
Submitted:
01 October 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
Cisplatin (CDDP), an important chemotherapeutic agent, could result in potential hepatotoxicity, but the precious molecular mechanisms remain unclear. In this study, the protective effect of ellagic acid (EA) on CDDP exposure-induced hepatotoxicity and underlying molecular mechanisms were investigated using a mouse model. Mice were randomly divided into control, CDDP model, EA100 (i.e., EA 100 mg/kg/day), and CDDP plus EA 25, 50, and 100 mg/kg/day. Mice in all CDDP-treated groups were intraperitoneally injected with CDDP 20 mg/kg/day for 2 days. In all EA co-treatments, mice were orally administrated with EA for 7 days. Our results found that, compared to the control group, CDDP treatment resulted in liver dysfunction, oxidative stress, and hepatocyte necrosis, which are effectively revised by EA supplementation in a dose-dependent manner. Meanwhile, EA supplementation inhibited CDDP-induced the elevation of caspases-9 and -3 activities in the liver tissues of mice. Furthermore, EA supplementation significantly downregulated CDDP exposure-induced the increases of NF-κB, IL-1β, TNF-α, and IL-6 proteins and mRNAs, while further upregulated the expression of Nrf2, and HO-1 proteins and mRNAs. Taken together, our results reveal that EA supplementation could ameliorate CCDP-induced liver injury in mice via the opposite regulation of Nrf2/HO-1 and NF-kB pathways.
Keywords:
Ellagic acid
; cisplatin
; hepatotoxicity
; Nrf2/HO-1 pathway
; NF-κB pathway
1. Introduction
Cisplatin (cis-diamminedichloroplatinum II, CDDP) has been one of anticancer-based drugs and is widely used for treating human malignancies, such as sarcomas, malignant epithelial tumors, lymphoma, and germ cell tumors [1]. In clinical practices, CDDP uses usually cause the certain toxic damage and adverse effects, such as nephrotoxicity, neurotoxicity, hepatotoxicity, and reproductive toxicity [1,2,3,4]. These unwanted adverse effects usually limit its clinical application and outcomes. Therefore, the investigations on the molecular mechanisms of CDDP-caused toxic adverse effects and developing the effective protective agents are important and urgent.
It is known that CDDP can promote oxidative stress and crosslink with purine bases of DNA, finally result in mitosis and apoptotic cell death in cancer cells [5]. These actional mechanisms also contributed to explain the toxic effects of CDDP in normal cells or tissues [5]. In mammalian cells, liver is the main organ that involves the most metabolic reactions. CDDP could rapidly reach a higher concertation in the liver of patients and caused acute or chronic liver injury [5]. A series of studies have demonstrated that CDDP exposure could induce oxidative stress, inflammatory response, and apoptotic cell death in the livers of mice or rats [6,7]. Multiple signaling pathways, including cytochrome P450 2E1 (CYP2E1) pathway, nuclear factor erythroidderived 2-like 2 (Nrf2) pathway, mitochondrial apoptotic pathway, mitogen-activated protein kinase (MAPK) pathway, nuclear factor-κB (NF-κB) pathway, have been demonstrated to patriciate in regulating CDDP-induced hepatotoxicity [8,9,10,11]. Recently, several studies found that some natural product or medical plant extracts (including protocatechuic acid vitamin C, xanthorrhizol, 7-hydroxycoumarin, zingerone, Schisandra chinensis bee pollen extract, and Sonchus cornutus extract) could offer the effective protections against CDDP exposure-induced liver toxicity in mice or rats via targeting these mentioned pathways [9,10,11,12,13,14,15]. As so far, the discovery and development of protective agents from natural products against CDDP-caused adverse effects have received widespread attentions in worldwide.
Ellagic acid (EA), a bioactive polyphenolic compound, has been isolated from various gallnuts and fruits, such as black currants, pomegranate, raspberries, and mango [16]. It has been reported that EA possessed various pharmacological activities, such as anti-oxidative stress, anti-inflammatory, anticancer, antibacterial, immune-regulation, and anti-aging activities [17,18,19,20,21,22,23]. Qi et al found that oral administration of EA treatment at various dose (i.e., 50, 100, or 200 mg/kg/day for 3 weeks) could improve paraquat exposure-caused inflammatory cells infiltration, oxidative stress, apoptotic cell death in the livers of pigs by improving the gut microbial profile [24]. In another study, Aslan and colleagues found that EA supplementation could effectively attenuate chronic exposure of carbon tetrachloride (CCl4)-induced liver and kidney injuries in rats via the inhibition of oxidative stress through upregulating Nrf2 pathway and downregulating NF-κB pathway [25,26]. To date, the underlying molecular mechanisms of EA protecting against CDDP-induced toxic adverse effects remain unclear. Therefore, this current study is aimed to investigate the interventional effects of EA supplementation on CDDP exposure-induced liver toxicity and the potential molecular mechanisms using a mouse model.
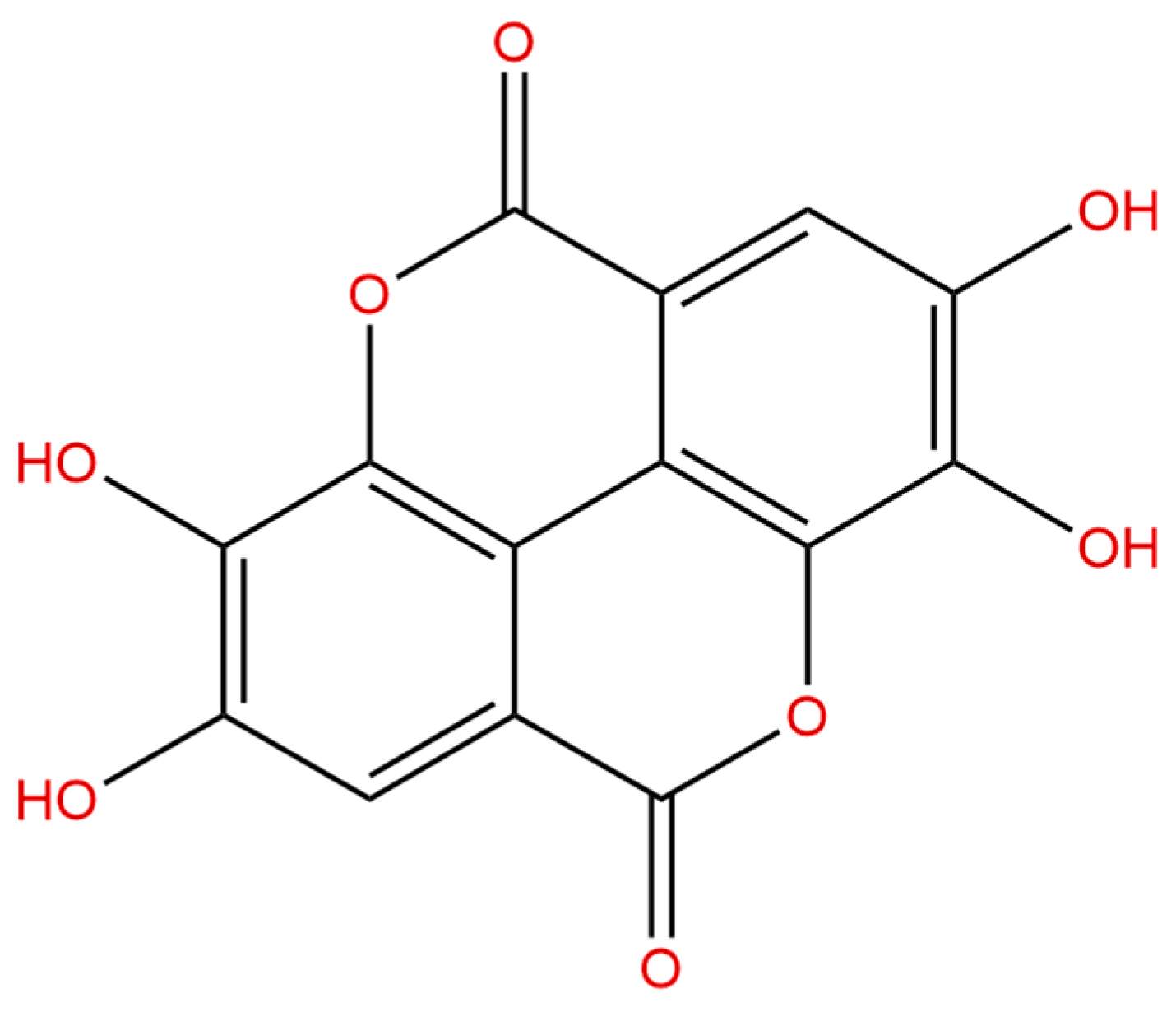
Figure 1.
The chemical structure of ellagic acid (EA).
2. Materials and Methods
2.1. Drugs and Reagents
Carboxyl methyl cellulose sodium (CMC-Na) and CDDP (CAS number: 15663-27-1, purity ≥ 99%) were provided by Sigma (Shanghai, China). Ellagic acid (CAS number: 476-66-4, purity ≥ 96%; EA) was purchased from Aladdin (Shanghai, China). All other regents are at least analytically pure.
2.2. Animals and Treatments
In the current study, all animal experiments were performed according to the regulations of Management of Experimental Animals of China Agricultural University and approved by the Institutional Animal Care and Use Committee of China Agricultural University (Approved number AW02303202-2-14). C57BL/6 mice (8-week-old, male, 20-22 g) were provided by Beijing Weitong Lihua Technology Co., Ltd (Beijing, China). Mice have one weeks’ adaptive period prior to the treatments. During experiments, all mice were fed in a standard environment (the relative humidity, 55 ± 5%; the room temperature, 22 ± 3 °C; and the light–dark cycle, 8:00 am-8:00 pm).
A total of forty-eight mice were randomly divided into control, CDDP model, EA100 (i.e., EA 100 mg/kg/day), and CDDP plus EA 25, EA50, and EA100 groups (n=8 in each group) and the detail treatments were showed as follows:
(1) control group. Mice were intraperitoneally (i.p.) injected with the equal of saline for 2 days and orally administrated with the equal volume of 0.5% CMC -Na for 7 days.
(2) model group. According to Lu et al study with a minor revision [8], CDDP was dissolved in saline at the final concentration of 2 mg/mL. At the first 2 days, mice were i.p. injected with CDDP at the final dose of 20 mg/kg/day (i.e., the accumulated dose is 40 mg/kg), after 2 h, then were treated with administrated intragastrically with the equal volume of 0.5% CMC-Na (i.e., 0.1 mL/10 g body weight). On the third day, mice were alone administrated intragastrically with 0.5% CMC-Na for additional 5 days.
(3) EA 100 group. EA was dissolved in 0.5% CMC-Na and prepared as a suspension at the final concentration of 10 mg/mL. Mice were i.p. injected with the equal of saline and administrated intragastrically with EA at the final dose of 100 mg/kg/day for 7 days.
(4)-(6) CDDP plus EA 25, EA50 or EA100 groups. EA was dissolved in 0.5% CMC-Na and prepared as a suspension at the final concentration of 2.5, 5 and 10 mg/mL, respectively. At the first 2 days, mice were i.p. injected with CDDP at the final dose of 20 mg/kg per day (i.e., the accumulated dose is 40 mg/kg), after 2 h, mice were administrated intragastrically with EA at the final doses of 25, 50 or 100 mg/kg per day (0.1 mL/10 g body weight), respectively. On the third day, mice were continually administrated intragastrically with EA at the final doses of 25, 50 or 100 mg/kg per day for additional 5 days.
At 12 h after the last dose of EA, mice were anesthetized and euthanized via the high dose of pentobarbital sodium (i.e., 80 mg/kg body weight, i.p.). The blood of each mouse was collected for biochemical analysis and the liver tissue was collected for the histopathological examination and the measurement of protein and gene expression.
2.3. Measurement of Aspartate Aminotransferase (AST) and Alanine Aminotransferase (ALT)
The fresh blood of each mouse was collected. Then, samples were centrifuged at 3000 × g for 15 min at the room temperature. The serum AST and ALT levels of each sample were tested using an automatic analyzer (Hitachi 7080, Tokyo, Japan) with the standard AST and ALT diagnostic kits (Kehua Biological engineering Company, Shanghai, China), according to our previous study [27].
2.4. Histopathological Analysis
Four livers in each treatment group were selected for histopathological examination and the detail protocols were accorded to the previous study [28]. In brief, liver tissues were fixed in the 10% neutral formalin solution for at least 48 h. Then, tissues undergo dehydration, permeability, paraffin embedding, and sectioning. Finally, hematoxylin-eosin (H&E) staining was performed. The histological parameters of hepatocyte necrosis, and inflammation were scored, according to Taghizadeh et al study, i.e., as 0, normal (there was no observed pathology damage); 1, mild (Minor damage with hepatocyte necrosis, and inflammatory cell infiltration); 2, moderate (pathology damage degree between score 1 and score 3); or 3, severe (Numerous liver cell necrosis and inflammatory cell infiltration) [29]. In each slice, 15 images were analyzed and the average value is calculated.
2.5. Measurements for Malondialdehyde (MDA) Level, and Catalase (CAT), and Superoxide Dismutase (SOD) Activites
The commercial MDA (catalogue number: A003-1-2), CAT (catalogue number: A007-1-1), and SOD kits (catalogue number: A001-3-2) (Nanjing Jiancheng, Nanjing, China) were employed to measure the MDA levels and the CAT, and SOD activities in the livers of mice, respectively, according to the instructions of kits.
2.6. Measurements of Inflammatory Markers
The commercial TNF-a, IL-1β and IL-6 ELISA kits were provided by R&D Systems (Minneapolis, MN, United States) and they were employed to measure these inflammation markers in the livers of mice, according to the instructions of kits.
2.7. Measurements of the Activities of Caspase-9 and Caspase-3
The relative activities of caspases-9 and -3 in the livers were determined using the commercial caspases-9 and -3 kits, according to the instructions (Beyotime, Haimen, China). In brief, each liver tissue sample was lysed using 0.5 mL lysis buffer (provided by the kits) for 15 min at 4 °C. Then, the lysate of each sample was collected using a refrigerated centrifuge (it sets up at 12,000 × g for 15 min; 4 °C). Finally, the supernatants were used for measurements. A BCA™ protein assay kit (Beyotime, Haimen, China) was employed to measure the protein concentration of each sample. The final activities of caspases and -3 of each sample were normalized to its protein level.
2.8. Quantitative RT-PCR
The total RNA isolations were carried our using a Total RNA Isolation Kit (Vazyme Biotech Co., Ltd., Nanjing, China), according to the instructions. The detail protocols for the cDNA synthesis, quantitative RT-PCR, and primer information were showed in the Suppl. Method and Suppl. Table 1, respectively.
2.9. Western Blotting
To analyze the expression of targeted protein, Western blotting was carried out, followed to the detail protocols in our previous study [30]. In brief, 20 mg of liver tissue of each sample were crushed and lysed in an ice-cold RIPA buffer with various protein inhibitors (i.e., 0.5 mM Na3VO4, 50 mM NaF, and 1 μg/mL PMSF). An automatic low-temperature crusher (Seville Company, Wuhan, China) was employed with the 30 W work condition (each cycle: 5 s ultrasonication and 3 s pause; 6 cycles). After ultrasonication, samples were centrifuged using a refrigerated centrifuge (it set up at 12,000 rpm for 15 min; 4 °C) and the supernatants of each sample were isolated and corresponding protein concentrations were quantified using a BCA™ protein assay kit. 20 μg protein of each sample were loaded and separated using the commercial SDS-PAGE Precast Gel (Beyotime, Haimen, China). The primary antibodies, including rabbit against Nrf2, HO-1 (1:1000 dilution; Proteintech, Chicago, United States), and NF-κB (1:1000 dilution; Santa Cruz, CA, United States) antibodies, and mouse against β-actin anybody (1:1000 dilution; Santa Cruz, CA, United States), were used. The blots were visualized using the Tanon Chemiluminescent Imaging System (Shanghai, China) and the protein expression in each gel were analyzed using the ImageJ software (NIH, MD, USA).
2.10. Molecular Docking Analysis
The binding affinities and modes of interaction between EA with Nrf2, Keap1, and NF-kB proteins were performed using AutodockVina 1.2.2 software. The molecular structure of EA was retrieved from PubChem Compound. The 2D coordinates of mouse Nrf2 (Uniport: Q60795), Keap1 (Uniport: Q9Z2X8), and NF-kB (Uniport: P25799) proteins were downloaded via the PDB website. Finally, molecular docking studies were carried out using an Autodock Vina 1.2.2 software (http://autodock.scripps.edu/).
2.11. Statistical Analysis
All data in the current study are analyzed using a GraphPad 9.0 software (Dotmatics Company, Boston, MA, USA). The results were presented as mean ± standard deviation (S.D.). The statistical analysis between any two groups was carried our using 1-way analysis of variance (ANOVA) with the Tukey’s multiple comparisons post hoc test. A p- value less than 0.05 was set as the significant difference.
3. Results
3.1. EA Supplementation Decreases the Levels of Serum ALT and AST
There was no mouse died in the period of experiment. Compared to the untreated control group, the marked liver dysfunction was detected in the CDDP model group with significantly increased serum ALT (increased to 48.7 U/L; p<0.001) and AST (increased to 78.7 U/L L; p<0.001) levels (Figure 2). EA supplementation could significantly attenuate CDDP exposure-induced liver dysfunction. In the CDDP plus EA 25, 50, and 100 mg/kg/day groups, the ALT levels were decreased to 42.7 U/L, 31.3 U/L (p<0.001), and 29.8 U/L (p<0.001), respectively; and the AST levels decreased to 67.8 U/L, 53.3 U/L (p<0.001), and 48.8 U/L (p<0.001) (Figure 2), respectively. Compared to the untreated control group, the levels of serum ALT and AST had no marked changes in the in the EA alone treatment group (Figure 2).
3.2. EA Supplementation Attenuates CDDP Exposure-Induced Histopathological Changes in the Livers
Compared to the untreated control group, CDDP exposure caused marked histopathological injury. As shown in Figure 3, the marked hepatocyte necrosis, inflammatory cell infiltration, and central venous congestion were observed in the liver tissues of CDDP-treated mice. Correspondingly, the SQSs were significantly increased to 2.50 (p<0.001). EA co-treatments at 50 and 100 mg/kg per day for 7 days significantly reduced CDDP exposure-induced liver injury, the SQSs were significantly decreased to 1.25 (p<0.05) and 0.75 (p<0.01), respectively. There was no marked histopathological injury observed in the EA 100 alone treatment group, compared to that in the vehicle-treated control group.
3.3. EA Supplementation Attenuates CDDP Exposure-Induced Liver Oxidative Stress Damage
Compared to the vehicle-treated control group, CDDP alone treated mice showed an elevated MDA levels (increased to 2.48 mmol per mg protein; p<0.001) and decreased CAT and SOD activities (decreased to 83.3 U per mg protein and 79.3 U per mg protein, respectively, both p<0.001). EA co-treatments markedly attenuated CDDP exposure-induced the increases of MDA levels and the decreases of SOD and CAT activities in the livers. In the CDDP plus EA50 and EA 100 groups, the MDA levels were significantly decreased to 2.21 mmol per mg protein and 1.92 mmol/mg protein (p<0.01 or 0.001), respectively; the CAT activities were significantly increased to 97.8 U per mg protein and 108.4 U per mg protein (p<0.01 or 0.001), respectively; and the SOD activities were significantly increased to 100.3 U/mg protein and 106.1 U per mg protein (both p<0.001), respectively (Figure 4).
3.4. EA Supplementation Attenuates CDDP Exposure-Caused Inflammatory Response in the Livers of Mice
The levels of IL-1β, TNF-a, and IL-6 proteins in the livers of CDDP-treated mice were significantly upregulated to 90.8 pg per mg protein, 24.1 pg/mg protein, and 74.3 pg per mg protein (all p<0.001) (Figure 5), respectively. EA co-treatments significantly decreased these inflammatory markers. I.e., in the CDDP plus EA 50 and CDDP plus EA 100 groups, the levels of IL-1β protein in the livers were significantly decreased to 69.3 pg per mg protein and 51.2 pg/mg protein ( p<0.01 or 0.001) (Figure 5A), respectively; the levels of TNF-a protein in the livers were significantly decreased to 16.2 pg/mg protein and 13.6 pg per mg protein ( p<0.01 or 0.001) (Figure 5B), respectively; and the levels of IL-6 protein in the livers were significantly decreased to 56.2 pg per mg protein and 47.8 pg per mg protein (both p<0.001) (Figure 5C), respectively. Compared to the vehicle-treated control group, the levels of IL-1β, TNF-a, and IL-6 proteins in the livers of mice had no marked change (Figure 5).
3.5. EA Supplementation Attenuates CDDP Exposure-Induced the Activation of Casapses in the Livers
As shown in Figure 6, caspases-9 and -3 activities in the livers of CDDP-treated mice were significantly increased to 3.1- and 3.2-fold (both p<0.001), respectively (Figure 6), compared to those in the vehicle-treated mice. Oral administration of EA at 50 or 100 mg/kg per day for 7 days could effectively attenuate CDDP exposure-mediated the upregulation of caspases-9 and -3 levels in the livers of mice. In the CDDP plus EA 50 group, the activities of caspases-9 and -3 were significantly decreased to 1.6- (p<0.001) and 1.9 -fold (p<0.001), respectively; and in the CDDP plus EA 100 group, the activities of caspases-9 and -3 were significantly decreased to 11.2- (p<0.001), and 1.4-fold (p<0.001), respectively.
3.6. EA Supplementation Upregulates the mRNA Expressions of Nrf2, and HO-1 Genes and Downregulates the mRNA Expressions of NF-kB, IL-1β, TNF-α, and IL-6 Genes in the Livers of Mice
The mRNA expressions of NF-kB, IL-1β, TNF-α, IL-6, Nrf2, and HO-1 genes in the livers of mice were determined. Compared to those in the untreated control group, CDDP exposure markedly promoted all genes expression, (i.e., the expression of NF-kB, IL-1β, TNF-α, IL-6, Nrf2, and HO-1 mRNAs increased to 3.67-, 3.71-, 4.24-, 3.43-, 1.58-, and 2.29-fold, respectively; all p<0.001) (Figure 7). EA supplementation regulated these genes’ expression. Especially in the CDDP plus EA100 groups, the expression of NF-kB, IL-1β, TNF-α, and IL-6 mRNAs were decreased to 1.54-, 1.67-, 1.61-, and 1.74-fold (all p<0.001), respectively, while the expression of Nrf2 and HO-1 mRNAs were increased to 2.30, and 3.49- fold (both p<0.001), respectively, compared to those in the CDDP alone treated mice. Meanwhile, EA alone treatment significantly promoted the mRNA expressions of Nrf2 and HO-1 genes (i.e., increased to 1.75- and 2.04- fold, respectively, both p<0.01), and had a minor effect on the mRNA expressions of NF-kB, IL-1β, TNF-α, and IL-6 genes, compared to these in the livers of the vehicle-treated mice.
3.7. EA Supplementation Promotes the Expression of Nrf2, HO-1 Proteins and Inhibites the Expression of NF-kB Protein in the Livers of Mice
As shown in Figure 8, CDDP alone treatment significantly promoted the expression of Nrf2, HO-1, and NF-kB proteins in the livers of mice, i.e., they were increased to 1.88-(p<0.05), 1.98- (p<0.05), and 4.23-fold (p<0.001), respectively. EA co-treatments markedly promoted the expression of Nrf2 and HO-1 proteins, but inhibited the expression of NF-kB protein. In the CDDP plus EA50 and CDDP plus EA 100 groups, the expressions of Nrf2 in the levers of mice were significantly upregulated to 2.87-, and 4.00-fold (p<0.01 or 0.001), respectively; and the expressions of HO-1 protein were significantly upregulated to 3.04-, and 4.15-fold (p<0.05 or 0.001), respectively; while the expressions of NF-kB protein were significantly downregulated to 1.69-, and 1.62-fold (both p<0.001), respectively, compared to the CDDP model group.
3.8. Molecular docking analysis results of EA with Nrf2, Keap1, and NF-kB proteins
To evaluate the affinity of EA with Nrf2, Keap1, and NF-kB proteins, the molecular docking analysis were performed using a Autodock Vina v.1.2.2 software. The binding energy for each interaction and binding poses were shown in Figure 9. EA with Keap1, and NF-kB proteins had the lower binding energy (i.e., the binding energies are -9.2 and -9.0 kcal/mol, respectively) than that is with Nrf2 protein (the binding energy is -6.4 kcal/mol), indicating that EA had the higher stable binding with Keap1, and NF-kB proteins then Nrf2 protein (Figure 9A). We further found that the binding of EA with Keap1 (Figure 9B), and NF-kB (Figure 9C) is via the visible hydrogen bonds and strong electrostatic interactions.
4. Discussion
Liver toxicity is one of unwanted adverse effects in patients during CDDP treatments [15]. In the present study, our results found that intraperitoneal CDDP administration at 20 mg/kg/day for 2 days (the cumulative dose is 40 mg/kg) could result in marked liver injury in mice, the elevated AST and ALT levels were detected in the serum (Figure 2). In addition, marked histopathology changes, including hepatocyte necrosis, inflammatory cell infiltration, central venous congestion in CDDP-treated liver tissues were detected (Figure 3). Furthermore, we found that CDDP exposure-induced liver injury involved the induction of inflammatory response and oxidative stress (Figure 3, Figure 4, Figure 5, Figure 6, Figure 7 and Figure 8). These findings are in line with the previous studies [15,31], indicating the liver toxicity model caused by CDDP were built successfully.
EA is a plant phenolic compound and could be isolated from various fruits, such as strawberries, pomegranate, and almonds [24]. EA possessed various biology activities, including anti-aging, anti-inflammation, anti-oxidant, and anti-infection [17,18,19,20,21,22,23]. Several previous studies had illustrated that EA supplementation could improve drugs (such as doxorubicin hydrochloride and fluoxetine) or toxins (such as CCl4, lipopolysaccharide [LPS]/d-galactosamine, hexavalent chromium, and alcohol)-induced cytotoxicity and liver toxicity by inhibiting oxidative stress, apoptosis, and inflammatory response in vitro or in vivo [25,32,33,34,35]. Consistently, our current data showed that EA supplementation could effectively revise CDDP exposure-induced the upregulation of lipid peroxidation, caspase activities, and inflammatory response in the livers of mice, and these protections might be partly dependent on the regulation of NF-κB pathway and Nrf2/HO-1 pathway (Figure 3, Figure 4, Figure 5, Figure 6, Figure 7 and Figure 8).
Excessive ROS production induced by CDDP could directly damage intracellular macromolecules (such as DNA, lipids, proteins,) or subcellular organelles (such as mitochondria, endoplasmic reticulum, and lysosome), which finally could induce cell death [4,10,16,23]. This finding was considered as a key molecular basis in the toxic process [36]. It has been demonstrated that inhibition of ROS production and oxidative stress via the antioxidant supplementation could partly attenuate CDDP-induced cell apoptosis and ototoxicity [37]. Our current results showed CDDP exposure significantly upregulated MDA levels, and significantly downregulated SOD and CAT activities in the livers of mice (Figure 4). The antioxidant enzymes SOD and CAT could directly catalyze superoxide anion and hydrogen peroxide (H2O2) form the non-toxic substances in eukaryotic cells, and plays a vital role in the process of anti-oxidative stress [26]. MDA is one of most important products of membrane lipid peroxidation and its increases indicated the occurrence of lipid peroxidation [25]. These evidences indicated that CDDP exposure could result in oxidative stress damage in the liver tissues of mice via the inhibition of antioxidant enzymes’ activities. Moreover, our data showed that EA co-treatment at the final doses of 50 and 100 mg/kg/day for 7 days could partly abolish the increase of MDA and the decreases of SOD and CAT activities in the livers of mice treated with CDDP (Figure 4). Similarly, Zhang et al found that EA co-treatment (at 100 mg/kg/day for 5 days) could significantly reduce the production of MDA and upregulate the total antioxidant capacity and the activities of GSH-PX in the jejunum tissues, following to protect against of diquat (a toxic pesticide) exporesure-caused mice [21]. In another study, Zhao et al found that EA supplementation at the doses of 50 and 100 mg/kg/day for 4 weeks could significantly chronic ethanol exposure-induced liver injury via upregulating CAT, SOD, and GSH-PX activities [38]. Taken together, these evidences supported that inhibition of lipid peroxidation and upregulation of antioxidant enzyme activities may play a critical role in the protective effects of EA against CDDP-induced liver toxicity. Mitochondria are the target of ROS, and dysfunctional mitochondria could elevate the release of cytochrome C (CytC), then in cascade to activating caspases-9 and -3 and initiating cell apoptosis [39]. Our data also found that EA co-treatment does-dependently downregulated the activities of caspases-9 and -3 in the livers of CDDP-treated mice (Figure 5). In short, these data indicated that EA supplementation could attenuate CDDP exposure-induced liver toxicity through inhibiting caspases activation-mediated cell apoptosis.
It is reported that CDDP exposure could induce the production of various pro-inflammatory factors and chemokines, including high mobility group box-1 (HMGB1), TNF-α, IL-6, and IL-1β, chemokine ligand 2 (Ccl2), Ccl7, and C-X-C motif Ccl2 (Cxcl2)[31]. Several studies also found that CDDP administration significantly upregulated the expression of NF-κB, a critical transcriptional factor in inflammatory response [40]. NF-κB could transcriptionally activate the expression of various pro-inflammatory genes, such as L-1β, IL-6, and TNF-α [40]. Our data showed that CDDP administration significantly upregulated the mRNAs and proteins’ expressions of these above-mentioned genes (i.e., NF-κB, IL-1β, IL-6, and TNF-α) and EA co-treatment effectively inhibited their expressions (Figure 5, Figure 7 and Figure 8). Several studies had demonstrated that EA is a potential inhibitor of NF-κB [25,41]. For example, EA supplementation could inhibit LPS-induced NF-κB activation in U937 monocytic cells and THP-1 differentiated macrophages, and it also inhibited the expression of NF-κB protein in the livers of mice [25,41]. Taken together, these evidences indicated that EA co-treatment could partly improve CDDP exposure-induced inflammatory response via the inhibition of NF-κB signaling pathway.
Nrf2 is a “house-keeper” transcription factor in response to stress against oxidative stress and inflammatory damage [42]. In the normal condition, Nrf2 protein mainly locates in the cytoplasm by binding with Kelch-like ECH-associated protein 1 (Keap1) at the sites of ETGE and DLG motifs [42]. Under the oxidative stress condition, the interaction of Nrf2 and Keap1 was disrupted and Nrf2 entered nucleus, then binding with ARE and transcriptionally activated antioxidant genes, such as CAT, GSH-PX, and HO-1, which usually play the protective role [43]. A recent study showed that Nrf2 knockout in mice significantly exacerbated CDDP exposure-induced acute kidney injury, demonstrating that Nrf2 played a protective role in the process of CDDP-induced toxic effects [44]. There are multiple studies found that EA co-treatment could promote the expression of Nrf2 and protect against drugs or toxins exposure-induced toxic events in vitro and in vivo [20,25,45,46]. EA has bene considered as a potential activator of Nrf2 [19]. For example, Gu et al found that EA pre-treatment (at 20 mg/kg, oral administration) significantly inhibited the acute liver injury caused by LPS/D-galactosamine co-exposure in mice through increasing the expression of Nrf2 and HO-1 proteins and downregulating the expression of NF-κB and TNF-α proteins [33]. In line with these previous published studies, our current data found EA co-treatmen significantly upregulate the mRNAs and proteins’ expression of Nrf2 and HO-1 genes in the livers of mice (Figure 7 and Figure 8). These evidences indicated that the activation of Nrf2/HO-1 pathway partly contributed the protective effect of EA against CDDP exposure-induced liver toxicity in mice. In addition, it has been reported that Nrf2 could partly inhibit NF-κB activates via its downstream gene HO-1 [47]. Consistently, a recent study showed that pharmacology inhibition of Nrf2 reduced the protective effect of EA on IL-1β-induced oxidative stress and inflammatory response in C28/I2 human chondrocytes [20]. Therefore, the activation of Nrf2 pathway by EA supplementation may also contribute to its inhibitory effect on the expression of NF-κB. The precious molecular mechanisms are still required in the further study. In addition, our results of molecular docking analysis showed that EA with Keap1, and NF-kB proteins had the lower binding energy than Nrf2 protein, indicating that EA had the higher stable binding with Keap1, and NF-kB proteins then Nrf2 protein (Figure 9). These evidences further supported our assumes.
Importantly, EA has the great safety, and has been used in food production as a food additive [48,49]. Tasaki and colleagues found that the no-observed-effect level (NOEL) of EA is male and female F344 rats is up to 3011 and 3254 mg/kg body weight/day, respectively[48]. Similar NOEL was also detected in safety assessment of urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of ellagic acid [49]. These evidences indicated that EA may be a potential agent against CDDP-induced liver toxicity and further commercial development could be considered.
5. Conclusions
In a short, our current results reveal that EA supplementation could effectively ameliorate CDDP exposure -caused liver toxicity through inhibiting oxidative stress and inflammatory response. The potential molecular mechanisms may be associated with the inhibition of NF-κB signaling pathway and the activation of Nrf2/HO-1 signaling pathway (Figure 10). Our current study highlighted that EA is a potential candidate against CDDP-induced acute liver injury during anti-cancer treatment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. The primer sequences of quantitative RT-PCR were presented in Suppl. Table 1.
Author Contributions
Conceptualization, C.D. and J.S.; methodology, Q.M, X.Z., Y. L, and M. L.; software, X.Z. and S.T.; validation, C.D. S.T., and J.S.; formal analysis, Y.L. and C.D.; project administration, C.D. and J.S.; writing—review and editing, C.D. and J.S.; funding acquisition, C.D., and J.S. investigation, X.Z., B.X.; and Y.L.; writing—original draft preparation, C.D. All authors have approved this version for publishing.
Funding
This study was supported by the National Natural Science Foundation of China (Award number 32102724) And the Special Fund Management Office for Basic Research Business Expenses of China Agricultural University (2023TC028).
Conflicts of Interest
All authors declare that there were no any conflicts of interest in this manuscript.
References
- Hill, N.O. Cis-platinum for cancer. The New England journal of medicine 1979, 301, 47. [CrossRef]
- Tang, C.; Livingston, M.J.; Safirstein, R.; Dong, Z. Cisplatin nephrotoxicity: new insights and therapeutic implications. Nature reviews. Nephrology 2023, 19, 53-72. [CrossRef]
- Squillace, S.; Niehoff, M.L.; Doyle, T.M.; Green, M.; Esposito, E.; Cuzzocrea, S.; Arnatt, C.K.; Spiegel, S.; Farr, S.A.; Salvemini, D. Sphingosine-1-phosphate receptor 1 activation in the central nervous system drives cisplatin-induced cognitive impairment. J Clin Invest 2022, 132. [CrossRef]
- Nofal, A.E.; Okdah, Y.A.; Rady, M.I.; Hassaan, H.Z. Gum Acacia attenuates cisplatin toxic effect spermatogenesis dysfunction and infertility in rats. International journal of biological macromolecules 2023, 240, 124292. [CrossRef]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer 2021, 21, 37-50. [CrossRef]
- Gong, C.; Qian, L.; Yang, H.; Ji, L.L.; Wei, H.; Zhou, W.B.; Qi, C.; Wang, C.H. Hepatotoxicity and pharmacokinetics of cisplatin in combination therapy with a traditional Chinese medicine compound of Zengmian Yiliu granules in ICR mice and SKOV-3-bearing nude mice. BMC complementary and alternative medicine 2015, 15, 283. [CrossRef]
- Al-Malki, A.L.; Sayed, A.A. Thymoquinone attenuates cisplatin-induced hepatotoxicity via nuclear factor kappa-β. BMC complementary and alternative medicine 2014, 14, 282. [CrossRef]
- Lu, Y.; Cederbaum, A.I. Cisplatin-induced hepatotoxicity is enhanced by elevated expression of cytochrome P450 2E1. Toxicological sciences : an official journal of the Society of Toxicology 2006, 89, 515-523. [CrossRef]
- Elhady, S.S.; Abdelhameed, R.F.A.; Mehanna, E.T.; Wahba, A.S.; Elfaky, M.A.; Koshak, A.E.; Noor, A.O.; Bogari, H.A.; Malatani, R.T.; Goda, M.S. Metabolic Profiling, Chemical Composition, Antioxidant Capacity, and In Vivo Hepato- and Nephroprotective Effects of Sonchus cornutus in Mice Exposed to Cisplatin. Antioxidants (Basel) 2022, 11. [CrossRef]
- Habib, S.A.; Suddek, G.M.; Abdel Rahim, M.; Abdelrahman, R.S. The protective effect of protocatechuic acid on hepatotoxicity induced by cisplatin in mice. Life sciences 2021, 277, 119485. [CrossRef]
- Hong, K.O.; Hwang, J.K.; Park, K.K.; Kim, S.H. Phosphorylation of c-Jun N-terminal Kinases (JNKs) is involved in the preventive effect of xanthorrhizol on cisplatin-induced hepatotoxicity. Arch Toxicol 2005, 79, 231-236. [CrossRef]
- Bhattacharyya, S.; Mehta, P. The hepatoprotective potential of Spirulina and vitamin C supplemention in cisplatin toxicity. Food & function 2012, 3, 164-169. [CrossRef]
- Mohamed, H.E.; Badawy, M.M.M. Modulatory effect of zingerone against cisplatin or γ-irradiation induced hepatotoxicity by molecular targeting regulation. Applied radiation and isotopes : including data, instrumentation and methods for use in agriculture, industry and medicine 2019, 154, 108891. [CrossRef]
- Huang, H.; Shen, Z.; Geng, Q.; Wu, Z.; Shi, P.; Miao, X. Protective effect of Schisandra chinensis bee pollen extract on liver and kidney injury induced by cisplatin in rats. Biomed Pharmacother 2017, 95, 1765-1776. [CrossRef]
- Sami, D.H.; Soliman, A.S.; Khowailed, A.A.; Alruhaimi, R.S.; Hassanein, E.H.M.; Kamel, E.M.; Mahmoud, A.M. The protective effect of 7-hydroxycoumarin against cisplatin-induced liver injury is mediated via attenuation of oxidative stress and inflammation and upregulation of Nrf2/HO-1 pathway. Environ Sci Pollut Res Int 2023, 30, 80181-80191. [CrossRef]
- Kim, D.H.; Sim, Y.; Hwang, J.H.; Kwun, I.S.; Lim, J.H.; Kim, J.; Kim, J.I.; Baek, M.C.; Akbar, M.; Seo, W.; et al. Ellagic Acid Prevents Binge Alcohol-Induced Leaky Gut and Liver Injury through Inhibiting Gut Dysbiosis and Oxidative Stress. Antioxidants (Basel) 2021, 10. [CrossRef]
- Azam, F.; Khan, M.A.; Khan, A.; Ahmad, S.; Zofair, S.F.F.; Younus, H. In silico and in vitro studies on the inhibition of laccase activity by Ellagic acid: Implications in drug designing for the treatment of Cryptococcal infections. International journal of biological macromolecules 2022, 209, 642-654. [CrossRef]
- Yoganathan, S.; Alagaratnam, A.; Acharekar, N.; Kong, J. Ellagic Acid and Schisandrins: Natural Biaryl Polyphenols with Therapeutic Potential to Overcome Multidrug Resistance in Cancer. Cells 2021, 10. [CrossRef]
- Wang, Q.; Botchway, B.O.A.; Zhang, Y.; Liu, X. Ellagic acid activates the Keap1-Nrf2-ARE signaling pathway in improving Parkinson’s disease: A review. Biomed Pharmacother 2022, 156, 113848. [CrossRef]
- Zhu, W.; Tang, H.; Li, J.; Guedes, R.M.; Cao, L.; Guo, C. Ellagic acid attenuates interleukin-1β-induced oxidative stress and exerts protective effects on chondrocytes through the Kelch-like ECH-associated protein 1 (Keap1)/ Nuclear factor erythroid 2-related factor 2 (Nrf2) pathway. Bioengineered 2022, 13, 9233-9247. [CrossRef]
- Zhang, X.; Wang, S.; Wu, Y.; Liu, X.; Wang, J.; Han, D. Ellagic Acid Alleviates Diquat-Induced Jejunum Oxidative Stress in C57BL/6 Mice through Activating Nrf2 Mediated Signaling Pathway. Nutrients 2022, 14. [CrossRef]
- Aishwarya, V.; Solaipriya, S.; Sivaramakrishnan, V. Role of ellagic acid for the prevention and treatment of liver diseases. Phytotherapy research : PTR 2021, 35, 2925-2944. [CrossRef]
- Kang, E.H.; Kown, T.Y.; Oh, G.T.; Park, W.F.; Park, S.I.; Park, S.K.; Lee, Y.I. The flavonoid ellagic acid from a medicinal herb inhibits host immune tolerance induced by the hepatitis B virus-e antigen. Antiviral research 2006, 72, 100-106. [CrossRef]
- Qi, M.; Wang, N.; Xiao, Y.; Deng, Y.; Zha, A.; Tan, B.; Wang, J.; Yin, Y.; Liao, P. Ellagic acid ameliorates paraquat-induced liver injury associated with improved gut microbial profile. Environmental pollution (Barking, Essex : 1987) 2022, 293, 118572. [CrossRef]
- Aslan, A.; Gok, O.; Erman, O.; Kuloglu, T. Ellagic acid impedes carbontetrachloride-induced liver damage in rats through suppression of NF-kB, Bcl-2 and regulating Nrf-2 and caspase pathway. Biomed Pharmacother 2018, 105, 662-669. [CrossRef]
- Aslan, A.; Gok, O.; Beyaz, S.; Ağca, C.A.; Erman, O.; Zerek, A. Ellagic acid prevents kidney injury and oxidative damage via regulation of Nrf-2/NF-κB signaling in carbon tetrachloride induced rats. Mol Biol Rep 2020, 47, 7959-7970. [CrossRef]
- Dai, C.; Liu, M.; Zhang, Q.; Das Gupta, S.; Tang, S.; Shen, J. Nootkatone Supplementation Attenuates Carbon Tetrachloride Exposure-Induced Nephrotoxicity in Mice. Antioxidants (Basel) 2023, 12. [CrossRef]
- Dai, C.; Zhang, X.; Lin, J.; Shen, J. Nootkatone Supplementation Ameliorates Carbon Tetrachloride-Induced Acute Liver Injury via the Inhibition of Oxidative Stress, NF-κB Pathways, and the Activation of Nrf2/HO-1 Pathway. Antioxidants (Basel) 2023, 12. [CrossRef]
- Taghizadeh, F.; Hosseinimehr, S.J.; Zargari, M.; Karimpour Malekshah, A.; Mirzaei, M.; Talebpour Amiri, F. Alleviation of cisplatin-induced hepatotoxicity by gliclazide: Involvement of oxidative stress and caspase-3 activity. Pharmacology research & perspectives 2021, 9, e00788. [CrossRef]
- Dai, C.; Zhang, Q.; Shen, L.; Sharma, G.; Jiang, H.; Wang, Z.; Shen, J. Quercetin Attenuates Quinocetone-Induced Cell Apoptosis In Vitro by Activating the P38/Nrf2/HO-1 Pathway and Inhibiting the ROS/Mitochondrial Apoptotic Pathway. Antioxidants (Basel) 2022, 11. [CrossRef]
- Gong, S.; Feng, Y.; Zeng, Y.; Zhang, H.; Pan, M.; He, F.; Wu, R.; Chen, J.; Lu, J.; Zhang, S.; et al. Gut microbiota accelerates cisplatin-induced acute liver injury associated with robust inflammation and oxidative stress in mice. J Transl Med 2021, 19, 147. [CrossRef]
- Devipriya, N.; Sudheer, A.R.; Srinivasan, M.; Menon, V.P. Effect of Ellagic Acid, a Plant Polyphenol, on Fibrotic Markers (MMPs and TIMPs) during Alcohol-Induced Hepatotoxicity. Toxicol Mech Methods 2007, 17, 349-356. [CrossRef]
- Gu, L.; Deng, W.S.; Liu, Y.; Jiang, C.H.; Sun, L.C.; Sun, X.F.; Xu, Q.; Zhou, H. Ellagic acid protects Lipopolysaccharide/D-galactosamine-induced acute hepatic injury in mice. Int Immunopharmacol 2014, 22, 341-345. [CrossRef]
- Beigi, T.; Safi, A.; Satvati, M.; Kalantari-Hesari, A.; Ahmadi, R.; Meshkibaf, M.H. Protective role of ellagic acid and taurine against fluoxetine induced hepatotoxic effects on biochemical and oxidative stress parameters, histopathological changes, and gene expressions of IL-1β, NF-κB, and TNF-α in male Wistar rats. Life sciences 2022, 304, 120679. [CrossRef]
- Cuevas-Magaña, M.Y.; Vega-García, C.C.; León-Contreras, J.C.; Hernández-Pando, R.; Zazueta, C.; García-Niño, W.R. Ellagic acid ameliorates hexavalent chromium-induced renal toxicity by attenuating oxidative stress, suppressing TNF-α and protecting mitochondria. Toxicology and applied pharmacology 2022, 454, 116242. [CrossRef]
- Gentilin, E.; Simoni, E.; Candito, M.; Cazzador, D.; Astolfi, L. Cisplatin-Induced Ototoxicity: Updates on Molecular Targets. Trends in molecular medicine 2019, 25, 1123-1132. [CrossRef]
- Kim, K.H.; Lee, B.; Kim, Y.R.; Kim, M.A.; Ryu, N.; Jung, D.J.; Kim, U.K.; Baek, J.I.; Lee, K.Y. Evaluating protective and therapeutic effects of alpha-lipoic acid on cisplatin-induced ototoxicity. Cell Death Dis 2018, 9, 827. [CrossRef]
- Zhao, L.; Mehmood, A.; Soliman, M.M.; Iftikhar, A.; Iftikhar, M.; Aboelenin, S.M.; Wang, C. Protective Effects of Ellagic Acid Against Alcoholic Liver Disease in Mice. Front Nutr 2021, 8, 744520. [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science (New York, N.Y.) 1998, 281, 1309-1312. [CrossRef]
- Fu, Y.; Xiang, Y.; Wang, Y.; Liu, Z.; Yang, D.; Zha, J.; Tang, C.; Cai, J.; Chen, G.; Dong, Z. The STAT1/HMGB1/NF-κB pathway in chronic inflammation and kidney injury after cisplatin exposure. Theranostics 2023, 13, 2757-2773. [CrossRef]
- Rønning, S.B.; Voldvik, V.; Bergum, S.K.; Aaby, K.; Borge, G.I.A. Ellagic acid and urolithin A modulate the immune response in LPS-stimulated U937 monocytic cells and THP-1 differentiated macrophages. Food & function 2020, 11, 7946-7959. [CrossRef]
- Bello, M.; Morales-González, J.A. Molecular recognition between potential natural inhibitors of the Keap1-Nrf2 complex. International journal of biological macromolecules 2017, 105, 981-992. [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxidants & redox signaling 2018, 29, 1727-1745. [CrossRef]
- Hu, J.; Gu, W.; Ma, N.; Fan, X.; Ci, X. Leonurine alleviates ferroptosis in cisplatin-induced acute kidney injury by activating the Nrf2 signalling pathway. Br J Pharmacol 2022, 179, 3991-4009. [CrossRef]
- Xiao, Y.; Huang, R.; Wang, N.; Deng, Y.; Tan, B.; Yin, Y.; Qi, M.; Wang, J. Ellagic Acid Alleviates Oxidative Stress by Mediating Nrf2 Signaling Pathways and Protects against Paraquat-Induced Intestinal Injury in Piglets. Antioxidants (Basel) 2022, 11. [CrossRef]
- Ding, X.; Jian, T.; Wu, Y.; Zuo, Y.; Li, J.; Lv, H.; Ma, L.; Ren, B.; Zhao, L.; Li, W.; et al. Ellagic acid ameliorates oxidative stress and insulin resistance in high glucose-treated HepG2 cells via miR-223/keap1-Nrf2 pathway. Biomed Pharmacother 2019, 110, 85-94. [CrossRef]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-κB Response Pathways in Drug-Induced Toxicity. Frontiers in cell and developmental biology 2021, 9, 809952. [CrossRef]
- Tasaki, M.; Umemura, T.; Maeda, M.; Ishii, Y.; Okamura, T.; Inoue, T.; Kuroiwa, Y.; Hirose, M.; Nishikawa, A. Safety assessment of ellagic acid, a food additive, in a subchronic toxicity study using F344 rats. Food Chem Toxicol 2008, 46, 1119-1124. [CrossRef]
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety assessment of Urolithin A, a metabolite produced by the human gut microbiota upon dietary intake of plant derived ellagitannins and ellagic acid. Food Chem Toxicol 2017, 108, 289-297. [CrossRef]
Figure 2.
The effects of EA supplementation on serum ALT (A) and AST (B) levels of CDDP-treated mice. Results are presented as mean ± S.D. (n = 8). Compared between 2 groups, *** p < 0.001. EA, ellagic acid.
Figure 2.
The effects of EA supplementation on serum ALT (A) and AST (B) levels of CDDP-treated mice. Results are presented as mean ± S.D. (n = 8). Compared between 2 groups, *** p < 0.001. EA, ellagic acid.
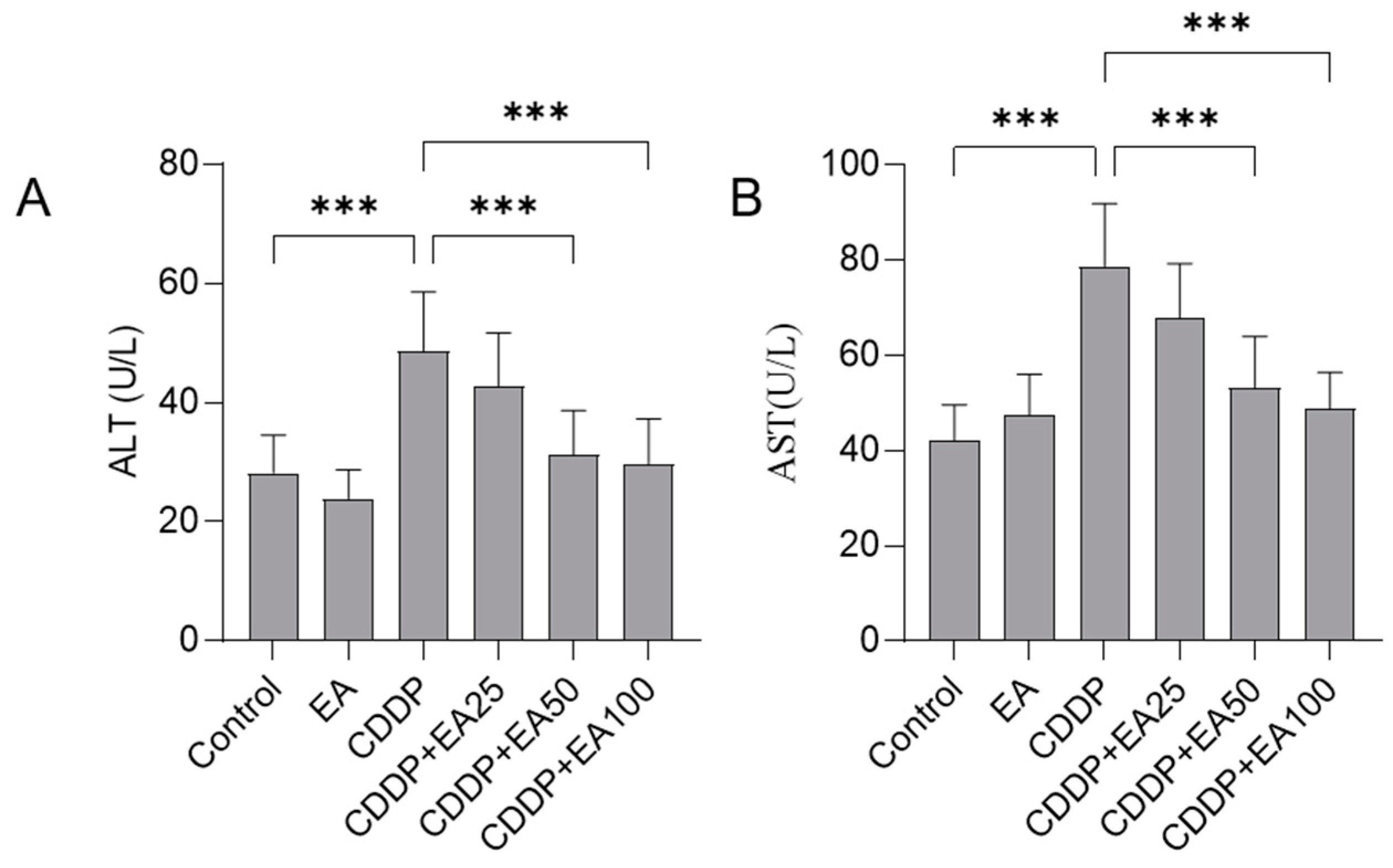
Figure 3.
EA supplementation attenuates CDDP-induced pathological injury in the livers. Representative H&E images (A) and the semi-quantitative scores (SQSs) (B) were presented. Results are presented as mean ± S.D. (n = 4). Compared between 2 groups, *p< 0.05, and *** p < 0.001. Scale bar: 50 μm. EA, ellagic acid; CDDP, cisplatin. Yellow arrow, inflammatory cell infiltration; Black arrow, hepatocyte necrosis.
Figure 3.
EA supplementation attenuates CDDP-induced pathological injury in the livers. Representative H&E images (A) and the semi-quantitative scores (SQSs) (B) were presented. Results are presented as mean ± S.D. (n = 4). Compared between 2 groups, *p< 0.05, and *** p < 0.001. Scale bar: 50 μm. EA, ellagic acid; CDDP, cisplatin. Yellow arrow, inflammatory cell infiltration; Black arrow, hepatocyte necrosis.
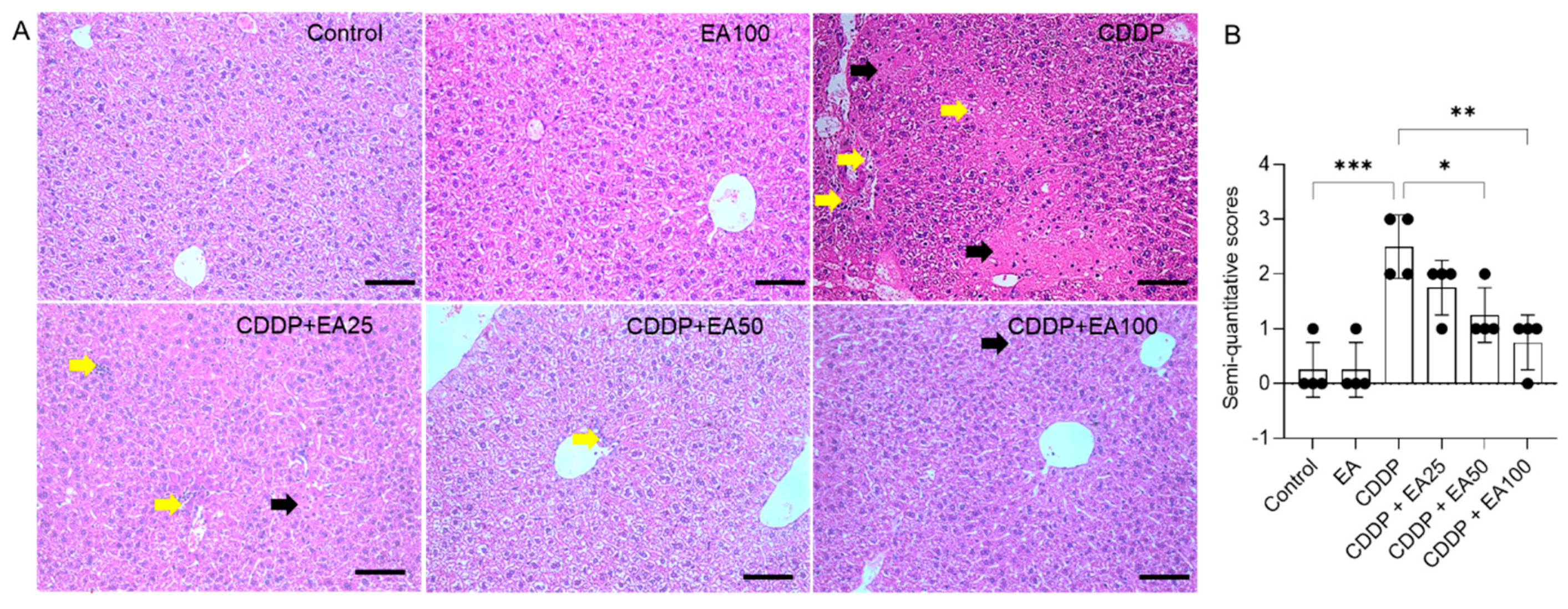
Figure 4.
The changes of the MDA levels (A), and CAT (B), and SOD (C) activities in the livers of mice. Results are shown as mean ± S.D. (n = 8). Compared between 2 groups, ** p < 0.01, or *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
Figure 4.
The changes of the MDA levels (A), and CAT (B), and SOD (C) activities in the livers of mice. Results are shown as mean ± S.D. (n = 8). Compared between 2 groups, ** p < 0.01, or *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.

Figure 5.
The levels of IL-1β (A), TNF-α (B), and IL-6 proteins (C) in the livers of mice. Results are presented as mean ± S.D. (n = 8). Compared between 2 groups, * p < 0.05, ** p < 0.01, or *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
Figure 5.
The levels of IL-1β (A), TNF-α (B), and IL-6 proteins (C) in the livers of mice. Results are presented as mean ± S.D. (n = 8). Compared between 2 groups, * p < 0.05, ** p < 0.01, or *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
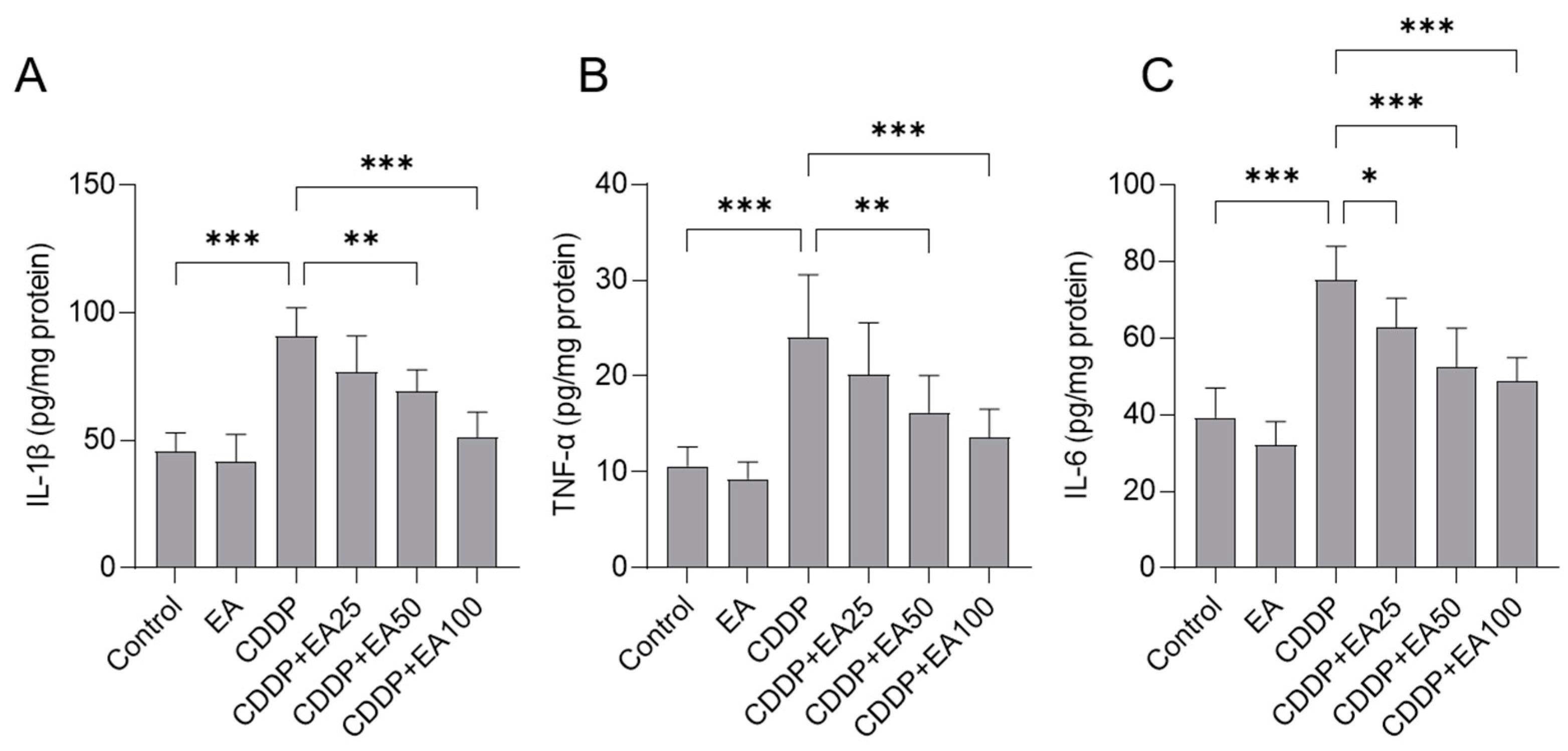
Figure 6.
Caspases-9 (A) and -3 (B) activities in the livers of mice. Results are presented as mean ± S.D. (n = 8). Compared between 2 groups, *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
Figure 6.
Caspases-9 (A) and -3 (B) activities in the livers of mice. Results are presented as mean ± S.D. (n = 8). Compared between 2 groups, *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
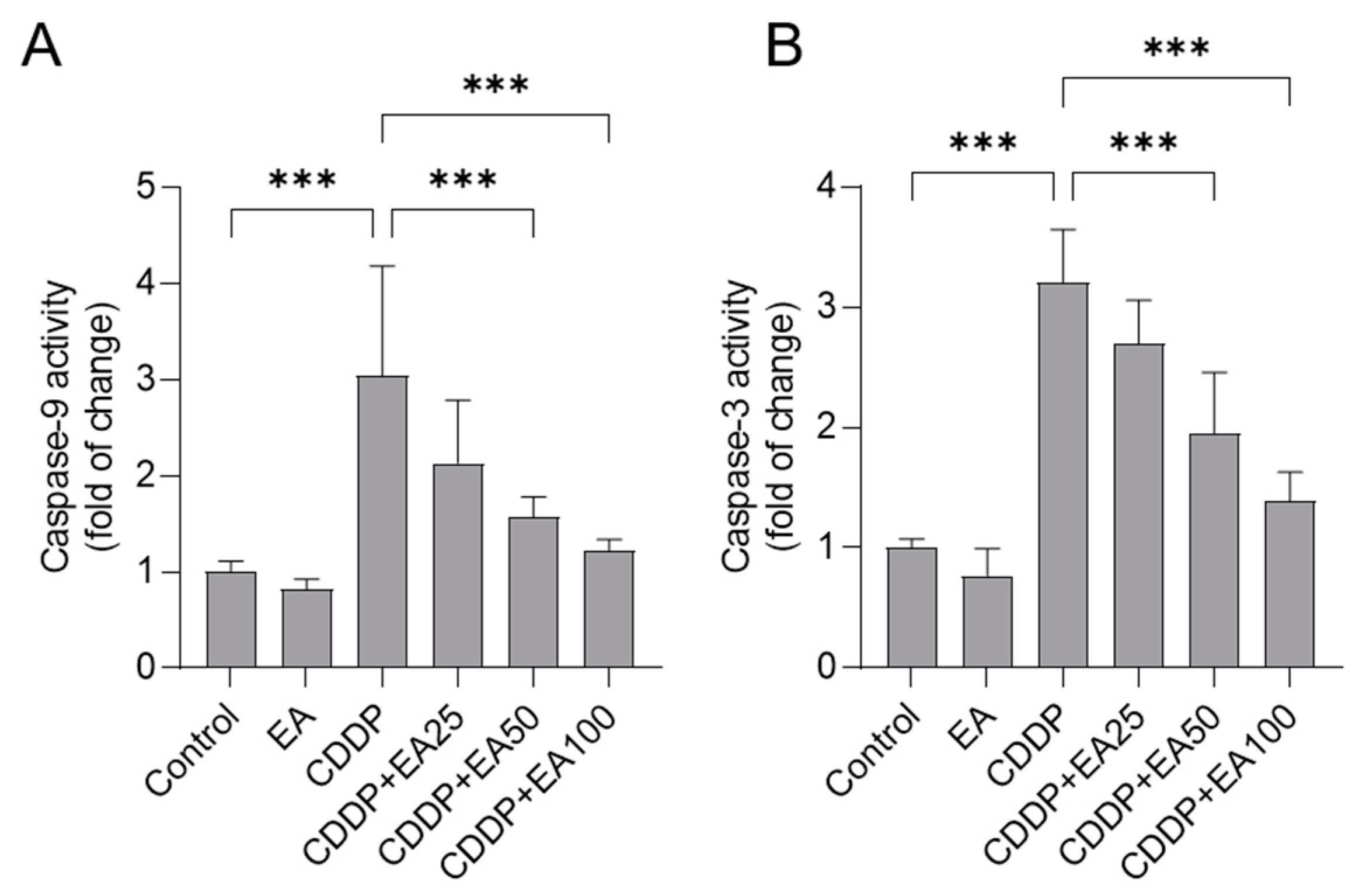
Figure 7.
The relative mRNA levels of NF-κB (A), IL-1β (B), TNF-α (C), IL-6 (D), Nrf2 (E), and HO-1 (F) in the livers of mice. Results are presented as mean ± S.D. (n = 6). Compared between 2 groups, * p < 0.05, ** p < 0.01, and *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
Figure 7.
The relative mRNA levels of NF-κB (A), IL-1β (B), TNF-α (C), IL-6 (D), Nrf2 (E), and HO-1 (F) in the livers of mice. Results are presented as mean ± S.D. (n = 6). Compared between 2 groups, * p < 0.05, ** p < 0.01, and *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
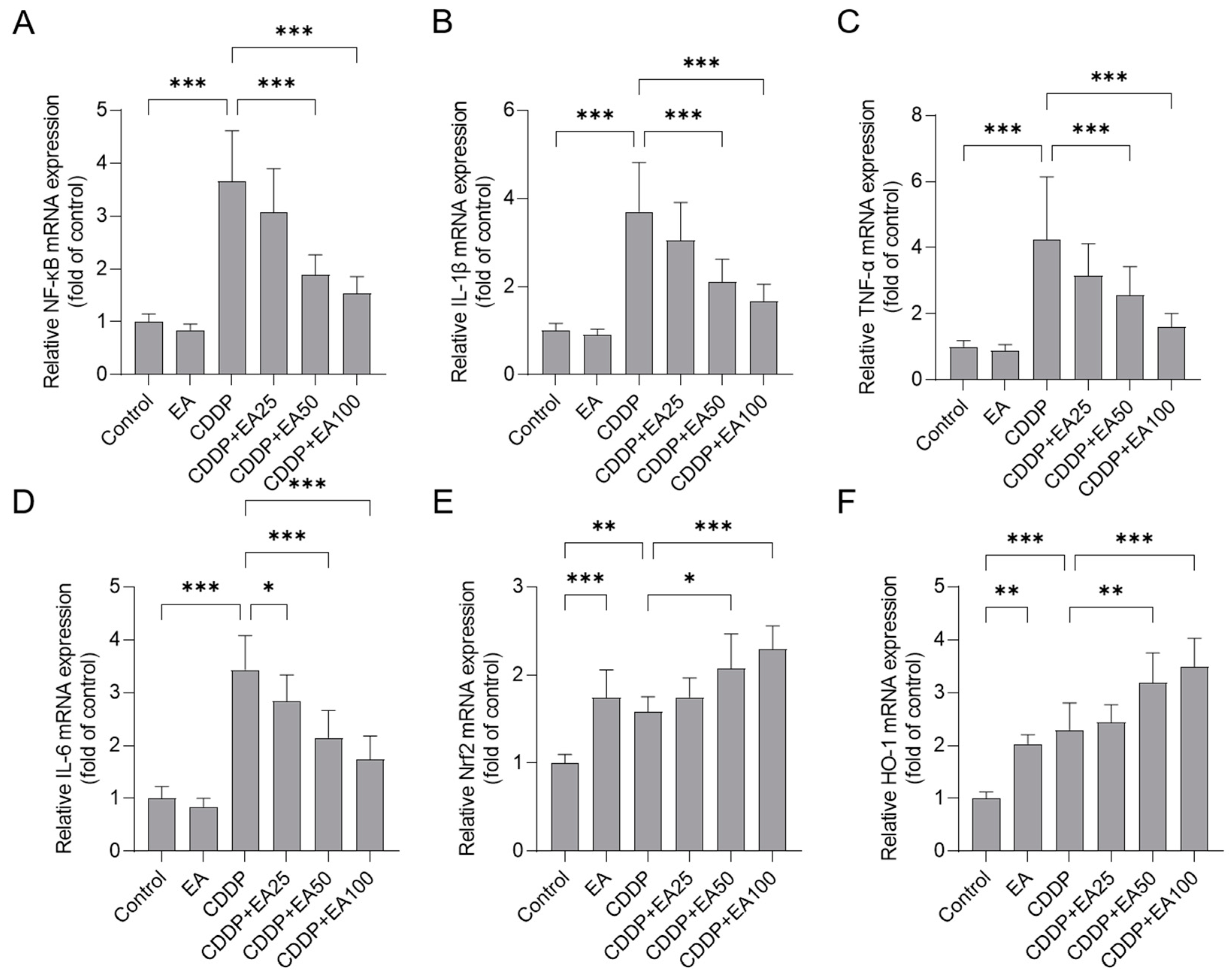
Figure 8.
Western blotting analysis of Nrf2, HO-1, and NF-κB proteins in the livers of mice. (A), the representative images of Western blotting. (B), the results of quantitative analysis (n = 4). Compared between 2 groups, * p < 0.05, ** p < 0.01, and *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
Figure 8.
Western blotting analysis of Nrf2, HO-1, and NF-κB proteins in the livers of mice. (A), the representative images of Western blotting. (B), the results of quantitative analysis (n = 4). Compared between 2 groups, * p < 0.05, ** p < 0.01, and *** p < 0.001. EA, ellagic acid; CDDP, cisplatin.
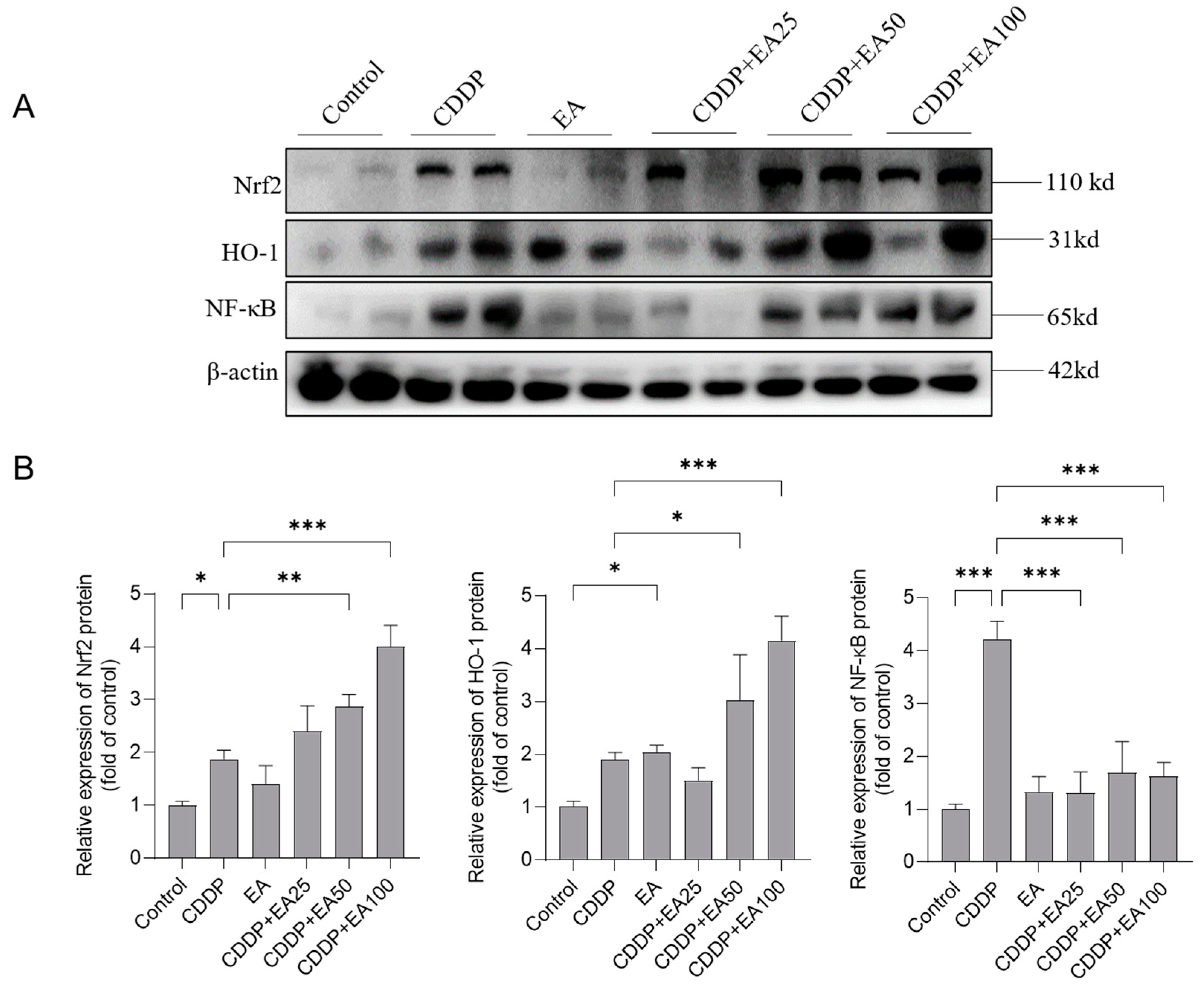
Figure 9.
Binding mode of EA with Keap1, Nrf2, and NF-κB by molecular docking. (A) The binding energy of EA with the potential targets. B and C, The binding models and corresponding 2D figures of interactions of EA with Keap1 (B) and NF-κB (C) proteins.
Figure 9.
Binding mode of EA with Keap1, Nrf2, and NF-κB by molecular docking. (A) The binding energy of EA with the potential targets. B and C, The binding models and corresponding 2D figures of interactions of EA with Keap1 (B) and NF-κB (C) proteins.
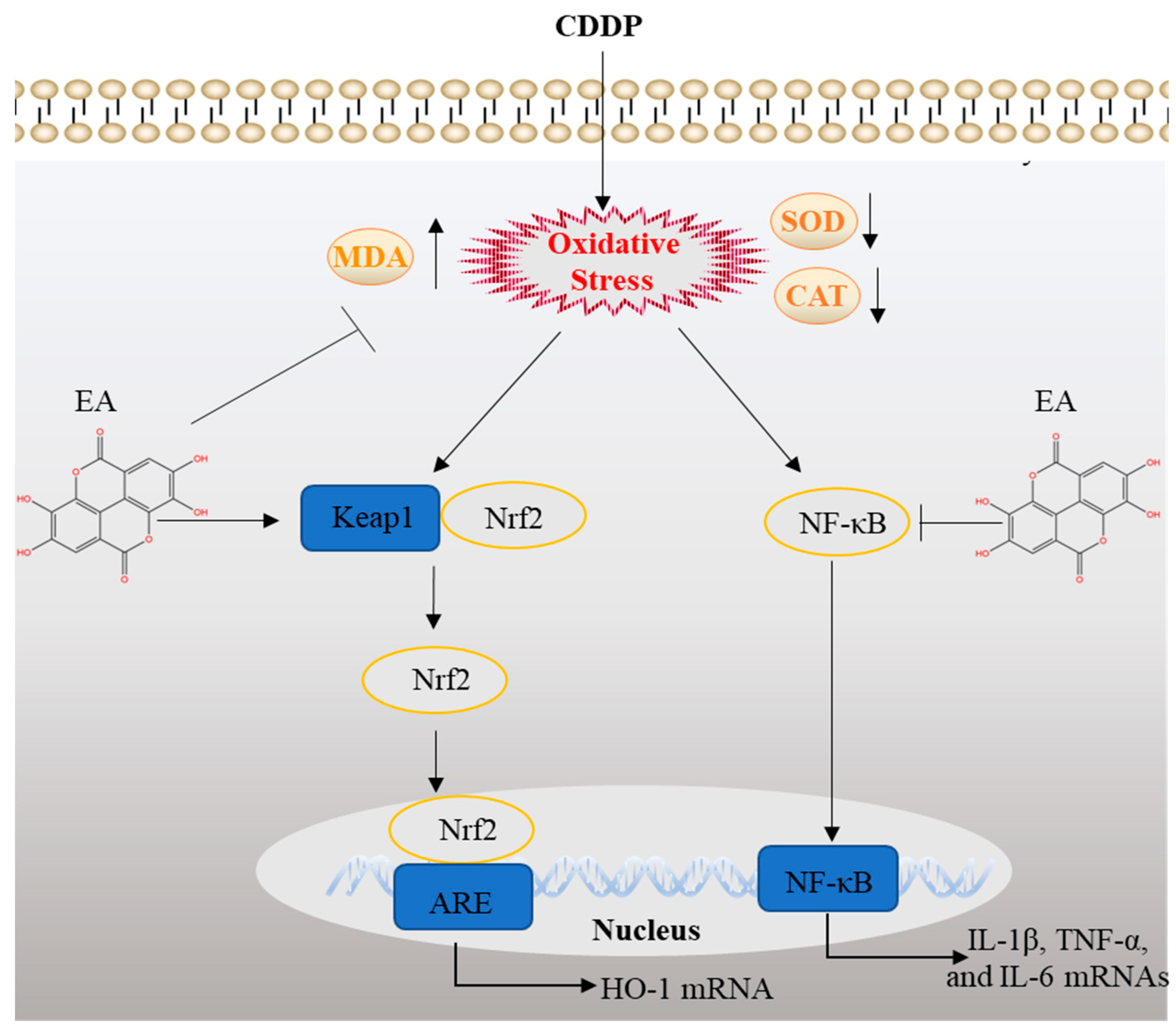
Figure 10.
Schematic diagram of the proposed mechanisms of EA protecting against CDDP exposure-caused liver toxicity. EA, ellagic acid; CDDP, cisplatin.
Figure 10.
Schematic diagram of the proposed mechanisms of EA protecting against CDDP exposure-caused liver toxicity. EA, ellagic acid; CDDP, cisplatin.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



