
Submitted:
01 October 2023
Posted:
01 October 2023
You are already at the latest version
Abstract
Neonatal diffuse cutaneous mastocytosis (NDCM) is defined as the infiltration of the epidermis by a clonal proliferation of mast cells, observed at birth, without initial signs of systemic involvement. The typical driver mutation is located in the KIT gene. We report a rare case of a boy, born at term, presenting already at birth with generalized subcutaneous nodules, on the face, scalp, trunk, back, hands, and feet. The spleen, liver and inflammatory markers were normal at birth. Tryptase was significantly elevated. Bone marrow biopsy showed no mast cell involvement at age 2 months. A punch biopsy at age 2 months revealed CD117-positive cells diffusely infiltrating the skin, with subsequent DNA NGS sequencing for the formalin fixed paraffin embedded tissue (FFPE) identifying the pathogenic NM_000222.3:c.1504_1509dup; p.(Ala502_Tyr503dup) variant in the KIT gene previously associated with cutaneous mastocytosis. At 2 years follow-up, he had splenomegaly and multiple cervical, and inguinal adenopathy while the skin nodules persisted especially on the scalp with accompanying pruritus. He received oral and local sodium cromoglycate, oral antihistamines, antibiotic cream for skin infection, and iron supplementation, however compliance to treatment was relatively low. The prognosis is difficult to predict, as he developed systemic involvement, failure to thrive and mild psychomotor delay. A case aggregation of NDCM reported in the literature was performed to provide a comprehensive overview of this rare pathology, to better understand the prognosis. NDCM is a life-threatening disease with severe complications. Almost half had severe complications, such as mast hepatosplenomegaly, adenopathy, bacterial infections, mast cell leukaemia, and systemic involvement.
Keywords:
Neonatal diffuse cutaneous mastocytosis
; KIT gene
; case report
; systemic mastocytosis
; review
1. Introduction
Cutaneous mastocytosis has been classified into three main types: diffuse cutaneous mastocytosis (DCM), urticaria pigmentosa (UP), and solitary mastocytoma [1]. Of them, DCM is the rarest and the most severe form. Neonatal diffuse cutaneous mastocytosis (NDCM) is a very rare form of the disease, characterized by the infiltration of the epidermis by a clonal proliferation of mast cells observed in the perinatal period, initially without signs of systemic involvement. The typical driver mutation for cutaneous mastocytosis is located in the KIT gene [2,3]. However, few reported cases have identified a molecular variant in the KIT gene [4,5,6,7]. The prognosis is poorly understood, as the disease has significant variability [1,2,3]. Thus, we aimed to present a case with NDCM and perform a literature review to provide a comprehensive overview of this rare pathology to better understand the prognosis.
2. Case report
A male infant, without any prenatal evaluations, was born at term (40 weeks of gestation), birth weight of 3800 grams, length at birth of 51 cm, and an APGAR score of 10. The mother had been smoking daily during pregnancy. His family history was unremarkable, he had healthy parents, a sister with vitiligo, and a brother with asthma. He is the mother’s fifth child. The patient and his family lived in a rural environment. The educational level of the mother was low, she could not read or write, while the father had only completed primary school.
2.1. At birth
He presented generalized subcutaneous nodules, on the face, scalp, trunk, back, hands, and feet. The palms and soles were spared of nodules (Figure 1, panels A, B and C). At age 1 month he showed the persistence of the cutaneous nodules, while the spleen and liver were normal in size. The Darier sign was positive, suggesting mast cell involvement. Clinical, imagistic (ultrasound), and laboratory work-up, including bone marrow biopsy and skin punch biopsy at age 2 months were performed to understand the etiology of this unusual presentation.
2.2. At 2 months of age
The patient weighed 4.9 kilograms, with a body length of 53 centimetres (Figure 2). The abdominal ultrasound showed no signs of visceral malformations, nor organomegaly. The ECG examination revealed a minor right bundle branch block. Blood count was in the normal range. Inflammatory markers had normal levels. He had elevated IgE levels, of 29.5 (normal below 15 UI/ml). Tryptase was significantly elevated at 540 μg/L (normal <11 μg/L). Bone marrow biopsy showed no mast cells. Cardiac echocardiography showed a medium-sized atrial septal defect (ASD) ostium secundum type of 6.7 mm, a left-to-right shunt, right-sided dilatation of the heart chambers, and stenosis of the pulmonary arteries. Pulmonary hypertension was also detected. Thus, a diagnosis of a congenital heart defect was established. Bone marrow aspiration cytology interpretation and flow cytometry evaluation demonstrated hypercellular bone marrow with no mastocyte involvement at age 2 months. A punch biopsy revealed CD117-positive cells diffusely infiltrating the skin, thus the diagnosis of NDCM was raised (Figure 1, panels D-K). Recommendations for oral and local sodium cromoglycate and oral antihistamines were made. Treatment started 1 month later due to difficulties in access to the drug. The patient’s mother was advised that the baby needs urgent medical attention in case of skin redness, diarrhea and/or vomiting with acute onset, or general malaise as they could represent a sign of systemic degranulation by the mastocytes. Additionally, a list of possible triggers for mastocyte degranulation was provided to the mother.
Subsequent DNA analysis, using Illumina Whole Exome Next Generation Sequencing was performed from FFPE–punch skin biopsy. Genomic DNA was quantified using the Qubit 4.0 fluorimetric Assay (Thermo Fisher Scientific) and sample integrity, based on the DIN (DNA integrity number), was assessed using a Genomic DNA ScreenTape assay on TapeStation 4200 (Agilent Technologies). Libraries were prepared from 100 ng of total DNA using the NEGEDIA Exome clinical grade sequencing service (Next Generation Diagnostic srl) which included library preparation, target enrichment using the Agilent V7 probe set and quality assessment. Sequencing was completed on a NovaSeq 6000 sequencing system using a paired-end, 2x150 cycle strategy (Illumina Inc.). The raw data were analyzed by Next Generation Diagnostics srl Whole Exome Sequencing pipeline (v1.0) which involves a cleaning step by UMI removal, quality filtering and trimming, alignment to the reference genome, removal of duplicate reads and variant calling (8-11). Variants were annotated using the Ensembl Variant Effect Predictor (VEP) tool5 (v. 104) (12). Variant filtering included variants with coverage>50X, population frequency < 0.1% in gnomAD, impact on protein, various in silico prediction models, and biological databases (Clinvar, COSMIC v94, ONKOKB). The median coverage of the sample was 197X, with 98.3% of bases covered >50X. DNA sequencing identified a pathogenic (13), heterozygous variant in the KIT gene NM_000222.3:c.1504_1509dup; p.(Ala502_Tyr503dup), previously associated with cutaneous mastocytosis (2). Therefore, the diagnosis of NDCM was confirmed.
2.3. At age 4 months
The patient weighed 5.7 kilograms with a body length of 61 centimetres (Figure 2). His bloodwork showed iron deficiency and anaemia. Cardiac echocardiography showed a diminished ASD of 6.3 mm and additional mitral and tricuspid valve regurgitation. Treatment included iron and calcium supplementation, antihistaminic (cetirizine), and local application and oral disodium cromoglycate.
2.4. At age 1 year and 2 months
The patient presented to the hospital with respiratory symptoms compatible with pneumonia. He weighed 8.5 kilograms, with a body length of 70 cm (Figure 2). Bacterial infection of the nodular lesions of the scalp, and secondary occipital and cervical adenopathy were present. Abdominal ultrasound examination revealed an enlarged spleen (splenomegaly) of 7.9 cm (normal 6.13±0.13cm). Additionally, right iliac fossa adenopathy and right and left inguinal adenopathy were noted. He had elevated inflammatory markers (C Reactive Protein 54 mg/l) and reactive leukocytosis. He had normal levels of IgA, IgG and IgM. He received antibiotic treatment, with subsequent improvement.
2.5. At age 1 year and 8 months
The patient presented to the hospital due to fever and ear pain. His weight was 9.5 kilograms and his body length of 78 cm (Figure 2). Bilateral otitis externa and adenotonsillar hypertrophy were diagnosed. Inflammatory markers were mildly elevated (11.5mg/l). Bone marrow biopsy could not be performed, due to sedation constraints during respiratory infection. A chest X-ray revealed bilateral perihilar nodular lung opacities with a tendency to confluence. Ultrasound of the soft tissues demonstrated bilateral cervical adenopathy of approximately 11 mm, relatively hypoechoic on both sides of the neck. Abdominal ultrasound showed a liver with a homogeneous echostructure and normal size. The spleen was enlarged, measuring 8.6 cm (normal 6,45±0.17 cm). The neurological examination showed delayed motor development, as he could maintain orthostatism independently only for a few seconds and could take steps with bilateral support. He did not have cerebellar or extrapyramidal signs. The psychological examination showed a mild delay in cognitive acquisitions, as expressive language included three words with a sense of communication, while receptive language was appropriate.
2.6. At age 2 years
The mother came with the boy for an outpatient care consultation yet refused hospital admittance despite the significant medical worry for the later-cervical tumor of more than 10 cm in diameter. At his age, he was still breastfeeding alongside eating various solid foods. The nodes had diminished on the face, yet the scalp had hundreds of nodules, apparently causing pruritus, as there were multiple scratching lesions that were infected (Figure 1, panels L, M and N). At the time of that consultation, the patient was not receiving the cromoglycate treatment. He was subsequently lost to follow-up.
3. Literature review
A case aggregation of NDCM reported in the literature was performed to provide a comprehensive overview of this rare pathology [6,7,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. The literature review revealed a total of 31 cases of NDCM, including the present case, reported from 1957 to 2020 are shown in summary in Table 1 and with details in supplementary Table 1 [6,7,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. The male-to-female ratio of these cases is 20:11. The most common symptom was diffuse erythema and multiple nodules on the skin. Almost half of the cases had systemic involvement (defined by the involvement of another organ/system other than skin). Laboratory work-up for diagnosis was complex. Elevated blood and/or urine markers (serum tryptase, serum histamine, and urine N-methylhistamine) were observed in half of the cases. Bone-marrow biopsy showed involvement, in half of cases where it was performed (7/31). Skin biopsy was performed in most cases (27/31), yet special stains (Toluidine Blue and Giemsa most used) were used only in 12 cases, while immunohistochemistry (IHC) was performed in 8 cases. In the literature, variants in the KIT gene reported in NDCM cases were identified in only 4 other cases, as follows: three cases p.(Asp816Val), and dup A502Y503 in one case [4,5,6,7]. Some reported cases of NDCM had negative molecular genetic testing results (5 cases), while for the majority genetic testing was not performed (Table 1). The main treatment involved antihistamines and/or cromoglycate in most cases (23/31) with systemic corticoids for systemic manifestations (8 cases), 1 case with Vitamin K and 1 case with Iron preparation. The longest follow-up reported was 27 years, and the vital status at follow-up was mostly alive. However, data extracted from literature reports cannot accurately show disease progression.
4. Discussion
NDCM is a rare form of cutaneous mastocytosis that presents a wide range of clinical manifestations, diagnostic findings, and treatment approaches. This discussion aims to compare the findings from a recent case with those from a comprehensive review of 31 cases in the literature.
The recent case shows a male patient, consistent with the male predominance observed in the literature [4,5,6,7,13,16,17,18,19,22,23,26,28,29,30]. The positive Darier sign in the present case aligns with most of the reviewed cases [5,6,7,14,15,16,17,20,21,23,25,28,29,30,31,32], indicating the commonality of this feature in NDCM. Clinical presentation and severity of disease show significant variability between the 31 reviewed cases. Systemic involvement was absent in the present case in the first year, yet developed after the age of 1 year, consistent with the literature where systemic involvement was reported in cases by Shah et al [4], Heide et al [5], Waters et al [17], Allison et al [19], Burgoon et al [20], Harrison et al [22]. However, cases with and without systemic involvement were reported, showing variability in the presentation of NDCM.
Diagnostic markers (serum tryptase, serum and urine histamine, urine N-methylhistamine) in the present case were elevated blood tryptase, similarly with cases reported by Heide et al [5], Chaudhary et al [6], Jenkinson et al [7], Mann et al [26], Walker et al [27], Duckworth et al [28], Koga et al [29], Lange et al [30], Ghiasi et al [31], Folch et al [33], and Turnbull et al [34] that reported elevated markers. However, urine N-methylhistamine was not performed in this case. The bone marrow biopsy in the present case did not show involvement, aligning with most of the reviewed cases [4,6,20,27,31].
The special stains used in the histologic evaluation of biopsies, particularly Toluidine Blue, in the present case are consistent with the literature (6,14,15,20,21,23,27,29,31,34). However, ultrastructural analysis was not conducted in the present case, similar to most of the reviewed cases, as this type of analysis is largely inaccessible and not necessary for diagnosis [14,15]. IHC in the present case showed diffuse positivity for CD117 (c-KIT), a finding that aligns with 4 cases in the literature [30,32,34]. The identification of the KIT - c.1504_1509dup p.(Ala502_Tyr503dup), as to our knowledge, was not reported before in the literature [6,7,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. A small number (4 out of 31) of cases in the literature reported a KIT pathogenic variant. Specifically, the reported KIT variants were p.(Asp8l6Val) in 3 cases and KIT dup A502Y503 in one case [4,5,6,7]. Some authors have only tested the p.(Asp8l6Val) variant, without KIT gene sequencing, therefore this approach could explain the negative results. In a study by Bibi et al. 2014, 75% of pediatric patients had variants in KIT, supporting the assumption that childhood mastocytosis is a clonal disease [1]. The authors noted that the spectrum of the disease and variants are limited compared to those seen in adults [1]. With further characterization of molecular causes in NDCM, future genotype-phenotype correlations could be proposed.
The treatment in the present case included Cromoglycate and antihistamines, aligning with most of the cases in the literature [4,5,6,16,25,29,30,32,33,34]. Although Cromoglycate is the mainstream medication for managing the symptoms of NDCM, the treatment efficiency in preventing complications is limited in some cases. Further research is needed to provide improved therapeutic approaches. The present case developed complications, including splenomegaly and cervical adenopathy, after 2 years of follow-up. This aligns with the literature, where complications were reported in cases by Shah et al [4], Heide et al [5], Chaudhary et al [6], Willemze et al [14], Sethuraman et al [16], Waters et al [17], Yasuda et al [18], Allison et al [19], Burgoon et al [20], Klaber et al [21], Fernandes et al [25], Lange et al [30], Ghiasi et al [31], and Folch et al [33]. In terms of follow-up duration, the present case had a follow-up of 2 years, which is within the range observed in the literature. The follow-up durations in the literature varied widely, from as short as 8 days to as long as 27 years [14]. This wide range underscores the chronic nature of NDCM and the need for long-term monitoring and management.
5. Conclusion
The present case provides additional insights into the variability of clinical presentation, diagnosis, and management of this precocious and severe form of neonatal diffuse cutaneous mastocytosis. The variant c.1504_1509dup in the KIT gene has not been reported before, in association with this form of disease. The case aggregation of reported affected individuals in the literature provides a comprehensive overview of this rare pathology. Most of the affected individuals had severe complications, such as mast hepatosplenomegaly, adenopathy, bacterial infections and/or mast cell leukaemia. Systemic involvement was observed in almost half. A gene-to-phenotype correlation cannot be made with this limited number of cases. Several cases presented did not have genetic testing performed at all, or limited genetic testing was performed only for the p.(Asp8l6Val) variant. With further characterization of molecular causes in diffuse cutaneous mastocytosis, future genotype-phenotype correlations could be proposed. Additionally, the skewed male-to-female ratio (2:1), raises the possibility that sex differences could play a role in pathogenesis. Despite existing treatment with Cromoglycate, more effective therapeutic strategies are needed.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1.
Author Contributions
E.G.O, M.B, M.P, and A.C.E were involved in patient diagnosis. M.B. was involved in treatment management. E.G.O and A.C.E drafted the manuscript. All authors revised the material.
Funding
This research received no funding.
Institutional Review Board Statement
The study involving human participants was reviewed and approved by the Ethics Committee of the Emergency Hospital for Children “Louis Turcanu” Timisoara. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin (patient’s mother) for the publication of any potentially identifiable images or data included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding authors. The data is not publicly available due to privacy or ethical restrictions.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Bibi S, Langenfeld F, Jeanningros S, Brenet F, Soucie E, Hermine O et al. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am. 2014; 34:239–262. [CrossRef]
- Chatterjee A., Ghosh J., Kapur R. Mastocytosis: a mutated KIT receptor-induced myeloproliferative disorder. Oncotarget. 2015; 6: 18250-18264. [CrossRef]
- Frieri M, Quershi M. Pediatric Mastocytosis: A Review of the Literature. Pediatr Allergy Immunol Pulmonol. 2013 Dec 1;26(4):175-180.5. [CrossRef]
- Shah PY, Sharma V, Worobec AS, Metcalfe DD, Zwick DC. Congenital bullous mastocytosis with myeloproliferative disorder and c-kit mutation. Journal of the American Academy of Dermatology. 1998 Jul 1;39(1):119-21. [CrossRef]
- Heide R, Zuidema E, Beishuizen A, Den Hollander JC, Van Gysel D, Seyger M et al. Clinical aspects of diffuse cutaneous mastocytosis in children: two variants. Dermatology. 2009 Nov 1;219(4):309-15. [CrossRef]
- Chaudhary N, Shapiro N, Bhutada A, Rastogi S. c-KIT-positive fatal diffuse cutaneous mastocytosis with systemic manifestations in a neonate. Journal of Pediatric Hematology/Oncology. 2019 Jul 1;41(5):e338-40. [CrossRef]
- Jenkinson HA, Lundgren AD, Carter MC, Diaz LZ, Levy ML. Management of a neonate with diffuse cutaneous mastocytosis: case report and literature review. Pediatric Dermatology. 2019 Jul;36(4):486-9. [CrossRef]
- Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014 Aug 1;30(15):2114-20. [CrossRef]
- Smith T, Heger A, Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome research. 2017 Mar 1;27(3):491-9. [CrossRef]
- Freed D, Aldana R, Weber JA et al. The Sentieon Genomics Tools–A fast and accurate solution to variant calling from next-generation sequence data. BioRxiv. 2017 Mar 10:115717. [CrossRef]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome biology. 2016 Dec;17(1):1-4. [CrossRef]
- Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine. 2015 May;17(5):405-23. [CrossRef]
- Willemze R, Ruiter DJ, Scheffer E, Van Vloten WA. Diffuse cutaneous mastocytosis with multiple cutaneous mastocytomas: report of a case with clinical, histopathological and ultrastructural aspects. British Journal of Dermatology. 1980 May 1;102(5):601-7. [CrossRef]
- Oku T, Hashizume H, Yokote R, Sano T, Yamada M. The familial occurrence of bullous mastocytosis (diffuse cutaneous mastocytosis). Archives of dermatology. 1990 Nov 1;126(11):1478-84. [CrossRef]
- Sethuraman G, Handa S, Radotra B, Kumar B. Diffuse cutaneous mastocytosis with bullae or bullous mastocytosis: a question of semantics. Pediatric Dermatology. 1999 Sep;16(5):409-11. [CrossRef]
- Waters WJ, Lacson PS. Mast cell leukemia presenting as urticaria pigmentosa: report of a case. Pediatrics. 1957 Jun 1;19(6):1033-42. [CrossRef]
- Yasuda T, Kukita A. A fatal case of purely cutaneous form of diffuse mastocytosis. Proc XII Int Cong Dermatol. 1962;2:1558-61.
- Allison J. Skin mastocytosis presenting as a neonatal bullous eruption. Australasian Journal of Dermatology. 1967 Jun;9(1):83-5. [CrossRef]
- Burgoon CF, Graham JH, McCaffree DL. Mast cell disease: a cutaneous variant with multisystem involvement. Archives of dermatology. 1968 Dec 1;98(6):590-605. [CrossRef]
- Klaber M. Diffuse cutaneous mastocytosis in mother and daughter. Proc R Soc Med 1976;69:16–18.
- Harrison PV, Cook LJ, Lake HJ et al. Diffuse cutaneous mastocytosis: a report of neonatal onset. Acta Derm Venereol 1979;59:541–543.
- Enomoto, Kusakabe, Matsumura, Kuno, Tamai, Kiyokane. Diffuse cutaneous mastocytosis responding to cyproheptadine. Clinical and experimental dermatology. 1999 Jan 1;24(1):16-8. [CrossRef]
- Hannaford R, Rogers M. Presentation of cutaneous mastocytosis in 173 children. Australasian journal of dermatology. 2001 Feb;42(1):15-21. [CrossRef]
- Fernandes EI, de Faria BC, Cartell A, dos Santos BA, Cestari TF. Systemic mastocytosis in childhood: report of 3 cases. J Pediatr (Rio J). 2002 Mar 1;78(2):176-80.
- Mann C, Sepp N, Simma B. Congenital cutaneous mastocytosis. The Journal of pediatrics. 2004 Jul 1;145(1):134. [CrossRef]
- Walker T, Von Komorowski G, Scheurlen W, Dorn-Beineke A, Back W, Bayerl C. Neonatal mastocytosis with pachydermic bullous skin without c-Kit 816 mutation. Dermatology. 2005 Nov 1;212(1):70-2. [CrossRef]
- Duckworth AK, Bhatti A, Barnes C. Diffuse cutaneous mastocytosis in fraternal twins. International journal of dermatology. 2009 Feb;48(2):170-2. [CrossRef]
- Koga H, Kokubo T, Akaishi M, Iida K, Korematsu S. Neonatal onset diffuse cutaneous mastocytosis: a case report and review of the literature. Pediatric dermatology. 2011 Sep;28(5):542-6. [CrossRef]
- Lange M, Niedoszytko M, Nedoszytko B, Łata J, Trzeciak M, Biernat W. Diffuse cutaneous mastocytosis: analysis of 10 cases and a brief review of the literature. Journal of the European Academy of Dermatology and Venereology. 2012 Dec;26(12):1565-71. [CrossRef]
- Ghiasi M, Ghanadan A, Jesri SB, Sotudeh S, Ramyar A. Diffuse cutaneous mastocytosis: report of a severe case with fatal outcome. Dermatology Online Journal. 2011 Mar 1;17(3). [CrossRef]
- Park MN, Kim GA, Chey MJ, Shim GH. A case of diffuse cutaneous mastocytosis in a newborn. Korean Journal of Perinatology. 2014 Jun 1;25(2):105-9.
- Folch BS, Almaraz RL, González RS, de Las Heras BM. Diffuse cutaneous mastocytosis. Presentation of 3 cases and therapeutic management review. Anales de Pediatr a (English Edition). 2016;5(84):286-8. [CrossRef]
- Turnbull L, Calhoun DA, Agarwal V, Drehner D, Chua C. Congenital mastocytosis: Case report and review of the literature. Cureus. 2020 Sep 21;12(9). [CrossRef]
- (WHO Anthro app Software for assessing growth of the world's children and adolescents. Geneva: WHO, 2009 (https://www.who.int/tools/growth-reference-data-for-5to19-years).
Figure 1.
Clinical presentation at age 1 month with generalized subcutaneous nodules, including the face, scalp, hands, and feet (Panels A, B, C). Darier’s sign was positive, suggesting mast cell degranulation (ovals Panel A). Punch biopsy of two skin nodules (Panel C arrows indicating the site of the punch biopsy), showed a thin epidermis and diffuse infiltration of mast cells in the superficial dermis with extension in the deep dermis, some forming aggregates around adnexal structures and blood vessels. Panel E shows aggregates of mast cells in the superficial dermis, panel F arrows show bundles of collagen, and panel G oval and arrow indicating perivascular aggregation of mast cells (Panels D-K). Toluidine Blue showed mast cells with intracytoplasmic purple granules (Panel H). The mast cells were diffusely positive for CD117 (Panels I-K). Panel L to N clinical presentation at the age of 2 years. Panel L shows aggregates of nodules on the scalp, panel M shows the thorax and abdomen with less visible signs of nodules, and panel N legs and feet with visible plaque-like lesions showing hyperkeratosis.
Figure 1.
Clinical presentation at age 1 month with generalized subcutaneous nodules, including the face, scalp, hands, and feet (Panels A, B, C). Darier’s sign was positive, suggesting mast cell degranulation (ovals Panel A). Punch biopsy of two skin nodules (Panel C arrows indicating the site of the punch biopsy), showed a thin epidermis and diffuse infiltration of mast cells in the superficial dermis with extension in the deep dermis, some forming aggregates around adnexal structures and blood vessels. Panel E shows aggregates of mast cells in the superficial dermis, panel F arrows show bundles of collagen, and panel G oval and arrow indicating perivascular aggregation of mast cells (Panels D-K). Toluidine Blue showed mast cells with intracytoplasmic purple granules (Panel H). The mast cells were diffusely positive for CD117 (Panels I-K). Panel L to N clinical presentation at the age of 2 years. Panel L shows aggregates of nodules on the scalp, panel M shows the thorax and abdomen with less visible signs of nodules, and panel N legs and feet with visible plaque-like lesions showing hyperkeratosis.
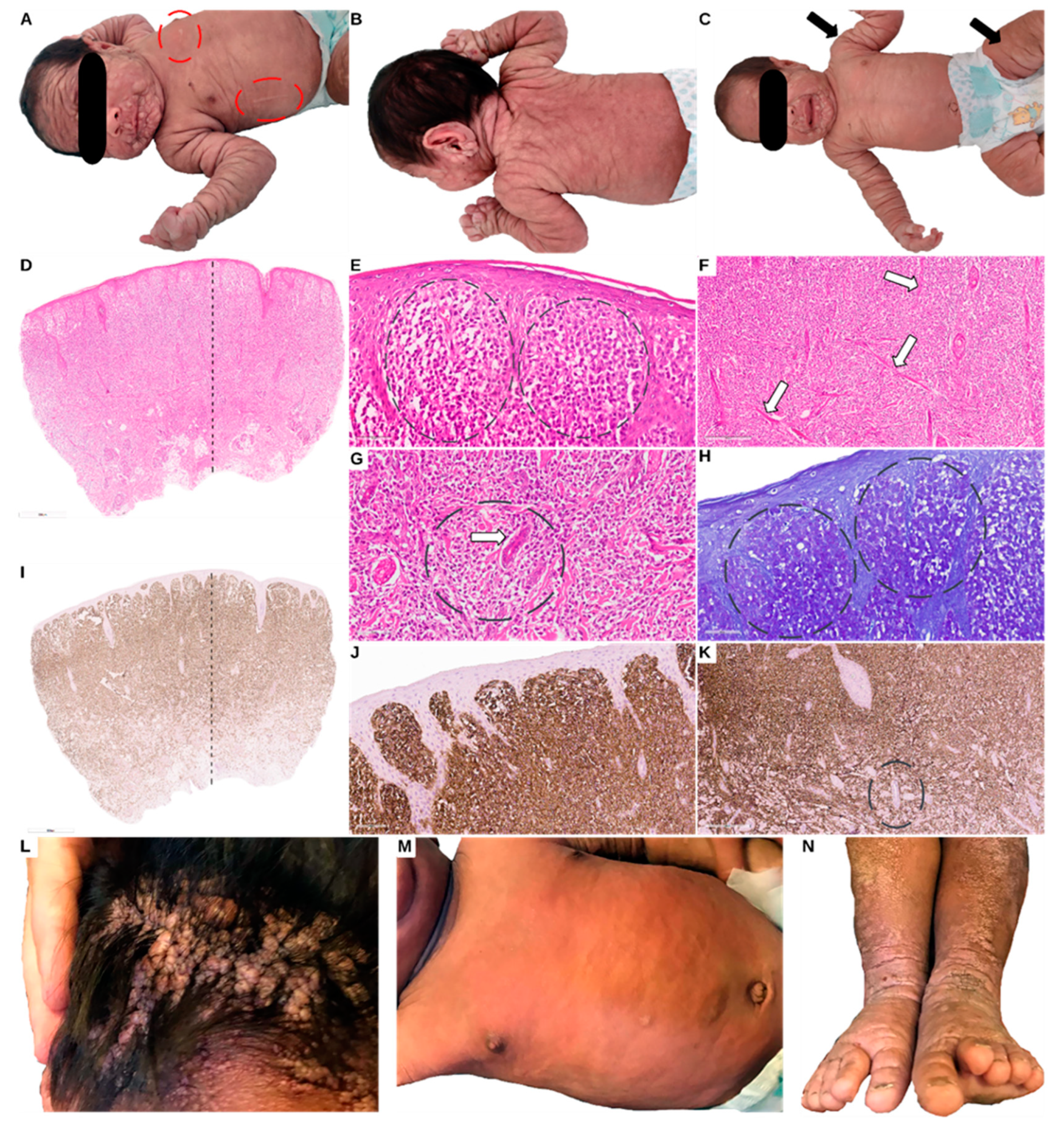
Figure 2.
Plotted growth parameters of the patient, using the World Health Organization reference (35).
Figure 2.
Plotted growth parameters of the patient, using the World Health Organization reference (35).
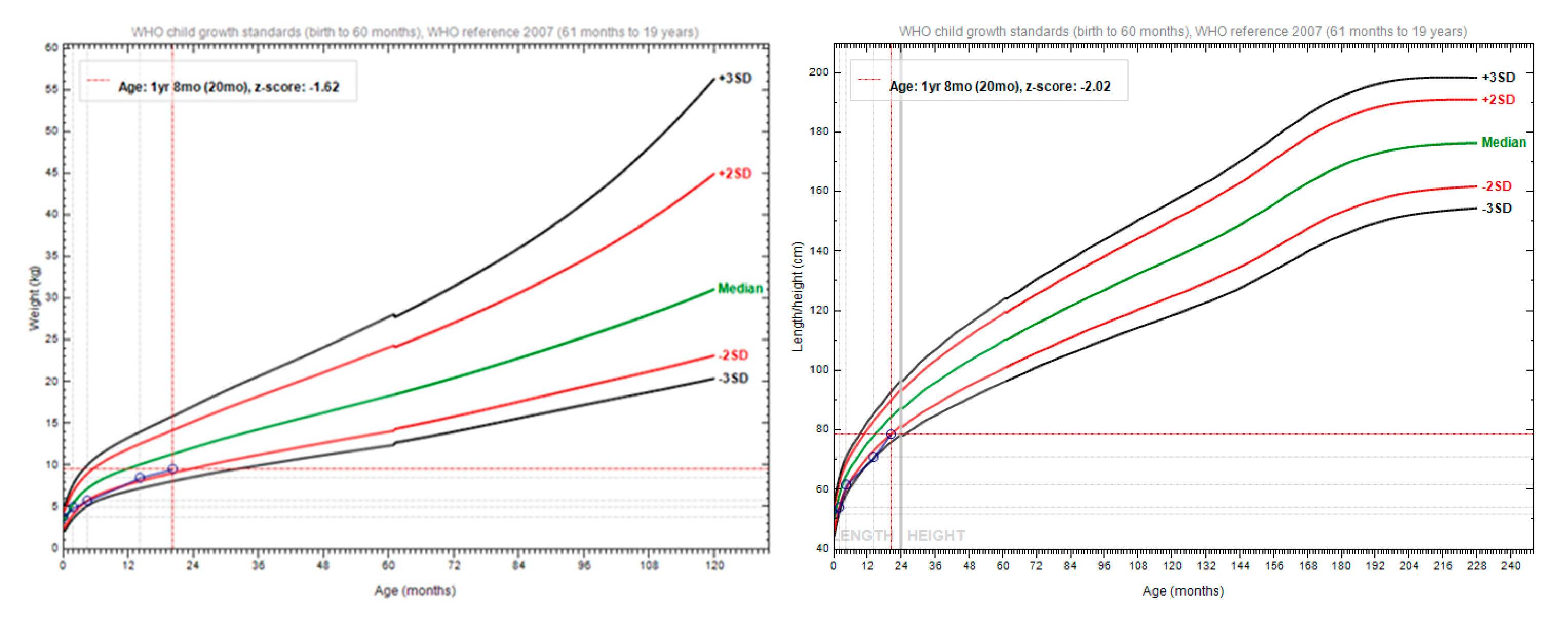

Table 1.
Literature review: summary of 31 cases of NDCM reported in the literature reported from 1957 to 2020. NA - not available. Minus (-) no information was found.
Table 1.
Literature review: summary of 31 cases of NDCM reported in the literature reported from 1957 to 2020. NA - not available. Minus (-) no information was found.
| Case aggregation of DCM from the literature | Present case | |
| Number of cases | 31 | 1 |
| Sex (Male/Female) | 19 M, 11 F, 1 NA | M |
| Darier sign (positive, negative or no information) | 20 Positive, 1 Negative, 10 and NA | Positive |
| Systemic involvement (SI) | 9 had SI, 13 without SI, and 9 NA | Yes |
| Positive (elevated) blood and/or urine markers | 15 had elevated markers, 7 had normal markers, and 9 NA |
Yes, elevated serum tryptase |
| Bone-marrow biopsy (BMB) | 7 had BMB with 4/7 with involvement, 13 without BMB, and 11 NA |
Yes - no involvement |
| Biopsy | 27 had a biopsy performed (skin the most common), 2 without biopsy, 2 NA |
Yes - skin |
| Special stains | 12 had special stains, (Toluidine Blue and Giemsa most commonly used), 8 had no special stains, 11 NA | Toluidine Blue |
| Ultrastructural analysis (UA) | 2 had UA, 26 without UA, and 3 NA | No |
| IHC | 5 had IHC for CD117, 3 had IHC for Tryptase, 9 without IHC, 14 NA | CD117 (c-kit) - diffuse positivity |
| Variant testing | KIT - p.(Ala502_Tyr503dup) - present case, 3 cases with KIT p.(Asp8l6Val), KIT dup A502Y503 | KIT - c.1504_1509dup p.(Ala502_Tyr503dup) |
| Treatment | The main treatment was antihistamines and/or cromoglycate (23 cases) with systemic corticoids for systemic manifestations (8 cases), 1 case with Vitamin K and 1 case with Iron preparation, and 6 cases with NA | Cromoglycate |
| Follow-up | The longest follow-up was 27 years | 2 years |
| Complications | Complications were reported in 17 cases with the most common spleen and/or hepatomegaly, 10 had none reported, 4 NA | Splenomegaly and cervical adenopathy |
| Vital status at follow-up (alive or dead) |
24 were alive at follow-up, 6 were dead with 1 death of unknown cause |
Alive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



