
Submitted:
03 October 2023
Posted:
04 October 2023
You are already at the latest version
Abstract
Developmental Coordination Disorder (DCD) is a neurodevelopmental condition characterized by non-progressive central motor impairments. Mild movement disorder features have been observed in DCD. Until now, the etiology of DCD remains unclear. Recent studies suggested a genetic substrate in some patients with DCD, but comprehensive knowledge about associated genes and underlying pathogenetic mechanisms is still lacking. In this study, we first identified genes described in literature in patients with a diagnosis of DCD according to the official diagnostic criteria. Second, we exposed the underlying pathogenetic mechanisms of DCD, by investigating tissue- and temporal gene expression patterns and brain-specific biological mech-anisms. Third, we explored putative shared pathogenetic mechanisms between DCD and frequent movement disorders with a known genetic component, including ataxia, chorea, dystonia, and myoclonus. We identified 12 genes associated with DCD in literature, which are ubiquitously expressed in the central nervous system throughout brain development. These genes are involved in cellular processes, neural signaling and nervous system development. There was a remarkable overlap (62%) in pathogenetic mechanisms between DCD-associated genes and genes linked with movement disorders. Our findings suggest that some patients might have a genetic etiology of DCD, which could be considered part of a pathogenetic movement disorder spectrum.
Keywords:
Developmental Coordination Disorder
; DCD
; Genetics
; Movement Disorders
; Pathogenetic Spectrum.
1. Introduction
Developmental Coordination Disorder (DCD) is one of the most common neurodevelopmental disorders [1], affecting about 5% of children above the age of 5 years [2,3] and often persisting into adulthood in 30-70% of cases [4]. The symptoms manifest as non-progressive central motor impairments, including motor apraxia, clumsiness, impaired limb coordination and gait instability [2,3,5]. Frequently accompanying non-motor deficits are poor executive functioning, attention deficit/hyperactivity disorder (ADHD), autism-spectrum disorder (ASD), specific language impairment and learning disabilities, including dyscalculia and developmental dyslexia, among others [3,6,7]. DCD can be diagnosed when all the following diagnostic criteria are met: a) the acquisition of motor skills is delayed for the child’s age; b) the symptoms start early in the development and significantly affect the child’s daily activities; c) neurological disorders that could be explanatory for the phenotype have been excluded, including movement disorders, hypotonia, muscle weakness, visual impairment and moderate to severe intellectual disability (ID) [2,3,5]. The clinical identification of DCD is often challenging [8,9,10], as mild features of movement disorders, including ataxia, dystonia and/or chorea, could be observed [6,11], and the motor phenotype of DCD may resemble physiologically immature motor features of young, typically developing children [3,12].
Despite the identification of putative risk factors for DCD, including male sex, prematurity, and perinatal oxygen perfusion problems [2,13,14,15], the underlying etiology remains unknown. Recently, the hypothesis of a genetic substrate of DCD was suggested by studies on family aggregation [16] and heritability of DCD, estimated to ≥70% in monozygotic twins [17,18]. This genetic hypothesis is further supported by the high prevalence of comorbid neurodevelopmental disorders (NDDs) in DCD, including ADHD (about 50%) and ASD (about 47%) [3,19,20,21]. In a recent copy-number variation (CNV) analysis in patients with DCD, rare CNVs in genomic loci were identified encompassing genes previously associated with ADHD and ASD, among other NDDs [22]. These findings suggest a shared genetic etiology between DCD and its comorbid NDDs [15,19,23]. So far, two genome-wide association studies performed in children with phenotypes resembling DCD failed to identify major risk genes for DCD [24,25]. In mice with DCD-like phenotypic traits, a recent quantitative trait locus analysis identified candidate genes correlating with impaired murine gait and coordination [26]. Until now, these findings have not been replicated in patients with DCD [26]. Therefore, despite the strong indications for genetic underpinnings, comprehensive knowledge about the associated genes and underlying pathogenetic mechanisms of DCD is still lacking.
Based on the current evidence, in the present study we hypothesized a genetic substrate in a subgroup of patients with DCD. To further investigate this, we firstly aimed at identifying all genes reported in literature in patients with a diagnosis of DCD according to the official diagnostic guidelines [2,3]. Secondly, we aimed at exposing the underlying temporal- and tissue gene expression patterns and brain-specific biological mechanisms of DCD-associated genes. Moreover, due to the possible clinical presence of mild movement disorder features in DCD, we hypothesized that DCD and movement disorders may share similar pathogenetic mechanisms. Accordingly, our third aim was to explore putative shared pathogenetic pathways between DCD and the most frequent pediatric movement disorders with a known genetic component, including ataxia, chorea, dystonia, and myoclonus [27,28].
2. Materials and Methods
2.1. First aim: Gene identification in literature
2.1.1. Comprehensive literature review and gene inclusion criteria
The first aim of our study was to identify all genes associated with DCD in literature, either described with single nucleotide polymorphisms (SNPs) or residing within loci where CNVs were reported. For this, we performed a comprehensive literature search using PubMed of the National Center for Biotechnology Information (Bethesda, MD: National Library of Medicine, US. Available online: https://pubmed.ncbi.nlm.nih.gov; accessed in May 2022) (Supplementary File SI). Prior to gene selection, we defined the following inclusion criteria, according to the official guidelines [2,3]. A gene was selected for inclusion when all of the following conditions were met: a) there was a clinical diagnosis of DCD (according to the DSM-4 or DSM-5 criteria); b) genetic variants (SNPs/ CNVs) were reported in studies using human genetic material; and c) the gene was not primarily associated with neurological conditions that are phenotypically explanatory for the coordination impairments, such as movement disorders, moderate to severe ID (defined in the ICD-10 as an IQ <50 [29]), neuromuscular, visual or vestibular disorders [3]. To verify whether genes were primarily associated with the abovementioned neurological conditions, we consulted PubMed and the Online Mendelian Inheritance in Man (OMIM; McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. Baltimore, MD. Available online: https://omim.org).
Moreover, for genes within a genetic locus where CNVs were associated with DCD in literature, the additional inclusion criterion of being associated with DCD-comorbid NDDs, such as ADHD or ASD, had to be met (Figure 1).
We retrieved genes from CNV-loci which were not reported in the original manuscripts using the exact cytogenetic coordinates in UCSC Genome Browser (Human Genome Browser, GRCh37/hg19; 2009. Available online: http://genome.ucsc.edu) [30]. For all CNVs described in literature, the reference genome build was GRCh37/ hg19.
2.2. Second aim: Analysis of pathogenetic mechanisms underlying DCD
2.2.1. Temporal gene expression analysis
As part of the second aim of our study, we first investigated the temporal expression patterns of the genes associated with DCD. According to the diagnostic criterion C of the DSM-5, the onset of DCD symptoms occurs in the early developmental period [2]. Using publicly accessible reads per kilobase per million (RPKM) RNAseq data from BrainSpan (Gencode v10, Atlas of the Developing Human Brain, https://www.brainspan.org/static/home), we explored whether the expression of genes associated with DCD was regulated during the development of the cerebellum, basal ganglia, and frontal cortex. These structures are the main anatomical regions involved in planning, coordination, and motor control [31,32], which are typically impaired features in DCD [2,3]. We reclassified 23 developmental stages available in BrainSpan into 7 phases (S1-S7, from 8 postconceptional weeks until the age of 19 years [33]; Supplementary Figure S1), as previously described [34,35].
2.2.2. Tissue gene expression analysis
Secondly, we explored the tissue expression patterns of the genes associated with DCD. Because DCD is a central cause of motor incoordination [36], we investigated whether the genes associated with DCD were specifically expressed in any central nervous system (CNS) structure, using the multi-gene query function of Genotype-Tissue Expression (GTEx) Portal (Analysis Release V8, available at: https://www.gtexportal.org) [37]. The available CNS structures included the amygdala, anterior cingulate cortex, caudate nucleus, cerebellum, frontal cortex, hippocampus, hypothalamus, nucleus accumbens, putamen, spinal cord, and substantia nigra. Publicly accessible RNAseq data was available as Transcript per Million (TPM).
2.2.3. Functional enrichment and biological pathway analysis in the DCD-associated gene co-expression network
Thirdly, we investigated the biological mechanisms underlying the genes associated with DCD. For this, we performed functional enrichment using MetaBrain (https://network.metabrain.nl) [38]. The resulting brain-specific DCD-associated gene co-expression network was procedurally enriched with 200 genes predicted to be functionally similar to the DCD-associated genes (hereafter referred as DCD-predicted genes; Supplementary File SII). Then, we performed biological pathway analysis using Metascape (version 3.5, available at: http://metascape.org) [39], where similar biological pathways are clustered together, as described elsewhere [35,39]. Afterwards, we used gProfiler (database version: Ensembl 104, Ensembl Genomes 51, Wombase ParaSite 15. Available online: https://biit.cs.ut.ee/gprofiler/gost) [40], ToppGene Suite (https://toppgene.cchmc.org/enrichment.jsp) [41] and MetaBrain to verify the reproducibility of these data. Throughout all analyses, we annotated the gene ontology (GO, http://geneontology.org/) [42], REACTOME (https://reactome.org/) [43], WikiPathways (https://www.wikipathways.org/) [44] and KEGG (https://www.kegg.jp/kegg/) [45] biological pathways with a statistically significant adjusted p-value of ≤10-5, corrected for multiple testing (Bonferroni). Clustered data was visualized through Cytoscape (https://cytoscape.org) [46].
We also investigated whether there was an overlap between the 200 DCD-predicted genes in the DCD-associated gene co-expression network (Supplementary File S2) and the genes from CNV-loci associated with DCD in literature. The presence of a gene in both lists would suggest a functional relationship with DCD.
2.3. Third aim: Analysis of putative pathogenetic overlap between DCD and ataxia, chorea, dystonia and/or myoclonus
2.3.1. Functional enrichment and biological pathway analysis in the shared DCD-associated/MD gene co-expression network
To explore a putative shared pathogenic background between DCD and the most frequent pediatric movement disorders with a known genetic component, including ataxia, chorea, dystonia and/or myoclonus [27,28], we generated a shared gene co-expression network for these five disorders (hereafter referred as DCD-associated/MD). For this purpose, we used the DCD-associated genes from literature, as well as ataxia, chorea, dystonia, and myoclonus genes from the Task Force on Genetic Nomenclature in Movement Disorders [47], enriched with 200 predicted genes per each group (Supplementary File S3). Then, we performed shared biological pathway analysis using the meta-analysis function of Metascape. We defined “overlapping” the biological pathways enriched for the genes associated with DCD and for at least one or more movement disorders (ataxia and/or chorea and/or dystonia and/or myoclonus).
To investigate whether the 200 DCD-predicted genes were linked with movement disorders, we compared these genes with established genes for ataxia, dystonia, myoclonus, chorea, spastic paraplegia, neurodegeneration with brain accumulation, and mixed movement disorders. We selected genes associated with these disorders based on gene lists from our hospital (available at: https://www.umcg.nl/-/afdeling/genetica/aanvragen-genoomdiagnostiek) [48], as well as gene lists from the Task Force on Genetic Nomenclature in Movement Disorders [47].
2.4. Statistical analyses
For all our statistical analyses and graphs, we used GraphPad Prism (version 9.4.0, for Windows; GraphPad Software, San Diego, California, USA, 2022. Available at: www.graphpad.com). For temporal gene expression analysis, we log10-transformed RPKM RNAseq data and performed both: 1) repeated measures one-way ANOVA, to compare the expression of the DCD-associated genes among the seven developmental stages within each structure (cerebellum, basal ganglia, and frontal cortex); and 2) repeated measures two-way ANOVA, to compare the average gene expression among the seven developmental stages between the cerebellum, basal ganglia, and frontal cortex. For tissue gene expression analysis, we normalized TPM RNAseq data into z-scores and performed a repeated measures one-way ANOVA to compare the average expression of the DCD-associated genes in all available nervous system structures. For both temporal- and tissue expression analyses, we corrected for multiple testing using Tukey’s test. Finally, we plotted these data in box-and-whisker plots and bar graphs. We set the significance level at α =.05.
3. Results
3.1. First aim: Gene identification in literature
3.1.1.12. genes associated with DCD in literature
We performed a comprehensive literature research in PubMed to identify genes previously associated with DCD. Nine articles out of 268 hits described genetic variants in three genes and 96 loci in patients with a clinical diagnosis of DCD (Supplementary Figure S2, Supplementary Files S4 and S5) [11,22,49,50,51,52,53,54,55]. According to our inclusion criteria (Figure 1), we selected three genes from two case reports of 23 patients with DCD (ABCC8, KCNJ11, KLF7) [11,49] and nine genes from seven CNV-loci reported in 10 patients with DCD in a CNV-analysis (CNTN4, CTNNA3, FHIT, GAP43, LSAMP, PTPRN2 RBFOX1, SHANK3, VIPR2) [22]. We reviewed the disease associations of these 12 genes and ascertained that none of them was primarily associated with a known neurological disorder affecting coordination (Supplementary Table S1). The nine genes from the seven CNV-loci were associated with DCD-comorbid NDDs. Altogether, these 12 genes thus matched the inclusion criteria and were selected for further in silico analyses (Table 1).
3.2. Second aim: Analysis of pathogenetic mechanisms underlying DCD
3.2.1. Ubiquitous expression of DCD-associated genes in brain throughout development
Using publicly available RNAseq data, we investigated both the temporal- and tissue-specific expression of the 12 DCD-associated genes. First, we analyzed their expression patterns during brain development in the cerebellum, basal ganglia, and frontal cortex across seven developmental stages (S1-S7, Supplementary Figure S1). The average log10 gene expression values of the 12 DCD-associated genes ranged from 4.36 to 6.79, varying per structure and per developmental stage (Supplementary Figure S3, Supplementary Table S2). Comparison of mean gene expression levels across the developmental stages within each structure and between the three anatomical structures showed no statistically significant difference after correcting for multiple comparisons (Supplementary Figure S3, Figure 2).
Second, we investigated whether the average expression of the 12 DCD-associated genes was specific for any of the available CNS structures. The mean gene expression levels ranged from -7.50e-007 to 8.33e-008 in the diverse CNS structures (Supplementary Table S3). There was no significant difference in average gene expression levels among the investigated CNS structures (Figure 3).
3.2.2. Three main biological themes in the DCD-associated gene co-expression network
To explore the biological relationship between the 12 DCD-associated genes, we generated a brain-specific DCD-associated gene co-expression network through functional enrichment and then performed biological pathway analysis. Using Metascape, 76 clusters were identified in the DCD-associated gene co-expression network comprising of 546 biological pathways (Supplementary File S6). Among the top 20 most significantly enriched clusters of biological pathways, we identified three main biological themes based on the parental biological term of each cluster (Table 2). The three themes were: 1) cellular processes, including cell junction organization, regulation of ion transport, cell-cell adhesion, and protein localization to membrane, among others; 2) neural signaling, including modulation of chemical synaptic transmission, synaptic signaling, and regulation of glutamatergic synaptic transmission; and 3) nervous system development, comprising of neuron projection development, L1CAM interactions, and brain development (Table 2, Supplementary File S6). Notably, similar biological pathways and themes were identified using different enrichment analysis programs (Supplementary File S6).
The brain-specific DCD-associated gene co-expression network contained 200 DCD-predicted genes (Supplementary File S2). We also investigated the overlap between the DCD-predicted genes and the genes in the DCD-associated CNV-loci (Supplementary File S5). Only one gene in the CNV-locus 16p11.2, TLCD3B, was present in the DCD-associated co-expression network.
3.3. Third aim: Analysis of putative pathogenetic overlap between DCD and ataxia, chorea, dystonia and/or myoclonus
3.3.1. Three main biological themes in the shared DCD/MD gene co-expression network
To investigate a putative shared pathogenetic background between DCD and pediatric genetic movement disorders, including ataxia, chorea, dystonia and/or myoclonus, we generated a shared DCD-associated/MD gene co-expression network. Here, we identified 2269 biological pathways that were enriched for genes associated with either DCD, ataxia, chorea, dystonia, myoclonus, or for several disorders simultaneously (Supplementary File S7). 543 biological pathways were enriched for the genes associated with DCD and for at least one or more movement disorders (ataxia and/or chorea and/or dystonia and/or myoclonus). These shared biological pathways were related to 1) neural signaling, such as modulation of chemical synaptic transmission, and trans-synaptic signaling, 2) nervous system development, including neuron projection-, axon-, pallium-, cerebral cortex-, telencephalon development, and 3) cellular processes, such as organelle localization, cellular homeostasis, and cellular transport, among others (Supplementary File S7). 206 of the 543 biological pathways (38%) were uniquely enriched for DCD-associated genes, including pathways related to 1) cellular processes, such as cell-cell interaction, cellular localization, homeostasis, and cellular metabolism, and 2) neurodevelopmental processes, such as dendrite development, postsynaptic density organization, and axonogenesis, among others (Supplementary File S7). The remaining 337 of the 543 biological pathways (62%) overlapped between DCD-associated genes and genes linked to one or more movement disorders (ataxia and/or chorea and/or dystonia and/or myoclonus) (Figure 4).
The largest overlap in biological processes was between DCD-associated genes and myoclonus genes (81 biological pathways) (Supplementary File S7). Moreover, we observed that 12 out of 200 DCD-predicted genes (6%) were established genes for ataxia, dystonia, myoclonus, and spastic paraplegia (Supplementary Table S4).
4. Discussion
To the best of our knowledge, this is the first study to comprehensively investigate and compare the pathogenetic mechanisms underlying DCD and those of several movement disorders. Our data shows the association of 12 genes with DCD in literature. These 12 DCD-associated genes are ubiquitously expressed in the central nervous system throughout brain development and are mainly involved in cellular processes, neural signaling and nervous system development. These results are supportive of a genetic substrate in a subgroup of patients with DCD. Furthermore, the underlying pathogenetic mechanisms of the DCD-associated genes overlap substantially (62%) with those of several movement disorders, including ataxia, chorea, dystonia and/or myoclonus. This implies that the genetic substrate of DCD could be regarded as part of a broader pathogenetic movement disorder spectrum.
The first aim of our study was to identify all genes associated with DCD in literature. By comprehensively reviewing the literature, we included 12 genes whose variants were reported in 33 patients with a clinical diagnosis of DCD according to the DSM-4/5 criteria [11,22,49]. These 12 genes were mainly associated with DCD-comorbid NDDs, such as ADHD and ASD, and with neurological conditions not primarily affecting coordination, such as epilepsy, depression, or schizophrenia. Interestingly, SNPs in ABCC8 and KCNJ11 were reported in patients with neonatal diabetes mellitus and DCD [49]. Although no causal correlation was found between their genotypes and neurodevelopmental outcomes, these patients received a clinical diagnosis of DCD according to the DSM-4 criteria [49]. Therefore, following our inclusion criteria, we included these two genes. Some other genes, such as KCNJ11, KLF7 and VIPR2, were previously associated with mild ID (IQ>55). Despite ID is an exclusion criterion for DCD, the official guidelines do not specify an IQ cut-off [2,3]. Instead, they indicate that DCD should not be diagnosed when the symptoms can be explained by moderate to severe ID, as defined by the ICD-10 [2,3]. This corresponds to an IQ<50 [29]. Therefore, because the abovementioned genes were associated with IQ>55, we included them in our list. Also, to ascertain that we did not select genes associated with a DCD-like phenotype, we defined the absence of a diagnosis of DCD according to the official DSM-4/5 criteria as an exclusion criterion. In fact, the association of DCD with genes reported with DCD-like phenotypes was unlikely, because these genes were associated with conditions that were exclusion criteria for DCD. This was, for instance, the case for COL6A1 [24], associated with Bethlem myopathy 1 (OMIM #158810) and Ullrich congenital muscular dystrophy 1 (OMIM #254090), and for IQSEC1 [25], associated with severe ID (OMIM #618687). As such, we did not include these genes in our list. Altogether, by strictly complying with the official diagnostic criteria for DCD [2,3], we are confident that the 12 included genes are representative of a possible genetic substrate in DCD. These results suggest the existence of a genetic subgroup among the putative etiological causes of DCD.
Our second aim was to expose the pathogenetic mechanisms underlying the 12 DCD-associated genes, through the analysis of temporal- and tissue gene expression and of brain-specific biological pathways. The ubiquitous expression of the 12 DCD-associated genes in the CNS during development suggests a role for these 12 genes in the pathogenesis of a central developmental motor disorder, such as DCD. Also, these results indicate the lack of a unique gene expression signature for the 12 DCD-associated genes. In fact, the temporal expression patterns of the 12 DCD-associated genes in the cerebellum, basal ganglia, and frontal cortex were similar to those of ataxia, dystonia and myoclonus genes in the cortico-basal-ganglia-cerebellar (CBGC) network [35,56,57]. In literature, the disruption of the CBGC network was described in both DCD [58,59,60,61,62] and in movement disorders, including ataxia, dystonia, and myoclonus [35,56,57,63,64,65,66]. Altogether, the analogous temporal expression patterns of DCD-associated genes and movement disorder genes, and the involvement of the CBGC network in both diagnostic groups suggest that DCD could be part of a pathogenetic spectrum of movement disorders.
In the unique DCD-associated gene co-expression network, we identified three main biological themes, including cellular processes, neural signaling and nervous system development. These generic biological processes were previously associated with ataxia, dystonia, and myoclonus [34,35,56,57,65]. Interestingly, two DCD-associated genes, CNTN4 and SHANK3, were present in the most significantly enriched pathways and had overlapping biological functions. As such, they may provide novel insights in the pathogenetic mechanisms of DCD. CNTN4 and SHANK3 belong to the contactin subgroup of the immunoglobulin superfamily and the SHANK family, containing multiple ankyrin repeats, respectively [67,68,69]. These genes encode neuronal cell adhesion molecules and scaffolding proteins that promote the modulation of neuronal activity, including glutamatergic synaptic excitability, nodal and paranodal organization, and various neurodevelopmental and cellular processes, such as neuron projection development, axono- and synaptogenesis, neurite outgrowth, synaptic growth and maintenance, and protein-protein interaction [67,70,71,72,73]. SHANK3-haploinsufficiency is reported in Phelan-McDermid syndrome, a rare condition characterized by ID, hypotonia, global developmental delay, and ASD, among other features [74]. Despite the known involvement of SHANK3 in this disorder, deletions of the 22q13 locus, associated with this phenotype, encompass a much larger group of genes [75]. Interestingly, both CNTN4 and SHANK3 have been associated with ASD [22,67,71,73,74] a very frequent DCD-comorbid NDD (about 47%) [3,20,21]. Moreover, motor coordination deficits and gait abnormalities were described in mice with biallelic deletions of either SHANK3 N-terminus or C-terminus [76,77]. Similarly, a quantitative trait locus analysis in BXD recombinant inbred lines of mice with DCD-like phenotypic traits proposed Cntn6/CNTN6, a gene adjacent to the region of chromosome 3 where CNTN4 is located [71], as a candidate gene for the regulation of murine coordination and postural control [77]. Altogether, these findings may indicate a putative novel role for contactins and ankyrins in the pathogenesis of DCD, which might be worth exploring in future studies.
In the DCD-associated gene co-expression network, we also explored the overlap between DCD-predicted genes and genes present in CNV-loci associated with DCD in literature. Based on our gene co-expression analysis, TLCD3B from the DCD-associated CNV-locus 16p11.2 may have a functional relationship with the 12 DCD-associated genes. Notably, we did not include TLCD3B in our analyses, because the gene was previously not directly associated with any DCD-comorbid NDD. As such, it did not meet our inclusion criteria. However, both deletions and duplications of the regions of the 16p11.2 locus encompassing TLCD3B were previously associated with various NDDs, such as DCD, ASD, language delay and different levels of ID [22,52,55], but also with movement disorders causing coordination impairments, including ataxia and dystonia [78]. Given that many genes are found in this locus, and that there was no direct association of TLCD3B with movement disorders, we consider TLCD3B a putative DCD-associated gene. TLCD3B encodes the TLC domain-containing protein 3B and functions as a ceramide synthetase, mediating stress responses and aiding survival of retinal cells [79]. Bulk expression of TLCD3B in the CNS is the highest in the cerebellum [37], indicating a possible crucial role for this gene in this brain structure. Recently, homozygous variants in TLCD3B were associated with cone-rod dystrophy 22, a retinal condition leading to progressive central vision loss [79]. Possibly, heterozygous variants in TLCD3B could induce a different phenotype than vision loss, such as coordination impairments. This should be investigated in future studies.
Our third aim was to explore a putative shared pathogenetic background between DCD and movement disorders, by analyzing a shared DCD-associated/MD gene co-expression network. We identified a 62% overlap in biological pathways between DCD-associated genes and genes linked to at least one or several movement disorders, such as ataxia, chorea, dystonia and/or myoclonus. These overlapping biological pathways were related to the same three biological themes of the unique DCD-associated gene co-expression network, including neural signaling, nervous system development and cellular processes. Interestingly, the remaining 38% of biological pathways enriched only for DCD-associated genes were related to similar biological themes, including cellular- and neurodevelopmental processes. Therefore, these findings indicate the absence of distinctive biological themes for the DCD-associated genes in relation to the investigated movement disorders. This suggests that the biological overlap between DCD and the abovementioned movement disorders could exceed the reported 62%. An overlapping pathogenetic substrate is also suggested by the fact that 6% (12/200) of the functionally DCD-predicted genes are established movement disorder genes, mainly linked with ataxia and myoclonus. Until now, neither the large biological overlap we observed between myoclonus- and DCD-associated genes, nor a putative phenotypic association between these two disorders were reported in literature. Although the presence of movement disorders is an exclusion criterion for DCD [2,3], mild clinical features of ataxia, dystonia, and/or chorea, have previously been described in patients with a diagnosis of DCD according to the official diagnostic criteria [6,11]. In these patients, the clinical distinction between DCD and mild movement disorders might lead to diagnostic delay. In perspective of our overlapping pathogenetic findings between the 12 DCD-associated genes and the investigated movement disorders, the question arises whether DCD exists as a unique diagnostic entity or as the milder end of a broader movement disorder spectrum. Based on our findings, we suggest considering the genetic subgroup of DCD as part of a pathogenetic movement disorder spectrum. This would have important diagnostic implications, such as the inclusion of mild movement disorder features as part of the motor phenotype in DCD. In future studies, we aim to further investigate the implications of this paper by thoroughly phenotyping a cohort of putative DCD patients. This may hopefully unravel the diagnostic conundrum existing between DCD and movement disorders. Additionally, in this cohort, genetic diagnostic analysis of the 12 DCD-associated genes might provide further evidence for an underlying genetic substrate of DCD.
We recognize several limitations to this study. First, the list of genes reported in literature in association with DCD was short, therefore our set of genes may be incomplete. So far, two GWAS were performed in patients with DCD-like phenotypes, but none in patients with a diagnosis of DCD. This is likely because diagnostic genetic testing is still not routinely performed in patients with an official diagnosis of DCD. We therefore hypothesize that new genetic associations or gene mutations will likely be exposed in the future. However, by comprehensively reviewing the literature and strictly complying with the diagnostic criteria for DCD, we are confident that our findings are representative of the current knowledge. Second, we used in silico analyses to expose the underlying pathogenetic mechanisms. We are aware that, although in silico analyses may be helpful to detect patterns of similarities within a large set of genes, these strategies might overlook specific gene characteristics, such as particular biological functions or molecular mechanisms.
In summary, in the present study, we aimed to explore the pathogenetic mechanisms underlying DCD-associated genes and compare them with those of several movement disorders. Our findings indicate a genetic substrate for a subgroup of patients with DCD and a substantial pathogenetic overlap with movement disorders, suggesting that DCD may belong to a broader pathogenetic movement disorder spectrum. These data have important diagnostic implications, such as the need for thorough phenotyping of DCD with the possible inclusion of mild movement disorder features, and the analysis of genetic variants in the 12 DCD-associated genes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1:Reclassification of seven developmental stages; Figure S2: Overview of the results of the literature research; Figure S3: Temporal gene expression of DCD-associated genes in different developmental stages of the cerebellum, basal ganglia, and frontal cortex; Table S1:Gene association to disease; Table S2: Descriptive statistics of temporal gene expression in different developmental stages of the cerebellum, basal ganglia and frontal cortex; Table S3: Descriptive statistics of tissue gene expression in different structures of the central nervous system; Table S4: Association of DCD-predicted genes with known movement disorders; File S1: Search string for PubMed; File S2: 200 genes predicted to be functionally similar to the DCD-associated genes (DCD-predicted genes); File S3: Shared DCD-associated/MD gene co-expression network; File S4: Literature research results; File S5: CNV-loci associated with DCD in literature; File S6: Biological pathway analysis results for the DCD-associated gene co-expression network; File S7: Biological pathway analysis results for the DCD-associated/MD gene co-expression network.
Author Contributions
Conceptualization M.G., F.V., D.A.S., D.S.V.; methodology, M.G., F.V., D.A.S., D.S.V.; formal analysis, M.G.; investigation, M.G.; data curation, M.G.; writing—original draft preparation, M.G.; writing—review and editing, M.G., F.V., D.A.S., D.S.V.; visualization, M.G., F.V., D.A.S., D.S.V.; supervision, F.V., D.A.S., D.S.V. All authors have read and agreed to the published version of the manuscript.
Funding
M. Garofalo received funding from the Junior Scientific Masterclass from the University of Groningen, University Medical Center Groningen. The APC were partially funded (10%) by the University of Groningen. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: 1) BrainSpan: https://www.brainspan.org/static/home; 2) Genotype-Tissue Expression (GTEx) Portal: https://www.gtexportal.org; 3) gProfiler: https://biit.cs.ut.ee/gprofiler/gost; 4) Gene panels from the University Medical Center Groningen: https://www.umcg.nl/-/afdeling/genetica/aanvragen-genoomdiagnostiek; 5) Metabrain: https://network.metabrain.nl; 6) Metascape: http://metascape.org; 7) ToppGene Suite: https://toppgene.cchmc.org/enrichment.jsp; 8) UCSC Genome Browser: http://genome.ucsc.edu.
Acknowledgments
We wish to thank Bente Hofstra for her help with the program Cytoscape.
Conflicts of Interest
D.A. Sival and D.S. Verbeek are members of the European Reference Network for Rare Neurological Diseases. The authors declare no conflict of interest.
References
- Francés, L.; Quintero, J.; Fernández, A.; Ruiz, A.; Caules, J.; Fillon, G.; Hervás, A.; Soler, C.V. Current state of knowledge on the prevalence of neurodevelopmental disorders in childhood according to the DSM-5: a systematic review in accordance with the PRISMA criteria. Child Adolesc. Psychiatry Ment. Health. 2022, 16, 27. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Neurodevelopmental Disorders. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed; American Psychiatric Association Publishing: Arlington, VA, United States, 2013; pp. 74–77. [Google Scholar]
- Blank, R.; Barnett, A.L.; Cairney, J.; Green, D.; Kirby, A.; Polatajko, H.; Rosenblum, S.; Smits-Engelsman, B.; Sugden, D.; Wilson, P.; et al. International clinical practice recommendations on the definition, diagnosis, assessment, intervention, and psychosocial aspects of developmental coordination disorder. Dev. Med. Child Neurol. 2019, 61, 242–285. [Google Scholar] [CrossRef] [PubMed]
- Tal Saban, M.; Kirby, A. Adulthood in Developmental Coordination Disorder (DCD): a Review of Current Literature Based on ICF Perspective. Curr. Dev. Disord. Reports. 2018, 5, 9–17. [Google Scholar] [CrossRef]
- Ip, A.; Mickelson, E.C.R.; Zwicker, J.G. Assessment, diagnosis, and management of developmental coordination disorder. Paediatr. Child Health. 2021, 26, 375–378. [Google Scholar] [CrossRef]
- Visser, J. Developmental coordination disorder: a review of research on subtypes and comorbidities. Hum. Mov. Sci. 2003, 22, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.; Röschinger, J.; Barck, K.; Büttner, G.; Hasselhorn, M. Learning Difficulties in Children with Symptoms of DCD And/or ADHD: Analyses from a Categorical and a Continuous Approach. Int. J. Disabil. Dev. Educ. 2022, 69, 69,1505–1521. [Google Scholar] [CrossRef]
- Hunt, J.; Zwicker, J.; Godecke, E.; Raynor, A. Assessing children to identify developmental coordination disorder: A survey of occupational therapists in Australia. Aust. Occup. Ther. J. 2023, 70, 420–433. [Google Scholar] [CrossRef]
- Dominguez-Vega, Z.T.; Dubber, D.; Elting, J.W.J.; Sival, D.A.; Maurits, N.M. Instrumented classification of patients with early onset ataxia or developmental coordination disorder and healthy control children combining information from three upper limb SARA tests. Eur. J. Paediatr. Neurol. 2021, 34, 74–83. [Google Scholar] [CrossRef]
- Tang, W.; van Ooijen, P.M.A.; Sival, D.A.; Maurits, N.M. 2D Gait Skeleton Data Normalization for Quantitative Assessment of Movement Disorders from Freehand Single Camera Video Recordings. Sensors. 2022, 22, 4245. [Google Scholar] [CrossRef]
- Lawerman, T.F.; Brandsma, R.; Maurits, N.M.; Martinez-Manzanera, O.; Verschuuren-Bemelmans, C.C.; Lunsing, R.J.; Brouwer, O.F.; Kremer, H.P.H.; Sival, D.A. Paediatric motor phenotypes in early-onset ataxia, developmental coordination disorder, and central hypotonia. Dev. Med. Child. Neurol. 2020, 62, 75–82. [Google Scholar] [CrossRef]
- Kuiper, M.J.; Brandsma, R.; Lunsing, R.J.; Eggink, H.; Ter Horst, H.J.; Bos, A.F.; Sival, D.A. The neurological phenotype of developmental motor patterns during early childhood. Brain Behav. 2019, 9, e01153. [Google Scholar] [CrossRef]
- van Hoorn, J.F.; Schoemaker, M.M.; Stuive, I.; Dijkstra, P.U.; Rodrigues Trigo Pereira, F.; Van der Sluis, C.K.; Hadders-Algra, M. Risk factors in early life for developmental coordination disorder:a scoping review. Dev. Med. Child. Neurol. 2021, 63, 511–519. [Google Scholar] [CrossRef]
- Hadders-Algra, M. Developmental Coordination Disorder: Is Clumsy Motor Behavior Caused By a Lesion of the Brain At Early Age? Neural Plast. 2003, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.; Sirigu, A. Developmental coordination disorder: core sensori-motor deficits, neurobiology and etiology. Neuropsychologia. 2015, 79, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Gaines, R.; Collins, D.; Boycott, K.; Missiuna, C.; DeLaat, D.; Soucie, H. Clinical expression of developmental coordination disorder in a large Canadian family. Paediatr. Child Health. 2008, 13, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Anckarsäter, H.; Lundström, S.; Kollber, L.; Kerekes, N.; Palm, C.; Carlström, E.; Langström, N.; Magnusson, P.K.E.; Halldner, L.; Bölte, S.; et al. The child and adolescent twin study in Sweden (CATSS). Twin Res. Hum. Genet. 2011, 14, 495–508. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Carlström, E.; Råstam, M.; Gillberg, C.; Anckarsäter, H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am. J. Psychiatry. 2010, 167, 1357–1363. [Google Scholar] [CrossRef]
- Martin, N.C.; Piek, J.P.; Hay, D. DCD and ADHD: A genetic study of their shared aetiology. Hum. Mov. Sci. 2006, 25, 110–124. [Google Scholar] [CrossRef]
- Ketcheson, L.R.; Pitchford, E.A.; Wentz, C.F. The Relationship Between Developmental Coordination Disorder and Concurrent Deficits in Social Communication and Repetitive Behaviors Among Children with Autism Spectrum Disorder. Autism Res. 2021, 14, 804–816. [Google Scholar] [CrossRef]
- Kopp, S.; Beckung, E.; Gillberg, C. Developmental coordination disorder and other motor control problems in girls with autism spectrum disorder and/or attention-deficit/hyperactivity disorder. Res. Dev. Disabil. 2010, 31, 350–361. [Google Scholar] [CrossRef]
- Mosca, S.J.; Langevin, L.M.; Dewey, D.; Innes, A.M.; Lionel, A.C.; Marshall, C.C.; Scherer, S.W.; Parboosingh, J.S.; Bernier, F.P. Copy-number variations are enriched for neurodevelopmental genes in children with developmental coordination disorder. J. Med. Genet. 2016, 53, 812–819. [Google Scholar] [CrossRef]
- Dewey, D. What Is Comorbidity and Why Does It Matter in Neurodevelopmental Disorders? Curr. Dev. Disord. Reports. 2018, 5, 235–242. [Google Scholar] [CrossRef]
- Mountford, H.S.; Hill, A.; Barnett, A.L.; Newbury, D.F. Genome-Wide Association Study of Motor Coordination. Front. Hum. Neurosci. 2021, 286, 669902. [Google Scholar] [CrossRef]
- Fliers, E.A.; Vasquez, A.A.; Poelmans, G.; Rommelse, N.; Altink, M.; Buschgens, C.; Asherson, P.; Banaschewski, T.; Ebstein, R.; Gill, M.; et al. Genome-wide association study of motor coordination problems in ADHD identifies genes for brain and muscle function. World J. Biol. Psychiatry. 2012, 13, 211–222. [Google Scholar] [CrossRef]
- Gill, K.; Rajan, J.R.S.; Chow, E.; Ashbrook, D.; Williams, R.W.; Zwicker, J.G.; Goldowitz, D. Investigating mouse motor coordination using quantitative trait locus analysis to model the genetic underpinnings of developmental coordination disorder. bioRxiv. 2022, 495138. [Google Scholar]
- Pérez-Dueñas, B.; Gorman, K.; Marcé-Grau, A.; Ortigoza-Escobar, J.D.; Macaya, A.; Danti, F. R.; Barwick, K.; Papandreou, A.; Ng, J.; Meyer, E.; et al. The Genetic Landscape of Complex Childhood-Onset Hyperkinetic Movement Disorders. Mov. Dis. 2022, 37, 2197–2209. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, R.; van Egmond, M.E.; Tijssen, M.A.; the Groningen Movement Disorder Expertise Centre. Diagnostic approach to paediatric movement disorders: a clinical practice guide. Dev. Med. Child. Neurol. 2021, 63, 63–252. [Google Scholar] [CrossRef] [PubMed]
- ICD-10 Version:2019. Available online: https://icd.who.int/browse10/2019/en (accessed on 22/05/2023).
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Gao, Z.; Davis, C.; Thomas, A.M.; Economo, M.N.; Abrego, A.M.; Svoboda, K.; De Zeeuw, C.I.; Li, N. A cortico-cerebellar loop for motor planning. Nature. 2018, 563, 113–116. [Google Scholar] [CrossRef]
- Leisman, G.; Braun-Benjamin, O.; Melillo, R. Cognitive-motor interactions of the basal ganglia in development. Front. Syst. Neurosci. 2014, 8, 16. [Google Scholar] [CrossRef]
- Wierenga, L.; Langen, M.; Ambrosino, S.; van Dijk, S.; Oranje, B.; Durston, S. Typical development of basal ganglia, hippocampus, amygdala and cerebellum from age 7 to 24. Neuroimage. 2014, 96, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Eidhof, I, van de Warrenburg, B.P.; Schenck, A. Integrative network and brain expression analysis reveals mechanistic modules in ataxia. J. Med. Genet. 2019, 56, 283–292. [CrossRef] [PubMed]
- Garofalo, M.; Vansenne, F.; Verbeek, D.S.; Sival, D.A. The Pathogenetic Basis for a Disease Continuum in Early- and Late-onset Ataxia-Dystonia supports a Unified Genetic Diagnostic Approach. Eur. J. Paediatr. Neurol. 2023, 43, 44–51. [Google Scholar] [CrossRef]
- Zwicker, J.G.; Missiuna, C.; Harris, S.R.; Boyd, L.A. Developmental coordination disorder: a review and update. Eur. J. Paediatr. Neurol. 2012, 16, 573–581. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020, 369, 1318–133. [Google Scholar] [CrossRef] [PubMed]
- de Klein, N.; Tsai, E.A.; Vochteloo, M.; Baird, D.; Huang, Y.; Chen, C.Y.; van Dam, S.; Oelen, R.; Deelen, P.; Bakker, O.B.; et al. Brain expression quantitative trait locus and network analysis reveals downstream effects and putative drivers for brain-related diseases. bioRxiv. 2023, 55, 377–388. [Google Scholar]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulous, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Kutmon, M.; Bohler, A.; Waagmeester, A.; Evelo, C.T.; Willighagen, E.L. Understanding signaling and metabolic paths using semantified and harmonized information about biological interactions. PLoS One. 2022, 17, e0263057. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N. S.; Wang, J. T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Lange, L.M.; Gonzalez-Latapi, P.; Rajalingam, R.; Tijssen, M.A.J.; Ebrahimi-Fakhari, D.; Gabbert, C.; Ganos, C.; Ghosh, R.; Kumar, K.R.; Lang, A.E.; et al. Nomenclature of Genetic Movement Disorders: Recommendations of the International Parkinson and Movement Disorder Society Task Force–An Update. Mov. Disord. 2022, 37, 905–935. [Google Scholar] [CrossRef]
- Genoomdiagnostiek UMCG. LAB-F0701 Genenlijst panels. Available online: https://www.umcg.nl/-/afdeling/genetica/aanvragen-genoomdiagnostiek (accessed on 13/03/2023).
- Busiah, K.; Drunat, S.; Vaivre-Douret, L.; Bonnefond, A.; Simon, A.; Flechtner, I.; Gérard, B.; Pouvreau, N.; Elie, C.; Nimri, R.; et al. Neuropsychological dysfunction and developmental defects associated with genetic changes in infants with neonatal diabetes mellitus: a prospective cohort study. Lancet Diabetes Endocrinol. 2013, 1, 199–207. [Google Scholar] [CrossRef]
- Bernier, R.; Steinman, K.J.; Reilly, B.; Stevens Wallace, A.; Sherr, E.H.; Pojman, N.; Mefford, H.C.; Gerdts, J.; Earl, R.; Hanson, R.; et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med. 2016, 18, 341–349. [Google Scholar] [CrossRef]
- Bernier, R.; Mudac, C.M.; Chen, Q.; Zeng, C.; Stevens Wallace, A.; Gerdts, A.; Earl, R. , Peterson, J.; Wolken, A.; Peters, A.; et al. Developmental trajectories for young children with 16p11.2 copy number variation. Am. J. Med. Genet. Part B. 2017, 174, 367–380. [Google Scholar] [CrossRef]
- De Cinque, M.; Palumbo, O.; Mazzucco, E.; Simone, A.; Palumbo, P.; Ciavatta, R.; Maria, G.; Ferese, R.; Gambardella, S.; Angiolillo, A.; et al. Developmental Coordination Disorder in a Patient with Mental Disability and a Mild Phenotype Carrying Terminal 6q26-qter Deletion. Front. Genet. 2017, 8, 206. [Google Scholar] [CrossRef]
- Coton, J.; Labalme, A.; Till, M.; Bussy, G.; Krifi Papoz, S.; Lesca, G.; Heron, D.; Sanlaville, D.; Edery, P.; des Portes, V.; et al. Characterization of two familial cases presenting with a syndromic specific learning disorder and carrying (17q;21q) unbalanced translocations. Clin. case reports. 2018, 6, 827. [Google Scholar] [CrossRef]
- Morris, C.A.; Mervis, C.B.; Paciorkowski, A.P.; Abdul-Rahman, O.; Dugan, S.L.; Rope, A.F.; Bader, P.; Hendon, L.G.; Velleman, S.L.; Klein-Tasman, B.P.; et al. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am. J. Med. Genet. Part A. 2015, 167, 2916–2935. [Google Scholar] [CrossRef]
- Hanson, E.; Bernier, R.; Porche, K.; Jackson, F.I.; Goin-Kochel, R.P.; Green Snyder, L.; Snow, A.V.; Stevens Wallace, A.; Campe, K.L.; Zhang, Y.; et al. The Cognitive and Behavioral Phenotype of the 16p11.2 Deletion in a Clinically Ascertained Population. Biol. Psychiatry. 2015, 77, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Sival, D.A.; Garofalo, M.; Brandsma, R.; Bokkers, T.A.; van den Berg, M.; de Koning, T.J.; Tijssen, M.A.J.; Verbeek, D.S. Early onset ataxia with comorbid dystonia: Clinical, anatomical and biological pathway analysis expose shared pathophysiology. Diagnostics. 2020, 10, 997. [Google Scholar] [CrossRef] [PubMed]
- van Noort, S.A.M.; van der Veen, S.; de Koning, T.J.; de Koning-Tijssen, M.A.J.; Verbeek, D.S.; Sival, D.A. Early onset ataxia with comorbid myoclonus and epilepsy: A disease spectrum with shared molecular pathways and cortico-thalamo-cerebellar network involvement. Eur. J. Paediatr. Neurol. 2023, 45, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Mariën, P.; Wackenier, P.; De Surgeloose, D.; De Deyn, P.P.; Verhoeven, J. Developmental Coordination Disorder: Disruption of the Cerebello-Cerebral Network evidenced by SPECT. Cerebellum. 2010, 9, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.; Fuelscher, I.; Williams, J. Neurophysiological Approaches to Understanding Motor Control in DCD: Current Trends and Future Directions. Curr. Dev. Disord. Reports. 2019, 6, 78–86. [Google Scholar] [CrossRef]
- Grohs, M.N.; Lebel, C.; Carlson, H.L.; Craig, B.T.; Dewey, D. Subcortical brain structure in children with developmental coordination disorder: A T1-weighted volumetric study. Brain Imaging Behav. 2021, 15, 2756–2765. [Google Scholar] [CrossRef]
- Biotteau, M.; Chaix, Y.; Blais, M.; Tallet, J.; Péran, P.; Alabaret, J.M. Neural Signature of DCD: A Critical Review of MRI Neuroimaging Studies. Front. Neurol. 2016, 7, 227. [Google Scholar] [CrossRef] [PubMed]
- Dewey, D.; Thompson, D.K.; Kelly, C.E.; Spittle, A.J.; Cheong, J.L.; Doyle, L.W.; Anderson, P. Very preterm children at risk for developmental coordination disorder have brain alterations in motor areas. Acta Paediatr. 2019, 108, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Sival, D.A.; van Noort, S.A.M.; Tijssen, M.A.J.; de Koning, T.J.; Verbeek, D.S. Developmental neurobiology of cerebellar and Basal Ganglia connections. Eur. J. Paediatr. Neurol. 2022, 36, 123–129. [Google Scholar] [CrossRef]
- Neychev, V.K.; Gross, R.E.; Lehéricy, S.; Hess, E.J.; Jinnah, H.A. The functional neuroanatomy of dystonia. Neurobiol. Dis. 2011, 42, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Nibbeling, E.A.R.; Delnooz, C.C.S.; de Koning, T.J.; Sinke, R.J.; Jinnah, H.A.; Tijssen, M.A.J.; Verbeek, D.S. Using the shared genetics of dystonia and ataxia to unravel their pathogenesis. Neurosci. Biobehav. Rev. 2017, 75, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Marapin, R.S.; van der Horn, H.J.; van der Stouwe, A.M.M.; Dalenberg, J.R.; de Jong, B.M.; Tijssen, M.A.J. Altered brain connectivity in hyperkinetic movement disorders: A review of resting-state fMRI. Neuroimage Clin. 2023, 37, 103302. [Google Scholar] [CrossRef]
- Oguro-Ando, A.; Zuko, A.; Kleijer, K.T.E.; Burbach, J.P.H. A current view on contactin-4, -5, and -6: Implications in neurodevelopmental disorders. Mol. Cell. Neurosci. 2017, 81, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yuan, M.; Lau, B.W.M.; Li, Y. SHANK family on stem cell fate and development. Cell Death Dis. 2022, 13, 880. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Penzes, P. Ankyrins: roles in synaptic biology and pathology. Mol. Cell. Neurosci. 2018, 91, 131–139. [Google Scholar] [CrossRef]
- Mercati, O.; Huguet, G.; Danckaert, A.; André-Leroux, G.; Maruani, A.; Bellinzoni, M.; Rolland, T.; Gouder, L.; Mathieu, A.; Buratti, J.; et al. CNTN6 mutations are risk factors for abnormal auditory sensory perception in autism spectrum disorders. Mol. Psychiatry. 2017, 22, 625–633. [Google Scholar] [CrossRef]
- Zuko, A.; Kleijer, K.T.E.; Oguro-Ando, A.; Kas, M.J.H.; van Daalen, E.; van der Zwaag, B.; Burbach, J.P.H. Contactins in the neurobiology of autism. Eur. J. Pharmacol. 2013, 719, 63–74. [Google Scholar] [CrossRef]
- Hortsch, M.; Nagaraj, K.; Godenschwege, T. The interaction between L1-type proteins and ankyrins-a master switch for L1-type CAM function. Cell. Mol. Biol. Lett. 2009, 14, 57–69. [Google Scholar] [CrossRef]
- Stevens, S.R.; Rasband, M.N. Anyrinks and neurological disease. Curr. Opin. Neurobiol. 2021, 69, 51–57. [Google Scholar] [CrossRef]
- Woike, D.; Wang, E.; Tibbe, D.; Hassani Nia, F.; Failla, A.V.; Kibæk, M.; Overgård, T.M.; Larsen, M.J.; Fagerberg, C.R.; Barsukov, I.; Kreienkamp, H.J. Mutations affecting the N-terminal domains of SHANK3 point to different pathomechanisms in neurodevelopmental disorders. Sci. Rep. 2022, 12, 902. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K; McDermid, H.E. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2012, 2, 186–201. [Google Scholar]
- Matas, E.; Maisterrena, A.; Thabault, M.; Balado, E.; Francheteau, M.; Balbous, A.; Galvan, L.; Jaber, M. Major motor and gait deficits with sexual dimorphism in a Shank3 mutant mouse model. Mol. Autism. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef]
- Steinman, K. J.; Spence, S.J.; Ramocki, M.B.; Proud, M.B.; Kessler, S.K.; Marco, E.J.; Green Snyder, L.; D'Angelo, D.; Chen, Q.; Chung, W.K.; et al. 16p11. 2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am. J. Med. Genet. Part A. 2016, 170, 2943–2955. [Google Scholar] [CrossRef]
- Bertrand, R.E.; Wang, J.; Xiong, K.H.; Thangavel, C.; Qian, X.; Ba-Abbad, R.; Liang, Q.; Simões, R.T.; Sampaio, S.A.M.; Carss, K.J.; et al. Ceramide synthase TLCD3B is a novel gene associated with human recessive retinal dystrophy. Genet. Med. 2021, 23, 488–497. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Gene inclusion criteria of the present study. Flowchart depicting the process of gene inclusion based on the inclusion criteria that we defined according to the official guidelines (DSM-5; Blank et al., Dev. Med. Child Neurol., 2019). First, we examined whether there was a clinical diagnosis of DCD according to the DSM-4 or DSM-5 criteria. We only included genes reported in patients with a clinical diagnosis of DCD. Genes reported in patients who were suspected of DCD or had a similar phenotype but no clinical diagnosis, were not included. Second, we screened for genetic variants that were reported in studies using human genetic material. Candidate genes from mice studies were therefore not included. Genetic variants were reported as either single nucleotide polymorphism (SNP) or copy-number variations (CNV) in a genetic locus. Third, we investigated the diseases associations of these genes. According to the official diagnostic criteria, we only included genes that were not primarily associated to any of the following conditions: movement disorders, moderate to severe ID (defined in the ICD-10 as an IQ <50), neuromuscular, visual or vestibular disorders. If all of the abovementioned criteria were met, then we included the gene for further in silico analysis. For genes residing within a CNV-locus, we further checked whether the gene was previously associated with DCD-comorbid neurodevelopmental disorders (NDDs), such as attention deficit/hyperactivity disorder or autism-spectrum disorder. If that was the case, then the gene was included for further in silico analysis. DCD= Developmental Coordination Disorder; SNP= single nucleotide polymorphism; CNV= copy-number variation; NDD= neurodevelopmental disorder.
Figure 1.
Gene inclusion criteria of the present study. Flowchart depicting the process of gene inclusion based on the inclusion criteria that we defined according to the official guidelines (DSM-5; Blank et al., Dev. Med. Child Neurol., 2019). First, we examined whether there was a clinical diagnosis of DCD according to the DSM-4 or DSM-5 criteria. We only included genes reported in patients with a clinical diagnosis of DCD. Genes reported in patients who were suspected of DCD or had a similar phenotype but no clinical diagnosis, were not included. Second, we screened for genetic variants that were reported in studies using human genetic material. Candidate genes from mice studies were therefore not included. Genetic variants were reported as either single nucleotide polymorphism (SNP) or copy-number variations (CNV) in a genetic locus. Third, we investigated the diseases associations of these genes. According to the official diagnostic criteria, we only included genes that were not primarily associated to any of the following conditions: movement disorders, moderate to severe ID (defined in the ICD-10 as an IQ <50), neuromuscular, visual or vestibular disorders. If all of the abovementioned criteria were met, then we included the gene for further in silico analysis. For genes residing within a CNV-locus, we further checked whether the gene was previously associated with DCD-comorbid neurodevelopmental disorders (NDDs), such as attention deficit/hyperactivity disorder or autism-spectrum disorder. If that was the case, then the gene was included for further in silico analysis. DCD= Developmental Coordination Disorder; SNP= single nucleotide polymorphism; CNV= copy-number variation; NDD= neurodevelopmental disorder.
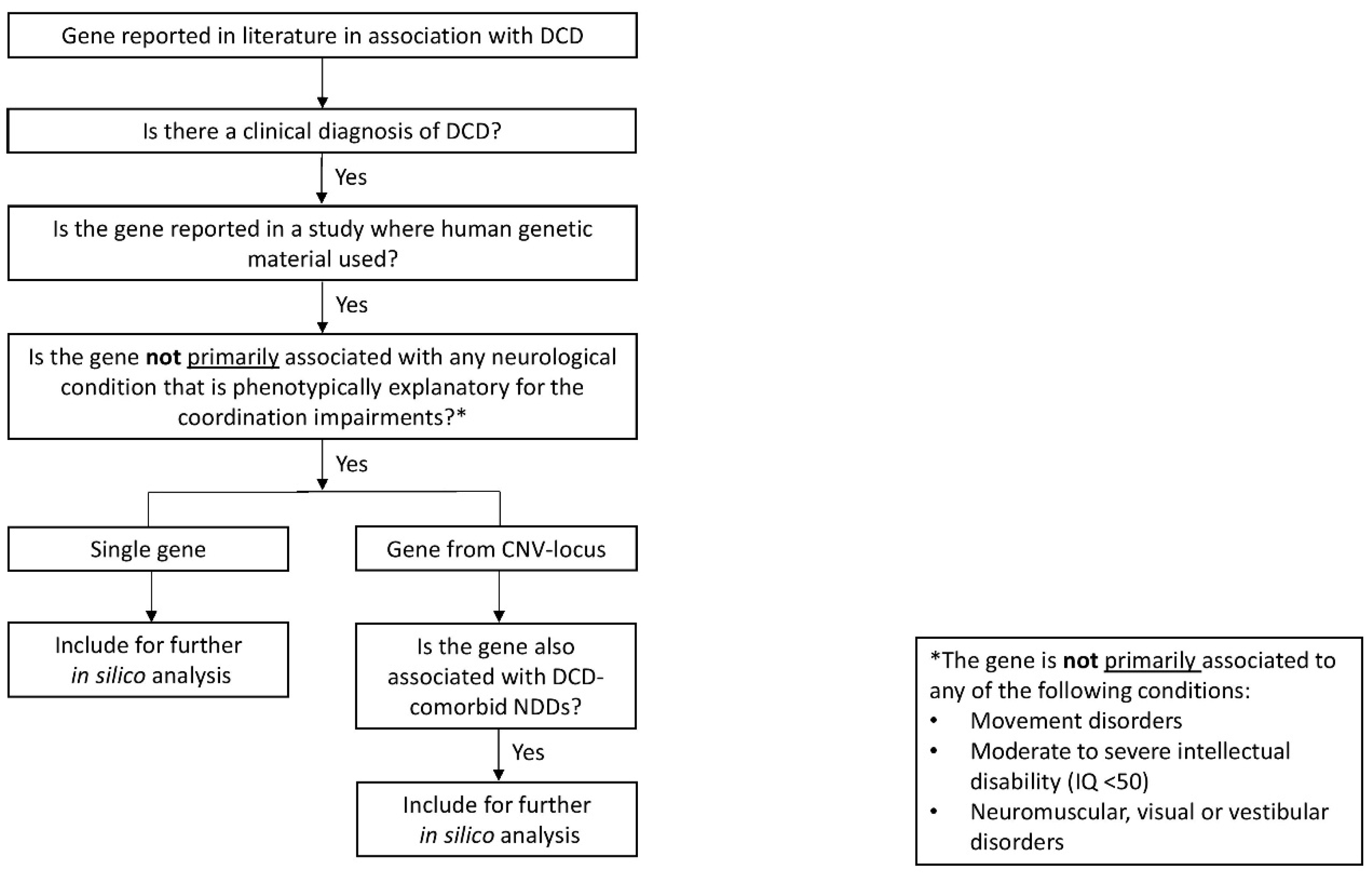
Figure 2.
Comparative temporal expression of the 12 DCD-associated genes across different developmental stages in the cerebellum, basal ganglia, and frontal cortex. Log10-transformed mean gene expression is depicted for each developmental stage (S1-S7) in the selected structures. Gene expression was available as reads per kilobase per million (RPKM). Comparison of average gene expression among the developmental stages between the cerebellum, basal ganglia, and frontal cortex showed no statistically significant difference after correcting for multiple comparisons. Mean, range, and standard deviation of each developmental stage in each of the three structures are reported in Supplementary Table S2. RPKM= reads per kilobase per million; S1= 8-13 postconceptional weeks; S2= 16-21 postconceptional weeks; S3= 24-37 postconceptional weeks; S4: 0-1 years; S5= 2-4 years; S6= 8-13 years; S7= 15-19 years.
Figure 2.
Comparative temporal expression of the 12 DCD-associated genes across different developmental stages in the cerebellum, basal ganglia, and frontal cortex. Log10-transformed mean gene expression is depicted for each developmental stage (S1-S7) in the selected structures. Gene expression was available as reads per kilobase per million (RPKM). Comparison of average gene expression among the developmental stages between the cerebellum, basal ganglia, and frontal cortex showed no statistically significant difference after correcting for multiple comparisons. Mean, range, and standard deviation of each developmental stage in each of the three structures are reported in Supplementary Table S2. RPKM= reads per kilobase per million; S1= 8-13 postconceptional weeks; S2= 16-21 postconceptional weeks; S3= 24-37 postconceptional weeks; S4: 0-1 years; S5= 2-4 years; S6= 8-13 years; S7= 15-19 years.
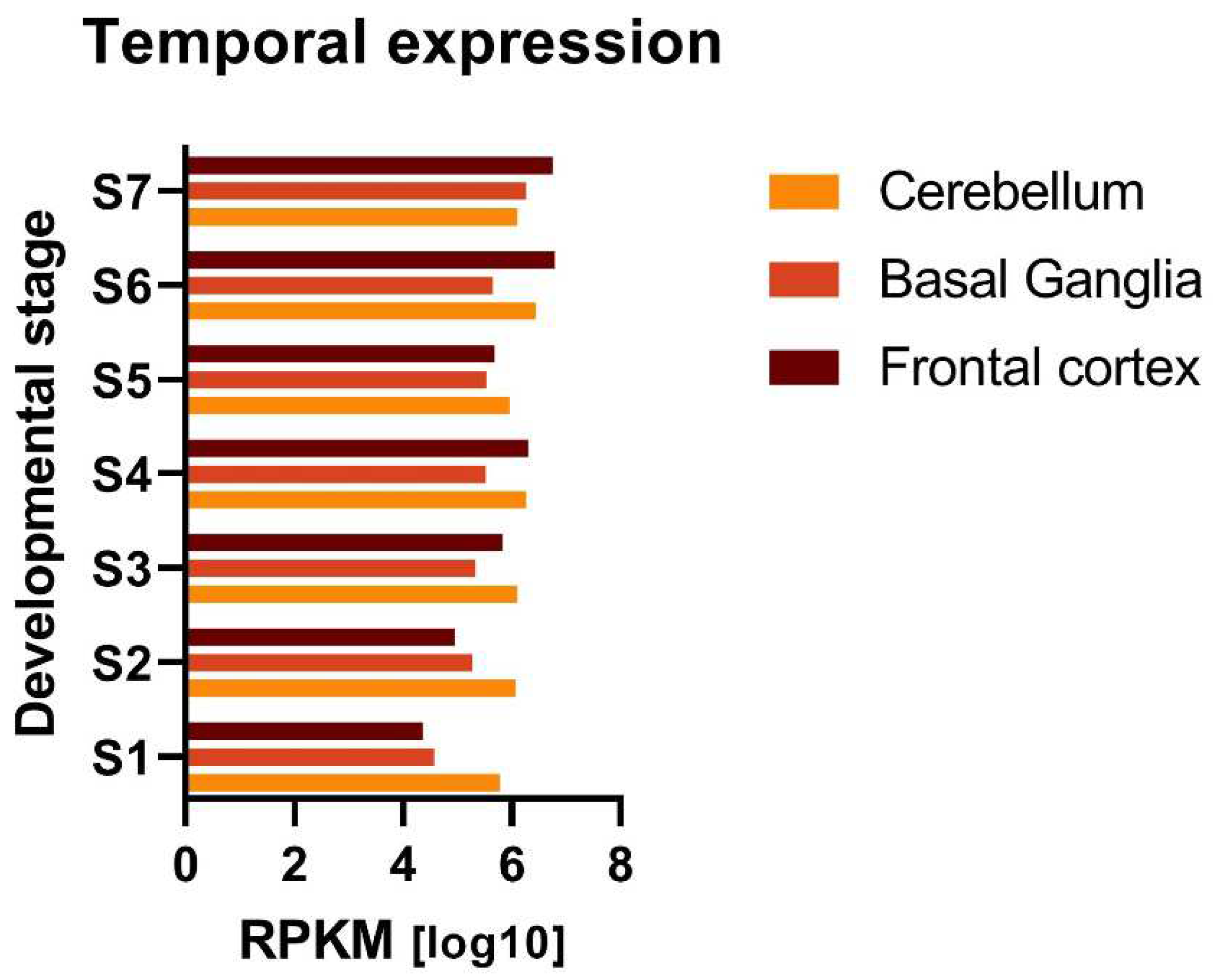
Figure 3.
Comparative tissue expression of the 12 DCD-associated genes in central nervous system structures. For each brain structure, the normalized mean gene expression values (z-score) are given as transcript per million (TPM). Comparison of average gene expression among the investigated brain structures showed no statistically significant difference. Mean, range, and standard deviation of the expression data for each brain structure are reported in Supplementary Table S3. TPM= Transcript per Million.
Figure 3.
Comparative tissue expression of the 12 DCD-associated genes in central nervous system structures. For each brain structure, the normalized mean gene expression values (z-score) are given as transcript per million (TPM). Comparison of average gene expression among the investigated brain structures showed no statistically significant difference. Mean, range, and standard deviation of the expression data for each brain structure are reported in Supplementary Table S3. TPM= Transcript per Million.
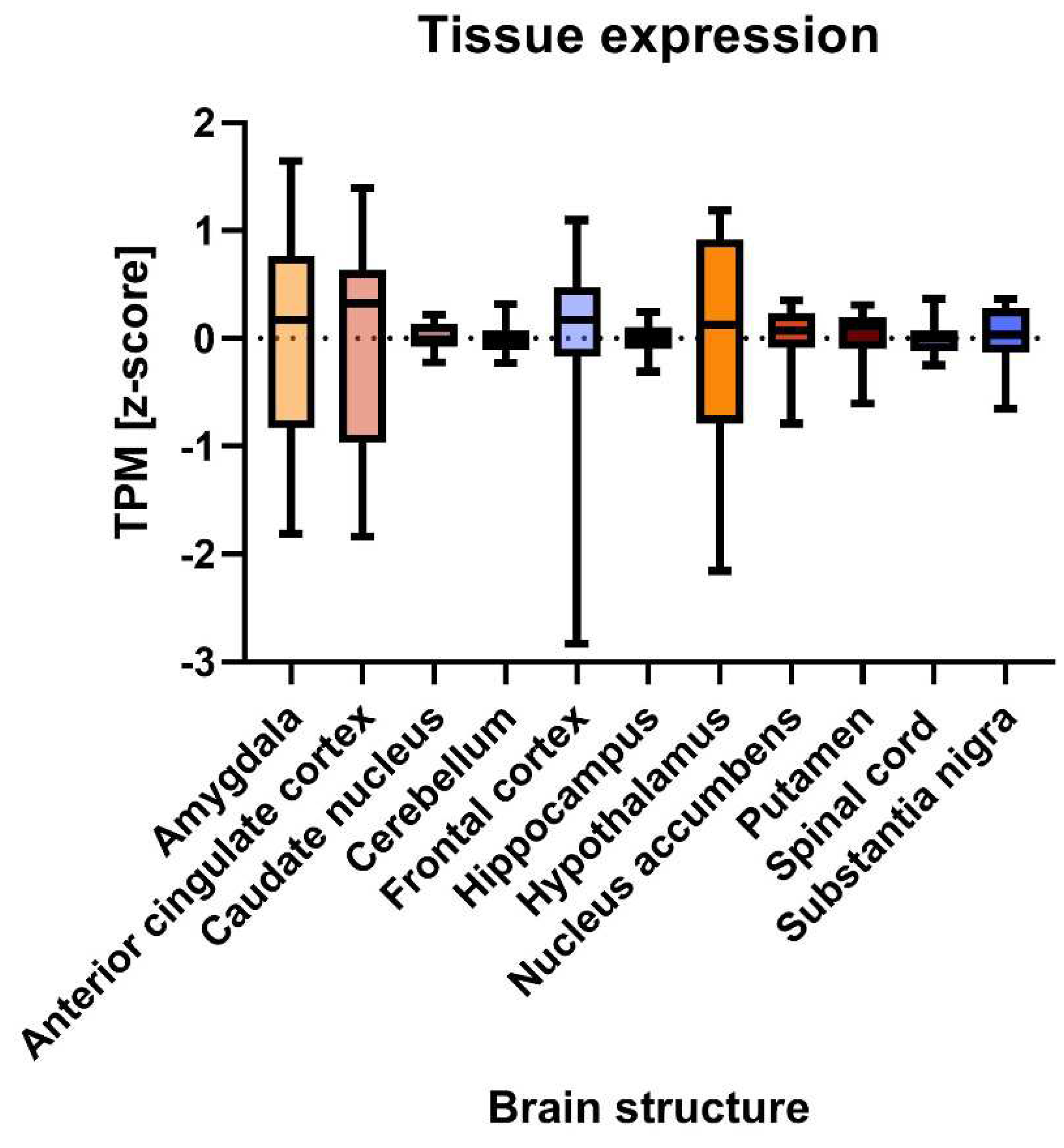
Figure 4.
Top significant biological pathways (depicted as clusters) enriched in the shared DCD-associated/MD gene co-expression network. Network plot of the top significant enriched biological pathways, depicted as clusters, in the shared DCD-associated/MD gene co-expression network. Similar biological pathways are grouped in clusters. Clusters are here represented as color-coded pie charts based on their enrichment for each gene group, where red= enriched for the ataxia genes, and blue= enriched for the chorea genes, green= enriched for the DCD-associated genes, purple= enriched for the dystonia genes, orange= enriched for the myoclonus genes. Clusters with similar biological functions are displayed in closer proximity to each other. The name of the cluster with the most significant p-value within each group is shown above.
Figure 4.
Top significant biological pathways (depicted as clusters) enriched in the shared DCD-associated/MD gene co-expression network. Network plot of the top significant enriched biological pathways, depicted as clusters, in the shared DCD-associated/MD gene co-expression network. Similar biological pathways are grouped in clusters. Clusters are here represented as color-coded pie charts based on their enrichment for each gene group, where red= enriched for the ataxia genes, and blue= enriched for the chorea genes, green= enriched for the DCD-associated genes, purple= enriched for the dystonia genes, orange= enriched for the myoclonus genes. Clusters with similar biological functions are displayed in closer proximity to each other. The name of the cluster with the most significant p-value within each group is shown above.

Table 1.
Final list of 12 genes reported in literature in patients with DCD.
| Gene | Genetic variant information | Clinical information |
|---|---|---|
| ABCC8 | See Supplementary Tables S3 and S7 of Busiah et al. (2013) [49] | 11 patients with nDM and DCD |
| CNTN4 | CNV (deletion) in locus 3p26.3, unknown inheritance [22] | 1 patient with isolated DCD |
| CTNNA3 | CNV (deletion) in locus 10q21.3, paternally inherited [22] | 1 patient with isolated DCD *1 1 patient with DCD and ADHD |
| FHIT | CNVs (deletion) in locus 3p14.2, unknown inheritance [22] | 2 patients with isolated DCD |
| GAP43 | CNV (deletion) in locus 3q13.31, de novo [22] | 1 patient with DCD and ADHD *2 |
| KCNJ11 | See Supplementary Tables S2 and S8 of Busiah et al. (2013) [49] | 11 patients with nDM and DCD |
| KLF7 | Not available [11] | 1 patient with DCD |
| LSAMP | CNV (deletion) in locus 3q13.31, de novo [22] | 1 patient with DCD and ADHD *2 |
| PTPRN2 | CNV (duplication) in locus 7q36.3, maternally inherited [22] | 1 patient with DCD, ADHD, and RD |
| RBFOX1 | CNV (deletion) in locus 16p13.3, maternally inherited [22] | 1 patient with isolated DCD *1 |
| SHANK3 | CNV (duplication) in locus 22q13.33, maternally inherited [22] | 1 patient with isolated DCD |
| VIPR2 | CNV (deletion) in locus 7q36.3, maternally inherited [22] | 1 patient with DCD and ADHD |
Footnote. 12 genes reported in literature in patients with a clinical diagnosis of DCD. These 12 genes matched all the inclusion criteria (Figure 1) and were therefore included in our in silico analyses. Further genetic information regarding the CNVs (chromosome, affected region of the locus, size of the CNV and genes found in that region) can be found in Supplementary File S5. The numbers in square brackets correspond to the references cited in the manuscript. nDM: neonatal diabetes mellitus; DCD: developmental coordination disorder; ADHD: attention deficit/ hyperactivity disorder; RD: reading disorder. *n Same patient as indicated by the same number.
Table 2.
Top 20 significant biological clusters enriched in the DCD-associated gene co-expression network.
Table 2.
Top 20 significant biological clusters enriched in the DCD-associated gene co-expression network.
| Biological cluster | Parental biological term | p-value (Log10) |
|---|---|---|
| Modulation of chemical synaptic transmission | Synaptic signaling | -26.82 |
| Synaptic signaling | Synaptic signaling | -21.91 |
| Cell junction organization | Cellular process – Cellular component organization | -15.87 |
| Neuronal system | Synaptic signaling – Chemical synaptic transmission | -14.69 |
| Behavior | Multicellular organismal process | -13.99 |
| Regulation of cell projection organization | Cellular process – Cellular component organization | -13.61 |
| Neuron projection development | Nervous system development | -12.77 |
| Regulation of ion transport | Cellular process – Transport | -9.48 |
| Protein localization to synapse | Cell process – Cellular localization | -8.94 |
| L1CAM interactions | Nervous system development – Axon guidance | -8.89 |
| Metal ion transport | Cellular process – Transport | -7.57 |
| Actin filament-based process | Cellular process | -7.00 |
| Cell-cell adhesion | Cellular process – Cell adhesion | -6.35 |
| Action potential | Biological regulation –Regulation of biological quality | -6.30 |
| Neuromuscular process | Nervous system process | -5.94 |
| Brain development | Nervous system development | -5.42 |
| Protein localization to membrane | Cellular process- Cellular localization | -5.32 |
| Calcium-ion regulated exocytosis | Cellular process – Export from cell | -5.32 |
| Regulation of glutamatergic synaptic transmission | Synaptic signaling | -4.91 |
| Locomotory behavior | Multicellular organismal process -Behavior | -4.78 |
Footnote. Top 20 most significantly enriched clusters of biological pathways for the 12 DCD-associated genes. In Metascape, biological pathways with similar biological functions are clustered together. The biological pathway with the highest p-value becomes the cluster representative, after which the cluster is named. The p-value for each cluster is expressed in log10, corrected for multiple testing. Parental biological terms are given for each cluster, indicating the broader terms of which the specific biological pathway (i.e., cluster representative) is part of, i.e., “cell junction organization” is a form of “cellular component organization”, which in turn is a “cellular process”. Based on the parental biological terms, we identified three main biological themes: 1) cellular processes, 2) neural signaling, and 3) nervous system development. The complete list of enriched clusters and biological pathways is shown in Supplementary File S6.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



