
Submitted:
01 October 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
This study assessed the associations between host genetic variants and subgingival microbiota in patients with metabolic syndrome (MetS); 103 patients with MetS underwent medical and periodontal examinations and had blood and subgingival plaque samples taken. DNA was extracted and processed (assessing a panel of selected single nucleotide polymorphisms (SNPs) first (hypothesis-testing), and then expanding to a discovery phase. The subgingival plaque microbiome from these patients was profiled. Analysis of associations between host genetic and microbial factors was performed, stratified for periodontal diagnosis. Specific SNPs within RUNX2, CAMTA1 and VDR genes were associated with diversity metrics, with no genome-wide associations detected for periodontitis severity or Mets components at p<10-7. Severe periodontitis was associated with pathogenetic genera and species. Some SNPs correlated with specific bacterial genera, as well as with microbial taxa, notably VDR (rs12717991) with Streptococcus mutans and RUNX2 (rs3749863) with Porphyromonas gingivalis. This is the first study exploring the role of host genotype variation in subgingival microbial dysbiosis in patients with MetS and periodontitis. In conclusion, variation in host genotypes may play a role in the dysregulated immune responses characterizing periodontitis and thus the oral microbiome, suggesting that systemic health-associated host traits further interact with oral health and microbiome.
Keywords:
metabolic syndrome
; periodontitis
; periodontal medicine
; cardiovascular disease
1. Introduction
“Infectogenomics” was introduced to define the effect of host genetic variants in influencing microbial colonization in a given echological niche[1]. Applying this concept to human diseases characterized by dysbiotic biofilms, “genetic dysbiosis” implies a host genome-driven imbalance between the integrity of barrier organs and their colonizing microorganisms[2,3].
In periodontitis, inflammation is thought to drive a progressive increase in the microbial diversity, leading in turn to perturbations in the microenvironment, such as increased availability of substrates favouring growth of Gram-negative bacteria[4]. The resulting dysbiosis predisposes to the activation of a host response cascade, causing periodontal tissue damage and, eventually, tooth loss. Data collected over the last 20 years provided evidence about the associations between host genetic variants and the presence and counts of specific bacteria in the subgingival niche. In this reggard, evidence was mainly based on analysis of a few specific candidate host genetic variants and a few specific candidate periodontopathogenic bacteria, analysed by checkerboard, culture or polymerase chain reaction PCR[5-8]. Only a handful of studies have employed a wider genome-wide approach[9] or a metagenomic analysis approach[10], so leaving us with a rather “restricted” view of potential associations between host genetic variants and subgingival dysbiosis.
The importance of integrating host genetic data for a better understanding of subgingival dysbiosis has been recently suggested[11]. In previous studies from our group[12,13] biomarkers of gingival crevicular fluid were shown to be associated with Mets, as well as left ventricular geometry with periodontitis. This latter finding further underscored the concept linking the burden of microinflammation on cardiovascular system[14]. Given the association between periodontitis and metabolic syndrome[15] and the potential effects of oral bacteria on gut microbiota[16,17] studying the effect of host genetic variants on the subgingival microbiota may also be particularly relevant to our understanding of connections between periodontitis and systemic health. Therefore, this study aimed to perform detailed analyses of associations between host genetic variants and subgingival microbiota in patients with Mets with or without periodontitis.
2. Results
Demographic and clinical characteristics of the 103 included subjects are reported in Table 1.
Patients were on average 58 years old, with a majority of males, and had an average BMI of nearly 32. The majority of patients (77%) were not regular dental attenders and oral hygiene habits (tooth brushing frequency, use of interdental cleaning tools) were not up to the standard required in patients at high risk of periodontal disease. Ten patients were classified as having no-mild periodontitis, 38 as moderate periodontitis and 55 as severe periodontitis[18]. Furthermore, 38 patients were diagnosed with caries (Table 2, Supplemental material).
2.1. Microbial diversity and metabolic syndrome
The MED pipeline analyzed 3,439,061 sequences after quality filtering and partitioned the sequences into 1264 nodes, which mapped to 301 human microbial taxa (HMT) at ≥ 98.5% identity. Severe periodontitis patients had a higher alpha diversity in the subgingival plaque microbiome as measured by Shannon, Simpson, Evenness and Chao1 indices compared to no/mild or moderate periodontitis with the differences not statistically significant (Figure 1A).
Among SNPs from the selected gene panel, 4 SNPs within 2 genes were significant at p<0.01 for alpha diversity: RUNX2 (rs1321081, rs3749863) and CAMTA1 (rs1193247, rs12407666). In addition, Evenness and Simpson indices were associated with SNPs in TRPS1 (rs1012478), GLT6D1 (rs57670611), KCNK1 (rs701223), VAMP3 (rs111692854) and VDR (rs731236) genes at the nominal p<0.05 significance (but none at the p<0.01 level). Supplemental material (Figure S1) shows the difference between patients with different Mets components.
No genome-wide associations at p <10-7 could be detected for alpha diversity metrics and periodontitis severity. The strongest genome-wide signals detected for periodontitis severity were at 6 closely associated loci within chromosome 15, downstream of the lincRNA RP11-209E8.1 and upstream of the WDR72 gene (p<0.0001; Figure 1C). The genome-wide variants associated with Shannon diversity were in genes CTD-3037G24.3 (rs62029200) and a SNP downstream of the lincRNA RP11-570K4.1 (rs4714409) at p<0.0001 (Figure 1C). Higher numbers of variants were associated with Simpson diversity than other alpha diversity metrics at p<0.0001: PGLYRP3, PGLYRP4, VAMP4, CEP135, CDH12, ZNF804B, RNU2-54P, FAM189A2, TACC2, MGMT, HBG2, TMEM132B, KIFC3 and ZNF701 (Figure 1C). Although no genome-wide signals for the different components of the metabolic syndrome were detected, variants in the following genes were associated with the components of metabolic syndrome at p<0.00001: (i) Waist circumference: PARD3B, CTD-2269F5.1, CSMD1, ADAMTSL3 (ii) Low HDL cholesterol: PPIL6, SMPD2, MICAL1, BAALC, ESRP1, DPY19L4 (iii) Hypertension: ANKS1B (iv) High fasting glucose: CNNM2, NT5C2, TEX2 and lincRNA RP3-390M24.1 (Supplemental Figure S2).
2.2. Genotype associations with microbial taxa
Phyla distributions in patients with different periodontal diagnosis showed predominance of Firmicutes in all three groups and phyla Fusobacteria, Spirochaetes and Synergistetes at a higher abundance in severe periodontitis compared to no-mild or moderate periodontitis (Supplemental Figure S3). No clear differences were observed when phyla distribution was studied across different genotypes (data not shown).
2.2.1. Genotype associations at the Genus level
Differences were observed at the genus level between severe and mild or moderate periodontitis (Aitchison; F=2.562; p=0.0015). Principal component analysis loadings revealed that no-mild or moderate periodontitis were associated with genera such as Actinomyces, Rothia, Corynebacterium, Granulicatella and Leptotrichia, whereas severe periodontitis was associated with Filifactor, Peptostreptococcus, Bacterioidetes, Fretibacterium, Treponema, Mogibacterium and Dialister (Fig. 2).
Figure 2.
A) Score plot of the first 2 principal components of CLR transformed 16S microbial genus abundance data from the MetS periodontitis patients. B) Loading plot from the principal components analysis showing microbial variables that most strongly influenced the first 2 principal components in plot A. C) Box and jitter plots of SNP variants from the selected gene panel that showed significant associations with relative abundance of different subgingival genera. Please note that each SNP was filtered for all alleles present at 10% MAF for the analysis, so for SNPs below this threshold the model was run for only two of the genotypes.
Figure 2.
A) Score plot of the first 2 principal components of CLR transformed 16S microbial genus abundance data from the MetS periodontitis patients. B) Loading plot from the principal components analysis showing microbial variables that most strongly influenced the first 2 principal components in plot A. C) Box and jitter plots of SNP variants from the selected gene panel that showed significant associations with relative abundance of different subgingival genera. Please note that each SNP was filtered for all alleles present at 10% MAF for the analysis, so for SNPs below this threshold the model was run for only two of the genotypes.
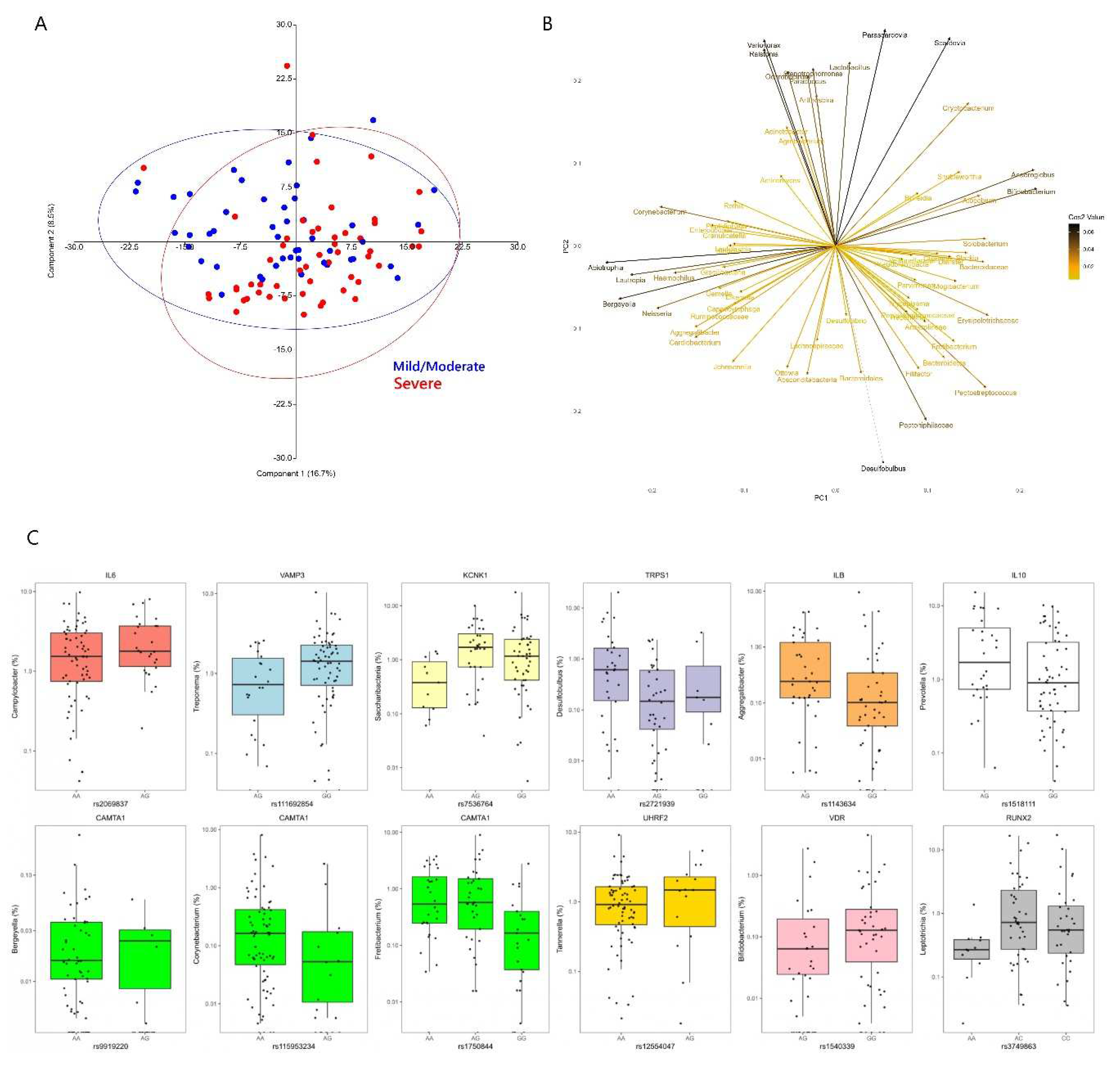
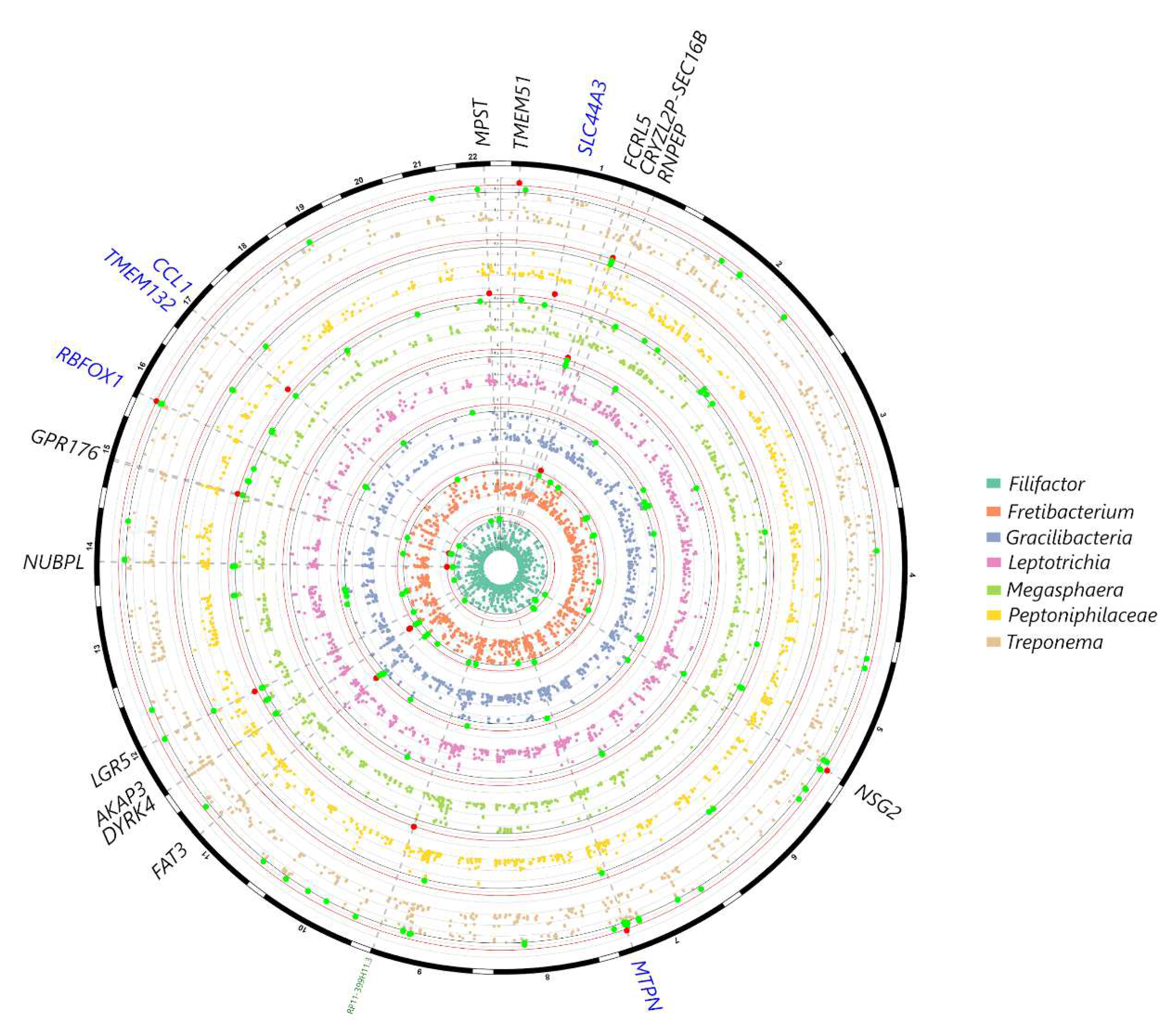
From the selected gene-SNP panel, IL6 was associated with Campylobacter, KCNK1 with Saccharibacteria, VAMP3 with Treponema, and several variants within CAMTA1 with Bergeyella, Fretibacterium, Actinomyces and Corynebacterium at p<0.001 (Figure 2). Associations were also observed with IL1B and Aggregatibacter, TRPS1 (Desulfobulbus), IL10 (Prevotella), UHRF2 (Tannerella), VDR (Bifidobacterium) and RUNX2 (Leptotrichia) at p<0.005 (Figure 2). In the genome-wide analysis, genera such as Filifactor, Gracilibacteria, Fretibacterium, Leptotrichia, Megasphaera and Treponema showed associations with several loci at p<10-5, however no significant associations could be detected at p<10-7. The gene-genus pairs that showed associations at p<10-5 included: NUBPL (Filifactor), FAT3 (Gracilibacteria), CAMTA1 and AKAP3 (Fretibacterium), FCRL5 (Leptotrichia), MPST, GPR176 (Megasphaera), SEC16B (Peptoniphilaceae) and TMEM51 (Treponema) (Fig. 3).
Figure 3.
Circular Manhattan plot showing subgingival genera in the MetS patients that showed the strongest signals with genetic markers in the genome-wide analysis, after correcting for age, sex, smoking frequency, BMI and population structure. Variants at p<10-3 for at least one trait are plotted, with signals at p<10-5 highlighted in red and markers at p<10-4 in green. Highlighted markers are annotated in the outer circle: SNPs within gene coding regions are in black, genes adjacent to exon SNPs are in blue, non-coding RNAs and other regulatory elements are annotated in green.
Figure 3.
Circular Manhattan plot showing subgingival genera in the MetS patients that showed the strongest signals with genetic markers in the genome-wide analysis, after correcting for age, sex, smoking frequency, BMI and population structure. Variants at p<10-3 for at least one trait are plotted, with signals at p<10-5 highlighted in red and markers at p<10-4 in green. Highlighted markers are annotated in the outer circle: SNPs within gene coding regions are in black, genes adjacent to exon SNPs are in blue, non-coding RNAs and other regulatory elements are annotated in green.
2.2.2. Genotype associations at the Species level
Consistent with the genus-level analysis, severe periodontitis was significantly different to no-mild or moderate periodontitis, when the microbial taxa were considered at the species-level (Aitchison; F: 2.136, p=0.0015). Taxa including classical ‘red complex’ species such as Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia and others such as Filifactor alocis, Fretibacterium fastidiosum, Fusobacterium nucleatum ssp nucleatum, Peptoniphilaceae [G-1] HMT 113, Lachnospiraceae [G-8] HMT 500, Peptostreptococcus stomatis and Dialister pneumosintes were associated with severe periodontitis (Figure 4). Taxa such as Granulicatella elegans, Gemella haemolysans, Neisseria flavescens, Rothia aeria, Streptococcus parasangunis clade 411 were associated with no-mild or moderate periodontitis (Fig. 4).
Figure 4.
A) Score plot of the first 2 principal components of CLR transformed 16S microbial species abundance data from the MetS periodontitis patients. B) Loading plot from the principal components analysis showing microbial variables that most strongly influenced the first 2 principal components in plot A. C) Box and jitter plots of SNP variants from the selected gene panel that showed significant associations with relative abundance of different microbial species in the subgingival plaque. Please note that each SNP was filtered for all alleles present at 10% MAF for the analysis, so for SNPs below this threshold the model was run for only two of the genotypes.
Figure 4.
A) Score plot of the first 2 principal components of CLR transformed 16S microbial species abundance data from the MetS periodontitis patients. B) Loading plot from the principal components analysis showing microbial variables that most strongly influenced the first 2 principal components in plot A. C) Box and jitter plots of SNP variants from the selected gene panel that showed significant associations with relative abundance of different microbial species in the subgingival plaque. Please note that each SNP was filtered for all alleles present at 10% MAF for the analysis, so for SNPs below this threshold the model was run for only two of the genotypes.
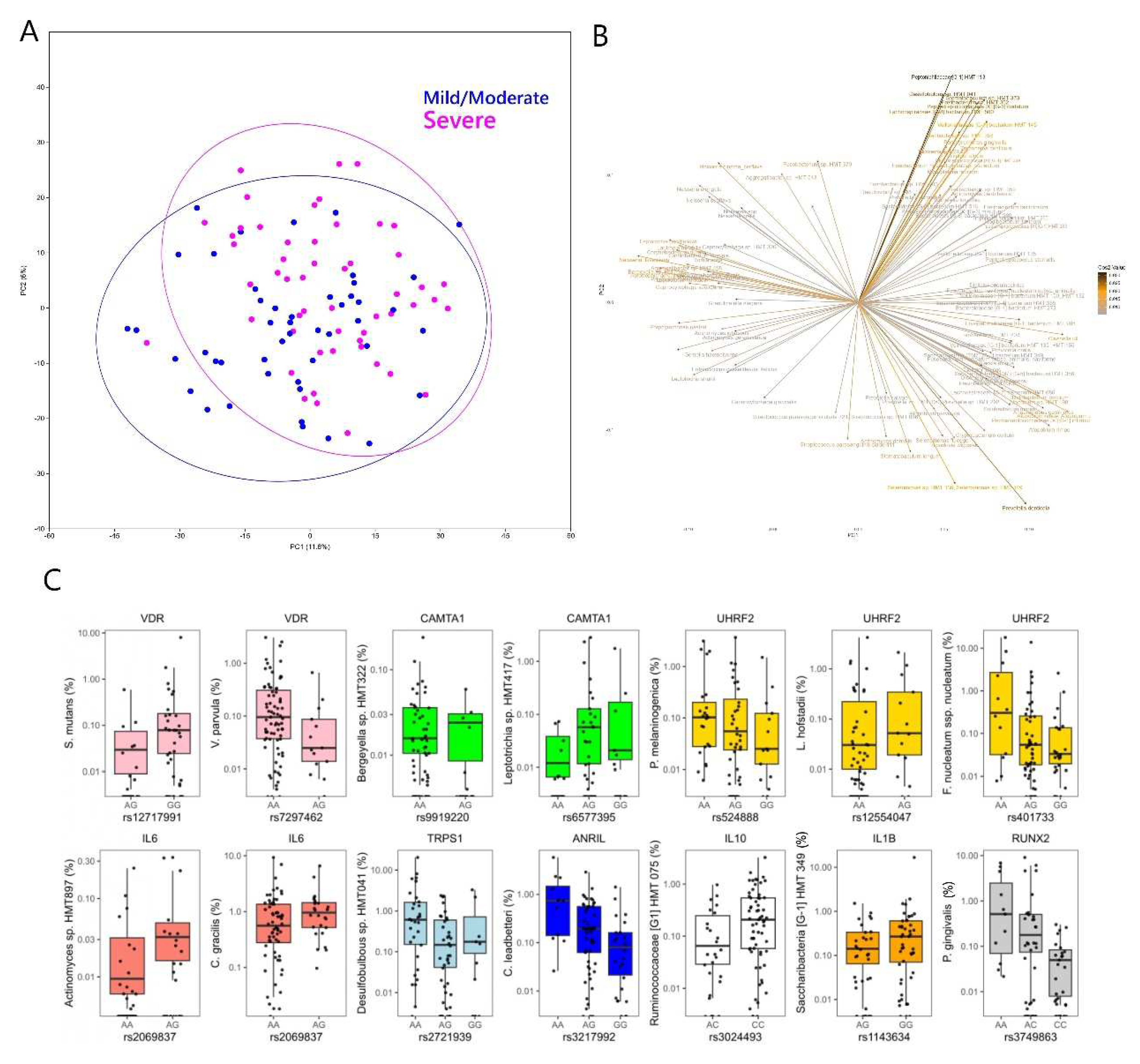
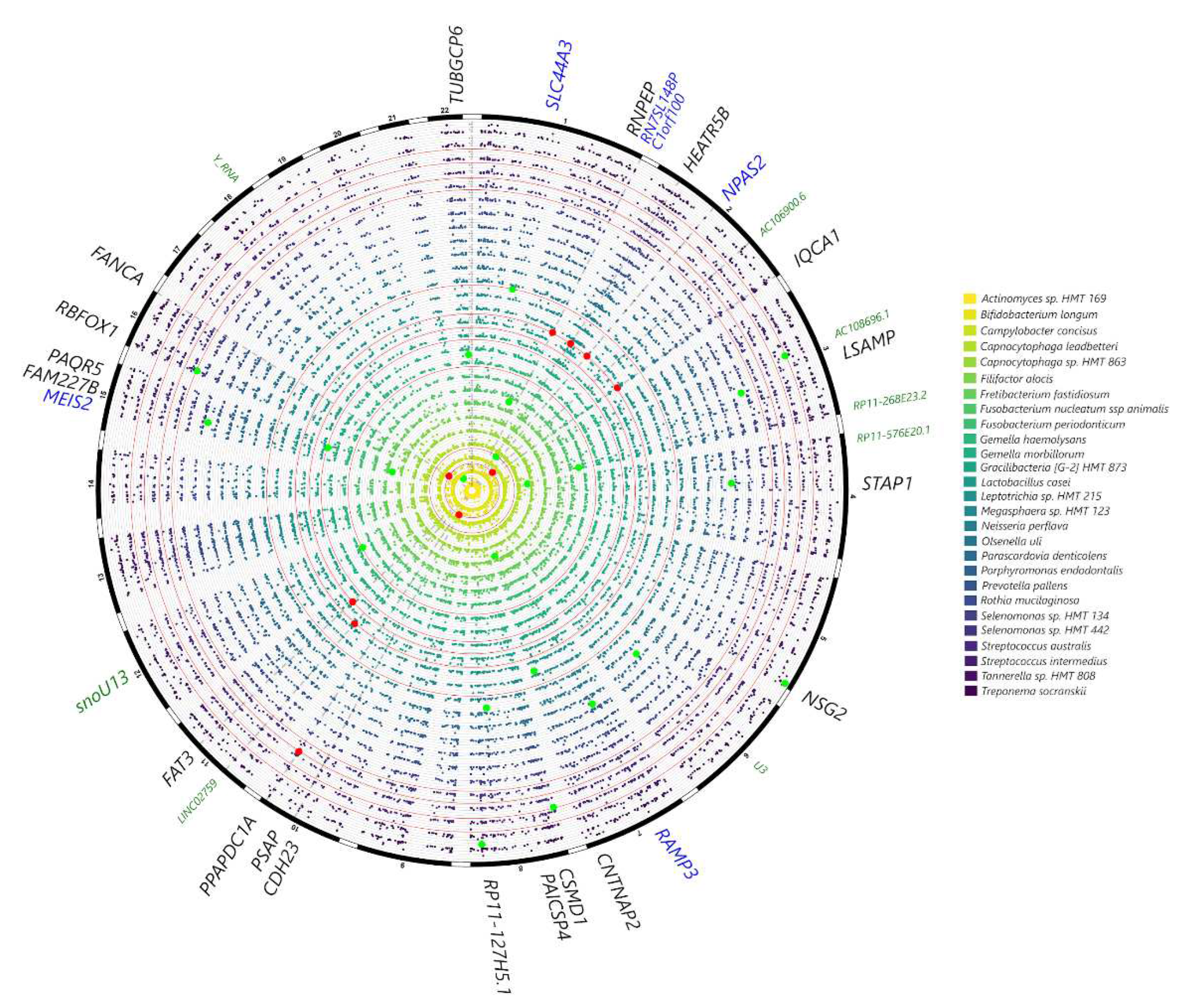
Among SNPs from the selected gene panel, associations were noted at p<0.0001 between VDR and rs12717991 and Streptococcus mutans (p<0.0001) and at p<0.001 between CAMTA1 and Bergeyella sp. HMT 332 and Leptotrichia sp., UHRF2 (Prevotella melaninogenica, Leptotrichia hofstadii), IL6 (Actinomyces sp. HMT 897), TRPS1 (Desulfobulbus sp. HMT 041). At p<0.005 further associations were noted between VDR and Veillonella parvula, UHRF2 (Fusobacterium nucleatum), IL6 (Campylobacter gracilis), ANRIL (Capnocytophaga leadbetteri), IL10 (Ruminococcaceae [G1] HMT 075), IL1B (Saccharibacteria TM7 [G1] HMT 349), RUNX2 (Porphyromonas gingivalis) (Figure 4). Although no genome-wide associations were detected (p<10-7), at p<10-6 the following gene-taxa associations were detected: Bifidobacterium longum (AC106900.6, CDH23, PSAP), Gracilibacteria [G-2] HMT 873 (FAT3), Lactobacillus casei (HEATR5B, IQCA1) and Selenomonas sp. HMT 442 (PPAPDC1A) (Fig. 5).
Figure 5.
Circular Manhattan plot showing subgingival microbial species in the MetS patients that showed the strongest signals with genetic markers in the genome-wide analysis, after correcting for age, sex, smoking frequency, BMI and population structure. Variants at p<10-4 for at least one trait are plotted, with signals at p<10-6 highlighted in red and suggestive associations at p<10-5 in green. Highlighted markers are annotated in the outer circle: SNPs within gene coding regions are in black, genes adjacent to exon SNPs are in blue, non-coding RNAs and other regulatory elements are annotated in green.
Figure 5.
Circular Manhattan plot showing subgingival microbial species in the MetS patients that showed the strongest signals with genetic markers in the genome-wide analysis, after correcting for age, sex, smoking frequency, BMI and population structure. Variants at p<10-4 for at least one trait are plotted, with signals at p<10-6 highlighted in red and suggestive associations at p<10-5 in green. Highlighted markers are annotated in the outer circle: SNPs within gene coding regions are in black, genes adjacent to exon SNPs are in blue, non-coding RNAs and other regulatory elements are annotated in green.
3. Discussion
This is the first study, to the best of our knowledge, reporting host genome-wide analysis and 16s subgingival microbiota in patients with MetS. This study broadly confirms that host genetic variants play a role in shaping the subgingival biofilm in periodontitis. Among target-studied SNPs, some showed associations with microbial diversity and with microbial species subgingivally. Measures of microbial diversity were associated with 7 of the 20 target genes. In particular, RUNX2 (rs1321081, rs3749863) and CAMTA1 (rs1193247, rs12407666) were associated with alpha-diversity, while SNPs in TRPS1 (rs1012478), GLT6D1 (rs57670611), KCNK1 (rs701223), VAMP3 (rs111692854) and VDR (rs631236) genes were associated with Evenness and Simpson indices, although only at the nominal p<0.05 significance. A different VDR SNP (rs12717991) showed a strong association with S. mutans and a slightly weaker association with Veillonella parvula. For incidence, S.mutans is notoriously a caries-associated bacterium, which appears to compete with periodontopathogenic bacteria, such as P.gingivalis[19]. Intriguingly, it has been shown that co-culture with V.parvula alters the physiology of S. mutans, giving it an advantage in surviving antimicrobial treatment[20,21]. These data suggest that a host genetic variant (such as this VDR SNP) may alter the physiology of the subgingival biofilm, potentially favoring the growth of bacteria, which in turn influences the biofilm differentiation and growth, along the lines of the keystone pathogen and IMPEDE theories[22]. Findings relative to the potential effect of the VDR gene variants in the subgingival microbiota are also in agreement with an association with alpha diversity in a recent study on twins[23] and, previously, with subgingival detection of P. gingivalis[24].
The associations between IL6 SNP (rs1800795) and genera Campylobacter and Actinomyces sp. HMT 897 confirm its possible involvement in shaping the subgingival microbiota. This has been previously described and suspected to be mediated by an increased production of IL-6, leading to an increased inflammatory cascade, favoring in turn the growth of specific microbes[6,25]. IL10 SNP (rs6667202) was associated with genera Prevotella and with species Ruminococcaceae [G1] HMT 075. This is interesting, as variants in this gene have recently emerged as potentially affecting the composition of the subgingival biofilm[3,23]. Other interesting associations emerging from this study are those between UHRF2 and both the genera Tannerella and species Prevotella melaninogenica, Leptotrichia hofstadii and F. nucleatum, between TRPS1 and Desulfobulbus, between RUNX2 and P.gingivalis and between CAMTA1 and several genera and species. Some of these bacteria are well-known periodontal pathogens, while others are oral health-associated. It is hard to speculate on specific mechanisms of associations at this stage, especially for genes whose involvement in periodontal pathogenesis is still somewhat unclear. A conserved noncoding element within CAMTA1 upstream of VAMP3 (rs10864294) seems to emerge as a potentially important locus in the association with the subgingival microbiome, given associations with alpha diversity, as well as with specific genera and species. Interestingly, gene variants in CAMTA1/VAMP3 have been suspected to be responsible for shared predisposition to both periodontitis and cardiovascular disease[26,27] which is relevant considering the nature of the present sample (MetS).
A second part of the analysis consisted of a discovery genome-wide analysis where no statistically significant signals emerged at p<10-7, consistent with other periodontal GWAS with no reported associations at this statistical level. This may in part be due to the small sample size of the present study. The closest associations with Shannon diversity data were found for CTD-3037G24.3 (rs62029200) and a SNP downstream of the lincRNA RP11-570K4.1 (rs4714409). Although other genes showed nominal associations with subgingival genera and species, such as genera Filifactor (gene NUBPL), Gracilibacteria (FAT3), Fretibacterium (CAMTA1, AKAP3), Leptotrichia (FCRL5), Megasphaera (MPST, GPR176), Peptoniphilaceae (SEC16B), Treponema (TMEM51), it is difficult to speculate as to what these putative associations mean. NUBPL is involved in assembly of mitochondrial Complex I and its expression in salivary glands is reported to be associated gamma delta T cell infiltration in primary Sjogren Syndrome[28]. A number of these genes, such as FCLR5, FAT3, AKAP3, RUNX2, CAMTA1, are also involved in immune response pathways, so it is plausible that these variants or identified genes could be involved in a dysfunctional host-microbial response in periodontitis, as the genera identified in this study to be associated with these genes previously recognized as being associated with periodontitis. Further, associations for the CSMD1 gene were found in the species-level GWAS and waist circumference (Figure 5, Supplemental Figure 2). For incidence, this gene is a regulator of the complement cascade highlighting potential interactions between Mets and periodontitis.
The robustness of the 16s microbial analysis is confirmed by the associations detected between periodontal status and subgingival genera and species. Genera such as Filifactor, Peptostreptococcus, Bacterioidetes, Fretibacterium, Treponema, Mogibacterium and Dialister were increased in severe periodontitis. Furthermore, taxa including classical ‘red complex’ species such as Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia and others with well-known associations with periodontitis, such as Filifactor alocis, Fretibacterium fastidiosum, Fusobacterium nucleatum ssp nucleatum, Peptoniphilaceae [G-1] HMT 113, Lachnospiraceae [G-8] HMT 500, Peptostreptococcus stomatis and Dialister pneumosintes, were associated with severe periodontitis (Fig. 4), while taxa such as Granulicatella elegans, Gemella haemolysans, Neisseria flavescens, Rothia aeria, Streptococcus parasangunis clade 411 were associated with no-mild or moderate periodontitis. This is in line with previous literature, as streptococci are well-known commensal bacteria, competing against periodontopathogenic bacteria, and usually highly abundant in healthy sites[29,30]. The role of Granulicatella in the periodontal biofilm is less clear, while G. adiacens has shown ability to co-aggregate with F. nucleatum[31]. Along with the classical periodontal pathogens described by Socransky and co-workers[32-34], the list of taxa associated with periodontitis in the present study includes some bacteria recently associated with periodontitis such as Fretibacterium[35], Dialister pneumosintes[36] and Filifactor alocis[37-39]. These associations are confirmatory, although novel in relation to this specific population of patients with minimal dental care, and all affected by Mets. The effect of the number of MetS components on subgingival microbiota was investigated, but only a tendency for association with microbial diversity was detected. A limitation of this analysis was the absence of diagnosis based on the 2018 classification of periodontal disease, which was not possible retrospectively in this population. The per-protocol AAP classification was instead used[18].
Some gene variants found in our periodontitis patients to link with components of Mets have recently been described as determinants in different metabolic disorders. In particular, PARD3 has been associated with signaling pathway related to diabetes[40]; CSMD1 affected BMI and blood lipid levels[41]; ADAMTSL3 was linked to metabolic impairment, especially for incipient diabetes, defined on the basis of both fasting and non-fasting blood glucose, and the distribution of lean body mass[42-44]; TEX2 was associated with diabetes and impaired lipid metabolism[45].
Periodontitis and MetS are clearly associated, possibly mediated by genetic, environmental and behavioural factors, as well as by bi-directional effects of dyslipidemia, reduced glucose tolerance, oxidative stress, molecular mimicry and dysbiosis[15]. This study shows that, even in MetS patients, host genetic variants are likely to influence the composition of the subgingival biofilm. Among tested SNPs (hypothesis-testing analysis), those in RUNX2, CAMTA1, VDR, IL6 and TRPS1 genes emerged as possibly the most likely to influence the subgingival microbiota in this patient sample, while no new SNPs clearly emerged as associated with the microbial outcomes at genome-wide analysis (hypothesis-generating). This adds to our understanding of infectogenomics but calls for further studies to elucidate pathogenic pathways leading to periodontal breakdown.
Strengths of this study are the ethnic homogeneity of the included subjects, the consistency of full mouth periodontal examinations carried out by a single calibrated examiner and the novelty of combining genome-wide host data with 16s microbial analysis, which should be considered the next step for the field of infectogenomics. The main limitation of the study is the absence of controls without the metabolic syndrome (with presence only of internal controls with MetS and no periodontitis).
Overall, some strenghts should be highlighted: 1) this is the first study to report analysis of GWAS and 16s subgingival plaque in patients with different degrees of periodontal disease, providing further evidence for infectogenomics effects on the subgingival biofilm; 2) this study suggests that systemic health-associated host traits may further interact with oral health and microbiome.
4. Materials and Methods
4.1. Study population
The study population has been reported before, including the finding that periodontitis may be associated with concentric left ventricular remodeling [13] and analysis of gingival crevicular fluid[46]. The analysis described in this paper investigated associations between host genetic variants and subgingival microbiota. The STROBE checklist was followed during the conduct and reporting of the study. In brief, 103 MetS patients attending the Department of Internal Medicine (Ospedale Cannizzaro and Ospedale Garibaldi), University of Catania, for outpatient examination signed informed consent to take part in the study and were included from July 2015 to July 2017. Ethics approval was obtained by the sponsor institution, University College London (reference 4242/01), and separately by the clinical centre in Catania (reference 1497/Cs, Comitato Etico Catania 1). The study was registered on clinicaltrials.gov (identifier NCT03297749).
Inclusion criteria:
- Caucasian ethnicity;
- Age 25- 75;
- Diagnosis of metabolic syndrome as defined by the revised NCEP ATP III (e.g. the presence of at least 3 of the following factors)[47]:
- 1.
- Waist circumference > 102 cm for men and > 88 cm for women;
- 2.
- High triglycerides: ≥ 150 mg/dL (1.7 mmol/L), or specific treatment for this lipid abnormality;
- 3.
- Low HDL cholesterol: < 40 mg/dL (1.03 mmol/L) in males, < 50 mg/dL (1.29 mmol/L) in females, or specific treatment for this lipid abnormality;
- 4.
- High blood pressure: systolic BP ≥ 130 or diastolic BP ≥85 mm Hg, or treatment of previously diagnosed hypertension;
- 5.
- High fasting plasma glucose: FPG ≥100 mg/dL (5.6 mmol/L), or previously diagnosed type 2 diabetes.
- Presence of at least 12 teeth.
Exclusion criteria:
- Pregnancy;
- Presence of infectious diseases such as hepatitis and HIV;
- Antibiotic pre-medication required for the performance of periodontal examination;
- Previous periodontal therapy within 6 months of the study visit.
4.2. Medical assessment and sampling
As described before[13], medical and smoking histories were recorded, body mass index (BMI), waist circumference and office blood pressure were taken and blood sampling was carried out for DNA extraction. Furthermore, intima-media thickness by B-mode real-time ultrasound, pulse wave velocity and echocardiographic data were collected. Patients’ dental history was investigated, including family history of periodontal disease, frequency of dental appointments, date of last appointment and previous treatment, reasons for tooth loss and frequency and type of tooth brushing. A six sites/tooth periodontal examination was carried out by a single calibrated examiner including full mouth plaque scores (FMPS)[48], full mouth probing pocket depth (PPD), clinical attachment level (CAL) bleeding on probing (FMBS) [48], tooth mobility and furcation involvement. Patients were classified as having periodontitis according to the criteria below[18]:
- healthy/mild periodontitis: < 2 sites on different teeth with CAL ≥ 4 mm or no sites with PPD ≥ 4 mm;
- moderate periodontitis: ≥ 2 sites on different teeth with CAL ≥ 4 mm or one site with PPD ≥ 4 mm;
- severe periodontitis: ≥ 2 sites on different teeth with CAL ≥ 6 mm and ≥ 1 site with PPD ≥ 4 mm.
Blood samples were taken by venipuncture in the antecubital vein of patients sitting in a semi-reclined position and were stored at -70° C until analysis.
Four subgingival plaque samples were taken from the disto-buccal surfaces of first molars. In the absence of these teeth, neighbouring teeth were chosen (second premolars, second molars, first premolars, canines in this order). The supragingival portion of the root surface of the site was carefully cleaned and the area was isolated from saliva before insertion of a sterile curette to the bottom of the pocket. After a single stroke each microbiological sample was extracted from the pocket and then pooled and placed into 1 ml of reduced transport fluid, which was then be placed in the laboratory freezer at –70°C for storage and analysis at the end of the study.
4.3. Genotyping, Imputation and Genome-wide association analysis
Study participants were genotyped from their peripheral blood cell DNA (Nucleon BACC2 kit, Nucleon Bioscience, Coatbridge, UK) using the Illumina Infinium Global Screening Array, which measures approximately 640,000 genetic markers. The genotyping was performed at the Queen Mary University of London (QMUL) Genome Centre. The genotype files were checked for sex assignment and filtered for >10% missing genotypes using the call rate calculations. A relatedness analysis was performed using PLINK v1.9 and the dataset trimmed at 0.1 relatedness cutoff[49]. The Sanger Imputation Server (https://imputation.sanger.ac.uk/) was used to impute the genotype files employing EAGLE2 for phasing, Positional Burrows-Wheeler Transform for imputation, and the Haplotype Reference Consortium version 1.1 as the reference panel[50-52]. Genotyped and imputed SNPs underwent quality control with PLINK: Multi-allelic variants, SNPs with an imputation INFO score below 0.3, genotype calls with a posterior probability <0.9, Minor Allele Frequency (MAF) <5%, a genotyping rate <90%, or a deviation from the Hardy-Weinberg equilibrium (P < 5.7 × 10−7) were removed. Participants with ≥10% missing genotypes were excluded. Genome-wide association tests were performed using the mixed linear model as implemented in the R package rMVP[53] on the imputed SNP dataset consisting of 3,919,976 markers that passed quality control. Factors such as sex, age, BMI, smoking status and three principal components of the population structure were incorporated as covariates. Significant SNPs were annotated, and their functions explored using SNPNexus[54].
4.4. Microbiome Profiling
The DNA from subgingival plaque samples were extracted using the MasterPure Gram-positive DNA purification kit (Epicentre, Madison, USA) with a lysozyme incubation step prior to extraction (ReadyLyse, Epicentre, Madison, USA). After screening for quality, the bacterial 16S rRNA gene region V3-V4 in the samples were amplified using Nextera primers (Forward – 5’ – TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG; Reverse – 5’ - GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC). The libraries were then multiplexed, barcoded and sequenced using the Miseq V3 600 cycle kit in the 300 bp paired-end read method on the Illumina Miseq platform (Illumina, USA) at the Genome Centre, Queen Marry University of London. The raw Illumina reads were quality filtered, trimmed, and merged with minimum overlap of 70 bases, Q30 check, a Q-score >20 for ambiguous bases recovered in the overlapping region and up to 2 ambiguous bases allowed in the overlap[55]. The sequences were aligned using Mothur (v.1.36.1) to the Silva 16S reference alignment (release 119)[56]. Minimum entropy decomposition (MED, v2.1) was used to partition the aligned sequences into nodes at 1-nt resolution with the minimum substantive abundance parameter at 200[57]. A BLAST search of the HOMD (v14.5) and NCBI bacterial 16S databases classified representative sequences of all MED nodes, which was used to map the nodes to the human microbial taxa (HMT) by per cent identities[58]. Nodes that mapped to multiple HMT with identical percent identities were assigned as separate taxa. The raw fastq files are deposited in the European Nucleotide Archive (Accession: PRJEB66327 & ERP151391).
Alpha diversity and multivariate statistical analyses were performed in the PAST software package (v3.22) on the HMT mapped abundance data. Interaction between periodontitis, components of Mets, and the subgingival microbiota were explored using permutational multivariate analysis of variance (PERMANOVA). Dissimilarities and distances used in the testing for differences included beta diversity indices such as Bray-Curtis, Horn, Jaccard, Kulczynski and Simpson[59].
4.5. Statistical analysis/power calculation
The sample size calculation was based on the pulse-wave velocity outcome resulting in a required sample size of 102 patients[13]. No sample size estimation was conducted for the host genome-microbial analysis. Data was entered in an MS Excel file and proofed for entry errors. The resulting database was locked and loaded in SPSS Version 23.0. Continuous, normally distributed variables are reported as means ± standard deviations (SD). Comparisons of continuous and categorical data between groups were analysed with ANOVA and Chi-square test, respectively. As previously reported, a dichotomous variable ‘severe periodontitis’ was given according to the criteria described above: no/mild/moderate periodontitis vs. severe periodontitis[18]. Data relative to all 3 groups (healthy-mild, moderate and severe) are also reported. Single nucleotide polymorphism (SNPs) calls from a panel of 20 available SNPs (Supplemental table S1) were extracted from the Infinium assay raw data using Illumina GenomeStudio Software (v2.0.5). The gene panel was selected based on previous associations with periodontitis or with subgingival microbes, previous systematic reviews of genes associated with periodontitis and with subgingival microbiota[60,61] complemented by GWAS and periodontal infectogenomics studies[9,62-68].
The initial analysis was hypothesis-testing, involving the associations between the selected gene panel SNPs and microbial outcomes. Due to multiple testing, a p value of <0.01 was considered statistically significant for these comparisons. Alpha diversity metrics and center log ratio transformed HMT mapped abundance data at the genus and species levels were tested against extracted loci filtered for MAF at 10%, using Generalized Linear Models as implemented in the MASS package in R (v4.3.1) and RStudio (v2022.07.1). A discovery analysis was then carried out on imputed genetic markers, using a p value of 5 x 10-7 as threshold for statistical significance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Table listing gene panel and their corresponding HG19 co-ordinates used to extract SNPs from the genotyping raw data. Figure S1: Box plots showing different alpha diversity metrics for patients that had 3,4 or 5 metabolic syndrome components. Majority of patients with 3 components had normal triglycerides and cholesterol levels.; Figure S2: Manhattan plots of gene variants against each component of the metabolic syndrome in the genome-wide analysis, after correcting for age, sex, smoking frequency, BMI and population structure. Variants at p<10-5 are annotated and highlighted in orange.; Figure S3: Stacked chart of the observed phyla distributions for all patients in this study, with the periodontitis diagnosis indicated in the x-axis.
Author Contributions
Conceptualization, L.N., A.S., R.A., F.P., N.D., L.M.; methodology, L.N., A.S., L.M.; software, A.S., R.A., N.D.; validation, L.N., A.S., F.P., N.D., L.M..; formal analysis, L.N., A.S., R.A.; investigation, L.N., A.S., R.A., A.D.P., F.P., N.D., M.R., L.M.; resources, L.N., F.P., N.D., L.M.; data curation, L.N., A.S., R.A., A.D.P., V.T., M.P., S.D.M., V.F., R.S., F.P., N.D., M.R., L.M.; writing—original draft preparation, L.N., A.S., R.A., N.D., M.R., L.M.; writing—review and editing, L.N., A.S., R.A., N.D., M.R., L.M.; visualization, M.R., L.M.; supervision, L.N., F.P., N.D., L.M.; project administration, L.N., F.P., N.D., L.M.; funding acquisition, L.N., F.P., N.D., L.M. All authors have read and agreed to the published version of the manuscript.
Funding
This study was also funded by the 2016-18 Research Plan of the University of Catania, Catania, Italy, Department of Clinical and Experimental Medicine (Project #A).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of University College London (reference 4242/01), and separately by the clinical centre (reference 1497/Cs).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
University College London (UCL) is gratefully acknowledged as the sponsor of the study. This study was also funded by the 2016-18 Research Plan of the University of Catania, Catania, Italy, Department of Clinical and Experimental Medicine (Project #A).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kellam, P.; Weiss, R. A. Infectogenomics: insights from the host genome into infectious diseases. Cell 2006 124(4), 695–697. [CrossRef]
- Nibali, L.; Henderson, B.; Sadiq, S. T.; Donos, N. Genetic dysbiosis: the role of microbial insults in chronic inflammatory diseases. Journal of oral microbiology 2014 6, 10.3402/jom.v6.22962. [CrossRef]
- Zoheir, N.; Kurushima, Y.; Lin, G. H.; Nibali, L. Periodontal infectogenomics: a systematic review update of associations between host genetic variants and subgingival microbial detection. Clinical oral investigations 2022 26(3), 2209–2221. [CrossRef]
- Hajishengallis, G.; Lamont, R. J. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Molecular oral microbiology 2012 27(6), 409–419. [CrossRef]
- Socransky, S. S.; Haffajee, A. D. Periodontal microbial ecology. Periodontology 2000 2005 38, 135–187. [CrossRef]
- Nibali, L.; Ready, D. R.; Parkar, M.; Brett, P. M.; Wilson, M.; Tonetti, M. S.; Griffiths, G. S. Gene polymorphisms and the prevalence of key periodontal pathogens. Journal of dental research 2007 86(5), 416–420. [CrossRef]
- Nibali, L.; Donos, N.; Brett, P. M.; Parkar, M.; Ellinas, T.; Llorente, M.; Griffiths, G. S. A familial analysis of aggressive periodontitis - clinical and genetic findings. Journal of periodontal research 2008 43(6), 627–634. [CrossRef]
- Nibali, L.; Pometti, D.; Tu, Y. K.; Donos, N. Clinical and radiographic outcomes following non-surgical therapy of periodontal infrabony defects: a retrospective study. Journal of clinical periodontology 2011 38(1), 50–57. [CrossRef]
- Divaris, K.; Monda, K. L.; North, K. E.; Olshan, A. F.; Reynolds, L. M.; Hsueh, W. C.; Lange, E. M.; Moss, K.; Barros, S. P.; Weyant, R. J. et al. Exploring the genetic basis of chronic periodontitis: a genome-wide association study. Human molecular genetics 2013 22(11), 2312–2324. [CrossRef]
- Ye, Y.; Carlsson, G.; Wondimu, B.; Fahlén, A.; Karlsson-Sjöberg, J.; Andersson, M.; Engstrand, L.; Yucel-Lindberg, T.; Modéer, T.; Pütsep, K. Mutations in the ELANE gene are associated with development of periodontitis in patients with severe congenital neutropenia. Journal of clinical immunology 2011 31(6), 936–945. [CrossRef]
- Zhang, S.; Yu, N.; Arce, R. M. Periodontal inflammation: Integrating genes and dysbiosis. Periodontology 2000 2020 82(1), 129–142. [CrossRef]
- Nibali, L.; Sousa, V.; Davrandi, M.; Liu, L. S.; Spratt, D.; Donos, N. Patterns of subgingival microbiota in different periodontal phenotypes. Journal of dentistry 2022 117, 103912. [CrossRef]
- Nibali, L.; Donos, N.; Terranova, V.; Di Pino, A.; Di Marca, S.; Ferrara, V.; Pisano, M.; Scicali, R.; Rabuazzo, A. M.; Purrello, F.; Malatino, L. Left ventricular geometry and periodontitis in patients with the metabolic syndrome. Clinical oral investigations 2019 23(6), 2695–2703. [CrossRef]
- Zanoli, L.; Ozturk, K.; Cappello, M.; Inserra, G.; Geraci, G.; Tuttolomondo, A.; Torres, D.; Pinto, A.; Duminuco, A.; Riguccio, G. et al. Inflammation and Aortic Pulse Wave Velocity: A Multicenter Longitudinal Study in Patients With Inflammatory Bowel Disease. J Am Heart Assoc. 2019 8(3), e010942. [CrossRef]
- Nibali, L.; Tatarakis, N.; Needleman, I.; Tu, Y. K.; D'Aiuto, F.; Rizzo, M.; Donos, N. Clinical review: Association between metabolic syndrome and periodontitis: a systematic review and meta-analysis. The Journal of clinical endocrinology and metabolism 2013 98(3), 913–920. [CrossRef]
- Lourenςo, T. G. B.; Spencer, S. J.; Alm, E. J.; Colombo, A. P. V. Defining the gut microbiota in individuals with periodontal diseases: an exploratory study. Journal of oral microbiology 2018 10(1), 1487741. [CrossRef]
- Olsen, I.; Yamazaki, K. Can oral bacteria affect the microbiome of the gut? Journal of oral microbiology, 2019 11(1), 1586422. [CrossRef]
- Page, R. C.; Eke, P. I. Case definitions for use in population-based surveillance of periodontitis. Journal of periodontology 2007 78(7 Suppl), 1387–1399. [CrossRef]
- Tu, Y.; Ling, X.; Chen, Y.; Wang, Y;, Zhou, N.; Chen, H. Effect of S. Mutans and S. Sanguinis on Growth and Adhesion of P. Gingivalis and Their Ability to Adhere to Different Dental Materials. Medical science monitor: international medical journal of experimental and clinical research 2017 23, 4539–5445. [CrossRef]
- Luppens, S. B.; Kara, D.; Bandounas, L.; Jonker, M. J.; Wittink, F. R.; Bruning, O.; Breit, T. M.; Ten Cate, J. M.; Crielaard, W. Effect of Veillonella parvula on the antimicrobial resistance and gene expression of Streptococcus mutans grown in a dual-species biofilm. Oral microbiology and immunology 2008 23(3), 183–189. [CrossRef]
- Liu, S.; Chen, M.; Wang, Y.; Zhou, X.; Peng, X.; Ren, B.; Li, M.; Cheng, L. Effect of Veillonella parvula on the physiological activity of Streptococcus mutans. Archives of oral biology 2020 109, 104578. [CrossRef]
- Van Dyke, T. E.; Bartold, P. M.; Reynolds, E. C. The Nexus Between Periodontal Inflammation and Dysbiosis. Frontiers in immunology 2020 11, 511. [CrossRef]
- Kurushima, Y.; Wells, P. M.; Bowyer, R. C. E.; Zoheir, N.; Doran, S.; Richardson, J. P.; Sprockett, D. D.; Relman, D. A.; Steves, C. J.; Nibali, L. Host Genotype Links to Salivary and Gut Microbiota by Periodontal Status. Journal of dental research 2023 102(2), 146–156. [CrossRef]
- Torrungruang, K.; Chantarangsu, S.; Sura, T.; Thienpramuk, L. Interplay between vitamin D receptor FokI polymorphism and smoking influences Porphyromonas gingivalis proportions in subgingival plaque. Journal of clinical periodontology 2020 47(8), 912–920. [CrossRef]
- Nibali, L.; Madden, I.; Franch Chillida, F.; Heitz-Mayfield, L.; Brett, P.; Donos, N. IL6 -174 genotype associated with Aggregatibacter actinomycetemcomitans in Indians. Oral diseases 2011 17(2), 232–237. [CrossRef]
- Schaefer, A. S.; Bochenek, G.; Jochens, A.; Ellinghaus, D.; Dommisch, H.; Güzeldemir-Akçakanat, E.; Graetz, C.; Harks, I.; Jockel-Schneider, Y.; Weinspach, K. et al. Genetic evidence for PLASMINOGEN as a shared genetic risk factor of coronary artery disease and periodontitis. Circulation. Cardiovascular genetics 2005 8(1), 159–167. [CrossRef]
- Aarabi, G.; Zeller, T.; Seedorf, H.; Reissmann, D. R.; Heydecke, G.; Schaefer, A. S.; Seedorf, U. Genetic Susceptibility Contributing to Periodontal and Cardiovascular Disease. Journal of dental research 2017 96(6), 610–617. [CrossRef]
- Li, N.; Li, Y.; Hu, J.; Wu, Y.; Yang, J.; Fan, H.; Li, L.; Luo, D.; Ye, Y.; Gao, Y.; Xu, H.; Hai, W.; Jiang, L. A Link Between Mitochondrial Dysfunction and the Immune Microenvironment of Salivary Glands in Primary Sjogren's Syndrome. Frontiers in immunology 2022 13, 845209. [CrossRef]
- Xie, H.; Hong, J.; Sharma, A.; Wang, B. Y. Streptococcus cristatus ArcA interferes with Porphyromonas gingivalis pathogenicity in mice. Journal of periodontal research 2012 47(5), 578–583. [CrossRef]
- Ho, MH.; Lamont, R.J.; Xie, H. Identification of Streptococcus cristatus peptides that repress expression of virulence genes in Porphyromonas gingivalis . Sci Rep 2017 7, 1413. [CrossRef]
- Karched, M.; Bhardwaj, R.G.; Asikainen, S.E. Coaggregation and biofilm growth of Granulicatella spp. with Fusobacterium nucleatum and Aggregatibacter actinomycetemcomitans BMC Microbiol 2015 15, 114. [CrossRef]
- Haffajee, A. D.; Socransky, S. S.; Smith, C.; Dibart, S. Relation of baseline microbial parameters to future periodontal attachment loss. Journal of clinical periodontology 1991 18(10), 744–750. [CrossRef]
- Haffajee, A. D.; Socransky, S. S.; Dibart, S.; Kent, R. L., Jr.Response to periodontal therapy in patients with high or low levels of P. gingivalis, P. intermedia, P. nigrescens and B. forsythus. Journal of clinical periodontology 1996 23(4), 336–345. [CrossRef]
- Socransky, S. S.; Haffajee, A. D.; Cugini, M. A.; Smith, C.; Kent, R. L., Jr. Microbial complexes in subgingival plaque. Journal of clinical periodontology 1998 25(2), 134–144. [CrossRef]
- Oliveira, R. R.; Fermiano, D.; Feres, M.; Figueiredo, L. C.; Teles, F. R.; Soares, G. M.; Faveri, M. Levels of Candidate Periodontal Pathogens in Subgingival Biofilm. Journal of dental research 2016 95(6), 711–718. [CrossRef]
- Ghayoumi, N.; Chen, C.; Slots, J. Dialister pneumosintes, a new putative periodontal pathogen. Journal of periodontal research 2002 37(1), 75–78. [CrossRef]
- Aruni, A. W.; Mishra, A.; Dou, Y.; Chioma, O.; Hamilton, B. N.; Fletcher, H. M.Filifactor alocis--a new emerging periodontal pathogen. Microbes and infection 2015 17(7), 517–530. [CrossRef]
- Ozuna, H.; Snider, I.; Belibasakis, G. N.; Oscarsson, J.; Johansson, A.; Uriarte, S. M. Aggregatibacter actinomycetemcomitans and Filifactor alocis: Two exotoxin-producing oral pathogens. Frontiers in oral health 2022 3, 981343. [CrossRef]
- Razooqi, Z.; Höglund Åberg, C.; Kwamin, F.; Claesson, R.; Haubek, D.; Oscarsson, J.; Johansson, A. Aggregatibacter actinomycetemcomitans and Filifactor alocis as Associated with Periodontal Attachment Loss in a Cohort of Ghanaian Adolescents. Microorganisms 2022 10(12), 2511. [CrossRef]
- Peña-Chilet, M.; Esteban-Medina, M.; Falco, M. M.; Rian, K.; Hidalgo, M. R.; Loucera, C.; Dopazo, J. Using mechanistic models for the clinical interpretation of complex genomic variation. Scientific reports 2019 9(1), 18937. [CrossRef]
- Ke, J.; Gao, W.; Wang, B.; Cao, W.; Lv, J.; Yu, C.; Huang, T.; Sun, D.; Liao, C.; Pang, Y. et al. Exploring the Genetic Association between Obesity and Serum Lipid Levels Using Bivariate Methods. Twin research and human genetics: the official journal of the International Society for Twin Studies, 2022 25(6), 234–244. [CrossRef]
- de Vries, P. S.; van Herpt, T. T.; Ligthart, S.; Hofman, A.; Ikram, M. A.; van Hoek, M.; Sijbrands, E. J.; Franco, O. H.; de Maat, M. P.; Leebeek, F. W. et al. ADAMTS13 activity as a novel risk factor for incident type 2 diabetes mellitus: a population-based cohort study. Diabetologia 2017 60(2), 280–286. [CrossRef]
- Zillikens, M. C.; Demissie, S.; Hsu, Y. H.; Yerges-Armstrong, L. M.; Chou, W. C.; Stolk, L.; Livshits, G.; Broer, L.; Johnson, T.; Koller, D. L., Kutalik, Z., Luan, J., Malkin, I., Ried, J. S., Smith, A. V., Thorleifsson, G., Vandenput, L., Hua Zhao, J., Zhang, W., Aghdassi, A. et al. Large meta-analysis of genome-wide association studies identifies five loci for lean body mass. Nature communications 2017 8(1), 80. [CrossRef]
- Zhu, Z.; Guo, Y.; Shi, H.; Liu, C. L.; Panganiban, R. A.; Chung, W.; O'Connor, L. J.; Himes, B. E.; Gazal, S.; Hasegawa, K. et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK Biobank. The Journal of allergy and clinical immunology 2020 145(2), 537–549. [CrossRef]
- Baca, P.; Barajas-Olmos, F.; Mirzaeicheshmeh, E. et al. DNA methylation and gene expression analysis in adipose tissue to identify new loci associated with T2D development in obesity. Nutr. Diabetes 12, 50 (2022). [CrossRef]
- Nibali, L.; Stephen, A.; Hagi-Pavli, E.; Allaker, R.; Pino, A. D.; Terranova, V.; Pisano, M.; Marca, S. D.; Ferrara, V.; Scicali, R.; et al. Analysis of gingival crevicular fluid biomarkers in patients with metabolic syndrome. Journal of dentistry 2022 118, 104065. [CrossRef]
- Grundy, S. M.; Brewer, H. B.; Jr, Cleeman, J. I.; Smith, S. C., Jr; Lenfant, C.; American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004 109(3), 433–438. [CrossRef]
- Guerrero, A.; Griffiths, G. S.; Nibali, L.; Suvan, J.; Moles, D. R.; Laurell, L.; Tonetti, M. S. Adjunctive benefits of systemic amoxicillin and metronidazole in non-surgical treatment of generalized aggressive periodontitis: a randomized placebo-controlled clinical trial. Journal of clinical periodontology 2005 32(10), 1096–1107. [CrossRef]
- Chang, C. C.; Chow, C. C.; Tellier, L. C.; Vattikuti, S.; Purcell, S. M.; Lee, J. J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 2015 4, 7. [CrossRef]
- Loh, P. R.; Danecek, P.; Palamara, P. F.; Fuchsberger, C.; A Reshef, Y.; K Finucane, H.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G. R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nature genetics 2016 48(11), 1443–1448. [CrossRef]
- Durbin R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics (Oxford, England) 2014 30(9), 1266–1272. [CrossRef]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A. R.; Teumer, A.; Kang, H. M.; Fuchsberger, C.; Danecek, P.; Sharp, K. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nature genetics 2016 48(10), 1279–1283. [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X. et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genomics, proteomics & bioinformatics 2021 19(4), 619–628. [CrossRef]
- Oscanoa, J.; Sivapalan, L.; Gadaleta, E.; Dayem Ullah, A. Z.; Lemoine, N. R.; Chelala, C. SNPnexus: a web server for functional annotation of human genome sequence variation (2020 update). Nucleic acids research 2020 48(W1), W185–W192. [CrossRef]
- Eren, A. M.; Vineis, J. H.; Morrison, H. G.; Sogin, M. L. A filtering method to generate high quality short reads using illumina paired-end technology. PloS one 2013 8(6), e66643. [CrossRef]
- Schloss, P. D.; Westcott, S. L.; Ryabin, T.; Hall, J. R.; Hartmann, M.; Hollister, E. B.; Lesniewski, R. A.; Oakley, B. B.; Parks, D. H.; Robinson, C. J. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 2009 75(23), 7537–7541. [CrossRef]
- Eren, A. M.; Morrison, H. G.; Lescault, P. J.; Reveillaud, J.; Vineis, J. H.; Sogin, M. L. Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. The ISME journal 2015 9(4), 968–979. [CrossRef]
- Chen, T.; Yu, W. H.; Izard, J.; Baranova, O. V.; Lakshmanan, A.; Dewhirst, F. E. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database: the journal of biological databases and curation 2010, baq013. [CrossRef]
- Hammer, Ø.; Harper, D. A. Past: paleontological statistics software package for educaton and data anlysis. Palaeontologia electronica 2001 4(1), 1.
- Nibali, L.; Zavattini, A.; Nagata, K.; Di Iorio, A.; Lin, G. H.; Needleman, I.; Donos, N. Tooth loss in molars with and without furcation involvement - a systematic review and meta-analysis. Journal of clinical periodontology 2016 43(2), 156–166. [CrossRef]
- Nibali, L.; Bayliss-Chapman, J.; Almofareh, S. A.; Zhou, Y.; Divaris, K.; Vieira, A. R. What Is the Heritability of Periodontitis? A Systematic Review. Journal of dental research 2019 98(6), 632–641. [CrossRef]
- Cavalla, F.; Biguetti, C. C.; Melchiades, J. L.; Tabanez, A. P.; Azevedo, M. C. S.; Trombone, A. P. F.; Faveri, M.; Feres, M.; Garlet, G. P. Genetic Association with Subgingival Bacterial Colonization in Chronic Periodontitis. Genes 2018 9(6), 271. [CrossRef]
- Rhodin, K.; Divaris, K.; North, K. E.; Barros, S. P.; Moss, K.; Beck, J. D.; Offenbacher, S. Chronic periodontitis genome-wide association studies: gene-centric and gene set enrichment analyses. Journal of dental research 2014 93(9), 882–890. [CrossRef]
- Shusterman, A.; Durrant, C.; Mott, R.; Polak, D.; Schaefer, A.; Weiss, E. I.; Iraqi, F. A.; Houri-Haddad, Y. Host susceptibility to periodontitis: mapping murine genomic regions. Journal of dental research 2013 92(5), 438–443. [CrossRef]
- Shusterman, A.; Munz, M.; Richter, G.; Jepsen, S.; Lieb, W.; Krone, B.; Hoffman, P.; Laudes, M.; Wellmann, J.; Berger, K. et al. The PF4/PPBP/CXCL5 Gene Cluster Is Associated with Periodontitis. Journal of dental research 2017 96(8), 945–952. [CrossRef]
- Munz, M.; Richter, G. M.; Loos, B. G.; Jepsen, S.; Divaris, K.; Offenbacher, S.; Teumer, A.; Holtfreter, B.; Kocher, T.; Bruckmann, C. et al. Meta-analysis of genome-wide association studies of aggressive and chronic periodontitis identifies two novel risk loci. European journal of human genetics: EJHG 2019 27(1), 102–113. [CrossRef]
- Munz, M.; Willenborg, C.; Richter, G. M.; Jockel-Schneider, Y.; Graetz, C.; Staufenbiel, I.; Wellmann, J.; Berger, K.; Krone, B.; Hoffmann, P. et al. A genome-wide association study identifies nucleotide variants at SIGLEC5 and DEFA1A3 as risk loci for periodontitis. Human molecular genetics 2017 26(13), 2577–2588. [CrossRef]
- Divaris, K.; Monda, K. L.; North, K. E.; Olshan, A. F.; Lange, E. M.; Moss, K.; Barros, S. P.; Beck, J. D.; Offenbacher, S. Genome-wide association study of periodontal pathogen colonization. Journal of dental research 2012 91(7 Suppl), 21S–28S. [CrossRef]
Figure 1.
A) Box and jitter plots of alpha diversity indices of the subgingival microbiota of mild-moderate and severe periodontitis MetS patients (no significant differences detected) B) Box and jitter plot of alpha diversity indices that showed associations with SNPs within RUNX2, CAMTA and the VDR genes. C) A Manhattan plot to compare associations between the subgingival microbiota alpha diversity measures and periodontitis severity after correcting for age, sex, smoking frequency, BMI and population structure. Suggestive associations highlighted in green.
Figure 1.
A) Box and jitter plots of alpha diversity indices of the subgingival microbiota of mild-moderate and severe periodontitis MetS patients (no significant differences detected) B) Box and jitter plot of alpha diversity indices that showed associations with SNPs within RUNX2, CAMTA and the VDR genes. C) A Manhattan plot to compare associations between the subgingival microbiota alpha diversity measures and periodontitis severity after correcting for age, sex, smoking frequency, BMI and population structure. Suggestive associations highlighted in green.
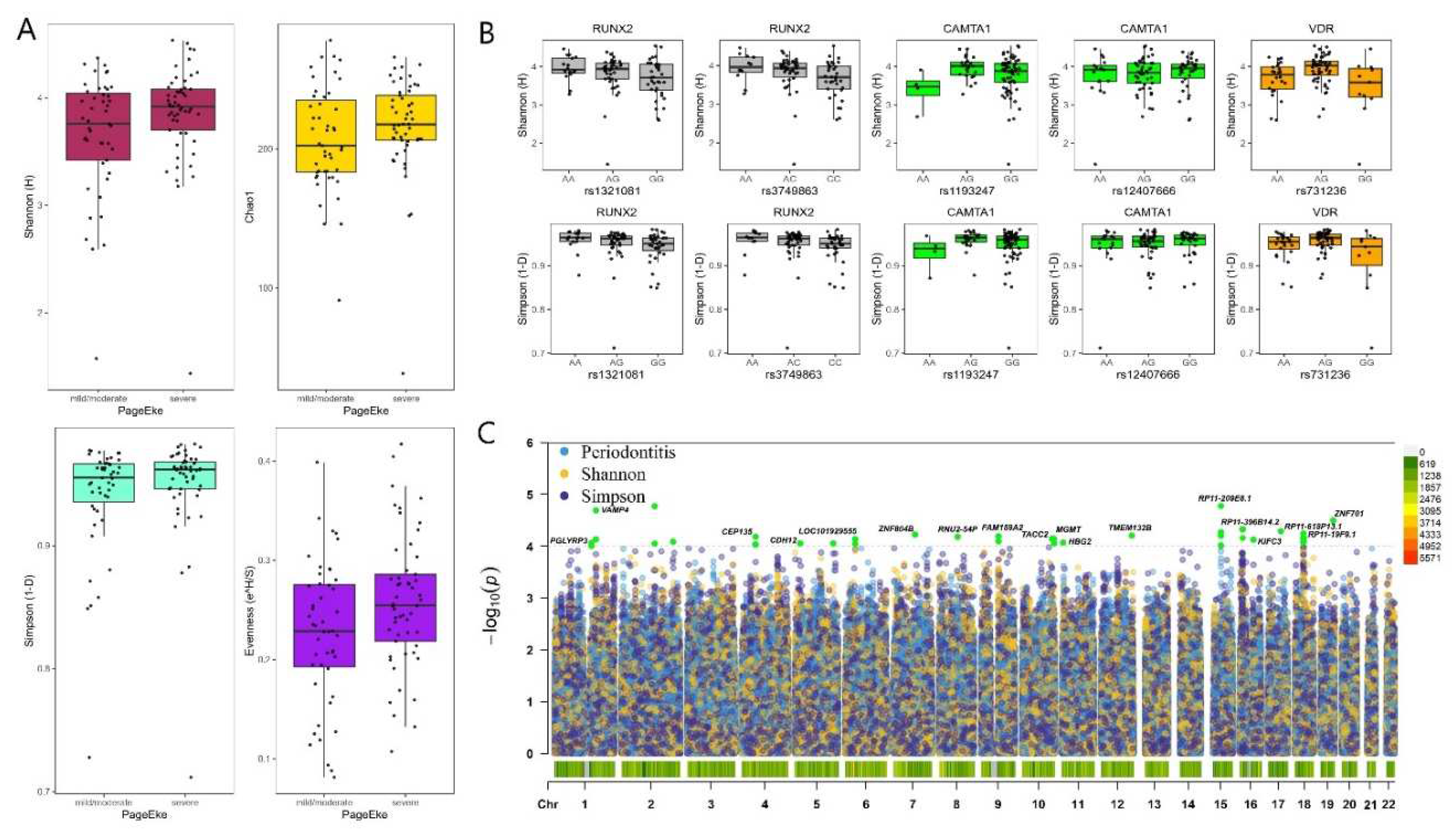

Table 1.
Demographics and dental history of included cases. BMI= body mass index.
| Average | ||
| Age | 58.12 ± 9.89 | |
| BMI | 31.88 ± 4.37 | |
| Frequency | ||
| Gender | Male | 65 (63.1%) |
| Female | 48 (36.9%) | |
| Smoking status | Non smoker | 67 (65.0%) |
| Current smoker | 28 (27.2%) | |
| Former smoker | 8 (7.8%) | |
| Frequency of dental visits | Never been | 2 (1.9%) |
| Only in case of problems | 79 (76.7%) | |
| 1/year | 12 (11.7%) | |
| >1/year | 10 (9.7%) | |
| Previous periodontal treatment | Yes | 3 (2.9%) |
| No | 100 (97.1%) | |
| Last dental visit | Never been | 2 (1.9%) |
| >10 years ago | 7 (6.8%) | |
| 1-10 years ago | 60 (58.3%) | |
| <1 year ago | 34 (33.0%) | |
| Tooth brushing frequency | <1/day | 6 (5.9%) |
| 1/day | 34 (33.0%) | |
| At least 2/day | 63 (61.1%) | |
| Type of toothbrush | None | 1 (1.0%) |
| Manual | 92 (89.3%) | |
| Electric | 10 (9.7%) | |
|
Use of interdental cleaning tools |
Daily-weekly | 14 (13.6%) |
| Never | 89 (86.4%) | |
Table 2.
Dental clinical parameters in all included subjects. DMFT= Decayed Missing Filled Teeth; FMPS= Full Mouth Plaque Score; FMBS= Full Mouth Bleeding Score; PPD= Probing Pocket Depth; CAL= Clinical Attachment Level.
Table 2.
Dental clinical parameters in all included subjects. DMFT= Decayed Missing Filled Teeth; FMPS= Full Mouth Plaque Score; FMBS= Full Mouth Bleeding Score; PPD= Probing Pocket Depth; CAL= Clinical Attachment Level.
| Average | Number | |
| Number of teeth (excluding third molars) | 22.77 ± 4.19 | 2345 |
| DMFT | 12.58 ± 6.07 | - |
| Decayed teeth | 0.63 ± 0.96 | 65 |
| Patients with caries detected | - | 38 (36.89%) |
| FMPS | 72.11 ± 22.51 | - |
| FMBS | 23.92 ± 19.62 | - |
| Average PPD | 2.44 ± 0.73 | - |
| Average CAL | 3.05 ± 1.12 | - |
| % PPDs 1-4mm | 93.17 ± 0.88% | - |
| % PPDs 5-6mm | 5.92 ± 0.75% | - |
| % of PPDs >6mm | 0.94 ± 0.23% | - |
| No-Mild periodontitis | - | 10 (9.7%) |
| Moderate periodontitis | - | 38 (36.9%) |
| Severe periodontitis | - | 55 (53.3%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



