
Submitted:
02 October 2023
Posted:
03 October 2023
You are already at the latest version
Abstract
Salmonella enterica includes enteric pathogens of zoonotic potential, possessing one of the largest pools of temperate phages in their genomes. One such class of phages previously called Podoviridae (Now genera placed directly under class Caudoviricetes temporarily) is the largest group of temperate phages that lysogenize clinically and economically important Salmonella enterica serovars. These phages are capable of generalized transduction and are well known for carrying foreign DNA (mobile genetic elements and resistance genes) to new bacterial species that play a major role in the evolution, pathogenicity, and host adaptability of Salmonella serovars. Here we report a novel species of podo viruses; Salmonella phage BIS08P22 (BIS08) that infects Salmonella enterica serovar Typhimurium strain SE-BS17 (Acc. NO: MZ503545). Phage BIS08 was viable only at biological pH and temperature (pH7 and 37 °C) and features 41,574 -base pair (bp) linear ds DNA genome with 47% GC content. It encodes 73 putative Open Reading Frames (ORFs), has a mosaic arrangement, and shares only 16 core genes with its closest homologs. BIS08 genome possesses only 52 % homology (genome-wide Nucleotide homology by VIRIDIC analysis) with members of the genus Lederbergvirus which is not sufficient to place it in the same genus. This phage has a unique terminase enzyme (DNA packaging motor) with no nucleotide and protein homology with any known member of the Lederbergvirus genus. We carried out a detailed phylogenetic and genome analysis of this novel phage and proposed its placement in a new genus. Our study also suggests a revision in the classification of the genus Lederbergvirus. We also performed in silico structural analysis of the BIS08 unique terminase enzyme comparing it to its close homologs.
Keywords:
Podo virus
; temperate phages
; Short tail phages
; novel phage terminase
; Lederbergvirus
1. Introduction
Bacteriophages are by far the most abundant entities on earth [1]. They infect bacteria and archaea. Based on their life cycles they can be categorized as temperate or virulent. Temperate bacteriophages do not immediately kill host bacterial cells and carry specialized enzymes such as integrases and site-specific recombinases [2]. These enzymes help in insertion of phage genome into bacterial chromosomes at specific sites as prophages a process called lysogenization [3], [4]. These prophages can replicate with bacterial genomes. Later they can excise from lysogen based on surrounding environment such as bacterial density, nutritional stress, and host growth status [5]. In contrast virulent phages take over host machinery produce their progeny and kill host bacterium. While virulent phages have gained popularity as therapeutic targets for MDR/XDR pathogenic bacterial strains [6], temperate phages are also well known to play a prominent role in bacterial evolution , adaptability, and pathogenicity. They are more beneficial to bacteria as they can provide additional factors that help them to survive and adapt to their hosts [7].
Temperate phages have been a subject of interest since long with the discovery of two major virulence factors encoded by phages. One corynephages that carry tox gene producing Diphtheria toxin for Corynebacterium Diphtheriae [8] and C1 phage that produce Botulin toxin (C and D type) for Clostridium botulinum [9]. With advancement in genome studies and analysis of pathogenic bacteria, temperate phages are now known to carry a wide array of virulence factors such as toxins (tox, stx1, stx2, ctxAB etc.)[10], proteins that help in bacterial invasion (SopE, sspH1) [11], enzymes that help bacteria to avoid oxidative stress [12], and serum resistance factors to name a few [13]. Temperate phages therefore play a critical role in pathogenicity of their host bacterium [14].
One such family of bacteriophages Podoviridae includes genus Lederbergvirus of temperate phages with short noncontractile tails and icosahedral heads. Podoviridae previously included 6 genera and a 2 subfamilies Autographivirinae and Picovirinae having 3 and 2 genera respectively (ICTV; https://ictv.global/report_9th/dsDNA/Podoviridae). In 2021 the International Committee of Taxonomy of Bacterial Viruses (ICTBV; https://ictv.global/sc/bacterial ) abolished the families Podoviridae, Myoviridae and Siphoviridae [15]. Now genera belonging to Podoviridae are directly placed under class Caudoviricetes not assigned to any family or order (https://ictv.global/taxonomy/visual-browser ) temporarily. Genus Lederbergvirus (previously called P22-like viruses) includes viruses capable of generalized transduction, hence they are important for host evolution and pathogenicity and have been extensively studied in the past [16], however, classification of Lederbergvirus genus has not been re-examined since ICTV adapted genomic taxonomy rather than protein similarity to account for phage gene synteny [17,18,19]. Podo viruses usually follow a rolling circle mode of DNA replication that produces long concatemers (REF). These concatemers are resolved into individual genomes that are packaged into preformed capsids using a powerful DNA translocation motor [20,21,22]. This DNA translocation motor is called terminase enzyme and has two subunits; the large and small terminase which are associated with portal protein[23].
Here we report the isolation, characterization, and phylogenetic analysis of a novel species of podo viruses Salmonella phage BIS08P22, that differs sufficiently from its close relatives of Lederbergvirus genus. We propose revision in classification of Lederbergvirus genus based on our analysis. Moreover, a detailed genomic analysis also indicates the presence of a unique terminase enzyme in this novel species. We performed in silico protein modeling of this terminase enzyme emphasizing its mode of action.
2. Materials and Methods
2.1. Bacterial Strain and Bacteriophage Isolation
Salmonella enterica serovar Typhimurium strain SE-BS17 was isolated and characterized in Molecular Virology Labs (MVL), CUI, Islamabad from retail poultry samples and its isolation procedure is published previously in [24] and [60]. This strain is also characterized by 16S rRNA typing (Macrogen, Seoul, Korea) and the resulting sequence is submitted to NCBI (GenBank Acc. No; MZ503545). SE-BS17 was used due to its high resistance profile carrying genes for beta lactamase (blaTEM-1) as well as extended spectrum beta lactamases (blaCTX-M). Salmonella phage BIS08P22 (BIS08) was isolated using standard protocols. Roughly one cm fragment of each intestine, stomach and spleen were pooled in a disposable petri plate. The sample was washed three times with PBS to remove any debris and then triturated into fine paste using scalpel blade. The triturated sample was mixed with 10 ml of buffered peptone water and incubated at 37 °C for 24 h. Next day the sample was spun down at 8000 rpm for 20 min to remove debris. The supernatant was filtered through 0.45 µm syringe filters and mixed with 200 µl of an overnight culture of SE-BS17 supplemented with 10 mM CaCl2 to facilitate phage binding and again incubated for 24 h at 37 °C. After incubation, 1 ml of the culture was spun down through 0.22 µm syringe filter and supernatant was tested on SE-BS17 by agar overlay method as described elsewhere [61]. Serial two-fold dilutions of phage lysate were prepared in Luria–Bertani (LB) broth. Meanwhile solidified agar plates were prepared by adding 15 mL of LB-agar in each plate. Later 2.5 mL of soft agar (0.5 % w/v) were dispensed in a Kimax glass tube and 250 µL of Se-BS17 overnight culture were added to it. This mixture was poured on top of agar plates, and they were left to solidify for 15 min. After they solidified one drop (10 µL) of phage lysate dilutions were spotted on to them and left to dry for 15mins. Later plates were inverted and kept in incubator at 37 °C for 24 h. Next day the dilution which produced clear zones of lysis against bacterial lawn was selected and processed further.
2.2. Purification and Large-Scale Amplification of Phage BIS08
Usually, the lysate obtained from an environmental sample is a mixture of phages. To ensure that we are dealing with progeny of one phage (pure culture) single plaque purification by pour plate method is employed as described by [62]. It is modification of agar overly method. Briefly, 25-30 µL of phage dilution which previously produced countable plaques and 250 µL of an overnight culture of SE-BS17 strain were added to 2.5 mL of soft agar (0.5% w/v). The mixture was gently poured on solidified LB agar plates, spread evenly to make a uniform layer, and allowed to solidify. Later plates were inverted and placed in incubator at 37 °C for 24 h. Next day a plate giving isolated and countable plaque was selected. One plaque was pierced by using a yellow tip cut from the edge and entire content of the plaque was suspended in 1 mL PBS (8 mM Na2HPO4, 2.7 mM KCl, 2 mM KH2PO and 137 mM NaCl) having pH 7.2 in 1.5 mL Eppendorf tube and gently rocked for 3h at room temperature. Later the contents were spun down, and supernatant was filtered and saved at 4 °C. This entire procedure was repeated 3 times to obtain pure cultures and the last phage lysate was labelled as master stock and subjected to large scale amplification. The phage was named Salmonella phage BIS08P22 (BIS08).
To increase the titer and get more volume the BIS08 was amplified in liquid cultures following standard protocols. Briefly, 1:100-fold dilution of SE-BS17 overnight culture was incubated in 500 mL LB medium at 37 °C and allowed to grow till mid log phase (OD600= 0.55). At this point the culture was inoculated with phage master stock obtained after single plaque purification ay MOI 1 and left without shaking for 15 min at 37 °C. Later the culture was left to grow at 37 °C overnight with continuous shaking at 200 rpm. Next day the culture was spun down at 8000 rpm (Centrifuge, Hermle, Germany ZK 496), at room temperate for 20 min. Supernatant was filtered through 0.22 µm syringe filter and saved at 4 °C. A concentrated sample of phage was prepared by PEG precipitation (Polyethylene Glycol 8000) (Sigma Aldrich Cat. No. 1546605). Ten percent PEG (w/v) was dissolved in phage lysate obtained after amplification. The PEG- Phage lysate was incubated at 4 °C overnight [63]. Next day the sample was spun down at 10,000 rpm (Hermle, Germany ZK496), for 40 mins at 4 °C. The supernatant was decanted, and Grey white PEG precipitate was dissolved in 2 mL PBS (pH 7.2) , filtered through 0.22 µm syringe filters and saved at 4 °C until further use. Later phage titer was calculated by agar- overlay method as described above.
2.3. BIS08 Stability and Growth Curve
The standard procedure of one-step growth curve as illustrated by Kropinski [64] was used to calculate phage latent period and burst size with few modifications. Briefly an overnight culture of SE-BS17 was diluted (1:100) in 10 mL LB medium and allowed to grow at 37 °C until mid-Log phase OD600 (0.55). At this stage the culture was inoculated with BIS08 at MOI 1 and left without shaking for 10 mins for phage adsorption. After this the entire culture (10 mL) was centrifuged at 10,000 rpm for 10 mins to spin down bacteria and adsorbed phages. Supernatant with free phage was decanted and titrated to determine the number of total un-adsorbed phages. The bacterial cells were suspended in 20 mL LB and allowed to grow at 37 °C with continuous shaking at 200 rpm. One mL aliquots were removed from this growing culture at every 5 min point and spun down immediately in a tabletop centrifuge at 10,000 rpm, filtered and stored on ice until titration. After 70 min, serial two-fold dilutions of all 14 time points were titrated on SE-BS17 using agar overlay method as described above. The experiment was performed in triplicates. The latent period, burst size as well as burst time was calculated using the following formula.
Burst size = C-B/ A-B = D/E
Where C= Phages after burst, B= Free phages , D= New phage released and A = Total applied phages and D= Number of infecting phages
BIS08 stability at different temperatures and pH conditions was also evaluated. For temperature stability test 1×106 pfu were suspended in 1mL PBS (pH 7.2) and incubated at 3 temperature points (37 °C, 60 °C and 80 °C) for 1h and then titrated using standard procedures. For pH stability testing Phosphate buffered saline at pH points 2,5,7,9, and 12 was prepared. 1×106 pfu of BIS08 were suspended in 1 mL of each PBS and incubated for 1 h at 37 °C. Later the phage was titrated using agar overlay method and serial two-fold dilutions [65].
2.4. BIS08 Growth Impact on SE-BS17
Salmonella Typhimurium strain SE-BS17 was used as host strain for BIS08 isolation and characterization. As temperate phages can have impact on fitness of hosts, a growth reduction assay was performed to evaluate the impact of BIS08 infection on SE-BS17 fitness as described by O’Flynn (REF) with few modifications. Briefly, a 1 mL of mid log phase culture of SE-BS17 having ~ 1×108 CFU/ mL were mixed with phage BIS08 at MOI10 (1× 109 pfu/ mL) and left without shaking for 15 min at 37 °C. After that the mixture was diluted in 20 mL of LB broth and left to grow at 37 C for 24 h. Every 2h 1 mL aliquot of the growing culture was removed, and dilution plating was performed to measure CFU/mL of SE-BS17. Experiment was performed in triplicates and an average of triplicates was plotted in graph. A standard curve of SE-BS17 strain was used to correlate Optical density values at various time points with colony forming units of SE-BS17 [66].
2.5. Transmission Electron Microscopy (TEM) Analysis
Transmission electron microscopy was carried out using PEG enriched sample. Roughly 5 µL of phage lysate (5 × 108 pfu/ mL) were incubated onto carbon- coated grids for 5 min at room temperature. Later grids were washed with same volume of distilled water and incubated with 2% Uranyl acetate solution for 2 min. Electron microscopy was carried out at National Institute of Biotechnology and Genetic Engineering (NIBGE) Faisalabad, Pakistan.
2.6. BIS08 Genome Sequencing
BIS08 DNA was extracted using 400 µL of PEG purified lysate having titer of 1×108 cfu / mL titer using Norgen Biotek Corp. Phage DNA extraction kit (Cat # 46800) using manufacturer’s instructions. Extracted DNA was subjected to sequencing using Illumina MiSeq platform at Massey Genome services (New Zealand). The phage DNA library was prepared by enzymatic digestion of phage DNA into random fragments (NexteraTM XT library kit_V2). Enrichment PCR was used to add barcoded illumine adaptors at each end of the fragments. Quality of phage DNA libraries was checked using LabChip® GX DNA high sensitivity kit, and Quant-iT dsDNA high sensitivity assay with PerkinElmer GX Touch HT Instrument and PerkinElmer Victor Plate Reader. Libraries were pooled and then loaded on to illumine MiSeq TM run. Sequencing was performed using MiSeq Micro 300 cycle kit V1 that resulted in 2(150) base paired end reads. FASTQMCF was used to remove PhiX control reads and adaptor sequences. Reads were trimmed at 0.01 error probability using Trimmomatic (Version 0.39). Quality of reads was checked using Fast QC (Version 0.11.5), Fast Q screen and SolexaQA++(37). Contigs were assembled from individual reads by Artimis software freely available at (http://sanger-pathogens.github.io/Artemis/Artemis/ ). Annotation of assembled genome was performed using GAMOLA version 2 and RAST (Freely available at https://rast.nmpdr.org/ ). Putative ORFs and genes of BIS08 genome were identified by HHpred freely available at https://toolkit.tuebingen.mpg.de/tools/hhpred as well as RAST. The annotated sequence of phage was named according to ICTV recommendations as “Salmonella phage BIS08P22” and is uploaded in NCBI under accession number OP377721.1.
2.6.1. In silico Terminase Analysis of the BIS08 Large and Small Terminase Subunits
A subset of terminase protein sequences, known as the large phage terminase protein SSBI08P22 and small phage terminase subunit, sequence SSBI08P22 (Nucleotide Accession number OP377721) identified in this study were modelled using the online tool Alphafold2 [29], [67]. The modelled enzymes are detailed and described, with the structural domains visualized using PyMOL Molecular Graphics System v2.0 [68]. Related confidence scoring (pLDDT and PTM) is listed where necessary. For the identified small protein, a stable quaternary structure was also explored using Alphafold2 using monomers of 6-10 subunits. Dali lite pair wise structural alignment [33], [36] of terminase structures was also conducted. PDBsum was also used to provide a pictorial depiction of the 3D structures developed in AlphaFold [37].
2.7. BIS08 Phylogenetic Analysis
Genome sequence of BIS08 was subjected to detailed phylogenetic analysis with its close homologs in NCBI. The genome was compared with phage as well as close prophage relatives in NCBI. Whole genome sequences of 24 close phage homologs of BIS08 and 5 close prophage homologs were selected by nucleotide BLAST of whole genome. These sequences were analyzed by VICTOR ( https://ggdc.dsmz.de/victor.php)[69] for their phylogenetic position using GBDP (Genome Blast Distance Phylogeny) method using settings recommended for prokaryotic viruses. The intergenomic distances produced were used to create a balanced minimum evolution tree by FASTME including SPR postprocessing with optimal formula D0. The midpoint rooted tree was visualized with ggtree. For genome wide comparison of BIS08 with these homologs VIRIDIC software was used. This software applies a traditional genome-based algorithm to measure intergenomic distances between whole genomes and is recommended by ICTV committee for bacterial viruses (International Committee for Taxonomy of Bacterial and Archaeal Viruses; ICTV, BAVS) [54]. Further phylogenetic placement of BIS08 was evaluated by protein based phylogenetic analysis of BIS08 using VIP Tree software freely available at https://www.genome.jp/viptree/ [70]. CoreGenes 5.0 software was used to analyse core genes shared by BIS08 with members of Lederbergvirus genus and with its close homologs (https://coregenes.ngrok.io/ )[71] .Protein sequence of two orthologs from BIS08 genome were selected and analyzed in ViPTree as well as VICTOR software. In order to identify the phage lifestyle in terms of Lytic or temperate cycles PhageLeads software was used. This software identifies recombination and integration tendency of a phage genome as well as virulence markers and resistance genes and is freely available at https://phageleads.dk/ [72]. Genome-wide comparison figures of BIS08 were obtained from ViPTree comparisons with close homologs selected based on SG score provided by computation of proteome distance by the software.
2.8. Statistical Analysis
Statistical analyses were performed using Origin 2019 (Origin Lab, Northampton, MA, USA). The statistical significance was determined using ANOVA followed by Tukey’s honest significant difference (HSD) test for specific comparisons. Statistical significance was reached at p < 0.05.
3. Results
3.1. Isolation and Charecterization of BIS08
Salmonella Typhumurium strain SE-BS17 (Acc. NO: MZ503545) was isolated and charecterized from poultry samples in a previous study carried out at MVL, CUI. This strain is multiple drug resistant and is published previously [24]. The strain was used as a host to isolate Salmonella phage BIS08P22 (BIS08). The BIS08 was isolated form retail poultry samples and produced mildly turbid plaques on SE-BS17 (Zones of lysis measuring 0.85 mm). The turbidity of plaques indicated its temperate nature which was later confirmed by sequence analysis. BIS08 host adsorption efficacy to mid log phase (0.4) culture was ~ 95 % after 15 mins of incubation at 37 °C . Maximum phage titer of 1 × 106 (in temperature stability assay) was observed at 37 °C. BIS08 was able to grow only at 37 °C and could not tolerate temperatures higher than that as no plaques were observed at 60 °C and 80 °C (Fig 1A). Same was true for pH tolerance as BIS08 produced maximum titer of 1.23 × 10 6 at pH 7 only. It did survive at pH2 and pH 9 however with significant drop in titers (2.3 × 101 and 7.1× 101) (Figure 1B). Phage has ~ 20 mins of latent period whereas first burst was observed at 25 mins albeit with very less increase in phage titer (2 × 103). At 30 mins however there was a sharp rise in phage titer as it nearly doubled to 2 × 106 (Fig.1 C). The burst size of BIS08 was 60 virions/ cell. To evaluate the impact of BIS08 infection on host growth, bacterial growth reduction assay was performed. It was observed that SE-BS17 when infected with BIS08 at MOI10 indeed grew significantly (p<0.0001, 24 h of incubation) slower (1.26 × 107 vs 2.6×108) than control (SE-BS17 without phage) (Fig.1 D). Transmission electron micrography indicated that BIS08 has icosa hedral head and a very short non-contractile tail a charecteristic of former family Podoviridae in class caudovirecetes. These phages are well known for specialized and generalized transduction and a few members are very well studied (Enterobacteria phage P22). (Fig.1E)
3.2. Genome analysis of Salmonella Phage BIS08
Salmonella phage BIS08P22 (BIS08) has 41572 bp genome that encodes for 73 Open Reasding Frmanes (ORFs). The genome has 47% GC content. Out of 73 ORFs, 58 ORFs used ATG as start codon whereas 4 ORFs (ORF 6, 8, 51 and 68, Figure 2) started with GTG and Only one started with TTG start codon (ORF 4). For ease of description the genome is diveded in to Structural module (I), Lysis Cassette (II) and Lytic to Lysogenic switch (III). Genome map of BIS08 is given in Figure 2.
3.2.1. Structural ORFs
BIS08 Structural ORFs had mosaic arrangements with different proteins having homology with different phages. Majority proteins are close to Lederbergvirus BTP1. This module can be divided into 3 main categories 1) tail spike and tail fiber proteins (ORF 5 and 20, color coded blue, Figure 2) that are distributed at start and end of structural module. Tail spike protein (1986 nt, and 662 AA, ORF 5) has maximum homology with only lederbergvirus BTP1 (97.63 % similarity with 100 % querry cover) for all other phages only 27 % nt sequence has 98 % homology rest did not match. This protein serves as adhesin to attach to the surface O- residues on Salmonella cell surface [25], [26]. Same was true for aminoacid sequence of ORF 5 that has maximum homology with BTP1 (96.07%) whereas all others have ≤ 70.9% homology. Second tail fiber protein (ORF 20, 405 nt, 135 AA) has maximum homology with Salmonella phage S18, SE16 and epsilon 34 (97.76% nt whereas 96% AA homology in BLASTp). 2) Capsid morphogenesis and stabilization proteins (ORFs 6-17). They include four ORFs (ORF 6, 7, 8 and 9) involved in DNA transfer to the host cell. These proteins are released from phage particle upon infection. This module also includes a head decoration/ assembly protein (ORF 10, 152 AA), A tail needle protein (ORF 11, 234 AA), a head closure Hc3 like protein (ORF 12, 473 AA), Head tail adaptor Ad3 like protein (ORF13, 167AA), A hypothetical protein (ORF 14, 187AA), Major capsid protein (ORF 15, 431 AA), and a head scffolding protein (ORF 16 , 304 AA). All these proteins had high similarity with Lederbergvirus BTP1 except for ORF 13, head-tail adaptor protein that had no homology with BTP1 and was 100 % identical to Enterobacteria phage ST104. 3) Third category includes proteins involved in DNA transport in to head such as a portal protein (ORF17, 726 AA) and DNA packaging macheniery Terminase large (ORF 18 487 AA), and small subunit (ORF 19, 153 AA) colored turquoise blue (Figure 2). Portal protein is conserved and have maximum homology with BTP1 like other structural proteins. However the most peculiar feature of BIS08 is presence of a unique terminase enzyme. Phage terminase large subunit (ORF 487AA, 1461 bp) of BIS08 had only one homolog in protein and nucleotide sequence that is Salmonella phage ST-35 (Accession No CP051279.1). All others memebers of Lederbergvirus genus had only 10 % querry cover and 33 % similarity index with BIS08 large terminase. Same was true for phage small terminase as it exhibits 66 % amino acid homology to three other members of unclassified Lederbergvirus genus and only one nucleotide homolog (100 %) Salmonella phage ST-35.
3.2.2. Phage Lysis Cassette
BIS08 like other podo-viruses possess Spannin-Holin-Lysozyme cassette for host cell lysis (ORF 28,29 and 30, Fig. 2, colored bright green). The lysis cassette comprises of an Rz- like spannin subunit (ORF 28, 462 bp, 154 AA) which has highest amino acid similarity with an unclassified Lederbergvirus Escherichia phage N7 (78%) whereas nucleotide sequence was similar (80 % query cover, 88.77% similarity) with Escherichia phage MSU52-L1. Usually, the spannin proteins are encoded as two-component proteins that span periplasmic space with Rz being inner membrane whereas R z1 is outer membrane protein of Gram-negative bacteria. These two proteins interact and cause membrane disruption that allows holin endolysin degradation of the cell wall. BIS08 spannin is one component spannin and Rz1 domain or its homolog is not present whereas this protein is present in two of the BIS08 close homologs ,BTP1 and epsilon 34 phages. The second component of the lysis cassette is Lysozyme R (ORF 29, 438 bp, 146 AA). And the third component is a holin (ORF30, 327bp, 108 AA). Both holin and endolysin are conserved to have high homology with Salmonella phage P22, and Epsilon 34 as well as several others.
3.2.3. Prophage Induction and Maintenance Proteins
Temperate phages usually possess proteins for integration in host genome and maintenance of the prophage. In BIS08 these genes are spread in three sections of the genome, A site-specific integrase (ORF 1, 1164 bp, 387 AA, colored red, Fig 2) responsible for integrating phage genome in host bacterium is present at the start of the genome. BIS08 integrase gene has only two hits in BLASTn Salmonella phage epsilon 34 and Salmonella phage SEN22 having 93 % and 84 % similarity respectively. Same was true for amino acid sequence where epsilon 34 and SEN22 had high similarity (97 % and 88 %) in BLASTp. The amino acid sequence of BIS08 integrase provided other hits in BLASTp but similarity index was less than 75 %. Three proteins involved in prophage induction and maintenance cI, cro, cII collectively called lytic to lysogenic switch proteins are present as one cluster (ORF 46, 47, 48, colored purple Fig 2.) in the second half of genome, and a fourth protein cIII is present upstream of this cluster (ORF 54). An important factor in temperate phage survival is timely switching from lytic to lysogenic phase. In BIS08 this switch comprises of cI (Lytic phase repressor), a cII and cIII proteins (involved in repressor activation, ORF 46 and 54, 70 AA and 57 AA respectively) and Cro proteins that suppress lysogeny and switch the mode to lytic growth.
3.2.4. Hypothetical and Characterized Proteins of BIS08
In total BIS08 encodes 72 ORFs, out of which 26 ORFs are hypothetical proteins (roughly 36 %) with no known functions (ORFs color coded ash-grey in genome map, Fig 2). Six proteins had no BLASTn homology and were present only in BIS08 (ORF 23, 24, 49, 50, 55, and 62, Fig 2). The other 20 ORFs had varying degree of similarity to phages from unclassified Lederbergvirus. An equal number of BIS08 proteins (26 ORFs) has BLAST homology with known proteins in database. These proteins are color coded Red in Figure 2. They include LPS modification enzymes for phage binding such as Bactoprenol-linked glucose transferase (ORF3, Fig 2) and an Acetyl transferase protein, involved in modification of surface O antigen (ORF 4, Fig 2, color coded red). Both proteins are present adjacent to tail spike protein (ORF 5, Fig 2) and have high homology with Salmonella phage MG40. An enzyme involved in fatty acid synthesis and degradation (KilA-N domain containing protein, ORF 27) is present adjacent to lysis cassette (Fig 2).
The genome also possesses a “nin region” like lambda phages between antitermination protein Q (ORF 32) and phage DNA replication protein O (ORF 44). This region includes 11 ORFs , half of which do not have any BLAST homology with known proteins and are listed as hypothetical proteins (ORF 35, 39-42). The proteins in the other half are related to lambda NinR region proteins [Ninz (ORF 33), NinG (ORF 34) and NinF (ORF 36), NinE (ORF37 and NinB (ORF 38)]. The function of majority “nin region” proteins are not known however NinB (orf) and NinG (rap) are known to influence lambda phage recombination when replication stops. These genes are present in epsilon 34 and other related phages of unclassified Lederbergvirus genus. The same region also has a highly conserved DNA helicase (ORF 43). Phage ORFs associated with control of lysogenic and lytic switching are present on right hand side of the genome. This region includes an anti-termination protein N (ORF 52) that activates lytic switch by suppressing transcriptional terminator activity and allowing production of phage proteins. This protein has high homology to Phage epsilon 34. Adjacent to this is ORF 53 that encodes HNH endonuclease having the highest similarity with Salmonella phage P6. This region also has a putative recombinase (ORF 56) and a P-loop containing ATpase that can catalyze diverse cellular functions such as DNA repair, signal transduction, membrane transport etc. This ORF has high similarity with Escherichia phage APC_ JM 3.2 (96 %). A putative ssDNA binding protein (ORF 58) with high similarity to Escherichia phage HK620 and a protein involved in repression of RecBCD pathway (ORF 59) with highest similarity to Salmonella phage SE1 are also present.
Although no virulence genes were identified in BIS08, it possesses putative eae gene (ORF 61) that has high homology with Salmonella phage PM43 only. This eae gene encodes effacement and attachment factor in Enteropathogenic E coli strains. This gene was smaller in size in BIS08 than E coli and Escherichia coli Phage vB_EcoS Sa179lw eae gene. Escherichia coli Phage vB_EcoS Sa179lw is one of the temperate phages whose presence in E. coli is implicated in pathogenicity by[27] . BIS08 eae gene has less homology (78 % query cover and 28 % similarity) to Escherichia coli Phage vB_EcoS Sa179lw. This gene may help bacterial host colonization [28]. In addition, BIS08 genome also possesses ea22 (ORF 64) and eaF (Orf 64) genes, both these genes are thought to promote lysogeny and regulation of lytic to lysogenic switch. BIS08 also carries eaA gene (ORF 68) whose product presumably allows the invasion of host restriction modification system.
3.2.5. BIS08 Terminase Analysis
BIS08 has a terminase enzyme (ORF 18 and 19) whose large and small subunit does not show any nucleotide homology with any member in Lederbergvirus genus and unclassified Lederbergvirus Division (un-LD) apart from ST-35.
A protein BLAST search of the amino acid sequence of terminase large subunit (TLs) indicated that only 10 percent amino acid sequence exhibit 33 % similarity with any member of Lederbergvirus genus and un-LD, however its amino acid sequence is identical to ST-35 TLs but did not appear in BLAST search as ST-35 is not annotated, only FASTA is submitted in NCBI. Same was true for the nucleotide sequence of TLs that was identical to ST-35 and had no other similarity in entire genus (3% QC and 99 % similarity). When compared in class Caudoviricetes using BLASTn rather than only Lederbergvirus genus it produced similar results. The terminase nucleotide sequence was present in several prophages whose sequence had no homology with BIS08 or ST-35, however from type of proteins these prophages possess they appear to be podo-viruses.
Nucleotide sequence of Terminase small subunit (TSs) had no similarity in Lederbergvirus genus, however when compared to Caudoviricetes class in general nucleotide BLAST it showed 99 % similarity with another phage SEN8 (Acc. No KM202159.1) from family Peduovirinae in addition to BIS08 and ST-35. The amino acid sequence of BIS08 TSs has similarity ranging from 66-36% with four other phages from un-LD (Acc. No URC09186.1, BEI45454.1, ABQ88402.1, URC09779.1, AZF93010.1, listed in order of decreasing similarity). To estimate terminase enzyme diversity in Lederbergvirus genus and un-LD we compared the TLs and TSs sequence from twenty selected accessions having varying degree of nucleotide similarity with BIS08 (Table S5) in VIRIDIC software. A maximum likelihood (MLA) phylogenetic tree using GDBP method was constructed using VICTOR single gene phylogeny (Figure 3A and B). A VIRIDIC heat map is given in Supplementary Figure S3. It was found that the genus has four distinct clades of TLs. Clade 3 (P22 like terminase) was most abundant (60 %). All close homologs of BIS08 possess this type of TLs. Clade 4 is found in 25 % of accessions (Figure 3A) whereas Sf6 and BIS08 make distinct clades (Clade 1 and 2). This analysis placed ST-35 and BIS05 as a separate lineage. Same was true for TSs phylogenetic analysis (Figure 3B). We performed a detailed in silico analysis of Protein domain prediction and structural modeling of BIS08 TLs and TSs to see its differences from other terminases in the genus.
3.2.5.1 Protein Modeling of BIS08 Terminase Small Subunit (TSs)
An initial protein BLAST of the BIS08 Terminase small subunit (TSs) amino acid sequence failed to identify a similar or previously published structure in the RCSB. To elucidate the apparently unique structure the online modelling tool Alphafold2 [29] was used first to gauge the secondary structure nature of the monomeric form and what higher order quaternary structure did the subunits make. Initial attempts to model the entire sequence as a monomer produced exceptionally poor scoring in the n-terminal and c-terminal domains (residues 1-14 and 126-151 respectively) and were omitted from the final monomer. This allowed for improved pLDDT values from 85.7 to 92.9 and a pTM score of 0.656. The output models superimposed well as per the scoring. The TSs sequence utilized to model the protein is shown in Figure 4A as is the secondary structure assignment and the monomer is shown Figure 4B. The n-terminal domain is formed by three short stacked alpha helices and a single long helix of 29 amino acids. The short stacked helical bundle forms a tight hydrophobic core at its center, maintained in part by leu25, Leu35, Phe38, Phe39, ile46, Phe56, Leu57 and Ile60. The central connecting domain is comprised of a long anti-parallel beta sheet loop (residues 81-106) followed by another alpha helix made in large part by the predominantly hydrophobic residues between T109-TAAIFWLKN-R119. A DALI structure search (Holm 2006) revealed some identity of our protein with the crystal structures of a bacteriophage Sf6 terminase small subunit from Lederbergvirus Sf6 (PDB codes 4dyq-B and 3hef-B; R.M.S.D 6.2 - 6.3 Å, [30], [31]). The superimposed structures are depicted in Figure 4C with most of the structural identity stemming from the short stacked helical bundle or N-terminal DNA-binding domains of the proteins.
To decipher the higher order quaternary structure of the protein Alphafold2 [32] was again used, this time using monomer numbers of six, eight and nine. Again, sequence length was modulated based on score and the ability of the program to complete the task successfully. Overall and much like the quaternary structure of the Sf6 terminase, a ring-like octameric assembly was necessary to make a complete ring for the BIS08 TSs (utilizing residues 15-151) and produced high confidence scoring for the top ranked complex (pLDDT=84.4, pTM=0.845 and ipTM=0.841; Figure 4D, lateral view, 4E top view at 90°). The pictured octamer is a highly interweaved structure, especially with respect to the c-terminal domain (residues 127-151) made up of two long β-sheets forming the neck of the protein and resembling and extended Beta-barrel. The main central body (residues 1-82) is formed by the aforementioned, N-terminal DNA binding domain and is capped by the long anti-parallel beta sheet loop (residues 81-106). The structure reveals a maximum diameter of approximately 90 Å and a height of 102 Å, while at the apex of the structure the diameter measures 23 Å and the base 48 Å.
3.2.5.2. Protein Modeling of BIS08 Terminase Large Subunit (TLSs)
A BLASTP search of the BIS08 Terminase Large subunit (TLS) produced no published proteins of significant similarity. Alphafold2 [32] analysis was subsequently utilized to elucidate the structure (pLDDT=91.7 pTM=0.621) which depicted two distinct protein domains (residues 1-275 and 276-486), with each subsequently reassessed independently in AlphaFold as well as being subjected to Dali homology searches online [33].
The N-terminal domain of the TLs protein had exceptional AlphaFold scoring (pLDDT=95.5, pTM=0.935) and subsequent DALI analyses (Table 1), described the protein as a Terminase that binds ATP/purine ribonucleoside triphosphate’s (GO:0005524/ GO:0035639). Like other members of this enzyme family, the BIS08 TLs AlphaFold structure possesses a central set of six parallel β-sheets which is flanked on one side by an additional set of two small anti-parallel β-sheets (residues 132-147) which are all sandwiched on either side by several α-helices (Figure 5A and 5B). Atop the enzyme are two additional alpha helices forming what is referred to as the Lid subdomain (residues 237-269) that sits atop the ATPase active site [34], a common feature for this enzyme group (Figure 5C). The top four enzymes produced exceptional R.M.S.D overlap with the modeled protein, however there were distinct differences in some secondary structure elements including any connective elongated loops, but in particular with the Lid domain of 3CPE (Sun 2008). One crystal structure (PDB code 4IEE) has a clearly defined ATP binding domain due to the presence of a non-hydrolysable analog Adenosine Triphosphate-γ-S (AGS) [35]. Its presence in part reveals several common features in ATP binding among the Terminases including a pyrophosphate binding domain present on the most central alpha helix and loop region of our model (residues 76-SGHGIGKSA-84). Other residues within four angstroms of terminal phosphate include Asn212 and Glu184. The adenosine moiety of ATP π-stacks with Trp30 and His179 of 4IEE. Given the probable loop movement in this region of BIS08, several candidates are possible for a similar interaction, including Tryptophan’s (30, 88 and 123) or Phe38 and Met243. Overall residues Asn212, Glu184, Gln45, Gly81, Lys82, Gly79, His78, Gly77 and Glu84, remain identical or mostly identical amongst the structures with the exemption of His78 (for Arginine) (Supplementary Figure S4).
A BLASTP search of the c-terminal domain of the BIS08 TLs protein (residues 276-469) was far more successful, showing homology with a number of Gene 2 proteins from Lederbergvirus Sf6 (PDB code 5c10; [40]) and Shigella phage Sf6 (PDB code 4IDH; [35]) possessing 33 % identity with our protein. Alphafold2 [32] analysis was subsequently utilized to elucidate the structure with exceptional scoring for the top ranked structure (pLDDT=95.9 pTM=0.927) and in some cases producing a sub two angstrom R.M.S.D identity with other published Terminase proteins following Dali homology searches (Table 2; [36]). Characteristic of this protein class, the structure consists of a central five stranded beta-sheet sandwiched between alpha helices ([40] ; Figure 6a and 6b) with residues 151-176 forming an extraneous Beta hairpin and alpha helical arrangement. Within this well characterized active site we observe three acidic Asp residues (181, 86 and 33 ) in TLs and a single catalytic Lys166, a residue that mediates metal cofactor Mg2+ binding in Terminase gp2 [40]. Amongst the homologous structures discovered in DALI only the Pseudomonas phage terminase protein (PDB code 8KDR-B; Dong 2023) and the Lederbergvirus Sf6 protein (PDB code 4IEE; [35]) possessed identically positioned active site residues.
3.3. Phylogenetic Analysis of BIS08P22
3.3.1. Comparative Genome Analysis with Prophages
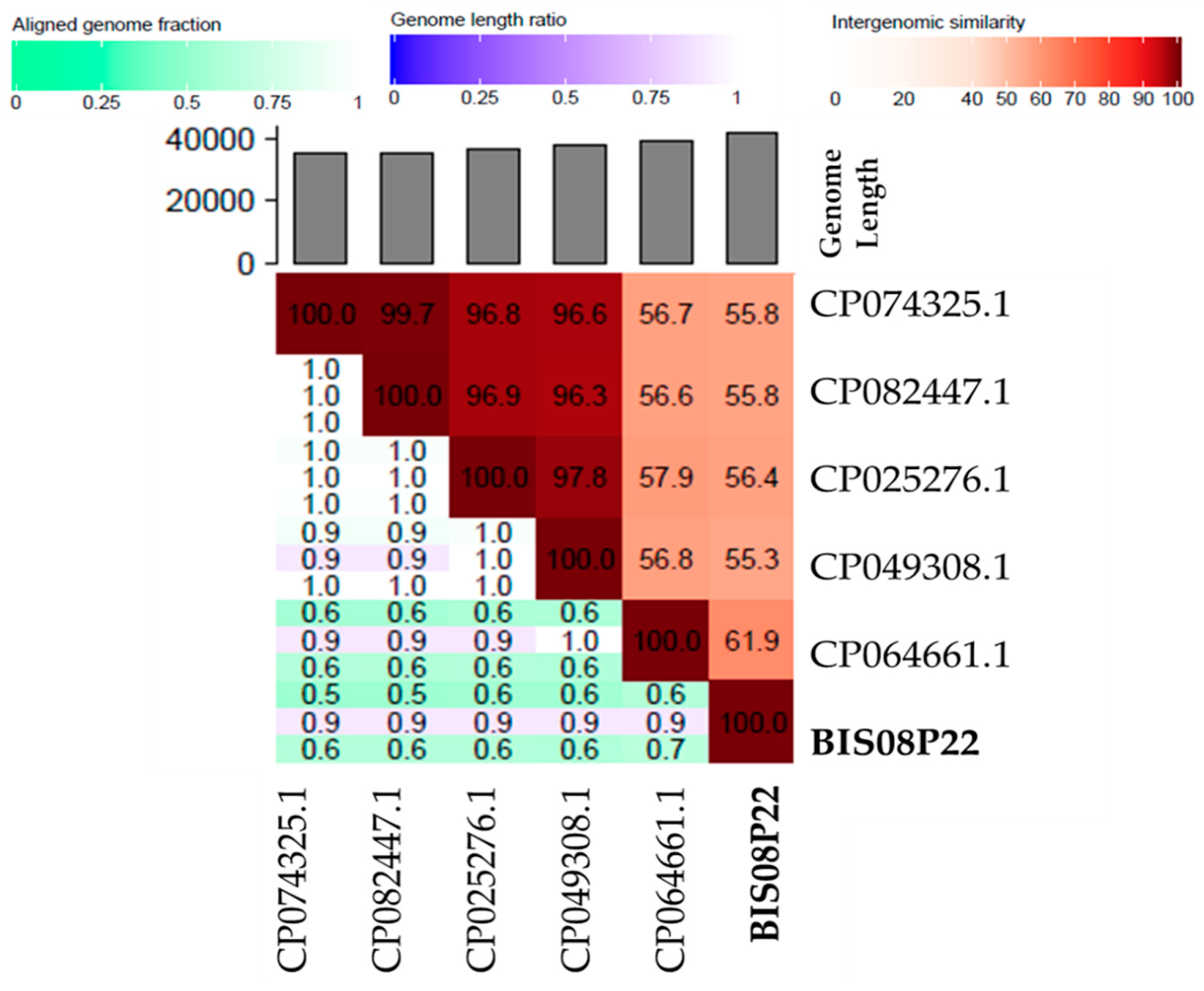
When BIS08 whole genome sequence is compared to other nucleotide sequences in general nucleotide BLAST it provides hits with several prophages albeit with less query coverage. BIS08 has highest homology with prophage sequence of Salmonella enterica species SJTUF14178 (Acc. No. CP064661.1) and SJTUF14170 (Acc. No. CP064663.1) with 63 % query cover and 98 % similarity and then with Salmonella enterica serovar Johannesburg strains CVM N58011 (Acc. No CP049308.1) and USDA-ARS-USMARC-60984 (Acc. No CP025276.1) with 54 % query cover and 95 % similarity index. Sequence of these prophages were extracted form the whole chromosome sequence in BLASTn. As per ICTV recommendations the sequence of these prophages and few others with less than 63 % query covers were subjected to VIRIDIC analysis using 95 % species and 70 % genus cutoff as standardized by ICTV. VIRIDIC analysis indicated that these prophages had maximum 61.9 % nucleotide homology with BIS08. Limited genome sequence homology within a few genes was detected which was pre-dominantly within the structural module of BIS08. This heat map indicates the overall similarity of BIS08 with its homologs is 61.9 %. It indicates that prophage CP074325.1, CP082447.1, CP025276.1, CP049308.1 are same species (≥ 95% similarity) and same genus cluster (≥ 70% similarity), whereas CP064661.1 was a different prophage and exhibited highest similarity (61.9%) to BIS08. However, both are different species in separate genera. The cluster table generated by VIRIDIC indicated that BIS08 is a new species and does not share genus cluster of these prophages in NCBI (Figure 3, Supplementary Table S1). Prophage accession numbers are given in Table S1.
Figure 3.
VIRIDIC heat map generated by using BIS08 close prophage homologs identified by general BLASTn.
Figure 3.
VIRIDIC heat map generated by using BIS08 close prophage homologs identified by general BLASTn.
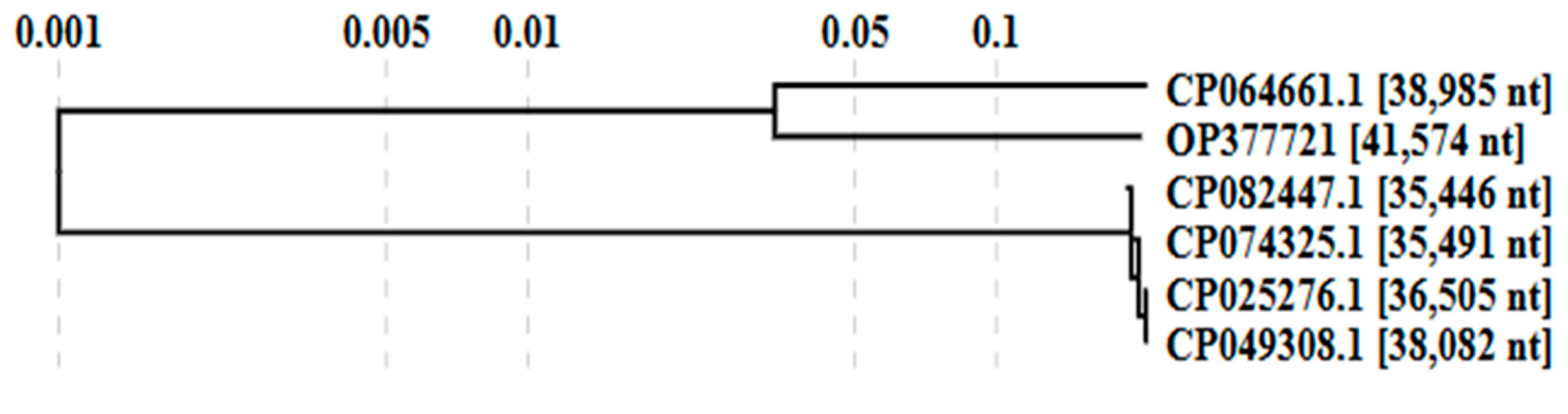
To evaluate relationship of BIS08 with its close prophage homologs a phylogenetic tree was constructed in VIP Tree software. It further validated results of VIRIDIC software and place BIS08 as a separate lineage (Figure 4). Genome wide comparison and alignment of various ORFs of BIS08 with its close prophage homologs is also produced by VIP tree and is given in Supplementary Figure S1.
Figure 4.
Protein sequence based (tBLASTx) phylogenetic tree of Salmonella phage BIS08P22 (OP377721) produced by VIP Tree software comparing protein sequence of close prophage homologs listed in Table S1.
Figure 4.
Protein sequence based (tBLASTx) phylogenetic tree of Salmonella phage BIS08P22 (OP377721) produced by VIP Tree software comparing protein sequence of close prophage homologs listed in Table S1.
3.3.2. Comparative Genome Analysis with Phage Homologs in NCBI
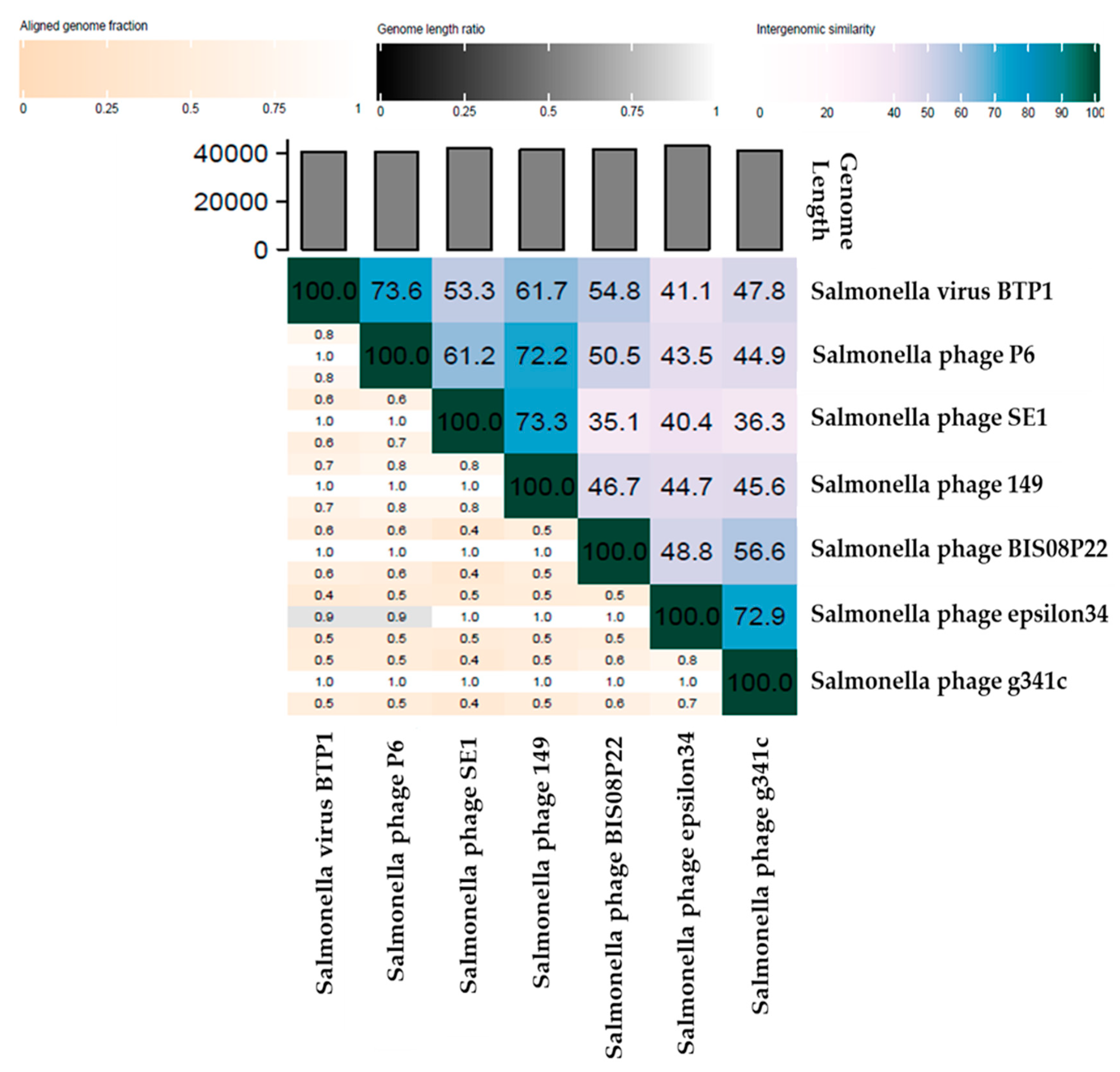
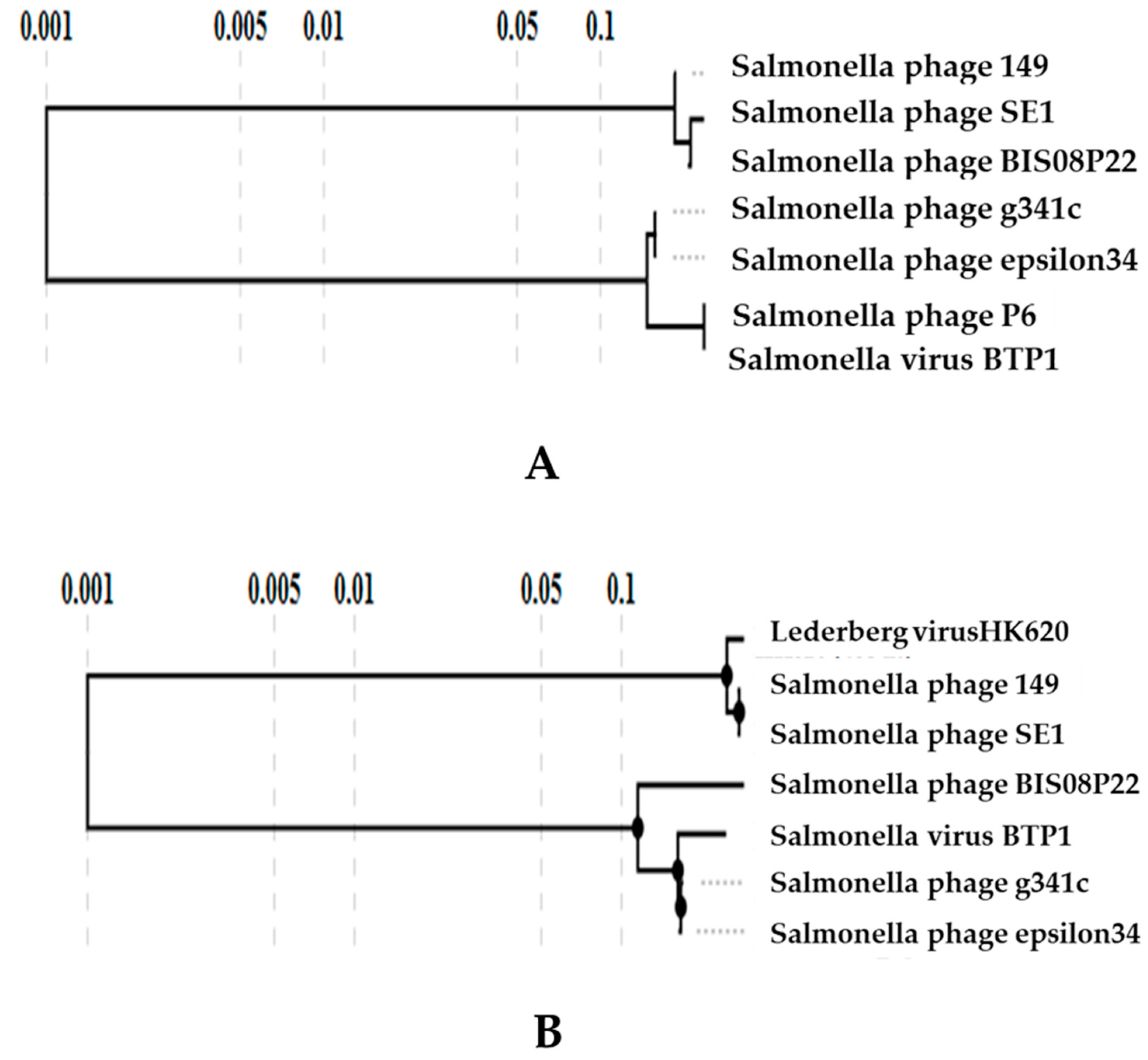
Sequence comparison of BIS08 in NCBI general nucleotide BLAST, depicted similarity with several prophage sequences (discussed in previous section). As mentioned earlier, morphological, and ultra-structural analysis of BIS08 via electron micrography indicated it is a podo-virus. Since class Podoviridae has been abolished by ICTV and genera are placed directly in class Caudoviricetes, therefore, to identify close phage homologs BIS08 genome was compared with class Caudoviricetes in BLASTn. It was observed that BLASTn produced similar hits with both genome comparison choices ; class Caudoviricetes or Lederbergvirus genus. BIS08 nucleotide sequence showed similarity with phages placed in “unclassified “division of Lederbergvirus division (un-LD). BIS08’s highest nucleotide similarity was observed with Salmonella phage P6 (OP265889.1) (50% QC and 96 % similarity), Salmonella phage g341c and Salmonella virus BTP1 (Acc. No. FJ000341.1 & NC_042346.1 respectively) (56-55 % QC and 94 %-95 % similarity). Other phages in genus Lederberg had even less query cover. This similarity was mainly restricted to the structural modules of these phages particularly capsid structure whereas tail spike gene was variable. The enzymes involved in DNA metabolism as well as integration and lysis cassettes had no nucleotide similarity. Comparison of genome wide nucleotide similarity by VIRIDIC indicated highest nucleotide similarity of BIS08 with Salmonella phage g341c (g341c)(56.6%), then with Salmonella phage BTP1 (BTP1) (54.8 %), Salmonella phage P6 (P6) (50.5 %), Salmonella phage epsilon34 (έ34)(48.8%), Salmonella phage S149 (S149)(46.7%), and Salmonella phage SE1 (SE1) (35.1%) in decreasing order (Figure 5). This analysis indicated that nucleotide similarity index of BIS08 was much less than 95% with any of its homologs in BLASTn therefore it is proposed to be classified as a new species. Moreover, as the similarity index was less than 70 % it is representing a new genus as per ICTV recommended criterion [43]. This was confirmed by the cluster table generated by VIRIDIC (Table S2). The analysis indicated that highest nucleotide similarity of BIS08 was observed with g341c and lowest with SE1. Therefore, BTP1 and P6 can be placed in one genus cluster but two different species. Whereas SE1 and 149 can be placed in one genus cluster , while g341c and έ34 make another genus.
Figure 5.
VIRIDIC heat map generated by using BIS08 close phage homologs in BLASTn. This heat map indicates the overall similarity of BIS08 with its phage homologs.
Figure 5.
VIRIDIC heat map generated by using BIS08 close phage homologs in BLASTn. This heat map indicates the overall similarity of BIS08 with its phage homologs.
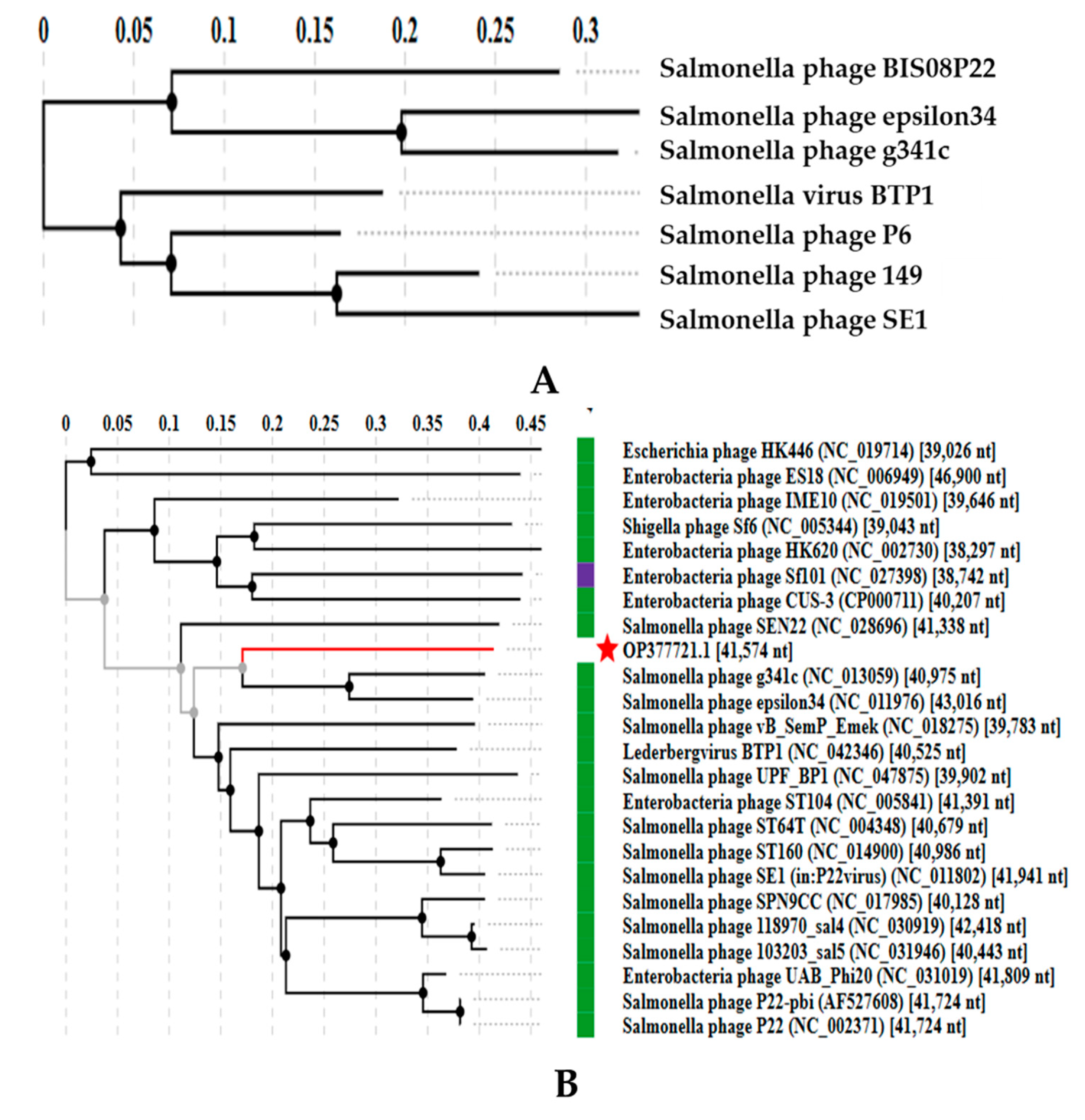
A phylogenetic tree to compare genome wide proteins ORFs of BIS08 with their closest homologs was generated by VIP tree (tBLASTx). This tree also placed BIS08 in a separate lineage (Figure 6A). Detailed alignment of various ORFs in these close homologs of BIS08 was produced by VIP tree software given in supplementary Figure S2. In addition, a phylogenetic tree between phage genomes through SG (similarity) score produced by VIP tree was also constructed (Figure 6B). Both phylogenetic trees place BIS08 in a separate group.
Figure 6.
A) A Maximum likelihood protein based phylogenetic tree (MLA) constructed by VIP software using genome wide similarities of BIS08 with closely related phages calculated by tBLASTx. B) A maximum likelihood tree produced through SG scores given by tBLASTx comparison of BIS08 homologs in Lederbergvirus genus.
Figure 6.
A) A Maximum likelihood protein based phylogenetic tree (MLA) constructed by VIP software using genome wide similarities of BIS08 with closely related phages calculated by tBLASTx. B) A maximum likelihood tree produced through SG scores given by tBLASTx comparison of BIS08 homologs in Lederbergvirus genus.
To further validate the phylogeny BIS08 core genes with closely related phages and with registered members of Lederbergvirus genus were determined using CoreGenes 5.0 software. There are six registered members of Lederbergvirus genus, including Lederbergvirus BTP1 (Acc. No NC_042346.1), HK620 (Acc. No. NC_ 002730.1), P22 (Acc. No. NC_ 002371.2), SE1Spa (Acc. No NC_011802.1 ), Sf6 (Acc. No NC_005344.1 ), and ST64T (Acc. No NC_004348.1). BIS08 shared 15 core genes out of 73 ORFs with compared phage genomes (Table S3) from unclassified Lederbergvirus division (~ 22 % of total ORFs) whereas it shared 12 core genes with registered ICTV members of Lederbergvirus genus (16.5% of total ORFs) (Table S4). We generated phylogenetic trees in VIP tree software using two orthologs from core genes (Table S3, and S4), Anti RecBCD protein (ORF 59) that promotes lytic life cycle by enhancing phage DNA damage (Figure 7A) and Rz spannin gene from lysis cassette (ORF 28). The result further emphasized BIS08 placement into a separate lineage (Figure 7B) .
Figure 7.
A) A Maximum likelihood tree based on protein sequence of anti-RecBCD protein (ORF 59) is produced with BIS08 close homologs. B) A maximum likelihood tree produced by using ortholog Rz (ORF 28) (a core gene identified by CoreGenes 5.0 software). Both these trees placeBIS08 in a separate lineage.
Figure 7.
A) A Maximum likelihood tree based on protein sequence of anti-RecBCD protein (ORF 59) is produced with BIS08 close homologs. B) A maximum likelihood tree produced by using ortholog Rz (ORF 28) (a core gene identified by CoreGenes 5.0 software). Both these trees placeBIS08 in a separate lineage.
3.3.3. Comparative Analysis of All Lederbergvirus Phages in Relation to BIS08
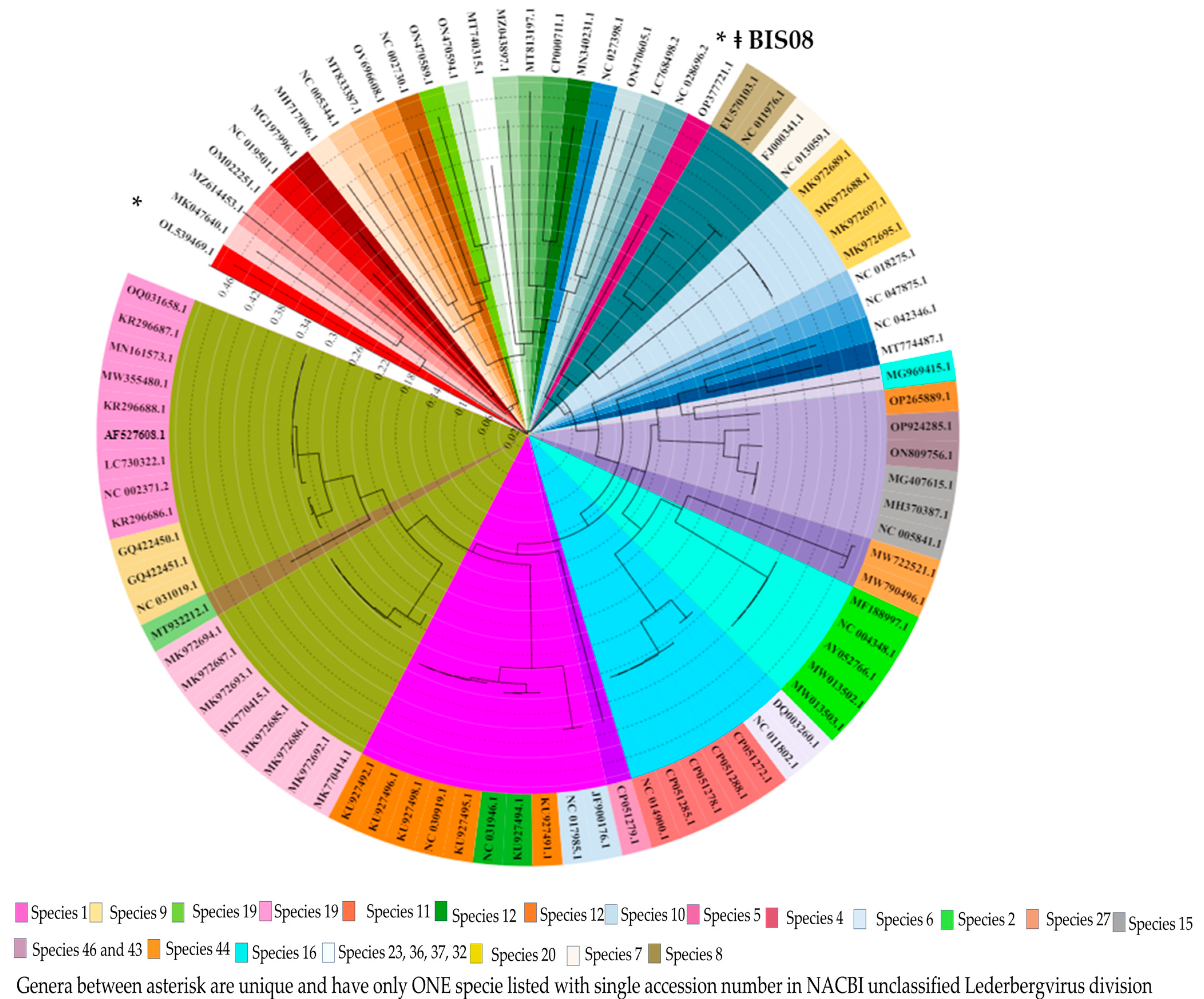
Lederbergvirus genus was initially placed in family Podoviridae and named P22-like phages in 1996. In 1998 the family Podoviridae was placed in order Caudovirales, in 1999 the name of genus was changed to P22-Like virus rather than phage and eventually renamed as Lederbergvirus genus after Esther Lederberg in 2018. In 2021 ICTV abolished family Podoviridae and order Caudovirales and now these genera are placed directly under class Caudoviricetes. Classification of this order has not been renewed since ICTV adopted fully genomic mode of classification. The phages placed in these genera were classified based on protein similarities, however the classification did not account for gene synteny which is a prominent factor in phage genome classification. Species demarcation criteria in Lederbergvirus genus is “no genome homology” between species. To put BIS08 in perspective of other species in Lederbergvirus genus and un-LD in NCBI we carried out detailed phylogeny of all the accession numbers listed in these two divisions. The analysis was carried out by VIRIDIC software and phylogenetic trees were constructed using VIP tree. There are 18 accession numbers issued to 6 registered species included in Lederbergvirus genus and 75 accession numbers are listed in un-LD. The phylogenetic tree is given in Figure 8.
According to VIRIDIC results BIS08 had maximum 56.6 % homology with any member in the two divisions. It further divided accession numbers present in both divisions into various genera and indicated that several species have no nucleotide similarity with registered members of the genus and with each other. Several phages in both divisions do not fulfill unique species criteria as genome homology is > 95% (Figure 8, Table S5), and majority species do not qualify to be placed in one genus as per ICTV recommendation of VIRIDIC nucleotide similarity ≥70% for phages in one genus. The heat map generated from this analysis and cluster table produced is given in Figure S5 and Table S5 respectively in supplementary material. Overall VIRIDIC analysis divided all phages (93 accessions in total) placed in these two divisions into 47 species clusters and 38 genus clusters (Figure S8, Table S5). The nucleotide similarity index was very low among different viruses placed in these two divisions. Using nucleotide sequence a protein-based tree was generated in VIP tree software and is given in Figure 8. This tree was in complete alignment with genera proposed by VIRIDIC as well as species demarcation by the software. The tree was annotated in iTOL software (freely available at https://itol.embl.de/ ). Accession numbers presented in phylogenetic tree are listed in the cluster table (Table S5) in order of appearance (clockwise). The tree indicates the evolutionary relation of these phages together with genera (indicated in different colors in inner circle). It was obvious from the alignment analysis of these phages (Figure S6, VIP tree) that nucleotide homology between different predicted genera was mainly confined to capsid structural module.
Figure 8.
A Maximum likelihood tree based on protein sequence of all accessions (93 in total) placed in Lederbergvirus genus and unclassified Lederbergvirus division is produced using tBLASTx calculations in VIP tree software. The tree is annotated in iTOL software [44]to indicate the genera and species identified / proposed by VIRIDIC software. Color coding in inner circle represents different genera, whereas color coding in accession numbers represents different species. In total 39 genera and 47 species were identified. The position of BIS08 is indicated with symbol ǂ and is placed in separate genus and a unique species.
Figure 8.
A Maximum likelihood tree based on protein sequence of all accessions (93 in total) placed in Lederbergvirus genus and unclassified Lederbergvirus division is produced using tBLASTx calculations in VIP tree software. The tree is annotated in iTOL software [44]to indicate the genera and species identified / proposed by VIRIDIC software. Color coding in inner circle represents different genera, whereas color coding in accession numbers represents different species. In total 39 genera and 47 species were identified. The position of BIS08 is indicated with symbol ǂ and is placed in separate genus and a unique species.
4. Discussion
Bacteriophage replication in host cells generally follows two distinct pathways, lytic or lysogenic life cycles. In former the host cell is lysed, and progeny virus particles are released, and phage DNA maintains its extrachromosomal status [45]. In later the phage genome carries genes (integrases and recombinases) that allow phage to insert its DNA in host genome. Although lytic phages are an extensive subject of research due to their utility in phage therapy the lysogenic or temperate phages also play major role in host evolution [46]. Temperate phages (P-22- like, P2 and Lambda-like phages) of former class Podoviridae have been extensively studied in past owing to their ability to carry generalized transduction and hence playing a strong role in bacterial evolution [47]. There is extensive evidence that resistance and pathogenicity markers as well as toxin genes are carried by these phages that enhance bacterial host pathogenicity [48]. ICTV recently abolished its classification of tailed bacteriophages that was based on virion shape (King et al. 2011) and adopted the genome-based classification [49]. Temporarily all genera in three classes (Sipho, Myo- and Podoviridae) are placed directly under class Caudoviricetes [15]. One such genus Lederbergvirus was previously called P22-like viruses and is well known for generalized transduction. Here we report isolation, detailed characterization, and genome analysis of a new species of temperate phage that has similarity with Lederbergvirus phages however it is distinct enough to constitute a new genus as per ICTV recommended genome-based classification criterion. We have also carried out genome-based classification analysis of all phages placed in Lederbergvirus and unclassified Lederbergvirus division indicating a need to revise classification.
BIS08 produced viable plaques only at biological pH and temperature (pH7.2 and 37 °C) which indicated its adaptability to host growth conditions. It was observed that three BIS08 prophage homologs preferred tRNAArg as integration site whereas the closest prophage with respect to genome similarity (CP064661.1) integrated at tRNAThr in Salmonella enterica serovar Typhimurium as indicated by BLASTn. It agrees with study that found Salmonella enterica prophages prefer 17 integration loci out of which six loci were tRNA genes coding for Arg, Thr, Ser, Pro, Met and Leu. It was observed that these are not randomly selected sites rather phages preferred evolutionarily conserved unique sites of these tRNAs as they occur only once at specific places in bacterial genomes and are indispensable [50], [51].
BIS08 exhibit typical podo virus morphology (hexagonal head and a short tail) having latent period of 25-30 mins and burst size of 60 virions/cell. The Lederberg viruses are reported to have high burst sizes of approximately 500 virions / cell (REF). In few reports P22 is reported to give smaller burst size of 34 virions/cell as well[52]. The phage infection also caused a delay in propagation of the host strain for extended period (14h post incubation at pH7, 37 °C) as indicated by bacterial growth when infected with BIS08 (Fig 1 D). Similar findings are reported by Gildea [53] where P22 delayed host strain Salmonella Typhimurium growth for 24 h.
ICTV recommends VIRIDIC software analysis for genome wide comparison of newly isolated bacteriophages. For a new phage to be placed in the existing genus it should have at least 70% similarity index in VIRIDIC, whereas 95 % similarity index is required to categorize it as an existing species [54]. For BIS08 the VIRIDIC analysis indicated maximum 56.8% similarity with any member of Lederbergvirus as well as unclassified Lederbergvirus division. Therefore, it cannot be placed in that genus. This fact was also ascertained by CoreGenes analysis where BIS08 shared only 12 genes with registered members of Lederbergvirus genus whereas only 15 genes with its close homologs (Table S3 and S4). Phylogeny of two CoreGenes also placed BIS08 as separate lineage. When we analyzed genome sequences of all phages placed in these two divisions it was observed that multiple accessions were assigned to the same species having ≥ 95% similarity. For example, nine accession numbers were assigned to Salmonella phage P22 under different phage names, however they had no difference in genome and were 100-99.9 % similar. Therefore, VIRIDIC placed them together as same species and same genus (Fig 8, Table S5). In total 93 phages in these two divisions were assigned to 47 species clusters and 39 different genera as per ICTV new classification criterion.
The similarities between Lederbergvirus phages were generally confined to their capsid structure module which is conserved between all short-tailed podo viruses. This module usually has 14 genes (P22,dedicated for capsid formation including a terminase (S and L) , portal, capsid scaffold, and major capsid proteins (3 genes) tail / head stabilization of procapsid (3 genes) DNA ejection and transfer proteins (4 genes) and tail spike protein [55], [56]. Sometimes an additions capsid decoration protein is also present (Epsilon 34 and phage L) making the total count of 15 proteins [57]. BIS08 capsid ,module (ORF 5-15) has 15 genes rather than 14. It agrees with the fact that phylogenetically BIS08 is closer to epsilon 34 rather than P22 originating from same ancestral clade (Figure 8 Acc. No NC_013059.1 ).The tail fiber however was different in nucleotide and amino acid sequence (only BTP1 has 100 similarity in both nt and AA, all others 27 % query cover for nt and 70% similarity in AA sequence) and clearly diverged from others (Figure S7).
A peculiar feature of BIS08 is its terminase gene (large and small) present as part of the structure module has no similarity with any member of the genus apart from ST-35 which is not annotated and is uploaded in NCBI as phage sequence isolated from human fecal metagenome analysis (Gen Bank acc. No CP051279.1). Lederbergvirus and other podo phages use a head full packaging mechanism. It requires packaging of DNA in a pre-formed procapsid structure. Three main components take part in this DNA filling. Phage portal protein and Phage terminase enzyme. Portal protein marks an opening at the procapsid entry site. Whereas terminase enzyme translocate the DNA into portal vortex by using ATP hydrolysis hence acting as powerful motors/ translocators. The terminase enzyme has two subunits Large and small. Small terminase interact with DNA keeping it in position on the vortex whereas large terminase stuff DNA and cleaves using ATPase and nuclease domains. These terminases are usually conserved between genera and can be used as core genes for viral taxonomy. BIS08 terminase was unique as it reflects no similarity in the Lederbergvirus genus as well as un-LD. Our terminase phylogenetic analysis indicated three major types of large terminases present in genus Lederbergvirus and un-LD. Type 1 is P22 like terminase (Clade 3and Clade 5 Figure 3A and B respectively) that is fully characterized and is most common type. Second type is Sf6 type (Lederbergvirus Sf6) it has also been characterized but is different from P22 terminases. Third type BIS08 and ST-35 type of terminase that makes a separate lineage and is not reported before. Our analysis indicated that small terminase of BIS08 makes an octameric ring like structure similar to Sf6 [31] rather than a nonameric ring of P22 terminase small subunit [58]. Despite structural analogy the two terminases were different and similarity was confined to N-terminal DNA binding domain of these proteins. The BIS08 large terminase has two distinct domains like Sf6 and P22 . The N-terminus ATPase domain of BIS08 has 6 staked parallel beta sheets in contrast to five beta sheets present in Sf6. Despite low nucleotide homology the large terminase exhibits functional similarity to Sf6.
Genome wide ORF comparison by alignment (Figure S6) indicated that phage similarity was confined to structure module in entire genus. Genes involved in DNA processing, recombination, anti-repression integration and lytic to lysogenic switching shared little to no homology. Overall phages in genus Lederbergvirus have similar genome organisation. All of them possess proteins involved in regulating lytic to lysogenic switch (cI, cro, cII or in some cases LexA family transcriptional regulator), bacterial cell lysis machinery having holin, endolysin and spannin subunits and a set of 12 proteins (Nin operon) starting from anti termination protein Q involved in transcription regulation. However similar arrangement can be found in lambda phages as well as other phages of former family Myoviridae [59]. Position of these genes is different in different phages of the genus.
Phylogenetic analysis of Lederbergvirus phages agreed with the VIRIDIC analysis. The species identified by VIRIDIC software were also confirmed by VIP tree created tBLASTx based phylogenetic tree. It is noteworthy that BIS08 was recognized as a new species as well as a separate genus by both analyses. First twenty-three accession numbers listed between asterisk in Figure 8 (Table S5) were all placed as unique species and genera by both analyses. Moreover, CoreGenes5 did not recognize any common genes between these genera. Revision of Lederbergvirus classification is therefore proposed on the basis of this analysis.
5. Conclusions
Salmonella phage BIS08P22 is a novel phage representing a new species as well as suggesting a new genus in class Caudoviricetes. Detailed analysis of BIS08 and other Lederberg viruses indicates a need to revise the classification of this genus. According to our analysis the accessions placed in Lederbergvirus genus and Un-LD can be divided into 39 genera and 47 species clusters. A taxonomic proposal is under preparation in consultation with ICTV correspondents. Our study provides an insight into a vaste diversity of P22-like phages that play a prominent role in modulating bacterial host pathogenicity.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1,.Figure S1: Alignment of Salmonella phage BIS08P22 various ORFs with close prophage homologs. These prophage sequences were extracted form chromosome sequences of their respective hosts in BLASTn. The scale on top left gives percentage homology depicted as rainbow of colors; Figure S2: Alignment of Salmonella phage BIS08P22 various ORFs with close Phage homologs. These phage sequences were extracted form NCBI nucleotide BLASTn. The scale on top left gives percentage homology depicted as rainbow of colors.; Figure S3: A VIRIDIC heat map generated for nucleotide sequence comparison of terminase gene in selected Accessions of the Lederbergvirus genus and unclassified Lederbergvirus division. The phage names are color coded as per Phylogenetic divisions given in main manuscript in Figure 3 that align with VIRIDIC data; Figure S4: The Potential ATP binding site of the BIS08 Terminase Large subunit present in the n-terminus. Residues (shown in line and stick formation) within 5 Å of the superimposed AGS of the large terminase of Sf6 (in blue; PDB code 4IEE) are shown and labelled. Residues in stick formation highlight common residues in near identical positions for the enzymes discovered during DALI analysis.; Figure S5: A heat map generated by VIRIDIC software using all accession numbers present in Lederbergvirus genus and unclassified Lederbergvirus division in NCBI. This map and cluster table provided in Table S5 provides an accurate relationship of these accession genomes. VIRIDIC divided these accessions in 38 genera and 39 species in total.; Figure S6: The genome wide ORF alignment produced by VIP tree software by using tBLASTx results for members of Lederbergvirus genus and unclassified Lederberg virus division phages. One member of each species was selected for this purpose. Moreover, the numbers on the left-hand side represent order of viruses in Table S5. Majority viruses classified by VIRIDIC software in separate genera share ORF homology to structural modules only.; Figure S7: VIP tree based phylogenetic tree of BIS08 tail spike protein (ORF5) produced by tBLASTx analysis. BIS08 and BTP1 make a separate lineage; Table S1: Table S1. VIRIDIC cluster table of BIS08 with its close prophage homologs ; Table S2. VIRIDIC cluster table for BIS08 with its close BLASTn Phage homologs; Table S3: The number of core genes identified by CoreGenes5.0 software in close homologs of BIS08; Table S4: The number of core genes identified by CoreGenes5.0 software shared by BIS08 with Lederbergvirus registered phage members (The number of homologs in each column is :12); Table S5: VIRIDIC cluster table for phages placed in Lederbergvirus genus and unclassified Lederbergvirus division.
Author Contributions
“Conceptualization, S.S. methodology, S.S, V.C.; software, S.S, V.C, I.A, TAB, E.A.; validation, S.J. N.B. A.B.; formal analysis, S.S. V.C; investigation, S.S. V.C. I.U, A.Y; resources, S.S.; data curation, S.S. A.B. A.T; writing—original draft preparation, S.S. V.C; writing—review and editing, S.S. V.C, S.J, N.B, X.X.; visualization, I.A.; supervision, S.S.; project administration, S.S.; funding acquisition, S.S All authors have read and agreed to the published version of the manuscript.”.
Funding
“This research received no external funding”.
Informed Consent Statement
Not applicable.
Data Availability Statement
The sequence data produced in this study is uploaded on NCBI with Accession Number OP377721.1.
Acknowledgements
Authors Ibrar Ahmad and Zarrin Basharat were employes of Alpha genomics a private firm and Authors Vince Carbone and Sofia Khanum are employes of AgResearch New Zealand however they have no claim on data produced in this research and they do not have any funding liability.
Conflicts of Interest
“The authors declare no conflict of interest.”
References
- J. E. S. Strange, P. Leekitcharoenphon, F. D. Møller, and F. M. Aarestrup, “Metagenomics analysis of bacteriophages and antimicrobial resistance from global urban sewage,” Sci Rep, vol. 11, no. 1, 2021. [CrossRef]
- Du Toit, “The language of phages,” Nat Rev Microbiol, vol. 15, no. 3, 2017. [CrossRef]
- Howard-Varona, K. R. Hargreaves, S. T. Abedon, and M. B. Sullivan, “MINI REVIEW Lysogeny in nature: mechanisms, impact and ecology of temperate phages Why study lysogeny?,” Nature Publishing Group, vol. 11, 2017.
- C. Groth and M. P. Calos, “Phage integrases: Biology and applications,” Journal of Molecular Biology, vol. 335, no. 3. 2004. [CrossRef]
- S. R. Casjens and J. H. Grose, “Contributions of P2- and P22-like prophages to understanding the enormous diversity and abundance of tailed bacteriophages,” Virology, vol. 496, 2016. [CrossRef]
- Z. Hibstu, H. Belew, Y. Akelew, and H. M. Mengist, “Phage Therapy: A Different Approach to Fight Bacterial Infections,” Biologics: Targets and Therapy, vol. 16. 2022. [CrossRef]
- E. Harrison and M. A. Brockhurst, “Ecological and Evolutionary Benefits of Temperate Phage: What Does or Doesn’t Kill You Makes You Stronger,” BioEssays, vol. 39, no. 12. 2017. [CrossRef]
- S. L. W. Zajdowicz and R. K. Holmes, “Phage Conversion and the Role of Bacteriophage and Host Functions in Regulation of Diphtheria Toxin Production by Corynebacterium diphtheriae,” 2016. [CrossRef]
- Y. Sakaguchi et al., “The genome sequence of Clostridium botulinum type C neurotoxin-converting phage and the molecular mechanisms of unstable lysogeny,” Proc Natl Acad Sci U S A, vol. 102, no. 48, 2005. [CrossRef]
- E. F. Boyd, M. R. Carpenter, and N. Chowdhury, “Mobile effector proteins on phage genomes,” Bacteriophage, vol. 2, no. 3, 2012. [CrossRef]
- S. Mirold et al., “Isolation of a temperate bacteriophage encoding the type III effector protein SopE from an epidemic Salmonella typhimurium strain,” 1999. [Online]. Available: www.pnas.org.
- P. L. Wagner and M. K. Waldor, “MINIREVIEW Bacteriophage Control of Bacterial Virulence,” Society, vol. 70, no. 8, 2002. [CrossRef]
- E. A. Miao and S. I. Miller, “Bacteriophages in the evolution of pathogen-host interactions,” Proceedings of the National Academy of Sciences of the United States of America, vol. 96, no. 17. 1999. [CrossRef]
- C. Wendling et al., “Tripartite species interaction: eukaryotic hosts suffer more from phage susceptible than from phage resistant bacteria,” BMC Evol Biol, vol. 17, no. 1, 2017. [CrossRef]
- Turner et al., “Abolishment of morphology-based taxa and change to binomial species names: 2022 taxonomy update of the ICTV bacterial viruses subcommittee,” Arch Virol, vol. 168, no. 2, 2023. [CrossRef]
- Y. N. Chiang, J. R. Penadés, and J. Chen, “Genetic transduction by phages and chromosomal islands: The new and noncanonical,” PLoS Pathog, vol. 15, no. 8, 2019. [CrossRef]
- P. J. Walker et al., “Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020),” Arch Virol, vol. 165, no. 11, 2020. [CrossRef]
- P. J. Walker et al., “Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021),” Arch Virol, vol. 166, no. 9, 2021. [CrossRef]
- Y. Zhu, J. Shang, C. Peng, and Y. Sun, “Phage family classification under Caudoviricetes: A review of current tools using the latest ICTV classification framework,” Frontiers in Microbiology, vol. 13. 2022. [CrossRef]
- E. Catalano, “Viral Genome Packaging Machines,” in Viral Genome Packaging Machines: Genetics, Structure, and Mechanism, 2005. [CrossRef]
- M. M. Susskind and D. Botstein, “Molecular genetics of bacteriophage P22,” Microbiological Reviews, vol. 42, no. 2. 1978. [CrossRef]
- Botstein, “Synthesis and maturation of phage P22 DNA. I. Identification of intermediates,” J Mol Biol, vol. 34, no. 3, 1968. [CrossRef]
- V. B. Rao and M. Feiss, “The bacteriophage DNA packaging motor,” Annual Review of Genetics, vol. 42. 2008. [CrossRef]
- S. Sattar et al., “Phenotypic characterization and genome analysis of a novel Salmonella Typhimurium phage having unique tail fiber genes,” Sci Rep, vol. 12, no. 1, 2022. [CrossRef]
- Seul, J. J. Müller, D. Andres, E. Stettner, U. Heinemann, and R. Seckler, “Bacteriophage P22 tailspike: Structure of the complete protein and function of the interdomain linker,” Acta Crystallogr D Biol Crystallogr, vol. 70, no. 5, 2014. [CrossRef]
- S. Steinbacher et al., “Phage P22 tailspike protein: Crystal structure of the head-binding domain at 2.3 Å, fully refined structure of the endorhamnosidase at 1.56 Å resolution, and the molecular basis of O-antigen recognition and cleavage,” J Mol Biol, vol. 267, no. 4, 1997. [CrossRef]
- Y. Te Liao, F. Liu, X. Sun, R. W. Li, and V. C. H. Wu, “Complete genome sequence of Escherichia coli phage vB_EcoS Sa179lw, isolated from surface water in a producegrowing area in northern California,” Genome Announc, vol. 6, no. 27, 2018. [CrossRef]
- E. Jerse and J. B. Kaper, “The eae gene of enteropathogenic Escherichia coli encodes a 94-kilodalton membrane protein, the expression of which is influenced by the EAF plasmid,” Infect Immun, vol. 59, no. 12, 1991. [CrossRef]
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov, and M. Steinegger, “ColabFold: making protein folding accessible to all,” Nat Methods, vol. 19, no. 6, 2022. [CrossRef]
- H. Zhao et al., “Crystal structure of the DNA-recognition component of the bacterial virus Sf6 genome-packaging machine,” Proc Natl Acad Sci U S A, vol. 107, no. 5, 2010. [CrossRef]
- H. Zhao, Y. N. Kamau, T. E. Christensen, and L. Tang, “Structural and functional studies of the phage Sf6 terminase small subunit reveal a DNA-spooling device facilitated by structural plasticity,” J Mol Biol, vol. 423, no. 3, 2012. [CrossRef]
- J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,” Nature, vol. 596, no. 7873, 2021. [CrossRef]
- L. Holm, “Using Dali for Protein Structure Comparison,” in Methods in Molecular Biology, 2020. [CrossRef]
- B. J. Hilbert, J. A. Hayes, N. P. Stone, C. M. Duffy, B. Sankaran, and B. A. Kelch, “Structure and mechanism of the ATPase that powers viral genome packaging,” Proc Natl Acad Sci U S A, vol. 112, no. 29, 2015. [CrossRef]
- H. Zhao, T. E. Christensen, Y. N. Kamau, and L. Tang, “Structures of the phage Sf6 large terminase provide new insights into DNA translocation and cleavage,” Proc Natl Acad Sci U S A, vol. 110, no. 20, 2013. [CrossRef]
- L. Holm, “DALI and the persistence of protein shape,” Protein Science, vol. 29, no. 1, 2020. [CrossRef]
- R. A. Laskowski, J. Jabłońska, L. Pravda, R. S. Vařeková, and J. M. Thornton, “PDBsum: Structural summaries of PDB entries,” Protein Science, vol. 27, no. 1, 2018. [CrossRef]
- S. Sun et al., “The Structure of the Phage T4 DNA Packaging Motor Suggests a Mechanism Dependent on Electrostatic Forces,” Cell, vol. 135, no. 7, 2008. [CrossRef]
- H. K. H. Fung et al., “Structural basis of DNA packaging by a ring-type ATPase from an archetypal viral system,” Nucleic Acids Res, vol. 50, no. 15, 2022. [CrossRef]
- H. Zhao et al., “Two distinct modes of metal ion binding in the nuclease active site of a viral DNA-packaging terminase: Insight into the two-metal-ion catalytic mechanism,” Nucleic Acids Res, vol. 43, no. 22, 2015. [CrossRef]
- S. Dong et al., “Structural basis of nucleosome deacetylation and DNA linker tightening by Rpd3S histone deacetylase complex,” Cell Res, 2023. [CrossRef]
- M. I. Daudén, J. Martín-Benito, J. C. Sánchez-Ferrero, M. Pulido-Cid, J. M. Valpuesta, and J. L. Carrascosa, “Large terminase conformational change induced by connector binding in bacteriophage T7,” Journal of Biological Chemistry, vol. 288, no. 23, pp. 16998–17007, 2013.
- M. Adriaenssens and J. Rodney Brister, “How to name and classify your phage: An informal guide,” Viruses, 2017. [CrossRef]
- Letunic and P. Bork, “Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation,” Nucleic Acids Res, vol. 49, no. W1, 2021. [CrossRef]
- “An investigation on the nature of ultra-microscopic viruses by Twort FW, L.R.C.P. Lond., M.R.C.S. (From the Laboratories of the Brown Institution, London) ,” Bacteriophage, vol. 1, no. 3, 2011. [CrossRef]
- V. S. Gummalla, Y. Zhang, Y. Te Liao, and V. C. H. Wu, “The Role of Temperate Phages in Bacterial Pathogenicity,” Microorganisms, vol. 11, no. 3. 2023. [CrossRef]
- E. V. Davies, C. Winstanley, J. L. Fothergill, and C. E. James, “The role of temperate bacteriophages in bacterial infection,” FEMS Microbiology Letters, vol. 363, no. 5. 2016. [CrossRef]
- Q. Chen et al., “Bacteriophage and Bacterial Susceptibility, Resistance, and Tolerance to Antibiotics,” Pharmaceutics, vol. 14, no. 7. 2022. [CrossRef]
- D. Turner, A. M. Kropinski, and E. M. Adriaenssens, “A Roadmap for Genome-Based Phage Taxonomy.,” Viruses, vol. 13, no. 3, Mar. 2021. [CrossRef]
- L.-M. Bobay, E. P. C. Rocha, and M. Touchon, “The adaptation of temperate bacteriophages to their host genomes.,” Mol Biol Evol, vol. 30, no. 4, pp. 737–751, Apr. 2013. [CrossRef]
- Y. Zhang, Y. Te Liao, A. Salvador, X. Sun, and V. C. H. Wu, “Prediction, Diversity, and Genomic Analysis of Temperate Phages Induced From Shiga Toxin-Producing Escherichia coli Strains,” Front Microbiol, vol. 10, 2020. [CrossRef]
- Jeon and J. Ahn, “Evaluation of phage adsorption to Salmonella Typhimurium exposed to different levels of pH and antibiotic,” Microb Pathog, vol. 150, 2021. [CrossRef]
- L. Gildea, J. A. Ayariga, B. K. Robertson, and R. Villafane, “P22 Phage Shows Promising Antibacterial Activity under Pathophysiological Conditions,” Archives of Microbiology & Immunology, vol. 06, no. 01, 2022. [CrossRef]
- C. Moraru, A. Varsani, and A. M. Kropinski, “VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses,” Viruses, vol. 12, no. 11, 2020. [CrossRef]
- Xiao et al., “Assembly and Capsid Expansion Mechanism of Bacteriophage P22 Revealed by High-Resolution Cryo-EM Structures,” Viruses, vol. 15, no. 2, 2023. [CrossRef]
- King, D. Botstein, S. Casjens, W. Earnshaw, S. Harrison, and E. Lenk, “Structure and assembly of the capsid of bacteriophage P22.,” Philos Trans R Soc Lond B Biol Sci, vol. 276, no. 943, 1976. [CrossRef]
- E. B. Gilcrease, D. A. Winn-Stapley, F. C. Hewitt, L. Joss, and S. R. Casjens, “Nucleotide sequence of the head assembly gene cluster of bacteriophage L and decoration protein characterization,” J Bacteriol, vol. 187, no. 6, 2005. [CrossRef]
- Roy, A. Bhardwaj, P. Datta, G. C. Lander, and G. Cingolani, “Small terminase couples viral DNA binding to genome-packaging ATPase activity,” Structure, vol. 20, no. 8, 2012. [CrossRef]
- G. Fermin, S. Mazumdar-Leighton, and P. Tennant, “Chapter 9 - Viruses of Prokaryotes, Protozoa, Fungi, and Chromista,” in Viruses, P. Tennant, G. Fermin, and J. E. Foster, Eds., Academic Press, 2018, pp. 217–244. [CrossRef]
- S. Sattar et al., “Genome Analysis and Therapeutic Evaluation of a Novel Lytic Bacteriophage of Salmonella Typhimurium: Suggestive of a New Genus in the Subfamily Vequintavirinae,” Viruses, vol. 14, no. 2, p. 241, 2022.
- H. Bao, H. Zhang, and R. Wang, “Isolation and characterization of bacteriophages of Salmonella enterica serovar Pullorum,” Poult Sci, 2011. [CrossRef]
- Y. E. Gencay, T. Birk, M. C. H. Sørensen, and L. Brøndsted, “Methods for isolation, purification, and propagation of bacteriophages of Campylobacter jejuni,” in Methods in Molecular Biology, 2017. [CrossRef]
- R. Yamamoto, B. M. Alberts, R. Benzinger, L. Lawhorne, and G. Treiber, “Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification,” Virology, vol. 40, no. 3, pp. 734–744, 1970. [CrossRef]
- M. Kropinski, “Practical Advice on the One-Step Growth Curve.,” Methods Mol Biol, vol. 1681, pp. 41–47, 2018. [CrossRef]
- M. L. Capra, A. Quiberoni, and J. Reinheimer, “Phages of Lactobacillus casei/paracasei: Response to environmental factors and interaction with collection and commercial strains,” J Appl Microbiol, 2006. [CrossRef]
- G. O’Flynn, A. Coffey, G. F. Fitzgerald, and R. P. Ross, “The newly isolated lytic bacteriophages st104a and st104b are highly virulent against Salmonella enterica,” J Appl Microbiol, 2006. [CrossRef]
- P. Bryant, G. Pozzati, and A. Elofsson, “Improved prediction of protein-protein interactions using AlphaFold2,” Nat Commun, vol. 13, no. 1, 2022. [CrossRef]
- L. C. Schrödinger, “The PyMOL molecular graphics system, version 1.3 r1.” August, 2010.
- P. Meier-Kolthoff and M. Göker, “VICTOR: genome-based phylogeny and classification of prokaryotic viruses,” Bioinformatics, vol. 33, no. 21, pp. 3396–3404, 2017.
- Y. Nishimura, T. Yoshida, M. Kuronishi, H. Uehara, H. Ogata, and S. Goto, “ViPTree: the viral proteomic tree server,” Bioinformatics, vol. 33, no. 15, pp. 2379–2380, 2017. [CrossRef]
- P. Davis, D. Seto, and P. Mahadevan, “CoreGenes5.0: An Updated User-Friendly Webserver for the Determination of Core Genes from Sets of Viral and Bacterial Genomes,” Viruses, vol. 14, no. 11, 2022. [CrossRef]
- Yukgehnaish et al., “PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach,” Viruses, vol. 14, no. 2, 2022. [CrossRef]
Figure 1.
Characterization of BIS08P22 A) Temperature stability assay indicating phage growth only at 37 °C, B) pH stability graph showing growth of BIS08 at pH7, and partial stability at pH9 C) One step growth curve indicating latent period of 20-25 mins and burst time 30 min. Burst size was calculated to be 60 virions / cell. D) Bacterial growth reduction assay to check impact of BIS08 infection on SE-BS17 growth; readings were taken for 24 h. SE-BS17 (Salmonella Typhimurium strain) was infected with BIS08 at MOI 10. Values are represented as average of triplicates for all experiments unless otherwise stated.
Figure 1.
Characterization of BIS08P22 A) Temperature stability assay indicating phage growth only at 37 °C, B) pH stability graph showing growth of BIS08 at pH7, and partial stability at pH9 C) One step growth curve indicating latent period of 20-25 mins and burst time 30 min. Burst size was calculated to be 60 virions / cell. D) Bacterial growth reduction assay to check impact of BIS08 infection on SE-BS17 growth; readings were taken for 24 h. SE-BS17 (Salmonella Typhimurium strain) was infected with BIS08 at MOI 10. Values are represented as average of triplicates for all experiments unless otherwise stated.
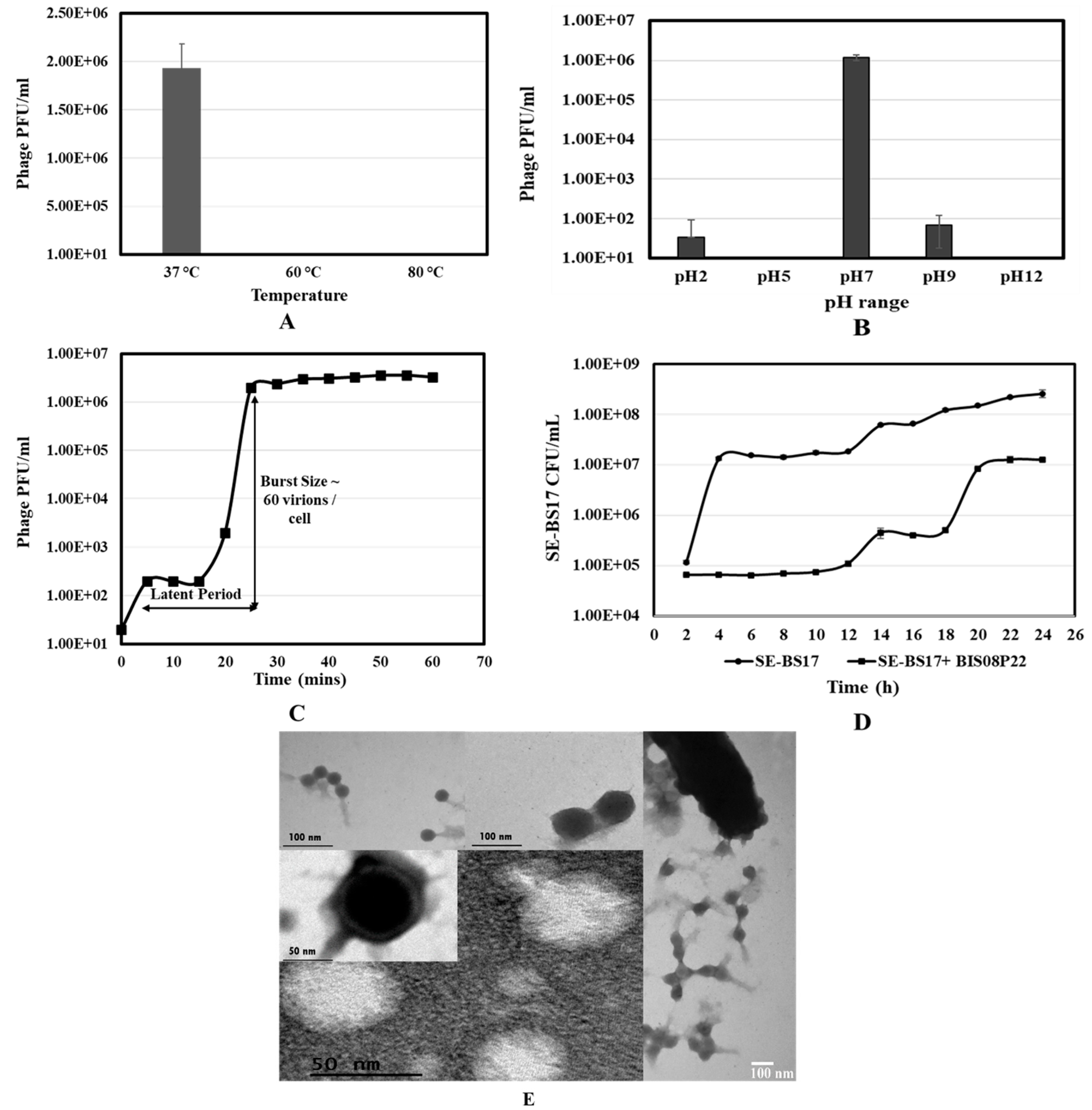
Figure 2.
Salmonella phage BIS08P22, genome map. The direction of arrows represents the strand polarity (+, -). Colour coding represents type of ORFs that are numbered acording to their position on genome and re illustrated in detail in 2.2. ORFs related to capsid morphology are colored black, whereas ORFs of tail morphology are colored blue. Hypothetical proteins are colored grey wereas proteins having BLASTp homology with known proteins in NCBI are colored red. Module II has lysis cassettee whereas module III includes integration cassette of BIS08. .
Figure 2.
Salmonella phage BIS08P22, genome map. The direction of arrows represents the strand polarity (+, -). Colour coding represents type of ORFs that are numbered acording to their position on genome and re illustrated in detail in 2.2. ORFs related to capsid morphology are colored black, whereas ORFs of tail morphology are colored blue. Hypothetical proteins are colored grey wereas proteins having BLASTp homology with known proteins in NCBI are colored red. Module II has lysis cassettee whereas module III includes integration cassette of BIS08. .
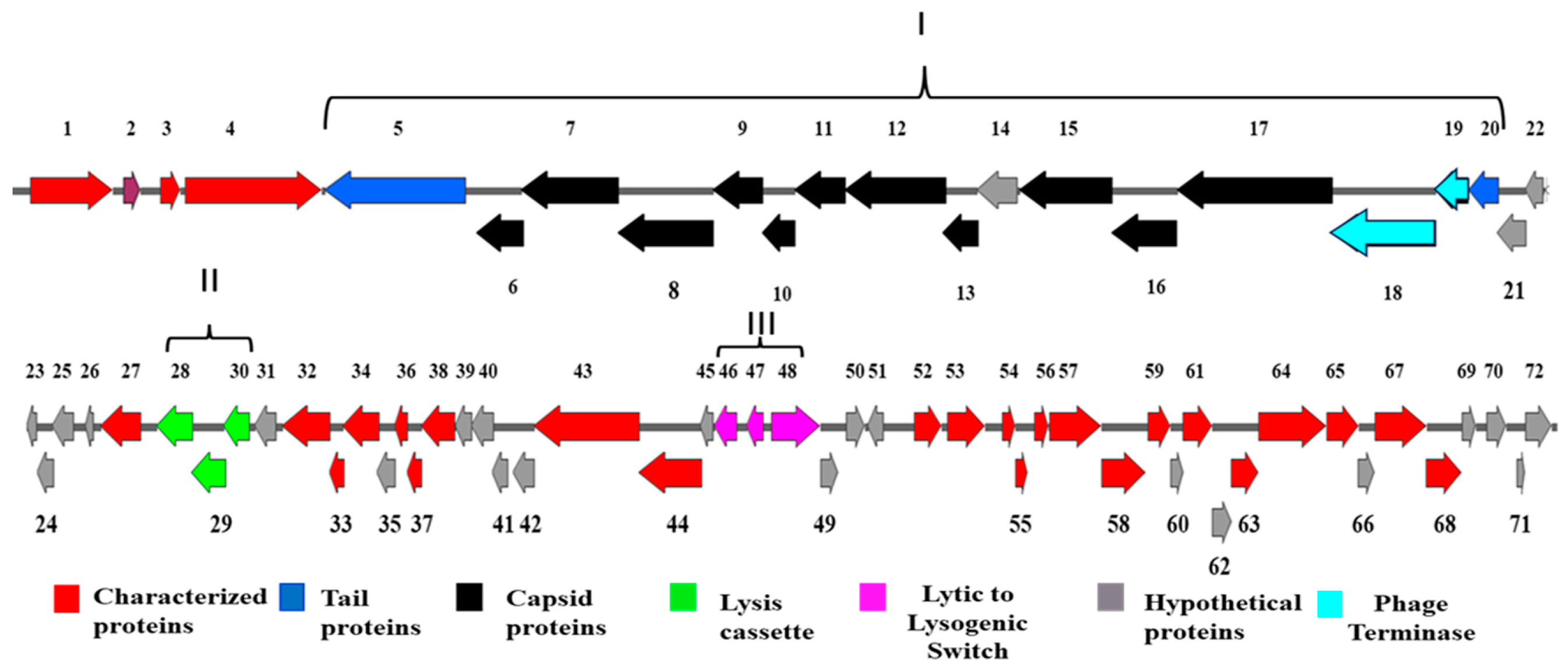
Figure 3.
A maximum liklihood tree of terminase Large subunit (TLs) nucleotide sequence (A) and terminase small subunit (TSs) nucleotide sequence (B) produced by VICTOR GDBP method.The tree was generated by providing nucleotide sequence of selected accessions from Ledebergvirus genus and un LD. Tree was annoted in iTol. Color coding represents 4 different clades in large terminase sequence, and 5 different clades in small termianse. BIS08 and ST-35 make a separate clade in both. .
Figure 3.
A maximum liklihood tree of terminase Large subunit (TLs) nucleotide sequence (A) and terminase small subunit (TSs) nucleotide sequence (B) produced by VICTOR GDBP method.The tree was generated by providing nucleotide sequence of selected accessions from Ledebergvirus genus and un LD. Tree was annoted in iTol. Color coding represents 4 different clades in large terminase sequence, and 5 different clades in small termianse. BIS08 and ST-35 make a separate clade in both. .
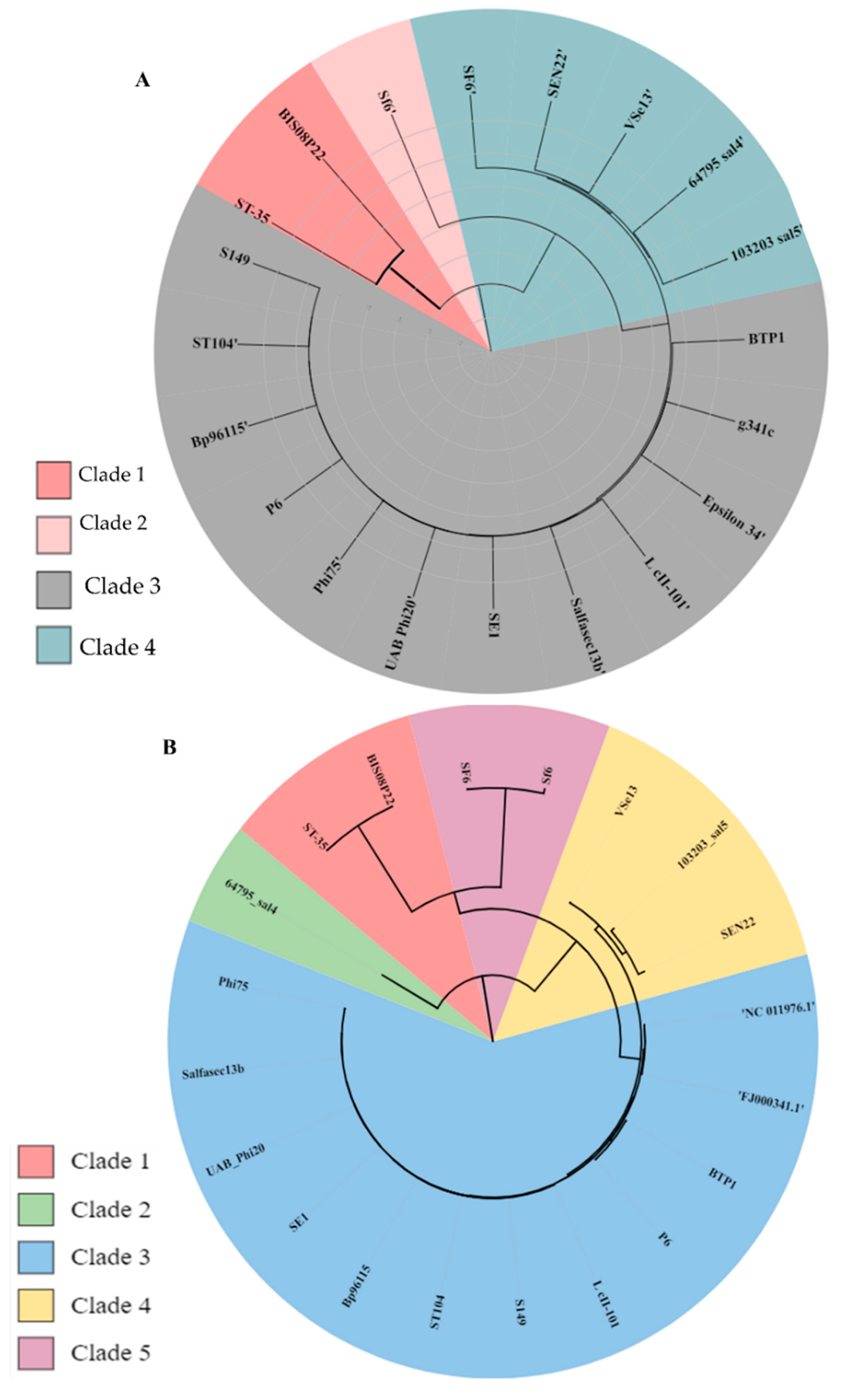
Figure 4.
A) PDBsum (ref) annotation of the sequence and secondary structure of the TSs monomer. B) The structure of the TSs monomer. The n and c terminal ends are labelled, and the short stacked helical bundle is indicated with an *. C) PDB structures of the Sf6 TSs from Lederbergvirus Sf6, PDB codes 4dyq-B (green) and 3hef-B (yellow) superimposed onto BIS08 TSs (magenta). D) The octameric form of TSs left and E) rotated 90 degrees right.
Figure 4.
A) PDBsum (ref) annotation of the sequence and secondary structure of the TSs monomer. B) The structure of the TSs monomer. The n and c terminal ends are labelled, and the short stacked helical bundle is indicated with an *. C) PDB structures of the Sf6 TSs from Lederbergvirus Sf6, PDB codes 4dyq-B (green) and 3hef-B (yellow) superimposed onto BIS08 TSs (magenta). D) The octameric form of TSs left and E) rotated 90 degrees right.
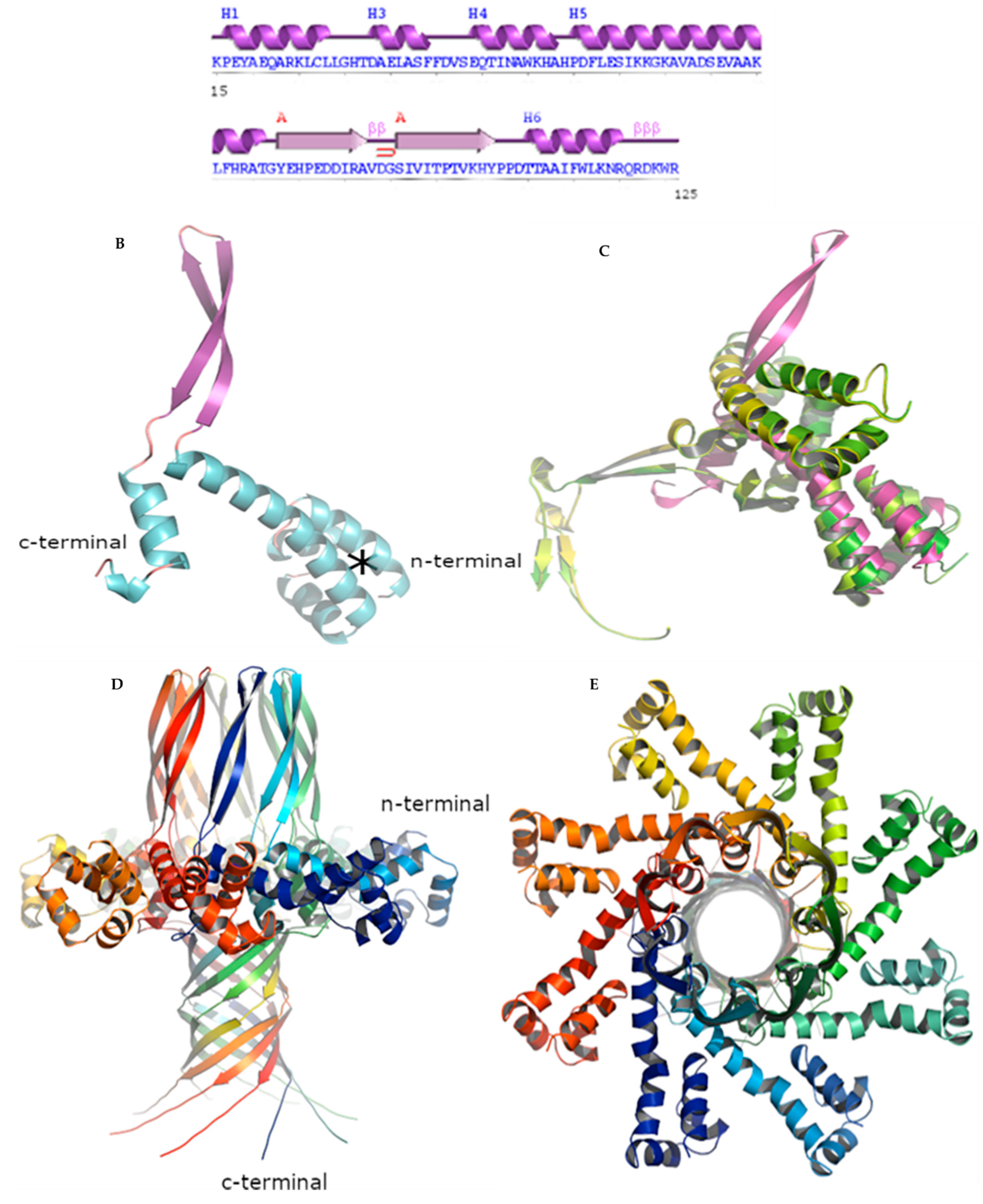
Figure 5.
A) The structure of the N-terminal ATPase domain of the BIS08 TLs (residues 1-275) determined in alpha fold. The n and c terminal ends are labelled as is the Lid subdomain is indicated with as *. b) A PDBsum [37] annotation of the secondary structure of the TLs monomer. Alpha helices are cylinders and beta sheets are shown as arrows. c) The N-terminal BIS08 ATPase (in blue) superimposed onto the ATPase domains identified in DALI. The crystal structure of the phage Sf6 large terminase (shown as light blue ribbons; PDB code 4IEE; [35]). The approximate domain of ATP binding is shown in relative positioning of non-hydrolysable analog Adenosine Triphosphate-γ-S (AGS). 3CPE [38], is purple, 4ZNK [34] is yellow and 6Z6D [39] is orange.
Figure 5.
A) The structure of the N-terminal ATPase domain of the BIS08 TLs (residues 1-275) determined in alpha fold. The n and c terminal ends are labelled as is the Lid subdomain is indicated with as *. b) A PDBsum [37] annotation of the secondary structure of the TLs monomer. Alpha helices are cylinders and beta sheets are shown as arrows. c) The N-terminal BIS08 ATPase (in blue) superimposed onto the ATPase domains identified in DALI. The crystal structure of the phage Sf6 large terminase (shown as light blue ribbons; PDB code 4IEE; [35]). The approximate domain of ATP binding is shown in relative positioning of non-hydrolysable analog Adenosine Triphosphate-γ-S (AGS). 3CPE [38], is purple, 4ZNK [34] is yellow and 6Z6D [39] is orange.
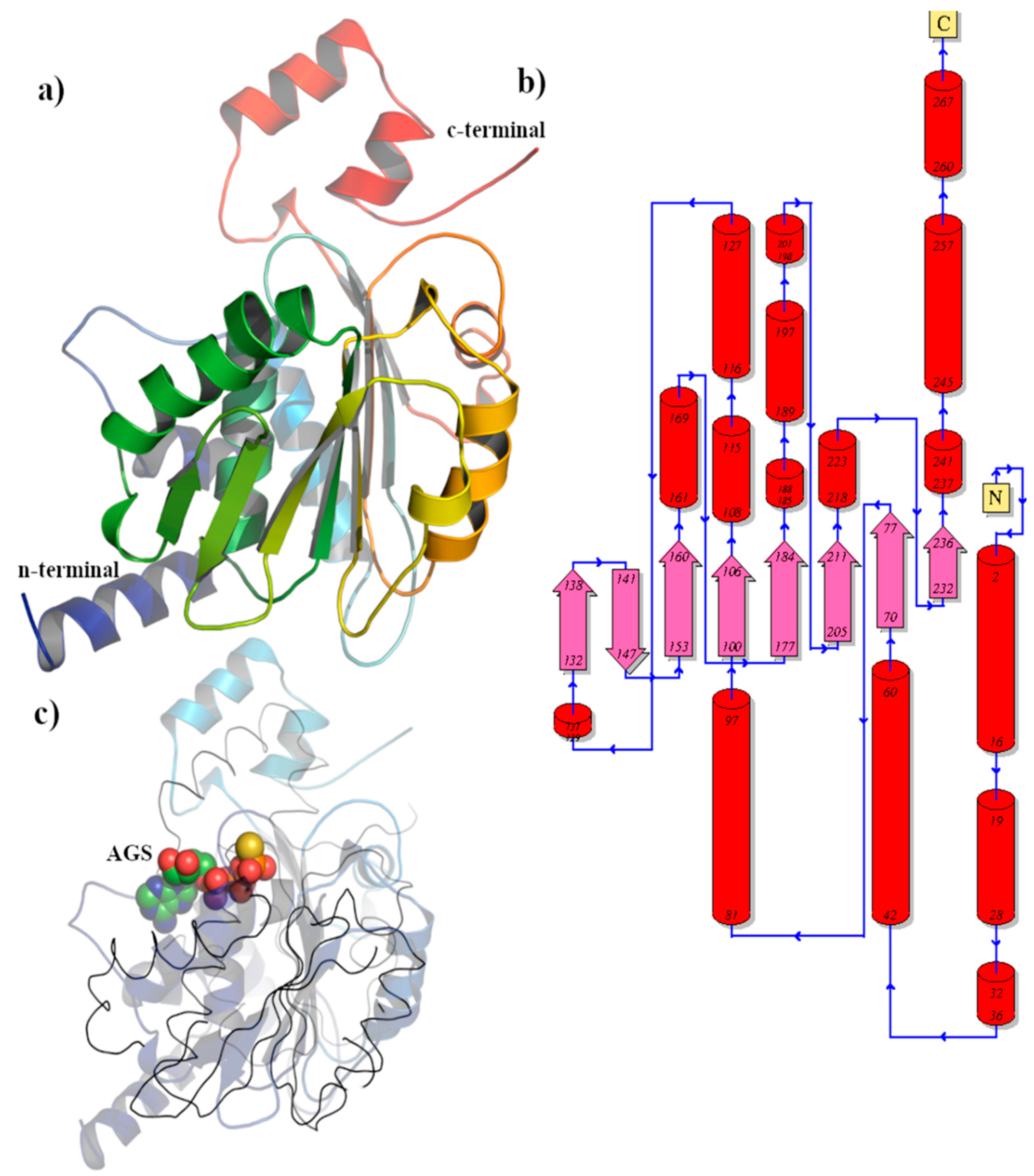
Figure 6.
a) A PDBsum (Laskowski 2018) annotation of the secondary structure of the c-terminal domain of the BIS08 Terminase Large subunit. Alpha helices are cylinders and beta sheets are shown as arrows. b) The AlphaFold structure of the C-terminal domain of the BIS08 Terminase protein (residues 276-469). The n and c terminal ends are labelled. c) The C-terminal BIS08 ATPase (in dark blue) superimposed onto the ATPase domains identified in DALI. These crystal structures include the Pseudomonas phage terminase protein (PDB cod 8kdr-B in yellow; [41]), Lederbergvirus Sf6 protein (PDB code 4IEE in pink; [35]), Byrnievirus HK97 terminase protein (PDB code 6Z6D in orange; [39]), Tequatrovirus T4 DNA packaging protein (PDB code 3CPE in green; [38]) and Escherichia phage T7 protein (PDB code 4BIL in light blue; [42]).
Figure 6.
a) A PDBsum (Laskowski 2018) annotation of the secondary structure of the c-terminal domain of the BIS08 Terminase Large subunit. Alpha helices are cylinders and beta sheets are shown as arrows. b) The AlphaFold structure of the C-terminal domain of the BIS08 Terminase protein (residues 276-469). The n and c terminal ends are labelled. c) The C-terminal BIS08 ATPase (in dark blue) superimposed onto the ATPase domains identified in DALI. These crystal structures include the Pseudomonas phage terminase protein (PDB cod 8kdr-B in yellow; [41]), Lederbergvirus Sf6 protein (PDB code 4IEE in pink; [35]), Byrnievirus HK97 terminase protein (PDB code 6Z6D in orange; [39]), Tequatrovirus T4 DNA packaging protein (PDB code 3CPE in green; [38]) and Escherichia phage T7 protein (PDB code 4BIL in light blue; [42]).
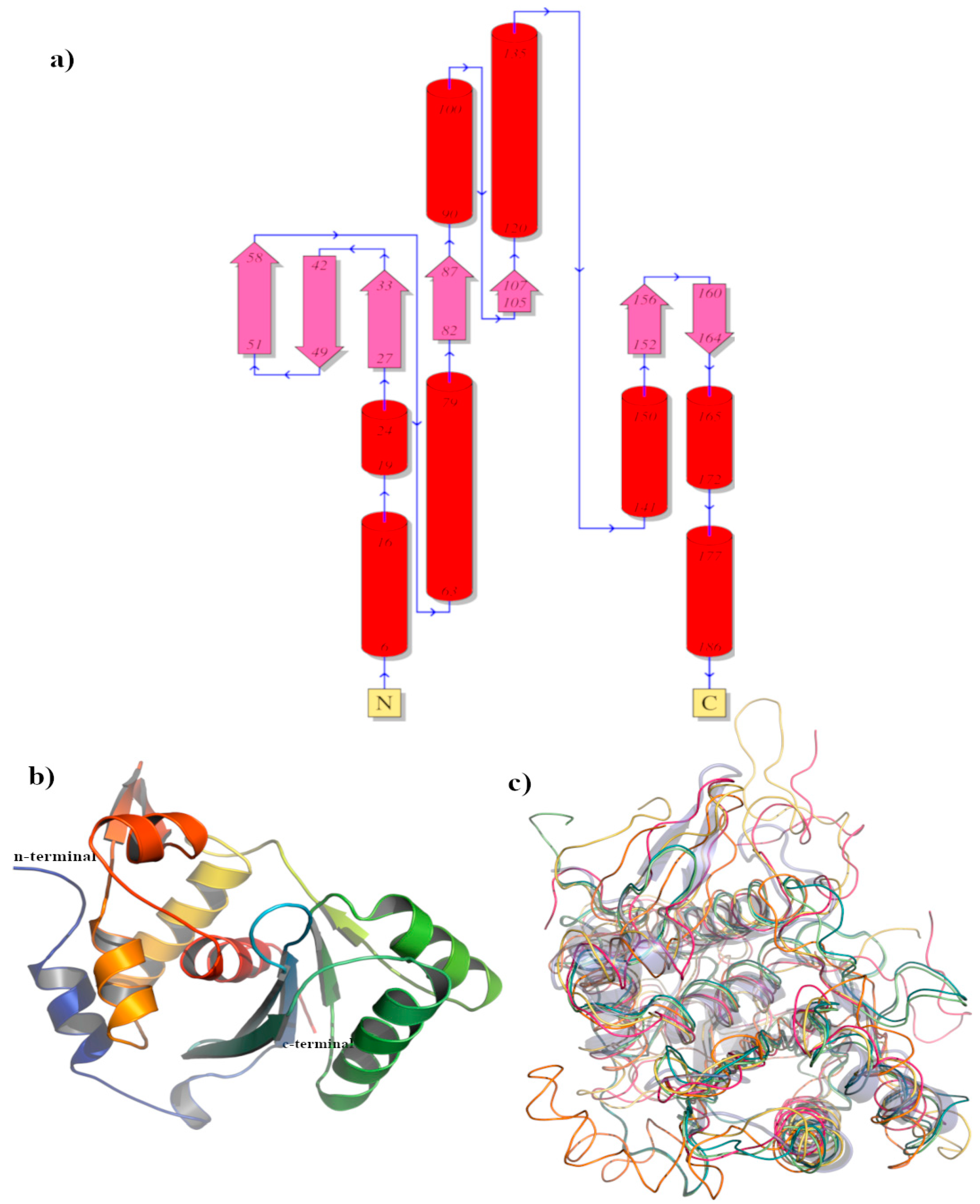

Table 1.
Dali-based [36] structural alignment of the N-terminal ATPase domain of the BIS08 TLs (Terminase Large subuni..t)
Table 1.
Dali-based [36] structural alignment of the N-terminal ATPase domain of the BIS08 TLs (Terminase Large subuni..t)
| Organism | CHAIN | Z-scorea | R.M.S.D.b | DESCRIPTION |
|---|---|---|---|---|
| Oshimavirus P7426 | 4ZNK-A | 20.9 | 2.8 | Phage terminase large subunit Gene Names: P74p84 EC: 3.6.4 (UniProt), 3.1.21 (UniProt) |
| Byrnievirus HK97 | 6Z6D-A | 20.1 | 3.1 | Terminase large subunit |
| Lederbergvirus Sf6 | 4IEE-A | 18.4 | 2.7 | Gene 2 protein |
| Tequatrovirus T4 | 3CPE-A | 18.3 | 3.1 | Large subunit terminase EC : 3.6.4 (UniProt), 3.1.21 (UniProt) |
a A measure of the statistical significance of the result relative to an alignment of random structures. bRoot-mean-square deviation (RMSD) of alpha-carbon atoms.
Table 2.
Dali-based structural alignment of the C-terminal (residues 276-469) domain of the BIS08 Terminase Large subunit.
Table 2.
Dali-based structural alignment of the C-terminal (residues 276-469) domain of the BIS08 Terminase Large subunit.
| Organism | CHAIN | Z-scorea | R.M.S.D.b | DESCRIPTION |
|---|---|---|---|---|
| Pseudomonas phage | 8DKR-B | 22.5 | 1.9 | Large terminase protein Gene names: vbpaeme217_00005 EC : 3.1 |
| Lederbergvirus Sf6 | 4IEE-A | 21.2 | 2.2 | Gene 2 protein |
| Byrnievirus HK97 | 6Z6D-A | 16.3 | 2.8 | Terminase large subunit |
| Tequatrovirus T4 | 3CPE-A | 16.0 | 2.6 | DNA packaging protein gp17 |
| Escherichia phage T7 | 4BIL-A | 16.0 | 2.6 | DNA maturase b, Gene Names: 19 EC: 3.6.4 (UniProt), 3.1.21 (UniProt) |
a A measure of the statistical significance of the result relative to an alignment of random structures. bRoot-mean-square deviation (RMSD) of alpha-carbon atoms.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



