Submitted:
30 September 2023
Posted:
02 October 2023
You are already at the latest version
Abstract
The evolutionary conserved STING-cGAS pathway represents one of the most important cytosolic DNA sensing systems that is activated in response to viral invasion and/or damaged integrity of the nuclear envelope. The key outcome of this pathway is the production of IFN, which subsequently stimulates hundreds of genes. In oncology, the situation is a complex one since this pathway may serve either anti- or pro-oncogenic roles in a context-dependent fashion. The prevailing understanding is that when the innate immune response is activated by sensing cytosolic DNA, such as from ruptured micronuclei, it results in the production of interferon which attracts cytotoxic cells to destroy tumors. However, in tumor cells that have adjusted to significant chromosomal instability, particularly in relapsed, treatment-resistant cancers, the cGAS–STING pathway often supports cancer progression. In these cells, the pathway initiates a non-canonical nuclear factor κB transcriptional response, fostering the epithelial-to-mesenchymal transition (EMT). Here, we review this intricate pathway in conjunction with cancer progression and development of new treatment modalities.
Keywords:
STING
; cGAS
; cGAMP
; interferon signaling
; cancer
; metastasis
; BRCA
; proton channel
1. Introduction
The cGAS-cGAMP-STING innate immunity pathway originally discovered in 2008-2013 [1,2,3,4,5,6,7,8] has gained a broad attention in respect to its role in tumorigenesis [9,10,11,12,13]. The number of articles on this topic have shown an explosive growth, including recently published excellent reviews [14,15,16,17,18,19,20,21]. The STING-cGAS pathway senses cytosolic DNA and becomes activated in response to viral invasion. Mechanistically, the STING-cGAS pathway (hereinafter referred to the canonical cGAS-cGAMP-STING-TVK1-IRF3,7 pathway) transduces a signal from Pathogen Associated Molecular Pattern (PAMP) molecules, e.g. fragments of immunogenic DNA, to specific effector proteins such as transcription factors IRFs and NF-kB that alter the expression of pro-inflammatory genes such as interferon, thereby eliciting a proper immune response using both autocrine and paracrine modes.
Theoretically, recognition of either foreign non-self (e.g. infections) signals or aberrations within the endogenous self (e.g. malignancies) signaling cues relies on the organism’s friend-or-foe recognition system that must be meticulously calibrated to fight against infections and tumors without inflicting harm to healthy tissues. This is why the pattern recognition receptors (PRRs) are fundamental to the innate immune system. Various forms of nucleic acids can be perceived as foreign or malignant. Whether it is naked DNA, modified or unmodified sequences, DNA coated with histones, DNA-RNA hybrids, RNA, small fragments, or cyclic dinucleotides, each can be seen as foreign elements to different extent.
There are more than 14 recognized DNA sensors in the human genome, with three playing primary roles [22]: 1) TLR9 is found in the endosomal membrane of B cells and plasmacytoid DCs. It detects external DNA and is particularly sensitive to unmethylated or hypomethylated CpG islands, 2) The AIM2 receptor resides in the cytosol of cells and operates against viruses and bacteria. Upon activation, it forms the inflammasome, which can activate caspases, leading to the release of IL-1β and IL-18 cytokines. This process also triggers pyroptosis, a specialized form of cell death. 3) Among the cytosolic DNA sensors, however, the cGAS system appears to be more widespread and important at most stages of infection (Figure 1). Perhaps not surprisingly, the cGAS-STING pathway was shown to intertwine with the DNA Damage response system because both systems deal with the fragmented DNA. Are there distinguishing characteristics between viral DNA and cellular DNA essential for initiating the cascade? Why is the cGAS-STING signaling not activated by cGAS in the nucleus, or by the cell's own DNA in the cytoplasm—of which there is an abundance during cellular aging? In this review, we discuss the mechanistic aspects of these interactions and their implications for cancer treatment.
2. Overview of the classical cGAS-cGAMP-STING-TBK1-IRF3 pathway: structure of key components and their molecular evolution
cGAS
This enzyme partakes in several physiological functions, predominantly in infection defense and associated inflammatory responses [23]. cGAS belongs to the large cGAS/DncV-like nucleotidyl transferase family with deep phylogenetic roots [24,25,26]. Enzymes of such activity are identified in both prokaryotes and eukaryotes, although bacterial counterparts produce a wide variety of cyclic di- and oligonucleotides
cGAS is able to interact with double-stranded DNA longer than 20 bp with sufficient affinity [5,27]. This length reflects the minimum DNA binding requirement for the cGAS monomer. However, for optimal activation, cGAS typically needs DNA strands longer than 45 bp to establish stable enzymatic complexes – dimers of DNA-cGAS (Figure 1). Binding of cGAS to DNA is also strongly enhanced when the DNA is bent. Such bending is influenced by HMGB1 protein (High Mobility Group B1) [28]. cGAS works in a sequence-independent fashion, yet there are exceptions, like its interaction with specific viral ssDNA structures in HIV [29].
Apart from dsDNA, RNA:DNA hybrids can also activate cGAS, a unique trait among nucleic acid sensors involved in IFN induction [30]. These hybrids, referred to as R-loops, also stimulate cGAS, and subsequently, IRF3, culminating in apoptosis, especially in BRCA1-mutated cancer cells [31,32].
On the contrary, complexes with dsRNA are formed lacking proper conformational change yielding no activation and thus no efficient induction of IFN, and cGAS in the cytoplasm may rest in transient low affinity complexes with RNA. Moreover, some RNAs are strong inhibitors of cGAS, for example, in hematopoietic stem cells circular RNA prevents cGAS activation by nuclear self dsDNA, maintaining the stem cells' dormancy [33]. Since some RNA viruses are also susceptible to cGAS-STING then it is tempting to speculate that RIG-I-like receptors are not the sole player in this field. One should anticipate further discoveries of regulatory roles of non-coding RNAs [34]. Recent studies indicate, however, that cytoplasmic RNAs do not activate cGAS per se but enhance its phase separation into liquid-liquid condensates [35,36]. Most likely, activation of cGAS by dsRNA is a result of reverse transcription, as it is in the case of retroviruses like HIV-1 [37].
Not only any unscheduled DNA release but other cellular stresses may be operational in terms of activation of cGAS. Perturbations of mRNA translations such as frameshifting are quite common during viral infections, indeed, a recent discovery describes activation of cGAS by pure ribosomes, and this may be reminiscent of ribosome collision in vivo, however, in this case activation of cGAS do not proceed to interferon secretion [38].
Structurally, human cGAS contains three distinct DNA binding sites within its C-terminal domain [23,39,29,40]. Its N-terminal domain, intrinsically disordered in the absence of DNA, appears to be non-essential for recognition of pathogenic or mitochondrial DNA [41,42,43,44,45], yet, it can additionally stabilize the cGAS-DNA complex [29,46]. This domain also mediates interactions with membranes, nuclear DNA, and is important for cGAS enrichment in centromeres. Additionally, cGAS has an NT core domain and a Mab21 domain, the latter of which contains a zinc-ribbon domain that's vital for DNA binding.
In the absence of DNA, cytoplasmic cGAS might be anchored to proteins such as G3BP1. This hints at a possible displacement by DNA in its pre-condensed state. Notably, G3BP1 augments the DNA binding capability of cGAS [47]
cGAMP
When cGAS is activated by DNA, it synthesizes the secondary messenger, 2',3'-cyclic CMP-AMP (2'3'-cGAMP) [2,48,49], a member of the cyclic dinucleotide (CDN) family. CDNs, important signaling molecules in prokaryotes with vast diversity, have only relatively recently been identified in animals. Mammalian cGAMP, primarily 2’,3’-cGAMP, is a mixture of isomers: 1) from 2’-OH of GMP to 5’-P-OH of AMP; and 2) from 3’-OH of AMP to 5’-P-OH-GMP.
STING
STING's activation mechanism by CDNs is conserved across various organisms. STING's direct engagement with various bacterial CDNs has been observed, and the effects of other CDNs in diverse organisms range from 3’,3’ UGA to 2’,3-cGAMP [50]. STING apparently originated early in bacteria, but the degree of sequence conservation is low (only 8 hyperconserved residues), moreover, bacterial STINGs bind c-di-GMP with high affinity preferring 3′–5′-linked cyclic dinucleotides in contrast to human STING, which works with mammalian cGAS product 2′–5′/3′–5′ cGAMP (2′,3′-cGAMP). Bacterial STING receptors are not activated by mammalian 2′,3′-cGAMP [51]. But mammalian STING is a quite unique adaptor protein, since it can also act as an independent pattern recognition receptor for bacterial cyclic dinucleotides [52,53].
Structurally, the product of the STING1 gene (a.k.a. ERIS, FLJ38577, MITA, MPYS, NET23, STING, TMEM173) is a multi-domain membrane protein featuring four transmembrane α-helices with only small loops exposed into the ER lumen, a cGAMP binding domain, and another cytoplasmic domain required for recruitment of downstream signalling molecules. Remember that mammalian STING is activated by binding either c[G(2',5')pA(3',5')p] or its isomer c[G(2',5')pA(2',5')p] [54], however, only 2'3'-cGAMP is considered to be fully functional in terms of STING stimulation [55]. Binding of cGAMP to STING leads to activation by increasing the level of its oligomerization: conversion from an inactive dimer to an active tetramer and higher-order oligomers [56]. Oligomers dissociate easily, but certain small molecules can adjust the stability of the oligomers[57]. In turn, this process of oligomer formation can be deregulated in various ways, for example, a single mutation can render STING perpetually active [58].
More specifically, in its resting state in the ER, STING exists as a dimer, complexed with proteins like TRAPβ, SEC61β, and STIM1. Upon binding to 2'3'-cGAMP, the wing-like structure of STING undergoes a remarkable movement, accompanied by the rotation of the ligand-binding domain, and this facilitates its translocation from ER to Golgi where binds to sulfated glycosaminoglycans [56,59,60,61] and may open a proton pore [62]. Activation by oligomerization of STING is also promoted by many other proteins including the mitochondrial nucleoid proteins HU, mitochondrial transcription factor A (TFAM), and HMGB1 (the highly mobile group box protein 1) [28] The extensive oligomerization of the cGAS-DNA complex [46] also gives rise to observable liquid-liquid condensation (phase separation), especially alongside STING oligomers when transitioning from the ER. This can be seen in cells exposed to viral DNA [63].
The transport of STING from the ER heavily relies on interactions with COPII, ARF GTPases, and palmitoylation at the transmembrane cysteine residue 91, a critical step for its assembly into multimeric complexes at the Golgi [64]. For effective downstream signaling, both the formation of STING oligomers and their transport are essential.
TBK1
The next indispensable player in the pathway is TANK-binding kinase 1 (TBK1), [50]: close to the Golgi, TBK1 phosphorylates STING's C-terminus, enabling its interaction with IRF3 and IRF7. Consequently, IRF3 or IRF7 get trapped, leading to its inevitable phosphorylation by TBK1, emphasizing TBK1's central role in the pathway. Additionally, when this complex is formed, TBK1 undergoes auto-phosphorylation. TBK1 inhibits this oligomerization process or in the cells treated with 3'-cGAMP as a way of regulation [65,66] TBK1 shows enhanced activity when STING is polyubiquitinated, a process mediated by E3 ubiquitin ligase – AMFR combined with INSIG1 [67].
Together with IκB kinase (IKK) TBK1 plays a crucial role in STING signaling, activating both interferon regulatory factor 3 (IRF3) and nuclear factor-κB (NF-κB). Therefore, STING's activation of TBK1 leads to the concurrent activation of both IRF3 and NF-κB pathways, akin to a fork in the signaling route [68].
IRF3, NF-κB and downstream signaling
Activation of cGAS, STING and TVK1 subsequently culminates in the activation of transcription factors such as IRF3, IRF7, and NF-κB, which govern the production of pro-inflammatory cytokines like IFN1-alpha, IFN1-beta, TNF-alpha, IL-6, IL-12, and IL-1beta [69,70]. Mechanistically, the phosphorylated IRF3 dimerizes, dissociates from the complex, and relocates to the nucleus, activating IFN type I, particularly IFN-β. The secreted IFN-1β modulates the activity of other cells, activating IFNAR1 and 2, which subsequently triggers the STAT pathway as an important part of antiviral defense. NF-κB's activation via STING involves various proteins, including NEMO, IKKβ, TBK1, TRIM32, and TRIM56.
Practically, in a straightforward model of cGAS-STING activation through dsDNA transformation, marked upregulation of pTBK1, pIRF3, and pSTING can be observed. In contrast, there might be a depletion of total STING. Additionally, among the IFN-responsive genes, a robust upregulation of IFNβ, OAS1, CXCL10, and ISG15 can be detected [71]. Transgene expression, when transfected with naked DNA, may be augmented by inhibiting cGAS-STING [72], also, electroporation efficiency can be enhanced by inhibition cGAS activity, as it boosts cell viability. [73]
The so-called non-canonical STING pathway is orchestrated by the DNA-binding protein IFI16, in conjunction with DNA damage response factors ATM and PARP-1 (Figure 2). In this case STING signaling engages also the tumor suppressor p53 and the E3 ubiquitin ligase TRAF6, which polyubiquitinates STING at K63), and all this sequence of events leads to the activation of NF-κB [74,75].
3. cGAS-STING in major DNA Damage Response pathways
It is pertinent to note that the cGAS-STING pathway is intricately intertwined with numerous signaling pathways, including those involved in DNA damage, rendering it susceptible to varied modes of regulation. For instance, in the event of DNA damage, the activation of the ATM/Chk2 pathway induces a replication checkpoint within the cell. Indeed, pharmacological inhibition of ATM imposes replication stress leading to an increased abundancy of DNA fragments in the cytosol and hence STING activation [76]. Moreover, STING is the major adaptor protein of the DNA sensors, since it converges the actions from cGAS, IFI16, ZBP1/DAI, DDX41, MRE11/Rad50, Ku 70 [77,1,52,78]. Thus STING engages in direct cross-talk with other pathways, for example, in myeloid cells MYD88 and STING can form a direct liaison [79] making the DNA-sensing network quite complex since MYD88 also interacts with β-catenin and activates aforementioned IRF3,IRF7 and NF-κB.
Efficient oncosuppressive function cGAS-STING in DNA damage response
While the absence of cGAS or STING does not directly induce carcinogenesis, it is clear that innate immunity precedes the priming of tumor antigen-specific CD8 T cells. The pathway serves among other means as a mechanism for numerous anti-cancer checkpoints. For example, cGAS knockout makes mice susceptible to colitis-associated colorectal cancer due to disruption of intestinal barrier integrity accompanied by decreased stem cell number, inflammation with abundant MDSC, STAT3 activation. Interestingly, normal intestinal epithelium appears to have cGAS in clear excess of STING [80].
A meta-analysis encompassing various cancers suggests that the presence of STING in tumors is most often associated with a favorable prognosis, correlating with increased disease-free survival/recurrence-free survival (DFS/RFS), but not with overall survival (OS) [81].
Senescence as the route out of cancer and the role of the STING pathway
Senescence and short-term inflammation is a powerful barrier to tumorigenesis, persistent inflammation is associated with tissue damage, and in established cancers, is linked to tumor growth and metastatic dissemination. This is confirmed by the observation that cGAS-triggered senescence occurs upon irradiation and oncogene activation in vivo [82] supporting the hypothesis that SASP controls tumorigenesis. cGAS is localized in the cytoplasm of non-dividing cells but enters the nucleus and associates with chromatin DNA during mitosis in proliferating cells [83]. Given that evading senescence can be a potential pathway to cancer, with the replicative crisis serving as a protective mechanism, it is not surprising that cGAS also senses telomeric damage. Specifically, extrachromosomal telomere repeats (ECTR) DNA is common to cancer cells that utilize the Alternative Lengthening of Telomeres (ALT) pathway. ECTRs in typical human fibroblasts activate the canonic cGAS-STING pathway, causing proliferation defects. Conversely, ALT cancer cells often have defective cytosolic DNA sensing. STING expression is reduced in ALT cancer cell lines and transformed ALT cells [84]. Mice with cGAS or STING knock-out (KO) show no SASP in response to IR or RasV12 (in normal mouse RasV12 cells enter SASP and are killed, whereas and in cGAS or STING KO these cells are retained) [85]. Recently, cGAS–STING pathway was found to play a significant role in driving inflammation related to aging through microglial intrinsic involvement in age-associated neurodegeneration. In mice, the activation of cGAS alone initiates a fundamental microglial gene expression program, which is common across various neurodegenerative diseases [86].
DDR and its links to carcinogenesis and tumor formation
Certain viruses, like parvoviruses, exploit DDR pathways. Given this, it is reasonable to hypothesize that antiviral defenses might counteract DDR. In support of this, cGAS has been found to inhibit DDR, specifically the HR pathway, though not NHEJ [87]. These disturbances can be categorized into various phases: presence of abnormal DNA in the nucleus, such as extrachromosomal DNA and extrachromosomal telomeric repeat DNA; loss of nuclear envelope integrity; formation of micronuclei; rupture of micronuclei; creation of DNA-containing vesicles; cytoplasmic chromatin fragments; presence of senescence-associated DNA in the cytosol; abundant formation of abasic DNA.
The phenomenon of the formation of a significant number of micronuclei is associated with escape from mitotic block, defects in chromosome segregation, and accumulation of damaged DNA fragments. In normal cells, any anomalies (exemplified by deletion of ARP2/3 [88]) lead to activation of p53 that halts cell cycle in G1 through CDKN1a and p21, and any significant DNA accumulated in cytosol induces cGAS-STING- and IRF3-associated interferon response. The so-called CAD (caspase-activated DNase) participates in micronuclei formation, as its inhibition reduces micronuclei formation and metastasis, while CAD activation, on the contrary, gives a more aggressive phenotype in a STING-dependent manner [89]. The survival of cancer cells with accumulated micronuclei is interesting in itself and is being actively studied. cGAS fully activates upon micronuclei rupture, however, specific conditions preceding this rupture can impact the activation process. For instance, if DNA transcription within the micronuclei is active, the activation of cGAS is less efficient [90]. Mitotic arrest can be induced by nocodazole, resulting in CIN with abundant micronuclei with aberrant chromosome segregation, whereas decreased p21 (caused by depletion of STING, TBK1, IRF3 or by overexpression of cGAS) inhibits these phenomena [91].
Homologous Repair (HR) deficiency triggers the cGAS-STING signaling, augmenting the survival prospects of such cancer cells. Although understanding the survival of HR-deficient cancer cells remains a complex puzzle, it is evident that other mutations, such as those in TP53BP1, MAD2L2, HELB, RIF1, are also important. Indeed, some damages to oncosuppressor genes both germline or somatic is a prerequisite to carcinogenesis. The canonic cGAS-STING is involved in DDR pathway in the situation of loss/insufficiency of oncosuppressors such as BRCA1 or BRCA2.
STING and BRCA1,2
The role of BRCA1 is paramount for DNA repair (including repair of transcription-associated DNA damage). When not degraded by exonucleases such as MRE11, single-stranded DNA from halted replication forks translocates into the cytosol and activates the cGAS-STING pathway, signaling a response to replication stress [32]. Interestingly, cGAS binds directly to the replication fork, decelerating replication but reinforcing its stability. Without cGAS, there is increased sensitivity to both radiation therapy and chemotherapy [94]. Fascinatingly, BRCA1-mutated cancer cells displayed accumulation of cytoplasmic RNA-DNA hybrids, products of R-loop processing. Such accumulation, sensed by cGAS and TLR3, is apparent when nuclear R-loop metabolism becomes unregulated. Notably, this event is prominent in cells with mutations in SETX and BRCA1. [31]
Equally intriguing is the role of the hyaluronan-mediated motility receptor (HMMR, also known as RHAMM). Its interaction with BRCA1 has ramifications not just for cell division but also for the polarization of certain epithelial cells, influencing cell division in the process. Mouse models showed how HMMR magnifies the pro-oncogenic effects of mutated BRCA1. This leads to micronuclei formation and subsequent cGAS-STING activation. Both canonical and non-canonical NF-κB signaling pathways get activated as a result [95].
It is important to distinguish between intrinsic and extrinsic tumor processes. While BRCA1 functions alongside PALB2, disrupting their interaction in a hepatocellular carcinoma mouse model led to heightened T-cell infiltration and enhanced PD-1 antibody response, in sync with both intrinsic and extrinsic cGAS-STING activation [96].
BRCA2 inactivity, similar to BRCA1 or FANCD2, escalates TNFα production, leading to augmented autocrine TNFα signaling. Absence of BRCA2 also coincides with the formation of micronuclei and activation of the ASK1 and JNK pathways. BRCA2-deficient TNBCs also may possess some intensification of cGAS-STING signaling [97].
It's worth emphasizing that essential genome integrity signaling operates independently of the canonical IFN pathways. This is highlighted by the fact that cGAMP reduces NAD+ levels, subsequently inhibiting PARylation and suppressing HR. On the other hand, activating the cGAS-STING-TBK1 axis prompts DDR induction through the autophosphorylation of ATM kinase. This action triggers the CHK2-p53-p21 signaling pathway, resulting in cell cycle arrest [98].
STING and mismatch repair deficiency
Deficiency in mismatch repair (MMR), as seen in MLH1 deletion, also leads to accumulation of cytosolic DNA and activation of cGAS-STING, yielding interferon-β production [99]. MMR deficient cancer cells respond better to ICB due to high cancer neoantigens level these need to inhibit STING pathway somehow.
STING and oncogenic infections
Chronic H. pylori infection can increase STING expression and activate its signaling in mice [100]. It's not clear how normal microflora affects this pathway or colorectal cancer progression, but interesting data links STING to colitis and (colitis associated cancer) – STING knock-out in mice reduces pyroptotic cell death and thus the animals are more prone to CAC [101] (compare this to the different results of cGAS knockout [80]).
4. Cross-talk of cGAS-STING with major cell death pathways
STING's role in cell death pathways is well-documented and reviewed [102]. Generally, STING activation per se does not induce cell death, either necrotic or PCD (programmed cell death), only some cells are susceptible like human myeloid cells [103]. In T cells, naïve B cells, and malignant B cells, STING or its agonists can trigger mitochondrial apoptosis, this was especially well documented in murine models of chronic lymphocytic leukemia and multiple myeloma [104].
Necroptosis is dependent on the presence of a basic level of IFN signaling, cGAS-STING plays a visible important role in necroptotic cell death in cancers, activation of necroptosis by RIPK3 or MLKL in macrophages is dependent on DNA sensing and is elevated in autoimmune conditions like systemic lupus erythematosus (SLE)) [105]. Vice versa, necroptotic signaling stimulates STING, whereas suppression of RIPK3 inhibits STING by redirecting STING to autophagosomes [106]. In the mouse, STING agonists induce necroptosis in primary macrophages in vitro in a STING and RIPK3-dependent mode linked to production of and IFN and TNF [107]
Pyroptosis, the cell death pathway that employs the NLRP3 inflammasome, and lysosomal cell death are promoted by STING after its translocation to the lysosome through an increase in lysosomal membrane permeabilizaton, especially in human myeloid cells we may speak about the major cGAS-STING-NLRP3 pathway that works as a default death program in these cells [103]. Also, SYK tyrosine kinase was singled out as the key mediator of pyroptosis and also a direct interactor of STING [101]. A significant recent finding is that STING activation causes a marked increase in pH within the Golgi, attributed to STING’s direct function as a proton channel. This channel activity is essential for subsequent NLRP3 inflammasome activation, leading to pyroptosis and IL-1beta secretion, which is distinct from lipidation and autophagy processes. [108,62].
STING promotes ferroptosis, especially noticeable in human pancreatic cancer cells. Several mechanisms might be at play, including those involving ROS or lipid peroxidation, for example, ferroptosis inducer erastin enhances accumulation of STING in mitochondria increasing mitochondrial fusion concomitantly with ROS increase and lipid peroxidation, and in xenografted mice STING KO renders these cells less prone to ferroptosis induction. [109]. In a broad context, the mechanism of such STING action may involve ROS that act through 4-hydoxy-nonenal promoting STING carbonylation at C88 preventing palmitoylation and translocation from ER thus ROS action is predominantly STING inhibiting, this may decrease both responses against pathogens or tumors [110]. Also, STING may promote ferroptosis in kidney, and at least one of the mechanism of such phenomena is STING-dependent ferritinophagy through interacting with nuclear receptor coactivator 4 (NCOA4), and the ferroptosis with increased ROS and lipid peroxidation may be controlled by increase of STING expression (positively) or blocked by STING antagonists [111].
STING contributes to autophagy, facilitating the removal of cytoplasmic DNA. This process results in a reduction of cGAS levels, while beclin 1 acts to inhibit DNA-cGAS. This intricate regulatory loop operates independently of IFN and TBK1 activation and is mediated by lipidated LC3 on the Endoplasmic Reticulum-Golgi Intermediate Compartment (ERGIC) and the Golgi, in a mechanism reliant on the COP-II complex and ARF GTPases. The ERGIC containing STING functions as a membrane source for LC3 lipidation, a critical phase in autophagosome formation. cGAMP triggers LC3 lipidation via a pathway dependent on WIPI2 and ATG5 but independent of ULK and VPS34-beclin kinase complexes.
In some cells the activation of cGAS relies on spleen tyrosine kinase (SYK), which phosphorylates Y214/215 in cGAS (or Y200/201 in mice) when cGAS forms a complex with V-ATPase [112]. Moreover, some cells activate the non-canonical STING pathway, leading to abundant secretion of the pro-cancer IL-6 (this also requires inhibition of ERK1/2 [113].
Interestingly, ATM modulates SRC to influence TBK1 and T1IFN. This aligns with SRC's established role in fostering PRR complexes with TBK1. It's possible that ATM serves to keep SRC activity in check, avoiding overactive inflammatory signaling [114].
A significant discovery has unveiled the STING-PKR-like endoplasmic reticulum kinase (PERK)-eIF2α pathway. This is a previously unidentified mechanism within the cGAS-STING pathway, which plays a crucial role in the innate immune regulation of cap-dependent messenger RNA translation. When cGAMP binds to STING, the latter, located at the ER, interacts and activates the ER-resident kinase, PERK, through their intracellular domains. This interaction occurs before the activation of TBK1-IRF3. Once activated, PERK phosphorylates eIF2α, establishing a translation program that prioritizes inflammation and survival [115]
In discussion of the regulation of the cGAS-cGAMP-STING-TBK1-IRF3 axis, we should stress again that STING knockout results in in heightened sensitivity to viral or bacterial pathogens. In oncology, however, this situation is much more complex and intricate.
5. Roles of cGAS-STING pathway in response to the radio- and chemotherapies
Radiotherapy elicits DNA damage and activates cGAS-STING
cGAS-STING pathway in response to radiotherapy that mechanistically activates dendrite cells (DC). In this schematic, a critical phase is marked by ionizing radiation (IR) provoking a surge in cGAMP secretion from tumor cells. This escalation subsequently induces the release of type I interferon in adjacent cells. This response, which is crucial to the adaptive immune response to radiation, relies predominantly on the cGAS-STING pathway rather than HMGB-1 release or MyD88 signaling [116]. cGAMP, acting through STING, initially induces TNFα-dependent hemorrhagic necrosis in a manner that is independent of T-cells. Subsequently, it aids in the CD8 T-cell-dependent containment of any residual disease, counteracting the tumor-promoting effects of M2 macrophages in pancreatic cancer [117].
Although radiation activates the cGAS/STING pathway through the generation of cytoplasmic DNA, ATM orchestrates signaling pathways to enhance TBK1's phosphorylation and activity, and consequently innate immunity, independently of cGAS/STING. Interestingly, ATM modulates SRC to influence TBK1 and T1IFN. This aligns with SRC's established role in fostering PRR complexes with TBK1 [114].
Chemotherapy and cGAS-STING
Pharmaceutical agents such as cisplatin and etoposide have the capability to induce leakage of nuclear DNA into the cytoplasm, inherently triggering STING-dependent cytokine production. Subsequently, these inflammatory cytokines undergo external amplification through a STING-dependent process, facilitated by infiltrating phagocytes that target the dying cells [118]. Furthermore, in tumor-bearing mice cGAMP increases the antitumor activity of 5-FU, and also reducing its toxicity [119].
In cancers deficient in homologous recombination (HR), it seems that the cGAS-STING axis fosters the survival of cancer cells. However, aberrations in this axis make the cells less responsive to PARP inhibitors (PARPi). Additionally, the cGAS-STING pathway holds a significant role in response to other DNA-damaging agents. For instance, cGAS-STING signaling amplifies the immune response induced by cisplatin in patients with bladder cancer. In a mouse xenograft tumor model, cGAS-STING signaling induction by cisplatin reduced bladder cancer cell growth and enhanced CD8+ T-cell and dendritic cell infiltration [120]. In BRCA1 deficient breast cancers, cisplatin also acts concomitantly with STING activation [121,97].
PARPi and cGAS-STING
cGAS co-localizes with PARP1, and by inhibiting the DNA Damage Response (DDR), cGAS prevents certain viruses from hijacking this mechanism. Notably, olaparib, a characteristic PARP inhibitor (PARPi), obstructs the interaction between PARP1 and cGAS [87]. In traditional fashion, PARPi induces accumulation of cytosolic DNA fragments due to unresolved DNA damage, stimulating the cGAS-STING pathway and amplifying the production of type I interferons. PARPi also improves anti-tumor immunity independently of a BRCA background [122], but olaparib in conjunction with STING agonists impedes murine BRCA1 deficient breast cancer model growth in wild type mice but shows no impact in STING-KO variants [123].
Certainly, the action of PARPi in BRCA2 deficient tumors correlate well with the immune cell infiltration, moreover, resistance to PARPi negatively correlates with decreased infiltration including production of pro-inflammatory cytokines like TNFα [124].
In a murine Brca1-deficient ovarian cancer model, the use of olaparib to inhibit PARP1 promotes antitumor immunity, especially when combined with PD-L blocking antibodies, and this action is STING-dependent [125]. Notably, in many cancers with DDR defects, such as NSCLC deficient in ERCC1 (excision repair cross-complementation group 1) or TNBC deficient in BRCA1, PARPi synergize with IFN-γ to induce PD-L1 expression on the cell surface, thereby amplifying the effectiveness of immune checkpoint inhibitors, ICI [126]. However, a contradictory observation arises in mouse and human ovarian cancer models. In this context, cells exhibit divergent sensitivities to olaparib when analyzed in vitro compared to in vivo, with the latter scenario revealing a progressive development of resistance. This resistance is tied to STAT3 activation in tumor cells, simultaneously occurring with the activity of pro-tumor M2-like TAMs, and the latter process is apparently STING-independent, while STING agonists promote reprogramming tumor-associated cells in favor of anti-tumor activity in the case of PARPi-resistant tumors [127].
Immune checkpoint blockade (ICB) and cGAS-STING pathway
ICB relies on the binding of specific monoclonal antibodies to inhibitory immune proteins PD-1, PD-L1, CTLA4, LAG3. Here, the cGAS-STING pathway is also important [21]. Also, DDR and ICB have a profound connection: deficiencies in DDR mechanisms can give rise to immunogenic cancer neoantigens, thereby amplifying the efficacy of ICB therapies [128].
6. Duality of the cGAS-STING pathway effects in cancer
As a rule, nucleosomal cGAS exerts a certain pro-oncogenic effect; since it inhibits HR (but not NHEJ) in the presence of double-stranded breaks through direct binding to PARP1 (via poly(ADP-ribose) preventing binding to replication fork-stabilizing protein Timeless. Genotoxic exposures such as etoposide, camptothecin and peroxide promotes BLK (B-lymphoid tyrosine kinase), also constitutive BLK phosphorylation of cGAS tyr215 seems to retain cGAS in the cytoplasm, and dephosphorylation of cGAS (by unknown phosphatases) in response to DNA damage leads to its nuclear translocation. In many models (immortalized human HCA2-TERT fibroblasts, A549 tumors, LLC in male C57BL/6 mice) to such extent that cGAS knockout inhibits growth concomitantly with inhibitions of DNA damage. [87]. This cGAS action presumably occurs independently of STING and also may promote growth of some tumors.
It is well known that tumors whose initiation depends on ATM or BRCA1 loss have relatively high levels of inflammation along with T-cell infiltration from the beginning of malignant growth, but this does not lead to the elimination of early tumors due to STING and IFN gene activation. Thus, STING knockout results in decreased neoangiogenesis, increased CD8+ T-cell infiltration, and restoration of therapeutic resistance to double immune checkpoint (ICB) blockade [129]. It is not unequivocally proven, but quite likely that nuclear cGAS can inhibit DNA repair, thereby contributing to oncogenesis at the level of the whole organism [87].
Prolonged activation and subsequent inflammation can lead to immune cell exhaustion, a phenomenon frequently observed in tumor environments. Although complete deactivation of the cGAS-STING pathway in primary tumors is rare, overstimulation of this pathway can paradoxically support cancer growth. This is exemplified by the activation markers such as TBK1 and IRF3 phosphorylation and heightened IFN-β and ISG mRNA expression. Specifically, in Triple Negative Breast Cancers (TNBCs), an overactive cGAS–IL-6–IL-6R signaling is linked to decreased survival [130].
Genotoxic stress in STING-positive cancers causes activation of this pathway, and in addition, some cells activate the non-canonical STING pathway, leading to abundant secretion of the immunosuppressive IL-6 (this also requires inhibition of ERK1/2 [113]. Perhaps, non-canonical signaling through STING is responsible the most for the pro-oncogenic modalities of STING action.
Chromosomal instability (CIN) manifests in tangible outcomes like aneuploidy. However, its root causes can vary significantly. For instance, 17 distinct CIN signatures have been pinpointed, potentially reflecting 17 different etiologies [131]. Therefore, understanding the right contexts that influence the effects either in pro- or contra tumor progression in any complex pathway such as cGAS-STING is premature and it will require many future efforts. The inherent duality of the role of cGAS- STING in high CIN cancers can be outlined as follows: cancer cells attract immune cells and die, but simultaneously activate motility and distant metastasis [132].
In certain models of triple-negative breast cancer (TNBC) cells exhibiting high CIN, the reduction of cGAS–STING levels resulted in a decrease of IL-6 production and reduced survival of cancer cells. Indeed, the production of cytokines like IL-6 is regulated by the non-canonical NF-κB signaling, moreover, this pathway likely disrupts the conventional STING–TBK1–IFN signaling, thus suggesting that STING activation may yield both anti- and pro-tumor effects depending on transient states of the evolving tumor [133].
7. Metastasis and cGAS-STING pathway
STING activation by cGAMP may curtail metastasis in specific models, such as the murine CT26 colorectal cancer, perhaps due to the curbed suppression of T-cells by MDSC-driven ROS and NOS [134]. In some other oncological contexts STING activation is also extremely effective in inhibiting metastasis, for example, in lung adenocarcinoma (LUAD), where migrated cancer cells may stay for a significant amount of time in a dormant state escaping the detection by NK and T cells. In one murine model STING activation has been shown to be extremely effective in suppressing the activation of such pre-disseminated cells [135].
Unfortunately, in high CIN cancers cGAS-STING somehow promotes motility and metastasis [132,136], most likely due to the aforementioned connection between STING activation and upregulation of NF-KB, which subsequently elevates the expression of EMT-associated transcription factors.
Epithelial-mesenchymal transition (EMT) in cancer is a complex and partially reversible phenomenon that mirrors developmental processes. It hinges on the transient adoption of migratory cell phenotypes contributing to the spread of the primary tumor and seeding metastases. Mechanistically, EMT is driven by the activation of a specific gene signature. However, the role of cGAS-STING in mediating interactions between tumor cells and their microenvironment, including EMT, remains largely elusive (reviewed in [137]).
It is important to note that cGAS/STING activity alone is inadequate to induce EMT or account for invasive behavior; moreover, at least in some models, the activation of NF-kappaB exhibits anti-tumor properties even when occurring alongside aneuploidy [138].
A recent study proposed a solution to the problem: it is CIN, rather than aneuploidy itself, that is a principal driver of cancer metastasis suggesting that chromosomes lagging due to CIN become entrapped outside the nucleus, creating micronuclei, and get detected by the cGAS/STING cytosolic DNA sensing pathway, which activates the non-canonical NF-κB signaling, resulting in an EMT and heightened cellular invasiveness [136].
Another metastasis-promoting role of STING activation is connected to tachyphylaxis, a diminished IFN responsiveness to repeated stimulation, which occurs during persistent activation of cGAS-STING signaling, concomitant with non-canonical NF-kappaB pathway induction. In human TNBC, tumors with high GAS and low STING displayed fewer tumor-infiltrating lymphocytes and were linked to decreased distant metastasis-free survival (DMFS), whereas tumors with low cGAS and high STING demonstrated a more favorable prognosis [139].
8. Pharmaceutical achievements in the regulation of the cGAS-STING pathway
For many tumors, STING agonists are the prime candidates for precision oncotherapy. In many mouse models, stimulating STING with small molecules has displayed promising outcomes. This activation leads to a decrease in tumor size, primarily due to increased T-cell infiltration, tumor vesicle disruption, and heightened apoptosis. More importantly, it enhances the immunogenic properties of tumors that were previously resistant to immunological interventions, reverting resistance to anti-PD-1 therapies. The resulting secreted IFN-1 has multiple effects: it activates antigen-presenting cells, including dendritic cells (DCs), it stimulates natural killer cells (NKs), it hinders the mobilization of M2 macrophages and MDSCs, it primes cytotoxic cells. Simple intratumoral administration of cGAMP works to some extent [140,141] , The early results, for example, with pancreatic cancer in mice, have demonstrated that cGAMP acts through STING T-cell-independent and TNFα-dependent hemorrhagic necrosis [117].
STING agonists and antagonists (Fig 3) have been extensively reviewed [142,143,144,145,146,147], so here we only emphasize that development of these small molecules is challenged by relatively large binding pocket of 700 Da in the STING molecule and differences in mouse and human STINGs. Also, it is rather difficult to minimize side effects such excessive stress in T cells with the cytokine storm, apoptosis of cancer-associated cells rather than production of interferon, as well as the induction of the tolerogenic enzyme IDO. There are other crucial points to consider, starting from the vast inter-species differences. For instance, DMXAA binds efficiently to mouse STING but not to human STING, leading to stark differences between preclinical and clinical results. Desired effects might be muted due to interference from other pathways. For instance, to achieve the intended effects of cGAMP or other STING agonists, it might be necessary to block pathways like JAK2-STAT3 [148]
Compound 53 (Figure 3) allowed to unveil an extremely interesting feature of STING, since it binds STING in its transmembrane area, inhibiting ion-channeling activity, a distinction from MSA-2 and diABZI [62]. This leads to a marked increase in Golgi pH levels, due to its intrinsic role as a proton channel, vital for the activation of the NLRP3 inflammasome and subsequent pyroptosis, also resulting in IL-1beta secretion, and is independent of lipidation and autophagy.
Stabilized cyclic dinucleotide compounds, which activate human STING, are in clinical trials for potential application in cancer immunotherapy, however, the toxicity limit of these agents may be tied to the initiation of necroptosis due to STING hyperactivation of a "shock-like" condition, simultaneously increasing the levels of TNF and various inflammatory cytokines. Yet, reported clinical trials showed that cGAMP-similar CDN analogs like ADU-S100 monotherapies are not effective either in oncohematology or in therapy of solid tumors and slightly more efficient in combination with pembrolizumab anti-PD1 (adaptive T cell blockade) – reviewed in 2022 by Samson and Ablasser [19].
Administration schedules
It was suggested quite early that anti-tumor STING activation in oncotherapies should be “acute and moderate” and not “persistent or extensive” [19]. This is now fully recognized: hit-and-run approaches are now preferred over prolonged treatments [149]. This ideas are in a sharp contrast to the experimentation with cycling regimes in combinations with PARPi and other drugs [150].
STING antagonists
Apparently, periodic inhibition and activation of the cGAS-STING pathway is also worth trying. Indeed, many small molecule STING inhibitors have been developed, including irreversible ones [151] and PROTACs [152]. While these inhibitors were primarily designed for treating auto-immune diseases, their potential in oncology should not be ignored.
Nanosystems for efficient delivery of cGAS-STING modulators
cGAMP and analogs work much better when aided by smart delivery vehicles. Since there is an excellent review [153], then among recent breakthroughs and improvements using these approaches the most interesting ones include polymerosomes made with matryoshka layers CDN encapsulated with PEI and biodegradable disulfide cross-lined polycarbonate and covered by PEG [154] so that it should unfold in the cytoplasm, polymerosomes [155], stable cyclic dinucleotide nanoparticles with a hydrophobic nucleotide lipid (3',5'-di(oleoyl-deoxycytidine) [156].
Optimization of liposome composition allowed to obtain CDNs stably incorporated into the liposomes; protect CDN from spontaneous degradation in blood; retaining activity ADU-100 [157]; and diABZI [158].
Other than through the use for the simple CDN delivery, some nanocomposites are believed to act through mitochondrial membrane damage concomitant with release of mtDNA and activation of cGAS-STING [159] these ultra-small Cu2−XSe (CS) nanoparticles on the organosilicon nanoparticles and covered with cancer cell membranes
STING as a drug
Here, STING mutants, such as N153S (first known from patients with the autoimmune disease STING-associated vasculopathy with onset in infancy) are especially valuable. In MC38 colorectal cancer cells, even an admixture of cells with such STING greatly increased the sensitivity of the whole population of cancer cells to ICB in vivo, achieving sensitization of “cold” tumors. This important discovery paves for the use of STING as a transgene in vehicles such as AAV, OVs and other [ 160].
Also, using nanoparticle delivery, the recombinant TM-truncated STING protein combined with cGAMP has been shown to effectively activate STING signaling both in vitro and in vivo [161].
9. Agonists of cGAS- STING pathway in distinctive cancer types and perspectives of personalization
It is important to realize tissue specificities for understanding some differences in tumor progression depending on the origin of the primary tumor as well as the tissues of the distant metastasis: cGAS is abundant in leukocytes, and have low expression level in the brain, liver, prostate, skeletal muscle, skin, intestine, testis, stomach, thus many of the described effects are pertinent for cGAS in infiltrating blood cells. STING is abundant in endothelial cells and cardiomyocytes, macrophages, DCs. These simple facts are important when considering perspectives of different STING pathway modulators depending on the tissue of tumor origin. Potential targets for STING modulators are within the tumor microenvironment, which is abundant in immune cells. These immune cells can subsequently exert effects on the tumor itself.
Gliomas
Reflecting on the introduction of various ways of modulating the cGAS-STING pathway in oncology, it is important to realize that in many tissues the level of STING expression is low, especially in most normal brain cells as well as in human glioblastoma cells (a classic example of an immunologically cold tumor). In contrast, it appears that blood vessels associated with glioblastoma are STING positive, so they may well respond to STING agonists, such as ADU-S100, by secretion of inflammatory cytokines. At least in some mouse models of glioblastoma (especially in GL261 cells, where both KRAS and p53 are mutated), ADU-S100 causes massive infiltration by inflammatory macrophages, neutrophils and NK, prolonging survival. It is possible that STING can also be successfully activated in human glioblastoma, where it is strongly expressed in tumor-associated blood vessels [162].
In other gliomas STING may also be important despite relatively low expression level. For example, STING may increase resistance to temozolomide (TMZ) in pediatric high grade glioma (pHGG) through interaction with GBP3 (TMZ upregulates it), inducing expression of p62 (Sequestosome 1), nuclear factor erythroid 2 like 2 (NFE2L2, NRF2), and O6-methlyguanine-DNA-methyltransferase (MGMT). In patients, GBP3 expression is high and positively correlated with STING, NRF2, p62, and MGMT expression in human glioblastoma tumors. IR increases in IFN-β levels and this can be blocked by STING inhibitors (GSK690693, H151).
IR also induce secretion of DAMPs (ATP and HMGB1). DAMP levels in H3.3-G34R and H3.3-WT mouse and human pHGG cells in response to IR was also reduced by these inhibitors. Therefore, cGAS/STING pathway affects stimulation of DAMPs released upon IR-induced DNA damage [163].
PARP inhibitor pamiparib appears to penetrate through BBB and STING agonist (diABZl) increases efficacy of the treatment by checkpoint CHK1/2 inhibition (together with 3.3-G34R pHGG and AZD7762) and in a murine model [164].
Pancreatic ductal adenocarcinoma (PDAC)
It is important to emphasize again that STING-positive cancers tend to be immunologically "hot" and STING-negative cancers tend to be "cold", with the clear exception of highly aggressive pancreatic ductal adenocarcinoma (PDAC) among other cold cancers, reflecting their nature that obviously does not fall under the dichotomous "temperature" classification [165]. This type of cancer often exhibits fibrosis/desmoplasia, characterized by infiltration of pancreatic stellate cells, fibroblasts, and immune cells, all contributing to a complex extracellular matrix. Interestingly, STING expression is prevalent in 90% of PDAC cases, whereas tumors expressing both cGAS and STING are associated with somewhat improved patient survival. Such double-positive tumors have strong infiltration by cytotoxic CD8+ cells [166]. This is presumably due to activation of the canonic cGAS-STING pathway, however, it is important to remember that there is also STING-PERK-eIF2α route that promotes fibrosis[115]
In PDAC, CD73 emerges as a promising therapeutic target. Its presence is linked with poorer patient survival. CD73, expressed on both tumor-associated macrophages (TAMs) and PDAC cells, influences myeloid cells to adopt an M2-like phenotype. These M2-like cells, when in proximity to PDACs, promote tumor growth and diminish the genotoxic stress induced by treatments like ionizing radiation (IR) or gemcitabine. Intriguingly, the absence of CD73 appears to stimulate the cGAS-STING pathway [167].
In PDAC cells with p53 mutations, the formation of micronuclei is linked to the mitotic phosphorylation of SUMO-specific protease 3 (SENP3). Within the DNA damage response (DDR), p53 activates SENP3, which in turn promote cellular senescence in positive correlation with longer survival outcomes for PDAC patients. Furthermore, SENP3 activity enhances anti-tumor immune responses, a mechanism that is facilitated by cGAS[168].
Melanoma
In melanoma research, the B16F10 murine syngeneic model, a classical "cold" cancer, is rather popular and it is well known to respond to STING agonists [169]. For immune checkpoint anti-tumor therapies, the cGAS-STING pathway is crucial. In experiments, mice lacking cGAS exhibited reduced B16 melanoma growth when treated with a PD-L1 antibody. In line with this, direct cGAMP delivery into the muscles hindered melanoma growth and prolonged the survival of the tumor-bearing mice, even though cGAMP should be rapidly degraded by the enzyme, it might not have sufficient time to exert its effect [170].
Oncohematology
Considering the potential effects or the interferon-independent actions of STING, these might be especially useful in oncohematology. For instance, a specific STING agonist, DMXAA, induces the expression of IFN-β and various inflammatory cytokines. This not only promotes dendritic cell maturation but also results in a remarkable proliferation of leukemia-specific T cells, extending survival in two AML models [171].
Personalization on the basis of genomic profiling
Importantly, in the human population there exist STING polymorphisms: R71H-G230A-R293Q (HAQ) in 20.4%, R232H in 13.7%, G230A-R293Q (AQ) in 5.2%, and R293Q in 1.5%. The R71H substitution has a notably lower intrinsic activity, the R232H and R293Q variant are poorly responsive to bacterial CDNs such as c-di-AMP and 3'3' cGAMP, and these variants are important for bacterial infections [172,173]. Therefore, personalized approaches [174] should be accordingly weighted in oncology. When developing a therapy using a specific agonist, it's crucial to identify the genetic variants of STING that the agonist can or cannot bind to, and the presence of a specific STING variant in a patient should be considered before determining the appropriate treatment.
Modulating cGAS-STING according to the tumor peculiarities
Emphasizing personalized treatments and acknowledging individual variances remain an important avenue [19]. Some STING agonists appear to function independently of T cells, despite the fact that STING agonist triggered type 1 interferon affects a variety of cells, including T-lymphocytes. Therefore, their tumor-reducing effects ought to have precise calibration to optimize their therapeutic impact [175] on innate signaling via APCs in the tumor microenvironment (TME), facilitating cross-priming of tumor antigen-specific CD8+ T cells.
Recent studies have revealed that ARID1A, an integral part of the highly conserved SWI/SNF chromatin-remodeling complex, facilitates RNF8-driven ubiquitination and subsequent degradation of Chk2. In cancers where ARID1A is deleted, there's an activation of the STING-mediated innate immune response. Consequently, tumors characterized by mutations or reduced expression of both ARID1A and ATM/Chk2 exhibited increased lymphocyte infiltration and were less aggressive. [176]. This observation aligns with the recognized synergy between lymphocyte mobilization and ATM deficiency [177].
For cancers with KRAS mutations, it is established that mtDNA release into the cytoplasm can activate STING, and suppression of STING has been linked to immune evasion in lung cancer cells with KRAS mutations [178].
EGFR enhances STING activity through phosphorylation [179], therefore, evaluating the influence of all recognized hyperactive EGFR mutations, especially those integral to oncogenesis, STING movement, and degradation, becomes paramount. Additionally, the assessment of pharmacologically relevant EGFR inhibitors is warranted.
For individualization and making choices on whether to include STING agonists in oncotherapeutic regimes or not, it is reasonable to suggest that test tumors for high CIN and proper selection of STING modulators should be done, but it is clear that the problem of practical measurements of CIN is rather difficult so surrogate measures such as aneuploidy should be routinely done with tumor biopsies. Moreover, considering the problem of the decline in IFN response with continued STING activation, it has been proposed that a certain group of patients might benefit more from inhibiting the cGAS-STING pathway to control the tumor's inherent chronic inflammation and its resulting immune suppression (characterized by high cGAS and low STING levels [139].
10. Perspectives for efficient synergies between cGAS-STING regulation and other anti-cancer therapeutic modalities
Interactions between different anti-cancer drugs
Complex combinations of STING agonists with diverse drugs should be explored. In TNBC contexts, for example, STING agonists improve CIP therapy outcomes (involving IL2 and anti-PD-1) in TNBC animal models, particularly amplifying NK activation – a critical factor in metastasis inhibition [180]. To enhance the specificity of their action, the focus should be on how STING can be delivered exclusively to targeted cells.
Combination of a systemic STING agonist MSA-2 (Figure 3) with osimertinib, a third-generation EGFR inhibitor, leads to a decline in advanced PE tumors, perhaps through the action of the STING on the tumor microenvironment [181].
Broad protein kinase inhibitors and autophagy inducers such as sorafenib promote degradation of cGAS, TBK1, and IRF3, thus inhibiting the recruiting of STING with TBK1 and IRF [182]. Perhaps such compounds may find future use in many cancers with excessive STING activation in proper combinations with other drugs and not only in renal and hepatocellular carcinomas.
In conjunction with certain cytotoxic therapies, Checkpoint Kinase 1 (Chk1) inhibition modifies the internal cellular environment but sometimes suppresses expected responses [183]. One proposed solution is supplement Chk1 inhibitors with STING agonists, and some strategies indicated potential synergy [184].
Cell-based therapies
While the inherent T cell receptors (TCRs) usually demonstrate low affinity for self or tumor antigens, there's a transformative approach. Extracting a patient's lymphocytes, they're reprogrammed with chimeric antigen receptors (CARs). These CARs are specially designed to firmly latch onto proteins that tumors exhibit. However, challenges persist: solid tumors emit signals that deter T cells and hide specific oncomarkers [185]. Perhaps STING agonists should be an ideal match with CAR-T cells, and now intensive studies are being carried out to validate this combination. Indeed, when combined with the STING agonists DMXAA or cGAMP, CAR-T cells significantly improved tumor management, perhaps due to a prolonged presence of CAR T cells within the tumor microenvironment [186,185].
Immunotherapies such as Oncolytic Viruses (OV)
Mechanistically, impaired STING pathway renders cancer cell more sensitive to direct viral oncolysis by definition, since that means that these cells are defenseless against DNA viruses and retroviruses. At least this is true with T-VEC (GM-CSF bearing HSV) in melanoma [187]. Therefore, OVs are prime candidates for modern oncotherapies against STING pathway impaired cancers. STING itself or STING agonistic sequences were used as transgenes in OVs, and this was also suggested independently [188-190].
Thus, the initial concept behind the use of oncolytic viruses (OVs) against cancer cells was rooted in the viruses' ability to selectively replicate within tumors, subsequently killing the tumor cells and disrupting their supporting vasculature. However, a more contemporary perspective posits that OVs function as a form of immunotherapy. This is often termed "immunogenic cell death," wherein virus-infected tumor cells, while dying, release damage-associated molecular patterns (DAMPs) that attract immune cells and stimulate antitumor responses. Additionally, these viruses can target the tumor-associated vasculature by replicating within endothelial cells.
Among poxviruses, engineered vaccinia virus has been extensively investigated as OVs for aggressive cancers. For example, genetic engineering gave a potent immune-activating variant of MVA (rMVA, MVA∆E5R-Flt3L-OX40L). Removing a specific gene and incorporating two others yielded a powerful anti-tumor response, contingent on several cellular pathways. The potential of integrating STING agonist transgenes has also been posited. [188]
Interestingly, killed virus in some settings produces excellent anti-cancer effects, underlining the immuno-activation as the predominant fashion of action of at least some OVs. For example, Inactivated Modified Vaccinia Virus in human or murine plasmacytoid dendritic cells (pDCs) induces type I IFN whereas live vaccinia virus fails. Here viral DNAs in the cytosol activated cGAS and downstream –STING, IRF3 and IRF7, IFN gene expression, and tumor death through the generation of antitumor CD8+ and CD4+ effectors [189].
It also important to remember that the activation of STING may impede the action of OVs (especially OVs with double stranded DNA genomes such as poxviruses, herpesviruses and adenoviruses) on cancer cells both in vitro and in vivo.
11. Conclusions
As a result of the incessant struggle of multicellular organisms against infections, the evolution of innate immunity has shaped cGAS-STING pathway so that it has rather complex relations with aging and oncogenesis. While this anti-pathogen sensor has been adeptly repurposed to detect malignant cells with abnormal nuclei, its functions in an aging and consequently cancer-prone body have become so convoluted that many cancers can co-opt elements of this pathway, distort its function, and use it against the immune system. Therefore, additional studies are required to properly employ modulators of this pathway in oncology. Particularly, it is crucial to highlight the challenges in extrapolating results from animal models to humans. Their limitations are exemplified by the fact that mouse dendritic cells do not produce IFN-λ1, a molecule prevalent in certain human cells [191].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Proteins engaged in cGAS-STING pathway regulation of importance for oncology.
Author Contributions
Conceptualization and writing—N.B.P.,I.A.N.,A.A.D.,O.N.S.; supplementary table—K.N.T., visualization—T.V.K; editing and funding acquisition—N.A.B. All authors have read and agreed to the published version of the manuscript.”
Funding
This work was financed by the Ministry of Science and Higher Education of the Russian Federation within the framework of state support for the creation and development of World-Class Research Centers ‘Digital Biodesign and Personalized Healthcare’ (No 75-15-2020-913).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Authors are grateful to Y.M. Rozenberg for critical reading of the manuscript.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING Is a Direct Innate Immune Sensor of Cyclic Di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS Produces a 2’-5’-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2’,5’)pA(3’,5’)p] Is the Metazoan Second Messenger Produced by DNA-Activated Cyclic GMP-AMP Synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef]
- Shi, X.; Wang, S.; Wu, Y.; Li, Q.; Zhang, T.; Min, K.; Feng, D.; Liu, M.; Wei, J.; Zhu, L.; et al. A Bibliometric Analysis of the Innate Immune DNA Sensing cGAS-STING Pathway from 2013 to 2021. Front Immunol 2022, 13, 916383. [Google Scholar] [CrossRef]
- Yoon, S.H.; Waters, C.M. The Ever-Expanding World of Bacterial Cyclic Oligonucleotide Second Messengers. Curr Opin Microbiol 2021, 60, 96–103. [Google Scholar] [CrossRef]
- Wein, T.; Sorek, R. Bacterial Origins of Human Cell-Autonomous Innate Immune Mechanisms. Nat Rev Immunol 2022, 22, 629–638. [Google Scholar] [CrossRef]
- Bose, D. cGAS/STING Pathway in Cancer: Jekyll and Hyde Story of Cancer Immune Response. Int J Mol Sci 2017, 18, 2456. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Song, Y.; Chu, Q.; Wu, K. Activating cGAS-STING Pathway for the Optimal Effect of Cancer Immunotherapy. J Hematol Oncol 2019, 12, 35. [Google Scholar] [CrossRef]
- Hoong, B.Y.D.; Gan, Y.H.; Liu, H.; Chen, E.S. cGAS-STING Pathway in Oncogenesis and Cancer Therapeutics. Oncotarget 2020, 11, 2930–2955. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.-C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA Damage Response in Immuno-Oncology: Developments and Opportunities. Nat Rev Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov 2021, 11, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.N.; Victorelli, S.G.; Salmonowicz, H.; Dasgupta, N.; Liu, T.; Passos, J.F.; Adams, P.D. Cytoplasmic DNA: Sources, Sensing, and Role in Aging and Disease. Cell 2021, 184, 5506–5526. [Google Scholar] [CrossRef] [PubMed]
- Vashi, N.; Bakhoum, S.F. The Evolution of STING Signaling and Its Involvement in Cancer. Trends Biochem Sci 2021, 46, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-M.; Qian, M.-J.; Yuan, T.; Chen, R.-H.; He, Q.-J.; Yang, B.; Ling, Q.; Zhu, H. cGAS and Cancer Therapy: A Double-Edged Sword. Acta Pharmacol Sin 2022, 43, 2202–2211. [Google Scholar] [CrossRef]
- Huang, R.; Ning, Q.; Zhao, J.; Zhao, X.; Zeng, L.; Yi, Y.; Tang, S. Targeting STING for Cancer Immunotherapy: From Mechanisms to Translation. Int Immunopharmacol 2022, 113, 109304. [Google Scholar] [CrossRef]
- Li, J.; Bakhoum, S.F. The Pleiotropic Roles of cGAS-STING Signaling in the Tumor Microenvironment. J Mol Cell Biol 2022, 14, mjac019. [Google Scholar] [CrossRef]
- Samson, N.; Ablasser, A. The cGAS-STING Pathway and Cancer. Nat Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef]
- Xu, Y.; Nowsheen, S.; Deng, M. DNA Repair Deficiency Regulates Immunity Response in Cancers: Molecular Mechanism and Approaches for Combining Immunotherapy. Cancers (Basel) 2023, 15, 1619. [Google Scholar] [CrossRef]
- Yum, S.; Li, M.; Frankel, A.E.; Chen, Z.J. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annual Review of Cancer Biology 2019, 3, 323–344. [Google Scholar] [CrossRef]
- Huérfano, S.; Šroller, V.; Bruštíková, K.; Horníková, L.; Forstová, J. The Interplay between Viruses and Host DNA Sensors. Viruses 2022, 14, 666. [Google Scholar] [CrossRef] [PubMed]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural Mechanism of Cytosolic DNA Sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Imler, J.-L. cGAS-STING: Insight on the Evolution of a Primordial Antiviral Signaling Cassette. Fac Rev 2021, 10, 54. [Google Scholar] [CrossRef]
- Kranzusch, P.J. cGAS and CD-NTase Enzymes: Structure, Mechanism, and Evolution. Curr Opin Struct Biol 2019, 59, 178–187. [Google Scholar] [CrossRef]
- Kuchta, K.; Knizewski, L.; Wyrwicz, L.S.; Rychlewski, L.; Ginalski, K. Comprehensive Classification of Nucleotidyltransferase Fold Proteins: Identification of Novel Families and Their Representatives in Human. Nucleic Acids Res 2009, 37, 7701–7714. [Google Scholar] [CrossRef] [PubMed]
- Kranzusch, P.J.; Lee, A.S.-Y.; Berger, J.M.; Doudna, J.A. Structure of Human cGAS Reveals a Conserved Family of Second-Messenger Enzymes in Innate Immunity. Cell Rep 2013, 3, 1362–1368. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS Senses Long and HMGB/TFAM-Bound U-Turn DNA by Forming Protein-DNA Ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef]
- Herzner, A.-M.; Hagmann, C.A.; Goldeck, M.; Wolter, S.; Kübler, K.; Wittmann, S.; Gramberg, T.; Andreeva, L.; Hopfner, K.-P.; Mertens, C.; et al. Sequence-Specific Activation of the DNA Sensor cGAS by Y-Form DNA Structures as Found in Primary HIV-1 cDNA. Nat Immunol 2015, 16, 1025–1033. [Google Scholar] [CrossRef]
- Ak, M.; T, S.; D, C.; M, G.; K, H.; M, G.; Av, K.; L, A.; Kp, H.; V, H. Cytosolic RNA:DNA Hybrids Activate the cGAS-STING Axis. The EMBO journal 2014, 33. [Google Scholar] [CrossRef]
- Crossley, M.P.; Song, C.; Bocek, M.J.; Choi, J.-H.; Kousorous, J.; Sathirachinda, A.; Lin, C.; Brickner, J.R.; Bai, G.; Lans, H.; et al. R-Loop-Derived Cytoplasmic RNA-DNA Hybrids Activate an Immune Response. Nature 2023, 613, 187–194. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.-J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.-L.; et al. SAMHD1 Acts at Stalled Replication Forks to Prevent Interferon Induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Xia, P.; Wang, S.; Ye, B.; Du, Y.; Li, C.; Xiong, Z.; Qu, Y.; Fan, Z. A Circular RNA Protects Dormant Hematopoietic Stem Cells from DNA Sensor cGAS-Mediated Exhaustion. Immunity 2018, 48, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-Viral Specificity of IFN-Induced Genes Reveals New Roles for cGAS in Innate Immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rong, M.; Lv, Y.; Zhu, D.; Xiang, Y. Regulation of cGAS Activity by RNA-Modulated Phase Separation. EMBO Rep 2023, 24, e51800. [Google Scholar] [CrossRef]
- Liu, C.; Yang, J.; Zheng, J.; Fu, J.; Wang, W.; Duan, L.; Qian, X.; Yang, Y. Phase Separation in cGAS-STING Signaling: Cytosolic DNA Sensing and Regulatory Functions. Chembiochem 2023, 24, e202300147. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Wan, L.; Juszkiewicz, S.; Blears, D.; Bajpe, P.K.; Han, Z.; Faull, P.; Mitter, R.; Stewart, A.; Snijders, A.P.; Hegde, R.S.; et al. Translation Stress and Collided Ribosomes Are Co-Activators of cGAS. Mol Cell 2021, 81, 2808–2822. [Google Scholar] [CrossRef]
- Guerra, J.; Valadao, A.-L.; Vlachakis, D.; Polak, K.; Vila, I.K.; Taffoni, C.; Prabakaran, T.; Marriott, A.S.; Kaczmarek, R.; Houel, A.; et al. Lysyl-tRNA Synthetase Produces Diadenosine Tetraphosphate to Curb STING-Dependent Inflammation. Sci Adv 2020, 6, eaax3333. [Google Scholar] [CrossRef]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep 2014, 6, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Chen, Z.J. DNA-Induced Liquid Phase Condensation of cGAS Activates Innate Immune Signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446. [Google Scholar] [CrossRef] [PubMed]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.P.F.; Puig Lombardi, E.; Herve, S.; De Silva, N.S.; Rookhuizen, D.C.; Zueva, E.; Goudot, C.; et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep 2019, 26, 2377–2393. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Cambier, S.; Gray, E.E.; Stetson, D.B. Tight Nuclear Tethering of cGAS Is Essential for Preventing Autoreactivity. Elife 2019, 8, e47491. [Google Scholar] [CrossRef]
- Xie, W.; Lama, L.; Adura, C.; Tomita, D.; Glickman, J.F.; Tuschl, T.; Patel, D.J. Human cGAS Catalytic Domain Has an Additional DNA-Binding Interface That Enhances Enzymatic Activity and Liquid-Phase Condensation. Proc Natl Acad Sci U S A 2019, 116, 11946–11955. [Google Scholar] [CrossRef]
- Liu, Z.-S.; Cai, H.; Xue, W.; Wang, M.; Xia, T.; Li, W.-J.; Xing, J.-Q.; Zhao, M.; Huang, Y.-J.; Chen, S.; et al. G3BP1 Promotes DNA Binding and Activation of cGAS. Nat Immunol 2019, 20, 18–28. [Google Scholar] [CrossRef]
- Hornung, V.; Hartmann, R.; Ablasser, A.; Hopfner, K.-P. OAS Proteins and cGAS: Unifying Concepts in Sensing and Responding to Cytosolic Nucleic Acids. Nat Rev Immunol 2014, 14, 521–528. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type-I Interferon Pathway. Science 2013, 339, 10–1126. [Google Scholar] [CrossRef]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide That Activates Human STING. Cell Rep 2013, 3, 1355–1361. [Google Scholar] [CrossRef]
- Morehouse, B.R.; Govande, A.A.; Millman, A.; Keszei, A.F.A.; Lowey, B.; Ofir, G.; Shao, S.; Sorek, R.; Kranzusch, P.J. STING Cyclic Dinucleotide Sensing Originated in Bacteria. Nature 2020, 586, 429–433. [Google Scholar] [CrossRef]
- Shu, C.; Yi, G.; Watts, T.; Kao, C.C.; Li, P. Structure of STING Bound to Cyclic Di-GMP Reveals the Mechanism of Cyclic Dinucleotide Recognition by the Immune System. Nat Struct Mol Biol 2012, 19, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.-D.; Sotelo-Troha, K.; von Moltke, J.; Monroe, K.M.; Rae, C.S.; Brubaker, S.W.; Hyodo, M.; Hayakawa, Y.; Woodward, J.J.; Portnoy, D.A.; et al. The N-Ethyl-N-Nitrosourea-Induced Goldenticket Mouse Mutant Reveals an Essential Function of Sting in the in Vivo Interferon Response to Listeria Monocytogenes and Cyclic Dinucleotides. Infect Immun 2011, 79, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-Function Analysis of STING Activation by c[G(2’,5’)pA(3’,5’)p] and Targeting by Antiviral DMXAA. Cell 2013, 154, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wu, J.; Chen, Z.J.; Chen, C. Molecular Basis for the Specific Recognition of the Metazoan Cyclic GMP-AMP by the Innate Immune Adaptor Protein STING. Proc Natl Acad Sci U S A 2015, 112, 8947–8952. [Google Scholar] [CrossRef]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.-C.; Zhang, X. Cryo-EM Structures of STING Reveal Its Mechanism of Activation by Cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef]
- Lu, D.; Shang, G.; Li, J.; Lu, Y.; Bai, X.-C.; Zhang, X. Activation of STING by Targeting a Pocket in the Transmembrane Domain. Nature 2022, 604, 557–562. [Google Scholar] [CrossRef]
- Tang, E.D.; Wang, C.-Y. Single Amino Acid Change in STING Leads to Constitutive Active Signaling. PLoS One 2015, 10, e0120090. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A Conserved PLPLRT/SD Motif of STING Mediates the Recruitment and Activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef]
- Ergun, S.L.; Fernandez, D.; Weiss, T.M.; Li, L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 2019, 178, 290–301. [Google Scholar] [CrossRef]
- Liu, B.; Carlson, R.J.; Pires, I.S.; Gentili, M.; Feng, E.; Hellier, Q.; Schwartz, M.A.; Blainey, P.C.; Irvine, D.J.; Hacohen, N. Human STING Is a Proton Channel. Science 2023, 381, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, L.; Shen, J.; Zhai, Y.; Jiang, Q.; Yi, M.; Deng, X.; Ruan, Z.; Fang, R.; Chen, Z.; et al. The STING Phase-Separator Suppresses Innate Immune Signalling. Nat Cell Biol 2021, 23, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with Covalent Small-Molecule Inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is an Endogenous High-Affinity Ligand for STING. Mol Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef]
- Tang, C.-H.A.; Lee, A.C.; Chang, S.; Xu, Q.; Shao, A.; Lo, Y.; Spalek, W.T.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.-C.A. STING Regulates BCR Signaling in Normal and Malignant B Cells. Cell Mol Immunol 2021, 18, 1016–1031. [Google Scholar] [CrossRef]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci Signal 2012, 5, ra20. [Google Scholar] [CrossRef]
- Jin, L.; Xu, L.-G.; Yang, I.V.; Davidson, E.J.; Schwartz, D.A.; Wurfel, M.M.; Cambier, J.C. Identification and Characterization of a Loss-of-Function Human MPYS Variant. Genes Immun 2011, 12, 263–269. [Google Scholar] [CrossRef]
- Chamma, H.; Guha, S.; Laguette, N.; Vila, I.K. Protocol to Induce and Assess cGAS-STING Pathway Activation in Vitro. STAR Protoc 2022, 3, 101384. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Fang, Y.; Lin, Z.; Yang, L.; Zheng, L.; Hu, H.; Yu, T.; Huang, B.; Chen, S.; Wang, H.; et al. Inhibition of cGAS-Mediated Interferon Response Facilitates Transgene Expression. iScience 2020, 23, 101026. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Zhang, C.-P.; Qiu, H.-Y.; Zhang, H.-X.; Chen, Q.-B.; Zhang, Y.-M.; Lei, X.-L.; Zhang, C.-X.; Yin, H.; Zhang, Y. Enhancement of the Viability of T Cells Electroporated with DNA via Osmotic Dampening of the DNA-Sensing cGAS-STING Pathway. Nat Biomed Eng 2023. [Google Scholar] [CrossRef] [PubMed]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS Cooperate in the Activation of STING during DNA Sensing in Human Keratinocytes. Nat Commun 2017, 8, 14392. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-Canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol Cell 2018, 71, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Raulet, D.H. The DNA Damage Response Arouses the Immune System. Cancer Res 2006, 66, 3959–3962. [Google Scholar] [CrossRef]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. C-Di-AMP Secreted by Intracellular Listeria Monocytogenes Activates a Host Type I Interferon Response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Liu, X.-Y.; Du, X.-X.; Jiang, Z.-F.; Su, X.-D. The Structural Basis for the Sensing and Binding of Cyclic Di-GMP by STING. Nat Struct Mol Biol 2012, 19, 728–730. [Google Scholar] [CrossRef]
- Chen, F.; Wu, R.; Liu, J.; Kang, R.; Li, J.; Tang, D. The STING1-MYD88 Complex Drives ACOD1/IRG1 Expression and Function in Lethal Innate Immunity. iScience 2022, 25, 104561. [Google Scholar] [CrossRef]
- Hu, S.; Fang, Y.; Chen, X.; Cheng, T.; Zhao, M.; Du, M.; Li, T.; Li, M.; Zeng, Z.; Wei, Y.; et al. cGAS Restricts Colon Cancer Development by Protecting Intestinal Barrier Integrity. Proc Natl Acad Sci U S A 2021, 118, e2105747118. [Google Scholar] [CrossRef]
- Kim, Y.; Cho, N.-Y.; Jin, L.; Jin, H.Y.; Kang, G.H. Prognostic Significance of STING Expression in Solid Tumor: A Systematic Review and Meta-Analysis. Front Oncol 2023, 13, 1244962. [Google Scholar] [CrossRef] [PubMed]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through cGAS Promotes Senescence. Nat Cell Biol 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS Is Essential for Cellular Senescence. Proc Natl Acad Sci U S A 2017, 114, E4612–E4620. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Shen, Y.-L.; Hsia, H.-Y.; Tiang, Y.-P.; Sung, T.-L.; Chen, L.-Y. Extrachromosomal Telomere Repeat DNA Is Linked to ALT Development via cGAS-STING DNA Sensing Pathway. Nat Struct Mol Biol 2017, 24, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING Drives Ageing-Related Inflammation and Neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Haarer, E.L.; Theodore, C.J.; Guo, S.; Frier, R.B.; Campellone, K.G. Genomic Instability Caused by Arp2/3 Complex Inactivation Results in Micronucleus Biogenesis and Cellular Senescence. PLoS Genet 2023, 19, e1010045. [Google Scholar] [CrossRef]
- Haimovici, A.; Höfer, C.; Badr, M.T.; Bavafaye Haghighi, E.; Amer, T.; Boerries, M.; Bronsert, P.; Glavynskyi, I.; Fanfone, D.; Ichim, G.; et al. Spontaneous Activity of the Mitochondrial Apoptosis Pathway Drives Chromosomal Defects, the Appearance of Micronuclei and Cancer Metastasis through the Caspase-Activated DNAse. Cell Death Dis 2022, 13, 315. [Google Scholar] [CrossRef]
- MacDonald, K.M.; Nicholson-Puthenveedu, S.; Tageldein, M.M.; Khasnis, S.; Arrowsmith, C.H.; Harding, S.M. Antecedent Chromatin Organization Determines cGAS Recruitment to Ruptured Micronuclei. Nat Commun 2023, 14, 556. [Google Scholar] [CrossRef]
- Basit, A.; Cho, M.-G.; Kim, E.-Y.; Kwon, D.; Kang, S.-J.; Lee, J.-H. The cGAS/STING/TBK1/IRF3 Innate Immunity Pathway Maintains Chromosomal Stability through Regulation of P21 Levels. Exp Mol Med 2020, 52, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Li, J.; Montrose, D.C.; Martinez, L.A. P53 Engages the cGAS/STING Cytosolic DNA Sensing Pathway for Tumor Suppression. Mol Cell 2023, 83, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Saha, S.; Bettke, J.; Nagar, R.; Parrales, A.; Iwakuma, T.; van der Velden, A.W.M.; Martinez, L.A. Mutant P53 Suppresses Innate Immune Signaling to Promote Tumorigenesis. Cancer Cell 2021, 39, 494–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, H.; Zhang, J.; Wang, Y.; Simoneau, A.; Yang, H.; Levine, A.S.; Zou, L.; Chen, Z.; Lan, L. cGAS Suppresses Genomic Instability as a Decelerator of Replication Forks. Sci Adv 2020, 6, eabb8941. [Google Scholar] [CrossRef]
- Mateo, F.; He, Z.; Mei, L.; de Garibay, G.R.; Herranz, C.; García, N.; Lorentzian, A.; Baiges, A.; Blommaert, E.; Gómez, A.; et al. Modification of BRCA1-Associated Breast Cancer Risk by HMMR Overexpression. Nat Commun 2022, 13, 1895. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Kang, Z.; Foo, T.K.; Shen, Z.; Xia, B. Disrupted BRCA1-PALB2 Interaction Induces Tumor Immunosuppression and T-Lymphocyte Infiltration in HCC through cGAS-STING Pathway. Hepatology 2023, 77, 33–47. [Google Scholar] [CrossRef]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 Deficiency Instigates cGAS-Mediated Inflammatory Signaling and Confers Sensitivity to Tumor Necrosis Factor-Alpha-Mediated Cytotoxicity. Nat Commun 2019, 10, 100. [Google Scholar] [CrossRef]
- Banerjee, D.; Langberg, K.; Abbas, S.; Odermatt, E.; Yerramothu, P.; Volaric, M.; Reidenbach, M.A.; Krentz, K.J.; Rubinstein, C.D.; Brautigan, D.L.; et al. A Non-Canonical, Interferon-Independent Signaling Activity of cGAMP Triggers DNA Damage Response Signaling. Nat Commun 2021, 12, 6207. [Google Scholar] [CrossRef]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-Tumor Immunity. Cancer Cell 2021, 39, 96–108. [Google Scholar] [CrossRef]
- Song, S.; Peng, P.; Tang, Z.; Zhao, J.; Wu, W.; Li, H.; Shao, M.; Li, L.; Yang, C.; Duan, F.; et al. Decreased Expression of STING Predicts Poor Prognosis in Patients with Gastric Cancer. Sci Rep 2017, 7, 39858. [Google Scholar] [CrossRef]
- Gong, W.; Liu, P.; Zhao, F.; Liu, J.; Hong, Z.; Ren, H.; Gu, G.; Wang, G.; Wu, X.; Zheng, T.; et al. STING-Mediated Syk Signaling Attenuates Tumorigenesis of Colitis-associated Colorectal Cancer Through Enhancing Intestinal Epithelium Pyroptosis. Inflamm Bowel Dis 2022, 28, 572–585. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, R.; Tang, D. The STING1 Network Regulates Autophagy and Cell Death. Signal Transduct Target Ther 2021, 6, 208. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-H.A.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.-C.A. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res 2016, 76, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Weindel, C.G.; Smirnova, I.; Tang, A.Y.; Ilyukha, V.; Sorokin, M.; Buzdin, A.; Fitzgerald, K.A.; et al. Constitutive Interferon Signaling Maintains Critical Threshold of MLKL Expression to License Necroptosis. Cell Death Differ 2019, 26, 332–347. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.; Liu, Q.; Li, X.; Yang, Y.; Wu, L.; Wu, X.; Zhao, Y.; Ren, J. RIPK3-MLKL Necroptotic Signalling Amplifies STING Pathway and Exacerbates Lethal Sepsis. Clin Transl Med 2023, 13, e1334. [Google Scholar] [CrossRef]
- Brault, M.; Olsen, T.M.; Martinez, J.; Stetson, D.B.; Oberst, A. Intracellular Nucleic Acid Sensing Triggers Necroptosis through Synergistic Type I IFN and TNF Signaling. J Immunol 2018, 200, 2748–2756. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy Induction via STING Trafficking Is a Primordial Function of the cGAS Pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef]
- Li, C.; Liu, J.; Hou, W.; Kang, R.; Tang, D. STING1 Promotes Ferroptosis Through MFN1/2-Dependent Mitochondrial Fusion. Front Cell Dev Biol 2021, 9, 698679. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox Homeostasis Maintained by GPX4 Facilitates STING Activation. Nat Immunol 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Jin, L.; Yu, B.; Wang, H.; Shi, L.; Yang, J.; Wu, L.; Gao, C.; Pan, H.; Han, F.; Lin, W.; et al. STING Promotes Ferroptosis through NCOA4-Dependent Ferritinophagy in Acute Kidney Injury. Free Radic Biol Med 2023, S0891-5849(23)00605-6. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Cao, L.-B.; He, W.-R.; Zhong, L.; Guo, Y.; Yang, Q.; Shu, H.-B.; Hu, M.-M. Endocytosis Triggers V-ATPase-SYK-Mediated Priming of cGAS Activation and Innate Immune Response. Proc Natl Acad Sci U S A 2022, 119, e2207280119. [Google Scholar] [CrossRef] [PubMed]
- Al-Asmari, S.S.; Rajapakse, A.; Ullah, T.R.; Pépin, G.; Croft, L.V.; Gantier, M.P. Pharmacological Targeting of STING-Dependent IL-6 Production in Cancer Cells. Front Cell Dev Biol 2021, 9, 709618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S.; Parsels, L.A.; Shi, J.; Ramnath, N.; Wahl, D.R.; et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res 2019, 79, 3940–3951. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Zhu, Y.; Zhang, Q.; Guan, H.; Liu, S.; Chen, S.; Mei, C.; Chen, C.; Liao, Z.; et al. A Non-Canonical cGAS-STING-PERK Pathway Facilitates the Translational Program Critical for Senescence and Organ Fibrosis. Nat Cell Biol 2022, 24, 766–782. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Baird, J.R.; Friedman, D.; Cottam, B.; Dubensky, T.W.; Kanne, D.B.; Bambina, S.; Bahjat, K.; Crittenden, M.R.; Gough, M.J. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res 2016, 76, 50–61. [Google Scholar] [CrossRef]
- Ahn, J.; Xia, T.; Konno, H.; Konno, K.; Ruiz, P.; Barber, G.N. Inflammation-Driven Carcinogenesis Is Mediated through STING. Nat Commun 2014, 5, 5166. [Google Scholar] [CrossRef]
- Li, T.; Cheng, H.; Yuan, H.; Xu, Q.; Shu, C.; Zhang, Y.; Xu, P.; Tan, J.; Rui, Y.; Li, P.; et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep 2016, 6, 19049. [Google Scholar] [CrossRef]
- Fu, G.; Wu, Y.; Zhao, G.; Chen, X.; Xu, Z.; Sun, J.; Tian, J.; Cheng, Z.; Shi, Y.; Jin, B. Activation of cGAS-STING Signal to Inhibit the Proliferation of Bladder Cancer: The Immune Effect of Cisplatin. Cells 2022, 11, 3011. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst 2017, 109, djw199. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 2019, 79, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bergholz, J.S.; Ding, L.; Lin, Z.; Kabraji, S.K.; Hughes, M.E.; He, X.; Xie, S.; Jiang, T.; Wang, W.; et al. STING Agonism Reprograms Tumor-Associated Macrophages and Overcomes Resistance to PARP Inhibition in BRCA1-Deficient Models of Breast Cancer. Nat Commun 2022, 13, 3022. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 Abrogation Triggers Innate Immune Responses Potentiated by Treatment with PARP Inhibitors. Nat Commun 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep 2018, 25, 2972–2980. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP Inhibition Enhances Tumor Cell-Intrinsic Immunity in ERCC1-Deficient Non-Small Cell Lung Cancer. J Clin Invest 2019, 129, 1211–1228. [Google Scholar] [CrossRef]
- Ding, L.; Wang, Q.; Martincuks, A.; Kearns, M.J.; Jiang, T.; Lin, Z.; Cheng, X.; Qian, C.; Xie, S.; Kim, H.-J.; et al. STING Agonism Overcomes STAT3-Mediated Immunosuppression and Adaptive Resistance to PARP Inhibition in Ovarian Cancer. J Immunother Cancer 2023, 11, e005627. [Google Scholar] [CrossRef]
- Kornepati, A.V.R.; Rogers, C.M.; Sung, P.; Curiel, T.J. The Complementarity of DDR, Nucleic Acids and Anti-Tumour Immunity. Nature 2023, 619, 475–486. [Google Scholar] [CrossRef]
- Bruand, M.; Barras, D.; Mina, M.; Ghisoni, E.; Morotti, M.; Lanitis, E.; Fahr, N.; Desbuisson, M.; Grimm, A.; Zhang, H.; et al. Cell-Autonomous Inflammation of BRCA1-Deficient Ovarian Cancers Drives Both Tumor-Intrinsic Immunoreactivity and Immune Resistance via STING. Cell Rep 2021, 36, 109412. [Google Scholar] [CrossRef]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. cGAS-STING Drives the IL-6-Dependent Survival of Chromosomally Instable Cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarró, L.; Couturier, D.-L.; Liu, L.; Schneider, M.; et al. A Pan-Cancer Compendium of Chromosomal Instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tijhuis, A.E.; Foijer, F. The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells 2019, 8, 1228. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, O.P.G.; Unterholzner, L. DNA Sensing in Cancer: Pro-Tumour and Anti-Tumour Functions of cGAS-STING Signalling. Essays Biochem 2023, EBC20220241. [Google Scholar] [CrossRef]
- Cheng, H.; Xu, Q.; Lu, X.; Yuan, H.; Li, T.; Zhang, Y.; Tan, X. Activation of STING by cGAMP Regulates MDSCs to Suppress Tumor Metastasis via Reversing Epithelial-Mesenchymal Transition. Front Oncol 2020, 10, 896. [Google Scholar] [CrossRef]
- Hu, J.; Sánchez-Rivera, F.J.; Wang, Z.; Johnson, G.N.; Ho, Y.-J.; Ganesh, K.; Umeda, S.; Gan, S.; Mujal, A.M.; Delconte, R.B.; et al. STING Inhibits the Reactivation of Dormant Metastasis in Lung Adenocarcinoma. Nature 2023. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Shen, R.; Liu, D.; Wang, X.; Guo, Z.; Sun, H.; Song, Y.; Wang, D. DNA Damage and Activation of cGAS/STING Pathway Induce Tumor Microenvironment Remodeling. Front Cell Dev Biol 2021, 9, 828657. [Google Scholar] [CrossRef]
- Vasudevan, A.; Baruah, P.S.; Smith, J.C.; Wang, Z.; Sayles, N.M.; Andrews, P.; Kendall, J.; Leu, J.; Chunduri, N.K.; Levy, D.; et al. Single-Chromosomal Gains Can Function as Metastasis Suppressors and Promoters in Colon Cancer. Dev Cell 2020, 52, 413–428. [Google Scholar] [CrossRef]
- Li, J.; Hubisz, M.J.; Earlie, E.M.; Duran, M.A.; Hong, C.; Varela, A.A.; Lettera, E.; Deyell, M.; Tavora, B.; Havel, J.J.; et al. Non-Cell-Autonomous Cancer Progression from Chromosomal Instability. Nature 2023, 620, 1080–1088. [Google Scholar] [CrossRef]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING Activation of Tumor Endothelial Cells Initiates Spontaneous and Therapeutic Antitumor Immunity. Proc Natl Acad Sci U S A 2015, 112, 15408–15413. [Google Scholar] [CrossRef]
- Ohkuri, T.; Kosaka, A.; Ishibashi, K.; Kumai, T.; Hirata, Y.; Ohara, K.; Nagato, T.; Oikawa, K.; Aoki, N.; Harabuchi, Y.; et al. Intratumoral Administration of cGAMP Transiently Accumulates Potent Macrophages for Anti-Tumor Immunity at a Mouse Tumor Site. Cancer Immunol Immunother 2017, 66, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhang, W.; Guo, H.; Wang, L.; Wu, H.; Ding, L.; Yang, B. Strategies Involving STING Pathway Activation for Cancer Immunotherapy: Mechanism and Agonists. Biochem Pharmacol 2023, 213, 115596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Huang, J.; Zhao, Y.; Wang, S.; Xu, J.; Yin, K. Targeting STING in Cancer: Challenges and Emerging Opportunities. Biochim Biophys Acta Rev Cancer 2023, 1878, 188983. [Google Scholar] [CrossRef] [PubMed]
- Vasiyani, H.; Wadhwa, B.; Singh, R. Regulation of cGAS-STING Signalling in Cancer: Approach for Combination Therapy. Biochim Biophys Acta Rev Cancer 2023, 1878, 188896. [Google Scholar] [CrossRef] [PubMed]
- Coderch, C.; Arranz-Herrero, J.; Nistal-Villan, E.; de Pascual-Teresa, B.; Rius-Rocabert, S. The Many Ways to Deal with STING. Int J Mol Sci 2023, 24, 9032. [Google Scholar] [CrossRef]
- Motedayen Aval, L.; Pease, J.E.; Sharma, R.; Pinato, D.J. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J Clin Med 2020, 9, 3323. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, R.; Cen, S.; Zhou, J. STING Modulators: Predictive Significance in Drug Discovery. Eur J Med Chem 2019, 182, 111591. [Google Scholar] [CrossRef]
- Suter, M.A.; Tan, N.Y.; Thiam, C.H.; Khatoo, M.; MacAry, P.A.; Angeli, V.; Gasser, S.; Zhang, Y.L. cGAS-STING Cytosolic DNA Sensing Pathway Is Suppressed by JAK2-STAT3 in Tumor Cells. Sci Rep 2021, 11, 7243. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA Sensing by the cGAS-STING Pathway in Health and Disease. Nat Rev Genet 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Ghonim, M.A.; Ibba, S.V.; Tarhuni, A.F.; Errami, Y.; Luu, H.H.; Dean, M.J.; El-Bahrawy, A.H.; Wyczechowska, D.; Benslimane, I.A.; Del Valle, L.; et al. Targeting PARP-1 with Metronomic Therapy Modulates MDSC Suppressive Function and Enhances Anti-PD-1 Immunotherapy in Colon Cancer. J Immunother Cancer 2021, 9, e001643. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Zhu, Z.; Johnson, R.L.; Zhang, Z.; Herring, L.E.; Jiang, G.; Damania, B.; James, L.I.; Liu, P. Development of VHL-Recruiting STING PROTACs That Suppress Innate Immunity. Cell Mol Life Sci 2023, 80, 149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ventura, C.J.; Fang, R.H.; Zhang, L. Nanodelivery of STING Agonists against Cancer and Infectious Diseases. Mol Aspects Med 2022, 83, 101007. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Guo, B.; Qiu, X.; Xia, Y.; Qu, Y.; Cheng, L.; Meng, F.; Zhong, Z. Polymersome-Mediated Cytosolic Delivery of Cyclic Dinucleotide STING Agonist Enhances Tumor Immunotherapy. Bioact Mater 2022, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang-Bishop, L.; Kimmel, B.R.; Ngwa, V.M.; Madden, M.Z.; Baljon, J.J.; Florian, D.C.; Hanna, A.; Pastora, L.E.; Sheehy, T.L.; Kwiatkowski, A.J.; et al. STING-Activating Nanoparticles Normalize the Vascular-Immune Interface to Potentiate Cancer Immunotherapy. Sci Immunol 2023, 8, eadd1153. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Deng, H.; Wu, L.; Wang, D.; Shi, L.; Qian, Q.; Huang, X.; Zhu, L.; Gao, X.; Yang, J.; et al. Supramolecular Cyclic Dinucleotide Nanoparticles for STING-Mediated Cancer Immunotherapy. ACS Nano 2023, 17, 10090–10103. [Google Scholar] [CrossRef]
- Ji, N.; Wang, M.; Tan, C. Liposomal Delivery of MIW815 (ADU-S100) for Potentiated STING Activation. Pharmaceutics 2023, 15, 638. [Google Scholar] [CrossRef]
- Zhang, J.; Cui, X.; Huang, Y.; Xu, X.; Feng, C.; Li, J. Anticancer Effect of STING Agonist-Encapsulated Liposomes on Breast Cancer. Molecules 2023, 28, 3740. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Z.; Huang, H.; Shen, G.; Ren, Y.; Mao, X.; Wang, L.; Li, Z.; Wang, W.; Li, G.; et al. Cancer Immunotherapy Based on Cell Membrane-Coated Nanocomposites Augmenting cGAS/STING Activation by Efferocytosis Blockade. Small 2023, e2302758. [Google Scholar] [CrossRef]
- Vornholz, L.; Isay, S.E.; Kurgyis, Z.; Strobl, D.C.; Loll, P.; Mosa, M.H.; Luecken, M.D.; Sterr, M.; Lickert, H.; Winter, C.; et al. Synthetic Enforcement of STING Signaling in Cancer Cells Appropriates the Immune Microenvironment for Checkpoint Inhibitor Therapy. Sci Adv 2023, 9, eadd8564. [Google Scholar] [CrossRef]
- He, Y.; Hong, C.; Yan, E.Z.; Fletcher, S.J.; Zhu, G.; Yang, M.; Li, Y.; Sun, X.; Irvine, D.J.; Li, J.; et al. Self-Assembled cGAMP-STINGΔTM Signaling Complex as a Bioinspired Platform for cGAMP Delivery. Sci Adv 2020, 6, eaba7589. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Knelson, E.H.; Jimenez-Macias, J.L.; Nowicki, M.O.; Han, S.; Panagioti, E.; Lizotte, P.H.; Adu-Berchie, K.; Stafford, A.; Dimitrakakis, N.; et al. STING Activation Promotes Robust Immune Response and NK Cell-Mediated Tumor Regression in Glioblastoma Models. Proc Natl Acad Sci U S A 2022, 119, e2111003119. [Google Scholar] [CrossRef]
- Xu, H.; Jin, J.; Chen, Y.; Wu, G.; Zhu, H.; Wang, Q.; Wang, J.; Li, S.; Grigore, F.-N.; Ma, J.; et al. GBP3 Promotes Glioblastoma Resistance to Temozolomide by Enhancing DNA Damage Repair. Oncogene 2022, 41, 3876–3885. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Banerjee, K.; Mujeeb, A.A.; Hartlage, C.S.; Núñez, F.M.; Núñez, F.J.; Alghamri, M.S.; Kadiyala, P.; Carney, S.; Barissi, M.N.; et al. H3.3-G34 Mutations Impair DNA Repair and Promote cGAS/STING-Mediated Immune Responses in Pediatric High-Grade Glioma Models. J Clin Invest 2022, 132, e154229. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.J.S.; Sojwal, R.S.; Gubatan, J.; Rogalla, S. The Tumor Immune Microenvironment in Pancreatic Ductal Adenocarcinoma: Neither Hot nor Cold. Cancers (Basel) 2022, 14, 4236. [Google Scholar] [CrossRef]
- Kabashima, A.; Matsuo, Y.; Ito, S.; Akiyama, Y.; Ishii, T.; Shimada, S.; Masamune, A.; Tanabe, M.; Tanaka, S. cGAS-STING Signaling Encourages Immune Cell Overcoming of Fibroblast Barricades in Pancreatic Cancer. Sci Rep 2022, 12, 10466. [Google Scholar] [CrossRef]
- Jacoberger-Foissac, C.; Cousineau, I.; Bareche, Y.; Allard, D.; Chrobak, P.; Allard, B.; Pommey, S.; Messaoudi, N.; McNicoll, Y.; Soucy, G.; et al. CD73 Inhibits cGAS-STING and Cooperates with CD39 to Promote Pancreatic Cancer. Cancer Immunol Res 2023, 11, 56–71. [Google Scholar] [CrossRef]
- Hu, G.; Chen, Y.; Yang, X.; Wang, Y.; He, J.; Wang, T.; Fan, Q.; Deng, L.; Tu, J.; Tan, H.; et al. Mitotic SENP3 Activation Couples with cGAS Signaling in Tumor Cells to Stimulate Anti-Tumor Immunity. Cell Death Dis 2022, 13, 640. [Google Scholar] [CrossRef]
- Francica, B.J.; Ghasemzadeh, A.; Desbien, A.L.; Theodros, D.; Sivick, K.E.; Reiner, G.L.; Hix Glickman, L.; Marciscano, A.E.; Sharabi, A.B.; Leong, M.L.; et al. TNFα and Radioresistant Stromal Cells Are Essential for Therapeutic Efficacy of Cyclic Dinucleotide STING Agonists in Nonimmunogenic Tumors. Cancer Immunol Res 2018, 6, 422–433. [Google Scholar] [CrossRef]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS Is Essential for the Antitumor Effect of Immune Checkpoint Blockade. Proc Natl Acad Sci U S A 2017, 114, 1637–1642. [Google Scholar] [CrossRef]
- Curran, E.; Chen, X.; Corrales, L.; Kline, D.E.; Dubensky, T.W.; Duttagupta, P.; Kortylewski, M.; Kline, J. STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep 2016, 15, 2357–2366. [Google Scholar] [CrossRef]
- Yi, G.; Brendel, V.P.; Shu, C.; Li, P.; Palanathan, S.; Cheng Kao, C. Single Nucleotide Polymorphisms of Human STING Can Affect Innate Immune Response to Cyclic Dinucleotides. PLoS One 2013, 8, e77846. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Moreno, J.S.; Hamann, L.; Shah, J.A.; Verbon, A.; Mockenhaupt, F.P.; Puzianowska-Kuznicka, M.; Naujoks, J.; Sander, L.E.; Witzenrath, M.; Cambier, J.C.; et al. The Common HAQ STING Variant Impairs cGAS-Dependent Antibacterial Responses and Is Associated with Susceptibility to Legionnaires’ Disease in Humans. PLoS Pathog 2018, 14, e1006829. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhao, J.; Li, Y.; Tomer, S.; Selvaraju, M.; Tien, N.; Sun, D.; Johnson, D.K.; Zhen, A.; Li, P.; et al. Structural and Biological Evaluations of a Non-Nucleoside STING Agonist Specific for Human STING A230 Variants. bioRxiv 2023. [Google Scholar] [CrossRef]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Anti-Tumor Immunity. Cell Rep 2018, 25, 3074–3085. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 Axis Promotes cGAS/STING Signaling in ARID1A-Deficient Tumors. J Clin Invest 2020, 130, 5951–5966. [Google Scholar] [CrossRef]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.-Y. ATM Inhibition Enhances Cancer Immunotherapy by Promoting mtDNA Leakage and cGAS/STING Activation. J Clin Invest 2021, 131, e139333–139333. [Google Scholar] [CrossRef]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov 2019, 9, 34–45. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Veleeparambil, M.; Kessler, P.M.; Willard, B.; Chattopadhyay, S.; Sen, G.C. EGFR-Mediated Tyrosine Phosphorylation of STING Determines Its Trafficking Route and Cellular Innate Immunity Functions. EMBO J 2020, 39, e104106. [Google Scholar] [CrossRef]
- Milling, L.E.; Garafola, D.; Agarwal, Y.; Wu, S.; Thomas, A.; Donahue, N.; Adams, J.; Thai, N.; Suh, H.; Irvine, D.J. Neoadjuvant STING Activation, Extended Half-Life IL2, and Checkpoint Blockade Promote Metastasis Clearance via Sustained NK-Cell Activation. Cancer Immunol Res 2022, 10, 26–39. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, Q.; Jiang, T.; Wang, W.; Zhao, J.J. Targeting Tumor-Associated Macrophages with STING Agonism Improves the Antitumor Efficacy of Osimertinib in a Mouse Model of EGFR-Mutant Lung Cancer. Front Immunol 2023, 14, 1077203. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liang, W.; Li, K.; Liao, X.; Chen, J.; Qiu, X.; Liu, K.; Qiu, D.; Qin, Y. Sorafenib Suppresses the Activation of Type I Interferon Pathway Induced by RLR-MAVS and cGAS-STING Signaling. Biochem Biophys Res Commun 2022, 623, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Wayne, J.; Brooks, T.; Landras, A.; Massey, A.J. Targeting DNA Damage Response Pathways to Activate the STING Innate Immune Signaling Pathway in Human Cancer Cells. FEBS J 2021, 288, 4507–4540. [Google Scholar] [CrossRef]
- Brooks, T.; Wayne, J.; Massey, A.J. Checkpoint Kinase 1 (Chk1) Inhibition Fails to Activate the Stimulator of Interferon Genes (STING) Innate Immune Signalling in a Human Coculture Cancer System. Mol Biomed 2021, 2, 19. [Google Scholar] [CrossRef]
- Smith, T.T.; Moffett, H.F.; Stephan, S.B.; Opel, C.F.; Dumigan, A.G.; Jiang, X.; Pillarisetty, V.G.; Pillai, S.P.S.; Wittrup, K.D.; Stephan, M.T. Biopolymers Codelivering Engineered T Cells and STING Agonists Can Eliminate Heterogeneous Tumors. J Clin Invest 2017, 127, 2176–2191. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING Agonist Promotes CAR T Cell Trafficking and Persistence in Breast Cancer. J Exp Med 2021, 218, e20200844. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res 2016, 76, 6747–6759. [Google Scholar] [CrossRef]
- Yang, N.; Wang, Y.; Liu, S.; Tariq, S.B.; Luna, J.M.; Mazo, G.; Tan, A.; Zhang, T.; Wang, J.; Yan, W.; et al. OX40L-Expressing Recombinant Modified Vaccinia Virus Ankara Induces Potent Antitumor Immunity via Reprogramming Tregs. J Exp Med 2023, 220, e20221166. [Google Scholar] [CrossRef]
- Dai, P.; Wang, W.; Yang, N.; Serna-Tamayo, C.; Ricca, J.M.; Zamarin, D.; Shuman, S.; Merghoub, T.; Wolchok, J.D.; Deng, L. Intratumoral Delivery of Inactivated Modified Vaccinia Virus Ankara (iMVA) Induces Systemic Antitumor Immunity via STING and Batf3-Dependent Dendritic Cells. Sci Immunol 2017, 2, eaal1713. [Google Scholar] [CrossRef]
- Kolyasnikova, N.M.; Pestov, N.B.; Sanchez-Pimentel, J.P.; Barlev, N.A.; Ishmukhametov, A.A. Anti-Cancer Virotherapy in Russia: Lessons from the Past, Current Challenges and Prospects for the Future. Current Pharmaceutical Biotechnology 2023, 24, 266–278. [Google Scholar]
- Pang, E.S.; Daraj, G.; Balka, K.R.; De Nardo, D.; Macri, C.; Hochrein, H.; Masterman, K.-A.; Tan, P.S.; Shoppee, A.; Magill, Z.; et al. Discordance in STING-Induced Activation and Cell Death Between Mouse and Human Dendritic Cell Populations. Front Immunol 2022, 13, 794776. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Paradigmatic outline of the cGAS-cGAMP-STING-TBK1-IRF3-IFN pathway. Note the 2:2 stoichiometry in cGAS-DNA complex. Created with Biorender.com.
Figure 1.
Paradigmatic outline of the cGAS-cGAMP-STING-TBK1-IRF3-IFN pathway. Note the 2:2 stoichiometry in cGAS-DNA complex. Created with Biorender.com.
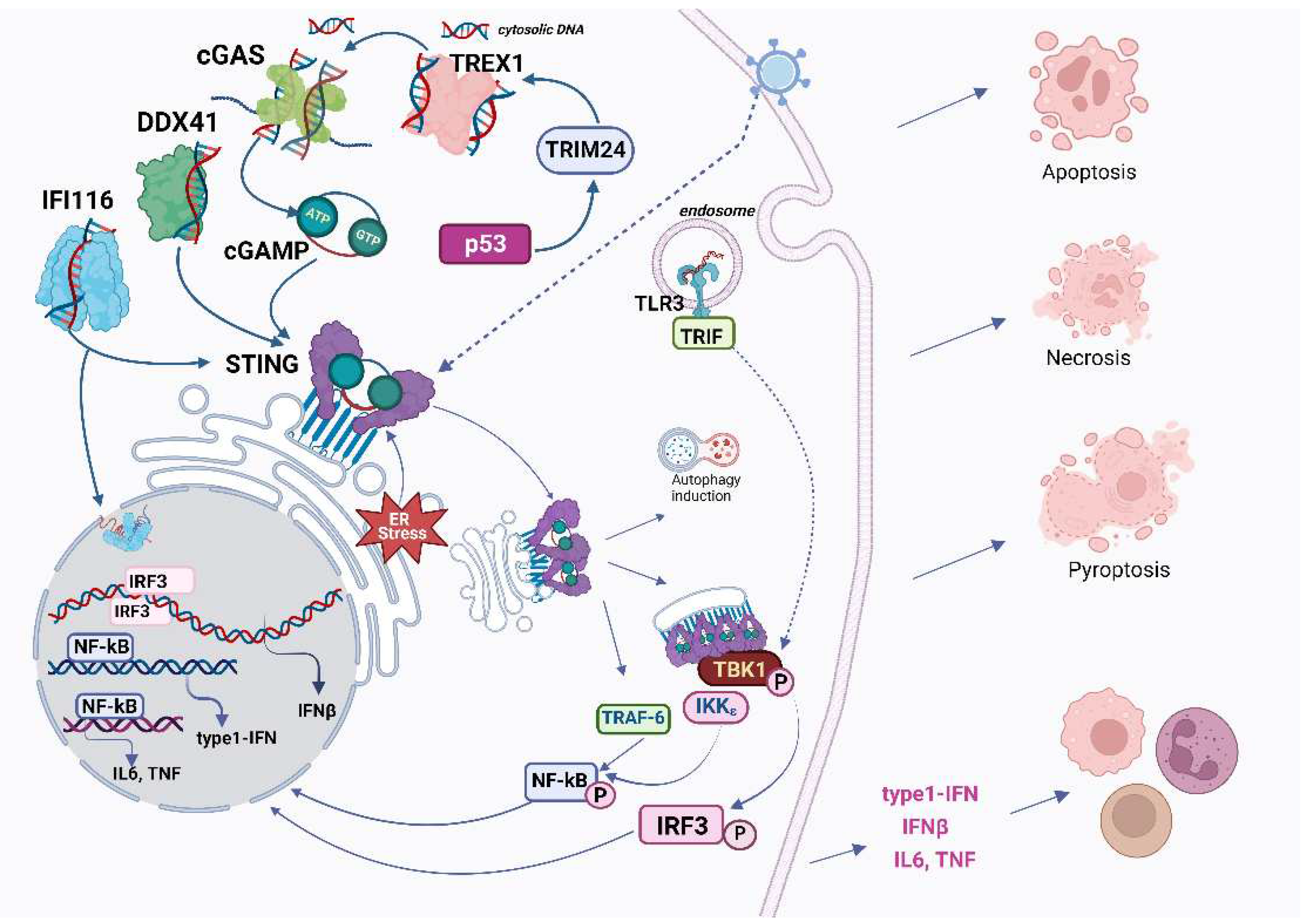
Figure 2.
cGAS-STING in prevention of carcinogenesis and its dualistic role in cancer progression. Created with Biorender.com.
Figure 2.
cGAS-STING in prevention of carcinogenesis and its dualistic role in cancer progression. Created with Biorender.com.
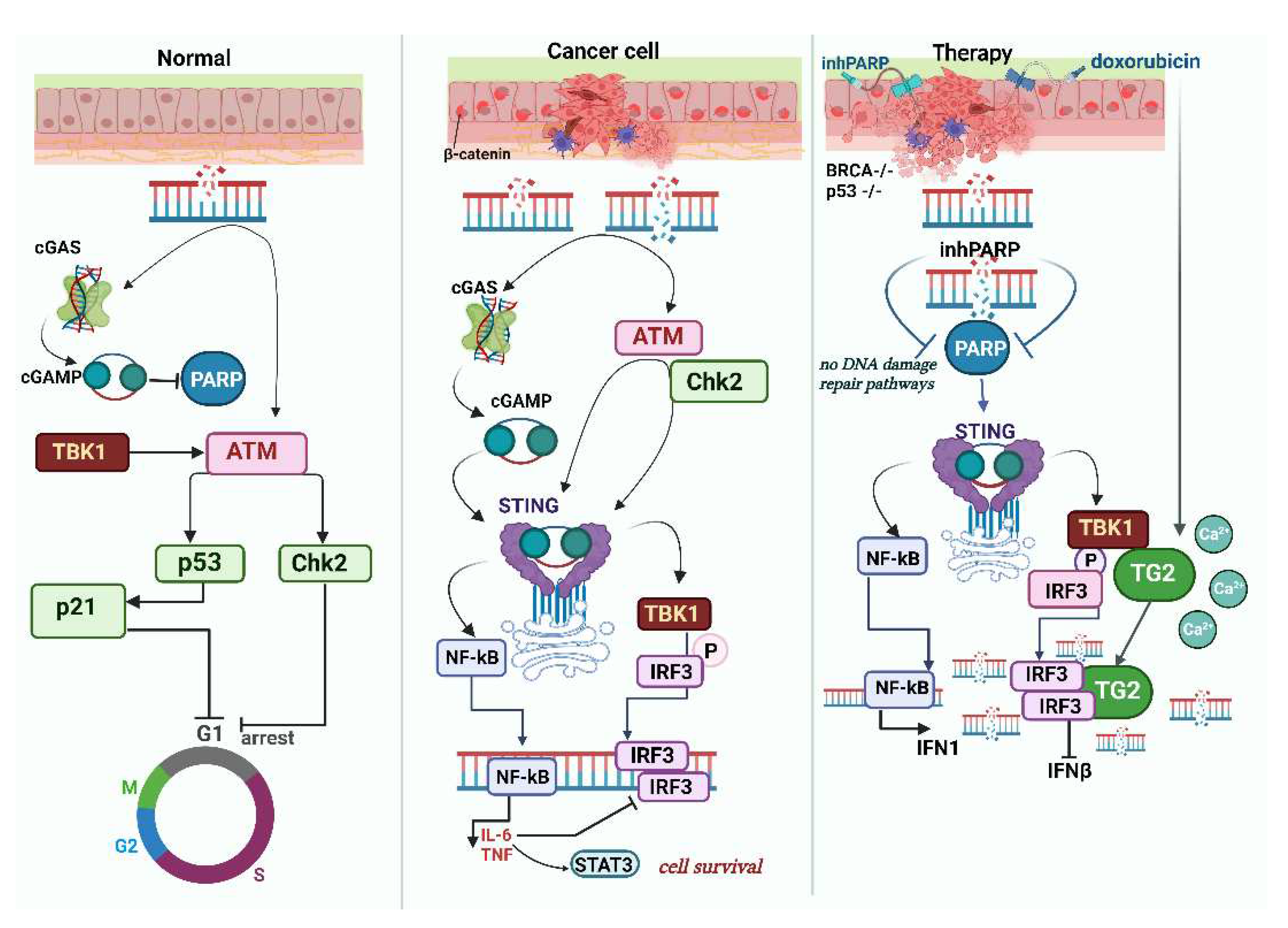
Figure 3.
Structural formulae of the most well studied cyclic dinucleotides and non-nucleotide STING agonists. ADU-S100 is especially efficient in murine cancer models like B16, CT26, 4T1. Compound 53 works in a different mode. Created with ACD/Chemsketch.
Figure 3.
Structural formulae of the most well studied cyclic dinucleotides and non-nucleotide STING agonists. ADU-S100 is especially efficient in murine cancer models like B16, CT26, 4T1. Compound 53 works in a different mode. Created with ACD/Chemsketch.
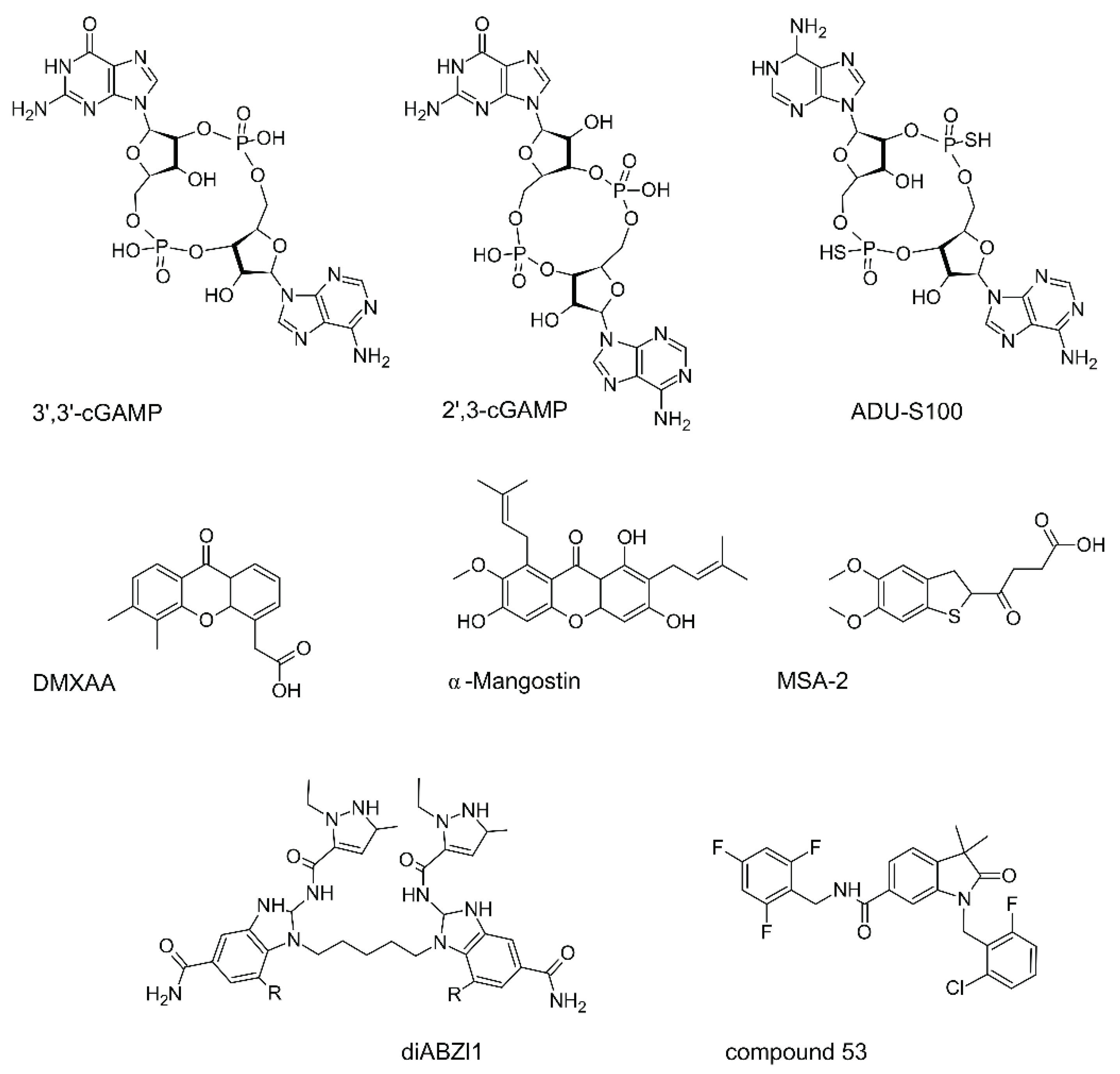
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



