Submitted:
30 September 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
Formulating a novel concept about the origin of human aging has been constrained by the dominance of a “classic theory” that was proposed nearly 70 years ago. Despite concern over the validity of some of its assumptions, the theory remained basic to our understanding of aging’s relationship with natural selection (NS). However, the logic upon which it rests was tested and subsequently challenged. The present theory describes the single cause of human aging consistent with Darwin’s evolutionary requirement for selection of adaptive traits. It describes an emergent property of the developmental program (DP), that is expressed upon completion of ontogenesis. It involves redundant expression of regulatory processes from the last stage of the DP. That mechanism subsequently preserves a non-aging, stable interval of unchanging NS during which reproductive fitness is achieved. Thereafter, loss of DP regulatory redundancy due to reliability limits, stochastic mutation accumulation, reproductive and a specific type of DNA damage, initiates aging which causes an inexorable decline in strength of NS to begin. It starts approximately a decade later than proposed in the classic theory. Since reproduction and aging are inextricably linked by the same emergent property, selection of that regulatory mechanism makes both traits products of NS.
Keywords:
aging vs survival
; DNA damage
; double-stranded breaks
; Gompertz-Makeham Law
; developmental regulatory redundancy
; tradeoffs vs emergent properties
; fecundity
; reproductive fitness
; redundancy reliabiiity
; programmed aging
Clarification and Correction
“Recursive”, meaning that each step taken in original research will feed into other steps, such that after completing an initial version, review and verification of its conclusions will sometimes reveal that they are inaccurate. Such is the case of my original theory [1] the publication of which preceded that of Giaimo and Traulsen, [2]. In my opinion, their report undermined the logic of the classic theory of aging upon which some significant assumptions of my first version were erroneously based. Upon reading the convincing data that aging precedes and causes decline of NS, not vice versa, this recursive version of my theory has been duly corrected.
The title of this essay has been copied and modified from Darwin’s famous text which omits aging’s origin. Lacking explanation of how aging could comply with laws of Natural Selection, the omission has been referenced as “Darwin’s Dilemma”. With all due respect for Darwin’s brilliance, I modestly offer this work with hopes that my theory will fill at least part of that void.
Darwin’s dilemma – Apparent conflict between natural selection (NS) and aging
According to Gould [3], Darwin recognized evolution as a fact involving change over time, and descent with modification from common ancestors. He also designated natural selection (NS) as “my theory” intended to explain how specific events of evolution occurred. Unlike evolution which is well-substantiated and supported by sound evidence, NS is supported only by observational evidence and the assumptions that “selection” requires individual benefit, heritable variation, differential survival, and reproduction.
Soon after its publication, the relevance and validity of Darwin’s theory [4] to the evolution and pervasive presence of aging was questioned [5]. Restriction of reproductive potential by aging was considered prima facie evidence that his description of NS mechanics was inadequate. However, Darwin remained confident, commenting that “My theory says there must be some hidden compensating (individual, theory conforming) benefit of aging so therefore there must be one” [6]. Goldsmith opined that Darwin’s theory, “was perhaps 99 percent correct” since the involvement of NS in the evolution of aging was seemingly indefensible except through unshakable confidence rather than with supporting evidence. Some biogerontologists feel that to explain the origin of aging would require controversial methods such as “group selection on a scale that goes beyond the theory of multilevel selection”[7,8]. Others agree that aging is programmed [9,10].
In defense of Darwin’s neglect of aging within the scope of NS theory, his intellectual focus was on diversity, adaptation, variation, competition, overproduction, and speciation, which are relevant to life and survival. His theory makes no mention of aging’s evolution nor its selection. This omission is not surprising since it is doubtful that Darwin gave much thought to a functional relationship between NS and intrinsic mortality, since his focus was on design, not deconstruction. However, he extolled death as an engine of evolutionary progress proclaiming that “from the war of nature, famine and death, the most exalted object which we are capable of conceiving, namely, the production of higher animals directly follows”. Thus, viewing death as beneficial to the progress of evolution may have strengthened Darwin’s confidence that aging, while seemingly contradictory to his theory, probably provides a hidden benefit that explains it’s omnipresence.
Also, molecular and cellular characteristics of traits associated with evolution were not known during his lifetime, making relevant issues of NS either vague or incomplete in his original composition. Thus, senescence received relatively little attention until the middle 20th century when Comfort [11] published reviews of the subject. Subsequently, additional, core mechanics were elucidated and documented, causing the modern theory to be far more complicated and detailed, but better understood than when it was conceived [12].
Finally, criticism of Darwin’s theory was unjustified because evolutionary developmental biology (evo-devo) was yet to be comprehensively described. A primary obstacle to explaining how individual benefit is compatible with the onset and progression of aging is the assumption that it’s evolution was independent of development. As a result, aging is routinely studied in isolation because its cause is assumed to be hidden within its phenotype. Alternatively, if Darwin’s “hunch” was correct, then the widespread occurrence of aging in eukaryotes may have resulted from its selection as an adaptive emergent property of the DP that initially facilitated reproductive fitness despite later causing aging. Thus, an emergent property of ontogenesis that makes fertility and mortality inseparable as a direct target of NS could explain the evolution of human aging in compliance with Darwinian principles of NS [13].
As a sidenote, it has been proposed that the traditionally held view of Darwin’s individual benefit requirement does not apply to aging because other aspects of evolution provide tenable explanations for its existence. These could include “non-selection”, random walk or neutral evolution which in theory hold that most molecular variation does not affect fitness and, therefore, its evolutionary fate is best explained by stochastic processes [14]. However, these mechanisms cannot create adaptations, making them inappropriate for inclusion in this theory.
Conditions and Restrictions
The preceding reference [13] is to an emergent property of ontogenesis that links development and aging. It is unique because it represents a single adaptive mechanism that was selected to facilitate successful reproduction and to cause aging. Due to the diversity of life forms on earth, the concept of aging’s evolution to be described herein is not intended for application in species besides human beings and other mammals that nurture their young. Different processes ensure death in other phylogenetic groups based upon the peculiarities of their unique evolutions [15]. Thus, aging’s presence in one form or another throughout Eucarya has few exceptions, but it undoubtedly occurs by differing terminal mechanisms depending upon each species’ unique lifespan evolutions [16].
Pioneers in evolutionary theories of aging
In 1930, the evolution of aging was introduced as a research topic by Ronald Fisher [17]. Subsequently, Brian Charlesworth [18] acknowledged “innovative thinkers” including Peter Medwar [19], George Williams [20], William Hamilton [21] and Thomas Kirkwood [22,23] who made significant contributions to the field by developing cogent theories of aging’s origin.
The central theories of aging that are currently accepted include Mutation Accumulation (MA) [19] and Antagonistic Pleiotropy (AP) [20], which are attributed to Medawar and Williams, respectively. The theories were later complemented by Disposable Soma Theory (DST) [23]. A synthesis of these theories that provides a contemporary framework for the evolution of aging is based upon acceptance of logic that the force of NS inexorably weakens as fecundity declines over time. This concept was mathematically formalized and further developed by Hamilton [21] Charlesworth [24] and Rose et al, [25] bringing greater credibility to the now “classic theory” of aging.
A thorough review that presents support of and challenges to these authors’ theoretical concepts of aging’s evolution was published by Johnson et al [26].
The following brief commentary contains selected topics that are intended to serve as background and introduction to a novel concept that conflicts with basic assumptions of the “classic theory”.
Criticism of the aging’s cause as proposed in the “classic theory”
The classic theory is currently favored by prominent biogerontologists despite its containing specific assumptions that are debatable. Those established assumptions impose significant constraints upon the development of alternative explanations that might expand our understanding of aging’s evolution [27].
Some questionable assumptions of the classic theory are that:
- An inherent quality of NS is “strength” or “force” that weakens with age because less reproductive potential remains. Eventually somatic neglect due to lack of oversight by NS allows mutation and pleiotropic gene accumulation which causes aging.
- Aging is multifactorial; no single cause/mechanism of its evolution exists.
- Senescence begins at the onset of peak reproductive probability (fecundity) during adolescence.
- An inexorable decline of NS strength begins upon completion of ontogenesis.
- Aging is the product of a tradeoff of limited energy resources favoring reproduction over survival.
The following few sections discuss these assumptions upon which the classic theory is based.
Declining fecundity weakens strength of NS allowing aging to occur by accumulated stochastic damage
“Evolution embraces chance and necessity, randomness and determinism, which are enmeshed by the creative if not a conscious process of NS” [28]
This quote from Darwin’s work, contradicts the classic theory’s central concept that aging results from neglect by NS. In effect the classic theory holds that unlike all other evolved traits, aging is different, emerging randomly without involvement of NS’s creative process in violation of Darwin’s concept of “design”. This is not to deny that random mutations occur throughout life and are the source of variation that can be selected and differentially inherited among generations. However, nothing can evolve without the influence of NS. Although Darwin never included aging in his Theory, he opined that because it exists as a universal trait of biological organisms, it must possess some hidden compensating benefit to have evolved. He was criticized for taking this position and for adamantly believing in the validity of his theory [6].
The reason that a central concept in Darwin’s theory was rejected is the nearly universal opinion that aging lacks individual benefit and thus, could not be selected. While seemingly logical, the assumption is based entirely upon the premise that aging per se evolved independent of other life history events, and that it is multifactorial. Also, a tenet of the classic theory is that the strength of NS wanes due to age-related declining fecundity. As a result, progressive weakening of NS over time contributes to random accumulation of mutations that are thought to represent the aging phenotype.
It is agreed that incorporation of mutations anytime in life without NS, would yield disorganization and chaos because most mutations are disadvantageous. However, unlike the aging phenotype which is “programmatic”, random accumulation of mutations without some sort of underlying informational compass would produce chaos. Thus, the existence of an aging phenotype, which is programmatic not chaotic, does not support the premise of the “classic theory” that it occurs due to random accumulation of mutations in the absence of NS.
Thus, while there is consensus among biogerontologists that NS neglect of aging plays a passive role in its origin, they generally agree that it has no selective influence in the process. However, evidence will be provided to support the possibility that one or more aspects of aging’s origins are selected due to related “hidden and theory conforming“ characteristics as envisaged by Darwin [6].
To scientifically account for the design of organisms due to natural processes, a subsidiary objective of Darwin’s work may have been the accumulation of evidence for common descent with diversification [28,29]. As part of the current theory, one consequence of such design in humans and other mammals that nurture their young is that the aging process is inseparable from NS as it affects final stages of development and acquisition of reproductive fitness.
Declining strength of natural selectionis caused by aging, not vice versa
Giaimo and Truelsen [2] recognized that some issues in the “classic theory” depend upon specific assumptions that are not necessarily satisfied. Perhaps the most important of these is the presumed inexorable decline of NS beginning with the onset of sexual maturation and persisting throughout the reproductive lifespan.
The authors proposed that since the strength of selection on fecundity and survival at a given age depends on the whole life history [30] i.e., on their complete age trajectories, then the age-specific selection force should typically change as the life history evolves. They proposed that selection forces may at times be stronger later in life than earlier. Walker [1] suggested that the post-ontogenetic, horizontal segment that is evident in the Gompertz-Makeham plot is a decade of human life reflecting that dynamic when compared with the preceding years of adolescence (Figure 1).
In contrast, the classic theory, identifies a declining pattern of NS force beginning during adolescence that is invariant to evolutionary change.
Abbreviations: NS -, NS+, NS 0/+ represents force of natural selection as decreasing, increasing or non-changing/slightly increasing, respectively
This figure was modified from that in Volume 54, Number 14 United States Life Tables, 2003 by Elizabeth Arias, Ph.D., Division of Vital Statistics is in the public domain and is allowed to be reproduced and modified with permission.
Giaimo and Traulsen [2] proposed that any interval of unchanging NS strength will be unstable and transient, and is eventually abandoned, which is consistent with the present theory. Thus, they concluded that a constant declining force of NS with age is not its intrinsic property nor a major contributor to the mechanism of aging as proposed in and implicitly assumed by the classic theory [21,31].
To the contrary, they found that aging causes NS to decline, not vice versa [Figure 2]
That finding when incorporated into the current hypothesis, undermines the validity of the classic theory. It provides the conditions whereby a single specific mechanism that was selected to complete development could also cause aging. Those conditions are that the inexorable decline in NS that purportedly begins in adolescence upon completion of ontogenesis, actually starts a decade later after reproductive fitness is achieved.
If then, the classic theory’s central premise that aging results from declining strength of NS is incorrect, and aging weakens the strength of selection, then we are back to the original question; “what causes aging?”
Aging is thought to be multifactorial
There is consensus among contemporary biogerontologists that aging is multifactorial [32,33]. This belief originated with Medawar and Williams, much of whose work constitutes the generally accepted, “classic theory” of aging.
Initially, Medawar [19] felt that the essential task of gerontologists was to identify and isolate the cause(s) of senescence. However, Williams [20] disagreed, opining that “if the assumptions made in the present study, re: Pleiotropy, Natural Selection, and the Evolution of Senescence, are valid” then finding a small number of causal factors among the multitude of its maladaptive effects is a “logical impossibility”. This opinion was derived from the aging phenotype, which displays “impressive diversity” that represents many maladaptive characteristics, some or all of which were thought to cause senescence. No single mechanism or pathway causal of age-associated, structural and functional decline was recognized. Thus, aging was presumed to be multifactorial based upon known negative effects of intrinsic as well as extrinsic/environmental damages on many levels of somatic organization [34]
Williams’ opinion dominated, and many contemporary biogerontologists agree that the specific mechanism of aging will never be discovered because “aging results from a myriad of causes” that change molecular structure and function, “such that counteracting one or several of them would make little difference” [35].
In support of the concept, Medawar created a thought experiment using test tubes from which he concluded that aging results from randomly occurring damage and could not have evolved. He concluded that constant, random breakage of glass tubes in his model sufficiently simulated lethal forces of nature to explain aging as resulting from a weakening strength of NS. However, some objections were raised about the validity of Medawar’s “test tube experiment” and to antagonistic pleiotropy theory that seemingly question the credibility of the submitted evidence [36].
Aging is not multifactorial
Current research efforts to identify the cause of aging focus upon its phenotype. The investigative approach may seem reasonable, but it is not. It promotes the erroneous assumption that the key to understanding aging’s cause exists within its process. I propose that assuming the cause of mammalian aging resides in its impressively diverse phenotype is folly. The aging phenotype represents erosion of form and function that has already begun, and the dynamic process of somatic deconstruction is proceeding. Generally, when seeking to identify a cause, one wouldn’t expect to find it within an effect. It is more logical that an action will precede an outcome. Thus is the case for the cause of human aging.
A good analogy is expecting to find the cause of ontogenesis within the phenotype of a developing organism. Like the aging phenotype, that of ontogenesis displays “impressive diversity” but does not contain its cause. Some might debate that comparing development and aging does not represent similar examples of cause and effect, but certainly there is no disagreement that fusion of gametes is the single origin/cause of the ontogenetic phenotype’s impressive diversity. Thus, fertilization is the initiating cause of development after which the organism will display a changing, staged, multifactorial phenotype until adulthood is reached. This comparison of phenotypes suggests that there is a single, mechanistic cause for aging that derives from a preceding, non-aging stage of life. The only difference between development and aging is that the former is the product of an evolved program. The latter is programmatic, resembling a program but not expressed according to a predictable plan or schedule. Aging results from damage to programmed regulatory maintenance of the soma beyond ontogenesis. Regulatory oversight continues to be expressed beyond ontogeny to sustain integrity of the final developmental stage until reproductive fitness is achieved following nurture and independence of progeny. While it is generally agreed that aging per se is devoid of individual benefit, when it is inextricably linked with the highest evolutionary priority of reproductive fitness, it is possible that a mechanism involving both traits was selected pursuant to constraints of Darwin’s theory. That adaptive evolutionary process will be described below in the section on the role of emergent properties in continuity of development and aging.
Programmatic appearance of the aging phenotype
A minor criticism of the classic theory is that while recognizing the programmatic nature of aging, it does not explain the origin(s) of that characteristic. Instead, it suggests that declining strength of NS is relevant to aging only through progressive neglect that allows mutation and pleiotropic gene accumulation to cause degenerative changes in the soma.
It is agreed that without NS, mutation would yield disorganization and decay because most mutations are disadvantageous. If so, how is the programmatic nature of aging explained? One explanation could be linkage to a degenerating, preexisting program, specifically that of development from which aging derives [28,29]. Without some sort of informational compass, such as the last stage of the preceding DP, the aging phenotype would not be programmatic. It would be chaotic.
In contrast, random damage to preexisting developmental regulatory mechanisms distributed throughout the soma would create similarities of aging phenotypes among individuals. If damage is random, affecting different parts of the soma but especially those of a putative, preexisting regulatory program, then the resulting phenotypes would bear some resemblance of pattern. Furthermore, epigenetic influences at their differing loci that suffer damage would allow the aging phenotype to be programmatic but variable even among closely related individuals.
Senescence does not begin upon sexual maturation
The classic evolutionary theory proposes that aging occurs because continued survival of individual organisms contributes less and less to “reproduction” as lifetime “fertility” declines. Consequently, natural selection is less effective at reducing mortality at older ages. This theory has been extended and qualified and is still the dominant paradigm for the evolution of aging [18,19]. In context of classic theory, fitness and fertility are often used interchangeably with fecundity, making their meanings when referring to reproductive events somewhat ambiguous.
Peak reproductive probability/potential or fecundity during adolescence was defined by Williams [20] as the onset of declining NS and initiation of senescence. He overlooked fitness, which involves relative success of different genotypes to transmit their genes to the next generation through an optimal balance of survival, fecundity and mating. These interactions vary importantly depending on the selective context [37]. Thus, the distinction between fitness and fecundity as they relate to reproduction is sometimes not recognized within interpretations of the classic theory, but fecundity seems to be specifically implied [38].
This is not a trivial point since the precise meaning of reproduction is significant when describing the time of senescence’s onset, the primary cause of aging and the onset/duration of NS decline. Classic theory proposes that inexorable decline in “strength” of NS occurs throughout the reproductive lifespan, beginning at puberty and continuing until fecundity is lost [39,40]. Choosing reproductive lifetime as the interval of declining NS seems reasonable because of its significance in evolution. Such decline would link the initial stages of reproductive potential or fecundity with the beginning of senescence and decline of natural selection. However, while seemingly reasonable, it is incorrect.
At issue with Williams’ assumption that NS declines and senescence begins after peak reproductive probability during the developmental program, is that “fecundity” does not have the evolutionary significance of reproductive fitness, which occurs in humans during the post-ontogenetic decade when nurture unto independence of progeny occurs. The result of reproductive fitness is generational variation, which is at the heart of evolution. An organism must survive until all reproductive stages are realized since survival without descent is evolutionarily meaningless. Thus, it is doubtful that inexorably declining NS occurs throughout the reproductive lifespan since that assumption embraces the evolutionary improbability of declining NS and increasing senescence before fitness and reproductive success are achieved (Figure 3).
The Medawar/Williams graph of declining NS shows it beginning upon reproductive maturation, thereby avoiding the horizontal phase of the Gompertz-Makham plot (Figure 3). It is possible that this deviation was used to conform with their theory that the inexorable decline in NS and the start of aging begin with the onset of fecundity and continue throughout the reproductive lifespan [41,42].
It is this author’s opinion that the basis for assuming that the onset of senescence and persistent decline in NS begin at peak reproductive probability was to provide support of and compliance with a major constraint of the classic theory that NS declines throughout the reproductive lifespan beginning at puberty. If instead, the more evolutionarily reasonable time of reproductive fitness/success were used to define aging’s onset and decline of NS, then the theory directly linking declining fecundity and waning strength of selection over the entire course of the reproductive lifespan as a passive cause of aging would be invalidated. The authors’ required conformity within their theory that declining fecundity initiates and exacerbates selection’s decline.
In fact, that element of the classic theory is incorrect since declining reproductive potential is not the cause of weakening strength of NS nor of aging. Based upon recent findings, it is aging that reduces strength of NS [2] not declining fecundity. Thus, the events and timing critical to validity of the classic theory conflict with the actual dynamics of reproductive events during development and aging as they relate to the declining strength of NS.
Thus, two questionable assumptions of the classic theory are that senescence begins at reproductive maturation during adolescence, and then initiates an inexorable decline in NS. This would produce an exponential reduction in human fecundity; the biologic capacity of men and women to reproduce irrespective of pregnancy or production of offspring, contrary to that which occurs after progeny are nurtured and become independent [43].
Inclusion of disposable soma in the classic theory
In the 1970s, the “Disposable Soma” theory (DST) of aging [44] was added to those of Medawar and Williams.
Kirkwood proposed that because resources are limited [33], most organisms will fare best if they restrict the energetic demands of somatic maintenance to favor those for reproduction. Thus, DST postulates that a trade-off between survival and reproduction allows mutations and other maladaptive cellular manifestations to accumulate over time due to declining strength of NS and insufficient energy for adequate repair [44,45]. However, as will be subsequently discussed, survival and aging are not the same. While limited energy resources can change the rate of aging to affect lifespan and even cause death, starvation does not initiate morpholysis nor drive its familiar Gompertzian pattern of progression.
History of linkage between development and aging
In 1825, Benjamin Gompertz reported that human death rates rise exponentially between 20 and 60 years of age [41]. The data that were based upon death and population records became known as the “law of mortality”.
A decade later Adolphe Quetelet, published the first research report on human development and aging [46]. He proposed that the life-course is a sequential two-phase model wherein growth and decline are continuous.
Subsequently, William Makeham expanded the original age range of mortality in the Gompertz law from 10 - 80 years [42,47]. An important aspect of Makeham’s contribution is that it included data from birth until 10 years of age; a range during which mortality decreases. This dynamic is relevant to the current theory, because it shows that aging need not always increase, but under certain conditions it can cease or stabilize. Since aging is associated with decreasing NS, the fact that it can intermittently and temporarily cease to change as during the horizontal, terminal segment of the Makeham parameter, is relevant to the mechanism of aging proposed in the current theory (Figure 1).
Quetelet’s growth and decline curve stressed “developmental” aspects of individual life that was the inverse of Gompertz’, which emphasized “aging” characteristics [48,49]. Both scientists professed that the course of human life consists of two sequential processes of change that include development and aging. The maximum developmental age of Quetelet’s report and the minimum for aging per Gompertz’s are the same. This commonality of apparent termination and origination of development and aging, respectively, raised the question of how is the transition from one process to the other explained [50]?
This question is not answered by the classic theory of aging. First of all, it doesn’t conform with Quetelet’s and Gompertz’ opinions that the maximum of development and minimum of aging occurs at 30 years old. The classic theory professes that after peak reproductive potential is achieved during puberty, fecundity continuously declines. This dynamic presumably initiates an inexorable weakening of NS that allows aging to proceed beginning during adolescence upon completion of ontogenesis at about 20 years of age. However, the evolutionary mandate for successful reproduction requires descent, which is not satisfied until reproductive fitness is reached following nurture and independence of progeny. These events generally take place during the post-ontogenetic decade of human life between 20 and 30 years of age, contradicting Medawar’s and Williams’s proposal that senescence begins and NS declines during adolescence. In fact, the beneficial changes that occur in the human body during the first decade of young adulthood, suggest that the developmental process is still underway for about ten years beyond completion of ontogenesis.
Yates et al [51] published thoughts on loss of integration and resilience as they affect biological senescence. In summary, a newborn organism is less stable than it will be at about 30 years of age, when many physiological processes are at peak capacity. Thereafter, a linear decline and loss of “reserve” occurs as aging begins and progresses to a point when stability reaches a minimum required for system autonomy [52] when death from “old age” occurs due to physiological losses that began at physical maturity and peak vitaltiy (Figure 3). Thus, the life trajectory in dynamic terms begins with growth, development, and differentiation, all of which are negentropic processes that are orderly. Upon their conclusion, entropic processes that destroy order become dominant causing death of the organism [53,54] (Figure 4).
This summary is consistent with the current theory, but it doesn’t explain how the transition from development to aging occurs.
How is the transition from development to aging explained?
Hayflick described the DP as “a cornerstone of modern biology within which a purposeful genetic program drives all biological processes that occur from conception to reproductive maturation. But, once reproductive maturation is reached, thought is divided as to whether the aging process is a continuation of the genetic program or whether it is the result of random losses in molecular fidelity” [55].
Some investigators feel that its continuation into adulthood is maladaptive, thereby, eventually causing aging [56,57,58,59]. They proposed several different ways that continuation of DP elements into adulthood could cause aging, but specific and convincing mechanistic details were not provided.
A significant problem with these theories is that they fail to explain how components of the DP continue beyond completion of the program and why NS doesn’t prevent their negative effects from being expressed. As the sentinel of evolution, NS should prevent aging from beginning upon completion of ontogenesis, which according to classic theory, occurs in humans at about 20 years of age. In contrast, the Gompertz law of mortality shows that aging, which presumably is coupled with declining NS, begins in humans at about 30 years of age and increases exponentially thereafter. If selection’s decline, promotes aging, then why would emergence and continuation of purported maladaptive events resulting from continued expression of the DP not be selected against about ten years before NS declines at the true onset of aging? Perhaps lack of selection against continued expression of developmental processes that cause aging resulted from oversight by a “shortsighted watchmaker”, i.e., inefficient selective surveillance of the post-ontogenetic phase of the human lifespan [57].
In contrast to theories proposing that aspects of the developmental program continue to negatively impact survival by causing aging, antagonistic pleiotropy theory assumes that senescence begins in the DP with the advent of reproductive maturation at puberty. The graphic presentations of Medawar’s and Williams’s theories, respectively, show an exponential weakening of NS beginning at 20 years of age in humans upon completion of the DP (Figure 2). The figure represents Williams’s comment [20] that senescence begins at peak reproductive probability and declining NS should also begin at that time. This suggestion conflicts with demographic aging data as presented in the Gompertz-Makeham plot, and also with the logic that aging should occur upon reproductive success, not a decade before.
It is somewhat illogical to assume that the strength if NS would decline before Darwinian fitness with its contribution to the gene pool of the next generation occurs. It is during this interval of time, between completion of ontogenesis and initiation of aging, that the mechanism which guarantees parental physical fitness for completion of the evolutionary obligation to reproduce occurs. It also ensures that aging and death will follow thereafter. So it is reasonable that some aspect of developmental regulation continues beyond reproductive success, since the adult soma though becoming increasingly fragile due to decaying regulatory control, would logically require some degree of maintenance throughout life in order to survive for as long as possible while aging. However, It seems that in discussion of the origins of aging, there is little mention of somatic maintenance regulation in young adults after completing the DP.
Actually, continuation of the DP into young adulthood is not maladaptive, but rather is essential for completion of the evolutionary task of reproduction when descent with modification upon which evolution is based, is satisfied [60,61].
Continuation of the DP as a prelude to aging
While it is generally agreed that expression of the DP is finite, Walker [1] recently proposed that ontogenesis and aging are functionally linked through “morphostasis”. It is a transient, non-aging stage of life defined by stable continuity and sameness through time as differentiated from morphogenesis/ontogenesis, which is characterized by creation and change [62]. Morphostasis is visually apparent as the horizontal, terminal, or “plateau” phase in graphs of the the Gompertz-Makeham law (Figure 5). This post-ontogenetic non-aging phase of development lasts in humans for about a decade between the ages 20 to 30 years. The transition from ontogenesis to morphostasis provides an important clue to solving the “Unsolved problem of biology”[18] that resides in behavior of the regulatory program for development as it continues unto morphostasis and aging as an emergent property of ontogenesis.
Ontogeny begins with the creation of a zygote and concludes during late adolescence with the emergence of a sexually mature young adult organism. The contemporary age threshold for the transition to adulthood within the DP is commonly recognized as 16 to 18 years, ranging from 14 to 21 years in different nations and even within regions of countries. Because sexual maturity can occur during ontogeny, it is possible for individuals to produce children at about 14 to 16 years of age, before completing their own DPs. However, when ontogenesis ends with creation of a reproductively competent young adult, its physical and functional development must continue to facilitate protection and nurturing of its progeny unto independence. As recognized by Williams [19], “The care of dependent offspring [nurturing] is as important to human reproduction as the production of gametes”. Thus, during the end of the parental DP and into morphostasis, progeny are protected and nurtured for about 12 to 14 years. (Figure 6)
To complete growth/development and achieve reproductive fitness, the last stage of parental ontogenesis links the DP with morphostasis. Then final physiological development of the parent, including that of muscle strength, reaction time, sensory abilities, and cardiac functioning occur. The reproductive system, motor skills, strength, and lung capacity all operate at their best as evidenced by the performance of professional athletes who are most accomplished during this decade. Also, many women have additional children during early-adulthood [63]. These attributes usually peak at a parental age of about 25 years and wane as 30 years of age is approached.
Thus, the young adult “grows” into its full adulthood when developmental potential is realized during the five or more years following completion of ontogenesis. During this decade of young adulthood the procreative value of new parents is realized through their demonstration of reproductive fitness which occurs at about 25 to 28 years of age. This important stage of development leading to reproductive fitness is sustained by an emergent property of ontogenesis that is a direct target of Darwinian selection. The significant consequences of the post-ontogenetic decade involving continued parental development and descent of genetic characteristics through independence of their progeny is essential for evolution. Without such descent, evolution is meaningless [13,64].
As parents’ approach 30 years of age, senescence begins and the strength of NS relevant to them declines due to progressive decay of the emergent process from the DP that evolved to complete physical development and reproductive fitness. Upon completion of the post-ontogenetic decade, aging begins with many subtle degenerative changes throughout the body. They include declining visual and auditory acuity, integumental qualitative decline such as thinning/ graying of hair and drying/wrinkling of the skin. Besides anatomical changes, evidence of physiological disorganization includes reduced response and recovery time following physical exertion. The immune system also becomes less adept at resisting disease, and reproductive capacity starts to decline [63]. These physical and functional defects are exacerbated during the following years, as frailty and vulnerability to chronic illness progress. Death eventually becomes inevitable.
The temporal intersection of development and aging proposed herein is inconsistent with the classic theory of aging that is based upon an inexorable decline in NS that purportedly begins at adolescence with attendant physical and functional defects of senescence following thereafter (Figure 3). That the strength of NS should decline before acquiring reproductive fitness as proposed in the classic theory seems evolutionarily inappropriate.
The present theory states that aging begins later in development than proposed in the classic theory. Rather than waning fecundity suppressing NS strength, aging begins upon attainment of reproductive fitness, i.e., when an organism demonstrates its ability to survive and transmit its genes to the next generation. Unlike fecundity, reproductive fitness/success requires nurturing of progeny before their independence. The majority of time during which it occurs in humans is within the post-ontogenetic interval of a decade of so, from parental ages of 20 to about 30 years.
The temporal relationships of reproduction and aging associated with the developmental process derive from primitive societies that evolved when human lifespan rarely exceeded 30 years. As a result, childbirth and rearing occurred early in life. This evolved history remains relevant today, and it is not unusual for some humans to procreate upon reaching sexual maturation during adolescence. However today, reproductive events typically occur later than they did in the past due to improved health conditions, increased life expectancy, pressures of society, personal choice and financial status. Nonetheless, the underlying biological mechanisms including the timing of sexual maturation and the onset of senescence remain the same as when they evolved in early humans.
Linking development to aging: Emergent properties involving developmental regulatory processes
Entities that acquire novel characteristics when becoming part of a larger system are said to be emergent properties. They arise from collaborative functioning of some of the system’s components. However, the emergent properties do not belong to any one part of it, but help adapt it to specific conditions and generally increase chances for survival [65,66]. Real world examples include schooling fish, flocking birds, and swarming locusts where the individuals are all part of the larger system that provides them with greater chances to avoid predation. Thus, the emergent property of each demonstrates a collaborative function, just as the soma is an emergent property of its individual molecular components, e.g. carbohydrates, lipids, proteins, and nucleic acids [67]. Similarly, homeostasis is an emergent property resulting from cooperative effects of integrated developmental regulatory processes. Emergent properties of ontogenesis promote further development, reproductive fitness, and aging.
Functionally collaborative regulatory processes within the DP direct gradual changes in size, shape, and function throughout the soma from conception to young adulthood. They translate genetic potentials into functioning mature systems, i.e., genotypes to phenotypes. Since the DP is a program, it is finite and cannot continue its dynamic expression beyond its last stage, when the parental soma first reaches adulthood. Because constructive stages of development are non-repeating, the local regulatory mechanisms during ontogenesis are modified appropriately for each developmental stage. Since the soma undergoes programmed change during ontogenesis, little regulatory redundancy occurs making their outcomes deterministic or predictable. However, when constructive stages of the soma end, physiological homeostasis continues as required for completion of reproduction, and to sustain homeodynamic organization/maintenance of the total organism for the rest of its life.
An adaptive process that links ontogenesis with the subsequent post-ontogenetic decade of young adulthood is selected to complete development and facilitate reproductive fitness during morphostasis. That evolutionary requirement is fulfilled by post-ontogenetic, redundant expression of developmental regulatory mechanisms that governed the last stage of the DP [60]. Thus, morphostasis is an emergent property of ontogenesis based upon redundancy of its developmental regulatory processes.
Continued redundant expression of the developmental regulatory mechanism beyond ontogenesis forgoes the need to evolve a novel program(s)/mechanisms for maintenance of the adult. Furthermore, this effect, brings stability and sustains a non-aging condition during the morphostasis decade. It allows final parental development to facilitate nurture of progeny unto their independence thereby achieving reproductive fitness before aging begins. Order resulting from redundant expression of regulatory oversight during morphostasis opposes entropy, despite the lack of ongoing somatic construction. Thus, a non-aging, negentropic state of organismal existence persists after the DP ends, so as to complete the act of reproduction.
Giaimo and Traulsen [2] proposed that during the progression of life, a transient phase occurs when NS does not decline and may even increase slightly. They proposed that such a period of non-aging could exist as an unstable equilibrium within the dynamic evolution of lifespan. Morphostasis satisfies those conditions since its non-aging state is transient, followed by the onset of aging and an inexorable decline in strength of NS.
The post-ontogenetic non-aging phase of development lasts in humans for about a decade between the ages 20 and 30 years [68]. It is visually evident on graphs of the Gompertz law of mortality as the horizontal, terminal, or “plateau” phase that completes the Makeham contribution (Figure 5).
Once parental physical development is completed and progeny sufficiently nurtured to make them independent, reproductive fitness is achieved. Thereupon, any further evolutionary purpose for continued organismal existence wanes.
Mechanisms central to survival and the origin of aging. Tradeoff vs “Unmaintainability”/Non-maintainability.
Tradeoffs
A Trade-off occurs when one trait increases or decreases causing the reciprocal effect in another [69]. Although the mechanistic basis for life-history trade-offs is unclear [70,71], they have traditionally been thought to result from competition between traits for limited resources available to an organism.
When seeking choices to meet or improve a certain goal, preferential traits need not be paired exclusively with only one reciprocally affected alternative. For example, limited energy may be traded off from reproduction to handle acute but recurring events such as protection of territory, to fight a rival, to migrate, etc. to seek new territory when in isolated and barren environments. So tradeoffs allow choice in resource allocation from many alternatives traits to that which is preferred. Or vice versa if reproduction is of higher priority than survival, then limited resources could be allocated for procreation rather than life extension, or to secure any of the previously mentioned alternatives. But there is no restriction on allocation which is determined by conditions at large. For instance, DST proposes that limited energy resources can be used to favor reproduction or survival. Allocation is not obligatory, but depends upon circumstance.
The presumption of limited resources makes their allocation a central factor in popular explanations for the evolution of aging. Examples of targets for adaptive resource allocation include reproduction, maintenance, somatic growth and DNA damage repair [72]. The most fundamental of tradeoffs is thought to involve survival and reproduction [73]. While limited resources are allocated differentially to the reproductive effort or to survival, they are presumably insufficient to maximally satisfy both needs.
Since reproduction is evolutionarily favored over survival, Kirkwood [44] proposed that a tradeoff of limited resources to support reproductive robustness in youth is chosen over somatic maintenance later in life, thereby causing aging [74,75]. However, if that presumption is valid, then celibacy could exclude the use of limited resources to support reproduction. However, that celibacy increases survival is questionable, but even if it did, aging would not be prevented. In fact, energetic processes are required for somatic maintenance, whether or not resources are preferentially used to support reproduction. Nonetheless, DST favors reproduction over “survival” or “longevity”, since the rate of aging can indeed be altered by abundant resources.
Other tradeoffs involving rates of aging between early and late life or between survival and reproduction are also popular theories [32,76,77].
Trade-offs can also occur between physiological traits and also genetic factors. These include pleiotropy, environmental factors, or combinations of these two types that result in negative interactions between traits [72,73,78].
Thus, survival can be affected by any number of environmental, life style, temperature, humidity, water, excessive predatory stress, etc. that can modulate the timing, frequency of reproduction and duration of survival, and also accelerate aging and cause death. Furthermore, survival or non-survival i.e., death by a multiple of causes can occur, but none has been shown to cause aging, which has a specific onset and progression. Nor do those factors that cause death do so in an age-related pattern described in the Gompertz law as exponentially increasing while causing the well described inexorable decline in strength of NS. In other words, they do not explain why under optimal conditions in the absence of the aforementioned stressors, aging occurs nonetheless [79].
Since survival is the process of continued existence despite risks of living e.g., accident, predation, natural disasters, extrinsic disease, and senescence, it can continue during aging which is a time-related process of somatic deconstruction. Thus, survival, which can be extended or shortened, is not aging, which once initiated, proceeds unto death.
It is difficult to explain the evolution of aging by tradeoff because an increase in one trait requires a decrease in another [69]. Thus, the word “survival” is cautiously employed instead of aging since it is illogical that the existence of a tradeoff of limited resources would increase fitness while decreasing aging. Since aging proceeds after sexual maturity, all that allocation of resources to survival can do is to affect its duration. A decrease in survival is the reduced rate of dying due to an energetically based decrease in maintenance, which can be reversed or accelerated based upon the allocation of available resources. For example starvation to the extent of emaciation as was reported in Nazi concentration camp prisoners certainly threatened their survival, but when they were refed, their physical health improved in many cases. In contrast, physiological dysregulation resulting from aging follows an irreversible programmatic progression. Thus, reduced survival is not reduced aging!
Despite the popularity and acceptance of these theories, their underlying molecular mechansms have been evasive. Only limited examples of antagonistically pleiotropic genes have been demonstrated [80]. However while examples of genes with antagonistic effects on life history traits are limited, circumstantial evidence in support of the aging-fitness tradeoff is extensive [73].
A confounding aspect of the classic theories is that they cannot explain the origin of aging without involving senescence in their explanation. For example, for accumulation of stochastic mutations to occur as part of the aging process, [19] the strength of NS must be in decline or else they would be selected against. Similarly, Williams clearly recognized senescence as a requirement for his theory of antagonistic pleiotropy, stating “It is necessary to postulate genes that have opposite effects on fitness at different ages”[20]. Finally, Kirkwood’s theory proposes allocation of resources in consideration of a tradeoff of survival for reproduction in youth over somatic maintenance “later in life”[23]. Resources that are allocated to favor reproduction over survival at different ages clearly recognize that lifespan is determinate, and that restricted energy can modulate the duration of life but cannot cause aging to evolve. Starvation for example can cause death, but not by the process of aging. In fact, starvation can occur while aging is proceeding because they are separable. Thus, while the classic theories describe processes by which aging rate can be altered by an effect on survival , they do not explain the how the evolution of aging occurred de novo, i.e., from a non-aging state.
A significant reason that the mechanism proposed in the current theory is different from the others is that they assume that an inexorable decline in NS due to waning fecundity makes aging inevitable. Thus, they all assume the involvement of senescence in the theory that is supposed to explain its origins. None of them meet that challenge to describe the cause of aging de novo.
These arguments suggest that aging cannot result from preferential allocation of resources to reproduction. Survival/longevity can be affected by such a trade-off, but not aging. Instead, utilization of energy resources is limited by aging, not vice versa. In other words, animals don’t “choose” to allocate limited resources to favor reproduction over aging; aging does the choosing,
So, while the popular notion that traditional trade-offs exist and can alter aging rate is valid, they are not sufficiently powerful to provide a general explanation nor even a specific mechanism for the evolution of aging in humans as described herein. If it is even possible to have a tradeoff that causes aging it, must be integrated with numerous other principles to do so [81].
Thus, the tradeoff theories discussed thus far are consistent with criticism of Darwin’s opinion that aging is not a product of NS. For example, generous allocation of resources to reproduction is not selected to restrict somatic maintenance and thereby cause aging! There is no resolution of Darwin’s dilemma to explain how aging could occur in compliance with mechanics of NS, through trade off since the theories don’t describe a latent individual benefit in the aging process, nor satisfactorily explain why and how it could be realized. These issues will be addressed below in the novel theory that links ontogenesis and morphostasis by a mechanism that solves the unsolved mystery of biology [19].
“Unmaintainability”
The most fundamental of tradeoffs involve allocation of limited resources for reproduction over survival [73], which has been mistakenly associated with the evolution of aging [23]. Presumably then, if resources were unlimited or their allocation not constrained, then aging would not occur. Alternatively, if the traded commodity were exclusively committed to promoting reproduction while subsequently causing aging, then no alternative intermediate would be possible to alter the joint relationship, making aging inevitable. In that case, the intermediate would play a committed and exclusive role in that relationship independent of either trait. Thus, I hesitate to describe the cause of aging definitively as a traditional tradeoff because if the current theory is valid, then unlike tradeoffs, there is no alternative to reproductive success since the immediate onset of aging must follow morphostasis, the life stage following completion of the DP, whether or not reproductive fitness is achieved.
If a mechanism is presumed to cause aging, it should do so directly, not through modulation of the rate of that which it is supposed to cause. That effect alone indicates that the processes proposed in classic theory are not the primary cause(s) of aging.
Sometimes trade-offs that are anticipated do not occur. Despite being seemingly obvious, they occasionally do not happen as assumed by simple conceptualizations that normally predict their occurrence. On those occasions, that which appears to be a trade-off does not fit the model. An example is the relationship between reproduction and aging, which unlike reproduction and survival is not a tradeoff.
Often a legitimate tradeoff that could explain the relationship between reproduction and survival is used to suggest that “evolution of aging” involves such a mechanism [23]. However, aging is the time-related deterioration of the physiological functions that are necessary for survival, thereby differentiating it from survival which is continuing to live or exist, typically in spite of disease, accidents, ordeal, difficult circumstances and importantly, even aging. Thus, survival represents the duration of living or lifespan, the mechanism of which is irrelevant to that which causes aging.
In a recent paper [82,83], the authors proposed that when variation occurs in one trait rather than another, the differential distribution can be explained in several ways. Only one is relevant to the current theory, but two are discussed to distinguish the classic theory from the current one.
In the first case, variation could have existed but evolved away, i.e., some organisms possessed a trait that was better adapted than an alternative. As a result, its frequency increased at a cost to the other, eventually resulting in only the presence of the better adapted trait. The theories begin with a non-aging phenotype that eventually displays aging properties following invasion by a limited number of genes. These dynamics are proposed in MA and AP theories, or by differential allocation of energy per DST. However, the tradeoffs do not restrict the outcome to be aging. To the contrary, “evolutionary tradeoffs between traits that are a consequence of the genetic background of organisms and their environment are not immutable and can be altered if some changes in the genetic background or the environment occur” [83]. Thus, organisms displaying such traits could evolve ageless phenotypes if the limited number of genes were less adapted or if the available energy were allocated differently. In other words, a non-aging condition could theoretically occur by chance. As a result, a tradeoff cannot serve as the mechanistic basis for aging, although it can certainly apply to longevity, ie. rates of aging.
Alternatively, “there are some complex organisms such as humans for which no variation could possibly exist so that they do not age”, i.e., aging cannot be avoided [82]. The authors argued that if the aging process were linked to the complexity of life through an emergent property that was “unmaintainable”, then numerous competing demands upon an organism’s regulatory complexity would make senescence inevitable.
It is undeniable that the origin of aging per se is antithetical to NS because it is generally accepted as lacking individual benefit. However, in the context of the current theory, aging shares a mechanism with reproductive fitness to which they are both inextricably linked. Under this special condition, selection of the mechanism subserving both fitness and aging can occur. If aging is inseparable from reproduction because they share the same mechanism which facilitates both traits, then aging cannot be traded. Under this unique circumstance, it may be considered adaptive and thus, selectable.
The process of physiological “unmaintainability” represents a pivotal mechanism of life history, since it provides the answer to Anderson’s question [50]; how is the transition from the end of development to the beginning of aging explained?
The only logical way to connect development with aging is to link the last ontogenetic stage to the following decade through redundancy of complex developmental regulatory processes. Regulatory redundancy dynamics link reproductive success with individual aging. When redundancy of regulatory processes that link ontogenesis to morphostasis progressively fail, then the non-aging phase of young adulthood ends, causing aging and the strength of NS to inexorably weaken. Thus, loss of regulatory redundancy represents the mechanism that initiates aging and causes it to exponentially proceed. The mechanism simultaneously regulates two life history traits in the same direction, independent of resource allocation. There is no loss component.
Reproduction is not complete when the DP ends. Although birth could occur during ontogenesis, the nurture of progeny, which is as important to reproduction as exchange of gametes must occur thereafter. This temporal perturbation of the reproductive sequence presents a significant challenge since biological systems require robustness [66] to maintain stability until reproduction is completed. In order to survive and reproduce, dynamic equilibrium must be maintained in biological systems despite ever-changing conditions [82]. Thus, morphostasis emerged from ontogenesis through the action of developmental regulatory redundancy.
The first occasion of reproductive success generally happens during the post-ontogenetic stage of morphostasis within an environment resulting from emergence of regulatory redundancy of the last stage of the DP. If an offspring were created during the DP, then its nurturing and independence could occur during morphostasis, but it is not necessary since the mechanism is not dependent upon successful reproduction. For example, childless couples experience “normal” survival/longevity by the same mechanism as those who bear and nurture children. Both examples also experience aging by the same mechanism. Thus, should pregnancy occur, developmental regulatory redundancy was selected to ensure the opportunity for reproductive fitness during the transient stage of morphostasis by preventing aging during nurture unto independence of progeny. However, it does not require fitness for aging to proceed
Redundant expression of the regulatory mechanism from the last stage of the DP is an emergent property of ontogenesis that creates and sustains morphostasis. However, the regulatory mechanism is unmaintainable because of constraints imposed by finite numbers of redundant units, reliability limits, reproductive mechanism damage, apoptosis-related DNA damage, and other biophysical or biochemical limitations. Such constraints make variation in such a mechanism impossible, thereby preventing the existence of any alternative [,83]. Because it sustains a non-aging stage of life, inevitable decay of “unmaintainable” developmental regulatory redundancy causes gradual physiological dysregulation that erodes somatic homeostasis, allowing progression of disorder as a primary cause of aging [65,66,84]. Aging then continues in parallel with the degrading mechanism whether or not reproductive success was achieved. Aging will always follow when degradation of the mechanism that creates and sustains the non-aging stage of morphostasis degrades below a critical mass and/or in those areas that are essential for continued life.
The regulatory mechanism doesn’t simply disappear in one step, but instead it degrades progressively as redundant units of its construct are progressively lost. The persistence of the degrading redundant mechanism during aging provides somatic maintenance, albeit waning as redundancy is lost. Nonetheless, it is sufficient to allow reproductive success to occur several times after aging begins, such as happens in humans and other such iteroparous animals. Thus, the mechanism that facilitates reproductive success is selected, not reproduction per se. This may seem a matter of splitting hairs, but it is meaningful because the mechanism, which is a product of NS, provides not only for fitness, but also for aging. Since the redundant mechanism inseparably links development and reproduction with aging and death, both are de facto products of natural selection. So, while there may be a tradeoff of resources between reproduction and survival (rate of aging) there is no tradeoff of resources for one or the other. Paradoxically, both are selected.
The unmaintainable emergent property cannot be selected against because it brings individual as well as population benefit of reproductive success and descent as well as aging and death. Accordingly, the current theory proposes that the functional relationship between reproduction and aging is not the result of a tradeoff.
The mechanism for the evolution of human aging proposed herein is unique because it describes one mechanism that sustains a non-aging environment conducive to final parental development and reproductive success and also explains the onset and progression of aging. Thus, aging is not multifactorial!
The concept of “unmaintainability” is consistent with Giaimo & Trualsan’s [2] proposal that while negative aging is impossible, a transient stage of non-aging involving an “unmaintainable” process that would eventually degrade to cause aging is tenable. Aging is the inevitable outcome of physical and biochemical constraints that prevent unending redundant developmental regulatory oversight from sustaining a non-aging condition. Thus, an emergent property involving the developmental regulatory process that is based upon redundancy cannot indefinitely keep the soma in a non-aging state because biophysical restrictions (reliability theory, entropy and loss of redundancy) and or biochemical constraints (reproductive effects, DNA damage and apoptosis) demands its eventual failure. Thus, loss of redundant regulatory elements that link the DP with aging prevents there from being an alternative, making unending survival an impossibility.
The seemingly paradoxical relationships between reproduction and aging that are linked by a single unmaintainable emergent property of ontogenesis explains how senescence can be a product of Darwinian selection. Those facts were not included in the initial report [1] and thus, did not explain how aging could be selected. Oversight of the important tradeoff in the original mechanistic theory is corrected to show that indeed mammalian aging meets the criteria for selection, providing a hidden compensating individual benefit as predicted by Darwin.
Section Summary
Humans age because morphostasis, a post-ontogenetic, temporary non-aging interval that results from developmental regulatory redundancy, an emergent property of ontogenesis, is unmaintainable. This stage is seen in the final segment of the Makeham parameter of the Gompertz-Makeham law of mortality. As redundancy of the developmental regulatory mechanism progressively fails, a point is reached when the number of redundant units cannot sustain the non-aging interval of morphostasis and aging begins. Its progression parallels the loss of redundant units whose effect on morphostasis stability wanes exponentially as more and more redundant units are lost. This is the single cause of aging in humans. Unlike tradeoffs, loss of the non-aging stage must cause aging because there is no alternative. Thus the key to understanding the origin of human aging is visably evident in morphostasis whose termination immediately initiates aging.
Mechanisms for loss of regulatory redundancy
Exponentially accelerating mortality remains one of the strongest generalizations of human aging. As described by the Gompertz model, exponentially increasing of mortality during aging prevails in all human populations living under all circumstances [85,86,87].
Historically, searches for a general biological theory to explain aging have failed because they require assumptions that would accommodate the diversity of species based upon their unique evolutions. While there may exist common underlying and basic processes that can be generalized, it is not possible to identify the unique, terminal mechanisms by which aging is expressed in all animals since species evolved different strategies [15]. For that reason, the current theory to explain the ultimate mechanism of aging is limited to humans and other iteroparous animals that nurture their young as the final stage of fulfilling the evolutionary mandate for reproductive success. Accordingly, a central assumption of this theory is that ontogenesis and aging are linked through a non-aging stage that evolved to accommodate details of the life history of humans and related mammals. The temporary maintenance of morphostasis, a non-aging interval between completion of the DP and onset of aging results exclusively from developmental regulatory redundancy, an emergent property of the last ontogenetic stage.
An important deficit of the classic theory is that it does not explain why an exponential increase in aging as described by the Gomperz law occurs, nor why late life mortality plateaus exist. These characteristics are recognized as true aspects of aging dynamics and should be better understood to answer relevant questions about aging’s evolution and its progression. However, their explanation has been elusive due to acceptance of Medawar’s and Williams’s original proposal that the cause of aging is multifactorial.
Kirkwood [88] explained that past research efforts to pursue specific single-aspect theories of aging have limited explanatory power. Because aging is presumed to be multifactorial, he suggested that there is a need to develop network theories in which the individual mechanisms interact with each other, using the methods of systems biology. He proposed that such an approach may make it possible to connect Gompertzian mortality patterns with the underlying primary mechanisms.
In disagreement, this author suggests that failure to explain the Gompertzian pattern of aging results from inability to apply reliability theory, a general concept of system failures that provides a means to understand many puzzling features of mortality and lifespan. Thus, a mechanism is impossible to identify due to unwavering acceptance of the multifactorial concept of aging that is embraced by the classic theory.
In contrast to that assumption, a basic concept of the current theory is that aging does not evolve de novo through stochastic accumulation of damage, but instead derives from continuity of ontogenesis with morphostasis involving redundant expression of the regulatory mechanisms required for completion of reproductive success. That link involving redundancy of the regulatory mechanisms that directed the last stage of the DP then allows explanation of the exponential progression of aging by Reliability theory as proposed by Gavrilov and Gavrilova [89].
Loss of redundancy due to specific types of molecular damage at functional regulatory sites as well as random damage to the soma, subserves emergence and progression of the programmatic aging phenotype. Application of reliability concepts in the current theory explain not only the exponential increase in mortality rate with age and the subsequent leveling off, but also the compensation law of mortality. During aging, each stage of somatic deconstruction corresponds to one of the organism's vitally important structures being damaged due to loss of redundancy. In complex organisms with many vital structures and significant redundancy, every occurrence of damage does not lead to death because of redundancy. This causes functional defects to accumulate and progressively advance the aging condition [Figure 7] . Eventually, all redundant elements are lost and aging stops, giving the appearance of a mortality plateau. Any subsequent assault on the soma causes death. Thus, aging is a direct consequence of systems redundancy that temporarily ensures increased reliability and lifespan of adult organisms until critical mass is lost.
Key: A: small boxes represent stages of development. The thin arrows represent minimal redundancy. B: A representative of multiple areas of the soma that are regulated by significant redundancy. C. The same representative area displaying loss of redundant units as a cause of aging’s initiation and progression. Figure based upon Gavrilov and Gavrilova [89]
System failure rates may contain both non-aging and aging terms as, for example, in the case of the Gompertz-Makeham law of mortality. The current theory proposes that during morphostasis, an emergent property of ontogenesis, regulatory redundancy sustains the final, age-independent Makeham component. During this phase of life, significant loss of redundancy does not occur until sufficient time has passed for reproductive success to have been achieved. Thereafter, during late stage morphostasis loss of redundancy begins, initiating aging which starts and progresses to display the characteristic Gompertzian profile. However, it is important to understand that fitness need not occur for aging to begin, since it is the mechanism to sustain morphostasis, which allows that evolutionary benefit to proceed in a non-aging environment, that was selected. Thus, as previously stated, aging will begin whether or not reproductive success occurs, since once the mechanism sustaining the transient non-aging state of morphostasis decays sufficiently, then aging must begin. Thus, the unmaintainable mechanism of regulatory redundancy, an emergent property of ontogenesis that temporarily suspends aging during morphostasis is selected. It is the regulatory redundancy process that is selected to allow time for completion of the reproductive act, even though its loss of redundant units causes aging to proceed.
Selection pressure to maintain the non-aging stage of organismal morphostasis declines once loss of redundant regulatory units falls below critical mass and aging begins. Aging then proceeds exponentially as increasing numbers of redundant regulatory components are lost. Resultant physiological dysregulation progressively erodes somatic homeostasis [65,66] (Figure 6). Despite loss of redundancy, developmental regulatory expression continues to provide maintenance of the soma to the extent possible during the post-ontogenetic decade and beyond, for the duration of life. Thus, iteroparous animals such as human beings, can continue to reproduce during middle age so long as sexual organs maintain function during the relatively slow aging process.
The failure rate of an organism initially grows exponentially with age following the Gompertz law. If the background, age-independent component of mortality also exists in addition to Gompertz function, we obtain the well-known Gompertz-Makeham law described earlier with the relevant non-aging component preceding and predicating the onset of aging and the Gompertz function. At advanced ages, the rate of mortality decelerates and asymptotically approaches an upper limit. The model explains not only the exponential increase in mortality rate with age and the subsequent leveling off, but also the compensation law of mortality [89].
Classic theory cannot provide these explanations of aging’s initiation and progression because application of reliability theory requires redundancy of multiple life sustaining traits that are distributed throughout the body as elements of the developmental regulatory process. If aging were multifactorial, this rationale would be invalid.
Effect of reproduction on longevity
Evolutionary theories of aging predict a trade-off of lifespan for fertility [20,23]. Studies using model organisms and data from contemporary human populations provide supporting evidence for this prediction [90]. Having more than four or five children increases the risk of certain diseases and shortens a woman's overall lifespan [91]. Numerous studies also describe an inverse correlation of lifespan with total amount of progeny as well as the age at primiparity [92]. Early maternal death is associated with early childbirth, and later death with later childbirth. From their findings the authors concluded that human life histories involve a trade-off between longevity and reproduction [75,93] .
Such studies are often cited in direct support of the DST, which emphasizes that energetic and metabolic costs associated with reproduction, lead to maternal deterioration, increased risk of disease, and higher mortality. While reproduction is a costly mechanism that alters metabolism of fat, investment of lipids in reproduction cannot be allocated to support somatic maintenance mechanisms [94]. Thus, this is an ancillary effect on somatic maintenance that is secondary to the primary relationship between aging and reproduction.
In contrast to DST, AP theory emphasizes a genetic trade-off, where genes that increase reproductive potential early in life increase risk of disease and mortality later in life. Such a trade-off would be expected even for women who have not given birth but who carry these gene variants. Several researchers have sought evidence for the trade-off between fertility and lifespan, with elusive results. A genetic correlation that is consistent with AP may underlie this relationship between reproductive age and lifespan, but the identity of a specific gene(s) has remained unknown.
In response to weakly supportive research findings, Blagosklonny [95] opined that the relationship between reproduction and aging lacks scientific consensus, and the relevant mechanisms are undiscovered.
Nonetheless, while a precise mechanism responsible for the recognized damping effect of reproduction on lifespan remains unclear, the theory remains popular from an evolutionary perspective.
The theory presented herein proposes a different approach to explaining not only the relationship of reproduction and longevity, but also of reproduction and aging. It is based upon the premise that developmental regulatory redundancy of the last stage of ontogenesis was selected to facilitate nurture of offspring and reproductive success. Damage through use of the redundant regulatory components that are directly associated with reproductive processes may cause their progressive loss relative to the number of reproductive events that occur. This explains why early reproduction and increased fertility, which is associated with loss of redundant regulatory elements, is associated with reduced lifespan and vice versa.
If loss of regulatory redundancy results from damage to the mechanism driving reproduction, then each pregnancy would initiate and/or exacerbate that loss. Since reproduction and survival are negatively correlated, then damage caused by the reproductive effort to the developmental regulatory mechanism that evolved to facilitate reproductive success would logically reduce relative lifespan. More pregnancies would generate more molecular damage thereby causing less redundancy. Every time the reproductive process proceeds, there is greater chance for damage to the reproductive component of the redundant regulatory mechanism, i.e., to the emergent property that evolved to ensure reproductive success. Thus, since it evolved in great part to facilitate reproductive success, then the more the mechanism is employed, the greater the chance of damage and loss of redundancy. This is one explanation for why reproduction decreases life span.
While the effect of reproduction to reduce survival is not universal, it is one of the better-established patterns in the biology of aging.
Effect of redundancy loss on differential programmatic characteristics and lifespan
Loss of redundancy in specific regulatory units among people in a population contributes to differential lifespan. The longest living individuals probably have either excessive redundancy in regulatory areas that resist intrinsic disease and thereby lessen the likelihood of their occurrence during aging, or else molecular damage to those sites is minimal or missing simply by luck. In addition, stochastic damage to non-regulatory physiological sites which are much more common, allow the aging phenotype to progress in its non-programmatic, diverse characteristics. Thus, specificity of aging phenotypes, e.g., represent damage to and loss or regulatory components that initiate it. This in turn decreases strength of NS allowing stochastic damage of regulatory sites and to those not related to regulation, to cause the seemingly multifactorial aging phenotype. Time of birth and frequency of reproduction which causes loss of regulatory redundancy also affects lifespan as previously described.
Programmatic nature of the aging phenotype
Classic theory recognizes the programmatic aging phenotype, but doesn’t attempt to explain how or why it occurs.
As redundancy wanes, DNA damage, specifically by double stranded breaks (DSBs), affect the regulatory mechanism causing it’s ability to sustain proper somatic maintenance to decline over the remaining course of life.
Random decay of specific redundant regulatory units derived from terminal developmental stages produce programmatic aging phenotypes. Since the developmental regulatory mechanism is an integral part of somatic construction and maintenance, then its decay during adulthood could represent not only the cause of aging, but also explain aging’s programmatic appearance. Stochastic random of damage of the regulatory units would produce an underlying albeit subtle, pattern of somatic deconstruction.
While random mutations occurring throughout the body can erode its maintenance in a general sense, those specifically affecting the regulatory processes of the last DP stage of one person would create an aging phenotype that may loosely resemble that of another, but not precisely. This occurs because the damaging mutations emerge randomly, affecting various parts of the soma, including the regulatory areas. Thus, stochastic damage to the regulatory process at different places that are distributed throughout the body cause variation in the aging phenotype even among identical twins and other closely related individuals. This effect accounts for the programmatic nature of the aging phenotype
DNA damage in relation to aging.
If a lifelong functioning regulatory mechanism evolved to ensure development, successful reproduction, and somatic maintenance during adulthood, it could like all other processes be vulnerable with advancing age to stochastic damage that degrades its efficiency. This is especially true because aging weakens strength of NS, increasing the chances for mutations and other forms of molecular damage to accumulate. This positive feedback of damage on aging while not being the primary cause of it, accelerates its expression and expands its phenotype. Furthermore, if the developmental regulatory mechanism is an integral part of somatic construction and maintenance, then its decay during adulthood represents not only the cause of aging, but also explains aging’s programmatic appearance. All reliability models consider aging as a progressive accumulation of random damage [96] and loss of regulatory redundancy that in turn, increases mortality which eventually “levels-off” as a function of increasing age.
A consensus of biogerontologists accepts the theory that accumulating macromolecular damage participates in creating aging phenotypes. DNA damage is expressed at physiological, cellular and molecular levels. These effects at different levels of somatic organization represent tertiary, secondary and primary contributors, respectively, to degenerative changes in form and function that collectively and increasingly degrade the ability of adult organisms to tolerate metabolic stress and to eventually die. DNA damage is assumed to have far-reaching effects that undoubtedly contribute to the so-called “pillars of aging” [97]. Thus, it is fairly certain that DNA damage contributes to senescence, epigenetic alterations, proteostatic stress, and mitochondrial dysfunction [98]. Genome rearrangements and structural variations that occur in somatic tissues and increase with age further support the concept that DNA damage is associated with aging [99,100].
Although erosion of genome integrity by DNA damage is favored as a basic cause of aging, there is little consensus regarding the type of damage that is primarily responsible. Thus, an important question in gerontology is whether any specific type predominates, and if so, which one? It has been suggested that the DNA double-stranded break (DSB) is a logical candidate [101].
DSB’s as well as base modifications, single-strand breaks (SSBs), double strand breaks (DSBs), and interstrand cross-links (ICLs) result from DNA damages that abnormally alter genetic structure and prevent the proper functioning of replication [102,103]. Most relevant to the mechanism of aging presented in the current theory are
DSBs that trigger apoptosis [104] that results in a faster depletion of stem cells and reduction of regulatory cell redundancy [105]. Other DNA lesions that trigger apoptosis include O6-methylguanine, base N-alkylations, bulky DNA adducts, DNA cross-links and DNA double-strand breaks (DSBs). DNA double-strand breaks (DSBs) are common, ultimate apoptosis-triggering lesions arising from primary DNA lesions during DNA replication. Thus, DNA replication is a necessary component of DNA damage-triggered apoptosis [106]. Repair of these lesions are important in preventing it [104].
The effect of cell autonomous events such as apoptosis would require less widespread DNA damage and might also explain aging’s programmatic phenotype by depletion of functional regulatory cells involved in maintenance of homeostasis during the reproductively important, first decade of young adulthood. It is of note that mutations of a general sort do not lead to apoptosis, so it is DSBs that are instrumental because natural mutations may not be eliminated leading to continued life but may give rise to abberant phenotypes including disease instead of normal aging [107]. Also, DSB damage to redundant regulatory mechanism will elicit epigenomic changes that contribute to the programmatic characteristics of aging phenotypes.
Aging could also result in part by cell non-autonomous mechanisms such as through the triggering of senescence, which can negatively impact the function of neighboring, undamaged cells through their Senescence-Associated Secretory Phenotype (SASP) [108,109]. Presumably, these effects would be independent of the programmatic effect resulting from loss of redundant regulatory elements. Thus, DNA damage is considered a potentially significant contributor to and universal participant in the aging process that can significantly contribute to loss of regulatory redundancy.
It is generally assumed that sufficient numbers and various types of molecular defects must accrue to negatively affect multiple aspects of cell biology. However, it is uncertain as to how many damaging events are needed to create the aging phenotype. Also, if DNA damage is randomly occurring, without some sort of informational compass, it would be difficult to explain why the aging phenotype is programmatic except as previously mentioned, i.e., damage to regulatory elements would cause programmatic aging profiles. However, redundancy of the regulatory system that evolved as an emergent property of the DP is the Achillies heel of maintenance since damage to components of the redundant system that are removed by apoptosis then contribute directly to initiation and progression of human aging. Thus, DNA damage that specifically reduces regulatory redundancy will erode fidelity of the whole body.
The occurrence of any type of DNA damage independent of other types may be insufficient to independently cause aging, except perhaps a specific type damage affecting regulatory cells that have broad reaching influence throughout the soma. Thus, for the most part, general DNA damage is a consequence of aging, and thus as presumed by this author to not be part of its causal mechanism.
Aging effects of double-stranded breaks (DSBs) in relation to loss of regulatory redundancy
The current theory proposes that aging is not caused by random accumulation of DNA damage in contrast to Medawar’s hypothesis [19]. It is caused by loss of redundancy of finite numbers of irreplaceable elements due to stochastic damage by DSBs that reduce the population by apoptosis, cellular senescence or pseudogenization. Other causes such as reliability limits to maintenance of redundant units have been previously discussed.
Nonetheless, the aging phenotype is complemented by random DNA damage, becoming progressively more complex and diverse as damage accumulates commensurate with age-related decline in strength of NS. The relevance of DSB-induced mutagenesis to the current Theory is its potential to reduce redundancy in the regulatory mechanism needed to achieve reproductive success and thereafter to sustain ordered somatic maintenance.
Apoptosis is removes damaged regulatory cells from the body, thus giving it a more specific, maladaptive role that is causally relevant to the mechanism of aging. Its damaging effect reduces the number of regulatory cells derived from the Developmental program that promote parental vitality and physical fitness during the post-ontogenetic period of life when nurturing of progeny unto independence occurs. In this way DSBs which can cause apoptosis may represent a primary contributor to loss of regulatory redundancy that directly initiates aging. There is increasing evidence that the various endpoints of DSB processing by different cells and tissues are part of the aging phenotype. For example, DSBs trigger the DNA damage response (DDR) that directs a cell to repair the break, undergo apoptosis, or become senescent. DNA damages can contribute to progressive decay and loss through apoptosis of irreplaceable cells within gene regulatory networks and may be a direct cause of aging [110,111].
Apoptotic cells form apoptotic bodies that are phagocytized by neighboring cells, without the release of cellular contents. To maintain organismal homeostasis, phagocytes engulf dead cells, which are then transferred to lysosomes, where their cellular components are degraded for reuse. As a result, damaged regulatory cells that undergo apoptosis would not accumulate with age, since once lost they are removed from the population of regulatory cells. This is one way that redundant regulatory elements can be reduced in number, i.e., apoptosis reduces their number after morphostasis is completed and reproductive fitness is achieved.
Experimental evidence of DSBs relevance to aging is demonstrated by the fact that their production in young knock out mice initiated the aging phenotype before it spontaneously emerged. However, because DSB’s also occur during aging, they may accelerate the process if they occur in the regulatory process, but their occurrence after the fact at sights other than the those containing the developmental regulatory process is coincidental of aging not causal of it [112].
Thus, genome structural changes resulting from DSB repair participate directly in the mechanism that slowly disrupts morphostasis to initiate senescence and subsequently contribute to accelerated aging (morpholysis). These facts are directly relevant to changes in regulatory signaling of the redundantly expressed, last morphogenetic module and thus, to the mechanism and onset of senescence. However, during youth and under normal physiological conditions, adverse outcomes of DSB processing are rare. In fact, they are beneficial when they efficiently remove irreversibly damaged cells or constructively participate in the developmental process, respectively [113,114].
The maladaptive effects of DSBs emerge later in life when associated with aging phenotypes such as atrophy, inflammation and immune senescence. However, these adverse conditions are consequences of senescence since they occur while aging is proceeding. More relevant to the present theory is that DSBs are intrinsic to various biological processes such as transcription and are key substrates for translocations, deletions and amplifications associated with aging and various cancers [115]. Induction of mutation, apoptosis, and/or cell growth varies in multi-cellular organisms, such that their balance and coordination may counter DNA damage to increase organismal survival [116].
It is unclear whether DSBs and their accumulating adverse effects ever rise to high enough levels to explain the observed functional loss and increased disease risk that are hallmarks of aging. This is important because DSB effects decrease redundancy exclusive of ancillary effects of general DNA damage to the soma and thus, would explain how relatively small numbers DSB damage is effective in causing aging throughout the body [101]. I presume that DNA damage doesn’t have to accumulate in great amounts since aging can be initiated by relatively fewer and more specific DSB damages to redundant regulatory components of the soma.
DSB damage induces epigenetic changes associated with aging
Since DSBs occur randomly, their effects on aging profiles will not be uniform since some damage may be more detrimental than others. This randomness in different loci whose genetic expression is determined by local epigenetic landscapes, accounts for the differences in aging phenotypes, even among identical twins.
DSBs compromise integrity of the epigenome. Epigenetic dysregulation is a hallmark of the aging process that jointly contributes to longevity through genetic impairment [117,118]. Changes in the activity of epigenetic enzymes during aging influence the expression of critical longevity genes whereas their alteration can drive large scale epigenetic changes in the genome. Single point mutations in epigenetic enzymes dramatically alter lifespan of lower organisms, suggesting that epigenetic modifications drive age changes. In light of the fact that many redundant enzyme systems exist in more complex higher organisms, it is possible that a this idea is too simplistic [119]. Alternatively, large-scale changes resulting from specific environmental factors or nutrient availability are more likely to be primary factors. Thus, it is generally accepted that cellular phenotypic diversity is explained by epigenetic influences that modify genetic expression.
Expression of regulatory genes is affected by their environments. Hence, expression of regulatory processes distributed throughout the body is modified by the epigenetic landscape within which they reside so that quantitative differences in stochastic damage to them will produce a differential aging phenotype even among closely related organisms.
Optimal gene expression is maintained through rounds of cell division due to key, stable epigenetic patterns of DNA methylation and chromatin-based regulation. However, when involving the developmental regulatory mechanisms, epigenetic profiles are more dynamic, sometimes following changes in gene expression rather than causing them, thereby not reflecting the stability of a true epigenetic state. This variation is of primary importance in the mechanism subserving the emergence of senescence from morphostasis, since structural alteration of the initiating genetic component affects subsequent outcome. Central to the current theory, is the importance of mechanistically distinguishing between stable epigenetic expression needed to maintain somatic homeostasis from its dynamic expression in the developmental regulatory mechanism during the various stages of morphogenesis, morphostasis and morpholysis. As an organism ages beyond adulthood, continuous and progressive loss of morphostasis regulatory redundancy exacerbates aging. As a result, efficacy of the mechanisms responsible for minimizing cellular damage declines during aging eventually resulting in an organism’s inability to maintain homeostasis. Indicative of this process are the many epigenetic marks such as DNA methylation, post-translationally modified histones, and chromatin that are changed during reprogramming and thereby dysregulated during aging. By not considering that a primary mechanism of aging also involves genetic and epigenetic elements that regulate morpholysis, epigenetic dysregulation during aging not preceding it, has emerged as a hallmark of the process [120]. The important difference between these two processes is that aging is driven by deterioration of the primary mechanism and thus, global epigenetic dysregulation is a consequence of the aging process, not a cause. Many of these age-related somatic changes can be ameliorated by increasing the levels of histones, strongly implicating direct transcriptional consequences of histone loss [121]. Genomic stability and chromatin structure are closely intertwined, the latter not only regulating accessibility of DNA damaging agents to the genome, but also participating in critical signaling roles for DNA lesions and their repair [122]. Since chromatin plays a critical role in regulating genomic stability and gene expression, it is possible that during aging such changes may be caused by the global alteration of chromatin structure as evidenced by loss of heterochromatin in human cells [121] to wide-spread reduction of histone levels during mitosis.
During the DP, regulatory gene expression changes as the epigenetic landscape is modified with each non-repeating stage of construction. These changes allow differential expression of the regulatory mechanism making it difficult for damage and errors to accrue at any specific stage. However, the lack of non-repeating stages during morphostasis and subsequent phases of life requiring that the regulatory mechanism become redundantly expressed, makes it vulnerable to damage and to an increased risk for the emergence of aging. When stages of somatic construction are complete, regulatory oversight of the adult soma must become redundant and appropriately expressed pursuant to the varying epigenetic conditions of the different body parts. Progressive stochastic damage erodes robust expression of these master regulatory mechanisms causing loss of redundancy that erodes functional and structural stability characteristic of senescence (Figure 8).
DSB-induced loss of regulatory redundancy affects epigenetic changes and accelerates aging
The premise that a redundant regulatory mechanism oversees morphogenesis and morphostasis but degrades to allow aging to proceed, is supported by published reports linking genome damage and epigenetic drift with aging. A study was designed to determine how experimentally induced DSBs in conjunction with correlative changes in the epigenome affect organismal aging. Mature adult mice ranging in age from 3 to 6 months were used in the model. The ages of mice selected for the study were considered mature, at “a life phase equivalent for humans ranging from 20 - 30 years” [123]. Thus, the study was begun in mice whose physiological ages were comparable to those of humans during the decade of morphostasis when non-repeating stages of the DP have ended, being replaced by redundant regulatory cycles prior to the onset of senescence .
The effect of reduced regulatory cells on aging was demonstrated in a transgenic mouse model that was created using the I-PpoI restriction enzyme to induce DSBs [124,125]. Non-mutagenic DSBs were created in non-coding regions of DNA at frequencies only a few-fold above normal background levels. It is presumed that the authors chose to cut ncDNAs because they are often transcribed to non-coding RNAs (ncRNAs) [126] which have been identified as regulators of chromatin structure and gene expression. Since ncDNA is regulatory, they are good candidates for simulating changes that occur during aging. Their experimental reduction mimics spontaneous loss of redundant regulatory units as predicted in the current theory.
Treatment caused premature organismal aging. After one month, treated mice displayed slight alopecia and loss of pigment on their feet, tails, ears and noses, compared to controls. These are common features of middle-aged, wild type mice. However, 4 to 6 months later, the mice displayed significantly accelerated physiological aging of skin, eye, muscle and brain. Transcriptional changes associated with acceleration of the epigenetic clock estimated the rate of aging to be about 50% faster than controls. By 10 months of age, gene expression patterns and DNA methylation in treated and control mice showed that dysregulation of skeletal muscle genes from significantly younger experimentally altered subjects was positively correlated with that of twenty-four-month-old wild types. Resulting molecular changes in the epigenome included histone modifications, DNA compartmentalization, smoothing of the epigenetic landscape, and loss of cellular identity.
These observations suggest that unlike other DNA damage, DSBs are independently capable of driving multiple age-related phenotypes as part of the aging mechanism. Also, the quality of DSB repair responses tends to degrade with age, causing increased genome structural variation in older mammals. Also, the diminishing quality of normally occurring DSB repair as well as that following long term exposure to agents causing their induction, is associated with premature aging. Thus, the experimental creation of DSBs in regulatory DNA from young adult mice, rapidly changed their epigenome; a change that was associated with initiation and acceleration of aging. Thus, the authors proposed that their findings did not support the generally held damage-based theories that aging is a random, multifactorial process, but instead that it is non-random and “potentially driven by a reproducible and predictable” mechanism.
Further evidence of DSB involvement in aging
Mice that were mutated in Ku70 or Ku80 proteins which are involved in repair of DSBs, displayed an early aging phenotype [112,126]. In this model, as well as in the one previously referenced, treatment by one factor alone initiated aging, caused it to accelerate, and can therefore be considered a single cause of aging.
The causal role of DNA damage in aging was further verified in two mouse models with “clean” DSBs that resulted from expression of a tetracycline-controlled SacI restriction enzyme or a I-PpoI restriction enzyme. Both models separately displayed symptoms of premature aging [101,128,129].
Finally, altering proper DSB processing and repair through use of non-specific, radiation-induced lesions in rodents and humans, as well as treatments with high doses of anti-cancer agents or by DSB enzymatic induction [50,133] can independently cause aging. These observations suggest that unlike other DNA damage, DSBs may be capable of driving multiple age-related phenotypes, as part of the aging mechanism. DSBs also promote epigenetic drift by triggering signals that recruit epigenetic regulators away from gene promoters to the DNA break site [130] and are consistently linked with accelerated aging if defectively repaired [131,132]. In contrast, efficient repair of DSBs or slowing of the rate of their damage was highly correlated with longer lifespan [133].
To summarize this brief review, DSBs and associated epigenetic changes that are directly related to expression of the regulatory mechanism for oversight of various life stages, e.g., ontogenesis, morphostasis and morpholysis, are different from those associated with somatic maintenance and function, especially during aging.
The proposed cause of aging is progressive degradation of the redundant developmental regulatory mechanism due to mis-repaired molecular damage [1] which causes loss of regulatory redundancy that mandates initiation of aging unto death.
The Theory
Aging results from a mechanism that evolved by means of NS to ensure reproductive success in humans and other mammals that nurture their young. In these animals, fecundity emerges during the developmental program, which ends when human beings are about 20 years old. However, reproductive fitness doesn’t occur until almost a decade later when progeny become independent of parental care. Since survival without descent is evolutionarily meaningless, parents must survive long enough to complete the required reproductive stage of nurture that makes their offspring capable of independence. To keep aging at bay until reproductive success is achieved, the somatic regulatory mechanism of the last DP stage is redundantly expressed to direct further parental development unto peak physical and functional vitality. The last reproductive stage of offspring nurturing is completed in humans as parents’ approach 30 years of age. At that point, regulatory redundancy is lost sufficiently to end the non-aging phase of morphostasis and initiate aging. This dynamic represents the single cause of aging, which stands in contrast to the popular notion that it is multifactorial.
Developmental regulatory redundancy and morphostasis are emergent properties of ontogenesis that cannot persist indefinitely because of reliability limits, stochastic mutations and reproduction-related damages. Thus, progressive loss of regulatory redundancy is an emergent property of morphostasis that causes aging. Physiological dysregulation resulting from loss of redundancy erodes homeostasis, causing aging to proceed exponentially.
Since reproductive fitness and aging are inseparably bonded by a single mechanism that is a product of natural selection, aging is adaptive in compliance with Darwin’s theory and consistent with his opinion that it provides a hidden, compensating benefit. The mechanism proposed in this theory explains the origin of individual as well as population aging thereby foregoing the need to explore questionable methods of group selection to explain the latter. Furthermore, because aging is inseparably linked with reproductive fitness, emergence of a hypothetical “Darwinian demon” [134] is impossible. Paradoxically, because its origins are directly linked with the DP, aging could be considered programmed.
Evolutionary perspective: Origin of human aging by means of natural selection
All populations, including those that breed slowly will increase in size if no limitations are imposed on their growth by nature. Those constraints take various forms in different populations, often including limited food supply, nesting sites, predators, disease, adverse climactic conditions and many more. Additionally, for sexual populations, mating opportunities are not available to all since candidates for copulation require “beneficial traits” to increase their chances of being “accepted”. Those differing, heritable traits bring variation to populations providing them the means to adapt to changing environmental conditions. Thus, through the process of NS, variations conducive to the survival and reproduction of individuals ensure continuation of their species consistent with the struggle for existence. Such “selection” by nature prevents geometric population increases that would occur in the absence of those checks.
Aging is not one of the limitations on population growth since most animals living in nature often die before reaching maturity due to one or more of the restrictive environmental factors. This may be the reason that a chapter on the evolution of aging is not included in Darwin’s classic book, “On the origin of species by means of Natural Selection”, which is focused upon survival and preservation of life in viable, reproductively competent organisms. A search of the literature reveals that the author had greater interest in the controversy over creation of life than in the process of its termination [56]. It seems that his only interest in death was that it generated variability thereby facilitating adaptation. It is obvious that the mechanism of aging would have held little interest to him at the time of his research.
Nonetheless, at the time when “Origin” was published, Darwin was asked how his theory of natural selection could explain the evolution of aging, to which he responded that it must provide some hidden, compensating benefit that makes it adaptive. This inability to provide an explanatory theory for the functional relationship between natural selection and aging was described by Goldsmith [6] as another of Darwin’s Dilemmas [135].
Following his life, Darwin's theory of evolution has been bolstered by many scientific advances including expanded fossil records, discovery of DNA, genetic replication processes, genome analysis, application of radioactive decay methods to geologic time dating, and observations of natural selection in the wild and in laboratories. But of course, there is no fossil record for aging nor current methods to specifically explain the process by which it evolved, leaving biogerontologists the only option of applying theory to provide tentative answers.
Senescence is the decrease in an organism’s ability to reproduce and the increase in the chances it will die as it ages. Explaining why senescence evolved at all has long been an issue for evolutionary theory, because any decrease in an organism’s ability to reproduce should be selected against, and senescence which occurs in all organisms and always results in death, completely ends reproductive potential.
Any theory intended to convincingly argue that aging evolved, must be of an explanatory type that has the potential to be tested since aging is explicable if not definitively explainable. As stressed by Lennox explicable, which means unambiguous, familiar or knowable, differs from explainable, wherein the question of cause or reason for being is deemed to be soluble. This theoretical approach was used by Darwin who performed thought experiments to determine the explanatory potential of NS, as opposed to providing direct supporting evidence of it [136].
There is scientific consensus that aging is non-adaptive for individual organisms and adaptive for populations. This dichotomy requires separate explanations of aging’s origin, which seems patently untenable and is a conundrum since populations are the sum of individuals. So, assuming that the aging mechanism is the same for individuals and populations, then how is it explained?
Presumably, nothing can evolve without the effect of NS, so if a trait such as aging wasn’t selected, we are left with the currently popular explanation of its evolution pursuant to the classic theory. It proposes that loss of NS strength allows accumulation of somatic damages characteristic of aging. Accordingly, aging originates and progresses as random damage resulting from neglect by NS accumulates. This popular concept is inconsistent with Darwin’s theory of evolution by NS. Alternatively, the current theory explains the evolution of aging as an inextricable part of an emergent property that evolved from the DP to ensure reproductive success and variability that is essential for evolution.
In an expansive discussion, Razeto-Barry & Frick [137] distinguished between the creative and non-creative views of NS. Per the non-creative view, NS merely eliminates traits while doing nothing to create new ones; the latter phenomenon is the result of mutation typical of MA and AP theories of aging.
In contrast, the creative view as presented in the current theory, sees NS as a force that makes probable combinations of mutations that are necessary for the development of at least some traits. While Razeto-Barry and Frick grant that NS cannot explain the origin of traits that arise by a single mutation, they argue that it can explain the occurrence of sequences of phenotypic changes that would otherwise be wildly unlikely to occur without selection causing the spread of changes prior to reaching the final one in the sequence [137]. This argument is relevant to the current theory wherein a sequence of events involving developmental regulatory redundancy that occurs upon completion of ontogenesis and is lost during morphostasis, promotes reproductive success and subsequently, aging.
Some NS philosophers have turned their attention to “mechanisms…to explain how a phenomenon comes about or how some significant process works” [138]. Individual parts/entities or activities/interactions that allow selection are specified by their unique roles in the selective process, whereas a mechanistic explanation works by associating those roles with specific phenomena [139]. In the case of human aging, the roles (redundancy of developmental regulatory processes and loss of redundancy) are associated with the specific phenomena of reproductive fitness and aging, respectively. Homeostasis is temporarily maintained through maintenance of a non-aging state by redundancy of the developmental regulatory mechanism during the post-ontogenetic stage of morphostasis. This stage is adaptive and thus selectable because it sustains a non-aging environment where final development of the parental generation can occur sufficiently to nurture offspring and achieve reproductive fitness. It also provides for somatic maintenance. But the number of functional regulatory units is finite so that redundancy once achieved is progressively lost due to physical and biochemical factors. Since regulatory redundancy temporarily sustains a non-aging state, the stability of morphostasis is progressively lost. This perturbation initiates aging from a previously non-aging state, weakens strength of NS, and erodes general somatic maintenance.
The current theory explains that aging is an essential part of such a mechanism involving regulatory redundancy, an emergent property of ontogenesis. It describes regulatory redundancy as an “unmaintainable” property that directs reproductive success during morphostasis, the non-aging phase of development. However, since regulatory redundancy is “unmaintainable”, it degrades thereafter initiating and driving aging. Thus, one way to secure explanatory status for selection of aging is to propose that it functions as part of a mechanism that is inextricably linked with reproduction.
To further the preceding explanation, aging may evolve by specific selection of reproduction that is inseparably bound to aging by a single damage component that is common to both. Abrams and Ludwig [140] assume a direct trade-off between mortality and fertility is mediated via a common ‘damage’ parameter. This concept is consistent with the current theory that aging is a product of NS, but not as a tradeoff. For example, if an unsustainable parameter that inseparably links reproductive success with aging is selected because it promotes fitness then the shared “damage” parameter explains the hidden, compensating benefit of aging as opined by Darwin.
Barros [141] argued that natural selection may be characterized as a two-level mechanism, with a population-level and an individual-level working together. That requirement is fulfilled in the current theory wherein individual and population aging emerge from a common mechanism that cannot be separated. The current theory proposes a mechanism that consists of two levels with each level working together. Since aging is the same whether occurring in one or more organisms its cause affecting individuals will similarly affect a population of those same individuals. Thus, the single mechanism of aging that involves linkage of development with aging through a common process explains individual and population aging thereby eliminating the need for another explanatory theory involving group selection for populations.
Multiple bouts of Darwin’s process involving different traits, acting sequentially or in concert, may then explain how the evolution of complex adaptations occur through the gradual evolution (change over time) of natural populations. This concept is relevant to aging which presumably is not a purposeful target of evolution but evolves by natural selection of the mechanism to which it is an inseparable part.
Aging is not selected per se. However, the mechanism to ensure reproductive success/peak fitness which mandates aging is selected. Thus, the mechanism is an essential component of fitness, but it also mandates aging, which comes as a hidden benefit (for evolution) in compliance with Darwinian law of NS
The mechanism of natural selection in this case overrides the need for selection of aging per se since it can’t occur de novo by evolution without its developmental component. The unifying mechanism makes fitness and aging inseparable. Thus, selection of reproductive success is also selection of aging. This is the root of the current theory
Is aging programmed?
Programmed death theory that originated nearly two centuries ago, [142] still has advocates, but their opinions are not widely shared. The concept that there is a limit to human survival based upon programmed control of death by aging is largely rejected by the scientific community. [143,144,145]. The reason for rejecting programmed death theory is that when aging is viewed as a separate trait, it has no individual benefit, thus being incapable of NS based upon Darwinian mechanics of evolution. As a result, some authors justify aging programs based upon the value of death to evolvability and demographics rather than to individual benefit [7,9,146,147,148,149].
These and other observations have suggested to some biologists that our understanding of aging is being constrained by restrictive evolutionary paradigms [146].
Mitteldorf proposed that several computational models have been created, but “evolution of an aging program requires group selection on a scale that goes beyond the theory of multilevel selection, a perspective that is already controversial”. So, he stated that the question of whether plausible models exist that can account for aging as a group-selected adaptation is central to our understanding of what aging is, where it comes from and, importantly, how “anti-aging medicine might most propitiously be pursued”. Kowald and Kirkwood reviewed computational models that evolve aging as an adaptation [143] and found fault with each of them. So based on theory alone, they endorsed the standing convention that aging must be understood in terms of trade-off models This author disagrees as has been previously argued in his discussion of tradeoffs, unmaintainablity and aging.
Paradoxically, the current theory was not created to defend an evolutionary basis for programmed aging, but upon greater scrutiny and interpretation of its ramifications, it could possibly provide more substantive support for the concept than it has ever received. Although this novel theory proposes that reproductive success is its primary objective, it makes the unique claim that a mechanism which evolved to facilitate reproduction is inseparable from that which causes aging thereafter. It is ironic that a program that evolved to enhance life through organismal development and reproduction will also cause aging and death. Because the developmental program is universally recognized as “a cornerstone of modern biology within which a purposeful genetic program drives all biological processes that occur from conception to reproductive maturation” [55] it can also rightly be considered an aging program because it always causes death, albeit presumably not purposefully. Thus, the developmental program can also be considered an aging program since development and aging are continuous functions that cause construction of new organisms while making their deconstruction inevitable, respectively. The reason theoreticians have had difficulty in providing a valid explanation for aging programs is because they do not begin their thinking with development, and thus can’t argue a specific cause for the evolution of aging.
Discussion and Conclusions
Aging occurs due to natural degenerative processes acting upon an emergent property of ontogenesis that ensures reproductive fitness. Since that redundant regulatory mechanism of the last ontogenetic stage preserves a non-aging somatic condition during acquisition of reproductive fitness, its progressive loss of supportive redundant regulatory units due to reproductive, physical (reliability theory) and biochemical (stochastic DSB DNA damage and apoptosis) damage requires loss of the transient, non-aging stage of morphostasis. Predictably loss of regulatory redundancy causes aging, an emergent property of morphostasis. Physiological dysregulation caused by decay of emergent properties such as redundant regulatory integrity has previously been suggested as a cause of aging [65].
Thus, if the assumption of the current theory is valid in its description of a mechanism for reproductive success that is inseparable from aging, then it is reasonable to assume that the mechanism alone was a product of NS. It is also possible that because loss of redundancy occurs during the latter half of the post-ontogenetic decade, when the strength of NS is still sufficiently strong to cause negative selection of the aging mechanism, it does not. If true, then the mechanism for aging was selected. Then it becomes questionable whether or not the emergent property of ontogenesis represents part of an evolved program to ensure not only reproductive success but aging and death as well. If so, then the theory not only resolves Darwin’s dilemma, but it also brings credibility to the programmed/adaptive theories of aging.
This process that is essential for resolving Darwin’s dilemma was inadvertently absent from my original version of “A mechanistic theory of development-aging continuity in humans and other mammals” [1], because the highly relevant findings of Gaimo and Trualeson [2] had not yet been published. That significant report showing that aging causes NS to decline, not vice versa, opened the horizons to alternative theories.
The historical significance of this current theory is that is resolves Darwin’s dilemma by showing that while aging may not be adaptive per se it provides hidden, compensating benefits, that justifies selection of the mechanism by which it is inextricably bonded to reproduction.
In conclusion, Darwin’s confidence in the validity of his Theory of NS was well founded!
Future directions
Gradual physiological dysregulation results in decay of homeostasis, a characteristic of aging. The cause as proposed in the current theory is loss of developmental regulatory redundancy, an emergent property of late morphostasis that continues during aging. While the emergent, processes may not be easily understood through clear metabolic pathways, it is possible that they can be precisely quantified and studied with small numbers of biomarkers [65,66].
Although regional epigenetic landscapes differentially affect expression of the regulatory mechanism, its genetic construct is the same throughout the soma. Thus, despite inevitable epigenetic modification, it may be possible to identify regulatory DNA that affects aging using redundancy of sequence to test the validity of the current theory. If so, future findings may provide a novel approach for application of aging research in medicine [144,146].
Author Contributions
RF Walker is the sole contributor and author of this Review/Theory
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
The author acknowledges Drs. S. Giaimo and A. Traulsen whose publication https://doi.org/10.1038/s41467-022-28254-3 was key to his understanding the origin of aging by natural selection.
Conflicts of Interest
The author declares no conflict of interest.
References
- Walker, R.F. A mechanistic theory of developmental aging continuity in humans and other mammals. Cells 2022, 119, 917. [Google Scholar] [CrossRef] [PubMed]
- Giaimo, S.; Traulsen, A. The selection force weakens with age because ageing evolves and not vice versa. Nat. Commun. 2022, 13, 686. [Google Scholar] [CrossRef]
- Gould, SJ. 1981. citing Darwin, Charles (1871). The Descent of Man, and Selection in Relation to Sex. pp. 152–153.
- Darwin, C. (1859) On the Origin of Species by Means of Natural Selection, John Murray (Pub), Albemarle St. London, pp 502.
- Thomson, K S. Marginalia: The meanings of evolution. American Scientist 1982, 70, 529–531. Bibcode:1982 AmSci.70..529T. JSTOR 27851662.
- Goldsmith, T.C. Darwin’ Dilemma, Chap.2. In The Evolution of Aging, 3rd ed.; Azinet Press Annapolis, Maryland, 2014, pp 31-33.
- Mitteldorf, J. Aging is a Group Selected Adaptation: Theory, Evidence, and Medical Implications. CRC Press 2016, 31. [Google Scholar]
- Okasha, S: (2015) Kin selection, group selection and altruism: a controversy without end?, Oxford University Press https://blog.oup.com/2015/01/kin-group-selection-controversy/. 2015.
- Mitteldorf, J.J. Demographic Evidence for Adaptive Theories of Aging. Biochemistry (Moscow), 2012, 77, 726–728. [CrossRef]
- Singer, MA. The Origins of Aging: Evidence that Aging is an Adaptive Phenotype. Curr Aging Sci. 2016, 9(2), 95–115. [Google Scholar] [CrossRef] [PubMed]
- Comfort, A. Biological aspects of senescence. BioI. Rev. 1954, 29, 284–329. [Google Scholar] [CrossRef]
- Gregory, T.R. Understanding Natural Selection: Essential Concepts and Common Misconceptions. Evo. Edu. Outreach 2009, 2, 156–175. [Google Scholar] [CrossRef]
- O’Conner, T. “Emergent Properties”, The Stanford Encyclopedia of Philosophy (Winter 2021 Edition), Edward N. Zalta (ed.), URL = https://plato.stanford.edu/archives/win.2021/entries/properties-emergent/.
- Duret, L. Neutral theory: The null hypothesis of molecular evolution. Nature Education 2008, 1:218.
- Libertini, G. Classification of phenoptotic phenomena. Biochemistry 2012, 77, 707–710. [Google Scholar] [CrossRef]
- Cohen, A. Aging across the tree of life: The importance of a comparative perspective for the use of animal models in aging, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, Volume 1864, Issue 9, Part A, 2018, Pages 2680-2689. [CrossRef]
- Fisher, R.A. The genetical theory of Natural Selection. 1930 Oxford University Press, Oxford:UK.
- Charlesworth, B. Fisher, Medawar, Hamilton and the Evolution of Aging. Genetics 2000, 156, 927–931. [Google Scholar] [CrossRef]
- Medawar, P.B. Medawar, P.B. An Unsolved Problem of Biology. (1952) H. K. Lewis, London. (Reprinted in Medawar, P.B. (1981) The Uniqueness of the Individual. 2nd Revised Edition, Dover, New York, 28-54.
- Williams, G. Pleiotropy, natural selection, and the evolution of senescence. Evolution 1957, 11, 398–411. [Google Scholar] [CrossRef]
- Hamilton, W.D. The moulding of senescence by natural selection. J. Theor. Biol. 1966, 12, 12–45. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L. Evolution of ageing. Nature 1977, 270, 301–304. [Google Scholar] [CrossRef]
- Kirkwood, T.B.; Holliday, R. The evolution of ageing and longevity. Proc. R. Soc. Lond. B Biol. Sci. 1979, 205, 531–546. [Google Scholar] [CrossRef]
- Charlesworth, B. Causes of natural variation in fitness: Evidence from studies of Drosophila populations. PNAS 2015, 112, 1662–1669. [Google Scholar] [CrossRef]
- Rose, M.R.; Rauser, C.L.; Benford, G.; Matos, M.; Mueller, L.D. Hamilton’s forces of NS after forty years. Evolution 2007, 61, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Maxim, N.; Shokhirev, B. Revamping the evolutionary theories of aging. Ageing Research Reviews, 2019; 55, 100947. [Google Scholar] [CrossRef]
- Roper, R.; Capdevila, P.; Salguero-Gómez, R. Senescence: Still an Unsolved Problem of Biology, 2019. [CrossRef]
- Ayala, F.J. Darwin's greatest discovery: Design without designer. PNAS 2007, 104 (suppl_1), 8567-8573. [CrossRef]
- AYALA, F.J. (2007) Darwin’s Greatest Discovery: Design Without Designer. In: National Academy of Sciences; Avise JC, Ayala FJ, editors. In the Light of Evolution: Volume I: Adaptation and Complex Design. Washington (DC): National Academies Press (US); 2007. 1. Available from: https://www.ncbi.nlm.nih.gov/books/NBK254313/.
- Charlesworth, B. Evolution in Age-Structured Populations.(2001) 2nd edn (Cambridge Univ. Press, 1994).
- Baudisch, A. Hamilton’s indicators of the force of selection. Proc. Natl Acad. Sci. USA 2005, 102, 8263–8268. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell, 2013, 153, 1194–121.
- Jin, K. . Modern Biological Theories of Aging. Aging and Disease. 2010, 1, 72–74. PMC 2995895.
- Liochev, S. Which Is the Most Significant Cause of Aging? Antioxidants, 4 (4), 2010, 793–810. [CrossRef]
- Bowles, J. Shattered: Medawar's test tubes and their enduring legacy of chaos. Med Hypotheses 2000, 54, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Pincheira-Donoso, D.; Hunt, J. Fecundity selection theory: concepts and evidence Biological Reviews 2015. [CrossRef]
- Lee, R.D. Rethinking the evolutionary theory of aging: Transfers, not births, shape senescence in social species. PNAS 2003, 100 9637-9642. [CrossRef]
- Zietsch, B.P.; Westberg, L.; Santtila, P.; Jern, P. Genetic analysis of human extrapair mating: heritability, between-sex correlation, and receptor genes for vasopressin and oxytocin. Evolution and Human Behavior 2015, 36, 2,, 130-136. doi.org/10.1016/j.evolhumbehav.2014.10.
- Smarr, M.M.; Sapra, K.J.; Gemmill, A.; Kahn, L.G.; Wise, L.A.; Lynch, C.D.; Factor-Litvak, P.; Mumford, S.L.; Skakkebaek, N.E.; Slama, R.; et al. Is human fecundity changing? A discussion of research and data gaps precluding us from having an answer. Hum Reprod. 2017, 1;32(3),499-504. [CrossRef]
- Gompertz, B. "On the Nature of the Function Expressive of the Law of Human Mortality, and on a New Mode of Determining the Value of Life Contingencies". Philosophical Transactions of the Royal Society 1825, 115, 513–585. [Google Scholar] [CrossRef]
- Makeham, W.M. On the Law of Mortality and the Construction of Annuity Tables. J. Inst. Actuaries and Assur. Mag. 1860, 8, 301–310. [Google Scholar] [CrossRef]
- Buck LGM.(2011) Fecundity and fertility. In: Buck Louis GM, Platt RW (eds). Reproductive and Perinatal Epidemiology. New York: Oxford University Press, 2011, 16 – 61.
- Kirkwood, T. (2017) The Disposable Soma Theory: Origins and Evolution. In: Shefferson RP, Jones OR, Salguero-Gómez R., editors. The Evolution of Senescence in the Tree of Life. Cambridge University Press; Cambridge: 2017.
- Kirkwood, T.B.L. Understanding ageing from an evolutionary perspective. J Int Med. 2008, 263, 2. [Google Scholar] [CrossRef]
- Quetelet, (1835) A Treatise on Man and the Development of His Faculties, William and Robert Chambers, Edinburgh, UK, 1835.
- Olshansky S., J.; Carnes, B.A. “Ever since Gompertz,” Demography, 1997, 34, 1, 1–15.
- J. E. Birren and J. J. F. Schroots, (2001) “The history of geropsychology,” in Handbook of the Psychology of Aging, J.E. Birren and K. W. Schaie, Eds., pp. 3–28, Academic Press, San Diego, Calif, USA, 5th edition, 2001.
- Schroots, J.J. On the dynamics of active aging. Curr Gerontol Geriatr Res 2012, 818564. [Google Scholar] [CrossRef]
- Anderson, JE. (1964) Developmental principles in childhood and maturity. In: Birren JE, editor. Relations of Development and Aging. New York, NY, USA: Arno Press; pp. 11–28.
- Yates, F.E.,; Benton, L.A.,; Rosen, R. Biological senescence: Loss of integration and resilience, Canadian Journal on Aging 1995, 14, (1), 106–130.
- Sehl, M.E.; Yates, F.E. Kinetics of human aging: I. Rates of senescence between ages 30 and 70 years in healthy people, J Gerontol. A 2001, 56, (5), B198– B208.
- Schroots, J.J.F. “On growing, formative change and aging,” in Emergent Theories of Aging, J.E. Birren and V. L. Bengtson, Eds., pp. 299–329, Springer, New York, NY, USA, 1988.
- J. J. F. Schroots and F. E. Yates, (1999) “On the dynamics of development and aging,” in Handbook of Theories of Aging, V.L. Bengtson and K. W. Schaie, Eds., pp. 417–433, Springer, New York, NY, USA.
- Hayflick, L. Biological Aging Is no Longer an Unsolved Problem. Annals of the New York Academy of Sciences. 2007, 1100, 1–13. [Google Scholar] [CrossRef] [PubMed]
- de Magalhães, J.P. Ageing as a software design flaw. Genome Biol. 2023, 24, 51. [Google Scholar] [CrossRef]
- de Magalhães, J.P. Programmatic features of aging originating in development: aging mechanisms beyond molecular damage? FASEB J. 2012; 26. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Aging is not programmed Cell Cycle. 2013, 12, 3736–3742. [CrossRef]
- Blagosklonny, M.V. Aging and Immortality: Quasi-Programmed Senescence and Its Pharmacologic Inhibition. Cell Cycle, 2006, 5, 2087–2102. [CrossRef]
- Kosova, G.; Abney, M.; Ober, C. Heritability of reproductive fitness traits in a human population. Proceedings of the National Academy of Sciences 2010, 107, 1772–1778. National Acad Sciences.
- de Magalhães JP,; Church GM. Genomes optimize reproduction: aging as a consequence of the developmental program. Physiology (Bethesda). 2005, 252-259. [CrossRef] [PubMed]
- Porpora, D. Porpora, D. (2015) Why Don’t Things Change? The Matter of Morphostasis. https://doi.org/10.1007/978-3-319-13773-5_9 In: Generative Mechanisms Transforming the Social Order. [CrossRef]
- Boundless Psychology. (2016). Physical development in adulthood. https://www.boundless.com/psychology/textbooks/boundless-psychology-textbook/human-development-14/early-and-middle-adulthood-74/physical-development-in-adulthood-287-12822/.
- Cabej, N. {2012} Ontogeny: The Workshop of Evolutionary Change, Editor(s):, Epigenetic Principles of Evolution, Elsevier, Pages 307-326. [CrossRef]
- Cohen, AA. Complex systems dynamics in aging: new evidence, continuing questions. Biogerontology. 2016, 17, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Kriete, A. Robustness and Aging-A Systems-Level Perspective. Biosystems 2013, 112, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Howe, J. Emergent Systems Are Changing the Way We Think 2017 https://www.aspeninstitute.org>blog-posts>emergent-S.
- DEFINED IN WIKTIONARY https://en.wiktionary.org/wiki/morphostasis.
- Garland, T Jr. Trade-offs. Current Biology 2014, 24, R60-R61.
- Garland, T. Jr.; Downs, C.J.; Ives, A.R. Trade-Offs (and Constraints) in Organismal Biology. Physiological and Biochemical Zoology 2022, 95, 82–112.
- Roff, D.A.; Fairbairn, D.J. The evolution of trade-offs: where are we? J. Evol. Biol. 2007, 20, 433–447. [Google Scholar] [CrossRef]
- Flatt, T. Life-history evolution and the genetics of fitness components in Drosophila melanogaster. Genetics 2020, 214, 3–48. [Google Scholar] [CrossRef]
- Zera, A.J.; Harshman, L.G. The physiology of life history trade-offs in animals. Annu. Rev. Ecol. Syst. 2001, 32, 95–126. [Google Scholar] [CrossRef]
- Saggere, R.M.S.; Lee, C.W.J.; Chan, I.C.W.; Durnford, D.G.; Nedelcu, A.M. A life-history trade-off gene with antagonistic pleiotropic effects on reproduction and survival in limiting environments. Proc. Roy. Soc. 2022. [Google Scholar] [CrossRef]
- Sotola, L. (2016). Disposable Soma Theory. In: Weekes-Shackelford, V., Shackelford, T., Weekes-Shackelford, V. (eds) Encyclopedia of Evolutionary Psychological Science. Springer, Cham. [CrossRef]
- Kirkwood, T.B.; Rose, M.R. Evolution of senescence: late survival sacrificed for reproduction. Philos. Trans. R Soc. Lond. B Biol Sci. 1991, 332, 15–24. [Google Scholar] [CrossRef]
- Rodrigues, M.A.; Flatt, T. Endocrine uncoupling of the trade-off between reproduction and somatic maintenance in eusocial insects. Current Opinion in Insect Science, 2016, 16, 1–8. [CrossRef]
- Lemaitre, J.F.; Berger, V.; Bonenfant,C.; Douhard, M.; Gamelon, M.; Plard, F.; Gaillard, J.M. Early-late life trade-offs and the evolution of ageing in the wild. Proceedings of the Royal Society B: Biological Sciences 2015, 282, 20150209. [CrossRef]
- Hughes, K.A.; Leips, J. Pleiotropy, constraint, and modularity in the evolution of life histories: insights from genomic analyses. Ann. NY Acad. Sci. 2017, 1389, 76–91. [Google Scholar] [CrossRef]
- Stefannacci, R.G. Changes in the body with aging. Overview of Aging, Merck Manual professional version 2022.
- Billard, B.; Vigne, P.; Braendle, C. A natural mutational event uncovers a life history trade-off via hormonal pleiotropy. Curr. Biol. 2020, 30, 4142-4154.e9. [CrossRef]
- Maklakov, A.A.; Chapman, T. Evolution of ageing as a tangle of trade-offs: energy versus function. Proc Biol Sci. 2019, 286, 20191604. [Google Scholar] [CrossRef]
- Wensink, M.J.; Cohen, A.A. The Danaid Theory of Aging. Front. Cell Dev. Biol. 2022, 9, 671208. [Google Scholar] [CrossRef] [PubMed]
- Bourrat, P.; Doulcier, G.; Rose, C.J.; Rainey, P.B.; Hammerschmidt, K. Tradeoff breaking as a model of evolutionary transitions in individuality and limits of the fitness-decoupling metaphor. eLife, 2022, 11, e73715. [CrossRef]
- Ferrucci, L. An Exciting Thought. J Gerontol Ser A. 2005, 60, 56. [Google Scholar] [CrossRef]
- Finch, C.E.; Crimmins, E.M. Inflammatory exposure and historical changes in human life-spans. Science. 2004, 305(5691), 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Robine, J.M. 2011 Age patterns of adult mortality. In: Rogers R, Crimmins E, editors. International Handbook of Adult Mortality. Springer; Dordrecht, The Netherlands: pp. 207–226.
- Beltrán-Sáncheza, H.; Crimmins, E.; Finch, C. Early cohort mortality predicts the rate of aging in the cohort: A historical analysis. J Dev Orig Health Dis. 2012, 3, 380–386. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L. Deciphering death: a commentary on Gompertz (1825) ‘On the nature of the function expressive of the law of human mortality, and on a new mode of determining the value of life contingencies’ Philos Trans R Soc Lond B Biol Sci. 2015, 370, 20140379. [CrossRef]
- Gavrilov, L.A.; Gavrilova, N.S. The Reliability Theory of Aging and Longevity. Journal of Theoretical Biology 2001, 213, 527–545. [Google Scholar] [CrossRef] [PubMed]
- Mace, R. University College London. "Increased Life Expectancy May Mean Lower Fertility." ScienceDaily. ScienceDaily, 17 February.
- Ryan, C.P.; Hayes, M.G.; Lee, N.R.; McDade, T.W.; Jones, M.J.; Kobor, M.S.; Kuzawa, C.W.; Eisenberg, D.T.A. Reproduction predicts shorter telomeres and epigenetic age acceleration among young adult women. Sci Rep. 2018, 8, 11100. [Google Scholar] [CrossRef]
- Descamps, S.; Boutin, S.; Berteaux, D.; Gaillard, J.M. "Best squirrels trade a long life for an early reproduction". Proc Biol Sci. 2006, 273, 2369–2374. [Google Scholar] [CrossRef]
- Westendorp, R.G.; Kirkwood, T.B. Human longevity at the cost of reproductive success. Nature. 1998, 396, 743–746. [Google Scholar] [CrossRef]
- Hansen, M.; Flatt, T.; Aguilaniu, H. "Reproduction, fat metabolism, and life span: what is the connection?" Cell Metab. 2013, 17, 10–19. [CrossRef]
- Blagosklonny, M.V. Why the disposable soma theory cannot explain why women live longer and why we age. Aging. 2010, 2, 884–887. [Google Scholar] [CrossRef]
- Gavrilov, L.A.; Gavrilova, N.S. 1991. section 5.6. “The limit of cell division: the key to the mechanism which determines life span?” pp 211-224 The Biology of Life Span: A Quantitative Approach. New York: Harwood Academic Publisher 385p.
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: linking aging to chronic disease. Cell. 2014, 159:709–713. [CrossRef]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The central role of DNA damage in the ageing process. Nature, 2021, 592, 695– 703.
- Alexander, P. The role of DNA lesions in the processes leading to aging in mice. Symp Soc Exp Biol. 1967, 21, 29–50. [Google Scholar] [PubMed]
- Gensler, H.L.; Bernstein, H. "DNA damage as the primary cause of aging". Q Rev Biol. 1981, 56, 279–303. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Molecular Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Köhler, K.; Ferreira, P.; Pfander, B.; Boos, D. The Initiation of DNA Replication in Eukaryotes. Springer, Cham. 2016, 443–460. [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006, 12, 440–50. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, J.; Palander, O.; Seifried, L.A.; Dick, F.A. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Molecular and Cellular Biology. 2012, 32: 900–12. [CrossRef]
- Kaina, B. DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem Pharmacol. 2003, 66, 1547–54. [Google Scholar] [CrossRef]
- Müllauer, L.; Gruber, P.; Sebinger, D.; Buch, J. ; Wohlfart, S,; Chott, A. Mutations in apoptosis genes: a pathogenetic factor for human disease. Mutat Res. 2001, 488, 211–31. [CrossRef]
- Kumari, R.; Parmjit, J. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype Frontiers in Cell and Developmental Biology, 2021, 9. [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010, 5 :99-118. [CrossRef]
- Vijg, J. From DNA damage to mutations: All roads lead to aging. Ageing Research Reviews, 2021, 68, 101316.
- Zhang,Y.; Herman, B. Ageing and apoptosis, Mechanisms of Ageing and Development, 2002, 123, 4, 245-260. [CrossRef]
- Gredilla, R.; Garm, C.; Stevnsner, T. "Nuclear and Mitochondrial DNA Repair in Selected Eukaryotic Aging Model Systems", Oxidative Medicine and Cellular Longevity. 2012, Article ID 282438, 12 pages. [CrossRef]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell, 2013, 155, 1104–1118. [CrossRef]
- Demaria, M.; Ohtani, N. ; Youssef, Sameh A.; Rodier, F.; Toussaint, W.; Mitchell, James R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, Martijn E. T.; Hoeijmakers, Jan H. J.; de Bruin, A.; Hara, E.; Campisi, J. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Developmental Cell 2014, 31; 722–733. [CrossRef]
- Nussenzweig, A.; Nussenzweig, M.C. Origin of chromosomal translocations in lymphoid cancer. Cell 2010, 41; 27-38.
- Toyoshima-Sasatani, M.; Imura, F.; Hamatake, Y.; Fukunaga, A.; Negishi, T. Mutation and apoptosis are well-coordinated for protecting against DNA damage-inducing toxicity in Drosophila. Genes Environ. 2023, 23;45(1), 11. [CrossRef]
- Samanta, S.; Rajasingh,S.; Cao,T.; Dawn,B.; Rajasingh, J. Epigenetic Dysfunctional Diseases and Therapy for Infection and Inflammation. Biochim Biophys Acta. 2017, 1863, 518–528. [CrossRef]
- Cournil, A.; Kirkwood, B. If You Would Live Long, Choose Your Parents Well. Trends in Genetics 2001, 17, 233–235. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.-H.; Chen, C.-C.; Li, W.; Tyler, J.K. Nucleosome Loss Leads to Global Transcriptional Up-Regulation and Genomic Instability during Yeast Aging. Genes & Development 2014, 28, 396–408. [CrossRef]
- Tsurumi, A.; Li, W. Global Heterochromatin Loss. Epigenetics 2012, 7, 680–688. [Google Scholar] [CrossRef]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the Genome Integrity Network. Nature reviews. Genetics 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Hagan, C. When are mice considered old? Jackson Lab Blog Post, Nov 06, 2017.
- Kim, J.; Sturgill, D.; Tran, A.D.; Sinclair, D.A.; Oberdoerffer, P. Controlled DNA Double-Strand Break Induction in Mice Reveals Post-Damage Transcriptome Stability. Nucleic Acids Research 2015, 44, e64–e64. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Yang, J.-H.; Bonkowski, M.S.; Amorim, J.A.; Ross, J.M.; Coppotelli, G.; Griffin, P.T.; Chew, Y.C.; Guo, W.; Yang, X.; et al. DNA Break-Induced Epigenetic Drift as a Cause of Mammalian Aging. bioRxiv 808659 2019. [CrossRef]
- Hainer, S.J.; Martens, J.A. Transcription of ncDNA. Transcription 2011, 2, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Li, H. ; Vogel, H,; Holcomb, V.B.; Gu, Y.; Hasty, P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer, Molecular and Cellular Biology 2007, 27, no. 23, pp. 8205–8214,.
- Kim, J.; Sturgill, D.; Tran, A.D.; Sinclair, D.A.; Oberdoerffer, P. Controlled DNA Double-Strand Break Induction in Mice Reveals Post-Damage Transcriptome Stability. Nucleic Acids Research 2015, 44, e64–e64. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Milholland, B.; de Bruin, A.; Curran, S.; Laberge, R.M.; van Steeg, H.; Campisi, J.; Maslov, A.Y.; Vijg, J. Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nature Communications 2015, 6, 6790. [Google Scholar] [CrossRef] [PubMed]
- Kane, A.E.; Sinclair, D.A. Epigenetic Changes during Aging and Their Reprogramming Potential. Critical reviews in biochemistry and molecular biology 2019, 54, 61–83. [Google Scholar] [CrossRef]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA Double-Strand Breaks: Production, Fidelity of Repair, and Induction of Cancer. Proc. Nat. Acad. Sci. 2003, 100, 12871–12876. [Google Scholar] [CrossRef]
- Harkema, L.; Youssef, S.A.; de Bruin, A. Pathology of Mouse Models of Accelerated Aging. Veterinary Pathology 2016, 53, 366–389. [Google Scholar] [CrossRef]
- Tian, X.; Firsanov, X.; Zhang, Z.; Cheng, Y.; Luo, L.; Tombline, G.; Tan, R.; Simon, M.; Henderson, S.; Steffan, J.; et al. SIRT6 Is Responsible for More Efficient DNA DoubleStrand Break Repair in Long-Lived Species. In Cell; 2019; Volume 177, pp. 622–638.e622. [Google Scholar]
- Leimar, O. "Evolutionary Change and Darwinian Demons". Selection. 2005, 2 (1–2),65–72. [CrossRef]
- Ludwig, R.J.; Welch, M.G. Darwin’s Other Dilemmas and the Theoretical Roots of Emotional Connection. Front Psychol. 2019, 10, 683. [Google Scholar] [CrossRef]
- Lennox, J. 1991 “Darwinian Thought Experiments: A Function for Just-so Stories.” In: Thought Experiments in Science and Philosophy, (Eds.) G. Massey, T. Horowitz.
- Razeto-Barry, P. ; Frick,R. “Probabilistic causation and the explanatory role of natural selection”, Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences 2011, 42, 344–355. [CrossRef]
- Machamer, P.; Darden, L.; Craver, C.F. Thinking about Mechanisms Philosophy of Science 2000, 67, 1–25, p2. [CrossRef]
- Havstad, Joyce C. Problems for Natural Selection as a Mechanism, Philosophy of Science 2011, 78, 522–523. [CrossRef]
- Abrams, P.A.; Ludwig, D. Optimality theory, Gompertz' law, and the disposable soma theory of senescence. Evolution 1995, 49, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Barros, D. Natural Selection as a Mechanism, Philosophy of Science 2019, 75, 306–322. [CrossRef]
- Weismann, A. Essays upon Heredity and Kindred Biological Problems. Science (Oxford) 1889, 14, 237–238. [Google Scholar] [CrossRef]
- Austad, S.N. Is aging programed? Aging Cell 2004, 3, 249–51. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L.; Melov, S. On the programmed/non-programmed nature of ageing within the life history. Curr. Biol. 2011, 21, R701– R707. [CrossRef]
- Kowald, A.; Kirkwood, T.B.L. Can aging be programmed? A critical literature review. Aging Cell 2016, 15, 986–998. [Google Scholar] [CrossRef]
- Mitteldorf, J. Can Aging be Programmed? Biochemistry (Mosc) 2018, 83, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, M.V.; Skulachev, V.P. Programmed Aging of Mammals: Proof of Concept and Prospects of Biochemical Approaches for Anti-aging Therapy. Biochemistry (Mosc) 2017, 82, 1403–1422. [Google Scholar] [CrossRef]
- Goldsmith, TC. Evolvability, Population Benefit, and the Evolution of Programmed Aging in Mammals. Biochemistry (Mosc) 2017, 82, 1423–1429. [Google Scholar] [CrossRef]
- Libertini, Giacinto. Non-programmed Versus Programmed Aging Paradigm. Curr. Aging Sci. 2015, 8, 56–68.
Figure 1.
Assuming that the force of NS is inversely correlated with mortality risk, a plot of The Gompertz-Makeham law shows that there are three segments of the Makeham function that indicate increasing, decreasing and non-changing intervals of NS strength. Mortality decreases between birth and about 10 years of age and increases during adolescence, from 10 to about 20 years thereafter. During the subsequent decade of 20 to 30 years of age, mortality is stable, neither decreasing nor increasing. Since the age-specific selection on survival at a given age typically changes as a function of the life history, then the strength of NS should vary throughout development. In fact, during ontogenesis, the strength of NS increases for the first decade, decreases in the second, and remains stable and unchanging for the final decade of morphostasis. An exponential increase in age-related deaths, which is correlated with an inexorable decline in the strength of NS, follows the occurrence of reproductive success about ten years after completion of the developmental program as depicted in the Gompertz function.
Figure 1.
Assuming that the force of NS is inversely correlated with mortality risk, a plot of The Gompertz-Makeham law shows that there are three segments of the Makeham function that indicate increasing, decreasing and non-changing intervals of NS strength. Mortality decreases between birth and about 10 years of age and increases during adolescence, from 10 to about 20 years thereafter. During the subsequent decade of 20 to 30 years of age, mortality is stable, neither decreasing nor increasing. Since the age-specific selection on survival at a given age typically changes as a function of the life history, then the strength of NS should vary throughout development. In fact, during ontogenesis, the strength of NS increases for the first decade, decreases in the second, and remains stable and unchanging for the final decade of morphostasis. An exponential increase in age-related deaths, which is correlated with an inexorable decline in the strength of NS, follows the occurrence of reproductive success about ten years after completion of the developmental program as depicted in the Gompertz function.
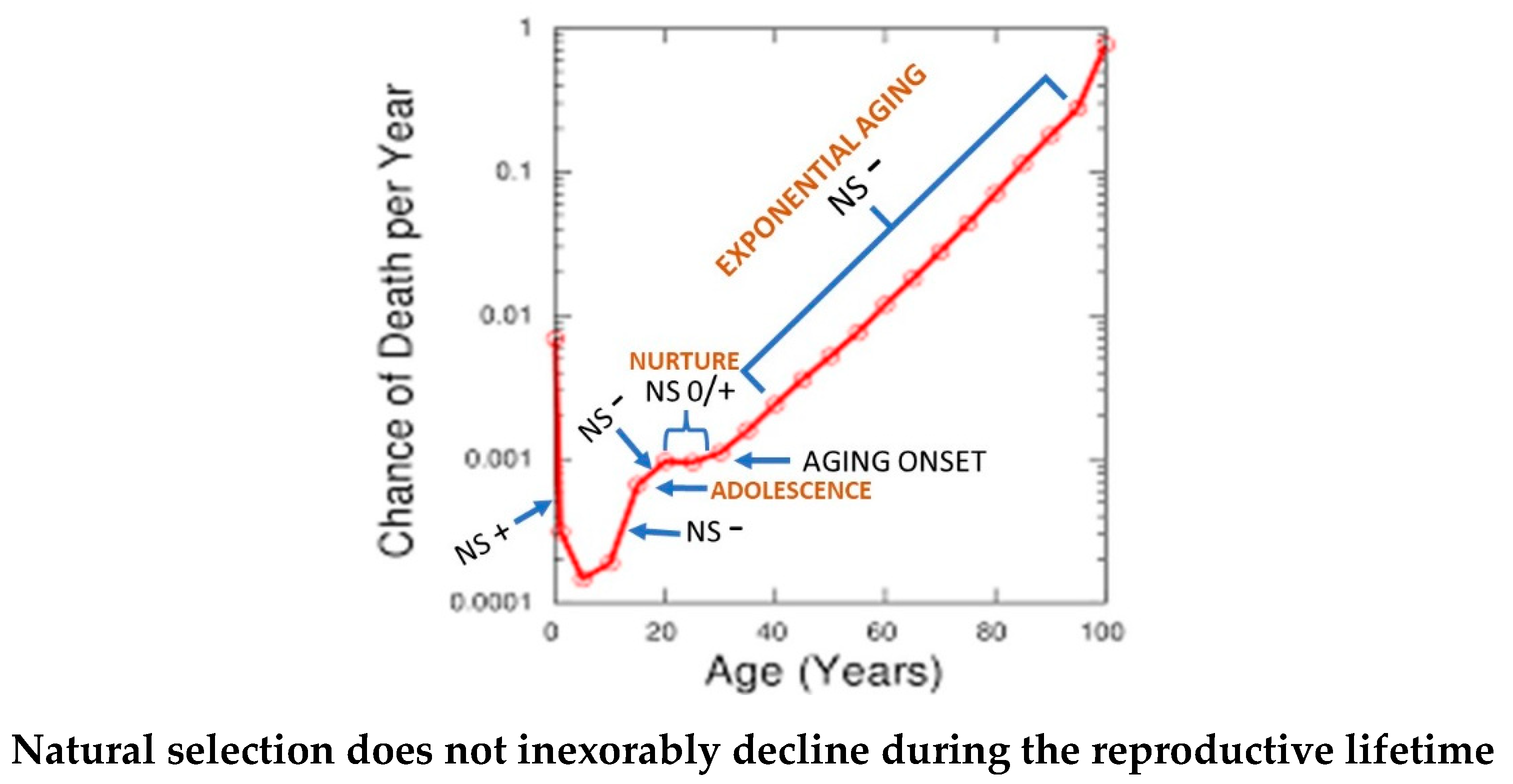
Figure 2.
Aging and resultant declining NS strength begin simultaneously in young adults that are about 30 years old. Thereafter, as aging proceeds, it decreases the probability of selection in parallel with advancing age and increasing physiological dysregulation. Thus, while age related weakening of NS allows accumulation of maladaptive mutations, physiological dysregulation, and other disruptive influences on somatic integrity, they do not cause aging, but rather are consequences of it.
Figure 2.
Aging and resultant declining NS strength begin simultaneously in young adults that are about 30 years old. Thereafter, as aging proceeds, it decreases the probability of selection in parallel with advancing age and increasing physiological dysregulation. Thus, while age related weakening of NS allows accumulation of maladaptive mutations, physiological dysregulation, and other disruptive influences on somatic integrity, they do not cause aging, but rather are consequences of it.
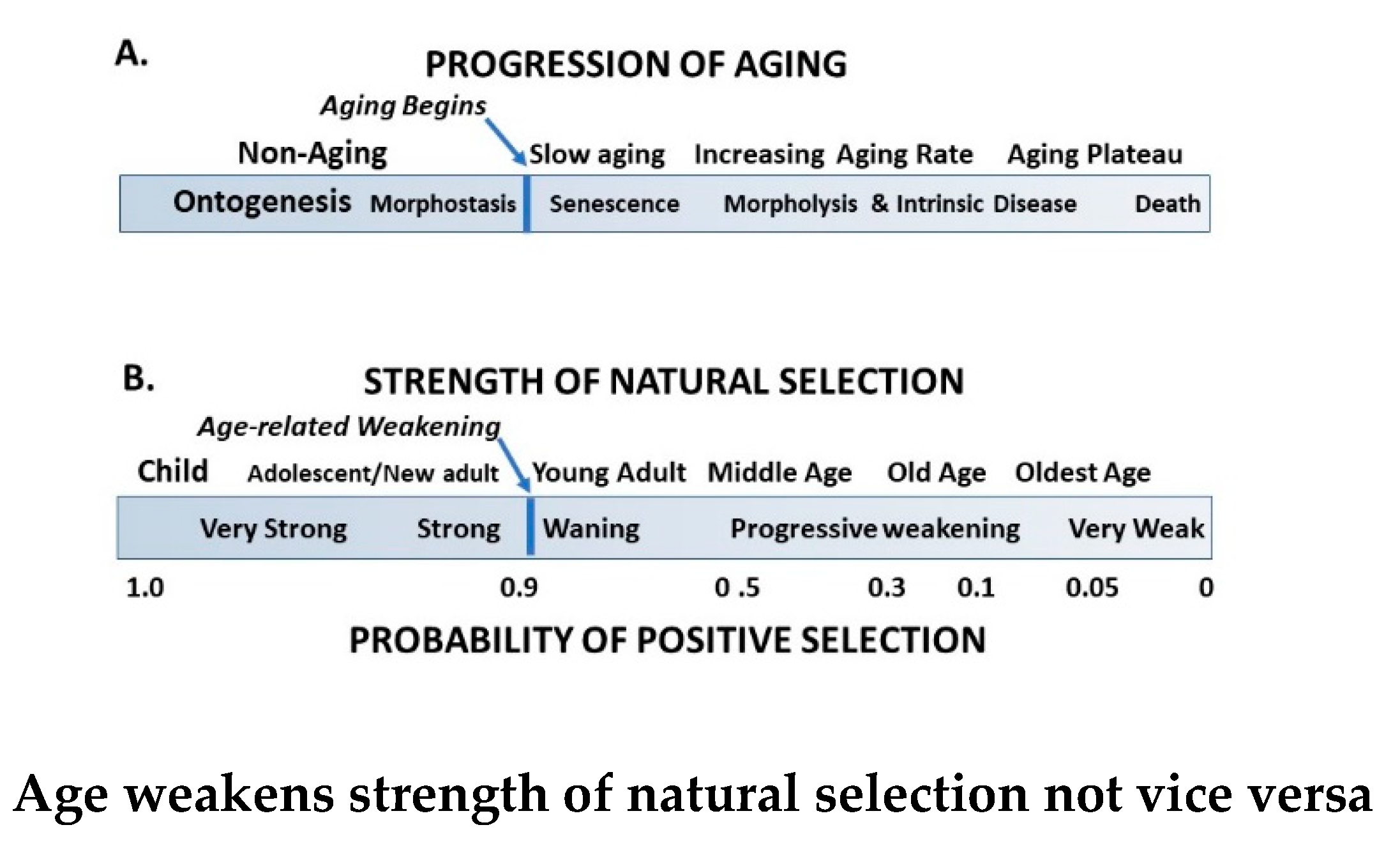
Figure 3.
Classic theory of aging proposes that the inexorable decline on strength of NS begins during adolescence which is about a decade before reproductive success is achieved. This is inconsistent with the significance of offspring nurturing unto their independence which according to Williams is as important as gamete production. Thus, to propose that the strength of natural selection declines before the occurrence of reproductive fitness is evolutionarily illogical. This figure is modified from Fabian D, Flatt. The Evolution of Aging. Nature Education Knowledge. 2011; 3, pursuant to Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Figure 3.
Classic theory of aging proposes that the inexorable decline on strength of NS begins during adolescence which is about a decade before reproductive success is achieved. This is inconsistent with the significance of offspring nurturing unto their independence which according to Williams is as important as gamete production. Thus, to propose that the strength of natural selection declines before the occurrence of reproductive fitness is evolutionarily illogical. This figure is modified from Fabian D, Flatt. The Evolution of Aging. Nature Education Knowledge. 2011; 3, pursuant to Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
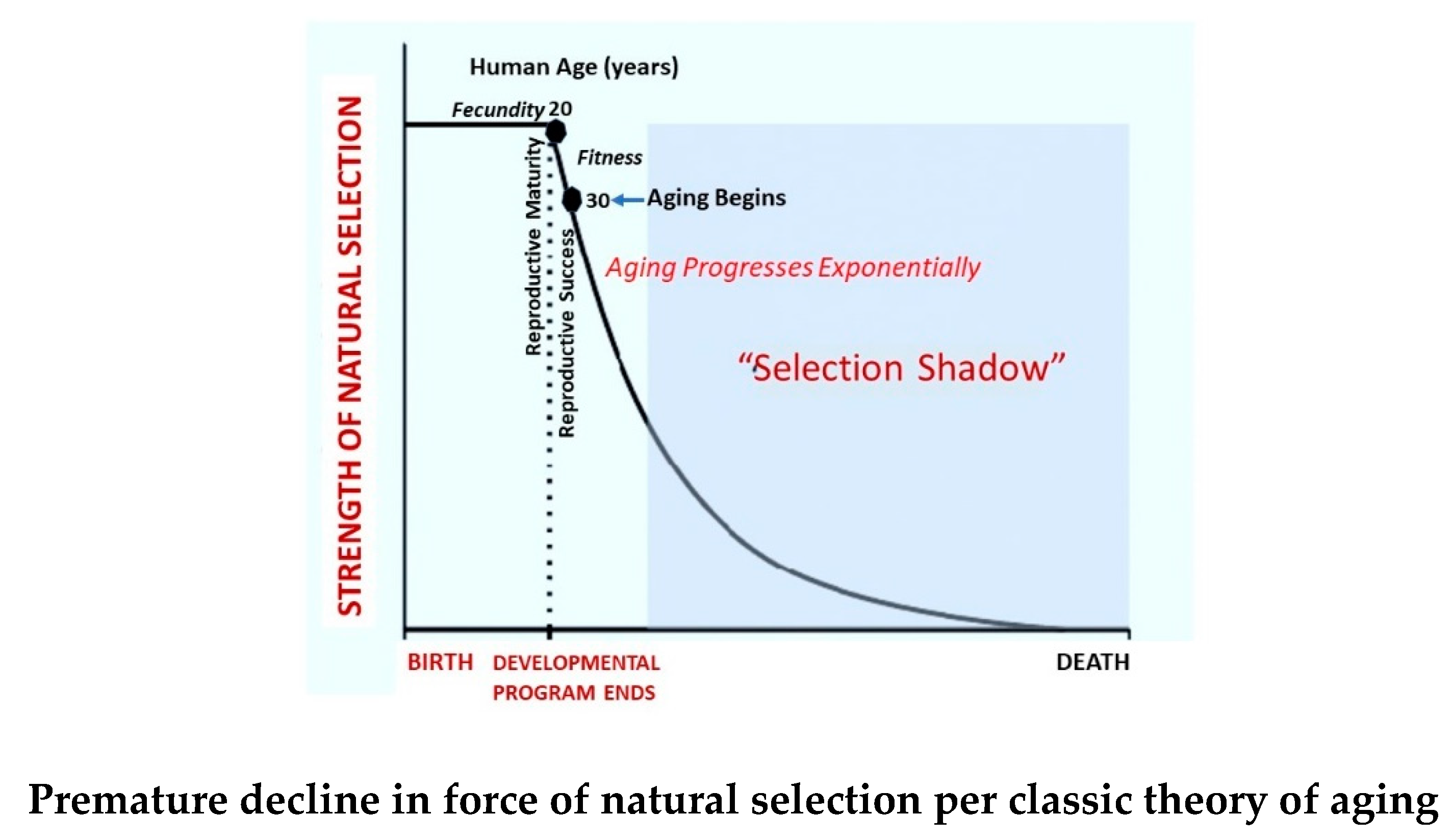
Figure 4.
The left vertical line indicates when humans reach maturity and achieve maximal stability at about 30 years old. During the decade of 20 – 30 years, many physiological processes are at peak capacity. After age 30, there occurs a linear decline and loss of reserve even when data are corrected to eliminate the influence of disease. Physiological losses progress to the point when the functional stability falls below the minimum required for system autonomy. The resultant effect is death from “old age”. Translated into dynamic terms, the life trajectory begins with growth, development, and differentiation, all of which are negentropic processes that are orderly. After maturity is reached, entropic processes become dominant, leading to a disorder of physiological processes and deconstruction of the soma. It is important to note that completion of development and onset of aging are continuous, not contiguous functions. Thus, the same soma that was previously non-aging becomes aging upon failure of the same mechanism that sustained it in a non-aging condition, i.e., both conditions are joined together in an uninterrupted sequence. In contrast, contiguous describes two or more things that abut each other but are not necessarily part of the same sequence. This figure was based upon reference [54] which is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Figure 4.
The left vertical line indicates when humans reach maturity and achieve maximal stability at about 30 years old. During the decade of 20 – 30 years, many physiological processes are at peak capacity. After age 30, there occurs a linear decline and loss of reserve even when data are corrected to eliminate the influence of disease. Physiological losses progress to the point when the functional stability falls below the minimum required for system autonomy. The resultant effect is death from “old age”. Translated into dynamic terms, the life trajectory begins with growth, development, and differentiation, all of which are negentropic processes that are orderly. After maturity is reached, entropic processes become dominant, leading to a disorder of physiological processes and deconstruction of the soma. It is important to note that completion of development and onset of aging are continuous, not contiguous functions. Thus, the same soma that was previously non-aging becomes aging upon failure of the same mechanism that sustained it in a non-aging condition, i.e., both conditions are joined together in an uninterrupted sequence. In contrast, contiguous describes two or more things that abut each other but are not necessarily part of the same sequence. This figure was based upon reference [54] which is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
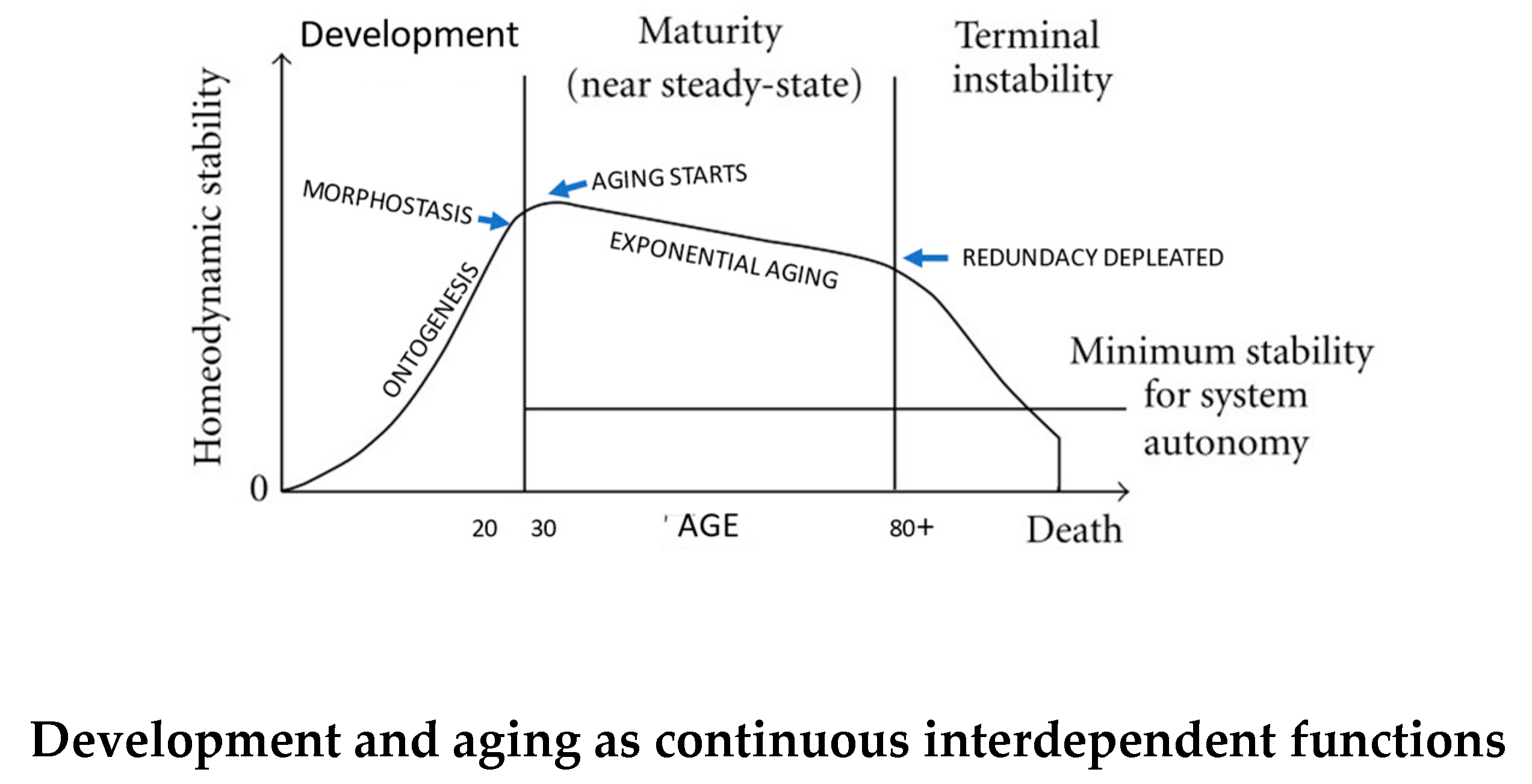
Figure 5.
Plot of the Gompertz-Makeham law of mortality showing the terminal, horizontal segment of the Makeham parameter called “morphostasis”. It represents the survival function of individuals during the age interval of 20 to 30 years showing the probability of living during its duration. It also represents the reliability function of redundancy within the developmental regulatory mechanism that provides stability of the final non-aging stage to facilitate achievement of reproductive success. Peak reproductive probability (fecundity) occurs before the end of ontogenesis, prior to the onset of morphostasis and occurrence of fitness. Hence, proposal of the classic theory that an inexorable decline in the strength of natural selection occurs during the developmental program is inconsistent with successful reproductive evolution. This figure was modified from that in Volume 54, Number 14 United States Life Tables, 2003 by Elizabeth Arias, Ph.D., Division of Vital Statistics is in the public domain and is allowed to be reproduced and modified with permission.
Figure 5.
Plot of the Gompertz-Makeham law of mortality showing the terminal, horizontal segment of the Makeham parameter called “morphostasis”. It represents the survival function of individuals during the age interval of 20 to 30 years showing the probability of living during its duration. It also represents the reliability function of redundancy within the developmental regulatory mechanism that provides stability of the final non-aging stage to facilitate achievement of reproductive success. Peak reproductive probability (fecundity) occurs before the end of ontogenesis, prior to the onset of morphostasis and occurrence of fitness. Hence, proposal of the classic theory that an inexorable decline in the strength of natural selection occurs during the developmental program is inconsistent with successful reproductive evolution. This figure was modified from that in Volume 54, Number 14 United States Life Tables, 2003 by Elizabeth Arias, Ph.D., Division of Vital Statistics is in the public domain and is allowed to be reproduced and modified with permission.
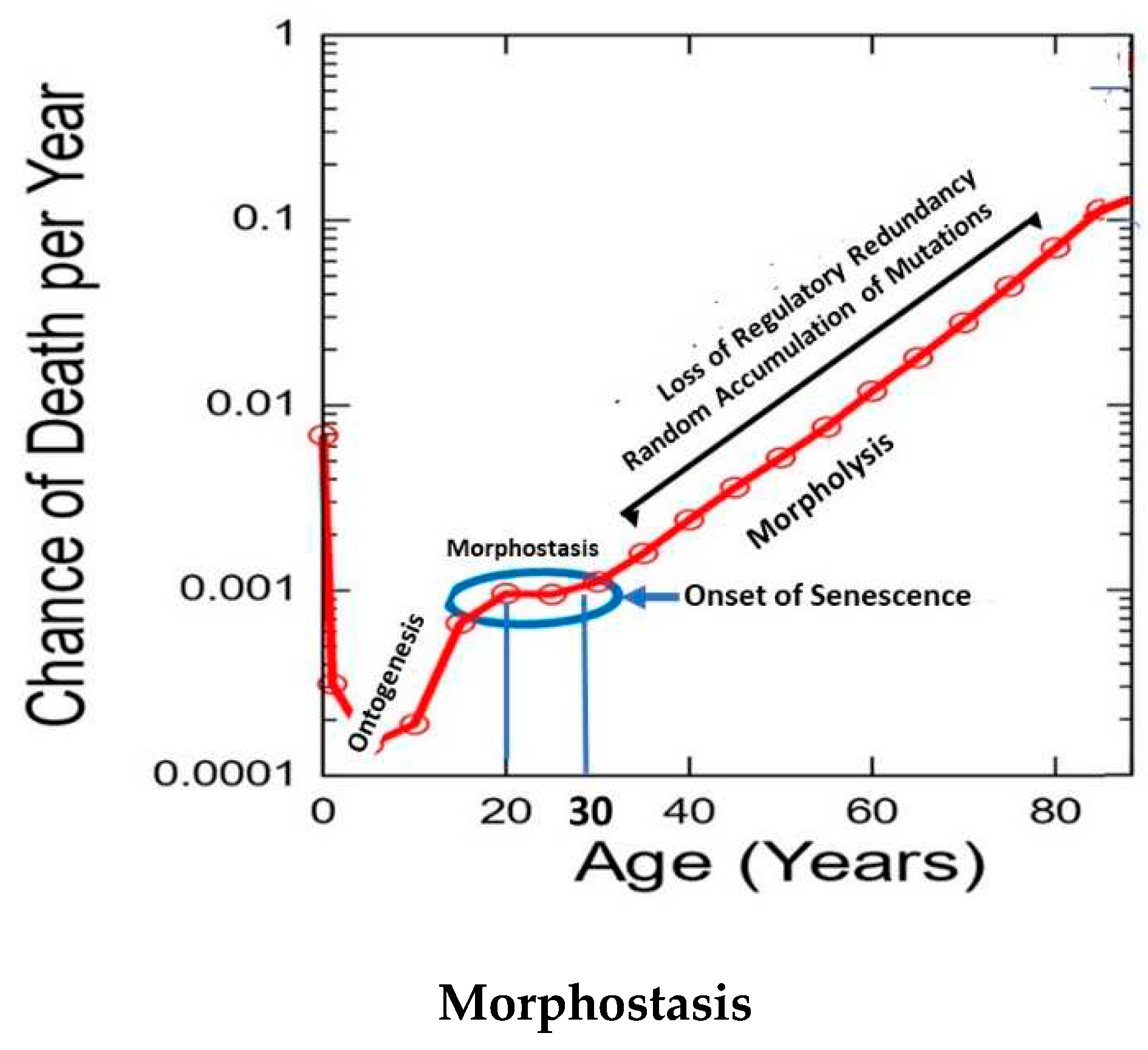
Figure 6.
Comparison of parental care of offspring from birth unto their independence. Upon completion of successful reproduction, parents reach the onset of aging, when progeny are weaned, capable of childbirth and subsequently for providing nurture unto independence of their own offspring. Thus the temporally overlapping sexual roles of parents and offspring provide for maintenance of life cycles and successful continuation of species.
Figure 6.
Comparison of parental care of offspring from birth unto their independence. Upon completion of successful reproduction, parents reach the onset of aging, when progeny are weaned, capable of childbirth and subsequently for providing nurture unto independence of their own offspring. Thus the temporally overlapping sexual roles of parents and offspring provide for maintenance of life cycles and successful continuation of species.
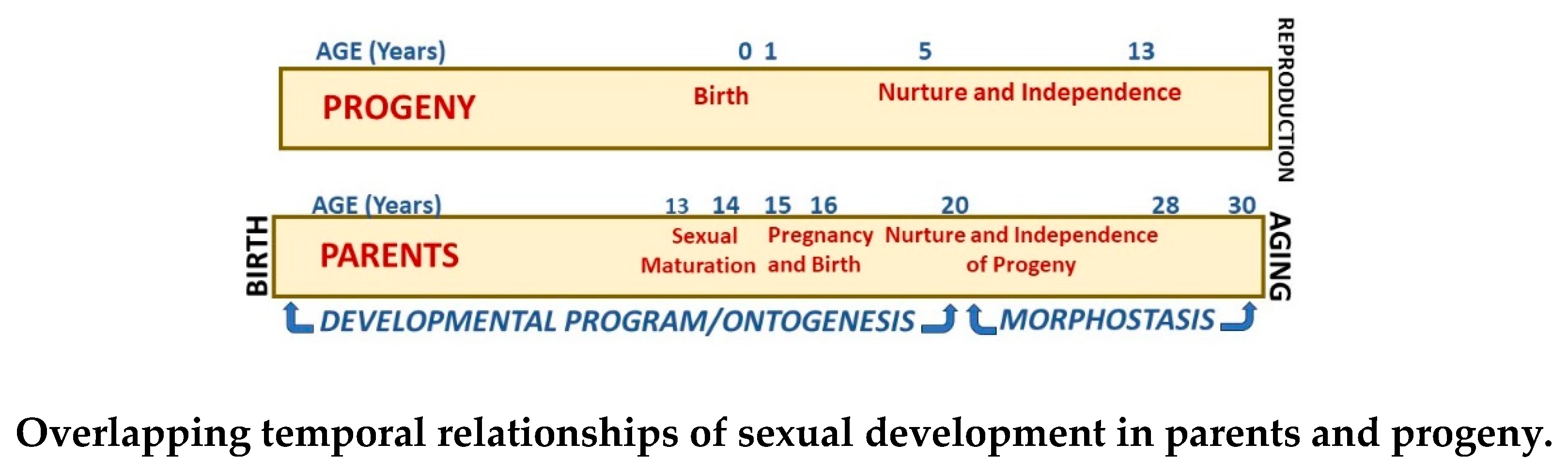
Figure 7.
Redundancy of developmental regulatory processes change throughout life. A. Multiples stages of somatic construction occur during ontogenesis causing their duration to be relatively brief. Thus, only minimal redundancy of regulatory processes for each succeeding stage are required to ensure successful progression of ontogenesis. However, the post-ontogenetic interval of morphostasis must be sustained in humans for about 10 years, or approximately one half of the time required for completion of the developmental program. B. To ensure the survival function of morphostasis, significant redundancy of the last developmental stage compared with those preceding it must occur to sustain the young adult soma allowing its completion of physical development, enhancement of vitality and fulfillment of its obligation to ensure reproductive success. C. As the post-ontogenetic decade approaches its end, loss of redundant regulatory units in areas that are vital to somatic maintenance, erode the reliability function required for sustaining morphostasis. Aging begins and proceeds in parallel with loss of regulatory redundancy.
Figure 7.
Redundancy of developmental regulatory processes change throughout life. A. Multiples stages of somatic construction occur during ontogenesis causing their duration to be relatively brief. Thus, only minimal redundancy of regulatory processes for each succeeding stage are required to ensure successful progression of ontogenesis. However, the post-ontogenetic interval of morphostasis must be sustained in humans for about 10 years, or approximately one half of the time required for completion of the developmental program. B. To ensure the survival function of morphostasis, significant redundancy of the last developmental stage compared with those preceding it must occur to sustain the young adult soma allowing its completion of physical development, enhancement of vitality and fulfillment of its obligation to ensure reproductive success. C. As the post-ontogenetic decade approaches its end, loss of redundant regulatory units in areas that are vital to somatic maintenance, erode the reliability function required for sustaining morphostasis. Aging begins and proceeds in parallel with loss of regulatory redundancy.
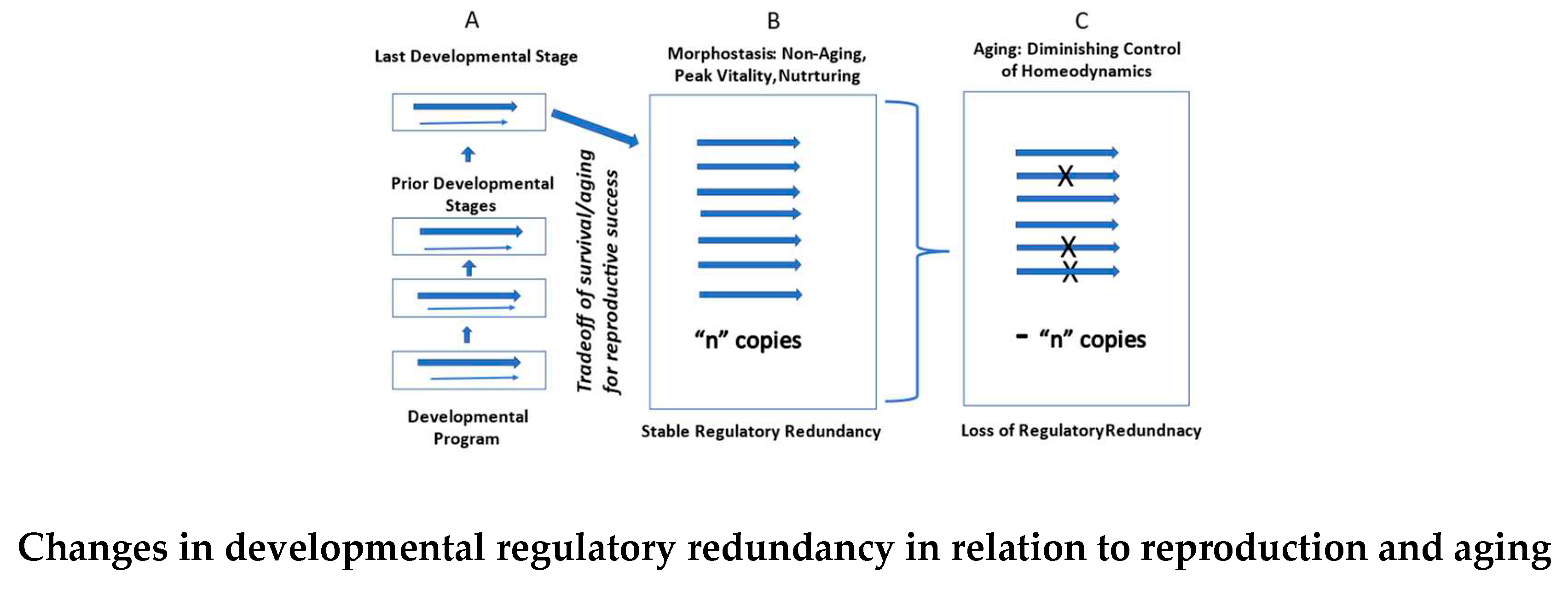
Figure 8.
DSB’s initiate a cascade of effects leading to apoptosis and/or cellular senescence. These damages in combination with other forms occur randomly throughout the soma contributing to the aging phenotype and pillars of aging. However, when they impact components of the organismal regulatory mechanism they reduce its redundancy and thus make a significant contribution to the onset and progression of aging. Reproduced and modified with permission from [101].
Figure 8.
DSB’s initiate a cascade of effects leading to apoptosis and/or cellular senescence. These damages in combination with other forms occur randomly throughout the soma contributing to the aging phenotype and pillars of aging. However, when they impact components of the organismal regulatory mechanism they reduce its redundancy and thus make a significant contribution to the onset and progression of aging. Reproduced and modified with permission from [101].
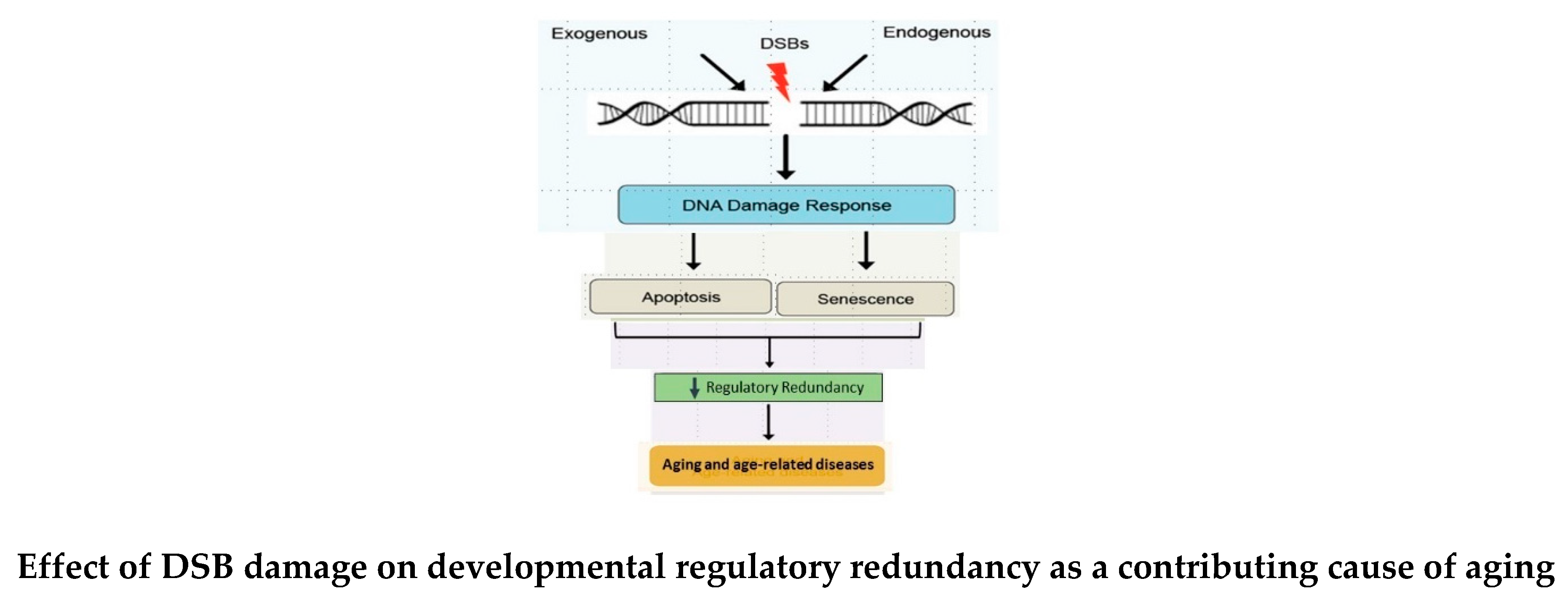
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



