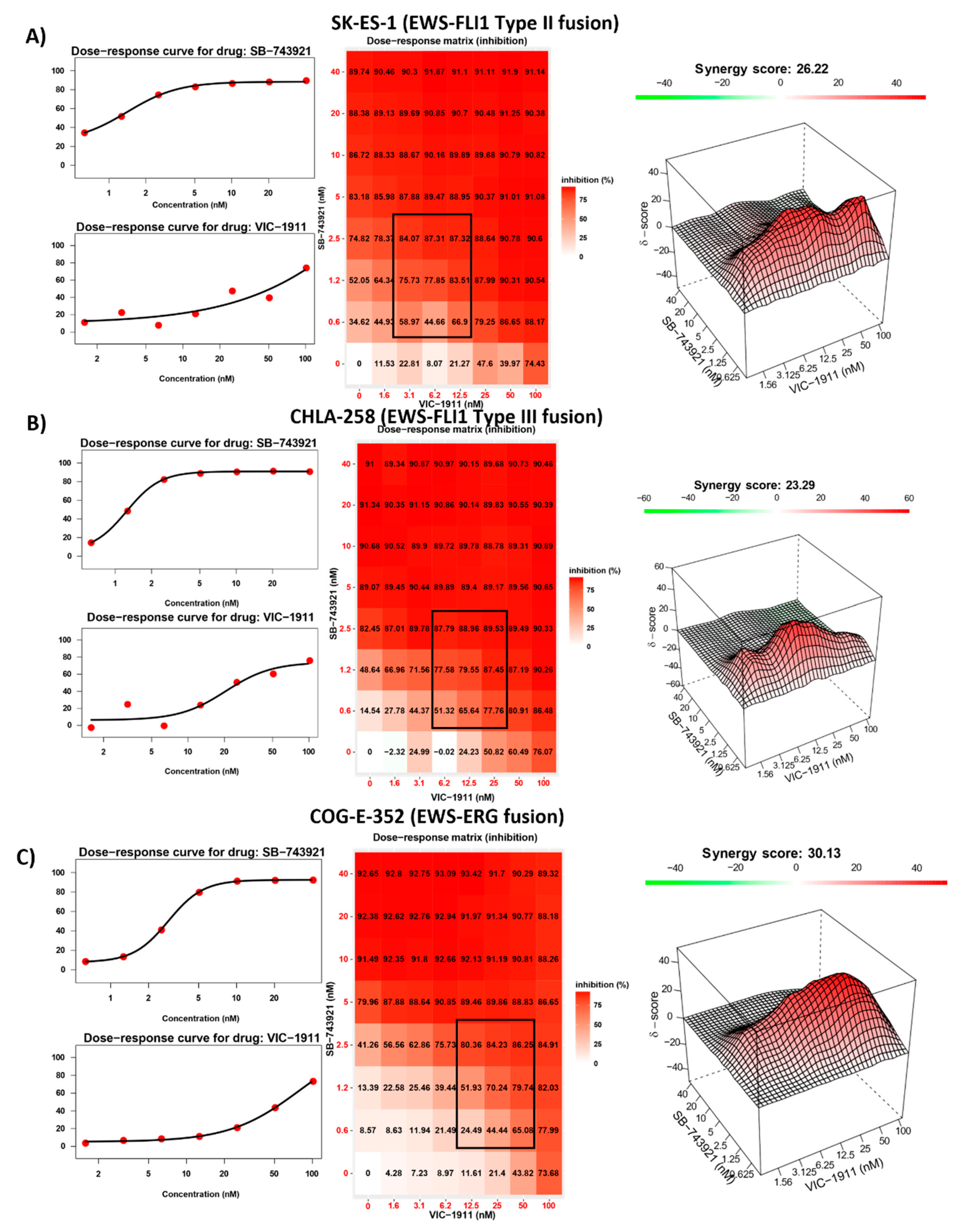
Figure 4.
Colony formation assay upon combination treatment indicates reduced tumorigenicity. SK-ES-1 and TC-71 cells were treated with single drugs and combination and were incubated for one-week post-treatment. Colonies formed were stained with crystal violet and images were taken by Celigo Imager A) Images of triplicates per each condition are represented for Control, SB-743921 (0.625 nM), SB-743921 (0.312nM), VIC-1911 (25 nM), VIC-1911 (12.5 nM) and combination groups SB-743921 (0.625 nM) +VIC-1911 (25 nM) and SB-743921 (0.625 nM) + VIC-1911 (12.5 nM). B) Plating efficiency of cells is represented, p≤0.01**, p-value**** ≤0.0001 is assessed by one-way ANOVA.
Figure 4.
Colony formation assay upon combination treatment indicates reduced tumorigenicity. SK-ES-1 and TC-71 cells were treated with single drugs and combination and were incubated for one-week post-treatment. Colonies formed were stained with crystal violet and images were taken by Celigo Imager A) Images of triplicates per each condition are represented for Control, SB-743921 (0.625 nM), SB-743921 (0.312nM), VIC-1911 (25 nM), VIC-1911 (12.5 nM) and combination groups SB-743921 (0.625 nM) +VIC-1911 (25 nM) and SB-743921 (0.625 nM) + VIC-1911 (12.5 nM). B) Plating efficiency of cells is represented, p≤0.01**, p-value**** ≤0.0001 is assessed by one-way ANOVA.
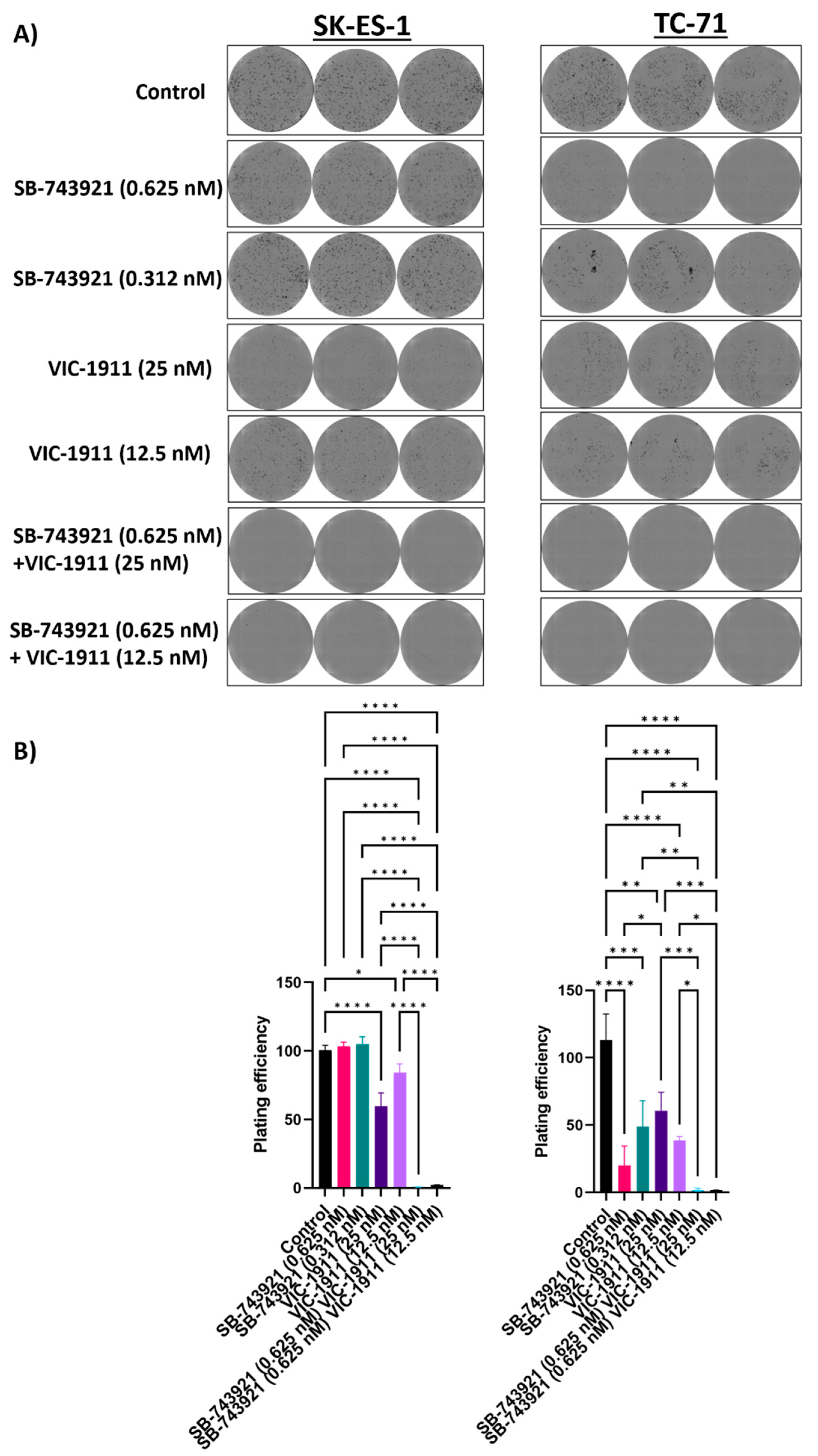
Figure 5.
Combination treatment enhances G2/M cell cycle arrest of EWS cells. Cell cycle analysis was performed on CHLA-10 and TC-71 cells upon drug treatments and cells were assessed for changes in cell cycle profile 24 hours post treatment after Propidium Iodide staining. Different phases of cell cycles are represented for different groups including, A) control, VIC-1911 (10 nM), VIC-1911 (20 nM), SB-743921 (0.5 nM), SB-743921 (1.0 nM) and combination treatment (SB-743921 1.0 nM + VIC-1911 10 nM & SB-743921 1.0 nM + VIC-1911 20 nM). B) Percentage of cells in G2/M phase and subG1 phase of cell cycle is represented by the bar graphs, p≤0.01**, p-value**** ≤0.0001 is assessed by one-way ANOVA.
Figure 5.
Combination treatment enhances G2/M cell cycle arrest of EWS cells. Cell cycle analysis was performed on CHLA-10 and TC-71 cells upon drug treatments and cells were assessed for changes in cell cycle profile 24 hours post treatment after Propidium Iodide staining. Different phases of cell cycles are represented for different groups including, A) control, VIC-1911 (10 nM), VIC-1911 (20 nM), SB-743921 (0.5 nM), SB-743921 (1.0 nM) and combination treatment (SB-743921 1.0 nM + VIC-1911 10 nM & SB-743921 1.0 nM + VIC-1911 20 nM). B) Percentage of cells in G2/M phase and subG1 phase of cell cycle is represented by the bar graphs, p≤0.01**, p-value**** ≤0.0001 is assessed by one-way ANOVA.
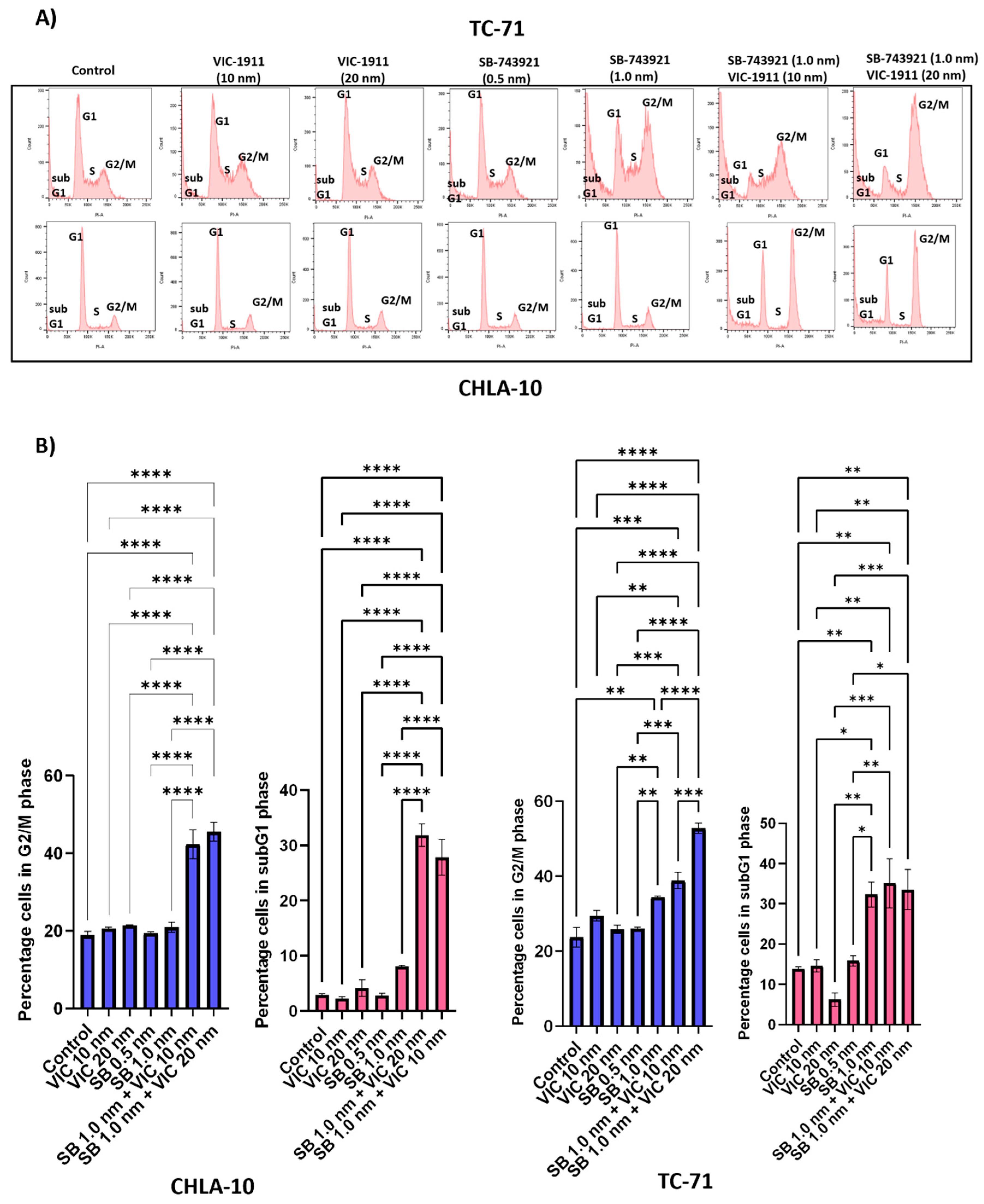
Figure 6.
Analysis of protein expression post-drug treatment. A) CHLA-10 and B) TC-71 cells treated with drugs were assessed for changes in protein expression 24 hours post-treatment via capillary electrophoresis-based Wes analysis. Increased protein levels of KIF11, p-KIF11Thr926 AURKA, and p-AURKAThr288 were observed for the drug combination group, whereas KIF15 levels were noticeably lower. Similarly, enhanced cleaved-PARP expression was observed with the combination treatment.
Figure 6.
Analysis of protein expression post-drug treatment. A) CHLA-10 and B) TC-71 cells treated with drugs were assessed for changes in protein expression 24 hours post-treatment via capillary electrophoresis-based Wes analysis. Increased protein levels of KIF11, p-KIF11Thr926 AURKA, and p-AURKAThr288 were observed for the drug combination group, whereas KIF15 levels were noticeably lower. Similarly, enhanced cleaved-PARP expression was observed with the combination treatment.
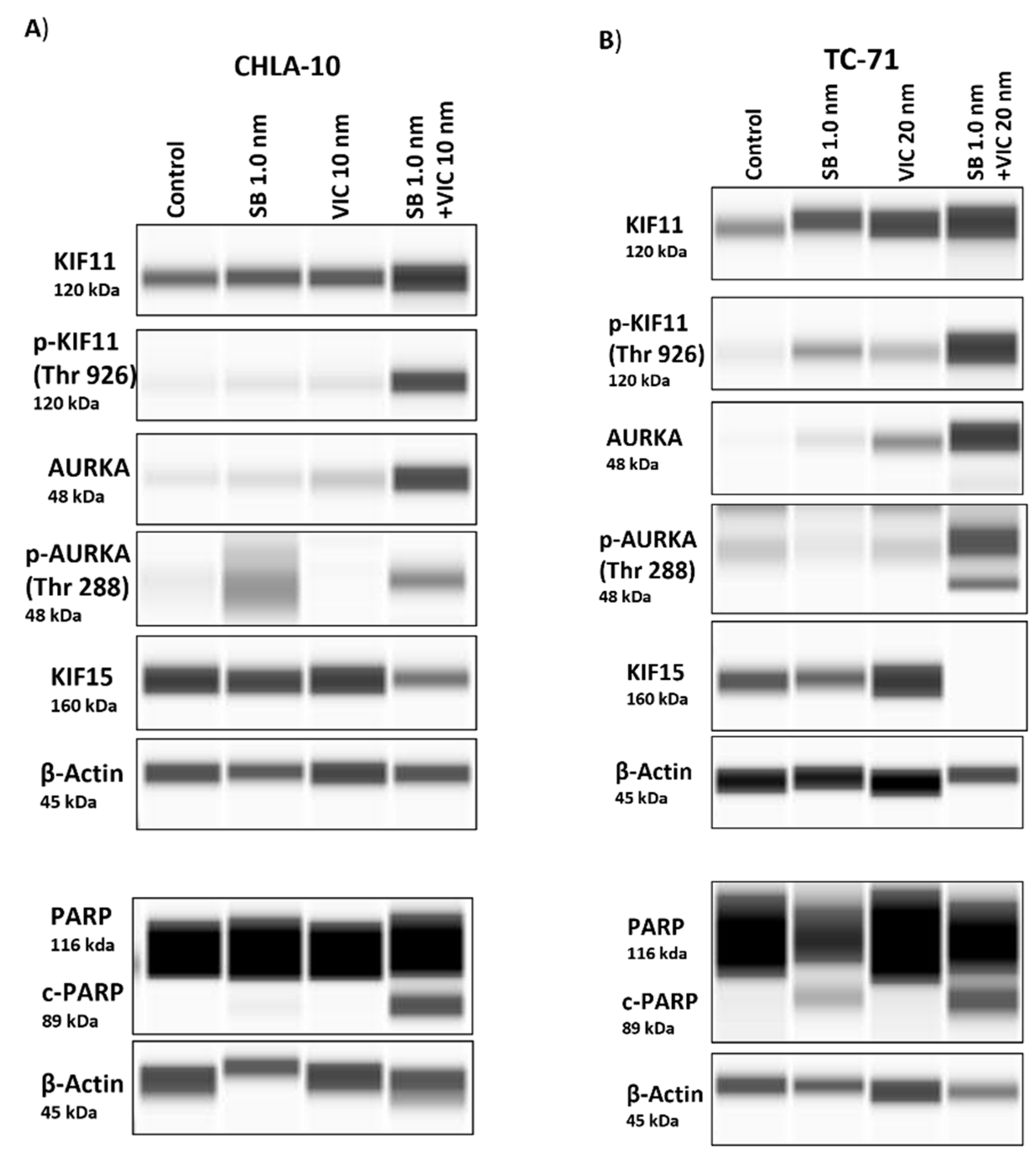

Table 1.
List of top 15 genes identified as essential for EWS tumor progression.
| Gene Rank | Gene ID | Gene symbol | Name | Pathway |
|---|---|---|---|---|
| 1 | 56992 | KIF15 | Kinesin family member 15 | Motor proteins |
| 2 | 1058 | CENPA | Centromere protein A | Mitosis, chromosome segregation and cytokinesis |
| 3 | 7153 | TOP2A | DNA Topoisomerase II alpha | Platinum drug resistance |
| 4 | 5502 | PPP1R1A | Protein phosphatase 1, regulatory (inhibitor) subunit 1A | Adrenergic signaling |
| 5 | 51361 | HOOK1 | Hook microtubule- tethering protein 1 | Vesicle trafficking |
| 6 | 6790 | AURKA | Aurora Kinase A | Oocyte meiosis |
| 7 | 5733 | PTGER3 | Prostaglandin E receptor 3 (subtype EP3) | Calcium signaling |
| 8 | 2619 | GAS1 | Growth-arrest-specific 1 | Membrane trafficking |
| 9 | 23306 | TMEM194A | Transmembrane protein 194A | Nuclear envelope stiffness |
| 10 | 1612 | DAPK1 | Death-associated protein kinase 1 | Autophagy |
| 11 | 29028 | ATAD2 | ATPase family, AAA domain containing 2 | Transcriptional activator |
| 12 | 783 | CACNB2 | Calcium channel, voltage dependent beta 2 subunit | MAPK signaling pathway |
| 13 | 9787 | DLGAP5 | Discs, large (Drosophila) homolog-associated protein 5 | Centrosome and spindle formation |
| 14 | 22974 | TPX2 | TPX2, microtubule-associated homolog (Xenopus laevis) | Regulation of kinetochore-microtubule interactions |
| 15 | 2956 | MSH6 | Muts homolog 6 | Mismatch repair |
1 List of top-15 genes identified to be essential for EWS tumor progression.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



